 Gurucharan Sudarshan1
Gurucharan Sudarshan1 Simy Weil1
Simy Weil1 Noa Rotem-Dai2
Noa Rotem-Dai2 Rivka Manor1
Rivka Manor1 Yariv Greenshpan2Oron Goldstein2,3
Yariv Greenshpan2Oron Goldstein2,3 Omri Sharabi2
Omri Sharabi2 Eliahu D. Aflalo1,4Rivka Ofir3,5
Eliahu D. Aflalo1,4Rivka Ofir3,5 Benyamin Rosental2,3
Benyamin Rosental2,3 Roi Gazit2,3,6
Roi Gazit2,3,6 Amir Sagi1,6*†
Amir Sagi1,6*†- 1Department of Life Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
- 2The Shraga Segal Department of Microbiology, Immunology, and Genetics, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer Sheva, Israel
- 3Regenerative Medicine and Stem Cell Research Center, Ben-Gurion University of the Negev, Beer Sheva, Israel
- 4Department of Life Sciences, Achva Academic College, Arugot, Israel
- 5Dead Sea and Arava Science Center, Central Arava Branch, Beer Sheva, Israel
- 6National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer Sheva, Israel
Crustacean cell line immortalization has gained a great deal of attention in recent decades for both scientific and applied reasons. Our goal in this study was to advance the state of art towards establishing an immortalized cell line by improving the proliferation rates of primary cells isolated from embryos of the giant freshwater prawn Macrobrachium rosenbergii by using a lentivirus expressing the Ras oncogene. The choice of Ras derived from its involvement in various cellular pathways, such as cell growth, differentiation, and survival, and its use as a tool for in-vitro immortalization, e.g., a specific mutated Ras (RasV12) was used to generate an arthropod cell line. Complementarily, in-silico screening of M. rosenbergii transcriptomic libraries for Ras expression indicated that Ras is already expressed at very early stages of embryo development. In the current study, we transduced primary M. rosenbergii embryonic cells with a lentivirus expressing RasV12 by using the white spot syndrome virus (WSSV IE1) promoter. Expression and sequencing (as followed by sequencing cDNA, confocal microscopy and FACS analysis) of the mutated Ras in the transduced cells confirmed that the lentivirus was successfully integrated into the genome. The lenti-MrRas transduction rate was 23% in the total primary cell population and more than 80% in a sub-population of cells with high granularity. Proliferation of lenti-MrRas transfected cells was enhanced to almost 1200% of the seeding density by the end of our experiment (18 days), which was double that of the control. We were thus successful in enhancing the longevity of embryonic primary cell cultures by ectopic expression of the mutated Ras protein, but the improvement was not sufficient for immortalization.
1 Introduction
According to the most recently available FAO figures, global crustacean aquaculture reached an enormous annual amount of 9.4 million tonnes (69.3 billion USDS/year), with shrimp being the second-most produced commodity (FAO, 2020). However, there are a number of problems associated with the thriving shrimp industry, especially the devastating effect of the industry on the environment and the susceptibility of cultured shrimp to severe viral infections, which can cause huge losses. A way forward to addressing these problems would be the establishment of crustacean cell culture systems for producing cultured seafood and for virology-related research. This idea fits well with the ongoing research program of our laboratory on the decapod crustacean Macrobrachium rosenbergii, commonly known as the giant freshwater prawn. M. rosenbergii is one of the best studied species of decapod crustaceans, with some of the most recent work in our laboratory producing a wide array of transcriptomic datasets (Sharabi et al., 2016; Abayed et al., 2019) and a sequenced genome (Levy et al., 2019). It is also a highly demanded aquaculture species, with annual global production reaching as much as 294,081 tonnes, valued at ~2.4 billion USD (FAO, 2020).
In the current study, we aimed to manipulate the cell cycle of an M. rosenbergii embryonic primary cell culture by using lentivirus transduction of the Ras oncogene to obtain cell proliferation and repetitive passages. Ras is a proto-oncogene that was first identified in studies on rats with sarcoma-causing viruses, hence the designation Ras (Cox and Der, 2010). It soon became known that Ras has a variety of functions as a normal component of cellular signal transduction involved in cell proliferation, differentiation, apoptosis, and transformation in vertebrates and invertebrates (Downward, 1997). Ras is expressed in the G1 phase of the cell cycle, thereby promoting the cell cycle from the G1 phase to the S phase (Downward, 1997). In mammals, Ras expression induces cyclin-D via the Raf/MAPK pathway, with the subsequent cyclin-D accumulation in the nucleus inducing cells to proliferate (Foster et al., 2010). In crustaceans, it was shown, for example, that Ras was upregulated in the crayfish Procambarus clarkii in response to bacterial infection under high temperature (Li et al., 2009).
It is also known that Ras mutates easily, with mutated Ras genes influencing the expression of various genes involved in cell signaling, growth, differentiation, and proliferation, and in tumor development (Gimple and Wang, 2019). Ras mutations are usually caused by point mutations on exon 2 codon 12 (our target of interest), exon 2 codon 13, and exon 3 codon 61 (Prior et al., 2012). Mutated Ras can cause constant expression of the Ras protein, which remains upregulated, leading, for example, to the proliferation of primary cells from Drosophila embryos (Simcox et al., 2008). Since RasV12 was used to induce cell proliferation and cell line generation in that arthropod species (Simcox et al., 2008), our working hypothesis was therefore that we could exploit this activated form of Ras for the same purpose in crustacean species, such as M. rosenbergii.
To date, there have been various attempts to create stable cell lines from crustacean cells (Jayesh et al., 2012), but, to the best of our knowledge, none has been successful. Previous attempts included electroporation and lipofection of immortalization markers in cells isolated from the hematopoietic tissue of Penaeus monodon (Thansa et al., 2018); the use of the viral oncogene human papillomavirus (HPV 16) in the hematopoietic tissue of Cherax quadricarinatus (Claydon and Owens, 2008); and the use of a tumor antigen gene from simian virus-40 to induce proliferation of cells from a primary cell culture in the lymphoid organ of Penaeus stylirostris (Tapay et al., 1995). Importantly, the hybridization of primary cells from the lymphoid organ of Penaeus monodon with Sf9 cells led to the establishment of a continuously proliferating hybridoma (PmLyO-Sf9) (Anoop et al., 2021).
Against the above background, we set out to induce the proliferation of primary cell cultures of M. rosenbergii embryonic cells by using ectopic expression of the Ras oncogene via lentivirus transduction. To this end, we mined the Ras gene in both transcriptomic and genomic libraries of M. rosenbergii (MrRas). A mutant version of MrRas (MrRasV12) was incorporated into a plasmid that included the white spot syndrome virus (WSSV) immediate early 1 gene (IE1) promoter (Li et al., 2009). The plasmid was packaged, and M. rosenbergii primary cell cultures were transduced by the mutated lenti-MrRas.
2 Materials and methods
2.1 Bioinformatic analyses
To mine the Ras gene from the M. rosenbergii genome, data from two transcriptomic libraries and the M. rosenbergii phased genome (Levy et al., 2019) – all of which were established in our laboratory – were used. One of the transcriptomic libraries includes various embryonic developmental stages (Abayed et al., 2019), and the other includes larvae, post larvae, and specific adult tissues (Sharabi et al., 2016). Homologs were mined from the two M. rosenbergii libraries by a tBLASTn search using the Ras amino acid sequence from Drosophila melanogaster (NP_476699.1; NCBI). mRNA sequences were aligned to the transcriptomic libraries, and putative protein sequences were compared using various NCBI BLAST algorithms (accessed during March and April 2021, https://blast.ncbi.nlm.nih.gov/Blast.cgi).
The genomic structure of MrRas was analyzed in silico by identifying introns and exons in the above transcriptomic and genomic libraries (Sharabi et al., 2016; Abayed et al., 2019). Phylogenetic analysis across a variety of species was performed using multiple alignments of Ras amino acid sequences from the following species: Mus musculus (NP_659124.2), Xenopus tropicalis (NP_001107338.1), Scylla paramamosain (ACY_66377.1), Gammarus pulex (EH_272032.1), 5 Dead Sea & Arava Science Center, Central Arava Branch, Israel. Homarus americanus (XP_042242987.1), Paralithodes camtschaticus (AAN_86143.1), M. rosenbergii (OP_654177), C. quadricarinatus (AEL_23042.1), Procambarus clarkii (AFV_09848.1), Daphnia magna (XP_032787339.1), Fenneropenaeus chinensis (AHA_93923.1), Marsupenaeus japonicus (AAK_14389.1), Penaeus monodon (QBA_57436.1), and Litopenaeus vannamei (XP_027229439.1). M. musculus and X. tropicalis were considered as out groups, while the remaining species were crustaceans. Sequences were obtained from the NCBI database, and the tree was generated using PhyML 3.0.
2.2 Animals
M. rosenbergii prawns were grown to maturity in artificial ponds at Ben-Gurion University of the Negev, Beersheba, Israel, as described previously (Levy et al., 2016). Thereafter, to ensure a constant supply of embryos for primary embryonic cell isolation, gravid females were obtained by housing four to six mature females with a blue claw male in 500-L tanks. The tanks were checked daily for the typical male reproductive guarding behavior to identify fertilized females, as previously described (Molcho et al., 2022).
2.3 Isolation and culture of primary cells from M. rosenbergii
Since the eggs are held on the abdomen of the female attached to the swimming legs, a disinfection procedure was carried out for surface sterilization of female prawns carrying developed eggs prior to cell isolation; the procedure involved submerging the animals in an aqueous solution of methylene blue (3 ppm) overnight as previously described (Molcho et al., 2022). Approximately 14 days following fertilization, embryos with visible eyes were removed with forceps, washed with a sterile saline solution (420 mOsmol) containing antibiotics and an antifungal reagent, namely, penicillin-streptomycin solution (1 mg/ml, Cat. No 03-031-1B, Biological Industries, Israel), tetracycline (25 µg/ml, Cat. No. T-9823, Merck, USA), and voriconazole (0.5 µg/ml, Cat. No. PZ0005, Merck, USA)—referred to here as saline PSTV. The separated eggs were further disinfected in saline PSTV with formalin (1% Cat. No. 252549, Sigma Aldrich, USA) for 1 min and an iodophor (1 ppm, Polydine, Dr. Fischer, Israel) for 3 min and then rinsed again in saline PSTV for 10 min. The eggs were homogenized in saline PSTV containing a protease and phosphatase inhibitor cocktail (Halt™ Protease Inhibitor, Cat. No. 87785, ThermoFischer Scientific, USA) under aseptic conditions. The homogenate, which consisted of dispersed cells, whole embryo tissue, and yolk protein, was passed through a 70-μm cell strainer to separate the egg envelopes and non-broken eggs from the dispersed cells. The filtrate was then pelleted by centrifugation at 850 g for 6 min at 18°C, suitable condition for isolation of prawn cells (Molcho et al., 2022). The residue remaining in the cell strainer was transferred to Eppendorf tubes, each containing 1 mg/ml collagenase type I and 1 mg/ml collagenase IV (Cat. No. C0130 and C5138 respectively, Sigma Aldrich, USA) and incubated for 30 min at room temperature. Afterwards it was centrifuged as above, and the pellet was washed with medium containing 10% fetal bovine serum (Cat. No. 04-127-1A, European grade, heat inactivated, South American origin FBS, Biological Industries, Israel) and combined with filtrated cell pellet. The total cell pellet was resuspended in 2 ml of saline PSVT, slowly poured into a percoll (Cat. No. 17-0891-01, GE Healthcare Bio-Sciences AB, Sweden) gradient adjusted with crustacean saline for osmolarity (100%-1.062 g/ml, 50%- 0.565 g/ml, 25%-0.282 g/ml and 12.5%-0.141 g/ml) and centrifuged again at 1000 g for 15 min at 18°C with reduced deceleration. The percoll gradient was used to separate the different embryonic cell types from the bulky yolk proteins (early embryonic nutrition). The yolk proteins appeared at 12.5% percoll while the cells migrated downwards the gradient and spread between 12.5% to 50% percoll (see Figure S1). All cell types were pooled and used for culture, transferred to a new 15 ml tube, centrifuged, and washed from percoll with upto 10 ml of saline PSVT. The cells obtained were reconstituted in 1 ml Optimem media with PSVT (Cat. No. 31985-047, Gibco, USA) The cells were visualized by trypan blue exclusion (Cat. No. 03-102-1B, Biological Industries, Israel) and counted with a hemocytometer. Cells were seeded at a concentration of 300,000 per well in a 96-well plate (Cat. No. 30096, SPL Life Sciences, South Korea) and cultured in DMEM: F12 (Cat. No. 01-170-1A, Biological Industries, Israel) supplemented with 10% FBS and PSVT (referred to below as the ‘growth medium’); the osmolarity of all commercial media used were adjusted to 420 mOsm with crustacean physiological saline (Frerichs, 1996). Culture plates were incubated at 28 °C with 5% CO2.
2.4 MrRas - pHAGE2 plasmid preparation
The full open reading frame (ORF) sequence of MrRas was amplified from cDNA previously prepared in our laboratory. For the insertion of the point mutation in the MrRas gene, guanine was replaced by thymine in the primer design in order to induce the desired change in the amino acid sequence at the 12th position from glycine to valine. We used the following forward primer F: ATGACGGAGTACAAGCTGGTGGTGGTGGGAGCGGTAGGCGT (where T indicates the replacement of guanine with thymine to induce a point mutation in the resulting PCR product). The reverse primer was designed to amplify the full MrRas ORF, including the stop codon, R: CTACAATATAATACACGGTCTTTTGGAC. The PCR product (561 bp) was ligated into the pGEM®-T Easy Vector Systems (Cat. No. A1360, Promega, USA) according to the manufacturer’s instructions. After cloning, mutated MrRas was sequenced using a MrRas reverse primer to validate the mutation. Mutated MrRas was cut from the pGEM®-T Easy vector and ligated into pHAGE2 (Mostoslavsky et al., 2005) to replace DsRed. The result was a novel plasmid consisting of the mutated MrRas mRNA sequence and the ZsGreen sequence under the WSSV IE1 promoter (Li et al., 2009); hereafter referred to as the ‘mutated MrRas plasmid’. This generated lentiviral plasmid, encoding the Zs Green protein under WSSV IE1 promoter, enabled delivery of the mutated MrRas gene into the genome of the cultured M. rosenbergii cells. A control pHAGE2 lentiviral plasmid encoding for ZsGreen and DsRed under the WSSV promoter was also generated, as described before (Rotem-Dai et al., 2021).
2.5 Lentivirus production using HEK 293T cells
Lentiviruses were produced in HEK293T cells cultured in DMEM (Cat. No. 01-055-1A, Biological Industries, Israel) supplemented with 10% FBS Biological Industries (Cat. No. 04-127-1A, European grade, heat inactivated, South American origin FBS, Biological Industries, Israel). Cells were grown to 90% confluence in 10-cm plates and then transfected with 10 µg of pHAGE2 vector and 3 µg of the packaging plasmids tat, rev, hgpm2 and vsvg, in a mass ratio of 1:1:1:2. The transfection was performed using jetPRIME® transfection reagent (Cat. No. 101000046, Polyplus, France) according to the manufacturer’s instructions. DMEM supplemented with 10% FBS was added for the following two days of culture, and on the third day, the culture medium containing the secreted lentiviruses was collected, filtered through a 0.45-µm filter, and either stored at −80 °C or concentrated by ultracentrifugation at 17,000 RPM for 90 min at 4 °C. Lentivirus pellets were suspended in DMEM supplemented with 10% FBS, aliquoted, and stored at −80 °C.
2.6 Incorporation of mutated lenti-MrRas into the genome of embryonic cells in primary culture
Embryonic cells seeded at a concentration of 300,000 cells per well in a 96-well plate were transduced with either 20 µl of mutated lenti-MrRas or lenti WSSV IE1 as the positive control (PC). Non-transduced negative control cells were grown in the same volume of growth medium. Forty-eight hours following transduction, 0.1 ml of growth medium was added. Seventy-two hours following transduction, a Beckman Coulter® Gallios™ flow cytometer (CytoFLEX S, USA) was used to measure the transduction rate of the lentivirus according to the expression of the reporter genes (DsRed or ZsGreen), followed by confocal microscopy (OLYMPUS FV10-268-098, Japan) analysis to detect the expression of fluorescent proteins. The presence of lentiviral sequences was also confirmed using PCR analysis.
2.7 Genomic DNA preparation, RNA isolation and PCR analysis
To confirm the transduction of mutated lenti-MrRas into the genome of treated embryonic cells, genomic DNA (gDNA) was isolated by pelleting the cells in 20 µl 0.2 N NaOH and heating for 70°C for 20 min in a Thermocycler Life Eco (Falc) (Molcho et al., 2022), and RNA was extracted using EZ_RNA kit (Cat. No. 20-410-100, Biological Industries, Israel). RNA was converted into cDNA by following the protocol of the qScript cDNA synthesis kit (Cat. No. 95047-100, Quantabio, USA). PCR was performed using MrRas primers (MrRas F: ATGACGGAGTATAAGCTGGTG, and MrRas R: CTACAATATAATACACGGTCTTTTGGAC, with a melting temperature of 57°C) using gDNA or cDNA as templates. PCR products from cDNA samples taken from control and mutated lenti-MrRas transduced cells were further validated by Sanger sequencing (Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University).
2.8 Confocal microscopy
Successful integration of mutated lenti-MrRas into the cell genome was validated by confocal microscopy showing the accumulation of ZsGreen reporter proteins localized inside the cytoplasm. Three days following transduction of M. rosenbergii primary embryonic cell cultures by the lentivirus, cells were transferred onto Fisherbrand™ SuperFrost Plus slides (Cat. No. J1800AMNZ, Fisher scientific, USA), fixed with 4% para-formaldehyde (Cat. No. 15710, ThermoFischer Scientific, USA) and stained with 4′,6-diamidino-2-phenylindole (DAPI) as a nucleic acid stain (Cat. No. 0100-20, DAPI Fluoromount-G® mounting medium, Southern Biotech). The slides were observed under an Olympus FV1000 laser confocal scanning biological microscope (OLYMPUS FV10-268-098, Japan) for fluorescent signals (DsRed and ZsGreen), and the signals were compared to those of DAPI-stained non-transfected control cells. Uninfected control cells were used as a reference for auto fluorescent cell signaling.
2.9 Fluorescence-activated cell sorting (FACS) analysis
For FACS analysis, cells were collected into tubes four days following transduction of M. rosenbergii embryonic primary cell cultures by lentiviruses, centrifuged at 850 g for 6 min and washed twice with saline. The cells were analyzed using a Beckman Coulter Gallios flow cytometer (CytoFLEX S, USA). The flow cytometry results were displayed using CyteExpert (CytExpert v2.5). Initially, the forward vs. side scatter gating of the cells was performed to separate the dead cells and debris from live cells by gating (Figure 6A). The ZsGreen protein was analyzed using two dimensional gates with forward scatter against FITC channel. This protocol enables the evaluation of the rate of transduction of mutated lenti-MrRas into the genome of embryonic cells.
2.10 Statistical analysis
Chi-square contingency analysis performed on proliferation rate data. Analysis performed using R software. p<0.01 considered statistically different.
3 Results
3.1 Mining of MrRas and the in-silico temporal and spatial patterns of MrRas
Using the Ras oncogene sequence from D. melanogaster, we identified a contig showing 84% similarity. Aligning the two translated and aligned sequences showed a high similarity between the D. melanogaster and the identified M. rosenbergii sequence was updated in the NCBI database (Accession No. OP_654177, Figure 1A). The expression pattern of MrRas in the embryo development library showed very high expression from day 1 to 17 of embryonic development (Figure 1B).
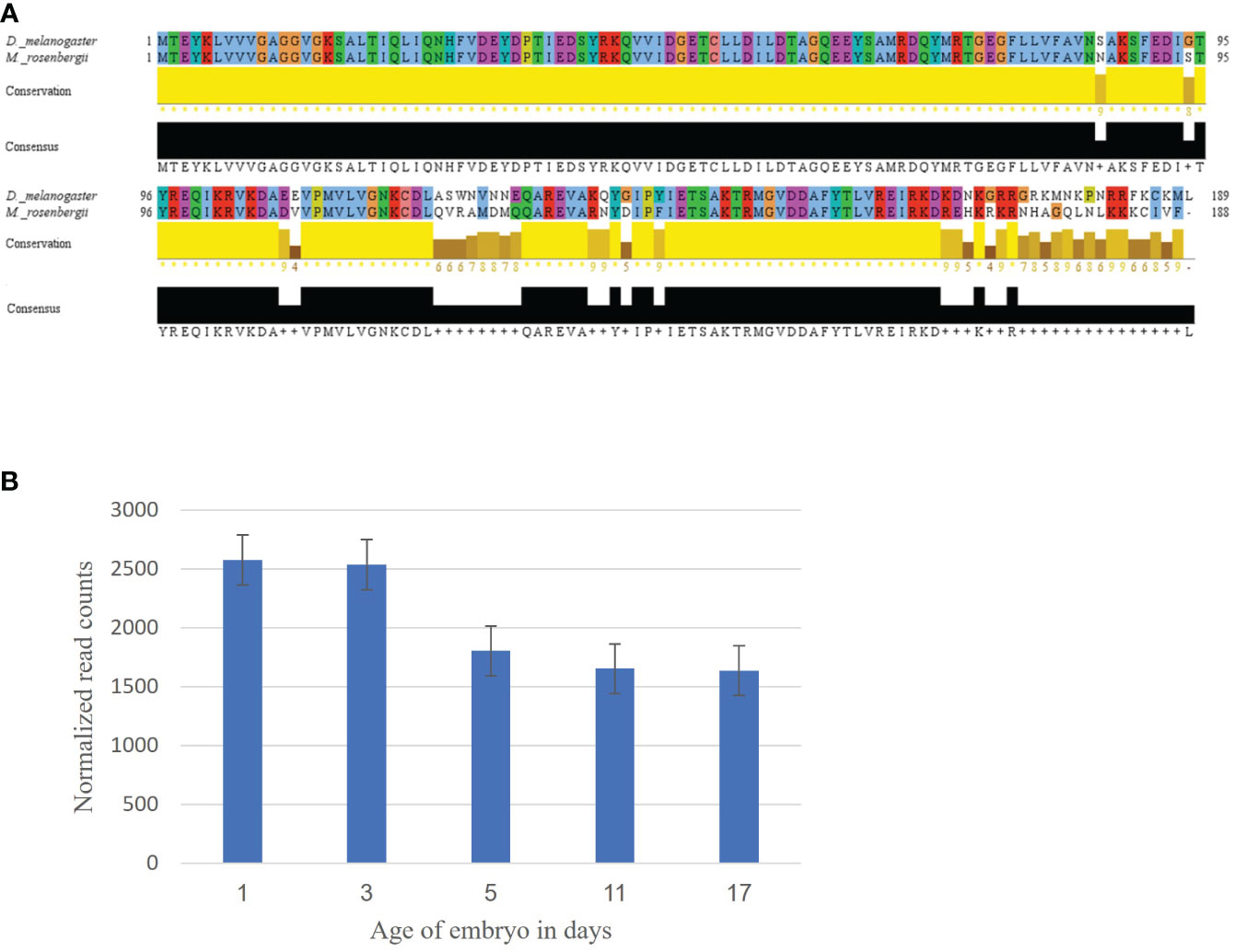
Figure 1 Mining of the Ras gene in M. rosenbergii. (A) Alignment of the Ras amino acid sequence from D. melanogaster with a putative M. rosenbergii Ras based on transcriptomic libraries. High conservation of the protein sequence alignment was found using Jalview (with default settings). For each position in the sequence alignment, the conservation rate and the consensus sequence are shown below. (B) MrRas expression pattern in embryos of different ages. The x-axis represents embryo development days, and the y-axis, normalized read counts. The error bars represent standard deviation.
3.2 MrRas genomic structure and phylogenetic analysis of amino acid sequences
According to the M. rosenbergii genome (Levy et al., 2019), the MrRas gene spans 15,308 bp and includes 5 introns and 6 exons (Supplementary Figure 2). The longest MrRas intron spans 8,032 bp, and the shortest, 196 bp. The longest and shortest exons span 180 bp and 51 bp, respectively. The main ORF spans 736 bp with the start codon, ATG, in the first exon at its 150th bp and the stop codon, TAG, in the last exon at its 15,278th bp (Supplementary Figure 2).
A phylogenetic tree based on the Ras amino acid sequences of 14 species (including 12 crustacean species) shows that MrRas bears the closest similarity to Paralithodes camtschaticus (the red king crab) (Figure 2). The phylogenetic analysis shows that Ras is highly conserved among vertebrates and crustaceans, and based on the outgroups, we can predict that Ras from different phyla has a common evolutionary origin (Figure 2).
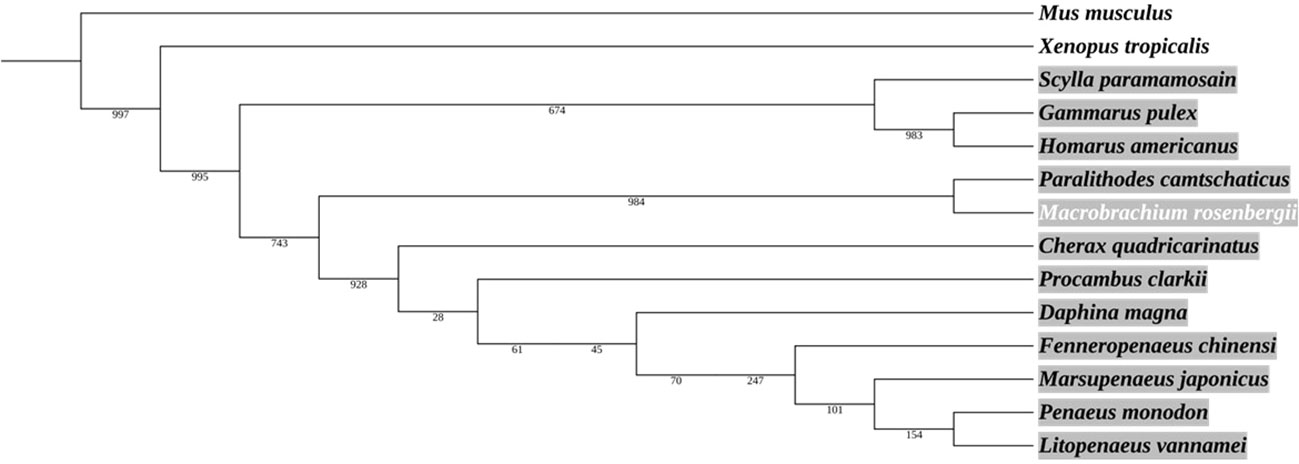
Figure 2 Phylogenetic analysis of Ras protein in crustaceans, including M. rosenbergii. The area marked in grey represents the crustaceans. The neighbor-joining tree was constructed according to the alignment of the entire Ras amino acid sequence. The numbers alongside the branches represent bootstrap values. The rodent Mus musculus and the amphibian Xenopus tropicalis served as outgroups.
3.3 WSSV IE1 - pHAGE2 lentiviral plasmid preparation
The constructed plasmid suitable for the M. rosenbergii cellular system the EF1α promoter of Homo sapiens was replaced by the WSSV IE1 promoter thereby producing a plasmid of 8259 bp (Figure 3A), which served as a positive control. For the mutated Ras gene, DsRed from the control plasmid was replaced by the sequence of mutated MrRas (561 bp), producing a plasmid of 8146 bp (Figure 3B). The plasmids were each packaged into a lentivirus for transduction into the host genome of the M. rosenbergii embryonic primary cell culture.
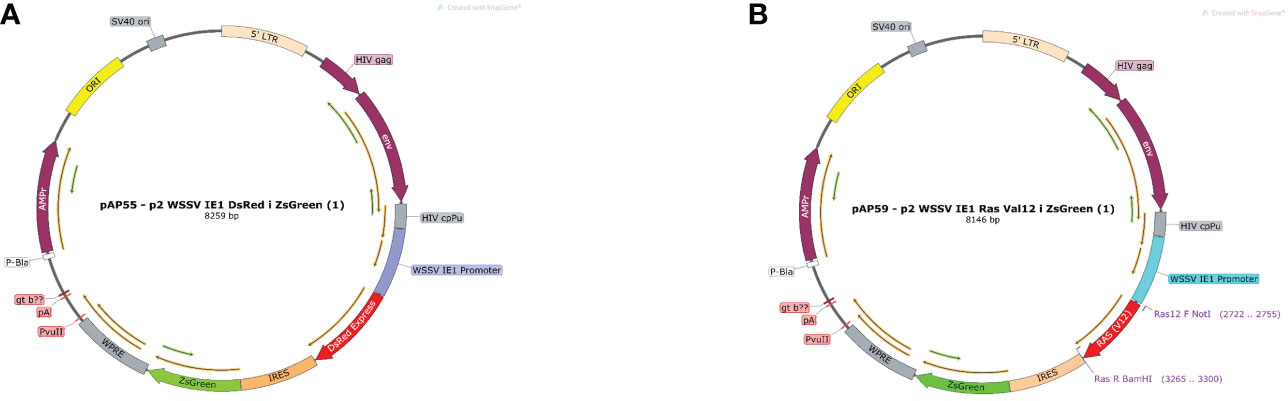
Figure 3 Plasmid map of pAP55 and pAP59. (A) Positive control plasmid with pAP 55 WSSV IE 1 promoter followed by two reporter genes DsRed and ZsGreen, indicated in red and green, respectively. (B) Plasmid pAP59; mutated MrRas is indicated in red, and the reporter gene ZsGreen, in green.
3.4 Lentiviral integration of mutated MrRas into an M. rosenbergii embryonic primary cell culture
To confirm transduction of the mutated lenti-MrRas into the genome of the transduced embryonic cells, RNA was extracted from both the control and treated cells 72 h after transduction. PCR amplifications were conducted with cDNA obtained from 1 μg RNA from each sample; the expression of the native MrRas, marked as control, and the mutated lenti-MrRas are shown in Figure 4A. Cells transduced with the mutated lenti-MrRas showed a prominent band in the gel compared to the untreated control, while the expression of MrActin (positive control) was similar in both control and treated cells. The control and mutated lenti MrRas were sequenced from the PCR product corresponding to the presented gel. were further confirmed by sequencing of the PCR products; the analysis, conducted in three repetitions, is presented in Figure 4B. In the PCR of the embryonic cells expressing mutated MrRas, the sequence shows T (thymine) instead of G (guanine, as is present in the untreated control cells expressing native MrRas). The putative deduced amino acid of each of three nucleotide codons is presented below the nucleotide alignments besides the mutated codon (Figure 4B).
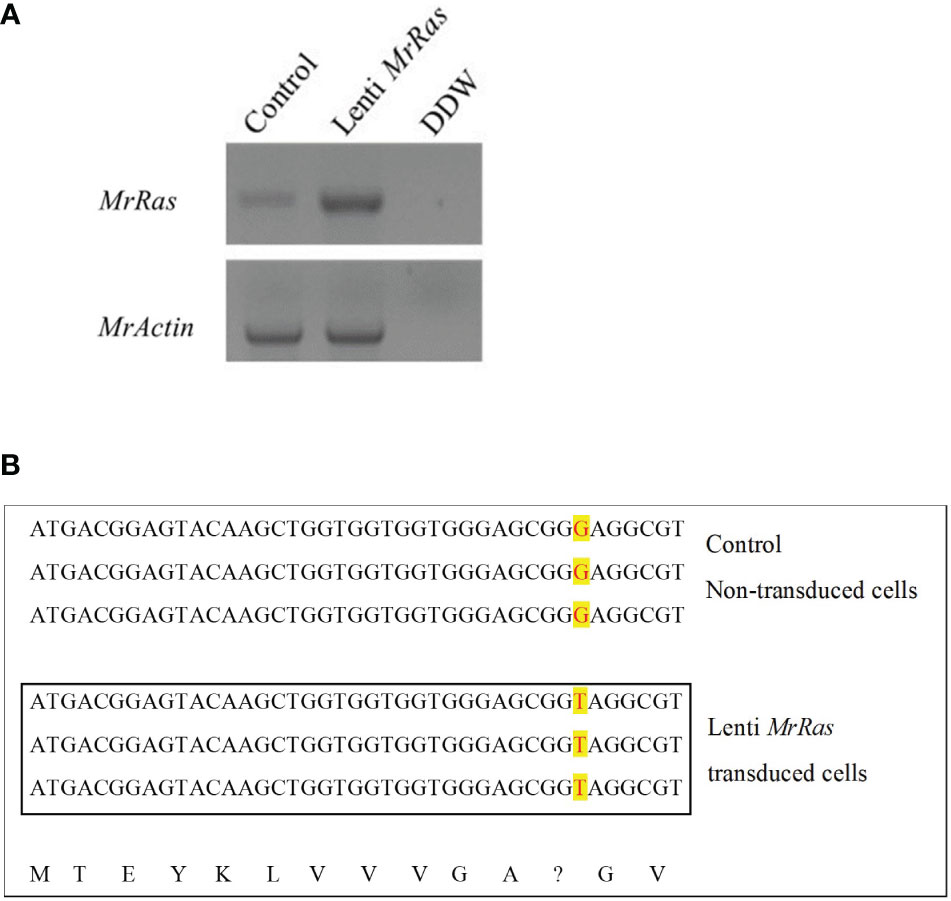
Figure 4 Integration of mutated lenti-MrRas into M. rosenbergii embryonic primary cell culture. (A) Gel of mutated Ras expression in transduced cells in culture (lenti-MrRas, top A). RNA extractions were performed after 18 days in culture. Control – non-transduced M. rosenbergii cells; DDW - doubly distilled water. MrActin served as the positive control (bottom). (B) Sanger sequencing of RNA isolated from control (top) and mutated lenti MrRas transduced cells (in box); the three lines in each box are biological replicates. The change in nucleotide from guanine to thymine may be seen in the mutated lenti-MrRas transduced cells compared to control cells (red over yellow background). The deduced amino acids from the sequences are presented in the bottom line. A question mark indicates the point mutation.
3.5 Expression of lentiviral particles in an M. rosenbergii embryonic primary cell culture
Full integration and constant expression of the mutated lenti-MrRas occurred three days post transduction as shown by (Rotem-Dai et al., 2021). In the present study, the expression of the reporter genes was validated using confocal microscopy. In the untreated control cells (Figures 5A-C) only staining with the DAPI nuclear counter stain could be observed (Figure 5A). The primary cell cultures transduced with the positive control, lenti WSSV IE1, expressed both reporter genes, DsRed and ZsGreen, as may be seen by the presence of both colors in the vicinity of the nuclei and cytoplasm (Figures 5D-F). The mutated lenti-MrRas transduced cells expressed only the ZsGreen fluorescent protein, as expected, since the mutated MrRas gene replaced the DsRed in the plasmid (Figure 5G–I). The presence of the reporter gene proteins in the cells three days post transduction indicated that successful transduction has occurred. Expression of mutated lenti-MrRas suggests successful integration and expression of mutated MrRas into the embryonic cell genome (Uchida et al., 2016).
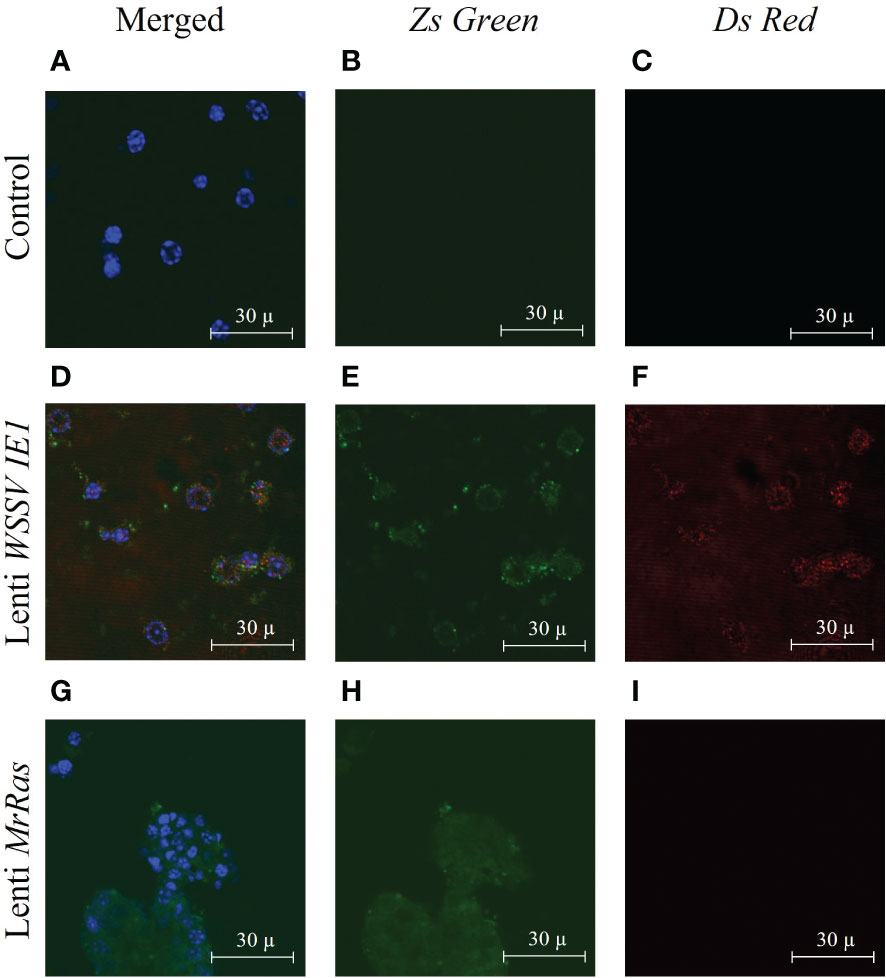
Figure 5 Expression of lentiviral particles in M. rosenbergii embryonic primary cell culture three days after transduction, as seen by confocal microscopy: (A-C) Representative images of control non-transduced cells, (D-F) Lenti WSSV IE1 transduced cells, and (G-I) mutated lenti MrRas transduced cells. Cultures were examined by confocal microscopy (using the appropriate lasers, 488 nm for ZsGreen and 546 nm for DsRed). The ‘Merged’ panel (A, D, G) on the left of the Figure shows additional nucleic acid staining performed with DAPI (and visualized with a 405 nm laser). Scale bar = 30 µm.
3.6 Transduction efficiency of mutated lenti-Mr Ras in primary culture of embryonic cells
Transfection efficiency was evaluated by flow cytometry (Figure 6) Forward vs. side scatter of the cells was performed to separate the dead cells and debris from live cells by gating (Figure 6A). The untreated cells demonstrated a 9.1% signal-positive population considered as auto-fluorescence and was thus taken as a background value (Figure 6B). According to the analysis of the reporter gene ZsGreen, the control plasmid WSSV IE 1 lentivirus showed 52% transduction (Figure 6C), as can be seen in the elevated FITC-axis in the two-dimensional FACS analysis. This finding indicates high transduction efficiency of the lentiviral vector, resulting in lentiviral reporter protein expression in M. rosenbergii embryonic primary cells. In cells transduced with mutated lenti-MrRas, the transduction rate (32%) was higher than that of the background value but lower than that of the positive control (Figure 6D). A specific subpopulation comprising 2.17% of the total events was equally transduced by both lentiviruses (Figure 7A, right-hand corner) and showed higher positive signals for FITC, whereas the untreated control cells were 14% positive for FITC (Figure 7B). The subpopulation cells treated with the positive control plasmid WSSV IE1 were 98% positive for FITC (Figure 7C), and those treated with mutated lenti-MrRas were 94% positive for FITC (Figure 7D). When the results for the control and mutated lenti-MrRas were overlaid to show the frameshift, it was evident that mutated lenti-MrRas showed a higher level of positive FITC signals compared to the control, which was due to autofluorescence. Figure 7E shows the frameshift towards the right in cells transduced with mutated lenti-MrRas, implying success in transduction.
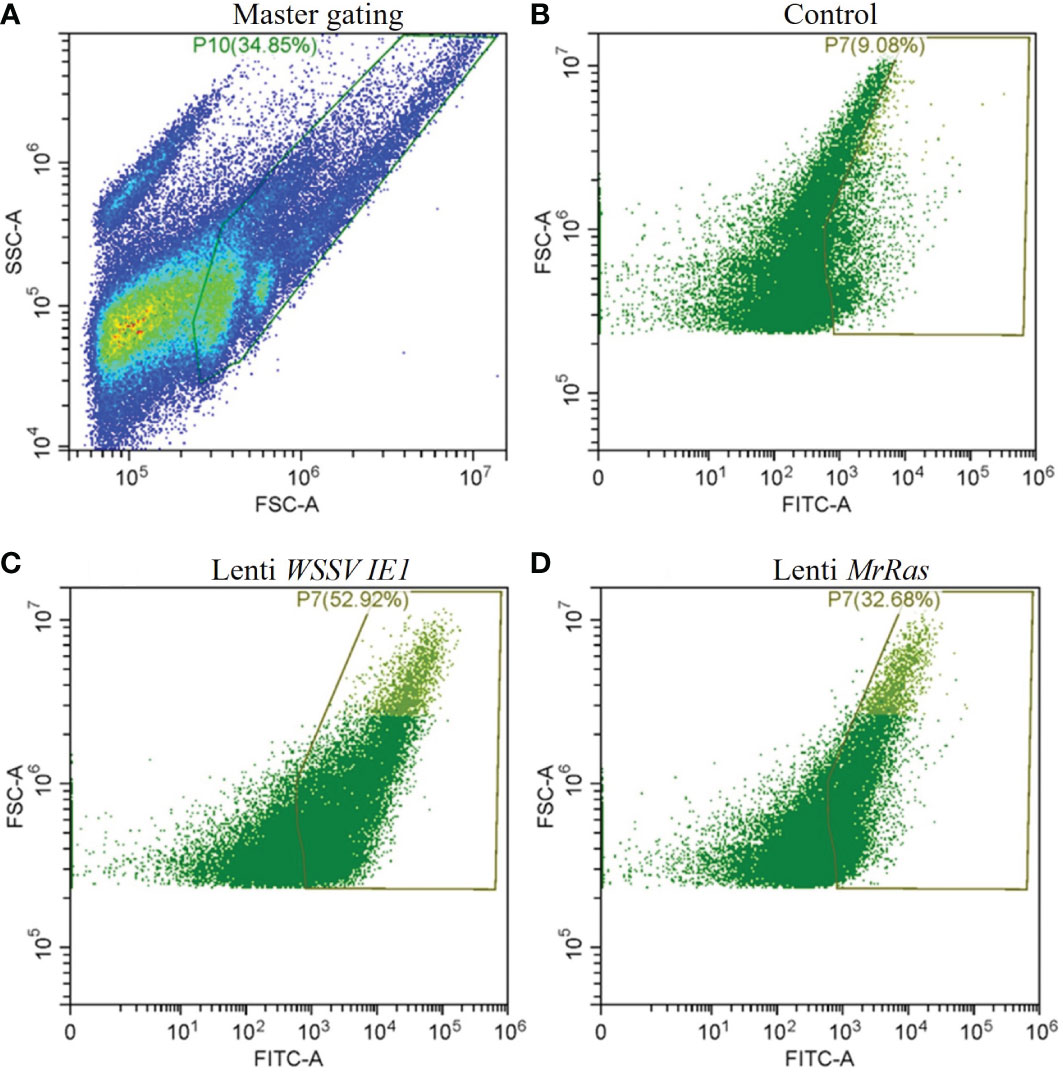
Figure 6 Transduction efficiency of the lentivirus platform in M. rosenbergii embryonic cells in primary culture analyzed via two-dimensional FACS analysis. (A) Gating strategies for primary M. rosenbergii embryonic cells. The primary scatter plot shows the entire events analyzed, gate consisting of live cells excluding debris. (B) Control cells express innate autofluorescence in FITC channel, and gates were based on a minimum threshold. (C) FITC signal of the positive control plasmid lenti WSSV IE1 transduced cells. (D) FITC signal of mutated lenti-MrRas transduced cells.
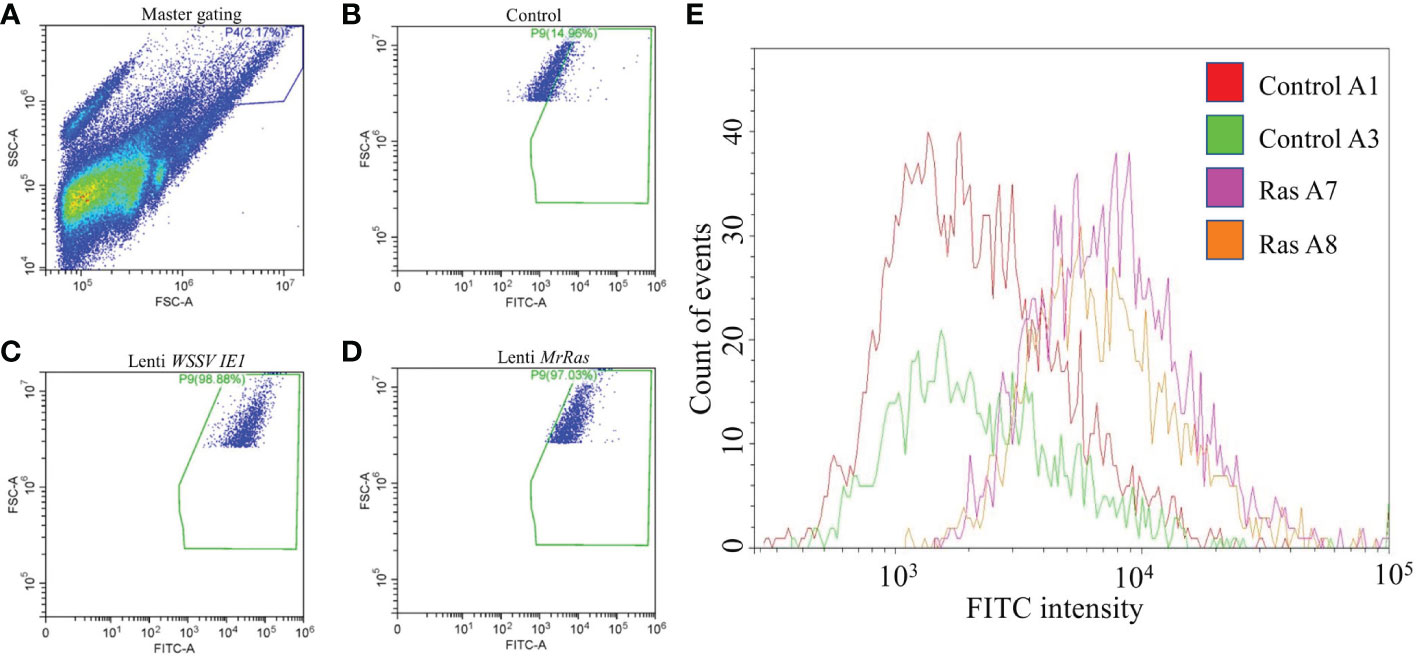
Figure 7 Depiction of a sub-population of cells exhibiting a higher rate of transduction. (A) The primary scatter plot shows entire events analyzed in control cells, gating on a specific population based on granularity and larger cell size. (B) FITC signal of the control cells expressed innate autofluorescence. (C) FITC signal of the positive control plasmid lenti WSSV IE1 transduced cells. (D) FITC signal of the mutated lenti MrRas transduced cells. (E) The traces for the positively transduced cells for FITC (purple and orange traces) were overlaid over the traces for the control (red and green).
3.7 The proliferation of M. rosenbergii embryonic primary cell cultures transduced with mutated lenti-MrRas
Cell transduced with mutated lenti-MrRas embryonic cells, and their respective controls were cultured for prolonged periods, i.e., up to 18 days. The culture represents the entire cellular population within the embryo. In the present culture conditions, only a few types of cells proliferate while others become quiescent. Higher proliferation rate is attributed to specific cell types within the culture and not the entire culture. This might reflect the time needed for the culture to fill the plates. Following seeding, the cells proliferated and filled the 96-well plates after 9 days in culture; thereafter, they were passaged into new plates and allowed to grow for another 9 days, at which stage they were designated either ‘control passage’ or ‘Ras passage’. The morphology of seeded embryonic primary cell cultures on the day of seeding is presented in Figure 8A (left); the morphologies and cell densities of the control culture and the mutated-lenti-MrRas-treated culture at the end of the experiment on day 18 are shown in the middle and right panels, respectively, of Figure 8A. The insertion of mutated lenti-MrRas into the genome of the cultured cells was detected only in the genomic DNA samples of the transduced cells, while the housekeeping gene MrActin was detected in both transduced and non-transduced control samples, confirming its presence in the cells also after a passage (Figure 8B). The control cultures reached 700% of their seeding density by the end of the experiment (Figure 8C). The novel mutated lenti-MrRas plasmid enhanced the natural proliferation index of the embryonic primary cell cultures in comparison to the control cultures (Chi-square test for association value 38,591.193, df=1, p<0.001): The transfected cells reached almost 1200% of their seeding density by the end of experiment 1, i.e., approximately double that of the control (Figure 8C). A replicate experiment showed the same trend, with increment in the density of the treated cultured was approximately 1.7-fold higher than that of the control (Figure 8C, Chi-square test for association value 30,456.877, df=1, p<0.001). Importantly, we did succeed in enhancing the proliferation potential of the embryonic primary cell cultures by shortening the doubling time of the culture by 22.5% in both experiments (detailed calculation is presented in the supplemental materials). However, none of our cultures reached a steady immortalized state.
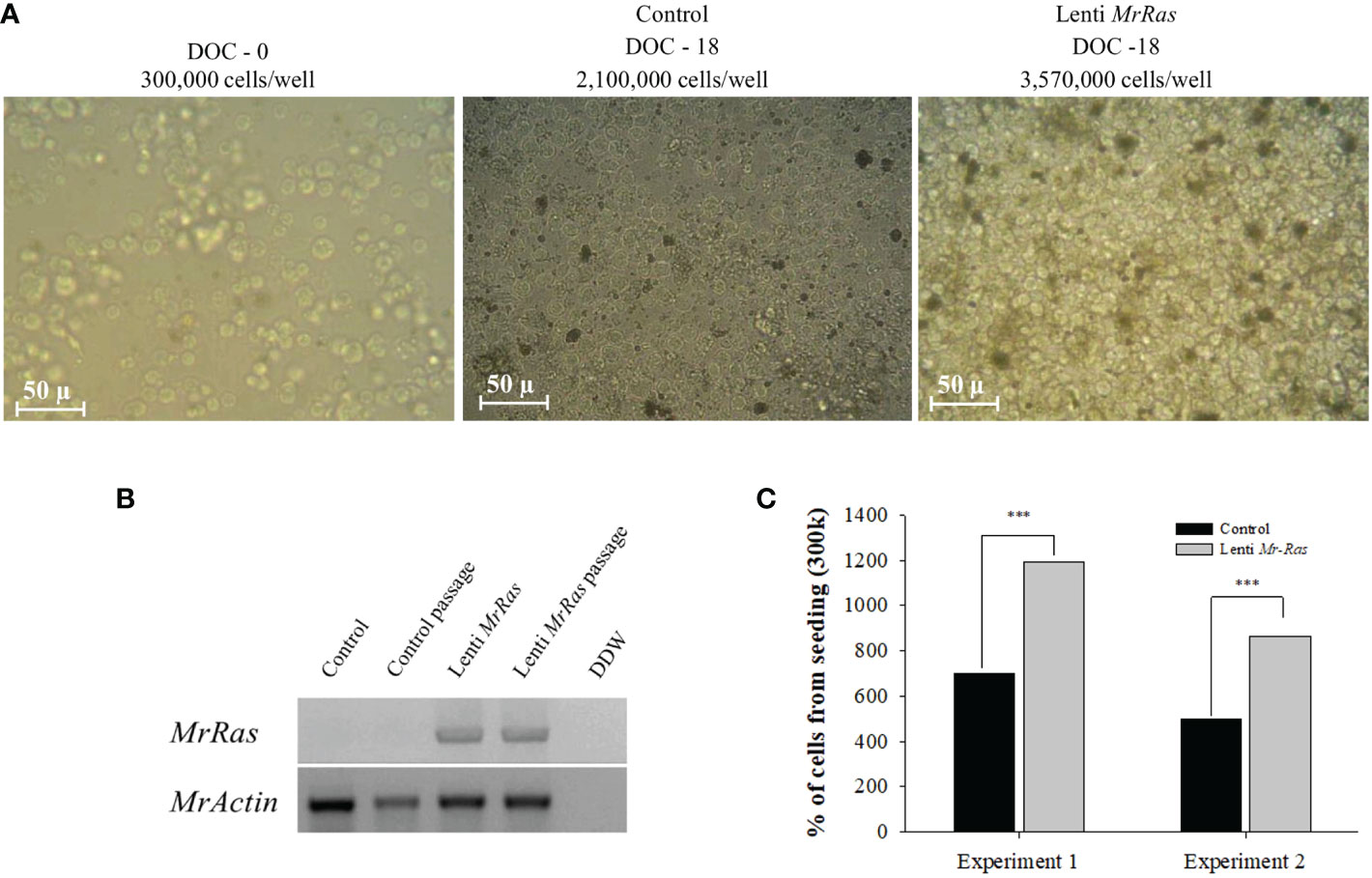
Figure 8 Proliferation in M. rosenbergii embryonic cell cultures and detection of the MrRas sequence in the genome of the transduced cells. (A) Representative light microscope images of cells seeded at a density of 300K cells/well (A, left); after 18 days in culture, control non-transduced cells (A, middle); and mutated lenti MrRas transduced cells (A, right). DOC = days of culture, scale bar = 50 µm. (B) Agarose gel of PCR products on gDNA template isolated from culture after 18 days in culture to confirm the presence of the genes in M. rosenbergii primary embryonic cells, including cells after one passage. (C) Percent of cells from seeding density in mutated lenti-MrRas transduced cells compared to control non-transduced cells. Statistical analysis was done using Chi-square contingency analysis performed on the proliferation rate data. Analysis performed using R software. *** refers to the statistical significant p value.
4 Discussion
The lack of an immortalized cell line constitutes a major obstacle to developing seafood cell agriculture and to conducting research on viral diseases in cultured crustaceans. Despite various attempts to immortalize crustacean primary cell cultures, there are no reports of success in the literature. In an attempt to promote proliferation and hence immortalization of M. rosenbergii primary embryonic cell cultures, we exploited a lentiviral platform using the WSSV promotor [previously developed for prawn cells in our laboratory (Rotem-Dai et al., 2021) for introducing the MrRas oncogene into the genome of these cell cultures. The rationale underlying this choice of experimental system for M. rosenbergii primary embryonic cell cultures was based on the induction of proliferation in mammalian cells using the oncogene RasV12 (Kumazaki et al., 2020) and in primary cells isolated from D. melanogaster (Dequéant et al., 2015). Support for this choice was obtained by the identification of Ras in M. rosenbergii and by an in-silico search that revealed MrRas expression in M. rosenbergii at early stages of embryonic development when cell proliferation is rapid, suggesting the possible involvement of MrRas in basic cellular functions at early developmental stages. Since Ras has a very important function in cell integrity and in various cellular functions, it is a highly conserved gene, being present in various species from yeasts to humans (Santos and Angel, 1989; Rotchell et al., 2001). Importantly, it has previously been identified in other decapod crustaceans, such as Procambarus clarkii and Penaeus japonicus (Huang and Chuang, 1998; Li et al., 2019; Liu et al., 2021). As in other species, the M. rosenbergii putative Ras amino acid sequence is also highly conserved and shares common ancestry with other decapod crustacean species as well as amphibians and rodents.
The experimental system used in the present study comprised an embryonic primary cell culture of developed embryos (at the10-12 days stage), which thus contained differentiated cells with native proliferative potential from all germ cell layers, including both dividing and non-dividing cells (Paris et al., 2021). On these cells, we tested the platform recently established in our laboratory by Rotem-Dai (Rotem-Dai et al., 2021), which was used for delivering foreign DNA to hematopoietic primary crustacean cell cultures. Due the ability of lentiviruses to transduce exogenic DNA in both dividing and non-dividing cells, this platform proved to be an efficient tool for the stable expression of the MrRas gene and its integration into the genome of M. rosenbergii embryonic primary cell cultures.
The plasmid used for transduction was designed to include a highly active decapod-specific viral promoter WSSV IE1, which can be transcribed by various transcription factors available in the recipient cells (Kao et al., 2020; Rotem-Dai et al., 2021). Since the lenti-WSSV IE1, which served as a positive control for the M. rosenbergii primary embryonic cell culture, had a similar 43.8% transduction rate to primary hematopoietic M. rosenbergii cells (Rotem-Dai et al., 2021), we concluded that this platform would also be efficient for the present system. We thus aimed to ectopically express mutated MrRas using our new stablished vector under the WSSV IE1 promotor in M. rosenbergii primary embryonic cell cultures. However, the quantitative data from FACS analysis showed that the total population of the embryonic primary cell culture was less susceptible to this mutated lenti-MrRas (23.6%) than to the lenti-WSSV IE1 (at 43.8%). Nonetheless, a specific subpopulation comprising 2.17% of the total primary embryonic cells was equally transduced by both viruses (mutated lenti-MrRas and lenti-WSSV IE1). The characteristics of this specific population could suggest a humoral system origin, due to features such as granulation and cell size, which are associated with immune cells in invertebrates. Previous studies have indeed shown subpopulations of cells containing granules, probably from the hematopoietic line of the heterogenous population of embryonic cells (Vadakke-Madathil et al., 2019). We note that the crustacean hematopoietic lineage consists primarily of semi-granular and granular cells (Söderhäll, 2016). The present study thus calls for further investigation of the different subpopulations in M. rosenbergii embryonic primary cell cultures, particularly their identification and proliferation rates.
The lentivirus was very successful in delivering the mutated MrRas into the genome, based on its ectopic expression of reporter genes, which was confirmed by sequencing. Nonetheless, despite the enhanced proliferation of cells in the culture (by 1.7-2 folds) and the shortening of the doubling time (by 22.5%), we were not successful in producing an immortalized M. rosenbergii cell line. The results did, however, show a significant improvement in the longevity of the cells due to the ectopic expression of the mutated Ras protein. The next steps to be taken in the development of such a culture will be to test cell viability after storing. There are grounds to believe that the percentage of viable cells will be on the high side, since heart explant cultures established from 2-month-old M. rosenbergii prawns exhibited 60% viability after storage in liquid nitrogen for one month (Goswami et al., 2010).
Comparable attempts to immortalize primary crustacean cells were recently reported by Anoop and his colleagues, who used a baculovirus to transduce Penaeus monodon primary lymphoid tissue with H-Ras V12 (Anoop et al., 2021). The transduction was successful, but it did not lead to proliferation of the cells. The reason could have been the age of the cells at the time of transfection, with age playing a key role in transduction and proliferation, and/or the addition of 5 mM sodium butyrate to the medium to enhance transgene expression (Anoop et al., 2021). Another attempt recorded in the literature involved the transduction of HPV E-6 and E-7 genes into C. quadricarinatus cells by lipofection, but, similarly to our case, immortalization was not achieved. The authors suggested that this was due to a lack of telomere maintenance (Claydon and Owens, 2008).
The present study has made an important step towards the immortalization of a cell line from M. rosenbergii primary embryonic cell cultures. Based on this study and on previous trials, we suggest that a multigenic approach should be adopted in future attempts to produce immortalized crustacean cell lines (Takahashi and Yamanaka, 2006). For instance, based on various cell cycle progression pathways and tumor formation, the use of RasV12 together with Myc could lead to success, since the Ras and Myc genes are closely related and share a similar downstream target in specific pathways (Rosalie et al., 1999). Our future work will seek to use multiplex platforms to obtain an immortalized M. rosenbergii cell line that could serve as a starter for a cellular crustacean aquaculture industry and could also be exploited in the viral diagnostic studies needed for crustacean aquaculture.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
GS wrote the article. SW, AS, and GS contributed to conception and design of the study. NR-D and RM design of the plasmid. OS, YG, and RG contributed to the lentivirus preparation. OG and BR supported with FACS analysis. EA contributed to cells images and figure design. RO consulted for cell culture. All participated in the review process. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the ministry of science and technology, Israel-Taiwan collaboration 00019994 and by ISRAEL SCIENCE FOUNDATION within the ISF-NSFC joint research program framework (Grant No. 2368/18).
Acknowledgments
We would like to thank Enzootic Ltd. and Colors Farm for the supply of embryos for the study. We would like to thank Prof. Ofer Ovadia, Dept. Life Sciences, BGU, for performing the statistical analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1100971/full#supplementary-material
Supplementary Figure 1 | Percoll density gradient of 100% (2 ml), 50% (3 ml), 25% (3 ml), 12.5% (2 ml) showing cells separated along the gradient, from yolk proteins on the upper fraction and debris and eggshell at the bottom of the lower fraction.
Supplementary Figure 2 | The structure of the MrRas gene. The total length of the MrRas gene is 15,308 bp. The blue horizontal bar indicates introns (size marked on top), and the yellow vertical bars indicate exons (size marked below). The start codon is indicated in a green balloon, and the stop codon is indicated in a red balloon.
References
Abayed F. A. A., Manor R., Aflalo E. D., Sagi A. (2019). Screening for dmrt genes from embryo to mature Macrobrachium rosenbergii prawns. Gen. Comp. Endocrinol. 282, 113205. doi: 10.1016/j.ygcen.2019.06.009
Anoop B. S., Puthumana J., Sukumaran V., Vazhappilly C. G., Kombiyil S., Philip R., et al. (2021). A novel approach of transducing recombinant baculovirus into primary lymphoid cells of penaeus monodon for developing continuous cell line. Mar. Biotechnol. 23 (4), 517–528. doi: 10.1007/s10126-021-10043-6
Claydon K., Owens L. (2008). Attempts at immortalization of crustacean primary cell cultures using human cancer genes. In Vitro Cell. Dev. Biol. Anim. 44 (10), 451–457. doi: 10.1007/s11626-008-9141-x
Dequéant M., Fagegaltier D., Hu Y., Spirohn K., Simcox A., Hannon G. J., et al. (2015). Discovery of progenitor cell signatures by time-series synexpression analysis during Drosophila embryonic cell immortalization. Pro. Ntl. Acm. Sci. 112 (42), 12974–12979. doi: 10.1073/pnas.1517729112
Downward J. (1997). Cell cycle: routine role for ras. Curr. Biol. 7 (4), R258–R260. doi: 10.1016/s0960-9822(06)00116-3
FAO (2020). “The state of world fisheries and aquaculture (SOFIA),” in Sus. Rome. Rome, Italy. doi: 10.4060/ca9229en
Foster D. A., Yellen P., Xu L., Saqcena M. (2010). Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1 (11), 1124–1131. doi: 10.1177/1947601910392989
Frerichs G. N. (1996). In vitro culture of embryonic cells from the freshwater prawn Macrobrachium rosenbergii. Aquaculture 143 (3), 227–232. doi: 10.1016/0044-8486(96)01281-1
Gimple R. C., Wang X. (2019). RAS: Striking at the core of the oncogenic circuitry. Front. Oncol. 9. doi: 10.3389/fonc.2019.00965
Goswami M., Lakra W. S., Rajaswaminathan T., Rathore G. (2010). Development of cell culture system from the giant freshwater prawn Macrobrachium rosenbergii (de man). Mol. Biol. Rep. 37 (4), 2043–2048. doi: 10.1007/s11033-009-9659-3
Huang C. F., Chuang N. (1998). Molecular cloning of ras cDNA from Penaeus japonicus (Crustacea, decapoda): Geranylgeranylation and guanine nucleotide binding. Gene 224 (1), 117–122. doi: 10.1016/S0378-1119(98)00528-9
Jayesh P., Seena J., Singh I. S. (2012). Establishment of shrimp cell lines: perception and orientation. Indian J. Virol. 23 (2), 244–251. doi: 10.1007/s13337-012-0089-9
Kao Z. N., Liu C. H., Liu W. J., Kumar R., Leu J. H., Wang H. C. (2020). Shrimp SIRT1 activates of the WSSV IE1 promoter independently of the NF-κB binding site. Fish Shellfish Immunol. 106, 910–919. doi: 10.1016/j.fsi.2020.08.034
Kumazaki M., Shimomura I., Kiyono T., Ochiya T., Yamamoto Y. (2020). Cell-type specific tumorigenesis with ras oncogenes in human lung epithelial cells. Biochem. Biophys. Res. Commun. 525 (2), 483–490. doi: 10.1016/j.bbrc.2020.02.113
Levy T., Rosen O., Eilam B., Azulay D., Aflalo E. D., Manor R., et al. (2016). A single injection of hypertrophied androgenic gland cells produces all-female aquaculture. Mar. Biotechnol. 18, 1–10. doi: 10.1007/s10126-016-9717-5
Levy T., Rosen O., Manor R., Dotan S., Azulay D., Abramov A., et al. (2019). Production of WW males lacking the masculine z chromosome and mining the macrobrachium rosenbergii genome for sex-chromosomes. Sci. Rep. 9 (1), 12408. doi: 10.1038/s41598-019-47509-6
Li R., Bai S., Yang D., Dong C. (2019). A crayfish Ras gene is involved in the defense against bacterial infection under high temperature. Fish Shellfish Immunol. 86, 608–617. doi: 10.1016/j.fsi.2018.11.062
Li F., Li M., Ke W., Ji Y., Bian X., Yan X. (2009). Identification of the immediate-early genes of white spot syndrome virus. Virology 385 (1), 267–274. doi: 10.1016/j.virol.2008.12.007
Liu Q. N., Tang Y. Y., Li Y. T., Zha X. H., Yang T. T., Zhang D. Z., et al. (2021). Differentially expressed genes in hemocytes of red swamp crayfish Procambarus clarkii following lipopolysaccharide challenge. Aquaculture 533, 735943. doi: 10.1016/j.aquaculture.2020.735943
Molcho J., Manor R., Shamsian M., Sudarshan G., Ofir R., Parker D., et al. (2022). On genome editing in embryos and cells of the freshwater prawn Macrobrachium rosenbergii. Aquaculture 558, 738391. doi: 10.1016/j.aquaculture.2022.738391
Mostoslavsky G., Kotton D. N., Fabian A. J., Gray J. T., Lee J. S., Mulligan R. C. (2005). Efficiency of transduction of highly purified murine hematopoietic stem cells by lentiviral and oncoretroviral vectors under conditions of minimal in vitro manipulation. Mol. Ther. 11 (6), 932–940. doi: 10.1016/j.ymthe.2005.01.005
Paris M., Wolff C., Patel N. H., Averof M. (2021). The crustacean model parhyale hawaiensis. Pre 147, 199–230. doi: 10.20944/preprints202106.0018.v1
Prior I. A., Lewis P. D., Mattos C. (2012). A comprehensive survey of ras mutations in cancer. Cancer Res. 72 (10), 2457–2467. doi: 10.1158/0008-5472.Can-11-2612
Rosalie S., Gustavo L., James D., Joseph N. R. (1999). Ras enhances myc protein stability. Mol. Cell 3 (2), 169–179. doi: 10.1016/S1097-2765(00)80308-1
Rotchell J. M., Lee J. S., Chipman J. K., Ostrander G. K. (2001). Structure, expression and activation of fish ras genes. Aquat. Toxicol. 55 (1), 1–21. doi: 10.1016/S0166-445X(01)00214-4
Rotem-Dai N., Weil S., Greenshpan Y., Abehsera S., Manor R., Ofir R., et al. (2021). Lentiviral-transduced ectopic expression of androgenic hormone in a crustacean hematopoietic primary cell culture. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.677679
Santos E., Angel N. R. (1989). Structural and functional properties of ras proteins. FASEB J. 3 (10), 2151–2163. doi: 10.1096/fasebj.3.10.2666231
Sharabi O., Manor R., Weil S., Aflalo E. D., Lezer Y., Levy T., et al. (2016). Identification and characterization of an insulin-like receptor involved in crustacean reproduction. Endocrinology 157 (2), 928–941. doi: 10.1210/en.2015-1391
Simcox A., Mitra S., Truesdell S., Paul L., Chen T., Butchar J. P., et al. (2008). Efficient genetic method for establishing Drosophila cell lines unlocks the potential to create lines of specific genotypes. PloS Genet. 4 (8), e1000142. doi: 10.1371/journal.pgen.1000142
Söderhäll I. (2016). Crustacean hematopoiesis. Dev. Comp. Immunol. 58, 129–141. doi: 10.1016/j.dci.2015.12.009
Takahashi K., Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4), 663–676. doi: 10.1016/j.cell.2006.07.024
Tapay L. M., Lu Y., Brock J. A., Nadala E. C. J., Loh P. C. (1995). Transformation of primary cultures of shrimp (Penaeus stylirostris) lymphoid (Oka) organ with simian virus-40 (T) antigen. Proc. Soc Exp. Biol. Med. 209 (1), 73–78. doi: 10.3181/00379727-209-43880
Thansa K., Rungsiwiwut R., Kitiyanant N., Taengchaiyaphum S. (2018). Optimisation of electroporation and lipofection protocols to derive the black tiger shrimp cell line (Penaeus monodon). Fish Shellfish Immunol. 81, 204–213. doi: 10.1016/j.fsi.2018.07.030
Uchida N., Green R., Ballantine J., Skala L. P., Hsieh M. M., Tisdale J. F. (2016). Kinetics of lentiviral vector transduction in human CD34(+) cells. Exp. Hematol. 44 (2), 106–115. doi: 10.1016/j.exphem.2015.10.003
Keywords: cell culture, lentivirus, Macrobrachium rosenbergii, proto-oncogene Ras, white spot syndrome virus
Citation: Sudarshan G, Weil S, Rotem-Dai N, Manor R, Greenshpan Y, Goldstein O, Sharabi O, Aflalo ED, Ofir R, Rosental B, Gazit R and Sagi A (2023) Enhanced proliferation in a prawn embryonic primary cell culture ectopically expressing mutated Ras. Front. Mar. Sci. 9:1100971. doi: 10.3389/fmars.2022.1100971
Received: 17 November 2022; Accepted: 13 December 2022;
Published: 09 January 2023.
Edited by:
Tomer Ventura, University of the Sunshine Coast, AustraliaReviewed by:
Jude Juventus Aweya, Jimei University, ChinaZhenkui Qin, Ocean University of China, China
Fei Zhu, Zhejiang Agriculture and Forestry University, China
Copyright © 2023 Sudarshan, Weil, Rotem-Dai, Manor, Greenshpan, Goldstein, Sharabi, Aflalo, Ofir, Rosental, Gazit and Sagi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amir Sagi, c2FnaWFAYmd1LmFjLmls
†ORCID: Amir Sagi, orcid.org/0000-0002-4229-1059