 Yajun Jing1,2†
Yajun Jing1,2† Ruizhe Guo2,3†Hongmin Wang2,3†
Ruizhe Guo2,3†Hongmin Wang2,3† Yantao Liang2,3,4*Yundan Liu2,3Yifei Feng2Qin Ma2
Yantao Liang2,3,4*Yundan Liu2,3Yifei Feng2Qin Ma2 Hongbing Shao2,3,4
Hongbing Shao2,3,4 Yeong Yik Sung4,5
Yeong Yik Sung4,5 Wen Jye Mok4,5
Wen Jye Mok4,5 Li Lian Wong4,5
Li Lian Wong4,5 Yu-Zhong Zhang2,6
Yu-Zhong Zhang2,6 Andrew McMinn2,3,7
Andrew McMinn2,3,7 Min Wang1,2,3,4,8*
Min Wang1,2,3,4,8* Jinyan Xing1*
Jinyan Xing1*- 1Department of Critical Care Medicine, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
- 2College of Marine Life Sciences, Ocean University of China, Qingdao, China
- 3Institute of Evolution and Marine Biodiversity, Frontiers Science Center for Deep Ocean Multispheres and Earth System, Center for Ocean Carbon Neutrality, Ocean University of China, Qingdao, China
- 4UMT-OUC Joint Academic Centre for Marine Studies, Qingdao, China
- 5Institute of Marine Biotechnology, Universiti Malaysia Terengganu, Kuala Terengganu, Malaysia
- 6State Key Laboratory of Microbial Technology, Marine Biotechnology Research Center, Shandong University, Qingdao, China
- 7Institute for Marine and Antarctic Studies, University of Tasmania, Hobart, TAS, Australia
- 8Haide College, Ocean University of China, Qingdao, China
Vibrio is a kind of common gram-negative bacteria, which is widely distributed in marine and estuarine environments. In the study, a novel marine phage vB_VhaP_PG11, infecting Vibrio hangzhouensis, was isolated from the offshore waters of Qingdao, China. vB_VhaP_PG11 is a double-stranded DNA phage. The whole genome proteomic tree shows that vB_VhaP_PG11 phage is related to two Vibrio phages, Vibrio phage 1.238.A._10N.261.52.F10 and Vibrio phage 1.245.O._10N.261.54.C7, but with low homology. Their amino acids identity with vB_VhaP_PG11 is 42.77 and 41.49% respectively. The prediction results of genome-blast distance phylogeny (GBDP) and the analysis gene-sharing network indicate that vB_VhaP_PG11 belongs to a new genus in Schitoviridae, named Qingschitovirus. The study of Vibrio phage vB_VhaP_PG11 provides basic information contributing to a better understanding of interactions between Vibrio phages and their hosts and helps analyze unknown viral sequences in the metagenomic database.
Introduction
Viruses are the most common life forms and play important roles in the ocean, with an abundance reaching 106 –109 virus-like particles (VLPs) per milliliter, i.e. approximately 10 times that of microorganisms (Fuhrman, 1999). Most of these viruses have the ability to infect bacteria, named, phages, which have a total global abundance of 1030–1032 (Wommack and Colwell, 2000; Ashelford et al., 2003; Suttle, 2005). Marine phages play crucial roles in regulating marine microbial populations, and community structure and affect global biogeochemical cycles (Breitbart, 2012). In marine environments, phages can infect about 5%–30% of heterotrophic bacteria and cyanobacteria (Middelboe, 2008). Microbial biomass can be shunted into dissolved organic matter (DOM) by viral lysis, which can also mediate the recycling of macro- and micro-elements, and influence marine biogeochemical cycling of sulfur, phosphorus, carbon, and other elements (Pratama and van Elsas, 2018). In addition, horizontal gene transfer (HGT) is very common in marine microorganisms and is effective in the spread of virulence factors. Broad-spectrum marine phages could act as vectors to promote HGT via transduction in natural environments (Jensen et al., 1998). The auxiliary metabolic genes (AMGs) in the genome of phages can also affect the metabolic processes of the infected cells and this is assumed to increase the adaptability of phages under pressure (Breitbart, 2012).
During past years, what we know about marine viral genomes and diversity has greatly expanded due to progress in the metagenome and metatranscriptome analysis (Mineta and Gojobori, 2016). However, most assembled environmental viral sequences belong to uncultured viruses, which can account for 40 - 90% of the total assembled viruses. These sequences are called viral dark matter (Krishnamurthy and Wang, 2017). The discovery of viruses from dark matter usually depends on the prediction of hypothetical viruses using comparisons with the reference virus gene and genome sequences (Santiago-Rodriguez and Hollister, 2022). The reference virus genomes are often derived from sequencing results of isolated viruses. Isolation of more phages will increase the comprehension of viruses, thus improving the ability to predict viruses that may exist in dark matter.
Vibrio is a genus of culturable, heterotrophic bacterium (Grimes et al., 2009) that is mainly distributed in marine environments and estuarine. Many species of marine Vibrio are supposed to be pathogens in aquaculture systems (Almeida et al., 2009). Some Vibrio cause disease in animals and humans, such as Vibrio cholerae, Vibrio mimicus, which causes gastroenteritis, and Vibrio vulnificus which causes parenteral infections in humans (Jing et al., 2020). In recent years, due to the characteristic ecological and physiological property, more and more marine Vibrio have been isolated (Loughran et al., 2022; Fahmy, 2022). Xu et al. isolated a novel Vibrio strain in 2009, named Vibrio hangzhouensis from sediments of the East China Sea. Vibrio hangzhouensis is a gram-negative motile bacillus with polar flagella. Cells are straight or slightly curved rods with rounded ends. None formed endospores (Xu et al., 2009). In recent years, more and more Vibrio phages have been isolated. Most of them have typical head-tail structures and belong to Caudoviricetes. As of December 2022, Vibrio phages belonging to Caudoviricetes comprised 68% of all Vibrio phages in the NCBI virus database. Nonetheless, knowledge of this important phage group is still inadequate, especially in consideration of their potential applications in phage therapy of pathogenic Vibrio species.
In the study, a novel phage named vB_VhaP_PG11 infecting Vibrio hangzhouensis has been isolated from the coastal waters of the Yellow Sea, Qingdao, China. Growth, genomic as well as phylogenetic analysis of phage vB_VhaP_PG11 are described. This study provides basic information for further understanding the genomic features of Vibrio phages.
Material and methods
Bacterial strain
The bacterial strain was isolated from seawater collected from Maidao, Qingdao, China (36.058°N, 120.428°E) in September 2020, which was provided by the laboratory of Prof. Yu-Zhong Zhang of Shandong University, China. The bacterial sample was cultured in 2216E medium and grown in an environment of 25°C. 16S rRNA gene sequence analysis was used to identify the molecular characteristics of the bacterial strain. BLAST search was used to test the homology of gene sequence (Chakravorty et al., 2007). The 16S rRNA gene sequence of the bacterial strain was similar to Vibrio hangzhouensis CN83 (Percent of identity at 99.37%) (Figure S1). The host strain was stored at -80°C in 2216E broth with 30% glycerol.
Preparation of phage
The 1 L sample seawater from Maidao, Qingdao was stored at 4°C until analysis. In order to filter out the phytoplankton and bacteria, the sample was filtered through a 0.22 μm pore-size membrane before the experiment. The double-layer agar method was adopted to isolate the phages in the sample. Briefly, 200 μL of logarithmic growth phase bacterial and 200 μL of seawater sample filtrate were mixed. The mixture was mixed with melted semi-solid medium (agar 7.5 wt.%) of 4.5 ml at 50°C and then poured onto the surface of the solid medium (agar 15 wt.%). The agar plate was cultured at 25°C and observed after 24h. When plaques appeared, they were picked out and placed in 1 mL SM Buffer (8 mM MgSO4·7H2O,100 mM NaCl, 50 mM Tris-Cl, pH=7.5), and then filtered the buffer onto a 0.22 μm PES Millipore filter. The phage was purified 3-5 times. We stored the purified phage solution in SM Buffer at 4°C (Jamalludeen et al., 2007; Guo et al., 2022).
One-step growth, thermal stability, and pH sensitivity
The 1 mL bacterial solution and 1 mL purified viral solution were cultured for 15 min with MOI 0.1 at 25°C. Samples were taken every 10 min. Viral abundance was determined by the double-layer agar method. The number of plaques formed in different periods was counted in order to draw the growth curve (Cai et al., 2019).
Ten vials of SM buffer were prepared with pH of 3 - 12, respectively, and sterilized at 121°C for 20 min (Liu et al., 2019). The phage solution (~108 PFU/mL) was diluted with SM buffer of different pH and placed for 2 h at optimum temperature. Phage solution with different pH was then mixed with host bacterial solution, which was in the logarithmic growth stage, with a concentration of 2 x107 CFU/mL. After 25 min infection, the double-layer agar was poured and cultured overnight at 25°C. The phage survival curve was drawn according to the number of plaques at each pH.
Several copies of the same phage solution were treated at - 20°C, 4°C, 25°C, 35°C, 45°C, 55°C, 65°C, and 75°C, respectively, for 2h (Zhang et al., 2020). The host bacterial solution in the logarithmic growth stage was mixed with the treated phage solution in a vortex. After 15 min infection, the mixture was poured onto the plate with melted semi-solid medium and cultured at room temperature overnight. The phage survival curve was drawn according to the plaques on the plate at different temperatures.
DNA extraction and genome sequencing of the phage
The phage genomic DNA was extracted using a viral DNA Kit (OMEGA) according to the manufacturer’s instructions. Purified phage genomic DNA was sequenced by the Illumina NovaSeq 6000 paired-end sequence method (2 × 150bp). The raw paired-end reads were trimmed and quality controlled by Trimmomatic with parameters (SLIDINGWINDOW:4:15 MINLEN:75) (version 0.36 http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic). ABySS (http://www.bcgsc.ca/platform/bioinfo/software/abyss) was used to perform genome assembly with multiple-Kmer parameters and to obtain the optimal results of the assembly. GapCloser software (https://sourceforge.net/projects/soapdenovo2/files/GapCloser/), using default parameters, was subsequently applied to fill up the remaining local inner gaps and correct the single base polymorphism for the final assembly results. The ORFs were analyzed by RAST (parameters: Domain: virus, Genetic Code: 11) (Aziz et al., 2008). All gene functions were predicted by BLASTp (parameters: evalue<1e-5) against the non-redundant (NR in NCBI) database (http://blast.ncbi.nlm.nih.gov/) to find homologs (Liu et al., 2017; Guo et al., 2022).
Bioinformatic and proteomic analysis
The bacteriophage proteomic tree was built by Virus Classification and Tree Building Online Resource (VICTOR) (Meier-Kolthoff and Göker, 2017) and genome alignments were conducted with ViPTree (Nishimura et al., 2017). Taxonomic information and genome sequences of viruses and their hosts were based on the NCBI virus database. All 86 viral genomes of Schitoviridae RefSeq were selected as reference sequences to build a whole-genome proteomic tree with vB_VhaP_PG11. The genome-blast distance phylogeny (GBDP) method was used to conduct pairwise comparisons of the nucleotide sequences (Meier-Kolthoff et al., 2013). The phylogenomic GBDP tree was inferred using the formula D6 and yielding average support of 54%. The numbers above branches are GBDP pseudo-bootstrap support values from 100 replications. The branch lengths of the resulting VICTOR trees are scaled in terms of the respective distance formula used. Taxon boundaries at the genus level were estimated by the OPTSIL program with an F value of 0.5 (Göker et al., 2009; Meier-Kolthoff et al., 2014). Genera based on ICTV classification were also shown by color ranges.
Network analysis
A gene content-based viral network analysis among genomes of vB_VhaP_PG11 and other members of Schitoviridae had been performed. All viral genomes of Schitoviridae from the NCBI virus database were selected. Viral clusters (VCs) were identified through ClusterONE with default parameters which were defined in the vConTACT 2.0 (–pc-inflation 1.2 –link-prop 0.3 –blast-evalue 1e-5) (Nepusz et al., 2012; Bin Jang et al., 2019). The classification of genera was based on the classification of ICTV. The network was visualized by Gephi version 0.9.6 (Xue et al., 2018).
Genome sequence accession number
The annotation results and related information have been submitted to GenBank. The complete genome sequence of phage vB_VhaP_PG11 is available in the GenBank under accession number OP745480.
Results and discussion
Biological characterization of vB_VhaP_PG11
Phage vB_VhaP_PG11, which was isolated from seawater collected from Maidao, Qingdao, China (36.058°N, 120.428°E), was able to infect Vibrio hangzhouensis. Phage vB_VhaP_PG11 lysed the host and formed clear plaques in the double-layer agar (Figure 1).
Figure 1 The plaques of the Vibrio phage vB_VhaP_PG11 on the lawn of host bacteria.
The viral one-step growth, temperature and pH stability were tested with the purpose of characterizing the biological features of the marine vibrio phage vB_VhaP_PG11. The one-step growth curve shows that the latent period of phage vB_VhaP_PG11 was 40 min, followed by a quick ascent (Figure 2A). The activity was greatly influenced by pH (Figure 2B). At pH 3 and 4, vB_VhaP_PG11 was almost completely inactivated. The activity increased with increasing pH. vB_VhaP_PG11 had the strongest ability to infect the host at a pH of 9. The activity of vB_VhaP_PG11 decreased slightly with an increase in pH. This suggests that it is sensitive to acidic environments but adapted to alkaline environments. In the temperature stability experiment, vB_VhaP_PG11 exhibited high activity at temperatures between - 20°C and 45°C, with an optimal temperature of 35°C. No infection activity infectious occurred when temperatures reached 55°C (Figure 2C).
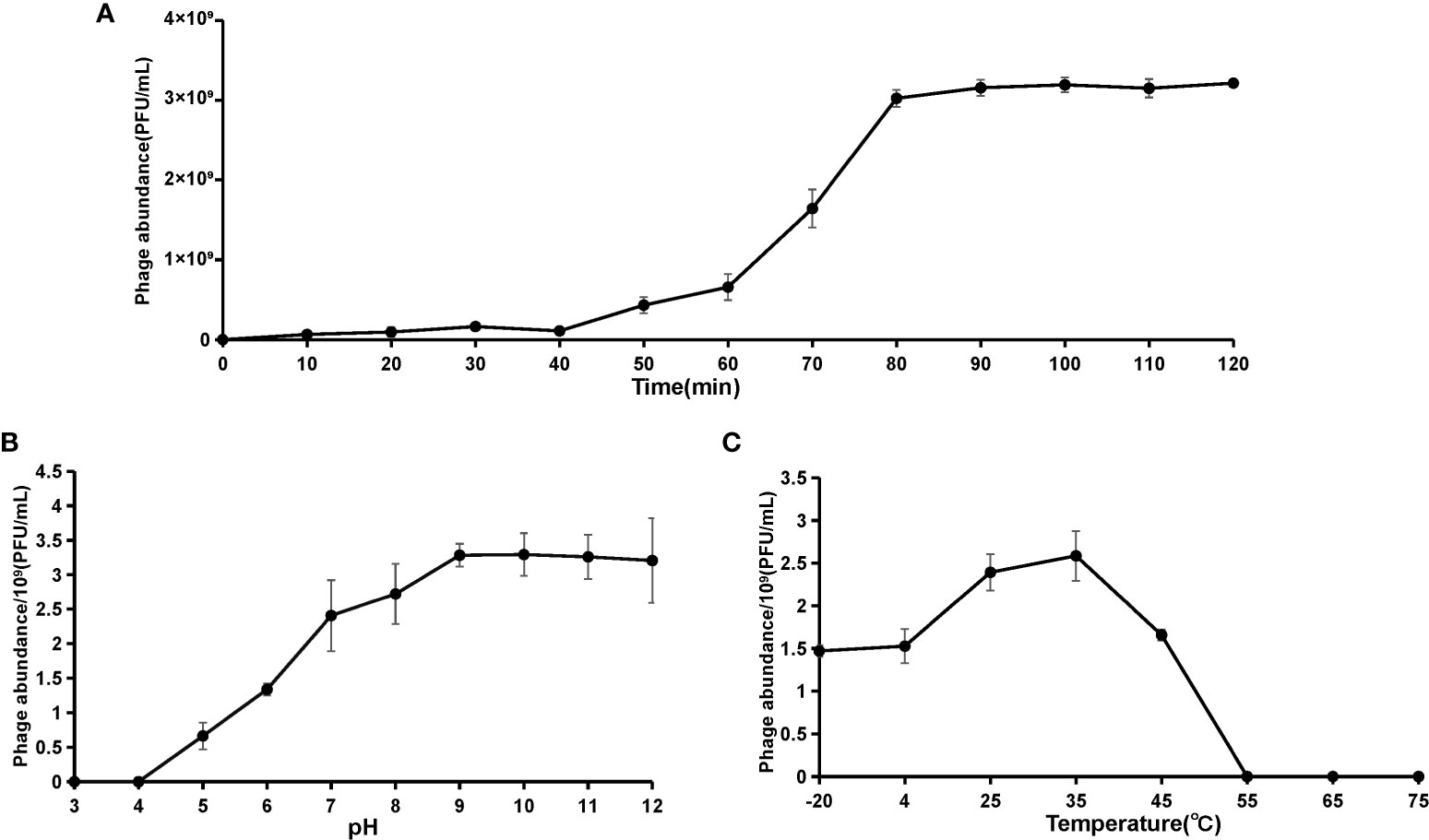
Figure 2 The curve of one-step growth (A), pH stability (B), and thermal stability (C) of Vibrio phage vB_VhaP_PG11. These experiments have been repeated three times, and the data are shown as mean ± SEM.
Genome sequencing and bioinformatic analysis
The genome of phage vB_VhaP_PG11 consists of a linear, double-stranded DNA molecule with a length of 71,843bp and a GC content of 41.4% and no tRNA genes. The NCBI blast analysis showed that vB_VhaP_PG11 has low similarity with any other known phages, which confirms it as a unique Vibrio phage species. The genome of vB_VhaP_PG11 was annotated using RAST (Aziz et al., 2008). A total of 108 ORFs were detected. Of these, 26 were in the positive strand and 82 were in the negative strand. The ORF functions were searched by BLASTp. Only 35 of 108 ORFs had known function, and the predicted ORFs were classified into four groups, including DNA replication, regulation, and nucleotide metabolism (ORF16, ORF19, ORF22-25, ORF28-30, ORF32, ORF33, ORF35, ORF36, ORF43, ORF46, ORF66, ORF69, and ORF101), phage packaging (ORF106 and ORF107), phage structure (ORF2, ORF3, ORF6-8, ORF10, ORF12, ORF13, ORF15, and ORF108) and one auxiliary metabolic gene (ORF53) (Table S1, Figure 3).
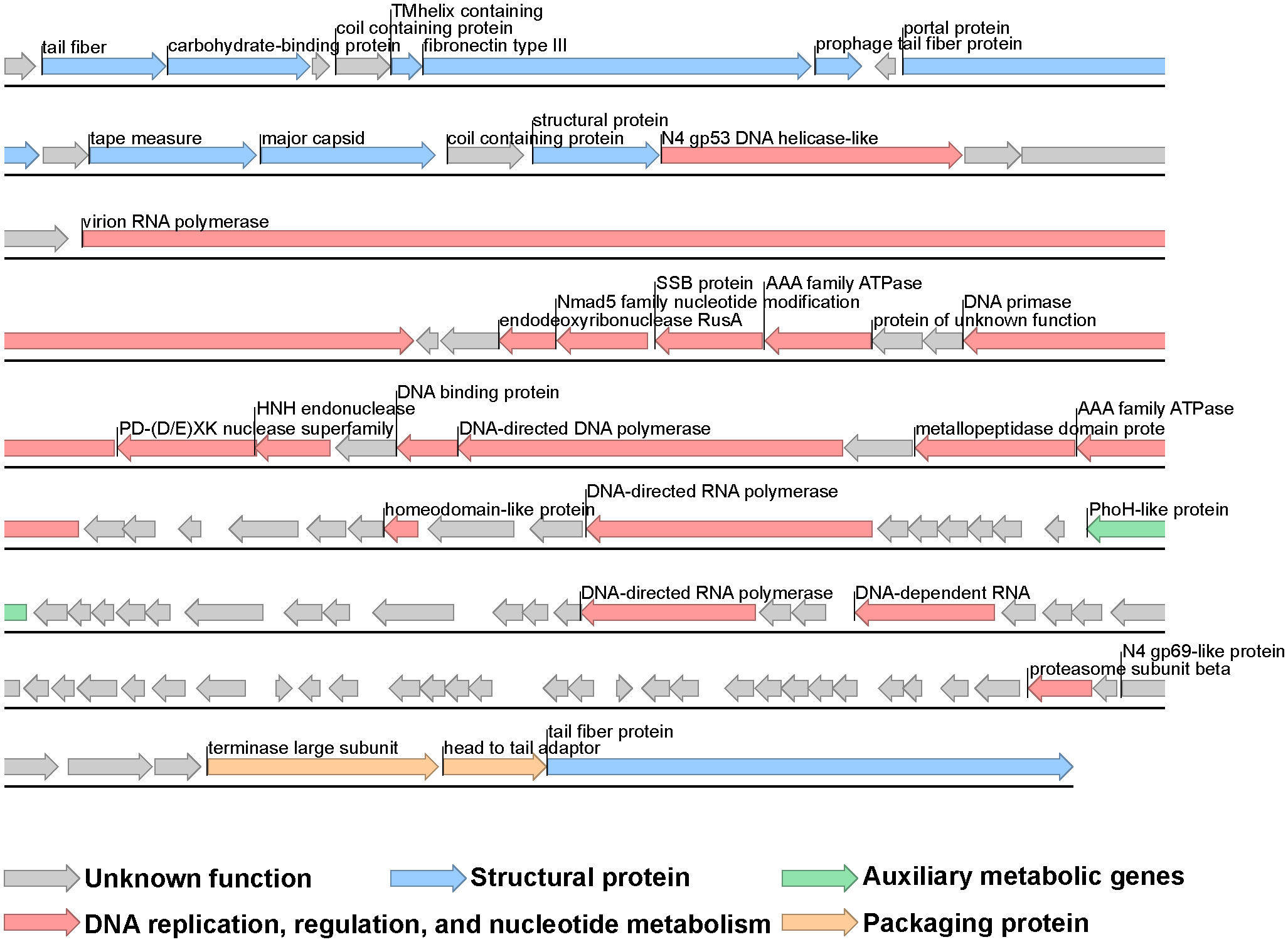
Figure 3 Genome map of Vibrio phage vB_VhaP_PG11. Putative functional categories are defined according to annotation and are represented by different colors. The length of each arrow represents the length of each gene.
The AMG phoH was detected in the vB_VhaP_PG11 genome, which belongs to a Pho regulon. This AMG plays an important role in affecting phosphate metabolism and uptake under phosphate-limiting conditions (Kim et al., 1993; Hsieh and Wanner, 2010; Luo et al., 2020). Phosphorus is the main element of nucleotide biosynthesis and together with nitrogen and silicon are the major limiting macronutrients in the ocean (Rohwer et al., 2000; Lindell et al., 2004; Sullivan et al., 2005; Paytan and McLaughlin, 2007; Sullivan et al., 2010; Kathuria and Martiny, 2011). Therefore, genes related to phosphorus acquisition, such as phoH, pstS, and phoA, may help the host acquire phosphorus during virus infection (Hsieh and Wanner, 2010).
Phage vB_VhaP_PG11 represents a novel viral cluster
According to the current classification standard for Schitoviridae by ICTV, a 95% DNA sequence identity is used as the standard for species classification within a genus. Each proposed new species is thus more than 5% different from other species on the DNA level. For the division of genera and subfamilies, 70% and 40% DNA sequence identity are required respectively (Wittmann et al., 2020). Based on this proposal of ICTV, a series of analyses were carried out.
The proteomic tree of the vB_VhaP_PG11 genome and reference sequences (RefSeq) belonging to the Schitoviridae family in the NCBI virus database was constructed by VICTOR and our results demonstrated that the closest relatives of phage vB_VhaP_PG11 are two Vibrio phages, named Vibrio phage 1.238.A._10N.261.52.F10 (NC_055735) and Vibrio phage 1.245.O._10N.261.54.C7 (NC_055736). The amino acid identity (AAI) between vB_VhaP_PG11 and Vibrio phage 1.238.A._10N.261.52.F10 and Vibrio phage 1.245.O._10N.261.54.C7 are 42.77 and 41.49%, respectively, less than 70% but higher than 40%, suggesting that vB_VhaP_PG11 may not belong to a known genus in Schitoviridae. The three phage genomes were aligned, and the comparative genomic analysis maps were drawn (Figure 4). The conserved genes in vB_VhaP_PG11 show a trend of aggregation and have very limited homology with the most similar phage. According to the prediction of OPTSIL (Figure 5), vB_VhaP_PG11 can be considered a new genus. The network analysis also demonstrated this. In the network analysis, nodes are clustered according to VC, colored according to the genus, and vB_VhaP_PG11 is classified as an “outlier”, not in any VC (Figure 6). These results suggest that phage vB_VhaP_PG11 is different from other marine Vibrio phages, and represents a new schitoviral genus, which we propose to name Qingschitovirus.
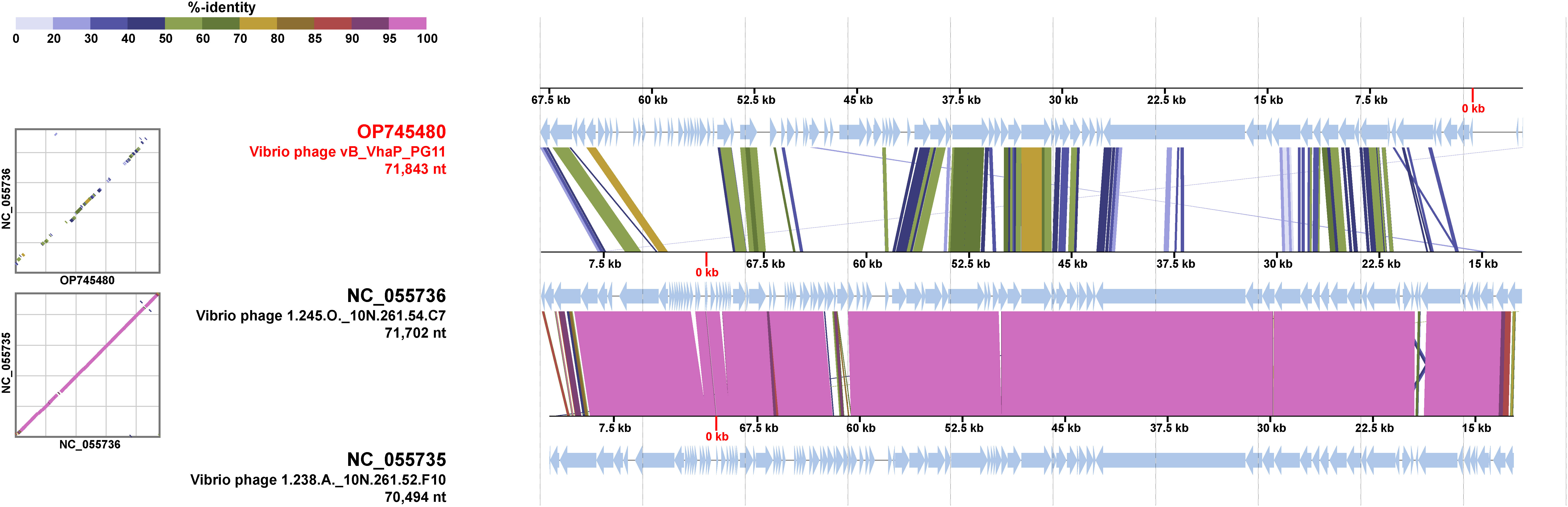
Figure 4 Genomic comparisons between Vibrio phage vB_VhaP_PG11, Vibrio phage 1.245.O._10N.261.54.C7, and Vibrio phage 1.238.A._10N.261.52.F10. The shading below each genome indicates sequence similarities between the genomes, with different colors representing the levels of similarity.
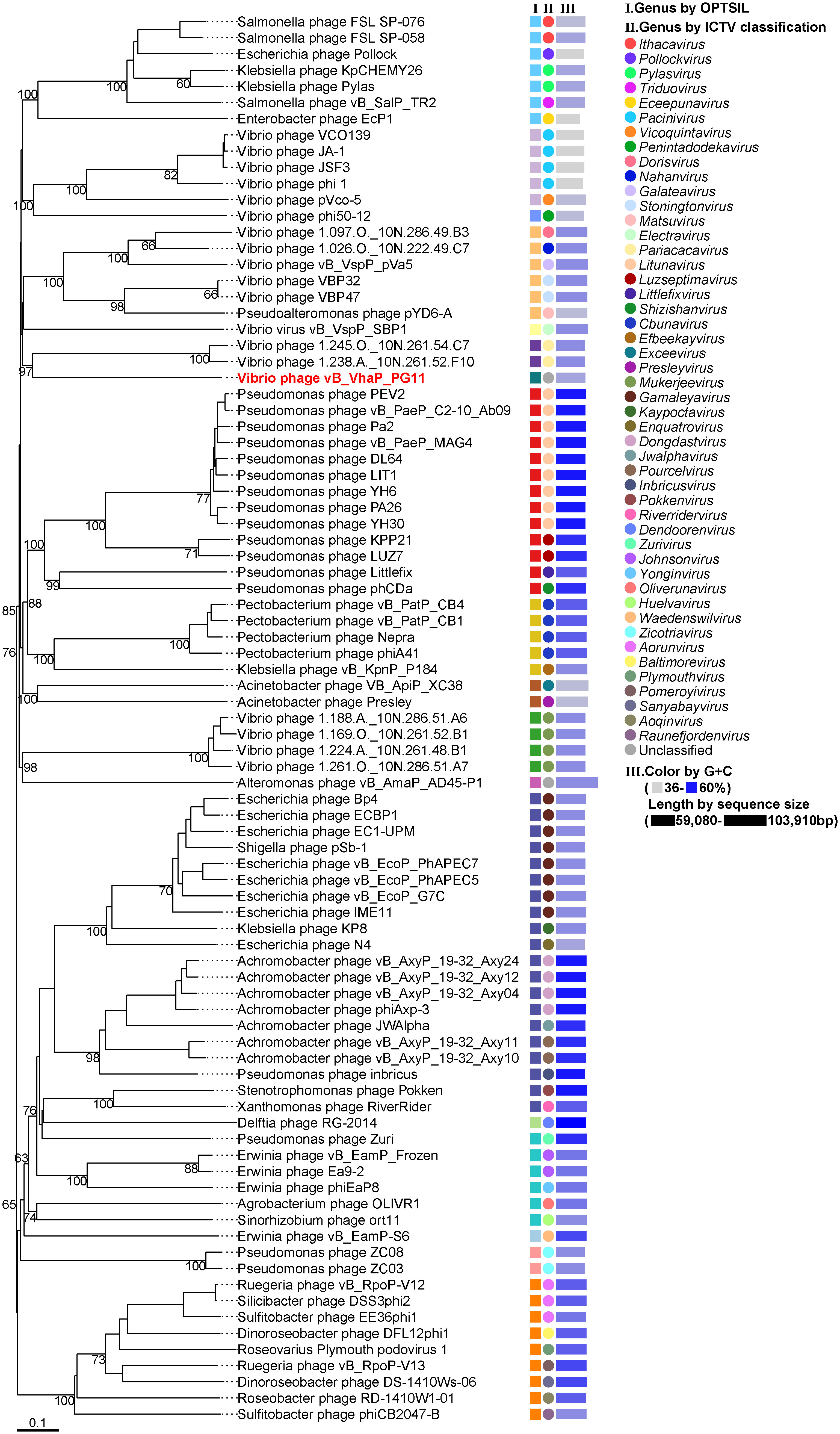
Figure 5 Whole-genome based proteomic tree of Vibrio phage vB_VhaP_PG11 and RefSeq of Schitoviridae from NCBI database and constructed by Virus Classification and Tree Building Online Resource (VICTOR) with the formula d6. The proteomic tree consists of 87 phage genomes. Three series of color boxes behind the tree indicate: I. Genera classified of all these phages classified by OPTSIL; II. Genera classified by ICTV; III. GC content and sequence size. GC content is represented by the color of the block, and the sequence size is represented by the length of the block.
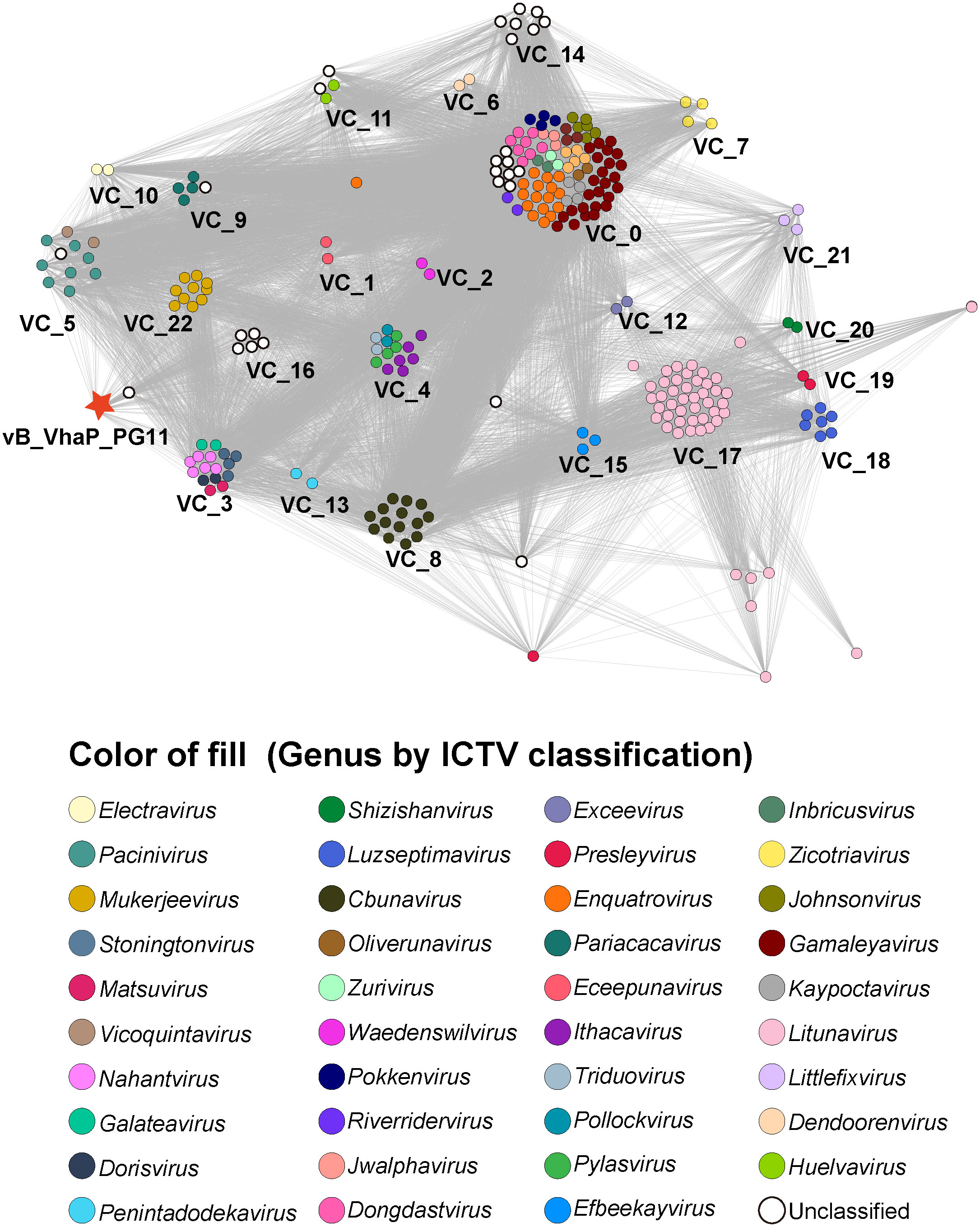
Figure 6 Network analysis based on genomes of Vibrio phage vB_VhaP_PG11 and all members of Schitoviridae from NCBI virus database. Each node represents a viral sequence and is colored according to genus. Viral clusters (VCs) are identified using ClusterONE with default parameters which are defined in the vConTACT 2.0.
Conclusion
Here, we isolated a novel schitovirus, phage vB_VhaP_PG11, infecting Vibrio hangzhouensis. Based on the genomic, comparative genomic, phylogenetic, and network analysis, phage vB_VhaP_PG11 represents a new viral genus, named Qingschitovirus. This study provides new data for studying the interaction between Vibrio and schitovirus and can be used as a reference genome to determine the taxonomic status of unknown schitoviruses in the metagenomes and metatranscriptomes. In the future, more phages in the Qingschitovirus should be isolated and will improve our understanding of the host range and ecological roles of this new viral genus.
Data availability statement
The data presented in the study are deposited in the GenBank repository, accession number OP745480 and OP788126.
Author contributions
YTL, MW, and JX: supervision, conceptualization and project administration. YJ: data analysis, writing and original draft. RG: validation, formal analysis, and software. HW: visualization and editing. YDL: methodology. YF and QM: investigation. HS: project administration. YS, WM, and LW: data curation. Y-ZZ: resources. AM: comments and revision. YTL, MW, JX, and AM: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the following funding sources: National Natural Science Foundation of China (No. 41976117, 42120104006, 42176111, 42188102 and 81701878), and the Fundamental Research Funds for the Central Universities (202172002, 201812002 and Andrew McMinn).
Acknowledgments
We thank for the support of the high-performance servers of Center for High Performance Computing and System Simulation, Pilot National Laboratory for Marine Science and Technology (Qingdao), the Marine Big Data Center of Institute for Advanced Ocean Study of Ocean University of China, the IEMB-1, a high-performance computing cluster operated by the Institute of Evolution and Marine Biodiversity, and the high-performance servers of Frontiers Science Center for Deep Ocean Multispheres and Earth System.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1092917/full#supplementary-material
References
Almeida A., Cunha A., Gomes N. C., Alves E., Costa L., Faustino M. A. (2009). Phage therapy and photodynamic therapy: low environmental impact approaches to inactivate microorganisms in fish farming plants. Mar. Drugs 7 (3), 268–313. doi: 10.3390/md7030268
Ashelford K. E., Day M. J., Fry J. C. (2003). Elevated abundance of bacteriophage infecting bacteria in soil. Appl. Environ. Microbiol. 69 (1), 285–289. doi: 10.1128/AEM.69.1.285-289.2003
Aziz R. K., Bartels D., Best A. A., DeJongh M., Disz T., Edwards R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Bin Jang H., Bolduc B., Zablocki O., Kuhn J. H., Roux S., Adriaenssens E. M., et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37 (6), 632–639. doi: 10.1038/s41587-019-0100-8
Breitbart M. (2012). Marine viruses: truth or dare. Ann. Rev. Mar. Sci. 4, 425–448. doi: 10.1146/annurev-marine-120709-142805
Cai L., Ma R., Chen H., Yang Y., Jiao N., Zhang R. (2019). A newly isolated roseophage represents a distinct member of siphoviridae family. Virol. J. 16 (1), 128. doi: 10.1186/s12985-019-1241-6
Chakravorty S., Helb D., Burday M., Connell N., Alland D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69 (2), 330–339. doi: 10.1016/j.mimet.2007.02.005
Fahmy N. M. (2022). Isolation and characterization of vibrio alginolyticus strain HAT3 causing skin ulceration disease in cultured sea cucumber holothuria atra (Jaeger, 1833). Egyptian J. Aquat. Res. 48 (1), 75–81. doi: 10.1016/j.ejar.2021.11.007
Fuhrman J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399 (6736), 541–548. doi: 10.1038/21119
Göker M., García-Blázquez G., Voglmayr H., Tellería M. T., Martín M. P. (2009). Molecular taxonomy of phytopathogenic fungi: a case study in peronospora. PloS One 4 (7), e6319. doi: 10.1371/journal.pone.0006319
Grimes D. J., Johnson C. N., Dillon K. S., Flowers A. R., Noriea N. F. 3rd, Berutti T. (2009). What genomic sequence information has revealed about vibrio ecology in the ocean–a review. Microb. Ecol. 58 (3), 447–460. doi: 10.1007/s00248-009-9578-9
Guo R., Zheng K., Luo L., Liu Y., Shao H., Guo C., et al. (2022). Characterization and genomic analysis of ssDNA vibriophage vB_VpaM_PG19 within microviridae, representing a novel viral genus. Microbiol. Spectr. 10 (4), e0058522. doi: 10.1128/spectrum.00585-22
Hsieh Y. J., Wanner B. L. (2010). Global regulation by the seven-component pi signaling system. Curr. Opin. Microbiol. 13 (2), 198–203. doi: 10.1016/j.mib.2010.01.014
Jamalludeen N., Johnson R. P., Friendship R., Kropinski A. M., Lingohr E. J., Gyles C. L. (2007). Isolation and characterization of nine bacteriophages that lyse O149 enterotoxigenic escherichia coli. Vet. Microbiol. 124 (1-2), 47–57. doi: 10.1016/j.vetmic.2007.03.028
Jensen E. C., Schrader H. S., Rieland B., Thompson T. L., Lee K. W., Nickerson K. W., et al. (1998). Prevalence of broad-host-range lytic bacteriophages of sphaerotilus natans, escherichia coli, and pseudomonas aeruginosa. Appl. Environ. Microbiol. 64 (2), 575–580. doi: 10.1128/AEM.64.2.575-580.1998
Jing C., Lu Q., YaLu Z., Lu G. (2020). Review on pathogenic vibrio and its phage control. J. Food Saf. Qual. 11 (24), 9288–9294.
Kathuria S., Martiny A. C. (2011). Prevalence of a calcium-based alkaline phosphatase associated with the marine cyanobacterium prochlorococcus and other ocean bacteria. Environ. Microbiol. 13 (1), 74–83. doi: 10.1111/j.1462-2920.2010.02310.x
Kim S. K., Makino K., Amemura M., Shinagawa H., Nakata A. (1993). Molecular analysis of the phoH gene, belonging to the phosphate regulon in escherichia coli. J. Bacteriol 175 (5), 1316–1324. doi: 10.1128/jb.175.5.1316-1324.1993
Krishnamurthy S. R., Wang D. (2017). Origins and challenges of viral dark matter. Virus Res. 239, 136–142. doi: 10.1016/j.virusres.2017.02.002
Lindell D., Sullivan M. B., Johnson Z. I., Tolonen A. C., Rohwer F., Chisholm S. W. (2004). Transfer of photosynthesis genes to and from prochlorococcus viruses. Proc. Natl. Acad. Sci. U.S.A. 101 (30), 11013–11018. doi: 10.1073/pnas.0401526101
Liu Z., Wang M., Meng X., Li Y., Wang D., Jiang Y., et al. (2017). Isolation and genome sequencing of a novel pseudoalteromonas phage PH1. Curr. Microbiol. 74 (2), 212–218. doi: 10.1007/s00284-016-1175-9
Liu Y., Zhao L., Wang M., Wang Q., Zhang X., Han Y., et al. (2019). Complete genomic sequence of bacteriophage P23: a novel vibrio phage isolated from the yellow Sea, China. Virus Genes 55 (6), 834–842. doi: 10.1007/s11262-019-01699-3
Loughran R. M., Emsley S. A., Jefferson T., Wasson B. J., Deadmond M. C., Knauss T. L., et al. (2022). Vibrio tetraodonis subsp. pristinus subsp. nov., isolated from the coral acropora cytherea at Palmyra atoll, and creation and emended description of vibrio tetraodonis subsp. tetraodonis subsp. nov. Antonie van Leeuwenhoek 115 (9), 1215–1228. doi: 10.1007/s10482-022-01766-0
Luo E., Eppley J. M., Romano A. E., Mende D. R., DeLong E. F. (2020). Double-stranded DNA virioplankton dynamics and reproductive strategies in the oligotrophic open ocean water column. Isme J. 14 (5), 1304–1315. doi: 10.1038/s41396-020-0604-8
Meier-Kolthoff J. P., Auch A. F., Klenk H. P., Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff J. P., Hahnke R. L., Petersen J., Scheuner C., Michael V., Fiebig A., et al. (2014). Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 9, 2. doi: 10.1186/1944-3277-9-2
Meier-Kolthoff J. P., Göker M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33 (21), 3396–3404. doi: 10.1093/bioinformatics/btx440
Middelboe M. (2008). Microbial disease in the sea: effects of viruses on carbon and nutrient cycling (Princeton, NJ, USA: Princeton University Press).
Mineta K., Gojobori T. (2016). Databases of the marine metagenomics. Gene 576 (2 Pt 1), 724–728. doi: 10.1016/j.gene.2015.10.035
Nepusz T., Yu H., Paccanaro A. (2012). Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 9 (5), 471–472. doi: 10.1038/nmeth.1938
Nishimura Y., Yoshida T., Kuronishi M., Uehara H., Ogata H., Goto S. (2017). ViPTree: the viral proteomic tree server. Bioinformatics 33 (15), 2379–2380. doi: 10.1093/bioinformatics/btx157
Paytan A., McLaughlin K. (2007). The oceanic phosphorus cycle. Chem. Rev. 107 (2), 563–576. doi: 10.1021/cr0503613
Pratama A. A., van Elsas J. D. (2018). The 'Neglected' soil virome - potential role and impact. Trends Microbiol. 26 (8), 649–662. doi: 10.1016/j.tim.2017.12.004
Rohwer F., Segall A., Steward G., Seguritan V., Breitbart M., Wolven F., et al. (2000). The complete genomic sequence of the marine phage roseophage SIO1 shares homology with nonmarine phages. Limnol. Oceanogr. 45 (2), 408–418. doi: 10.4319/lo.2000.45.2.0408
Santiago-Rodriguez T. M., Hollister E. B. (2022). Unraveling the viral dark matter through viral metagenomics. Front. Immunol. 13, 1005107. doi: 10.3389/fimmu.2022.1005107
Sullivan M. B., Coleman M. L., Weigele P., Rohwer F., Chisholm S. W. (2005). Three prochlorococcus cyanophage genomes: signature features and ecological interpretations. PloS Biol. 3 (5), e144. doi: 10.1371/journal.pbio.0030144
Sullivan M. B., Huang K. H., Ignacio-Espinoza J. C., Berlin A. M., Kelly L., Weigele P. R., et al. (2010). Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 12 (11), 3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x
Wittmann J., Turner D., Millard A. D., Mahadevan P., Kropinski A. M., Adriaenssens E. M. (2020). From orphan phage to a proposed new family–the diversity of N4-like viruses. Antibiotics 9 (10), 663. doi: 10.3390/antibiotics9100663
Wommack K. E., Colwell R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64 (1), 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Xue Y., Chen H., Yang J. R., Liu M., Huang B., Yang J. (2018). Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. Isme J. 12 (9), 2263–2277. doi: 10.1038/s41396-018-0159-0
Xu X. W., Wu Y. H., Wang C. S., Oren A., Wu M. (2009). Vibrio hangzhouensis sp. nov., isolated from sediment of the East China sea. Int. J. Syst. Evol. Microbiol. 59 (Pt 8), 2099–2103. doi: 10.1099/ijs.0.008698-0
Keywords: vibrio phage, genomic and comparative genomic analysis, phylogenetic analysis, schitoviridae, qingschitovirus
Citation: Jing Y, Guo R, Wang H, Liang Y, Liu Y, Feng Y, Ma Q, Shao H, Sung YY, Mok WJ, Wong LL, Zhang Y-Z, McMinn A, Wang M and Xing J (2023) Characterization and genome analysis of Vibrio phage vB_VhaP_PG11, representing a new viral genus. Front. Mar. Sci. 9:1092917. doi: 10.3389/fmars.2022.1092917
Received: 09 November 2022; Accepted: 28 December 2022;
Published: 17 January 2023.
Edited by:
Lusheng Xin, Yellow Sea Fisheries Research Institute (CAFS), ChinaReviewed by:
Liang Shen, Anhui Normal University, ChinaJun Sun, China University of Geosciences Wuhan, China
Copyright © 2023 Jing, Guo, Wang, Liang, Liu, Feng, Ma, Shao, Sung, Mok, Wong, Zhang, McMinn, Wang and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yantao Liang, bGlhbmd5YW50YW9Ab3VjLmVkdS5jbg==; Min Wang, bWluZ3dhbmdAb3VjLmVkdS5jbg==; Jinyan Xing, eGluZ2p5QHFkdS5lZHUuY24=
†These authors have contributed equally to this work