 Eun Soo Noh1†
Eun Soo Noh1† Byeong-chul Kang2†
Byeong-chul Kang2† Juyeon Kim3
Juyeon Kim3 Ji-Hyeon Jeon3,4
Ji-Hyeon Jeon3,4 Young-Ok Kim1Soon-Gyu Byun5Woo-Jin Kim5
Young-Ok Kim1Soon-Gyu Byun5Woo-Jin Kim5 Bo-Hye Nam1*
Bo-Hye Nam1*- 1Biotechnology Research Division, National Institute of Fisheries Science, Busan, South Korea
- 2D.iF Inc., Yongin-si 16954, Gyeonggi-do, South Korea
- 3Research and Development Center, Insilicogen Inc., Yongin-si 16954, Gyeonggi-do, South Korea
- 4Department of Biological Sciences, Sungkyunkwan University, Suwon, South Korea
- 5Aquaculture Industry Research Division, East Sea Fisheries Research Institute, National Institute of Fisheries Science, Gangneung, South Korea
Introduction
Major populations have adopted seafood-based diets worldwide, and overconsumption can lead to species extinction. Global warming and coastal sea-surface contamination lead to broken food chains by altering the sea environment. South Korea has a prevalent seafood culture, and is one of the biggest seafood importers and exporters in the world. The country constantly invests in aquaculture infrastructure to meet food requirements, increase production, and reduce marine hunting to preserve the marine ecosystem. The country also focuses on species that are not adapted to artificial aquaculture systems. In this study, we sequenced the genome of Gadus chalcogrammus (walleye pollock), a cold-water species with a deep-sea habitat (200–1,200 m depth) that requires temperatures of 1–10°C to survive (Bang et al., 2018). It is the second most commonly consumed fish in Korea, and is used worldwide in foods, such as surimi and roe (Anvari et al., 2018). Walleye pollock dominated the seafood market until the 1990s, but in the 2000s its market collapsed because of overfishing and the rise in sea-surface temperatures, which affected the cod ecosystem (Hwang et al., 2019; Kangsu et al., 2020). A decline in production led to fake labeling of other fish as walleye pollock. To control this malpractice, various molecular authentication systems, such as polymerase chain reaction (PCR) and other marker kits were introduced (Noh et al., 2019). Possibilities of artificial insemination to circumvent the unfavorable natural conditions were also explored to increase production in natural and aquaculture systems (Joo-Young and O-Nam, 2017). Various initiatives have attempted to breed this fish into aquaculture environments, but the reference genome to conduct genomic selection from the phenotype is missing. Only the mitochondrial genomes (Carr and Dawn Marshall, 2008; Sim et al., 2018) and partially assembled contigs are available for this fish, along with a few transcriptomes deposited in the National Center for Biotechnology Information (NCBI) database. Additionally, in the genus Gadus, only the genome for Gadus morhua is publicly available.
Significance of the Data
This Gadus chalcogrammus genome is another reference for molecular studies in the Gadus genus. It will be a valuable resource to conduct comparative analyses within the Gadus genus, and enhance the genomic selection process in molecular-assisted breeding.
Materials and Methods
Sample Collection and Genomic DNA Extraction
A single female fish (93 g) was obtained from the East Sea Fisheries Research Institute in March 2018, and maintained at 8 ± 0.5°C in aerated seawater. The abdominal muscle tissues were sampled aseptically and stored in liquid nitrogen for genomic DNA extraction. The complete experimental procedure from DNA isolation to sequencing was conducted by DNA Link, South Korea (www.dnalink.com), in accordance with the product protocol.
Genomic DNA Library Preparation and Sequencing
The concentrated genomic DNA (gDNA) (24 μg) from the given samples was prepared using the DNeasy Animal Mini Kit (Qiagen, Hilden, Germany). The completely isolated gDNA was quantified using an ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) and a Qubit fluorometer. The gDNA samples were then subjected to the following steps: fragmentation using the g-TUBE (Covaris, Woburn, MA, USA) to obtain >20-kb fragments; small-fragment filtration using 0.45X AMPure (Beckman Coulter, Brea, CA, USA); fragment end repair by ExoVII treatment; ligation of blunt adapters with double-stranded DNA fragments; attachment of primer and polymerase to SMRTbell templates (Template Prep Kit 1.0); and addition of magnetic beads. The impurities were washed out carefully using 1.0X AMPure, and only the double-stranded DNA fragments with blunt adapters were used for sequencing with P6-C4-chemistry (DNA sequencing Reagent 4.0) on the Pacific Biosciences (PacBio) sequencing platform, by capturing 1 × 240-minute-long videos of each SMRT cell. Similarly, the isolated gDNAs were also subjected to sequencing library preparation using stranded Illumina paired-end (PE) protocols (Illumina, San Diego, CA, USA). The fragmented libraries were subjected to size selection and sequencing on the Illumina HiSeq 2000 platform (Illumina).
Pre-processing and Genome Size Estimation
The Illumina DNA sequences were subjected to preprocessing steps; namely, adapter trimming, quality trimming (Q20), and contamination removal. The adapter and quality trims were conducted using Trimmomatic-0.32 functions (Bolger et al., 2014), and the microbial contamination was removed using CLCMapper v4.2.0 (www.qiagenbioinformatics.com) with an in-house database. The in-house database was constructed from the bacterial, viral, and marine meta-genomes (ftp://ftp.ncbi.nlm.nih.gov/genomes/GENOME_REPORTS/prokaryotes.txt, ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses/, and https://www.ncbi.nlm.nih.gov/bioproject/PRJNA13694, respectively). All preprocessed sequences from the PE library were subjected to genome size estimation using a k-mer-based method (Shin et al., 2018). The k-mer frequencies (k-mer size = 17) were obtained using the Jellyfish v2.0 method (Marçais and Kingsford, 2011), and the genome size was calculated using the given formulae: Genome coverage depth = (k-mer coverage depth × Average read length)/(Average read length – k-mer size + 1); and Genome size = Total base number/Genome coverage depth.
De novo Genome Assembly
Complete sequence reads were error-corrected using SMRT Analysis v2.3, and imported into a diploid-aware hierarchical genome assembler to construct the contigs from the long-sequence PacBio reads (FALCON) (Chin et al., 2016). The assembled contigs were further subjected to sequence polishing using the Quiver consensus method to reduce the base-calling errors (Chin et al., 2016). Finally, the assembled and polished contigs were assessed for completeness of the genome using BUSCO v5.0 (Simão et al., 2015). The reference BUSCO datasets are actinopterygii_odb10 and vertebrate_ odb10. The quality of the assembly was assessed by short-read mapping to the draft by BWA v0.7.15 (Li and Durbin, 2010) (Supplementary Figure 2).
De novo Repeat Region Prediction and Classification
The repeat regions were predicted using the de novo method, and classified into repeat subclasses. The de novo repeat prediction for G. chalcogrammus was conducted using RepeatModeler (www.repeatmasker.org/RepeatModeler/), which includes methods such as RECON (Bao and Eddy, 2002) (http://eddylab.org/software/recon/), RepeatScout (Price et al., 2005) (https://bix.ucsd.edu/repeatscout/), and TRF (Benson, 1999) (https://tandem.bu.edu/trf/trf.html). The modeled repeats were classified into subclasses by referencing the Repbase v20.08 database (www.girinst.org/repbase/) (Bao et al., 2015) and the repeats were masked using RepeatMasker v4.0.5 (www.repeatmasker.org) with RMBlastn v2.2.27+.
Gene Prediction and Annotation
The genes from the G. chalcogrammus draft genome were predicted using an in-house gene prediction pipeline, which includes three modules: an evidence-based gene modeler, an ab initio gene modeler, and a consensus gene modeler. Finally, the functional annotation processing was performed for the consensus genes. Initially, sequenced transcriptomes obtained from the two methods (Illumina [156.9 Gb] and Iso-Seq [75.6 MB]) were assembled with Trinity(v2.2.0) (Grabherr et al., 2011) and transdecoder v5.5.0 and the proteins sequence mapped to masked G. chalcogrammus draft genome. To train the ab initio and evidence-based gene modelers [including Exonerate v.2.2.0 (Slater and Birney, 2005), AUGUSTUS v.3.1 (Stanke et al., 2006), and GENEID v.1.3 (Blanco et al., 2002)], several genomes (Supplementary Table 4) were used for prediction. Finally, the transcript models and predicted models from the evidence-based and ab initio gene modelers were subjected to the consensus gene modeler to produce the final gene and transcript models. The consensus transcripts were then subjected to functional annotation from biological databases (NCBI-NR, Swiss-Prot, Gene Ontology, and KEGG Pathway) using OmicsBox v1.2 (Götz et al., 2008).
Preliminary Analysis Report
Initially, the genome size of G. chalcogrammus was estimated to be 683.61 Mb (Figure 1A) with 42 Gb of short-read sequences (Table 1A, Supplementary Table 2) and 629.66 Mb of representative contigs from 97 Gb of error-corrected long-read sequences (Supplementary Tables 1, 3). The contigs were then assembled into 116 scaffolds in the reference draft genome (Table 1B). The N50 of the assembled genome was 27,035,343 bases, and 245 Mb (38.89%) of the assembled contigs were covered by repeats, in which the long terminal repeat (LTR) elements dominated (34%). In total, 23,353 genes were predicted from the genome, with an average size of 9261.51 bases, and 90.4% completeness on the BUSCO score (Table 1C). Homologous sequences were found for 19,760 (84.61%) genes in GenBank, and 17,259 (73.90%) genes had Gene Ontology descriptions (Table 1D). The first genome published for the Gadus genus was G. morhua (gadMor1) in 2011, as an 832-Mb genome with an N50 of 2.3 kb (scaffold N50; 0.14 Mb) (Star et al., 2011). An improved version of the same genome (gadMor2) was published in 2017 with 116 kb (scaffold N50; 1.15 Mb) (Tørresen et al., 2017), and the third NCBI version (gadMor3) was 669 Mb with a contig N50 of 1.01 Mb (scaffold N50; 28.7 Mb) and 23 chromosomes. The gadMor3 genome was used as a reference to scaffold the contigs (N50: 3.6 Mb) with the RaGOO method (Alonge et al., 2019), and 167 scaffolds were obtained with an N50 of 27.03 Mb and 23 chromosomes. The complete workflow used in this study is illustrated in Supplementary Figure 1. Overall, this genome assembly improved significantly in fragmented assembly (Figures 1B–F) and BUSCO completeness score (Table 1B). However, there is conflict in chromosome number i.e, G. morhua have 23 chromosome and G. chalcogrammus has 22 chromosomes (Supplementary Table 5). Since, the contigs scaffold well with all G. morhua 23 chromosomes, this will be improved in future version of this genome assembly (Ishii and Yabu, 1985).
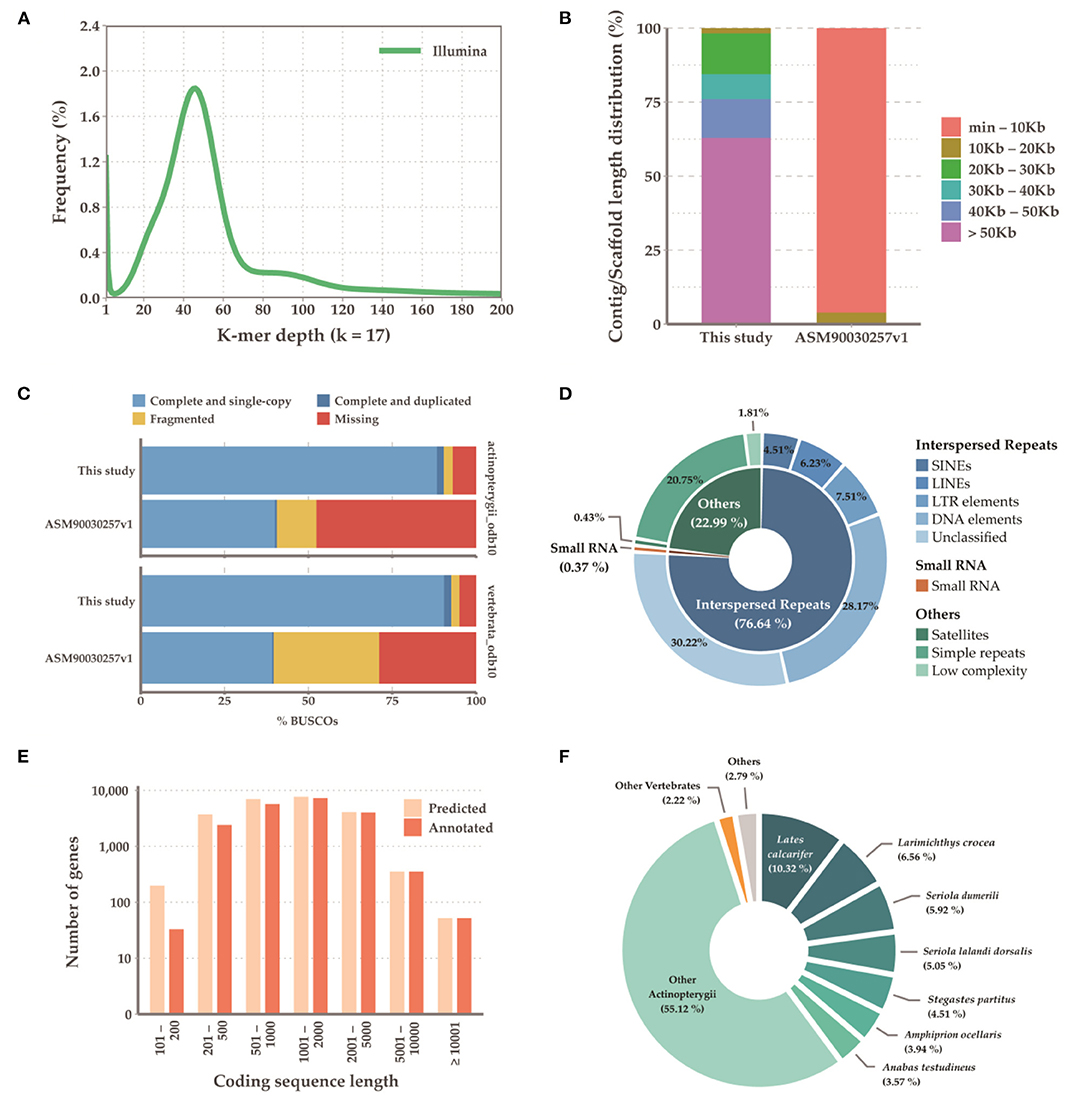
Figure 1. Summary of the sequencing results. (A) Genome size estimation with illumina short reads; (B) Contig length distribution between current assembly and ASM90030257v1; (C) BUSCO result summary with actinopterygii and vertebrate; (D) Repeat summaries; (E) Annotation summary of predicted genes; (F) Species distribution of mapped sequences in annotation process.
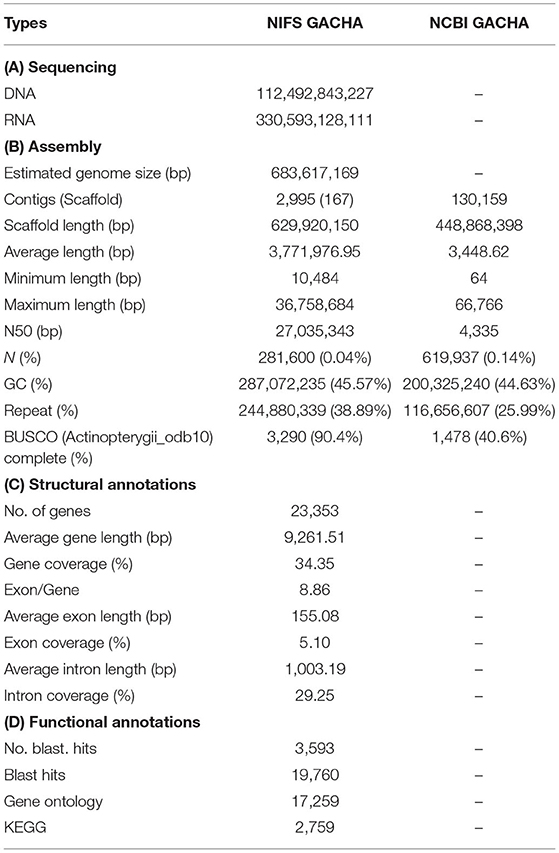
Table 1. Sequencing for annotation of the Gadus chalcogrammus draft genome.
Dataset Information to the User
The complete sequences generated in this study were deposited in the NCBI Sequence Read Archive under accession no. PRJNA736536. The assembled contigs and the annotation files (CDS, gff, repeats, and proteins) are available in the Figshare repository (https://figshare.com/s/2ff9e3a49a07c990a400) with all of the annotation details in a Readme file. The contig assembly of this draft genome was submitted to the NCBI Assembly database under accession no. JAHRIL000000000.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession: PRJNA736536]. The assembled contigs and the annotation files (CDS, gff, repeats, and proteins) are available in the Figshare repository (https://doi.org/10.6084/m9.figshare.14913921) with all of the annotation details in a Readme file. The contig assembly of this draft genome was submitted to the NCBI Assembly database under accession no. JAHRIL000000000.
Author Contributions
EN, B-cK, JK, and J-HJ: genome assembly and annotations. EN and B-HN: manuscript preparation. Y-OK, S-GB, and W-JK: sampling and sequencing. EN, B-cK, and B-HN: funding and modeling the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Collaborative Genome Program of the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (MOF) (No. 20180430) and the National Institute of Fisheries Science (R2022044).
Conflict of Interest
JK and J-HJ was employed by the company Insilicogen Inc. B-cK was employed by the company D.iF.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.744941/full#supplementary-material
References
Alonge, M., Soyk, S., Ramakrishnan, S., Wang, X., Goodwin, S., Sedlazeck, F. J., et al. (2019). RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224. doi: 10.1186/s13059-019-1829-6
Anvari, M., Smith, B., Sannito, C., and Fong, Q. (2018). Characterization of rheological and physicochemical properties of Alaska walleye pollock (Gadus chalcogrammus) roe. J. Food Sci. Technol. 55, 3616–3624. doi: 10.1007/s13197-018-3287-7
Bang, M., Kang, S., Kim, S., and Jang, C. J. (2018). Changes in the biological characteristics of walleye pollock related to demographic changes in the east sea during the late 20th century. Marine Coastal Fisheries 10, 91–99. doi: 10.1002/mcf2.10004
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11. doi: 10.1186/s13100-015-0041-9
Bao, Z., and Eddy, S. R. (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276. doi: 10.1101/gr.88502
Benson, G (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Blanco, E., Parra, G., and Guigó, R. (2002). “Using geneid to identify genes,” in Current Protocols in Bioinformatics (John Wiley & Sons, Inc.). doi: 10.1002/0471250953.bi0403s00
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Carr, S. M., and Dawn Marshall, H. (2008). Phylogeographic analysis of complete mtDNA genomes from Walleye Pollock (Gadus chalcogrammus Pallas, 1811) shows an ancient origin of genetic biodiversity. DNA Sequence 19, 490–496. doi: 10.1080/19401730802570942
Chin, C. S., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Hwang, D.-W., Shim, K., and Lee, C. I. (2019). Concentrations and risk assessment of heavy metals in tissues of walleye pollock (Gadus chalcogrammus) captured from the Northeastern Coast of Korea. J. Food Prot. 82, 903–909. doi: 10.4315/0362-028X.JFP-18-379
Ishii, K., and Yabu, H. (1985). Chromosomes in three species of gadidae (Pieces). Nippon Suisan Gakkaishi 51, 25–28. doi: 10.2331/suisan.51.25
Joo-Young, S., and O-Nam, K. (2017). The RNA/DNA ratio of mature eggs and the vitality of refrigerated sperm according to gonadal maturation and elapsed time after capturing pollock (Theragra chalcogramma) caught in perilla nets on the east coast. J. Korean Fisheries Sic. Sock. 50, 296–301. doi: 10.5657/KFAS.2017.0296
Kangsu, S., Chung-Il, L., and Hae-Kun, J. (2020). Long term changes in sea surface temperature around habitat ground of walleye pollock (Gadus chalcogrammus) in the East Sea. J. Korean Soc. Marine Environ. Safety 26, 195–205. doi: 10.7837/kosomes.2020.26.2.195
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Noh, E. S., Park, Y. J., Kim, E. M., Park, J. Y., Shim, K. B., Choi, T.-J., et al. (2019). Quantitative analysis of Alaska pollock in seafood products by droplet digital PCR. Food Chem. 275, 638–643. doi: 10.1016/j.foodchem.2018.09.093
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Shin, G.-H., Shin, Y., Jung, M., Hong, J.-M., Lee, S., Subramaniyam, S., et al. (2018). First draft genome for red sea bream of family sparidae. Front. Genetics 9:643. doi: 10.3389/fgene.2018.00643
Sim, H. K., Yu, J. N., and Jin, D. H. (2018). The complete mitochondrial genome of Gadus chalcogramma and phylogenetic analysis. Mitochondrial DNA B Resour 3, 454–455. doi: 10.1080/23802359.2018.1462118
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Slater, G. S. C., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31. doi: 10.1186/1471-2105-6-31
Stanke, M., Schöffmann, O., Morgenstern, B., and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62. doi: 10.1186/1471-2105-7-62
Star, B., Nederbragt, A. J., Jentoft, S., Grimholt, U., Malmstrøm, M., Gregers, T. F., et al. (2011). The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210. doi: 10.1038/nature10342
Keywords: walleye pollock, Gadus chalcogrammus, genome, Gadus, aquaculture
Citation: Noh ES, Kang B-c, Kim J, Jeon J-H, Kim Y-O, Byun S-G, Kim W-J and Nam B-H (2022) Draft Assembled Genome of Walleye Pollock (Gadus chalcogrammus). Front. Mar. Sci. 9:744941. doi: 10.3389/fmars.2022.744941
Received: 21 July 2021; Accepted: 17 January 2022;
Published: 16 February 2022.
Edited by:
Hui Zhang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Vita Gancitano, National Research Council (CNR), ItalyOle K. Tørresen, University of Oslo, Norway
Shengyong Xu, Zhejiang Ocean University, China
Copyright © 2022 Noh, Kang, Kim, Jeon, Kim, Byun, Kim and Nam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo-Hye Nam, bmFtYm9oeWUmI3gwMDA0MDtrb3JlYS5rcg==
†These authors have contributed equally to this work and share first authorship