
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 23 December 2022
Sec. Marine Biotechnology and Bioproducts
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1092731
This article is part of the Research Topic Omics-based identification, characterization, and validation of marine bioactive peptides View all 6 articles
 Ruihan Li1,2,3†
Ruihan Li1,2,3† Yu Huang1,2†
Yu Huang1,2† Chao Peng1,2
Chao Peng1,2 Zijian Gao4
Zijian Gao4 Jie Liu5
Jie Liu5 Xiaoting Yin6,7,8,9
Xiaoting Yin6,7,8,9 Bingmiao Gao10,11
Bingmiao Gao10,11 Tatiana V. Ovchinnikova12
Tatiana V. Ovchinnikova12 Limei Qiu6,7,8*
Limei Qiu6,7,8* Chao Bian1,2,3*
Chao Bian1,2,3* Qiong Shi1,2,3*
Qiong Shi1,2,3*Individual cone snail (Conus sp.) contains thousands of bioactive peptides, but there are limited studies on its antimicrobial peptides (AMPs). Here, we investigated AMPs along with AMP-derived genes in the representative Chinese tubular cone snail (C. betulinus) by integration of our previously published multi-omics (genomics, transcriptomics, and peptidomics) data. We identified a total of 466 putative AMP-derived genes from the genome, and most of them were annotated as histones. While at the mRNA level, only 77 AMP-derived genes were confirmed with transcriptomic evidence, among them Ubiquicidin, cgUbiquitin, Ap, and VK10 were the most abundant transcripts in the venom gland. In addition, 30 AMPs were further validated by peptidomics data. After in silico analysis including 3D modeling, 11 putative AMPs were chemically synthesized for an in vitro assessment, and eight peptides were proved with good antifungal activity. In summary, this work systematically characterized the AMP repertoire in C. betulinus. This valuable genetic resource provides a solid foundation for in-depth therapeutic applications and drug development.
Cone snails (Conus sp.) are a group of carnivorous marine mollusks known for their beautiful shells and special cocktail of venoms. With ~1,000 species listed on the world register of marine species (WoRMS, accessed on 29 September 2022, available online at http://www.marinespecies.org), they preferably live in intertidal zones of tropical and subtropical regions over the world (Tucker and Tenorio, 2013; Gao et al., 2017). These slow-moving predators can produce venoms with hundreds of peptides named conotoxins in each species, to prey on a variety of animals including worms, snails, and/or fishes (Gao et al., 2017; Gao et al., 2018a). With over 100 different conotoxins per Conus species, there are over 70,000 natural conotoxins estimated recently (Terlau and Olivera, 2004; Gao et al., 2017). In the past decades, conotoxins have become powerful tools for pharmacological and neuroscience research (Peng et al., 2016; Gao et al., 2018b). They are also attractive resources of new drugs due to their novel chemical structures, strong biological activity and high target selectivity (McGivern, 2007). Except conotoxins, antimicrobial peptides (AMPs) also serve as potential drugs, and many of them have been investigated with pharmaceutical details.
AMPs, the first-line of defense against exogenous pathogens, are considered as an alternative for antibiotics because of their broad-spectrum antimicrobial activity, prompt synthesis after infection and being less prone to resistance (Pasupuleti et al., 2012; Zhang and Gallo, 2016; Lewies et al., 2019). The complex multi-component venom contains various bioactive peptides including AMPs, which have been identified in diverse toxic animals such as spiders, snakes, scorpions and bees for promising therapeutic drug developments (de Barros et al., 2019; Tang et al., 2020; Khalil et al., 2021; Tawfik et al., 2021). AMPs in cone snails seem to be less attentional (Yang et al., 2001; Periyasamy et al., 2012; De León-Nava et al., 2016; Bernáldez-Sarabia et al., 2019; Younis et al., 2019; Ebou et al., 2021) compared to other animals. A previous study reported that a O1-superfamily conotoxin extracted from Californian cone (Californiconus californicus) venom can inhibit the growth of bacteria Mycobacterium tuberculosis (Bernáldez-Sarabia et al., 2019). Another type of conotoxin, namely ω-conotoxin MVIIA with identification from several cone snails as cyclic analogs, has shown antimicrobial activity against various bacteria (Yang et al., 2001; Hemu and Tam, 2017). With the explosion of multi-omics data, a recent review reported a proteotranscriptomic-based strategy to identify AMPs from cone snails (Ebou et al., 2021).
Although more than 3,000 AMPs have been identified from animals, fungi, plants, and bacteria to date (Wang et al., 2016; Haney et al., 2017), less attention has been paid to cone snails except for few reports such as those mentioned above. Here, we performed a multi-omics investigation on AMP/AMP-derived genes in the representative Chinese tubular cone snail (C. betulinus; dominant in the South China sea), with the main aim to lay a good foundation for in-depth development of AMPs as potential marine drugs.
A total of 466 putative AMPs or AMP-derived genes (Supplementary Table 1; each AMP-derived gene corresponding to one AMP) were identified from the coding sequences (CDS) of the previously published C. betulinus genome (Peng et al., 2021). Among them, 65 are widely distributed in the 35 partially assembled C. betulinus pseudochromosomes (Chr1~Chr35; Figure 1). Interestingly, 13 and 12 histone-derived AMP-containing genes are colonized in Chr29 (Figure 1B) and Chr18 (Figure 1C), respectively.
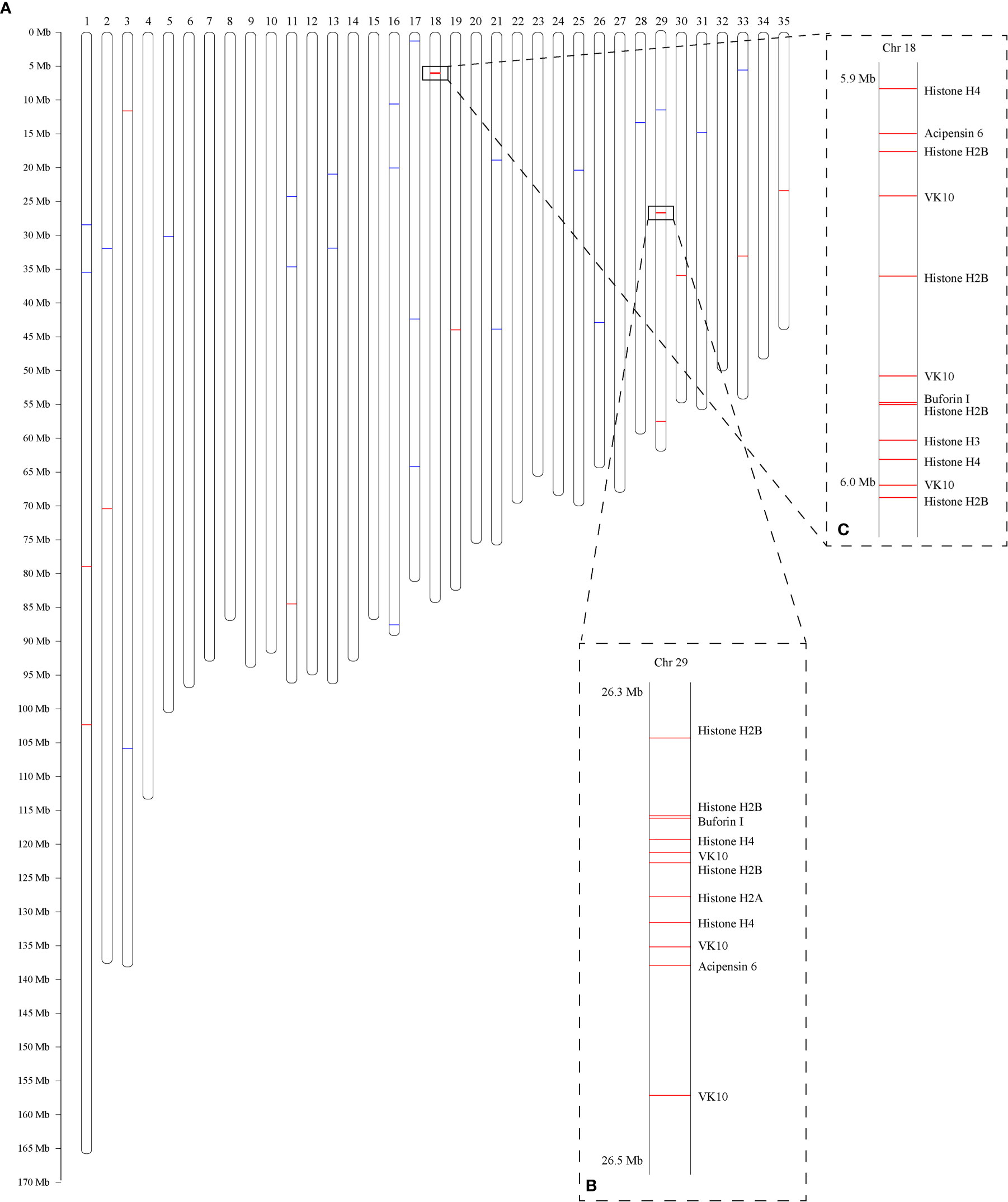
Figure 1 Genome-wide distribution of AMP-derived genes in C. betulinus. Numbers at the top represent the chromosome IDs, which are denoted with “Chr” + serial number. (A) Predicted 35 pseudochromosomes. Red bars represent histone-derived AMP-containing genes, and blue bars represent non-histone AMP-derived genes. (B) Colony of 13 histone-derived AMP-containing genes in the Chr29. (C) Colony of 12 histone-derived AMP-containing genes in the Chr18.
These putative AMPs can be divided into 13 classes (Figure 2A; Supplementary Table 1), in which histone and acipensin were the top two categories, representing almost 94.6% (441 out of 466) of all the identified AMPs. The histone class includes four types of histones (H2A, H2B, H3 and H4) and several renamed AMPs (such as buforin I, buforin II, hipposin and VK10). However, the acipensin class, also derived from histones, has been separated to form a new group (Shamova et al., 2014). Gene ontology (GO) annotation shows that these AMP-derived genes were enriched into “Human diseases” and “Cellular processes” terms, including a series of subclasses such as “Immune disease”, “Substance dependence”, “Cancer: overview”, “Infectious disease: bacterial” and “Cell growth and death” (Figure 2B). In addition, all the putative AMPs in the acipensin group were predicted to be acipensin 6 (Ac6), a fragment with 62-65 amino acids derived from the histone H2A (Shamova et al., 2014). More details of another representative yellowfin tuna GAPDH-related antimicrobial peptide (YFGAP), derived from the N-terminus of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; (Seo et al., 2012), are provided as a typical example in Figure 3.
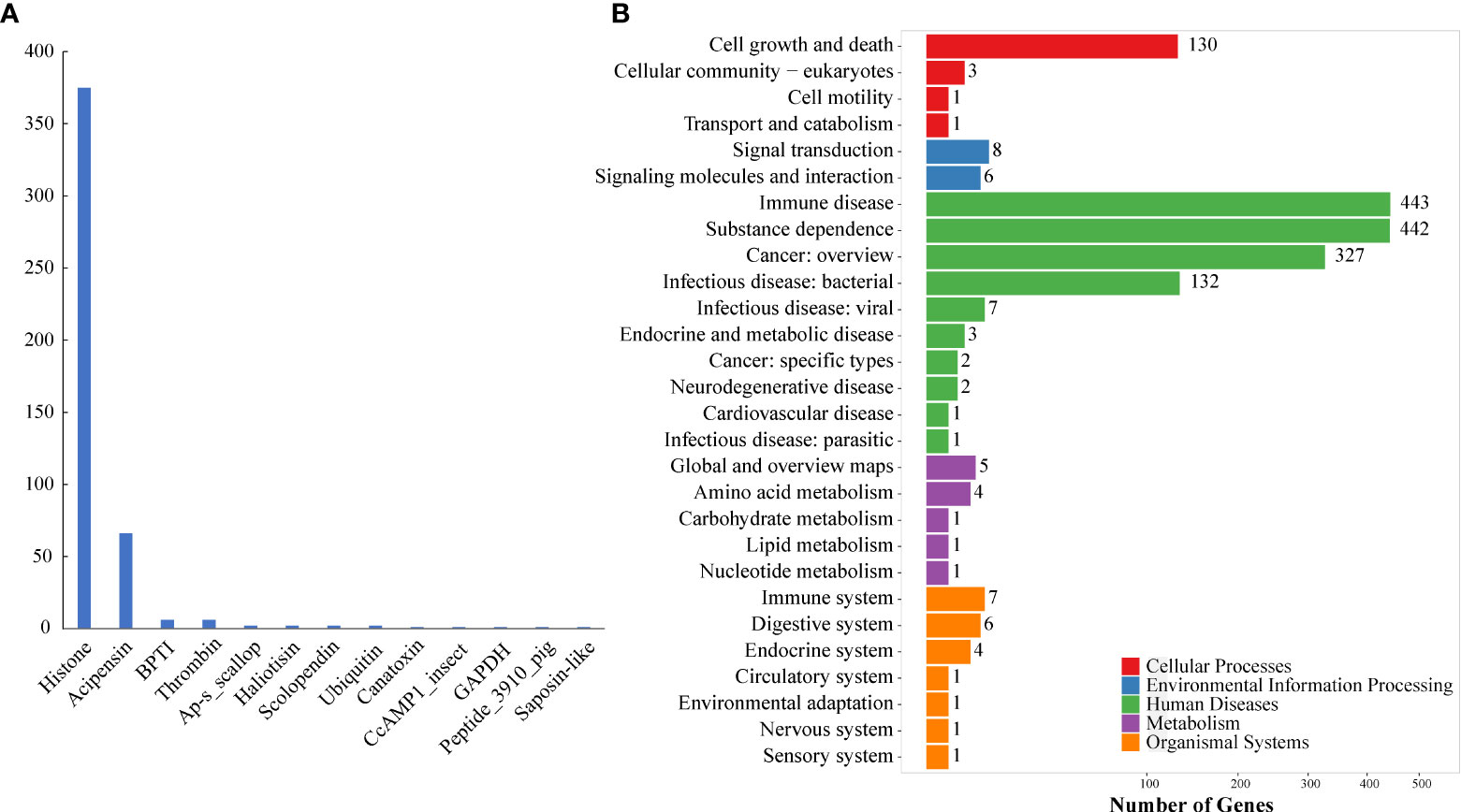
Figure 2 Classification and KEGG annotation of AMPs identified from the genome. (A) AMP classification. A total of 466 putative AMP/AMP precursors were categorized into 13 classes. The y-axis represents numbers of identified AMPs, while the x-axis represents the classified AMP groups. Histone (375) and acipensin (66) are the top two classes. (B) KEGG annotation of the identified AMP/AMP precursors. These genes were clustered into 111 KEGG pathways. Red represents “Cellular processes”, blue represents “Environmental information processing”, green represents “Human diseases”, purple represents “Metabolism”, and orange represents “Organismal systems”.
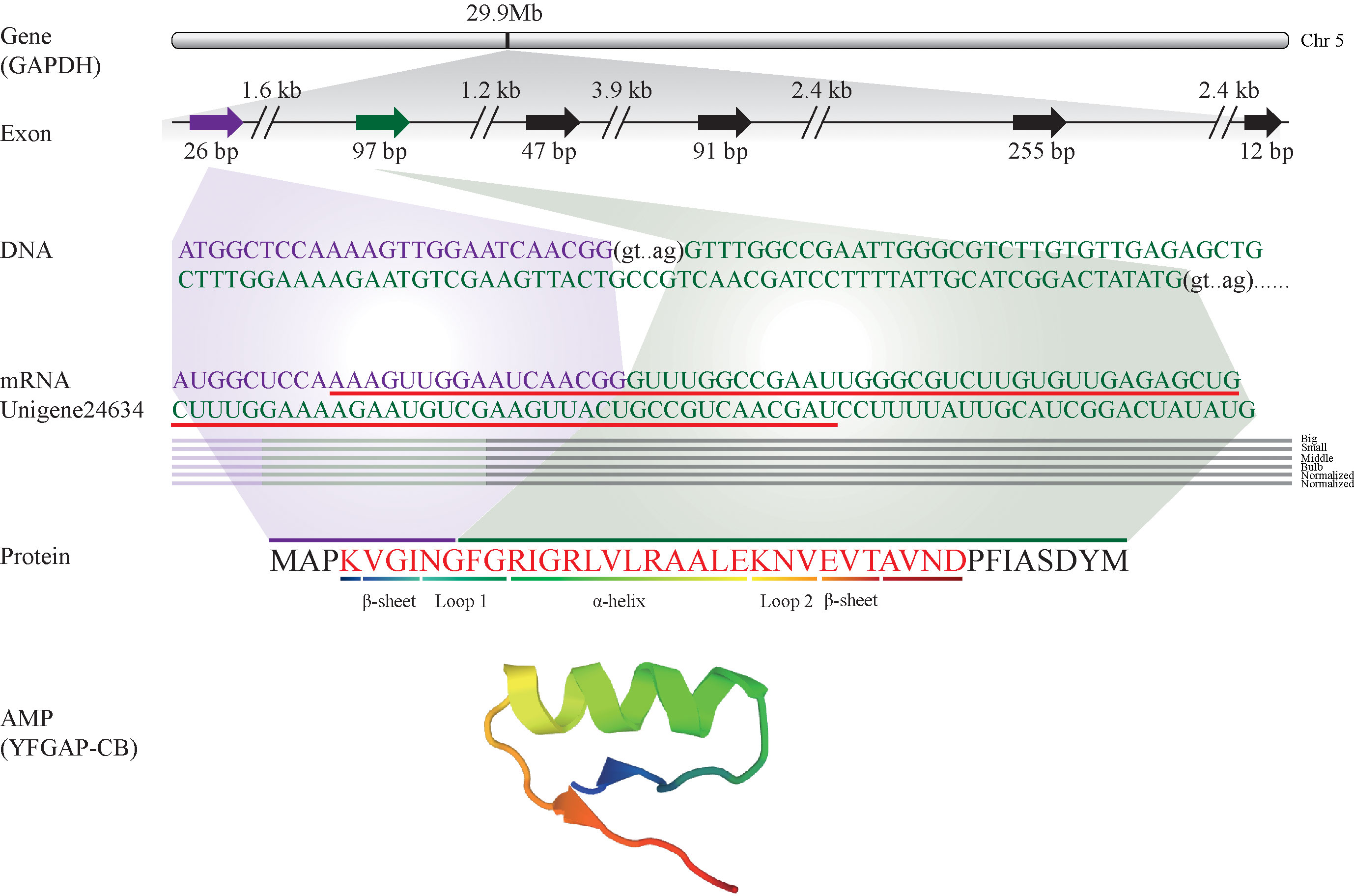
Figure 3 Identification and characterization of the representative YFGAP-CB and its derived gene (GAPDH) from the multi-omics sets of C. betulinus. YFGAP-CB is derived from the first and second exons of GAPDH gene (shadowed in purple and green, respectively) on Chr5. Double slashes represent introns. Red line highlights the nucleotide sequence with antimicrobial activity, which encodes the corresponding residues (in red) with different 3D structures.
Histone-derived AMPs is an important group, usually penetrating microorganism cell membrane and binding DNAs to inhibit their transcriptions (Parseghian and Luhrs, 2006; Kawasaki and Iwamuro, 2008; Doolin et al., 2020). Therefore, we paid more attention to the detailed number of histone genes in the whole genome of C. betulinus. Interestingly, a total of 103, 89, 115, and 96 histone genes were identified as histones H2A, H2B, H3 and H4, respectively. Compared to human, there are almost three times of histone genes in C. betulinus (see Table 1). This abundant histone gene resource may provide C. betulinus with high immunity against exogenous pathogenic microorganisms.
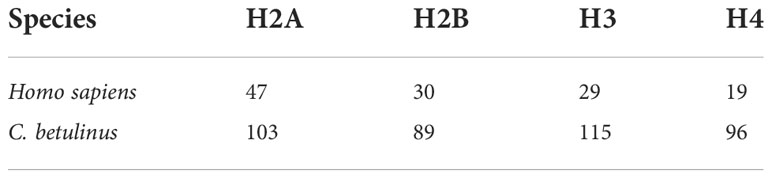
Table 1 Number of histone-derived genes in the genomes of human and C. betulinus.
From the five transcriptome datasets of C. betulinus published by us before (Peng et al., 2016), we identified 77 putative AMP-derived genes with transcriptomic evidence, of which 30, 50, 39, 51 and 44 transcripts were detected in the five (Big, Middle, Small, Bulb and Normalized) samples, respectively (Figure 4A; Supplementary Table 2). These AMPs could be divided into 19 classes (more than the 13 classes identified from the genomic data; see more details in the above section 2.1). As expected, BPTI (bovine pancreatic trypsin inhibitor), histone, thrombin, and scolopendin were the most abundant (Figure 4A).
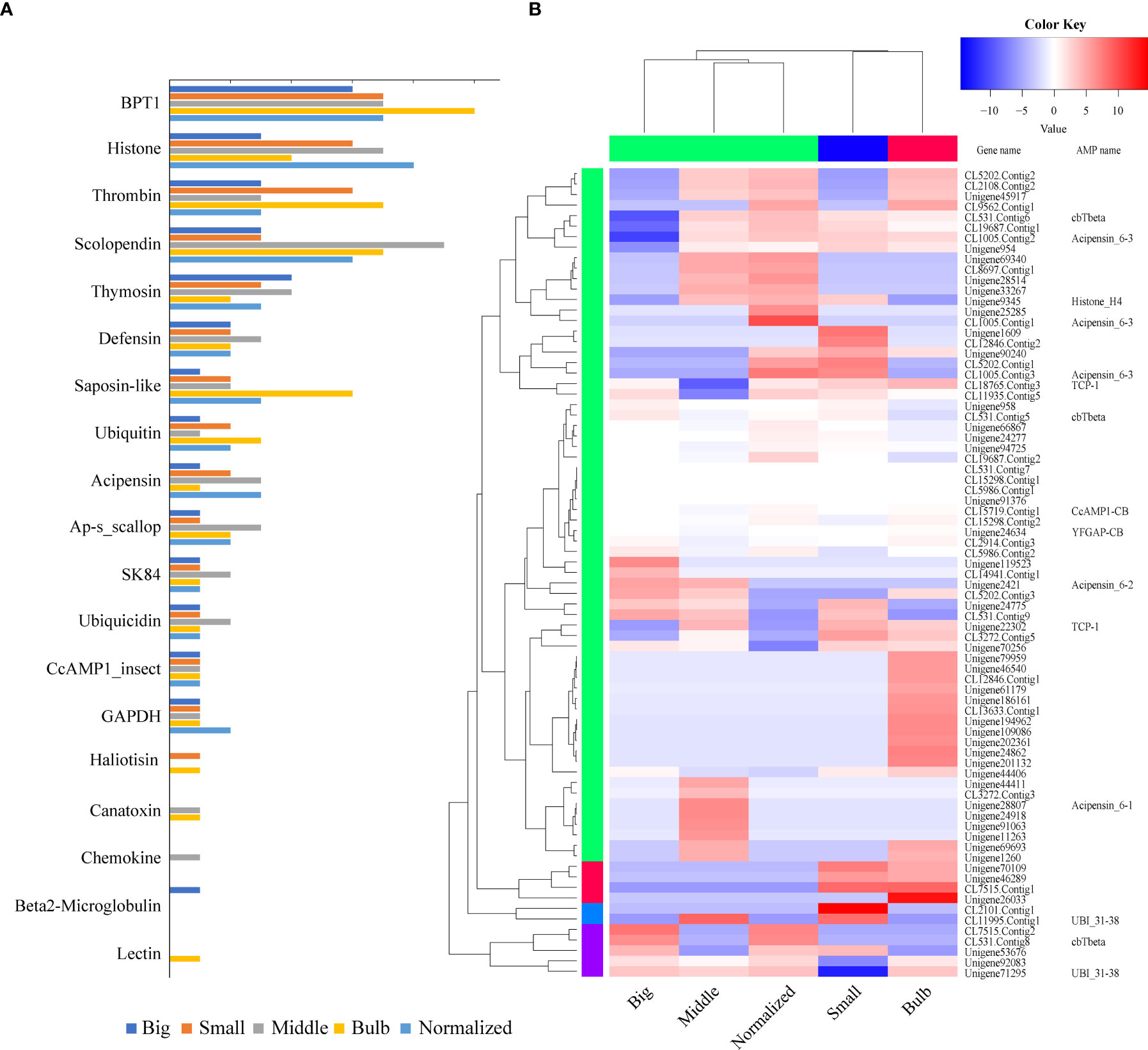
Figure 4 Tissue distribution and transcription of AMP-derived genes. (A) AMP distribution in the five transcriptomes. The y-axis represents the classified AMP groups, while the x-axis represents numbers of identified AMP/AMP precursors. BPTI, histone, thrombin, and scolopendin are the top four classes. (B) Various transcription patterns of AMP-derived genes in the five transcriptomes. Red color represents a relative high transcription level, while blue color represents a relative low transcription level.
Transcription level of each AMP-containing gene was calculated based on the routine FPKM (Fragments Per Kilobase per Million mapped reads) values (Figure 4B). The top 20 highly expressed AMP/AMP precursors in five samples were listed in Supplementary Table 3. Ubiquicidin (UBI), cgUbiquitin (an AMP isolated from the gill of Pacific oyster Crassostrea gigas; Seo et al., 2013), Ap (an AMP isolated from Argopecten purpuratus hemocytes; Arenas et al., 2009), and VK10 (a histone-derived AMP isolated from Varanus komodoensis; Bishop et al., 2017) were the most highly transcribed AMPs.
Our investigation on the AMP-containing genes revealed remarkable differences between the genome and transcriptome datasets (possibly due to the high intraspecific differences), which is similar to those previous findings of conotoxins (Peng et al., 2021). There were 40 shared AMP-derived genes with both genomic and transcriptomic evidence, of which 30 overlaps were further supported by corresponding peptides in the proteomic data. Nine genes were annotated as histones, and others included spondin-1, Filamin family, tryptase-2, Cyclin-dependent kinase family, Glyceraldehyde-3-phosphate dehydrogenase, and so on (Supplementary Table 4). A KEGG (Kyoto Encyclopedia of Genes and Genomes) cluster analysis of these 30 genes showed that “Human diseases” was still the most functional category (Supplementary Figure 1), suggesting an important role of these AMP precursors in immune and disease-resistance.
Physicochemical properties of these putative AMPs obtained from the above multi-omics data were analyzed by in silico methods. Finally, 11 peptides (see detailed sequences in Supplementary Table 5) were selected for chemical synthesis, because they were (1) positively charged cationic; (2) with isoelectric point over 8.0; (3) with CTDD and PAAC values (AMPml software, SR04248862021, China) over 0.5; (4) with α helices, β sheet, loop or extended structures; (5) with less than five Cysteines; and (6) no longer than 50 (short than 30 is preferred) in length. Among them, ten were novel AMP sequences that have never been reported before; another sequence (Histone_H4), previously reported in oyster Crassostrea virginica (Dorrington et al., 2011), was at the first time identified from cone snails. Based on 3D modeling, the 11 synthesized AMPs can be divided into three classes (Figure 5) according to their cationic secondary structures (Jenssen et al., 2006; Nguyen et al., 2011; Bin Hafeez et al., 2021): (1) nine peptides have one or several α helices; (2) one peptide (YFGAP-CB) has a mixed structure of α helix and β sheet; (3) one peptide (CcAMP1-CB) presents a β-sheet conformation. Transcription levels of these AMP-derived genes in the five examined samples (Peng et al., 2016) were calculated (Supplementary Table 6) for comparison.
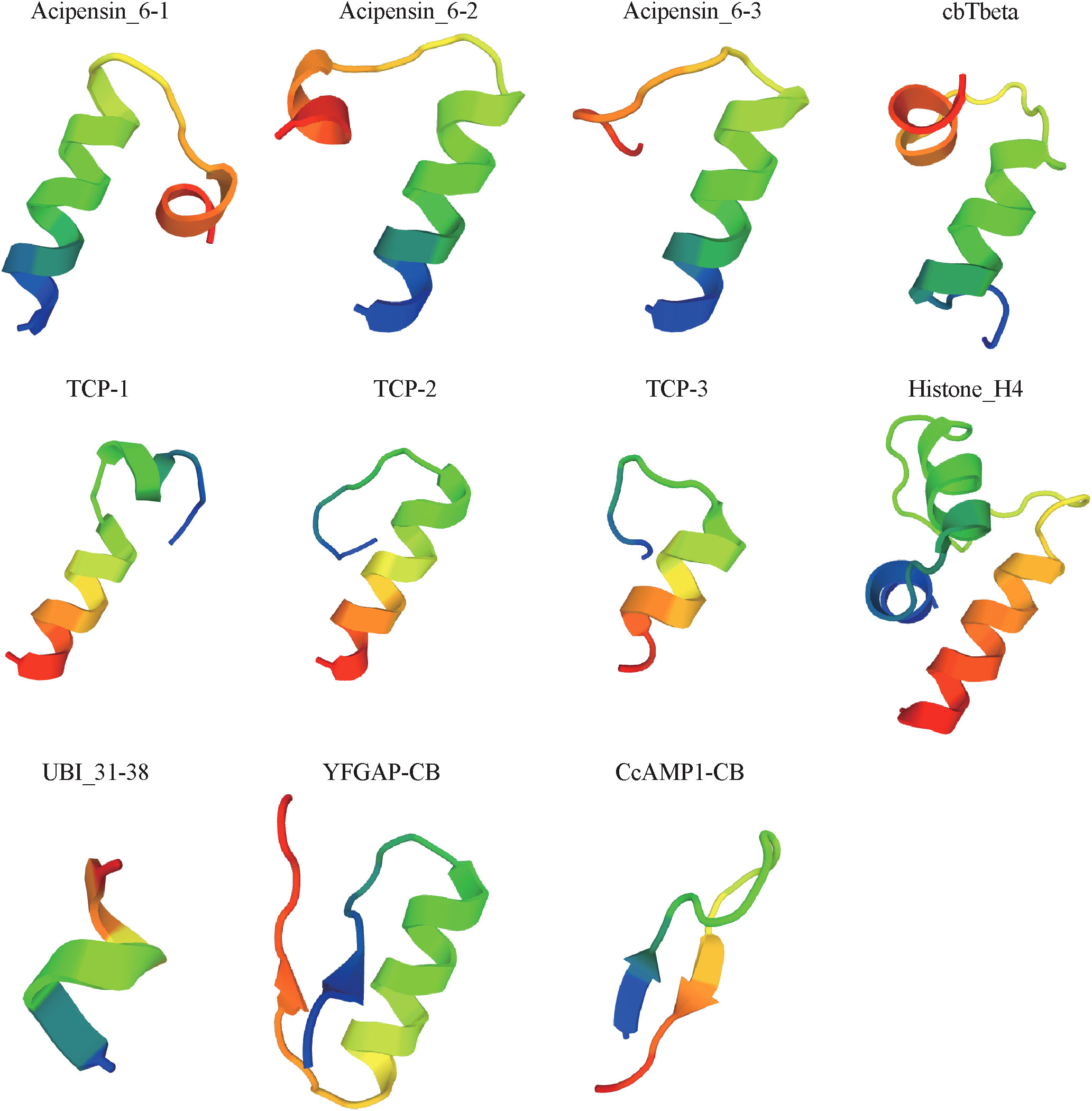
Figure 5 Predicted 3D structures of the 11 synthesized peptides.
In vitro assessment of the 11 synthesized peptides shows that nine peptides exhibited inhibition on various microorganisms (Table 2; Figure 6; Supplementary Figure 2), except for TCP-2 and TCP-3 (without antimicrobial activity). Interestingly, all except one (YFGAP-CB) of these nine peptides greatly inhibited growth of the yeast Pichia pastoris. The only non-antifungal AMP (YFGAP-CB) instead showed inhibition activity against gram-negative bacteria Escherichia coli (Figure 6A). In addition, three antifungal AMPs (cbTbeta, Histone_H4 and TCP-1; Figures 6C, F, H) had inhibitory effects on growth of different bacteria as well. For example, Histone-H4 specifically displayed the strongest inhibitory effects on gram-positive bacteria in our present study, such as significant inhibition (p<0.01) towards Streptococcus dysgalactiae (176 µM; Figure 6D) and Micrococcus luteus (88 µM; Figure 6E); however, CbTbeta also inhibited S. dysgalactiae but only at the highest concentration of 572 µM (p<0.01; Figure 6B). By contrast, TCP-1 presented antimicrobial abilities against gram-negative E. coli significantly, with the minimal concentration of 97.5 µM (p<0.01; Figure 6G). These results together illustrate that our synthesized peptides had a more general ability of antifungi, as well as specifically inhibited growth of gram positive or negative bacteria.

Table 2 Nine synthesized AMPs exhibited antimicrobial activity.
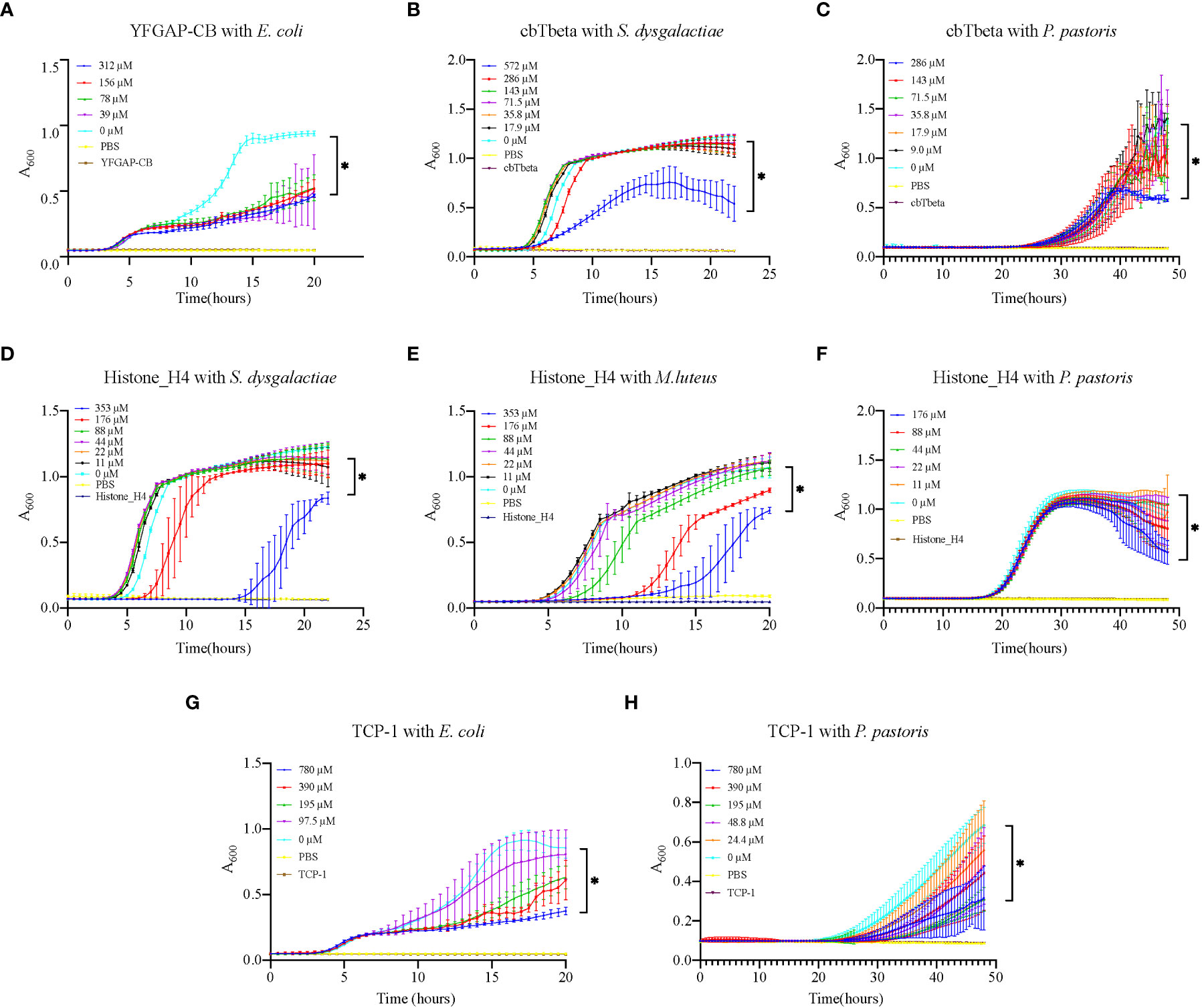
Figure 6 Growth inhibition of various microorganisms by synthesized AMPs. (A) E. coli growth inhibition by YFGAP-CB. (B) S. dysgalactiae and (C) P. pastoris growth inhibition by cbTbeta. (D) S. dysgalactiae, (E) M. luteus and (F) P. pastoris growth inhibition by Histone-H4. (G) E. coli and (H) P. pastoris growth inhibition by TCP-1. Three control groups were used in the assay: “0 µM” represents only microorganisms, “PBS” represents only PBS, and each peptide name represents only single peptide. * denotes significant differences (p<0.01) between the treatment groups and the control group.
With the wide application of AMPs in immunity and disease resistance, it is necessary to identify AMPs quickly and efficiently. High-throughput prediction of AMPs from genome and transcriptome datasets is one of the practical effective methods for discovery of novel AMPs sequences (Amaral et al., 2012; Houyvet et al., 2018; Ebou et al., 2021). By employing multi-omics analysis, we were able to obtain sequences of those potential AMP-derived genes, and further to illustrate their expression patterns in different individuals and various tissues of the same Conus species. Finally, nine predicted and synthesized peptide sequences from C. betulinus were verified with antimicrobial activity.
We identified 466 AMP-containing genes from the published genome, of which 441 were histone-derived; 77 AMP-containing genes were identified from the transcriptomes, of which 11 were histone-derived; 30 AMP-containing genes were validated from the venom proteome, of which 9 were histone-derived. The difference of histone-derived genes among these multi-omics data is possibly due to intraspecies differences as discussed before (Peng et al., 2021). Nevertheless, it also implies that some of these histone-derived genes may be silent in the venom duct and bulb at certain developmental stages, suggesting that these genes seem to perform the basic function of nucleosome formation to bind DNA in cell nucleus (Smith, 1991; Khorasanizadeh, 2004). However, as an innate defense mechanism, histone-derived AMPs were still an important category in C. betulinus, which was also verified in the subsequent antimicrobial activity assessment. To sum up, identification of active peptides (such as AMPs) besides conotoxins from cone snails could become a new strategy for the discovery of novel marine drugs.
Based on a recently published chromosome-level genome of another cone snail (C. ventricosus) (Pardos-Blas et al., 2021), we assembled the available scaffold-level genome of C. betulinus (Peng et al., 2021) to pseudochromosomes with the major aim to investigate genome-wide distribution of AMP-containing genes. A total length of 2.9 Gb was assembled to construct 35 pseudochromosomes (84.0% of the original genome; Supplementary Table 7), and the number of protein-coding genes was 20,366 (89.7% of the original protein set; Supplementary Table 8). Interestingly, only 13.9% (65 out of 466) of the identified AMP-containing genes could be located on 19 pseudochromosomes, accounting for about half of all chromosomes (Figure 1). The majority (88.0%, 22 out of 25) of non-histone AMP genes could be identified on various chromosomes, consistent with the 89.7% for location ratio in the entire gene set. However, most histone-derived AMP-containing genes were located on those unplaced scaffolds, which are worthy of in-depth investigations.
Nevertheless, both the Chr29 (Figure 1B) and Chr18 (Figure 1C) carried the most hits of 13 and 12 genes, respectively. As expected, the majority of these genes belong to the histone-derived AMP precursors. They aggregately distributed within the two regions of less than 200 kb. Since histone genes are clustered mainly onto chromosomes 1 and 6 in human, and chromosomes 3, 11 and 13 in mouse (Marzluff et al., 2002), we are therefore confident about the similar aggregative distribution of histone genes in the genome of C. betulinus. Meanwhile, most AMP-containing genes in another Conus snail C. ventricosus were predicted as histones too, and only 111 genes in the total of 958 AMP-derived genes (11.6%) were mapped onto the assembled chromosome (Supplementary Figure 3), which is consistent with our result in C. betulinus (13.9%). This may be due to the aggregation of massive histone genes in some narrow regions, but the highly repeated sequences are difficult for sequencing and assembly, thereby potentially leading to missing of many histone genes during the construction of chromosome-level genome assemblies.
AMPs are usually positively charged and amphipathic, and the formation of their secondary structures is important for antimicrobial activity (Bin Hafeez et al., 2021). The active AMPs identified from C. betulinus were all positively charged with different length of α-helical or β-sheet conformations, which may affect their antimicrobial activities. In our present study, the special YFGAP-CB with a mixed structure showed specific antibacterial activity against gram-negative bacteria instead of yeast at a low concentration of 39 µM (Figure 6A). The structures of three acipensin 6 (Figure 5) present an α-helical conformation to inhibit growth of yeast. Due to the differences of primary sequence, peptides with similar secondary structure are not always present similar antimicrobial activity (Jenssen et al., 2006). For example, TCP-2 and TCP-3 both have a α-helical conformation that is similar as TCP-1, but they didn’t exhibit antimicrobial activity. The slight differences of hydrophobicity, hydrophobic moment, net charge or helicity may also affect the antimicrobial activity of peptides (Jindal et al., 2014; Schmidtchen et al., 2014).
UBI is one of the most highly transcribed AMPs in our present study. Both full-length UBI_1-59 and truncated UBI_31-38 showed antibacterial activity in mouse (Brouwer et al., 2006). Similarly, for some long AMPs, we collected the truncated fragments (TCP-1, UBI_31-38, cbTbeta and Histone_H4) from the full-length AMPs of C. betulinus according to the reports. Our experiment preliminarily confirmed the antimicrobial activity of these truncated peptides. Therefore, combining in silico physicochemical analysis and 3D-structure modeling, as well as trimming and retaining only the active fragments reasonably, those AMP sequences with antimicrobial activity can be well designed, and the problem for synthesis of long AMP sequences could be overcame.
Most of the previously reported Conus AMPs presented either antibacterial (Periyasamy et al., 2012; Bernáldez-Sarabia et al., 2019; Younis et al., 2019) or antiparasitic activities (De León-Nava et al., 2016), while only few such as an open-chain MVIIA were antifungal (Yang et al., 2001; Hemu and Tam, 2017). In our present study, eight of the nine active synthesized AMPs showed antifungal activity, suggesting a great potential to develop Conus AMPs as antifungal agents. In addition, four AMPs also showed antibacterial activity (Figure 6). TCP-1 and YFGAP-CB activated against gram-negative bacteria, while cbTbeta and Histone_H4 inhibited gram-positive bacteria. For practical applications, these AMPs identified in our present study are valuable for further development as antimicrobial drugs and antifungal agents.
Based on sequence alignment and analysis, we identified a total of 466 putative AMP-containing genes from the genome of C. betulinus. Among them, 441 are histone-derived (375 + 66; see Figure 2A). At the mRNA level, we identified 77 putative AMP-derived genes from five transcriptome datasets, and 30 of them were further validated by proteome data. Nine synthesized peptides were confirmed with antimicrobial activity, and all but one showed inhibition against the yeast (suggesting an antifungal activity). A few AMPs specifically inhibited the growth of gram positive or negative bacteria as well. In summary, multi-omics-data based identification of AMPs and AMP-derived genes can quickly predict potential AMP/AMP precursors in a high-throughput way, which provides an efficient approach to develop marine drugs.
The applied genome (NCBI accession ID: GCA_016801955.1) and venom proteome data of C. betulinus were collected from our recently published project (Peng et al., 2021). The transcriptome assemblies of five samples (Big: venom duct of a snail 10 cm in body length, Middle: venom duct of a snail 8.7 cm in length, Small: venom duct of a snail 6 cm in length, Bulb: venom bulb from the middle-sized snail and Normalized: a normalized transcriptome of another medium-sized snail) were retrieved from another report by us (Peng et al., 2016). Protein coding regions of each transcript were predicted using TransDecoder package (Haas et al., 2013). Transcription levels of unique genes in each tissue were calculated by FPKM (Mortazavi et al., 2008). All the protein coding sequences from the genome and the transcriptomes were translated into amino acids to establish a comprehensive protein set of C. betulinus, which was subsequently used as the reference for AMP identification from the peptidomics dataset in our recently published paper (Peng et al., 2021).
We collected a total of 2,927 putative AMP sequences from the public Antimicrobial Peptides Database (APD3; (Wang et al., 2016). Our genomic gene set was firstly used to build an index database for sequence alignment. Potential AMP sequences were predicted from the genome by employing Blast v2.2.26 (McGinnis and Madden, 2004) with TBLASTN algorithms (e-value at 1e-5). The alignment hits with less than 50% aligned length were removed. The aligned regions containing potential AMP sequences were captured for further verification by using AMPml V1.2.7 (2021, China, 2021SR0424886; https://github.com/flystar233/AMPml), a software integrates machine-learning algorithms and amino-acid feature descriptors. Putative AMP sequences with values less than 0.5 in CTDD and PAAC models were filtered out. The same pipeline was applied to the transcript sets of five transcriptomes for the prediction of AMPs. Finally, we obtained nucleotide sequences for the putative AMPs and AMP-derived genes from the genome and transcriptome datasets. They were then classified based on GO annotations from the APD3 database, and functionally annotated based on the KEGG database (Kanehisa et al., 2017).
Human histone protein sequences were download from NCBI (Benson et al., 2007) as queries to predict histone genes in the C. betulinus genome, including 47, 30, 29, and 19 sequences of H2A, H2B, H3, and H4 along with their corresponding variants (Supplementary Table 9). These protein sequences were aligned to the C. betulinus genome assembly using TBLASTN (e-value: 1e-5) and manually checked.
In addition, transcription levels of all AMP-derived genes in each transcriptome set were compared and plotted using the ggplot2 package (Wickham, 2016) in R software v3.5.1 (https://www.R-project.org/). The heatmap was plotted in R script using the heatmap.2 function.
We collected the CDS sequences for AMP-derived genes from the five transcriptomes, and subsequently aligned them onto the assembled genome of C. betulinus by TBLASTN (e-value: 1e-5). Subsequently, those AMP sequences that were common in both genome and transcriptome datasets were identified to build an index database for alignment with the proteome data. The final list of shared AMPs among the three levels of omics datasets was generated by Blastp, and those sequences with aligned length of over 50% and similarity of over 80% were considered reliable.
Physicochemical traits were examined by Expasy’s ProtParam tool (Gasteiger et al., 2005) (https://web.expasy.org/protparam/), including molecular weight, isoelectric point, net charge and grand average of hydropathicity. 3D structures were built by PEP-FOLD3 (Lamiable et al., 2016), a de novo predicting approach fitted for short peptides. We manually collected 11 putative AMPs from the multi-omics datasets (Supplementary Table 5). Four candidate peptides (TCP-1, UBI_31-38, cbTbeta and Histone_H4) were truncated according to those verified active AMP truncations that were reported in previous studies, such as 31-38 residues of UBI (Brouwer et al., 2006), and 25-amino-acid thrombin-derived C-terminal peptide (TCP-25) (Petruk et al., 2020; Dahlman et al., 2021).
The 11 putative AMPs were chemically synthesized. All the peptides were assembled in insoluble resins based on Fmoc solid-phase peptide synthesis (Kent, 1988) at GL Biochem Ltd. (Shanghai, China). Generated peptide products were subsequently purified using Kromasil 100-5-C18 columns (4.6 mm × 250 mm) based on an analytical HPLC system and then were eluted with 1mL/min acetonitrile gradient. Purity of all peptides were over 95% (determined by HPLC-MS/MS; see Supplementary Figure 4) and stored in sterile deionized water at -80°C.
These synthesized peptides (2 mg each) were dissolved in phosphate buffer saline (PBS) and serial two-fold diluted multiple concentrations for antimicrobial assaying against E. coli, S. dysgalactiae, M. luteus and P. pastoris as previously reported (Jia et al., 2016). Each strain was incubated to the logarithmic phase before centrifugation and then washed by PBS for three times. All microorganisms were resuspended in PBS (104 CFU/mL). Subsequently, we mixed 10 µL of bacterial suspensions and 10 µL of PBS-dissolved synthesized peptides respectively and incubated them at 37° for 2 h. Three control groups were used in the assay, including only microorganisms, only peptides, and only PBS. In the final antimicrobial assays, we dispensed the mixtures into 96-well plates, and each well contained 200 µL of LB medium. All plates were incubated at 37° in a microplate reader (Biotek, Winooski, VT, USA). OD600 values were measured per 0.5 h after a shake with ten seconds for a total of 20 h (bacteria) or 48 h (fungus) to generate the growth curves. There were three parallel wells for each peptide, and the experiments were repeated at least twice to obtain reliable results.
All data were analyzed with the Statistical Package for Social Sciences (SPSS; IBM, Armonk, NY, USA) v26.0 and GraphPad Prism version 9.0.0 for Windows (GraphPad Software, San Diego, CA, USA; www.graphpad.com), and presented as mean ± standard deviation (n = 3). Paired two-tailed Student’s t-test and multiple comparisons were performed to test significant differences among various groups.
We obtained a pairwise whole-genome alignment between a scaffold-level genome of C. betulinus (Peng et al., 2021) with a recently published chromosome-level genome of C. ventricosus (Pardos-Blas et al., 2021) using Lastz v1.1 (Harris, 2007). Pseudochromosomes of C. betulinus were subsequently constructed after recombining the aligned regions of each chromosome. Localization of those identified AMP-derived genes on various pseudochromosomes were thereafter performed according to the one-to-one correspondence.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Conceptualization, QS; validation, QS, CB and YH; formal analysis, RL, JL and ZG; performed the experiments, XY and LQ; investigation, RL and YH; data curation, CP, BG and YH; writing—original draft preparation, RL, YH, XY and CP; visualization, RL; writing—review and editing, QS, CB, LQ, TO and BG; supervision, QS; project administration, YH and RL; funding acquisition, QS. All authors contributed to the article and approved the submitted version.
This research was funded by Shenzhen Science and Technology Program (Nos. JSGG20201102145601004, GJHZ20190819152407214), and Dapeng Special Program for Industrial Development (No. KJYF202001-17).
We thank Miss Yunhai Yi, a PhD student at University of Groningen, Netherlands, for her technical assistance. Mr. Tengfei Xu, the author of AMPml V1.2.7, is grateful for his assistance in using the software.
Author JL was employed by company BGI Genomics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1092731/full#supplementary-material
Amaral A. C., Silva O. N., Mundim N. C., de Carvalho M. J., Migliolo L., Leite J. R., et al. (2012). Predicting antimicrobial peptides from eukaryotic genomes: in silico strategies to develop antibiotics. Peptides 37 (2), 301–308. doi: 10.1016/j.peptides.2012.07.021
Arenas G., Guzmán F., Cárdenas C., Mercado L., Marshall S. H. (2009). A novel antifungal peptide designed from the primary structure of a natural antimicrobial peptide purified from Argopecten purpuratus hemocytes. Peptides 30 (8), 1405–1411. doi: 10.1016/j.peptides.2009.05.019
Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Wheeler D. L. (2007). GenBank. Nucleic Acids Res. 35 (Database issue), D21–D25. doi: 10.1093/nar/gkl986
Bernáldez-Sarabia J., Figueroa-Montiel A., Dueñas S., Cervantes-Luévano K., Beltrán J. A., Ortiz E., et al. (2019). The diversified O-superfamily in Californiconus californicus presents a conotoxin with antimycobacterial activity. Toxins 11 (2), 128. doi: 10.3390/toxins11020128
Bin Hafeez A., Jiang X., Bergen P. J., Zhu Y. (2021). Antimicrobial peptides: an update on classifications and databases. Int. J. Mol. Sci. 22 (21), 11691. doi: 10.3390/ijms222111691
Bishop B. M., Juba M. L., Russo P. S., Devine M., Barksdale S. M., Scott S., et al. (2017). Discovery of novel antimicrobial peptides from Varanus komodoensis (Komodo dragon) by large-scale analyses and de-novo-assisted sequencing using electron-transfer dissociation mass spectrometry. J. Proteome Res. 16 (4), 1470–1482. doi: 10.1021/acs.jproteome.6b00857
Brouwer C. P., Bogaards S. J., Wulferink M., Velders M. P., Welling M. M. (2006). Synthetic peptides derived from human antimicrobial peptide ubiquicidin accumulate at sites of infections and eradicate (multi-drug resistant) Staphylococcus aureus in mice. Peptides 27 (11), 2585–2591. doi: 10.1016/j.peptides.2006.05.022
Dahlman A., Puthia M., Petrlova J., Schmidtchen A., Petruk G. (2021). Thrombin-derived c-terminal peptide reduces Candida-induced inflammation and infection in vitro and in vivo. Antimicrobial Agents Chemotherapy 65 (11), e01032–e01021. doi: 10.1128/AAC.01032-21
de Barros E., Gonçalves R. M., Cardoso M. H., Santos N. C., Franco O. L., Cândido E. S. (2019). Snake venom cathelicidins as natural antimicrobial peptides. Front. Pharmacol. 10. doi: 10.3389/fphar.2019.01415
De León-Nava M. A., Romero-Núñez E., Luna-Nophal A., Bernáldez-Sarabia J., Sánchez-Campos L. N., Licea-Navarro A. F., et al. (2016). In vitro effect of the synthetic cal14.1a conotoxin, derived from Conus californicus, on the human parasite Toxoplasma gondii. Mar. Drugs 14 (4), 66. doi: 10.3390/md14040066
Doolin T., Gross S., Siryaporn A. (2020). Physical mechanisms of bacterial killing by histones. Adv. In Exp. Med. And Biol. 1267, 117–133. doi: 10.1007/978-3-030-46886-6_7
Dorrington T., Villamil L., Gómez-chiarri M. (2011). Upregulation in response to infection and antibacterial activity of oyster histone H4. Fish Shellfish Immunol. 30 (1), 94–101. doi: 10.1016/j.fsi.2010.09.006
Ebou A., Koua D., Addablah A., Kakou-Ngazoa S., Dutertre S. (2021). Combined proteotranscriptomic-based strategy to discover novel antimicrobial peptides from cone snails. Biomedicines 9 (4), 344. doi: 10.3390/biomedicines9040344
Gao B., Peng C., Chen Q., Zhang J., Shi Q. (2018a). Mitochondrial genome sequencing of a vermivorous cone snail Conus quercinus supports the correlative analysis between phylogenetic relationships and dietary types of Conus species. PloS One 13 (7), e0193053. doi: 10.1371/journal.pone.0193053
Gao B., Peng C., Yang J., Yi Y., Zhang J., Shi Q. (2017). Cone snails: a big store of conotoxins for novel drug discovery. Toxins 9 (12), 397. doi: 10.3390/toxins9120397
Gao B., Peng C., Zhu Y., Sun Y., Zhao T., Huang Y., et al. (2018b). High throughput identification of novel conotoxins from the vermivorous oak cone snail (Conus quercinus) by transcriptome sequencing. Int. J. Mol. Sci. 19 (12), 3901. doi: 10.3390/ijms19123901
Gasteiger E., Hoogland C., Gattiker A., Duvaud S. E., Wilkins M. R., Appel R. D., et al. (2005). “Protein identification and analysis tools on the ExPASy server,” in The proteomics protocols handbook. Ed. Walker J. M. (Totowa, NJ: Humana Press), 571–607.
Haas B. J., Papanicolaou A., Yassour M., Grabherr M., Blood P. D., Bowden J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8 (8), 1494–1512. doi: 10.1038/nprot.2013.084
Haney E. F., Mansour S. C., Hancock R. E. (2017). Antimicrobial peptides: an introduction. Methods Mol. Biol. 1548, 3–22. doi: 10.1007/978-1-4939-6737-7_1
Harris R. S. (2007). Improved pairwise alignment of genomic DNA (State College, PA, USA: The Pennsylvania State University).
Hemu X., Tam J. P. (2017). Macrocyclic antimicrobial peptides engineered from ω-conotoxin. Curr. Pharm. Design 23 (14), 2131–2138. doi: 10.2174/1381612822666161027120518
Houyvet B., Zanuttini B., Corre E., Le Corguillé G., Henry J., Zatylny-Gaudin C. (2018). Design of antimicrobial peptides from a cuttlefish database. Amino Acids 50 (11), 1573–1582. doi: 10.1007/s00726-018-2633-4
Jenssen H., Hamill P., Hancock Robert E. W. (2006). Peptide antimicrobial agents. Clin. Microbiol. Rev. 19 (3), 491–511. doi: 10.1128/CMR.00056-05
Jia Z., Zhang H., Jiang S., Wang M., Wang L., Song L. (2016). Comparative study of two single CRD c-type lectins, CgCLec-4 and CgCLec-5, from pacific oyster crassostrea gigas. Fish Shellfish Immunol. 59, 220–232. doi: 10.1016/j.fsi.2016.10.030
Jindal M., Le C., Mohd Yusof M., Sekaran S. (2014). Net charge, hydrophobicity and specific amino acids contribute to the activity of antimicrobial peptides. J. Health Trans. Med. 17 (1), 1–7. doi: 10.22452/jummec.vol17no1.1
Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45 (D1), D353–d361. doi: 10.1093/nar/gkw1092
Kawasaki H., Iwamuro S. (2008). Potential roles of histones in host defense as antimicrobial agents. Infect. Disord. Drug Targets 8 (3), 195–205. doi: 10.2174/1871526510808030195
Kent S. B. (1988). Chemical synthesis of peptides and proteins. Annu. Rev. Biochem. 57, 957–989. doi: 10.1146/annurev.bi.57.070188.004521
Khalil A., Elesawy B. H., Ali T. M., Ahmed O. M. (2021). Bee venom: from venom to drug. Molecules 26 (16), 4941. doi: 10.3390/molecules26164941
Khorasanizadeh S. (2004). The nucleosome: from genomic organization to genomic regulation. Cell 116 (2), 259–272. doi: 10.1016/S0092-8674(04)00044-3
Lamiable A., Thévenet P., Rey J., Vavrusa M., Derreumaux P., Tufféry P. (2016). PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 44 (W1), W449–W454. doi: 10.1093/nar/gkw329
Lewies A., Du Plessis L. H., Wentzel J. F. (2019). Antimicrobial peptides: the achilles' heel of antibiotic resistance? Probiotics Antimicrobial Proteins 11 (2), 370–381. doi: 10.1007/s12602-018-9465-0
Marzluff W. F., Gongidi P., Woods K. R., Jin J., Maltais L. J. (2002). The human and mouse replication-dependent histone genes. Genomics 80 (5), 487–498. doi: 10.1006/geno.2002.6850
McGinnis S., Madden T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32 (Web Server issue), W20–W25. doi: 10.1093/nar/gkh435
McGivern J. G. (2007). Ziconotide: a review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat 3 (1), 69–85. doi: 10.2147/nedt.2007.3.1.69
Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 5 (7), 621–628. doi: 10.1038/nmeth.1226
Nguyen L. T., Haney E. F., Vogel H. (2011). The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 29 (9), 464–472. doi: 10.1016/j.tibtech.2011.05.001
Pardos-Blas J. R., Irisarri I., Abalde S., Afonso C. M. L., Tenorio M. J., Zardoya R. (2021). The genome of the venomous snail Lautoconus ventricosus sheds light on the origin of conotoxin diversity. GigaScience 10 (5), giab037. doi: 10.1093/gigascience/giab037
Parseghian M. H., Luhrs K. A. (2006). Beyond the walls of the nucleus: the role of histones in cellular signaling and innate immunity. Biochem. Cell Biol. 84 (4), 589–595. doi: 10.1139/o06-082
Pasupuleti M., Schmidtchen A., Malmsten M. (2012). Antimicrobial peptides: key components of the innate immune system. Crit. Rev. Biotechnol. 32 (2), 143–171. doi: 10.3109/07388551.2011.594423
Peng C., Huang Y., Bian C., Li J., Liu J., Zhang K., et al. (2021). The first Conus genome assembly reveals a primary genetic central dogma of conopeptides in C. betulinus. Cell Discovery 7 (1), 11. doi: 10.1038/s41421-021-00244-7
Peng C., Yao G., Gao B. M., Fan C. X., Bian C., Wang J., et al. (2016). High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. GigaScience 5, 17. doi: 10.1186/s13742-016-0122-9
Periyasamy N., Arularasan S., Gayathri S. (2012). Antibacterial activity of the tissue extracts of Conus betulinus and Conus inscriptus Linnaeus 1758 (Mollusca: Gastropoda) from nagapattinam, southeast coast of India. Asian Pacific J. Trop. Dis. 2, S914–S919. doi: 10.1016/s2222-1808(12)60291-6
Petruk G., Petrlova J., Samsudin F., Giudice R. D., Bond P. J., Schmidtchen A. (2020). Concentration- and pH-dependent oligomerization of the thrombin-derived c-terminal peptide TCP-25. Biomolecules 10 (11), 1572. doi: 10.3390/biom10111572
Schmidtchen A., Pasupuleti M., Malmsten M. (2014). Effect of hydrophobic modifications in antimicrobial peptides. Adv. Colloid Interfac. 205, 265–274. doi: 10.1016/j.cis.2013.06.009
Seo J.-K., Lee M. J., Go H.-J., Kim G. D., Jeong H. D., Nam B.-H., et al. (2013). Purification and antimicrobial function of ubiquitin isolated from the gill of pacific oyster, Crassostrea gigas. Mol. Immunol. 53 (1), 88–98. doi: 10.1016/j.molimm.2012.07.003
Seo J. K., Lee M. J., Go H. J., Park T. H., Park N. G. (2012). Purification and characterization of YFGAP, a GAPDH-related novel antimicrobial peptide, from the skin of yellowfin tuna, Thunnus albacares. Fish Shellfish Immunol. 33 (4), 743–752. doi: 10.1016/j.fsi.2012.06.023
Shamova O. V., Orlov D. S., Balandin S. V., Shramova E. I., Tsvetkova E. V., Panteleev P. V., et al. (2014). Acipensins - novel antimicrobial peptides from leukocytes of the Russian sturgeon Acipenser gueldenstaedtii. Acta Naturae 6 (4), 99–109. doi: 10.32607/20758251-2014-6-4-99-109
Smith M. M. (1991). Histone structure and function. Curr. Opin. Cell Biol. 3 (3), 429–437. doi: 10.1016/0955-0674(91)90070-F
Tang X., Yang J., Duan Z., Jiang L., Liu Z., Liang S. (2020). Molecular diversification of antimicrobial peptides from the wolf spider lycosa sinensis venom based on peptidomic, transcriptomic, and bioinformatic analyses. Acta Biochim. Biophys. Sin. 52 (11), 1274–1280. doi: 10.1093/abbs/gmaa107
Tawfik M. M., Bertelsen M., Abdel-Rahman M. A., Strong P. N., Miller K. (2021). Scorpion venom antimicrobial peptides induce siderophore biosynthesis and oxidative stress responses in escherichia coli. mSphere 6 (3), e00267–e00221. doi: 10.1128/mSphere.00267-21
Terlau H., Olivera B. M. (2004). Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev. 84 (1), 41–68. doi: 10.1152/physrev.00020.2003
Tucker J., Tenorio M. (2013). Illustrated Catalog of the Living Cone Shells. (Wellington, FL. USA: MDM Press), 517.
Wang G., Li X., Wang Z. (2016). APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44 (D1), D1087–D1093. doi: 10.1093/nar/gkv1278
Wickham H. (2016). “Data analysis,” in ggplot2: Elegant graphics for data analysis (Berlin, Germany: Springer-Verlag New York Press), 189–201.
Yang J.-L., Lu Y.-A., Wu C., Tam J. P. (2001). “Antimicrobial and chemotactic activities of ω-conotoxin cyclic analogues,” in Peptides: The wave of the future (Dordrecht, the Nethelands: Springer Netherlands Press), 487–488.
Younis S., Taj S., Rashid S. (2019). Structural studies of Staphylococcus aureus aortase inhibiton via conus venom peptides. Arch. Biochem. Biophysics 671, 87–102. doi: 10.1016/j.abb.2019.06.003
Keywords: Conus betulinus, antimicrobial peptide, multi-omics, in silico prediction, in vitro assessment
Citation: Li R, Huang Y, Peng C, Gao Z, Liu J, Yin X, Gao B, Ovchinnikova TV, Qiu L, Bian C and Shi Q (2022) High-throughput prediction and characterization of antimicrobial peptides from multi-omics datasets of Chinese tubular cone snail (Conus betulinus). Front. Mar. Sci. 9:1092731. doi: 10.3389/fmars.2022.1092731
Received: 08 November 2022; Accepted: 30 November 2022;
Published: 23 December 2022.
Edited by:
Bin Wu, Zhejiang University, ChinaReviewed by:
Yueying Li, University of Oklahoma, United StatesCopyright © 2022 Li, Huang, Peng, Gao, Liu, Yin, Gao, Ovchinnikova, Qiu, Bian and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Limei Qiu, cWl1bGltZWlAcWRpby5hYy5jbg==; Chao Bian, YmlhbmNoYW9AZ2Vub21pY3MuY24=; Qiong Shi, c2hpcWlvbmdAZ2Vub21pY3MuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.