 Xiaolin Guo1Xixi Li1Feng Zhao1Danwen Liu1
Xiaolin Guo1Xixi Li1Feng Zhao1Danwen Liu1 Zujing Yang1Moli Li1Yajuan Li1Huilan Wei1
Zujing Yang1Moli Li1Yajuan Li1Huilan Wei1 Hao Wang1
Hao Wang1 Zhenkui Qin1*Zhifeng Zhang1,2
Zhenkui Qin1*Zhifeng Zhang1,2 Zhenmin Bao1,2
Zhenmin Bao1,2- 1Ministry of Education Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 2Key Laboratory of Tropical Aquatic Germplasm of Hainan Province, Sanya Oceanographic Institution, Ocean University of China, Sanya, China
Mollusca is the second largest animal phylum and represents one of the most evolutionarily successful animal groups. Mulinia lateralis, a small bivalve, is a promising model organism to facilitate studies of mollusc development. However, because of the lack of published genomic and transcriptomic resources, integrated research on the formation of larval shells in this species, which is a representative developmental process of molluscs and of great importance for larva survival, is hindered. In this study, the blastula, gastrula, trochophore larva, and D-shaped larva of M. lateralis were utilized for generating a comprehensive full-length transcriptome through Pacific BioSciences (PacBio) isoform sequencing (Iso-seq) and Illumina RNA-Seq. A total of 238,919 full-length transcripts with an average length of 3,267 bp and 121,424 annotated genes were obtained. Illumina RNA-Seq data analysis showed that 4,512, 10,637, and 17,829 differentially expressed genes (DEGs) were obtained between the two adjacent developmental stages. Functional annotation and enrichment analysis revealed the specific function of genes in shell biomineralization during different developmental stages. Twelve genes that may be involved in the formation of the larval shell of M. lateralis were identified, including insoluble shell matrix protein-encoding gene 1 (ISMP1), ISMP2, ISMP5, chitin synthase, tyrosinase, chitin-binding protein, collagen and pu14 involved in shell matrix deposition, and carbonic anhydrase, solute carrier family 4 member 8 (slc4a8), EF-hand, and a calmodulin coding gene C-2442 participated in ion transportation. In addition, calcium ion binding function, calcium signaling pathway, and endocrine and other factor-regulated calcium reabsorption pathways were significantly enriched. Weighted gene correlation network analysis (WGCNA) identified two modules related to biomineralization and larval shell formation, and slc4a8 and ring finger protein 41 (rnf41) were key hub genes that may be involved in this process. Moreover, it could be implied that the process of ion transport occurs earlier than the deposition of the shell matrix. This work provided a clear view of the transcriptome for M. lateralis and will be valuable in elucidating the mechanisms of larval shell formation as well as other developmental processes in molluscs.
Introduction
Molluscs play a crucial ecological role in the marine environment, including creating marine soft bottom, filtering harmful algae, and providing winter food for seabirds (Tan and Zheng, 2020). A pair of hard shells is the most significant characteristic of bivalve molluscs, which are firstly secreted by the shell gland or mantle and then solidified through the biomineralization process. Exploration of the genes and pathways involving in shell formation could provide us insights into the biomineralization regulatory mechanisms as well as uncover the fitness of bivalves in carbon dioxide acidifying seawater environment.
Biomineralization is a common physiological process in metazoans from sponges to vertebrates and from intracellular to extracellular (Gilbert et al., 2022). Shell formation is a complicated process and a typical biomineralization phenomenon in molluscs, and most related studies focus on bivalves (Yarra et al., 2021). During the development process, bivalves generally form two types of shells, larval shell and adult shell (Gilbert et al., 2022), and the major developmental steps occur rapidly within the first days of life to produce the larval shell (prodissoconch I (PD I)) (Yarra et al., 2021). The first shell formation process usually begins at the end of gastrulation and forms a shell-forming region (Marin et al., 2012). The dorsal population of cells in this region invaginates into the blastocoel to form the shell gland, after which cells in the shell gland gradually move outward, evaginating to form the shell field (Kurita et al., 2009). The shell field then grows over the larval body until the first larval shell finishes (Ramesh et al., 2017). The larval shell of bivalves plays a crucial role in their survival, which eventually differentiates from the adult shell-secreting organ mantle (Li et al., 2020). Although a moderate threefold increase of calcium content occurs in the trochophore larva, the first shell of a bivalve trochophore larva is not calcified (Ramesh et al., 2017), and this type of larval shell is designated as the initial non-calcified shell (InCaS) to distinguish it from the calcified larval shell (Huan et al., 2013). InCaS contains only organic components, which are mainly composed of polysaccharides and proteins, functions in crystal nucleation and growth, and provide the organic template for Ca2+ deposition during biomineralization (Falini et al., 2003; Marin et al., 2008; Arroyo et al., 2020). The larval shell begins to calcify in the D-shaped larva stage, and the calcified PD I shell forms during this period (Mouza et al., 2006). At the same time, an important part of the adult shell hinge forms in the posterior middle of the larval shell (Ansell, 1962).
Although the shell field can be discriminated in the gastrula, the larval shell does not appear until the later stage of the trochophore larva (Mouza et al., 2006; Huan et al., 2013; Ramesh et al., 2017; Ramesh et al., 2019). Generally, studies exploring embryonic shell development focus on two larval stages, the trochophore larva and D-shaped larva (Huan et al., 2013; Gonzalez-Socoloske et al., 2014; Ramesh et al., 2017; Li et al., 2020). Reported studies have identified several genes and many shell matrix proteins (SMPs) that are proven to be related to shell formation, such as decapentaplegic (dpp), tyrosinase (tyr), carbonic anhydrase (CA), and engrailed (Miyamoto et al., 1996; Nederbragt et al., 2002; Huan et al., 2013). In Pacific oysters (Crassostrea gigas), tyr gene plays a role in larval shell biogenesis (Huan et al., 2013). Miglioli et al. (2019) found that tyr is involved in the calcification process during larval development in Mediterranean mussels (Mytilus galloprovincialis). Bicarbonate transporters are reported to be key factors in the calcification of marine animals. The solute carrier 4 (SLC4) is responsible for supplying HCO3- to the site of calcification in the braching coral (Stylophora pistillata) (Zoccola et al., 2015). In blue mussels (Mytilus edulis), SLC family members SLC26 and SLC4 have been proven to be involved in the process of adult shell repair (Yarra, 2018). Some important transcription factors have also been found in C. gigas to function during the formation of larval shells, such as GATA2/3, BMP2/4, and SOXC homologs, which involve the regulation of membrane biogenesis, cell proliferation, and biomineralization (Nederbragt et al., 2002; Liu et al., 2015; Liu et al., 2017).
Although genetic resources have been exploited in recent years in molluscs (Liu et al., 2021), the available information is still limited in the second-largest animal phylum that molluscs represent. The full-length transcriptome is an alternative complete data resource for species without a reference genome (Hoang et al., 2017). With the continuous development of nucleic acid sequencing technology, the Pacific BioSciences (PacBio) single-molecule real-time (SMRT) sequencing, as the third-generation sequencing technology, has been utilized to generate full-length transcripts without assembly. Furthermore, the combined analysis of second- and third-generation sequencing could overcome the shortages of both technologies and is widely used currently (Grabherr et al., 2011; Cao et al., 2020; Luo et al., 2020; Liao et al., 2022). In bivalves, Zeng et al. (2022) published the first report of full-length transcriptome regarding bivalve gonads in freshwater pearl mussels (Hyriopsis schlegelii), and Liao et al. (2022) used a full-length transcriptome to reveal the tissue-specific gene expression profile of mangrove clam (Geloina erosa).
To investigate the first steps of shell formation during early developmental stages, Mulinia lateralis, which is naturally distributed in estuarine benthic and intertidal zones along the Atlantic coast from Canada to the Caribbean Sea (Walker and Tenore, 1984), was selected. M. lateralis is a typical dioecious species with a small size and short generation interval of 2 months, and artificial breeding of M. lateralis in the laboratory is easy to achieve, which provides a promising model species of bivalve studies (Guo and Allen, 1994; Cripe, 2006; Yang et al., 2021). Up to now, neither transcriptomic nor genomic information has been published in M. lateralis. In order to have a more comprehensive understanding of the larval shell genesis, we combined the long-read SMRT sequencing and the short-read RNA-Seq technologies in the present study and established an integrated transcriptome of M. lateralis with embryos/larvae of four early developmental stages (blastula, gastrula, trochophore larva, and D-shaped larva). Our study aims to generate a reference transcriptome dataset with functional annotation, explore gene expression patterns during early developmental stages, and identify possible genes as well as pathways involved in the initial shell formation. To the best of our knowledge, this is the first comprehensive transcriptome of M. lateralis, which provides a valuable genetic resource to facilitate studies on larval shell formation and the early development of molluscs.
Materials and methods
Sample collection and RNA preparation
Sexually mature M. lateralis adults were selected from the culture system of MOE Key Laboratory of Marine Genetic and Breeding, and spawning was induced with drying in the shade followed by 26°C thermal shock method. Fertilized eggs of M. lateralis were incubated in a 15-L tank with filtered seawater at 22°C. After removal of abnormal embryos and larvae, samples of four developmental stages, blastula (3 h post fertilization (hpf)), gastrula (5 hpf), trochophore larva (8 hpf), and D-shaped larva (14 hpf) were collected (Figure S1), immediately frozen in liquid nitrogen, and preserved at −80°C until RNA extraction.
The total RNA of 12 samples, with three parallel of each stage, was extracted individually by phenol–chloroform extraction (Liu et al., 2015). The extracted RNAs were analyzed with 1.2% agarose gel electrophoresis and NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). RNA concentration was measured using a Qubit 2 fluorometer (Invitrogen, Carlsbad, CA, USA), and integrity was assessed using the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). Then, the quantified RNA samples were subsequently used for cDNA library construction.
PacBio Iso-seq library construction, sequencing, and preprocessing
Equal amounts of RNAs from these four developmental stages of M. lateralis were pooled together for the construction of PacBio isoform sequencing (Iso-seq) library. The library was prepared according to the Isoform Sequencing protocol (Iso-Seq) using the Clontech SMARTer PCR cDNA Synthesis Kit (Clontech, Mountain View, CA, USA) and the BluePippin Size Selection System (Sage Science, Beverly, MA, USA) as described by Pacific Biosciences (PN 100-092-800-03). PacBio library was sequenced on the PacBio Sequel platform (Pacific Biosciences, Menlo Park, CA, USA).
Sequence data were processed using the SMRTlink 5.1 software (Hon et al., 2020). The subreads were processed by the Circular Consensus Sequence (CCS) algorithm to obtain the CCS (min_length 50, max_drop_fraction 0.8, no_polish TRUE, min_zscore −9,999.0, min_passes 2, min_predicted_accuracy 0.8, max_length 15,000). CCSs were then classified into full-length and non-full-length reads using pbclassify.py (ignore polyA false and minSeqLength 200). All fasta files produced were then fed into the cluster step for isoform-level clustering (ICE), followed by final Arrow polishing (hq_quiver_min_accuracy 0.99, bin_by_primer false, bin_size_kb 1, qv_trim_5p 100, and qv_trim_3p 30) (Cao et al., 2020). To further improve the sequencing accuracy, additional nucleotide errors in consensus reads were corrected using the Illumina RNA-Seq data of the present study with the software LoRDEC (Salmela and Rivals, 2014). Any redundancy in consensus reads was removed by CD-HIT (-c 0.95, -T 6, -G 0, -aL 0.00, -aS 0.99) (Fu et al., 2012) to obtain the final transcripts, which were then considered as the reference transcriptome (designated as ref) for the subsequent analysis.
Gene functional annotation and structural analysis
The obtained non-redundant transcript sequences were functionally annotated using the following databases: National Center for Biotechnology Information (NCBI) non-redundant protein (NR), EuKaryotic Orthologous Groups (KOG), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), NCBI nucleotide sequences (NT), Protein Family (Pfam), and Gene Ontology (GO). The first four database annotations were performed using diamond v0.8.36 with an e-value threshold of 1e−5 (Buchfink et al., 2015). The ncbi-blast-2.7.1+ with an e-value threshold of 1e−5 was utilized for NT database annotation, and the Pfam databases annotations were performed using Hmmscan of the HMMER 3.1 package (Nguyen et al., 2016). Script of protein annotation results based on Pfam database was used for GO annotations (Ashburner et al., 2000).
Predictive analysis of coding sequence (CDS) was performed using ANGEL v. 2.4 software, and bivalve species confident protein sequences were used for ANGEL training (Shimizu et al., 2006). Transcription factors were predicted using the AnimalTFDB 2.0 database with iTAK software (Tatusov et al., 2003). Coding–Non-coding index (CNCI) (Sun et al., 2013), Coding Potential Calculator (CPC) (Kong et al., 2007), Pfam-scan (Finn et al., 2016), Predictor of Long Non-coding RNAs, and Messenger RNAs based on an Improved k-mer scheme (PLEK) (Li et al., 2014) were integrated to identify long non-coding RNAs (lncRNAs) in the transcripts. Transcripts longer than 200 nt without coding potential were selected as lncRNA candidates.
Simple sequence repeats (SSRs) of the transcriptome sequences were identified using MISA 1.0 with default parameters (Beier et al., 2017). The minimum repeat time for core repeat motifs was set as 10 for mononucleotides, 6 for dinucleotides, and 5 for trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides.
Illumina library preparation and sequencing
Twelve RNA samples of M. lateralis embryos/larvae from four developmental stages were used for Illumina cDNA library construction using NEBNext Ultra™ RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) following the manufacturer’s instructions. Qualified libraries were applied to next-generation sequencing (NGS) using an Illumina Hiseq X Ten platform (Illumina, USA) to generate 150-bp paired-end sequence reads. Raw reads were firstly preprocessed for quality control to obtain the clean reads by removing reads containing adapters, ambiguous N nucleotides, and low-quality reads. Then, all clean reads obtained were mapped onto the PacBio Iso-Seq ref with bowtie2 (-end-to-end, -sensitive) (Langmead and Salzberg, 2012).
Identification and analysis of differentially expressed genes
The mapping results from bowtie2 were calculated by RSEM (Li and Dewey, 2011), and the read count values of each gene from each sample were converted into FPKM (expected number of fragments per kilobase of transcription sequence per mills base pairs sequence) to determine the expression level of each gene. Both heatmap and hierarchical cluster analyses were carried out using the R package “pheatmap”.
DESeq 1.10.1 package (Anders and Huber, 2010) was used for differential expression analysis of genes between two sequential developmental stages. Unigenes with false discovery rate (FDR)< 0.01 and |log2(FoldChange)| > 1 were considered to be the differentially expressed genes (DEGs).
Gene Ontology enrichment analysis of DEGs between different stages was implemented by the R package “GOseq” (Young et al., 2010), in which gene length bias was corrected. GO terms with a Benjamini and Hochberg (BH)-corrected p-value of less than 0.05 were considered significantly enriched. The enrichment analysis of the KEGG pathway was performed using KOBAS (2.0) (Xie et al., 2011). The p-value correction method and the threshold value were consistent with the above.
Weighted gene correlation network analysis
The R package “WGCNA” was utilized to construct the co-expression network (Langfelder and Horvath, 2008), and 25,664 annotated genes with FPKM values greater than 0.1 were selected. Firstly, samples were clustered to assess the presence of any obvious outliers. Then, the R function pickSoftThreshold was used to calculate the soft thresholding power β, and the connectivity between genes met a scale-free network distribution (scale-free R2 = 0.9) when the value of soft thresholding power β was set to 6. Hierarchical clustering and the dynamic tree cut function were used to detect modules, and gene significance (GS) and module membership (MM) were calculated to relate modules with clinical traits. The minimum module size was set to 30 (CutHeight = 0.99, minModuleSize = 30), and the modules with a higher correlation were merged (r< 0.25). The corresponding gene information of each module was extracted for further analysis. Lastly, Cytoscape 3.7.0 software was used to visualize the network of eigengenes.
Quantitative RT-PCR validation
Six unigenes related to larval shell formation (tyr, chitin synthase, CA, EF-hand, a calmodulin coding gene (designated as C-2442), and solute carrier family 4 member 8 (slc4a8)) were selected to confirm the RNA-Seq results with quantitative RT-PCR (RT-qPCR) according to their expression difference and functional annotation. RNA samples were isolated from M. lateralis of different developmental stages, and first-strand cDNAs were synthesized using PrimeScript™ RT reagent Kit (Takara, Dalian, China). Primers were designed with Primer Premier5 (Table S1), and Ef-1b was used as a reference gene (Li et al., 2019). PCRs were conducted using SYBR Green Real-Time PCR Master Mix (Takara, China) and the LightCycler 480 Real-time fluorescence quantitative PCR instrument (Roche, Basel, Switzerland). Data were analyzed using the 2−ΔΔCt method (Livak and Schmittgen, 2001) and presented as mean ± SD. All reactions were performed with three biological replicates and three technical replicates. Statistical analysis was tested using one-way ANOVA followed by Duncan’s test (SPSS 22.0, USA), and a statistically significant difference was set as p< 0.05.
Results and discussion
Establishment of full-length transcriptome of M. lateralis with PacBio Iso-Seq and Illumina RNA-Seq
Through PacBio Iso-Seq sequencing, a total of 6,623,650 subreads (15.11-Gb nucleotides) were obtained after initial quality control by removal of the adaptor reads and subreads<50 bp (Table 1). The subreads were then self-corrected, and a total of 337,922 CCSs were produced, of which 238,919 sequences were identified as full-length non-chimeric (FLNC) reads. After iterative clustering for error correction (ICE) and polishing using Arrow, 184,784 high-quality full-length consensus transcripts were generated. After the raw reads generated were trimmed and filtered by the Illumina RNA-Seq, more than 54,095,792 high-quality clean reads (Table 2) were generated for each sample with Q30 values greater than 92.18%. To build the full-length transcripts with higher accuracy and lower error rate, all full-length CCSs were corrected with Illumina RNA-Seq data by LoRDEC. At last, 121,424 unigenes, as the gene reference sequences, were obtained after the removal of redundant sequences by CD-HIT (Table 1). Then, the clean reads of each sample obtained by Illumina sequencing were mapped to the ref, and the mapping rate ranged from 64.64% to 84.75% (Table 2).
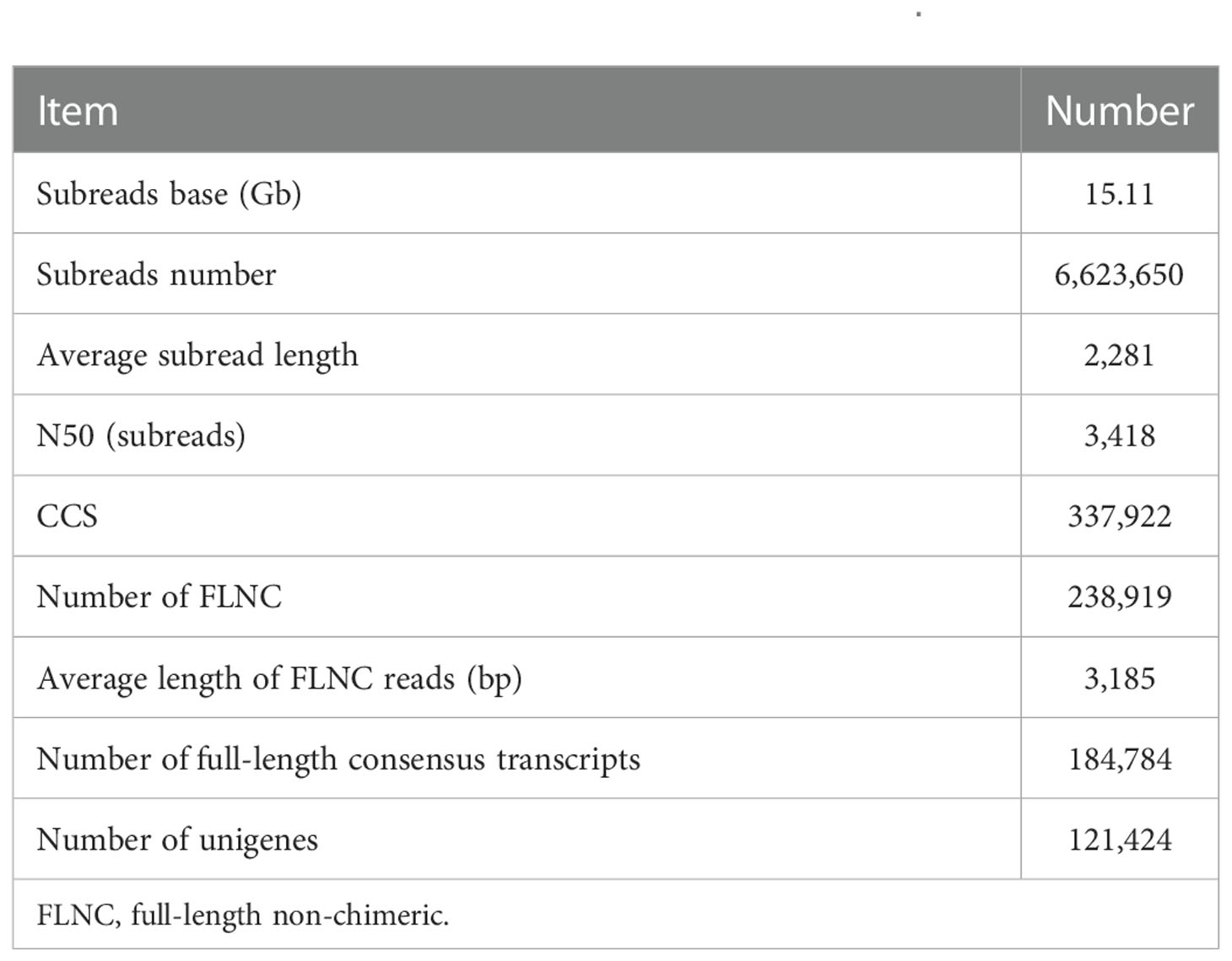
Table 1 Summary of PacBio Iso-Seq of Mulinia lateralis.
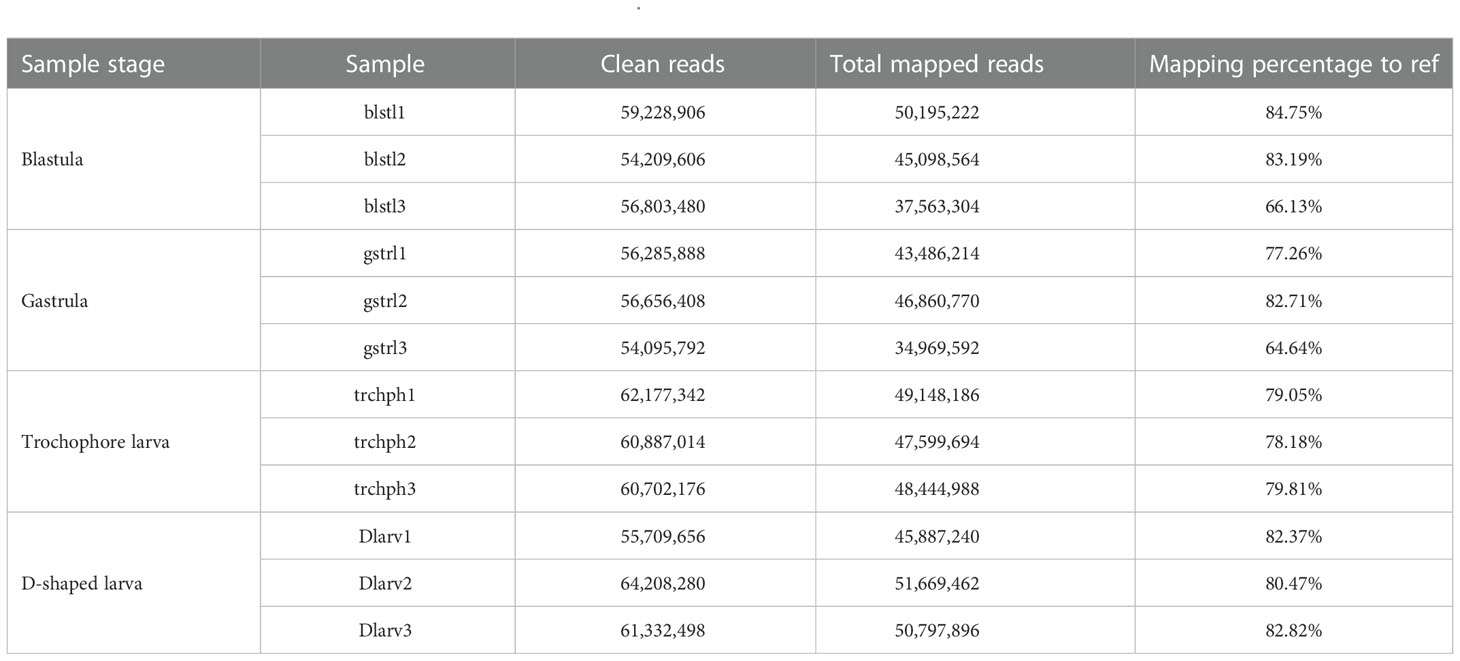
Table 2 Statistics of the Illumina RNA-Seq data of Mulinia lateralis.
This study combined the PacBio Iso-Seq and Illumina RNA-Seq technologies to generate a full-length transcriptome for M. lateralis. After correction, the average length of full-length consensus transcripts was 3,267 bp with an N50 of 3,879 bp (Table S2), which were much longer than that of the de novo assembled transcripts (Hu et al., 2019; Nie et al., 2021; Núñez-Acuña et al., 2022) and comparative with the full-length transcriptome of bivalve species recently reported (Liao et al., 2022; Zeng et al., 2022). The average mapping rate of Illumina sequences was greater than 75%, indicating that a good reference dataset was established. The results suggested the full-length transcriptome obtained in the present provided good genetic resources for M. lateralis.
Gene functional annotation of full-length transcripts
The identified 121,424 unigenes were annotated by searching against seven databases, and a total of 73,367 (60.42%) (Table S3) were annotated in at least one database, as well as 10,036 were annotated in all seven databases (Table 3, Figure 1A). According to the prediction by NR database, 62,786 unigenes were annotated in 642 homologous species, where the top 20 species were shown in Figure 1C. C. gigas, whose genome was the first to be described as a marine bivalve (Qi et al., 2021), was found to contain the highest number of homologous sequences (29,485, 46.96%) with M. lateralis. The second and third species were Lottia gigantea (5628, 4.63%) and Lingula anatina (3576, 2.95%), respectively. More than 60% of unigenes have been annotated in at least one database, which provided important information for studying the shell formation as well as other development processes of M. lateralis.
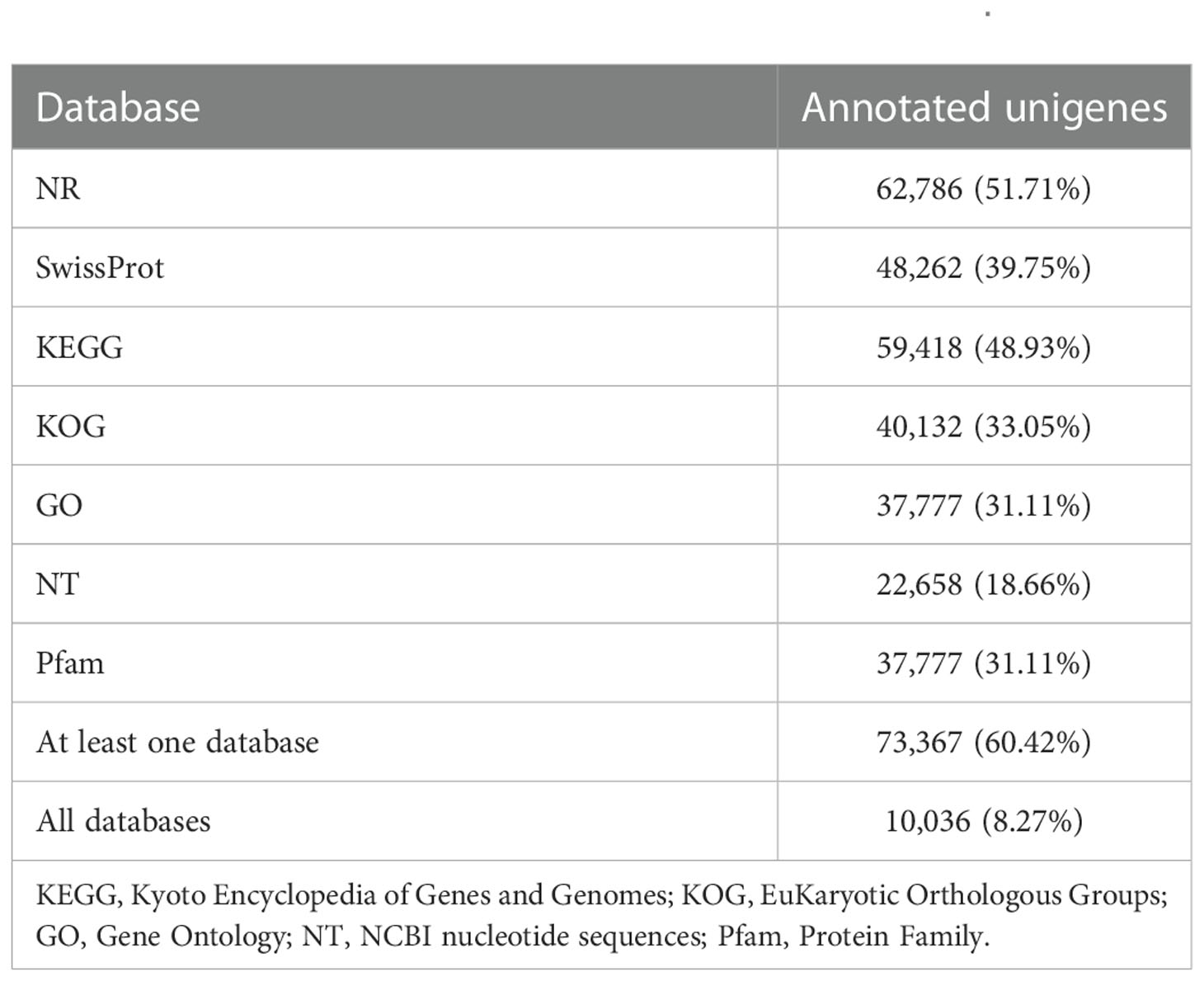
Table 3 Summary of unigenes annotation of Mulinia lateralis.
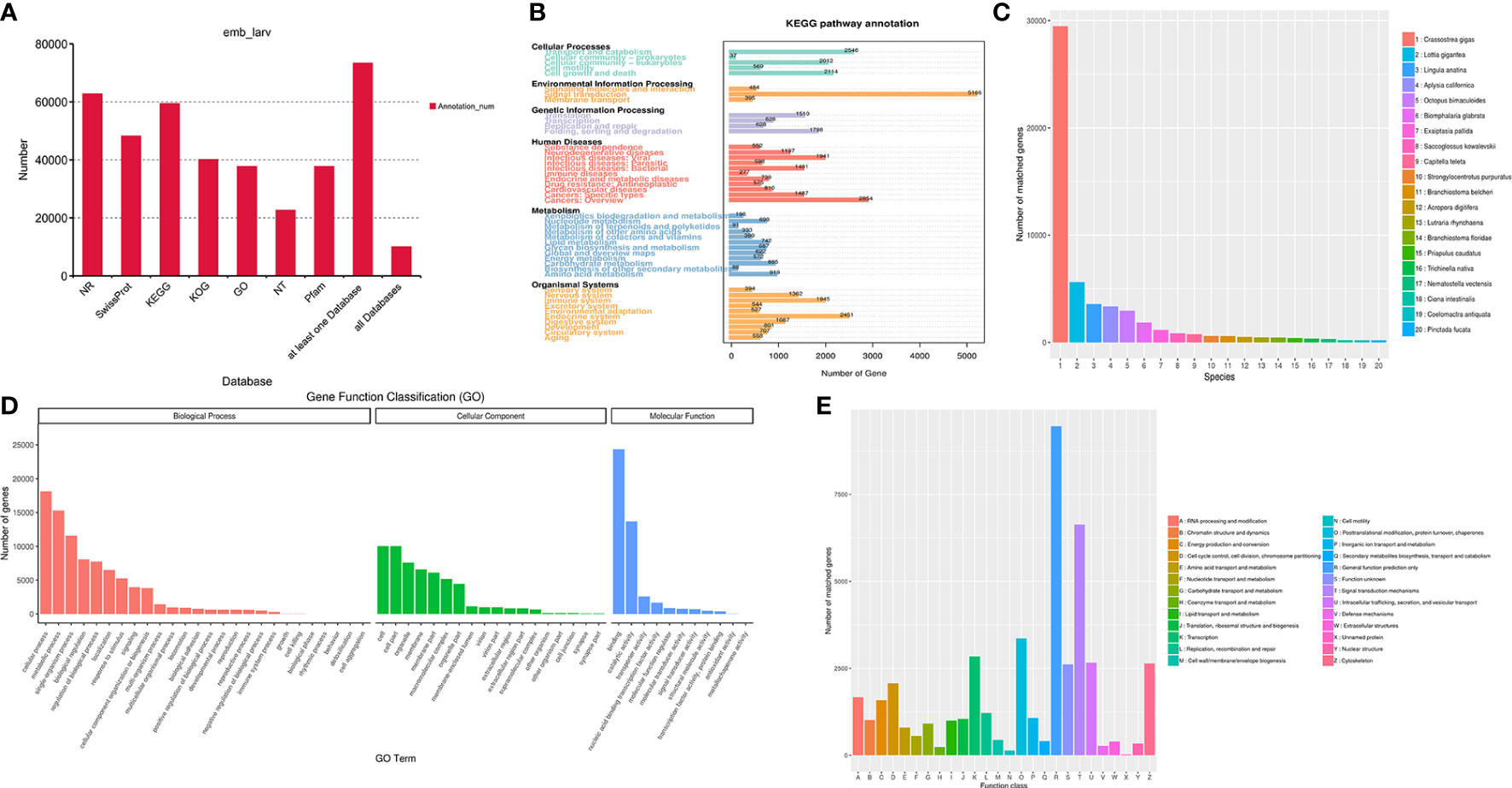
Figure 1 Functional annotation of Mulinia lateralis full-length transcripts. (A) Statistics of transcripts annotated in different databases. (B) KEGG pathway classification of annotated transcripts. (C) Distribution of the species with matched transcripts based on NR database. X-axis, different species ID; Y-axis, number of transcripts matched. (D) GO term classification of annotated transcripts. (E) KOG classification of the annotated transcripts. KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology; KOG, EuKaryotic Orthologous Groups.
There were 37,777 (31.11%) unigenes assigned to the three main GO categories and then categorized into 55 GO terms. The three most abundant terms in the biological process were “cellular process”, “metabolic process”, and “single-organism process”. Within the cellular component category, “cell part”, “cell”, and “organelle” were the prominent terms. “Binding”, “catalytic activity”, and “transporter activity” were dominant in the molecular function category (Figure 1D). The term “extracellular matrix” associated with the cellular component category is generally supposed to be related to shell formation genes (Song et al., 2022), and collagen and calcium-binding protein were recognized in this term.
A total of 59,418 (48.93%) transcripts were annotated in 1,280 pathways in the KEGG database. According to the KEGG classification, the unigenes were assigned to six level 1 KEGG categories (cellular processes, environmental information processing, genetic information processing, metabolism, organismal systems, and human diseases) and 45 level 2 subcategories (Figure 1B). The top five prominent subcategories were “Signal transduction”, “Cancers: Overview”, “Transport and catabolism”, “Endocrine system”, and “Cell growth and death”. Notably, it was reported that the “calcium signaling pathway”, “Wnt signaling pathway”, and “tyrosine metabolism pathway” probably play significant roles in the biomineralization process (Shi M. et al., 2013; Liu et al., 2015). Among these three pathways, calmodulin, tyr, β-catenin, TCF, and wnt5 are supposed to be related to calcium signal transduction and the shell formation process (Shi Y. et al., 2013; Gao et al., 2016; Zhang et al., 2021).
The KOG analysis demonstrated that 40,132 (33.05%) unigenes were classified into 26 functional clusters (Figure 1E). In M. lateralis, the most numerous five categories were “General function prediction only (R)”, “Signal transduction mechanisms (T)”, “Posttranslational modification, protein turnover, chaperones (O)”, “transcription (K)”, and “Intracellular trafficking, secretion, and vesicular transport (U)”. A total of 1,071 unigenes were assigned to “Inorganic ion transport and metabolism (P)”, 39 genes of which were annotated as members of the SLC4 family, a transporter family that can supply calcified substrates ( and ) to calcified sites through cross-epithelial transport (Ramesh et al., 2017). Moreover, 6,636 unigenes were assigned to the category “Signal transduction mechanisms (T)”, within which tyr, CA, calmodulin, and chitin synthase were reported to be involved in shell formation (Miyamoto et al., 1996; Miglioli et al., 2019; Wang et al., 2022).
Identification of TFs, SSRs, and lncRNAs
In this study, animalTFDB 2.0 was used to identify animal transcription factors in M. lateralis. A total of 3,015 transcripts were predicted as animal transcription factors (Table S4). The first three transcription factor families were the zf-C2HC transcription factor family (1177), Homeobox transcription factor family (249), and ZBTB transcription factor family (238) (Figure 2A).
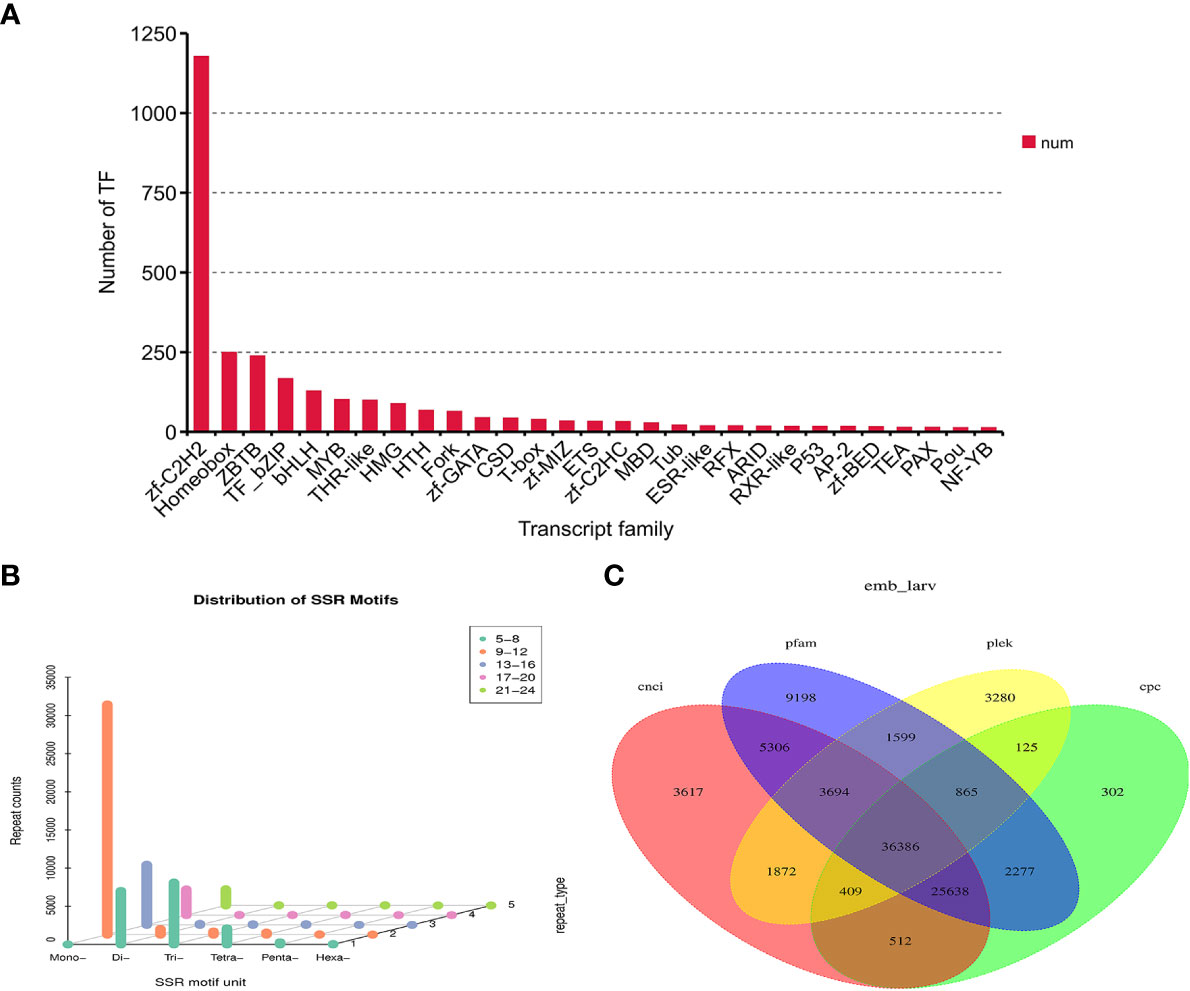
Figure 2 Identification of transcription factors (TFs), simple sequence repeats (SSRs), and long non-coding RNAs (lncRNAs) in Mulinia lateralis. (A) Prediction of transcription factor families. (B) Histogram distribution of different types of SSRs. (C) Venn diagram of lncRNAs predicted by CPC, CNCI, CPAT, and Pfam.
SSR is widely and evenly distributed in the genome of eukaryotes. The software MISA was used to search the SSR profiles in the unigenes of M. lateralis. A total of 39,049 SSR-containing unigenes were detected. In the four developmental stages, mono-nucleotide repeat motifs (9–12) were the most abundant, followed by di-nucleotide repeats and tri-nucleotide repeats (5–8) (Figure 2B).
Four computational approaches (CPC, CNCI, CPAT, and Pfam) were combined to identify lncRNAs. As is shown in Figure 2C, 36,386 unigenes were predicted as lncRNAs by all four methods, accounting for 29.97% of the total predicted lncRNAs.
Analysis of differentially expressed genes
To investigate the expression patterns of unigenes in M. lateralis, the Illumina clean reads were mapped back to the ref to determine their expression levels. The FPKM values of more than 95% of genes were ≤5, and approximately 1% of the genes were >15 (Table S5). A Venn diagram was drawn to exhibit the unigenes with FPKM > 0.1 in all four stages (30,178 unigenes) (Figure 3A), and the number of genes uniquely expressed in each stage was 776 (blastula), 1,974 (gastrula), 4,002 (trochophore larva), and 3,072 (D-shaped larva). There were 327 (blastula), 366 (gastrula), 413 (trochophore larva), and 485 (D-shaped larva) unigenes with high expression (FPKM > 60) in each of the four developmental stages. Specific to these highly expressed unigenes (FPKM > 60,619 genes), a hierarchical clustering analysis was performed with the normalized value of log10(FPKM + 1) (Figure 3B). The expression patterns of genes between the blastula and gastrula were similar, as most of the genes highly expressed in the blastula were also highly expressed in the gastrula. However, the genes highly expressed in the D-shaped larva were very low or even not expressed in the other three development stages (Figure 3B).
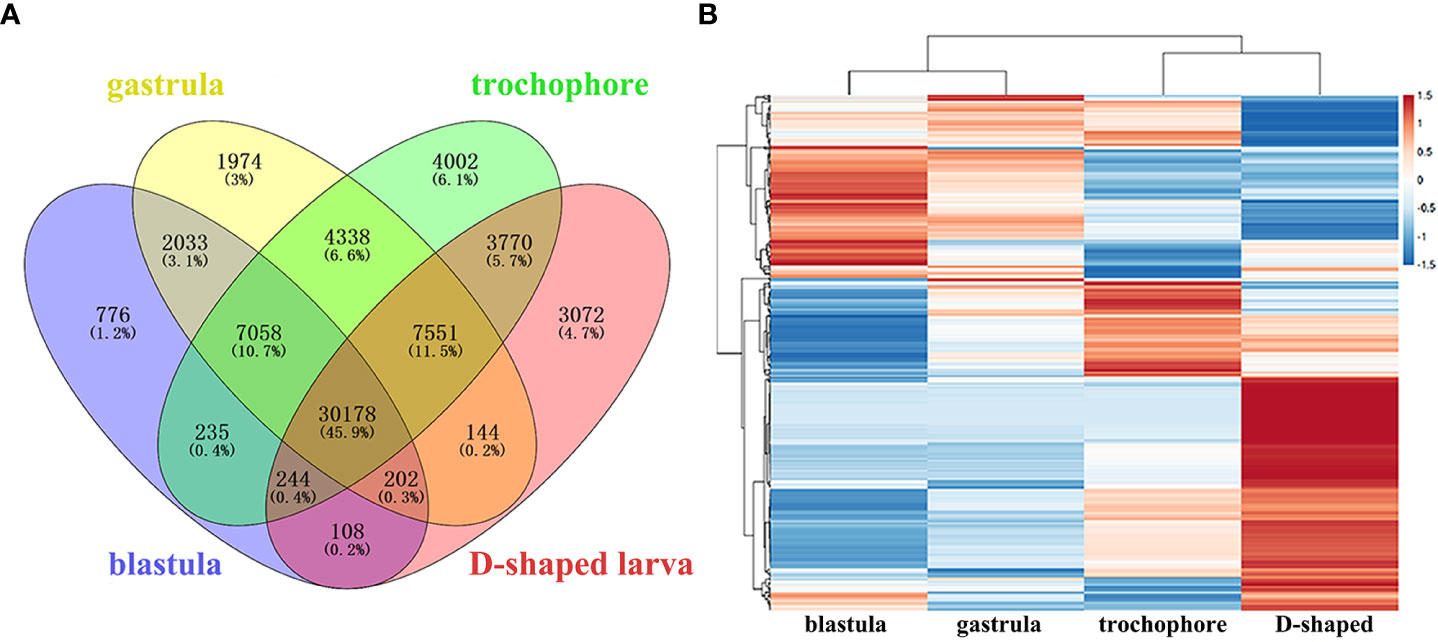
Figure 3 Analysis of gene expression in four development stages of Mulinia lateralis. (A) Venn diagram of unigenes expressed in four development stages. (B) Hierarchical clustering of the highly expressed unigenes in four development stages.
Pairwise comparisons between the two adjacent developmental stages (designated as b&g for the blastula and gastrula, g&t for the gastrula and trochophore larva, and t&d for the trochophore larva and D-shaped larva) were performed to identify the DEGs (Table S6). The maximal DEGs were observed between the trochophore larva and D-shaped larva (17,829), while the fewest existed between the blastula and gastrula, (4512). Moreover, 10,637 DEGs were identified between gastrula and trochophore larva (Table 4). Among those genes, tyr, CA, insoluble shell matrix protein-encoding gene 1 (ISMP1), and EF-hand, which play key roles in embryo shell development in bivalve molluscs (Zhang and He, 2011; Zhao et al., 2018), were identified to have significant differential expression between the trochophore larva and the D-shaped larva in M. lateralis.
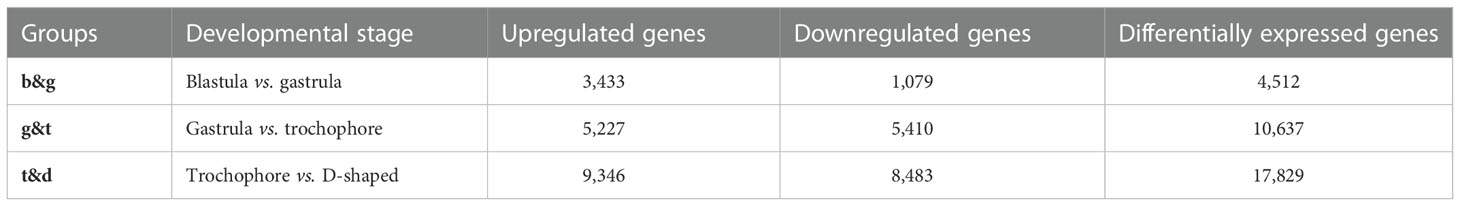
Table 4 Number of differentially expressed genes among developmental stages of Mulinia lateralis.
GO and KEGG enrichment analyses were used to mine DEGs related to shell formation. The most representative level 2 GO terms of DEGs from the three groups were “binding”, “cellular process”, and “metabolic process” (Table S7). In the “biological process” category, the top three GO terms for b&g were “organic cyclic compound metabolic process”, “cellular aromatic compound metabolic process”, and “heterocycle metabolic process”, which indicated that compound metabolic process may be very different between the blastula and gastrula. The DEGs in g&t were mainly assigned to “single-organism process”, “biological regulation”, and “regulation of biological process”. The most enriched GO terms for t&d were “single-organism process”, “single-organism cellular process”, and “localization”. In the “molecular function” category, more DEGs in g&t were enriched in “transition metal ion binding”, while DEGs of t&d were significantly enriched in “chitin binding” categories, as well as in “transcription factor activity”. In addition, 80, 172, and 254 DEGs in the b&g, g&t, and t&d groups, respectively, were involved in the “calcium ion binding function” GO category, some of which were involved in shell formation (EF-hand and calmodulin). The upregulated DEGs in the D-shaped larva such as collagen and calcium-binding proteins were involved in the “extracellular matrix”.
KEGG enrichment analysis showed that DEGs in b&g were mostly enriched in the pathways of “Huntington’s disease”, “Pathogenic Escherichia coli infection”, and “Phagosome” (Table S8, Figure 4A). “Metabolic pathways”, “Biosynthesis of secondary metabolites”, and “Protein processing in endoplasmic reticulum” were the top KEGG pathways in the g&t group (Table S8; Figure 4B). For the t&d group, the top three pathways were “Metabolic pathways”, “Cell cycle”, and “Biosynthesis of secondary metabolites” (Table S8; Figure 4C). Moreover, more DEGs in the t&d group were enriched into the “calcium signaling pathway” and “endocrine and other factor-regulated calcium reabsorption” pathways than those in the g&t group. Thirty DEGs enriched in the “calcium signaling pathway” were annotated as calcium/calcium-like protein-encoding genes, which are involved in the regulation of calcium transport and secretion (Li et al., 2004). A total of nine DEGs enriched in “endocrine and other factor-regulated calcium reabsorption” were annotated as solute carrier family 8 (slc8a), which encode proteins mediating Ca2+ extrusion in most cell types (Brini and Carafoli, 2011). The increasing genes involved in the calcium reabsorption pathway during larval development suggested that the trochophore larva was the key stage for the individuals to absorb calcium ions, which was coincident with the discoveries in mussels (Ramesh et al., 2017).
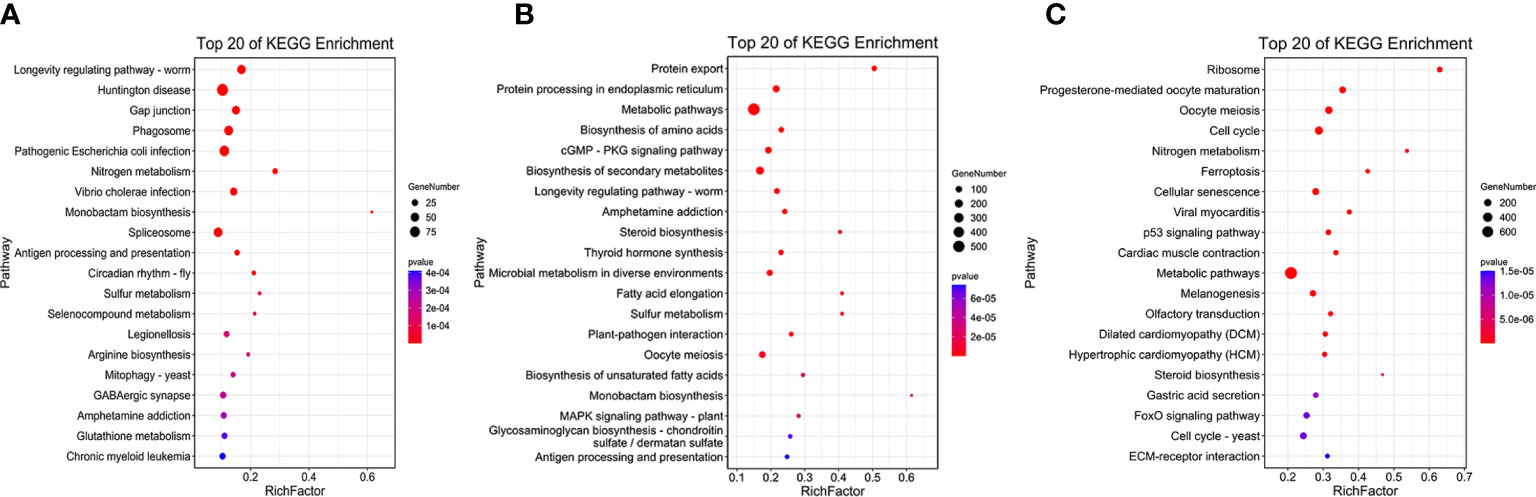
Figure 4 The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of tissue-specific expressed genes. (A) blastula vs. gastrula. (B) Gastrula vs. trochophore. (C) Trochophore vs. D-shaped.
Construction of the co-expression network
To construct and analyze the gene co-expression network, 25,664 annotated genes with FPKM values greater than 0.1 were utilized. With the soft thresholding power β of 6 and the minimum module size of 30, a total of 14 modules were identified through hierarchical clustering (Figure 5A). To explain the gene expression variation, module eigengenes (MEs) were calculated to represent each module. Relationships between the identified modules were mapped (Figure 5B), and the results indicated that gene expressions were relatively independent between modules (Figure 5D). With the connectivity analysis of eigengenes, these 14 modules could be clustered into two clusters (Figure 5C).
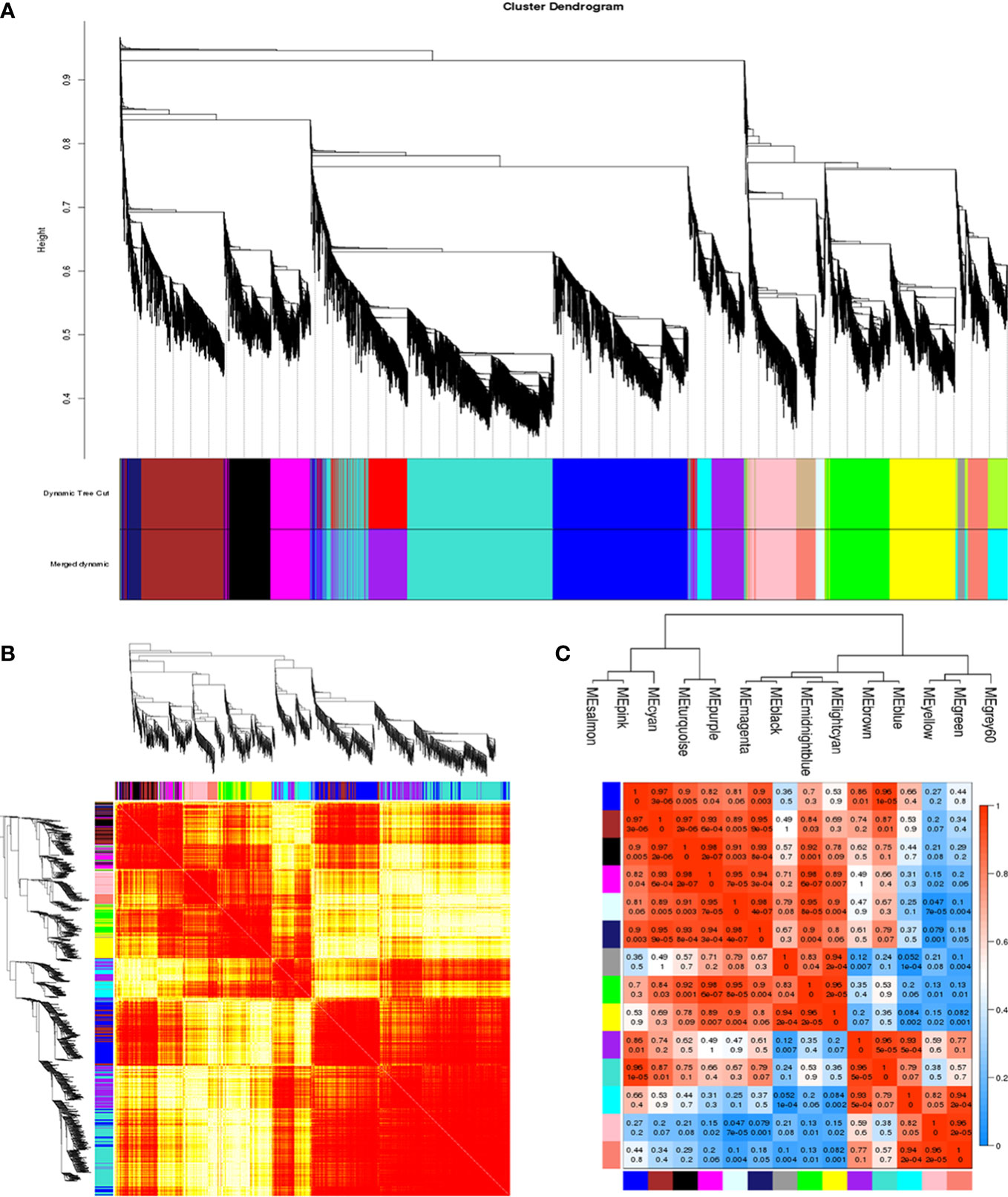
Figure 5 Construction of the co-expression network. (A) Clustering dendrogram of genes, with dissimilarity based on topological overlap, together with assigned module colors. (B) Clustering dendrogram of 14 module eigengenes. Heatmap depicts the topological overlap matrix (TOM) among all genes included in the analysis. Light color represents a low overlap. Progressively darker red color represents an increasing overlap. (C) Eigengene dendrogram. (D) Adjacency heatmap of module eigengenes. Red indicates a high correlation. Blue represents the opposite results.
The developmental stage (blastula, gastrula, trochophore larva, and D-shaped larva) was used as the traits to select the trait-related modules, and the association between the modules and traits was explored by measuring the correlation between ME values and features. The results indicated that pink and salmon modules were most positively correlated with D-shaped larva (r = 0.99, p = 4e–09; r = 0.89, p = 5e−04), and negative correlations were observed with the other three traits (Figure 6A). Meanwhile, a high degree of connectivity between pink and salmon modules was revealed (Figure 5C). In the pink module, insoluble matrix shell protein, tyr, and chitin-binding protein genes could be identified. Moreover, EF-hand and C-2442 were found in the salmon module. GO analysis of genes in the modules pink and salmon was conducted, and the results showed that the genes within the pink module were significantly revealed to have multiple functions associated with biomineralization, such as calcium ion binding and transmembrane transporter activity (Figure 6B), while the genes that belonged to the salmon module were significantly enriched in translation and protein metabolic process (Figure 6C).
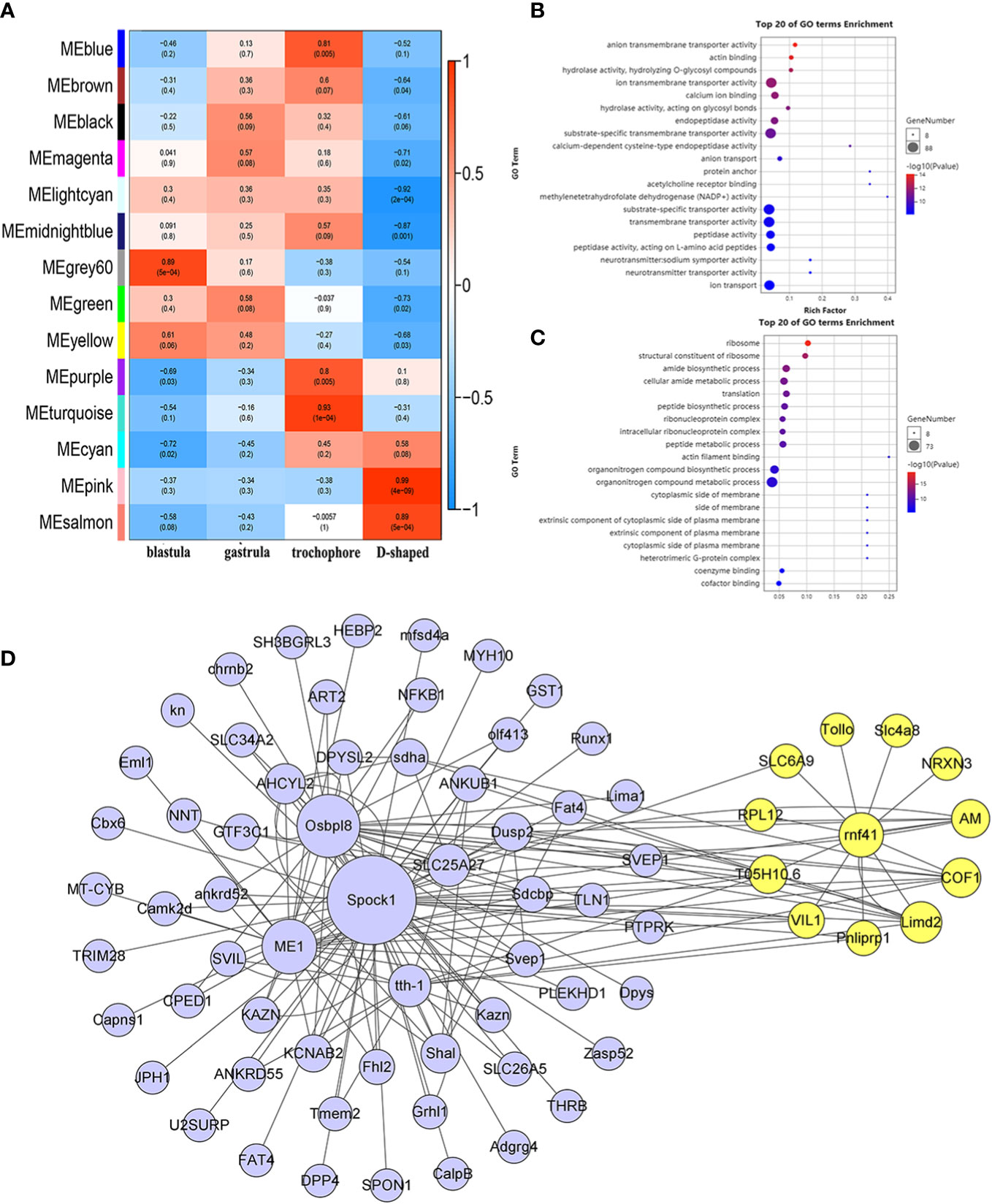
Figure 6 WGCNA of the annotated genes. (A) Module–trait associations. (B, C) GO analysis of the genes involved in the pink module (B) and salmon module (C). (D) The co-expression network of hub genes is in the pink module, and the yellow circles represent rnf41 and the closely related genes. WGCNA, weighted gene correlation network analysis; GO, Gene Ontology.
To further analyze the functions represented by the pink module, which exhibited the highest concentration in the D-shaped larva, we use a |GS| over 0.2 and an |MM| over 0.8 as cutoff criteria and identified 948 hub genes. According to the recommendation of the weighted gene correlation network analysis (WGCNA) official website, the top 85 genes with the highest weight value were selected, and the co-expression network was visualized using Cytoscape (Figure 6D). Among these hub genes, slc4a8, a bicarbonate transporter from the slc4 family, was identified, which is involved in calcium transport and shell repair, and calcification during blue muscle larval development (Ramesh et al., 2019). We found that rnf41 (E3 ubiquitin-protein ligase NRDP1) was highly connected with slc4a8 (weight = 0.6144), and these two genes may regulate the calcification process during the early shell formation (Fang et al., 2012).
Search for DEGs associated with biomineralization
To focus on the analysis of larval shell formation in M. lateralis, all DEGs were retrieved, and 12 candidate genes were obtained to be related to biomineralization. Among these 12 DEGs, 8 unigenes function in shell matrix deposition (ISMP1, ISMP2, ISMP5, chitin synthase, tyr, chitin-binding protein, collagen, and pu14), and 4 unigenes were related to ion transportation (CA, slc4a8, C-2442, and EF-hand).
In order to verify the genes that were putatively involved in the biomineralization process, we selected six unigenes, including two genes involved in matrix deposition (chitin synthase and tyr), two transporter genes (CA and slc4a8), and two calcium transporter genes (EF-hand and C-2442) for RT-qPCR analysis. As expected, the expression profiles of these genes showed similar trends with the RNA-Seq data (Figure 7).
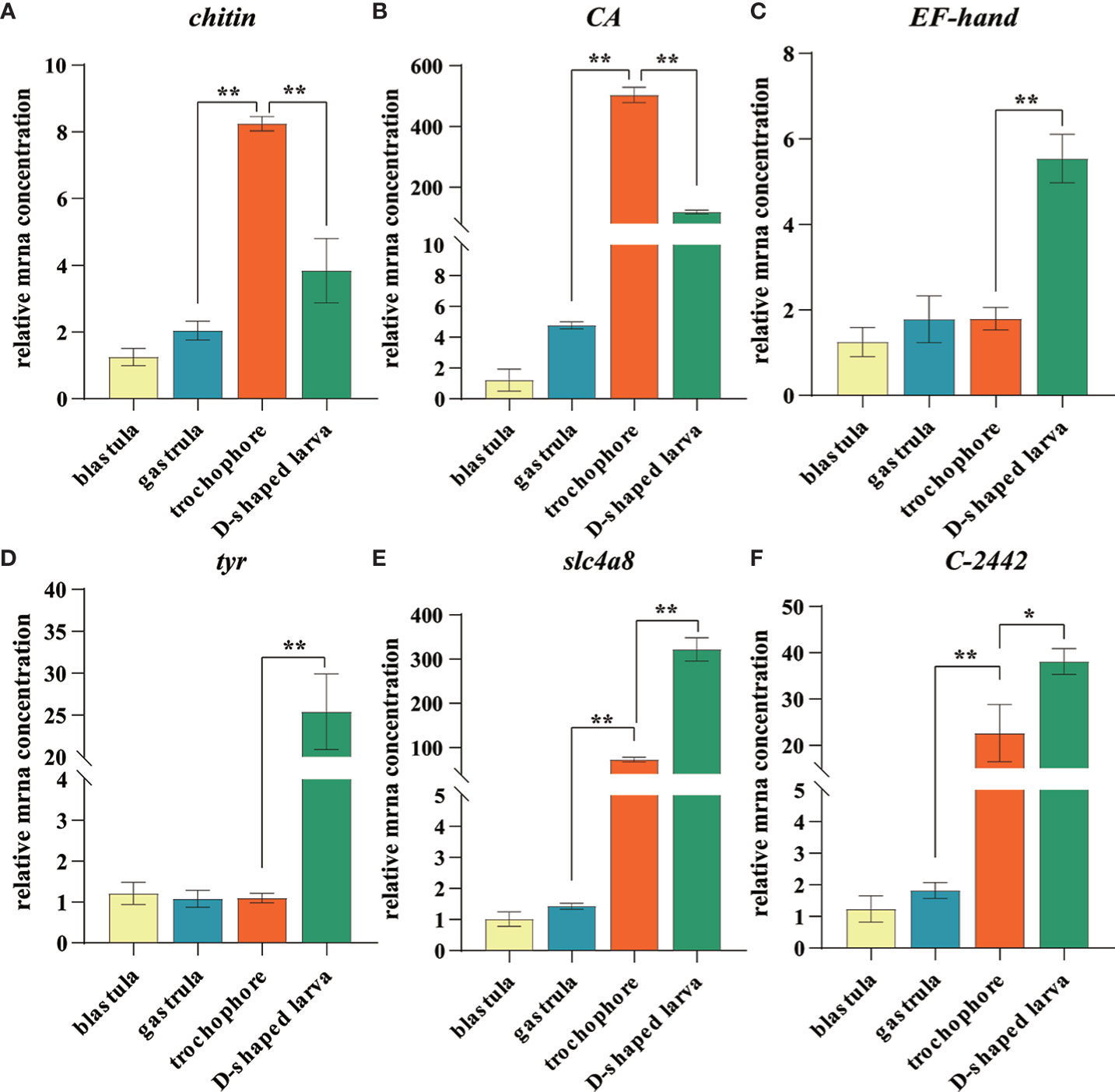
Figure 7 Expression level of identified larval shell formation genes among different developmental stages of Mulinia lateralis. (A–F) represents chintin synthase, carbonic anhydrase, EF-hand, tyrosinase, slc4a8 and C-2442 gene, respectively. Each gene with the lowest expression level was set as standard 1, and the expression levels in other stages were indicated as relative fold-change. All data were represented as mean ± SD (n = 3), *p< 0.05; **p< 0.001.
CA and chitin synthase exhibited similar expression patterns that considerably increased to the peak in trochophore larva and then dropped quickly in D-shaped larva. It is worth noting that the expression of CA dramatically increased to 503.8-fold that in the blastula. CA plays several essential roles in multiple physiological processes such as pH regulation, electrolyte balance, ionic transportation, carboxylation or decarboxylation reactions, and biocalcification, which is considered one of the most important enzymes in CaCO3 biomineralization due to its ability to catalyze the hydration of carbon dioxide and provides bicarbonate ions that react with Ca2+ to form CaCO3 (Supuran, 2011; Ramesh et al., 2017). It is proposed that molluscs usually absorb calcium first and CA catalyzes the hydration of CO2 reacting with calcium to facilitate its deposition at the stratum corneum formed by the shell matrix after calcium accumulates to a certain extent in the body (Evans, 2019; Sharker et al., 2021). Chitin synthases are crucial enzymes for the metabolism of chitin. Arthropod chitin forms a major part of the exoskeleton, and the supporting layer of the tubular trachea system and chitin synthase is a key factor to regulate most insect growth and molting (Jiang et al., 2021). In shellfish, chitin synthase has been proven to be involved in shell biosynthesis of trochophore larva chitin shell, which is then covered by the calcified shell at early D-shaped larva (Zhang et al., 2012; Liu et al., 2018). We observed that during the development of M. lateralis, the shell formation process was completed within 1 h. The obvious expression fluctuation suggested that CA and chitin synthase played a key role during the larval shell formation and calcium accumulation process in such a short time window.
slc4a8 and C-2442 began to upregulate in the trochophore larva and reached the peak in D-shaped larva. SLC4 family bicarbonate transporters mediate the co-transport of sodium and bicarbonate. In sea urchin embryos, the SLC4 family member regulates intra- and extracellular exchange of with Na+ and is fundamentally linked to the biological precipitation of CaCO3 (Hu et al., 2018). In molluscs, some SLC4A family members have been identified in mantle transcriptome data of C. gigas (Zhang et al., 2012), Pinctada fucata (Shi Y. et al., 2013), and Tridacna squamosa (Ip et al., 2017), but the function of these transporters and a description of the Ca2+ transporting mechanisms still need to be explored. C-2442 codes calmodulin, and the protein has been found to mediate the enhanced calcium deposition induced by CO2 exposure (Wang et al., 2022). The expression characteristics of both genes suggested that ion transportation mainly began at the trochophore larva and continuously provided a substrate for biomineralization.
EF-hand also encodes calmodulin, which is structurally conserved and functionally preserved throughout the animal kingdoms (Xu et al., 2017). It has been proven to be served as the calcium ion transporter during shell formation in a variety of shellfish (Huang et al., 2007; Sun et al., 2015; Jain et al., 2017). In molluscs, tyr is strongly expressed in the edge of the mantle, which is the main biomineralization tissue, and tyrosinase proteins have also been detected in the proteome of the shell prismatic layer and nacreous layer, indicating its key role in the shell matrix deposition (Huan et al., 2013; Miglioli et al., 2019; Ren et al., 2020; Zhu et al., 2021). Moreover, tyrosinase was confirmed to participate in the embryo shell ontogeny of the organic matrix in M. galloprovincialis and Hyriopsis cumingii (Miglioli et al., 2019; Ren et al., 2020). In M. lateralis, the expression pattern of these two genes was obviously upregulated in D-shaped larva. The results indicated that these genes may be involved in the formation of larval calcified shells. The characteristics of these genes suggested that the process of ion transport occurs earlier than the deposition of the shell matrix, and the trochophore larva and D-shaped larva were the key stages of larval shell formation. Further studies are needed to functionally validate the regulatory mechanisms of larval shell formation, and M. lateralis will provide a promising platform to facilitate the related research.
Conclusion
The present study provides new genetic resources of the comprehensive full-length transcriptome for M. lateralis, a promising model organism of bivalve molluscs. The results showed that a total of 238,919 high-quality non-redundant full-length transcripts were generated with an average length of 3267 bp, and 121,424 unigenes were annotated. The transcription factors (TFs), SSR, and lncRNAs of M. lateralis were predicted. In addition, 4512, 10,637, and 17,829 differentially expressed genes were obtained between the two adjacent developmental stages, of which 12 genes were identified to be involved in the formation of the larval shell, including ISMP1, ISMP2, ISMP5, chitin synthase, tyr, chitin-binding protein, collagen, pu14, CA, slc4a8, C-2442, and EF-hand. Our research provides valuable full-length transcriptome resources to M. lateralis and offers important clues for further exploration of the larval shell formation as well as other biological processes in molluscs.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The datasets generated for this study can be accessed through the Sequence Read Archive (SRA) database. The PacBio Iso-Seq transcriptome data were deposited under the accession number of SRR17545269. The Illumina RNA-Seq raw data were deposited as: blastula SRR17520086, SRR17520089, SRR17520090; gastrula SRR17520083, SRR17520084, SRR17520085; trochophore larva SRR17520080, SRR17520081, SRR17520082; D-shaped larva SRR17520079, SRR17520087, SRR17520088.
Author contributions
ZQ, ZZ, and ZB conceived and designed the study. XG and FZ performed the experiments. XG, XL, and DL participated in data analysis. ZY, ML, YL, HLW, and HW contributed to reagent/material/computer resource/sample culture and collection. XG and ZQ wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (32070516) and the Major Basic Research Projects of Shandong Natural Science Foundation (ZR2018ZA0748).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1111241/full#supplementary-material
References
Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1186/gb-2010-11-10-r106
Ansell A. D. (1962). The functional morphology of the larva, and the post-larval development of Venus striatula (da costa). J. Mar. Biol. Assoc. Uk 42, 419–443. doi: 10.1017/s0025315400001405
Arroyo L. R. G., Hernandez S. N. Y., Hernandez A. L., Rivera P. C. (2020). Ps19, a novel chitin binding protein from pteria sterna capable to mineralize aragonite plates in vitro. PloS One 15, e0230431. doi: 10.1371/journal.pone.0230431
Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., et al. (2000). Gene ontology: tool for the unification of biology. the gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Beier S., Thiel T., Münch T., Scholz U., Mascher M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Brini M., Carafoli E. (2011). The plasma membrane Ca²+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Csh. Perspect. Biol. 3, a004168. doi: 10.1101/cshperspect.a004168
Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cao M., Zhang M., Yang N., Fu Q., Su B., Zhang X., et al. (2020). Full length transcriptome profiling reveals novel immune-related genes in black rockfish (Sebastes schlegelii). Fish Shellfish Immunol. 106, 1078–1086. doi: 10.1016/j.fsi.2020.09.015
Cripe G. M. (2006). Contaminated sediment testing with the bivalve Mulinia lateralis: culture refinement for organism availability. Environ. Toxicol. Chem. 25, 1332–1336. doi: 10.1897/05-271R.1
Evans J. S. (2019). The biomineralization proteome: Protein complexity for a complex bioceramic assembly process. Proteomics 19, e1900036. doi: 10.1002/pmic.201900036
Falini G., Weiner S., Addadi L. (2003). Chitin-silk fibroin interactions: relevance to calcium carbonate formation in invertebrates. Calcif. Tissue Int. 72, 548–554. doi: 10.1007/s00223-002-1055-0
Fang D., Pan C., Lin H., Lin Y., Xu G., Zhang G., et al. (2012). Ubiquitylation functions in the calcium carbonate biomineralization in the extracellular matrix. PloS One 7, e35715. doi: 10.1371/journal.pone.0035715
Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., et al. (2016). The pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Fu L., Niu B., Zhu Z., Wu S., Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao J., Liu J., Yang Y., Liang J., Xie J., Li S., et al. (2016). Identification and expression characterization of three wnt signaling genes in pearl oyster (Pinctada fucata). Comp. Biochem. Physiol. B 196-197, 92–101. doi: 10.1016/j.cbpb.2016.03.003
Gilbert P. U. P. A., Bergmann K. D., Boekelheide N., Tambutté S., Mass T., Marin F., et al. (2022). Biomineralization: Integrating mechanism and evolutionary history. Sci. Adv. 8, eabl9653. doi: 10.1126/sciadv.abl9653
Gonzalez-Socoloske D., Taylor C. R., Thompson O. (2014). Distribution and conservation status of the antillean manatee (trichechus manatus manatus) in honduras. Lat. Am. J. Aquat. Res. 9, 123–131. doi: 10.5597/lajam00176
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guo X., Allen S. K. (1994). Sex determination and polyploid gigantism in the dwarf surfclam (Mulinia lateralis say). Genetics 138, 1199–1206. doi: 10.1093/genetics/138.4.1199
Hoang N. V., Furtado A., Mason P. J., Marquardt A., Kasirajan L., Thirugnanasambandam P. P., et al. (2017). A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genomics 18, 395. doi: 10.1186/s12864-017-3757-8
Hon T., Mars K., Young G., Tsai Y. C., Karalius J. W., Landolin J. M., et al. (2020). Highly accurate long-read HiFi sequencing data for five complex genomes. Sci. Data 7, 399. doi: 10.1038/s41597-020-00743-4
Huang J., Zhang C., Ma Z., Xie L., Zhang R. (2007). A novel extracellular EF-hand protein involved in the shell formation of pearl oyster. Bba Gen. Subj. 1770, 1037–1044. doi: 10.1016/j.bbagen.2007.03.006
Huan P., Liu G., Wang H., Liu B. (2013). Identification of a tyrosinase gene potentially involved in early larval shell biogenesis of the pacific oyster Crassostrea gigas. Dev. Genes. Evol. 223, 389–394. doi: 10.1007/s00427-013-0450-z
Hu Z., Song H., Yang M. J., Yu Z. L., Zhou C., Wang X. L., et al. (2019). Transcriptome analysis of shell color-related genes in the hard clam mercenaria mercenaria. Comp. Biochem. Physiol. D 31, 100598. doi: 10.1016/j.cbd.2019.100598
Hu M. Y., Yan J. J., Petersen I., Himmerkus N., Bleich M., Stumpp M. (2018). A SLC4 family bicarbonate transporter is critical for intracellular pH regulation and biomineralization in sea urchin embryos. Elife 7, e36600. doi: 10.7554/eLife.36600
Ip Y. K., Hiong K. C., Goh E. J. K., Boo M. V., Choo C. Y. L., Ching B., et al. (2017). The whitish inner mantle of the giant clam, Tridacna squamosa, expresses an apical plasma membrane Ca2+-ATPase (PMCA) which displays light-dependent gene and protein expressions. Front. Physiol. 8. doi: 10.3389/fphys.2017.00781
Jain G., Pendola M., Huang Y. C., Juan Colas J., Gebauer D., Johnson S., et al. (2017). Functional prioritization and hydrogel regulation phenomena created by a combinatorial pearl-associated two-protein biomineralization model system. Biochemistry 56, 3607–3618. doi: 10.1021/acs.biochem.7b00313
Jiang L. H., Mu L. L., Jin L., Anjum A. A., Li G. Q. (2021). RNAi for chitin synthase 1 rather than 2 causes growth delay and molting defect in Henosepilachna vigintioctopunctata. Pestic. Biochem. Physiol. 178, 104934. doi: 10.1016/j.pestbp.2021.104934
Kong L., Zhang Y., Ye Z. Q., Liu X. Q., Zhao S. Q., Wei L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Kurita Y., Deguchi R., Wada H. (2009). Early development and cleavage pattern of the Japanese purple mussel, Septifer virgatus. Zool. Sci. 26, 814–820. doi: 10.2108/zsj.26.814
Langfelder P., Horvath S. (2008). WGCNA: a r package for weighted correlation network analysis. BMC Bioinf. 9, 559. doi: 10.1186/1471-2105-9-559
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Liao X., Liu Y., Han T., Yang M., Liu W., Wang Y., et al. (2022). Full-length transcriptome sequencing reveals tissue-specific gene expression profile of mangrove clam Geloina erosa. Front. Physiol. 13. doi: 10.3389/fphys.2022.851957
Li H., Bai L., Dong X., Qi X., Liu H., Yu D. (2020). SEM observation of early shell formation and expression of biomineralization-related genes during larval development in the pearl oyster Pinctada fucata. Comp. Biochem. Physiol. D 33, 100650. doi: 10.1016/j.cbd.2019.100650
Li B., Dewey C. N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinf. 12, 323. doi: 10.1186/1471-2105-12-323
Liu G., Huan P., Liu B. (2015). A GATA2/3 gene potentially involved in larval shell formation of the pacific oyster. Crassostrea gigas. Dev. Genes Evol. 225, 253–257. doi: 10.1007/s00427-015-0511-6
Liu G., Huan P., Liu B. (2017). A SoxC gene related to larval shell development and co-expression analysis of different shell formation genes in early larvae of oyster. Dev. Genes Evol. 227, 181–188. doi: 10.1007/s00427-017-0579-2
Liu Z., Wang L., Yan Y., Zhang Y., Ge W., Li M., et al. (2018). D1 dopamine receptor is involved in shell formation in larvae of Pacific oyster Crassostrea gigas. Dev. Comp. Immunol. 84, 337–342. doi: 10.1013/j.dci.2018.03.009
Liu F., Li Y., Yu H., Zhang L., Hu J., Bao Z., et al. (2021). MolluscDB: an integrated functional and evolutionary genomics database for the hyper-diverse animal phylum Mollusca. Nucleic Acids Res. 49, D988–D997. doi: 10.1093/nar/gkaa918
Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Li S., Xie L., Zhang C., Zhang Y., Gu M., Zhang R. (2004). Cloning and expression of a pivotal calcium metabolism regulator: calmodulin involved in shell formation from pearl oyster (Pinctada fucata). Comp. Biochem. Physiol. B 138, 235–243. doi: 10.1016/j.cbpc.2004.03.012
Li Y., Zhang L., Li R., Zhang M., Li Y., Wang H., et al. (2019). Systematic identification and validation of the reference genes from 60 RNA-seq libraries in the scallop Mizuhopecten yessoensis. BMC Genomics 20, 288. doi: 10.1186/s12864-019-5661-x
Li A., Zhang J., Zhou Z. (2014). PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinf. 15, 311. doi: 10.1186/1471-2105-15-311
Luo W., Wu Q., Wang T., Xu Z., Wang D., Wang Y., et al. (2020). Full-length transcriptome analysis of Misgurnus anguillicaudatus. Mar. Genomics 54, 100785. doi: 10.1016/j.margen.2020.100785
Marin F., Le Roy N., Marie B. (2012). The formation and mineralization of mollusk shell. Front. Biosci. (Schol Ed) 4, 1099–1125. doi: 10.2741/s321
Marin F., Luquet G., Marie B., Medakovic D. (2008). Molluscan shell proteins: primary structure, origin, and evolution. Curr. Top. Dev. Biol. 80, 209–276. doi: 10.1016/S0070-2153(07)80006-8
Miglioli A., Dumollard R., Balbi T., Besnardeau L., Canesi L. (2019). Characterization of the main steps in first shell formation in Mytilus galloprovincialis: possible role of tyrosinase. P. R. Soc B-Biol. Sci. 286, 20192043. doi: 10.1098/rspb.2019.2043
Miyamoto H., Miyashita T., Okushima M., Nakano S., Morita T., Matsushiro A. (1996). A carbonic anhydrase from the nacreous layer in oyster pearls. Proc. Natl. Acad. Sci. U. S. A. 93, 9657–9660. doi: 10.1073/pnas.93.18.9657
Mouza M., Gros O., Frenkiel L. (2006). Embryonic development and shell differentiation in chione cancellata (bivalvia, veneridae): an ultrastructural analysis. Invertebr. Biol. 125, 21–33. doi: 10.1111/J.1744-7410.2006.00036.X
Nederbragt A. J., van Loon A. E., Dictus W. J. (2002). Expression of patella vulgata orthologs of engrailed and dpp-BMP2/4 in adjacent domains during molluscan shell development suggests a conserved compartment boundary mechanism. Dev. Biol. 246, 341–355. doi: 10.1006/dbio.2002.0653
Nguyen N. P., Nute M., Mirarab S., Warnow T. (2016). HIPPI: highly accurate protein family classification with ensembles of HMMs. BMC Genomics 17, 765. doi: 10.1186/s12864-016-3097-0
Nie H., Jiang K., Li N., Jahan K., Jiang L., Huo Z., et al. (2021). Transcriptome analysis reveals the pigmentation-related genes in two shell color strains of the Manila clam Ruditapes philippinarum. Anim. Biotechnol. 32, 439–450. doi: 10.1080/10495398.2020.1714635
Núñez-Acuña G., Fernandez C., Sanhueza-Guevara S., Gallardo-Escárate C. (2022). Transcriptome profiling of the early developmental stages in the giant mussel choromytilus chorus exposed to delousing drugs. Mar. Genomics 65, 100970. doi: 10.1016/j.margen.2022.100970
Qi H., Li L., Zhang G. (2021). Construction of a chromosome-level genome and variation map for the pacific oyster Crassostrea gigas. Mol. Ecol. Resour. 21, 1670–1685. doi: 10.1111/1755-0998.13368
Ramesh K., Hu M. Y., Thomsen J., Bleich M., Melzner F. (2017). Mussel larvae modify calcifying fluid carbonate chemistry to promote calcification. Nat. Commun. 8, 1709. doi: 10.1038/s41467-017-01806-8
Ramesh K., Yarra T., Clark M. S., John U., Melzner F. (2019). Expression of calcification-related ion transporters during blue mussel larval development. Ecol. Evol. 9, 7157–7172. doi: 10.1002/ece3.5287
Ren G., Chen C., Jin Y., Zhang G., Hu Y., Shen W. (2020). A novel tyrosinase gene plays a potential role in modification the shell organic matrix of the triangle mussel Hyriopsis cumingii. Front. Physiol. 11. doi: 10.3389/fphys.2020.00100
Salmela L., Rivals E. (2014). LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514. doi: 10.1093/bioinformatics/btu538
Sharker M. R., Sukhan Z. P., Sumi K. R., Choi S. K., Choi K. S., Kho K. H. (2021). Molecular characterization of carbonic anhydrase II (CA II) and its potential involvement in regulating shell formation in the pacific abalone, Haliotis discus hannai. Front. Mol. Biosci. 8. doi: 10.3389/fmolb.2021.669235
Shi M., Lin Y., Xu G., Xie L., Hu X., Bao Z., et al. (2013). Characterization of the zhikong scallop (Chlamys farreri) mantle transcriptome and identification of biomineralization-related genes. Mar. Biotechnol. 15, 706–715. doi: 10.1007/s10126-013-9517-0
Shimizu K., Adachi J., Muraoka Y. (2006). ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Bio.l 4, 649–664. doi: 10.1142/s0219720006002260
Shi Y., Yu C., Gu Z., Zhan X., Wang Y., Wang A. (2013). Characterization of the pearl oyster (Pinctada martensii) mantle transcriptome unravels biomineralization genes. Mar. Biotechnol. 15, 175–187. doi: 10.1007/s10126-012-9476-x
Song N., Li J., Li B., Pan E., Ma Y. (2022). Transcriptome analysis of the bivalve placuna placenta mantle reveals potential biomineralization-related genes. Sci. Rep. 12, 4743. doi: 10.1038/s41598-022-08610-5
Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41, e166. doi: 10.1093/nar/gkt646
Sun X., Yang A., Wu B., Zhou L., Liu Z. (2015). Characterization of the mantle transcriptome of yesso scallop (Patinopecten yessoensis): identification of genes potentially involved in biomineralization and pigmentation. PloS One 10, e0122967. doi: 10.1371/journal.pone.0122967
Supuran C. T. (2011). Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future. Med. Chem. 3, 1165–1180. doi: 10.4155/fmc.11.69
Tan K., Zheng H. (2020). Ocean acidification and adaptive bivalve farming. Sci. Total Environ. 701, 134794. doi: 10.1016/j.scitotenv.2019.134794
Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinf. 4, 41. doi: 10.1186/1471-2105-4-41
Walker R. L., Tenore K. R. (1984). Growth and production of the dwarf surf clam mulinia lateralis (Say 1822) in a Georgia estuary. Gulf Res. Rep. 7, 357–363. doi: 10.18785/grr.0704.07
Wang X., Li C., Lv Z., Zhang Z., Qiu L. (2022). A calcification-related calmodulin-like protein in the oyster crassostrea gigas mediates the enhanced calcium deposition induced by CO2 exposure. Sci. Total Environ. 833, 155114. doi: 10.1016/j.scitotenv.2022.155114
Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/molbev/msab153
Xu G., Cheng K., Wu Q., Liu M., Li C. (2017). Confinement alters the structure and function of calmodulin. Angew. Chem. Int. Ed. Engl. 56, 530–534. doi: 10.1002/anie.201609639
Yang Z., Huang X., Wang H., Pan H., Wang M., Ren Q., et al. (2021). Effects of microalgae diets and stocking density on larval growth, survival and metamorphosis of dwarf surfclam. Mulinia lateralis. Aquaculture 536, 736440. doi: 10.1016/j.aquaculture.2021.736440
Yarra T. (2018). Transcriptional profiling of shell calcification in bivalves (Edinburgh: University of Edinburgh).
Yarra T., Blaxter M., Clark M. S. (2021). A bivalve biomineralization toolbox. Mol. Biol. Evol. 38, 4043–4055. doi: 10.1093/molbev/msab153
Young M. D., Wakefield M. J., Smyth G. K. (2010). Goseq: Gene ontology testing for RNA-seq datasets. R Bioconductor 8, 1–25.
Zeng Q., Hu B., Blanco A. H., Zhang W., Zhao D., Martínez P., et al. (2022). Full-length transcriptome sequences provide insight into hermaphroditism of freshwater pearl mussel hyriopsis schlegelii. Front. Genet. 13. doi: 10.3389/fgene.2022.868742
Zhang G., Fang X., Guo X., Li L., Luo R., Xu F., et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54. doi: 10.1038/nature11413
Zhang L., He M. (2011). Quantitative expression of shell matrix protein genes and their correlations with shell traits in the pearl oyster Pinctada fucata. Aquaculture 314, 73–79. doi: 10.1016/j.aquaculture.2011.01.039
Zhang J., Yu P., Zhao Y., Zhou Q., Yang J., Hu Q., et al. (2021). Global analysis of transcriptome and translatome revealed that coordinated WNT and FGF regulate the carapacial ridge development of Chinese soft-shell turtle. Int. J. Mol. Sci. 22, 12441. doi: 10.3390/ijms222212441
Zhao R., Takeuchi T., Luo Y. J., Ishikawa A., Kobayashi T., Koyanagi R., et al. (2018). Dual gene repertoires for larval and adult shells reveal molecules essential for molluscan shell formation. Mol. Biol. Evol. 35, 2751–2761. doi: 10.1093/molbev/msy172
Zhu Y., Li Q., Yu H., Liu S., Kong L. (2021). Shell biosynthesis and pigmentation as revealed by the expression of tyrosinase and tyrosinase-like protein genes in pacific oyster (Crassostrea gigas) with different shell colors. Mar. Biotechnol. 23, 777–789. doi: 10.1007/s10126-021-10063-2
Keywords: Mulinia lateralis, full-length transcriptome, early development, larval shell formation, biomineralization
Citation: Guo X, Li X, Zhao F, Liu D, Yang Z, Li M, Li Y, Wei H, Wang H, Qin Z, Zhang Z and Bao Z (2023) Full-length transcriptome analysis provides insights into larval shell formation in Mulinia lateralis. Front. Mar. Sci. 9:1111241. doi: 10.3389/fmars.2022.1111241
Received: 29 November 2022; Accepted: 27 December 2022;
Published: 18 January 2023.
Edited by:
Yuehuan Zhang, South China Sea Institute of Oceanology (CAS), ChinaCopyright © 2023 Guo, Li, Zhao, Liu, Yang, Li, Li, Wei, Wang, Qin, Zhang and Bao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenkui Qin, cWluemtAb3VjLmVkdS5jbg==