 Ruzhuo Zhong
Ruzhuo Zhong Jianqiang Huang
Jianqiang Huang Yongshan Liao
Yongshan Liao Chuangye Yang
Chuangye Yang Qingheng Wang
Qingheng Wang Yuewen Deng
Yuewen Deng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 23 December 2022
Sec. Aquatic Physiology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1076804
The peanut worm (Sipunculus nudus) is an important economic and ecological fishery resource in China. To determine how S. nudus interacts with its culture environment, in this study, the diversity, composition, and interrelationships of bacterial communities in the intestine, coelomic fluid, and culture environment of S. nudus through high-throughput sequencing using the Illumina HiSeq sequencing platform. Alpha diversity analysis showed that the highest bacterial community richness and diversity were found in the sediments. Species annotation revealed that Proteobacteria dominated in all samples (48.92%–58.36%), and the other dominant phyla were highly variable, indicating a certain independence between the environment and the composition of the bacterial community of S. nudus. The coelomic fluid of S. nudus contained high concentrations of Bacillus and could be a source of potential probiotic bacteria for isolation and culture. Cluster and operational taxonomic unit Venn diagram analyses showed that the bacterial community composition in the intestine was more similar to that in the sediments compared with water. The predicted functional analysis of bacterial communities indicated that the functions of bacterial communities in the different surrounding environment were highly similar and involved in various aspects, such as membrane transport, amino acid metabolism, carbohydrate metabolism, and replication and repair. Our findings have important implications on the establishment of sustainable microecological regulation and management strategies for S. nudus culture and provide a reference for the development and utilization of probiotic bacteria.
The symbiotic associations between microorganisms and macroorganisms are ubiquitous and have been extensively studied. Microbial symbionts play key roles in the survival, homeostasis, and development of eukaryotic organisms (Taylor et al., 2007; Kamada et al., 2013; Foster et al., 2017; Bang et al., 2018), and the eukaryotic host provides a stable environment and constant nutrient supply for the associated bacteria. Previous studies have demonstrated that host-associated microbial community compositions are not stochastic but mostly determined by host phylogeny and the living environment (Zoetendal et al., 2001; Cárdenas et al., 2014; Brooks et al., 2016; Carrier and Reitzel, 2018). Unlike terrestrial animals, aquatic animals are directly exposed to water and even sediments, where microbes are the essential components of productivity, nutrient cycling, and water quality (Blancheton et al., 2013; Carbone and Faggio, 2016; Li TT et al., 2017). This information suggests that microbes play key roles in the material cycle and energy flow of aquaculture ecosystems (Moriarty, 1997). Therefore, establishing effective microbial and ecological strategies is essential for achieving sustainable aquaculture production and an in-depth and complete understanding of the characteristics of microflora in aquatic environments (Xiong et al., 2016). Numerous studies have considered the relationships between bacterial communities in aquatic animals and their cultured environment (Fan and Li, 2019; Fan et al., 2019; Sun et al., 2019; Musella et al., 2020; Sun et al., 2020; Zheng et al., 2021).
Peanut worm (Sipunculus nudus) is an edible marine sediment-inhabiting invertebrate (Li et al., 2015) that has a simple body structure; its digestive tract is bathed in coelomic fluid (Yang et al., 2020). It is frequently referred to as the “cordyceps of the sea” because of its delicious taste, abundant nutrients, and traditional Chinese medicine values (Zhang et al., 2011). Moreover, S. nudus is cultured along beaches with organisms by using nutrients from surface sediments without the need for a supplemental artificial diet (Adrianov and Maiorova, 2010). Hence, S. nudus is an important economic and ecological fishery resource in China. In recent years, studies on S. nudus have focused on its population genetics (Du et al., 2008; Du et al., 2009) and reproductive biology (Cao et al., 2020; Yang et al., 2020; Zhang et al., 2021). Although the intestinal microbiota of S. nudus has been studied in the past few years (Wang et al., 2018; Li et al., 2019), the relationship between S. nudus and its surrounding environment has seldom been investigated.
To address these issues, this study investigated and analyzed the bacterial communities in S. nudus (intestine and coelomic fluid) and its aquaculture environment (sediment and water). This work could help establish the relationship between different hosts and their associated intestinal microbe, improve the understanding of the health status and nutritional level of the hosts, and provide a reference for disease prevention in cultured aquatic animals.
Samples of S. nudus and its culture environment were collected from Zhanjiang Green Bay Aquatic Science and Technology Co., Ltd. (Zhanjiang, Guangdong, China). Healthy individuals with a similar size and undamaged body surface were collected randomly in the mudflat area, and their average weight was (12.24 ± 1.54) g. The body surface of the S. nudus was disinfected with 75% alcohol. The coelomic fluid samples were extracted with a 5 ml disposable sterile syringe (avoiding the internal organs of the S. nudus). Subsequently, sterilized scissors were used to dissections and remove the intact intestines of S. nudus. The intestinal were rinsed with sterile seawater three times, and then placed in sterile lyophilized tubes. The sediment samples (5 cm of the surface mud) were randomly in the S. nudus culture area were directly loaded into sterile freeze-storage tubes. 2 L seawater in the S. nudus culture area was prefiltered to remove large particles, then refiltered using a polycarbonate membrane with a pore size of 0.22 mm. Following filtration, the membrane was placed in a 2.0 mL sterile centrifuge tube. And all collected samples were immediately kept in liquid nitrogen and stored at -80°C for testing. Five intestinal samples (sample numbers I1, I2, I3, I4, and I5), five coelomic fluid samples (sample numbers Cf1, Cf2, Cf3, Cf4, and Cf5), five water samples (sample numbers W1, W2, W3, W4, and W5), and five sediment samples (sample numbers S1, S2, S3, S4, and S5) were included in the study.
The samples of S. nudus were placed in a 4°C refrigerator for 10 min for cryo-anesthesia. Subsequently, the body surface of the samples was disinfected using alcohol with 70% concentration, and the intact intestine was removed through dissection. The intestines were rinsed three times with sterile seawater then placed in sterile lyophilization tubes and marked. The samples were homogenized separately by using a tissue homogenizer, and the microbial genomic DNA was extracted from the intestine and coelomic fluid of S. nudus with the Stool DNA Kit (OMEGA, USA). The microbial genomic DNA was extracted from the water and sediment of the culture environment with the Water DNA Kit from OMEGA. All DNA extraction procedures were carried out in accordance with the instructions on the corresponding kits. The quality of the extracted microbial genomic DNA was tested via 1% agarose gel electrophoresis to ensure that the quality of the DNA could meet the conditions for subsequent PCR amplification. The concentration of the extracted microbial genomic DNA was detected using a NanoDrop ND-2000 ultra-micro spectrophotometer, and after passing the test, PCR amplification was performed on the primers of the highly variant region of the 16S rRNA gene V4. The primers used were 515F (5′GTGCCAGCMGCCGCGGTAA 3′) and 806R (5′GGACTACHVGGGTWTCTAAT 3′) (Hou et al., 2018; Aires et al., 2019; Wei et al., 2021). The PCR products were detected via agarose gel electrophoresis at 1% concentration, and the target bands were recovered and purified using the Qiagen cut-gel recovery kit. The recovered products were sent to Huada Gene (Wuhan) Co., Ltd., for high-throughput sequencing on the MiSeq platform.
Raw sequence data were processed using Barcode, FLASH, and Mallard. After quality filtering, the sequences were clustered by Mothur. The 97% similarity operational taxonomic unit (OTU) was used for taxonomic statistics, and the selected OTU representative sequence was compared and analyzed with the sequences in the database Greengene_2013_5_99 through the RDP classifier (v2.2) software to obtain the species annotation of each OTU in the sample. In the result analysis, each OTU was regarded as a kind of bacteria. QIIME (v1.80) software was utilized to perform a cluster analysis and obtain a cluster tree; the graph was drawn with R (v3.1.1). A principal component analysis (PCA) diagram was produced using R to analyze the differences in microbial community structure among the experimental samples. Bacterial community functions were predicted with the 16S rRNA sequencing data by using PICRUSt (Langille et al., 2013).
Alpha diversity index and COG abundance values were analyzed by one-way ANOVA using SPSS26.0 software, and statistical significance of differences between data was compared using Duncan’s comparison. A value of P < 0.05 was regarded as statistical significance.
After preprocessing the raw sequencing data and filtering the low-quality base sequences, a total of 1,221,571 valid sequences were obtained from the 20 intestine and coelomic fluid samples of S. nudus and samples from the water and sediments in the culture environment (anaverage of 61,078 sequences per sample). The coverage rate of each sample was higher than 99%, and the dilution curve reached a plateau (Figure 1), indicating that the probability of having undetected sequences in the samples was low and that the sequencing depth basically covered all species in the samples. Hence, the samples could effectively reflect the structure and diversity of the flora of S. nudus’ intestine and coelomic fluid and the water and sediments in the culture environment.
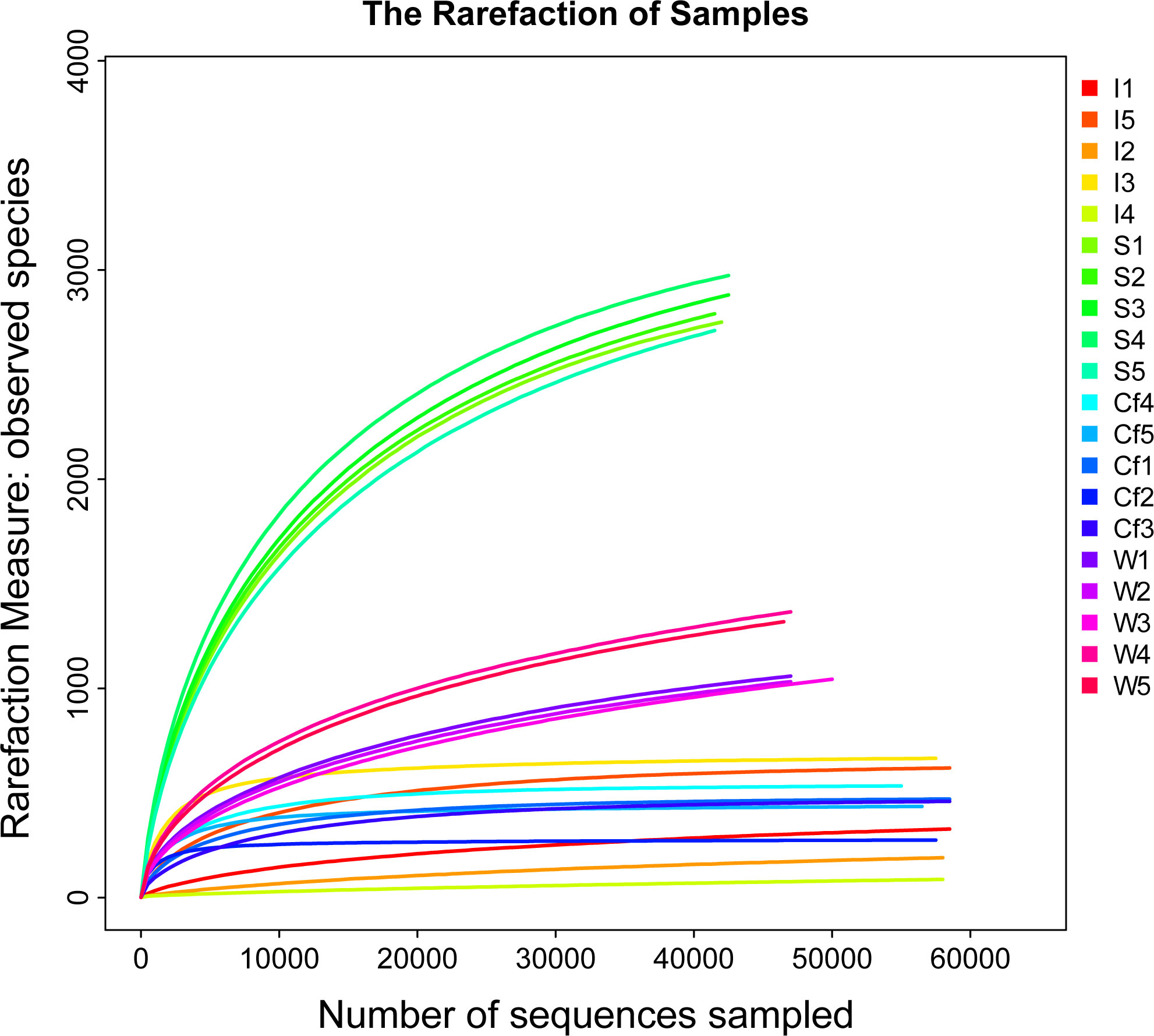
Figure 1 The rarefaction curves and rank abundance curve of different groups.
The effective sequences were clustered into 5416 OTUs, with 1081, 1157, 1960, and 3859 OTUs in the intestine, coelomic fluid, water, and sediments, respectively (Figure 2). Intestine and coelomic fluid had 336 OTUs, intestine and water had 404 OTUs, intestine and sediments had 521 OTUs, coelomic fluid and sediments had 372 OTUs, coelomic fluid and water had 381 OTUs, and sediment and water had 1419 OTUs.

Figure 2 Venn analysis in different samples.
Alpha diversity analysis was performed on each sample group. Chao and Ace indices were used to quantify the community richness in the samples, and Shannon and Simpson indices were used to calculate the community diversity in the samples (Figure 3). The results showed that the Chao, Ace, and Shannon indices increased sequentially in the intestine, coelomic fluid, water, and sediments of S. nudus, whereas the Simpson index increased inversely, indicating that the community richness and diversity of species were the highest in the sediments and the lowest in the intestine.
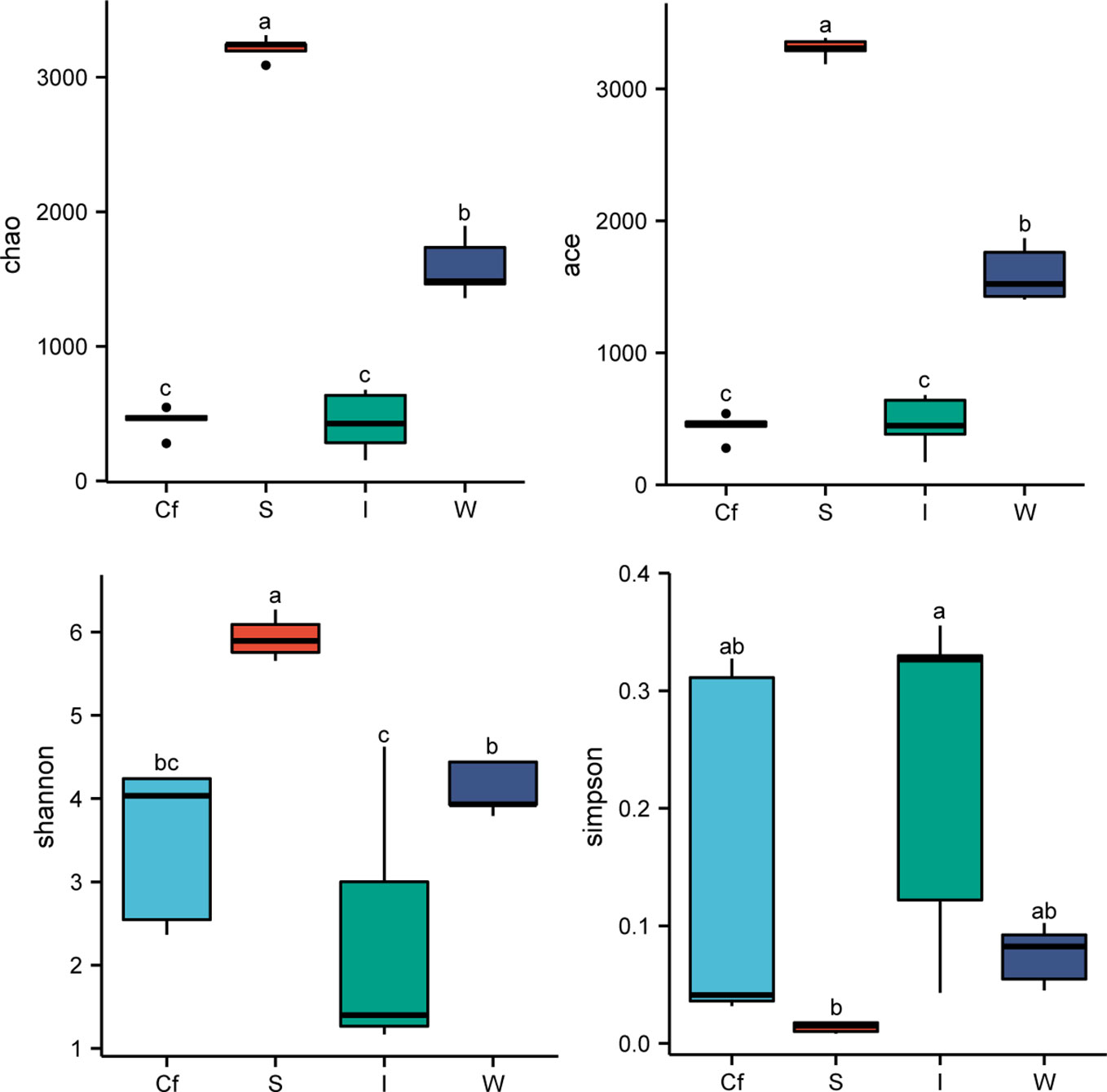
Figure 3 Alpha diversity index of water (W), sediments (S), coelomic fluid (Cf) and intestine (I) of S. nudus. Means with the same letters are not significantly different (P > 0.05).
The species annotation results showed that the detected bacteria belonged to 62 phyla, 41 classes, 68 orders, 71 families, 71 genera, and 28 species, and the composition of the flora of each sample varied considerably at different taxonomic levels (Table 1). The sediments possessed the most species at the phylum taxonomic level, and the intestine and coelomic fluid had the least.
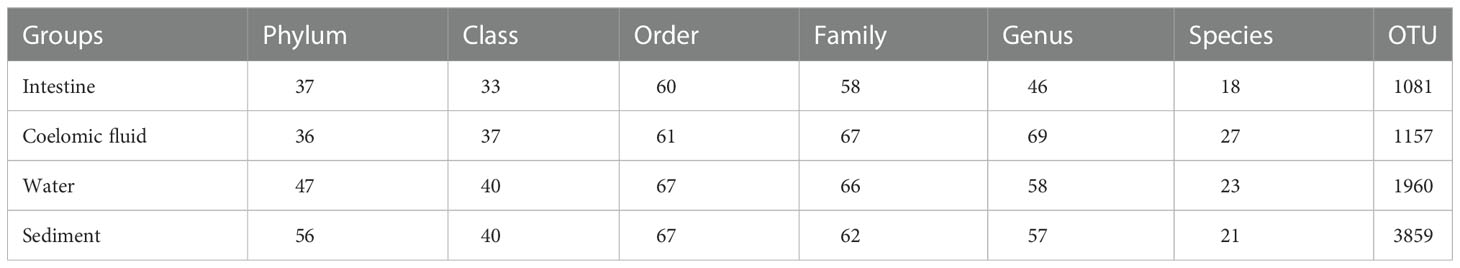
Table 1 Composition of bacterial communities in different samples.
The bacterial community composition and relative abundance at the phylum level are shown in Figure 4. Proteobacteria, with relative abundance ranging from 48.92% to 58.36%, was the most abundant phylum in all the samples, but the other dominant phyla varied considerably in the four sample groups. Bacteroidetes was the second most abundant phylum in the intestine and water, and its relative abundance ranged from 23.48% in the intestine to 35.15% in water; however, it was less abundant in the coelomic fluid and sediments, with relative abundance of 3.77% and 2.08%, respectively. Firmicutes was the second most abundant phylum in the coelomic fluid (20.74%) but was relatively scarce in the intestine, water, and sediments (0.79%, 0.21%, and 2.57%, respectively). Chloroflexi was the second most abundant phylum in the sediments (10.38%) but accounted for a relatively small proportion in the intestine, water, and coelomic fluid (0.86%, 0.24%, and 0.16%, respectively). Actinobacteria was the third most abundant phylum in coelomic fluid and water, with relative abundance ranging from 9.34% in coelomic fluid to 4.18% in water, but it was less abundant in the intestine and sediments (relative abundance of 1.13% and 0.58%, respectively). Spirochaetes was the third most abundant phylum in the intestine (11.71%), but it was rare in coelomic fluid, water, and sediments (0, 0.02%, and 1.01%, respectively). Euryarchaeota was the third most abundant phylum in the sediments (8.67%) but was scarce in the intestine, coelomic fluid, and water (0.02%, 0.11%, and 0.04%, respectively).
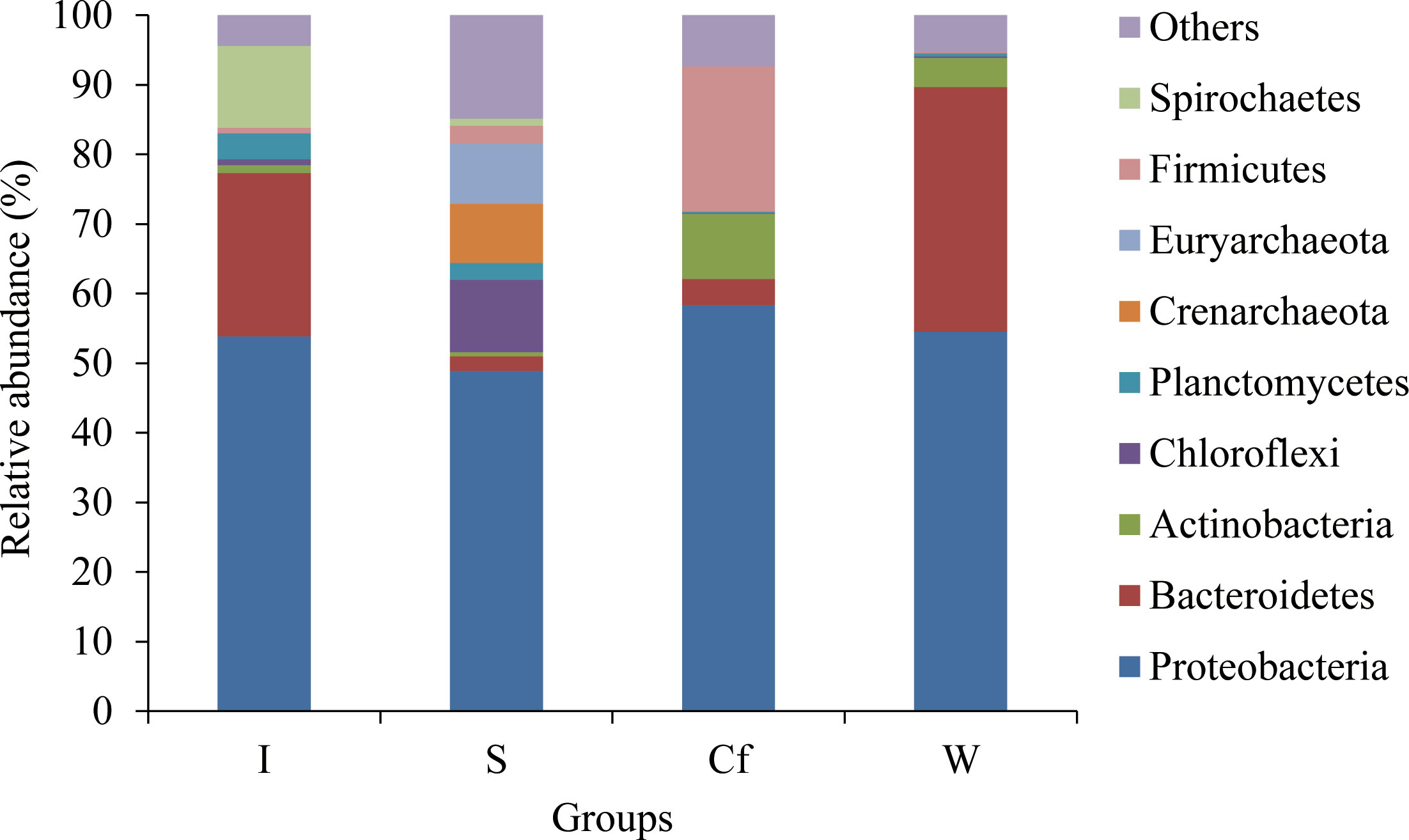
Figure 4 Relative abundance of phylum level bacterial communities in different samples.
To gain insights into the structure of the sample group flora, the top five OTUs occupying bacterial abundance in each sample group were selected for a tabular analysis (Table 2). The results showed that Deltaproteobacteria (23.85%) and Bacteroidales (22.90%) were the most abundant among the unknown bacteria in the intestine and could be considered biomarkers in the intestine. Vibrionales (15.33%) and Serratia (11.32%) were the most abundant among the unknown bacteria in the coelomic fluid and could be considered biomarkers in the coelomic fluid of S. nudus. Oceanospirillaceae (24.18%) was the most abundant among the unknown bacteria in water and could be considered biomarkers in water. By contrast, the relative abundance of bacteria detected in the sediments (mainly three unknown species of Helicobacteraceae) was low.
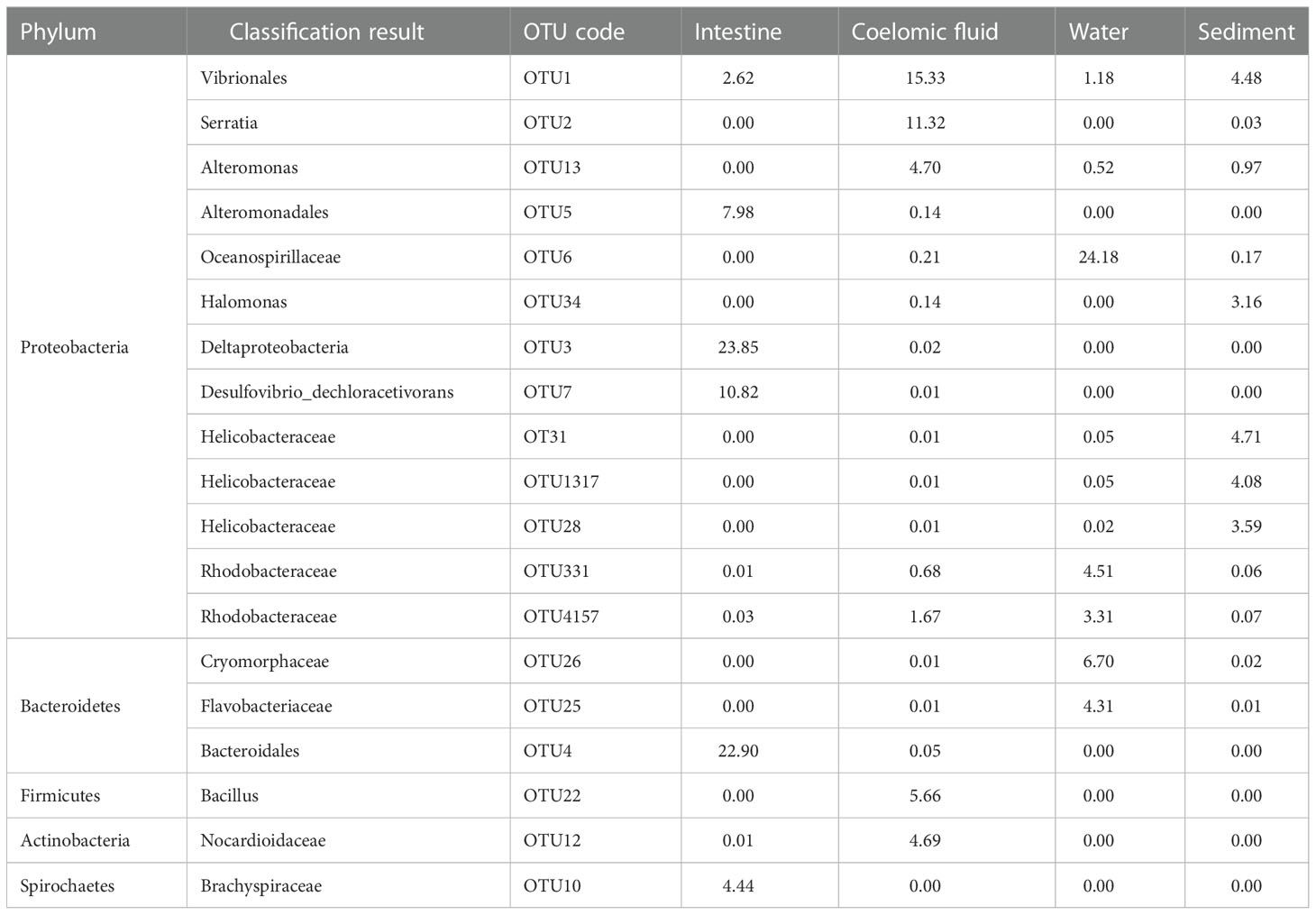
Table 2 Five most abundant bacterial OTUs in each sample.
Clustering analysis of all samples by QIIME software showed that the intestinal and coelomic fluid bacteria clustered into one group, indicating a similar bacterial community between the intestine and coelomic fluid (Figure 5). The branch lengths of the bacteria in the water and sediments, although different in length, also clustered into one group, indicating that the bacterial communities between water and sediments were also similar. The distance between the intestine and the sediments was shorter than that between the intestine and water, indicating that the bacterial communities in the intestine were more similar to those in the sediments. The PCA analysis showed the same results (Figure 6).
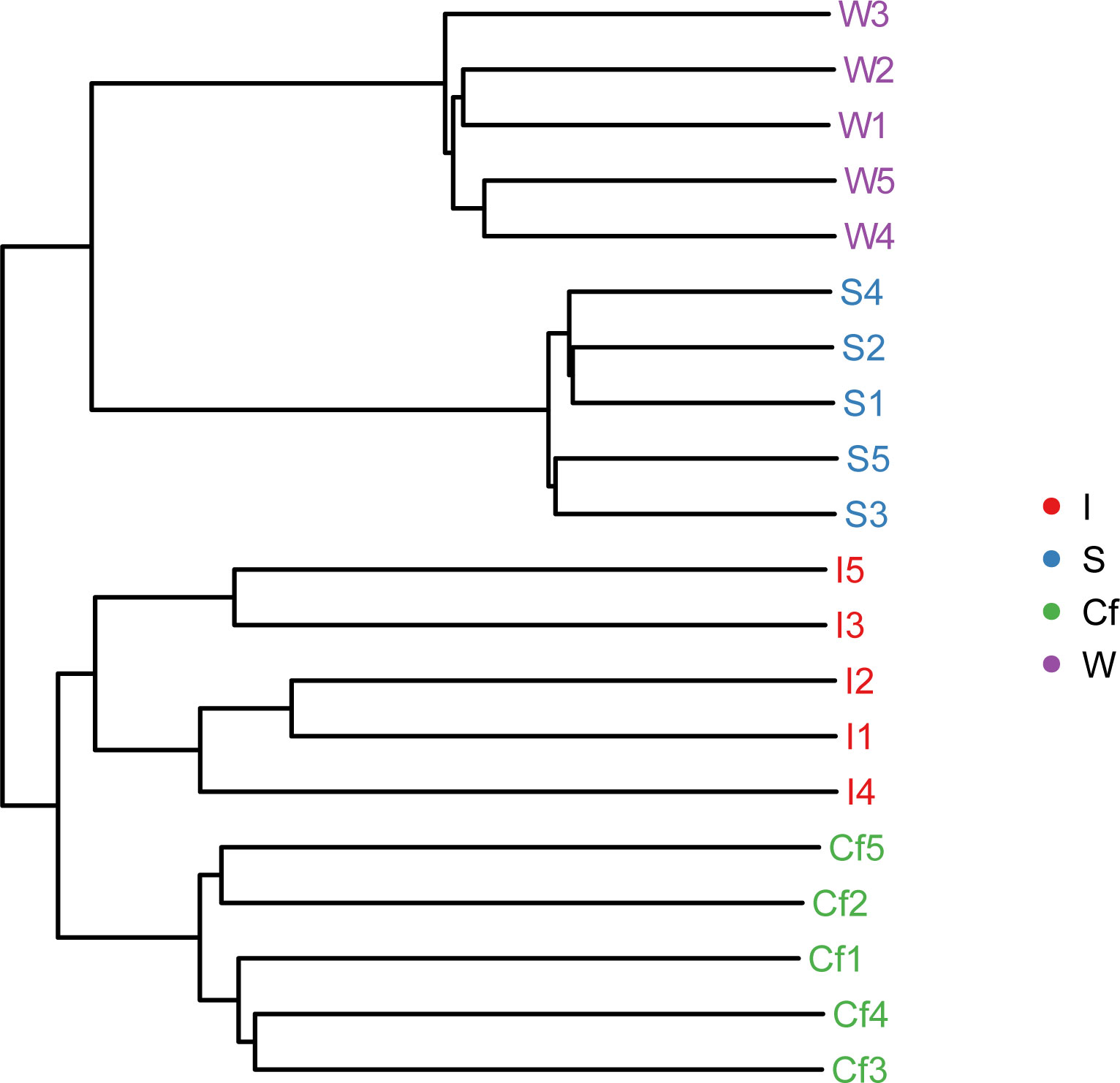
Figure 5 Unweighted UniFrac cluster tree of different samples (Samples with the same color in the graph indicate that they belong to the same group. The closer the samples are, the shorter the branch lengths are, indicating that the species composition of the two samples is more similar.).
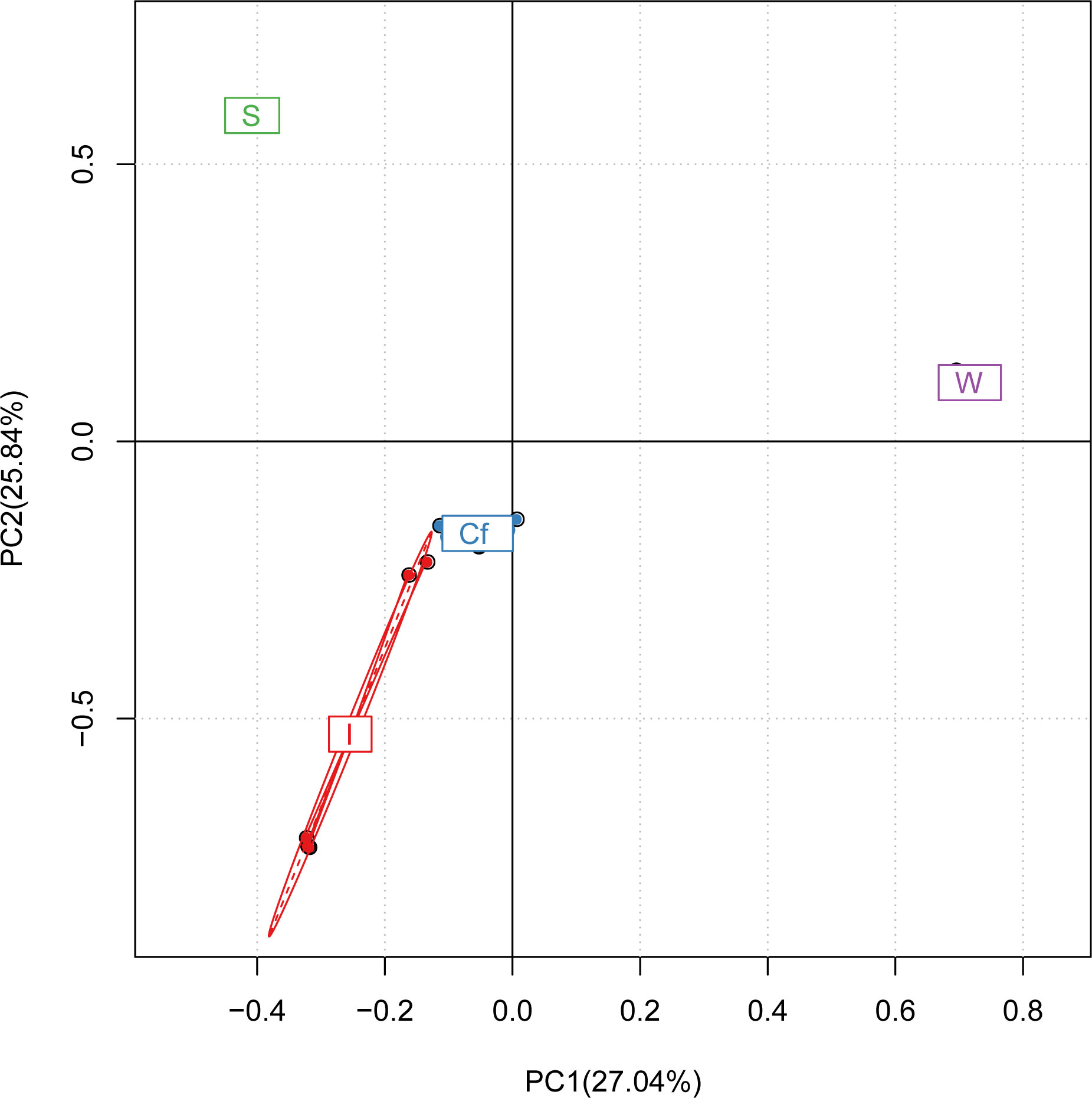
Figure 6 Principal component analysis of different samples (The closer the samples are, the more similar the composition of the samples).
The presumptive functions of the microbiota in S. nudus intestine and coelomic fluid and in the surrounding water and sediments in the cultured environment were illustrated using PICRUSt. The differences in COG abundance of the bacterial communities from the different samples determined based on the Clusters of Orthologous Group (COG) database are shown in Table 3. Compared with the taxonomic profiles, the functional profiles of all the groups were much more alike. All groups were enriched with functions related to membrane transport (relative abundance of 10.25%–12.74%), amino acid metabolism (relative abundance of 10.05%–11.65%), carbohydrate metabolism (relative abundance of 9.22%–10.56%), replication and repair (relative abundance of 6.66%–7.56%), and so on. These COG function classification results indicate that the S. nudus intestine and coelomic fluid microbial taxa maintained similar biological functions as those in the group at different locations or surrounding water and sediments in the cultured environment.
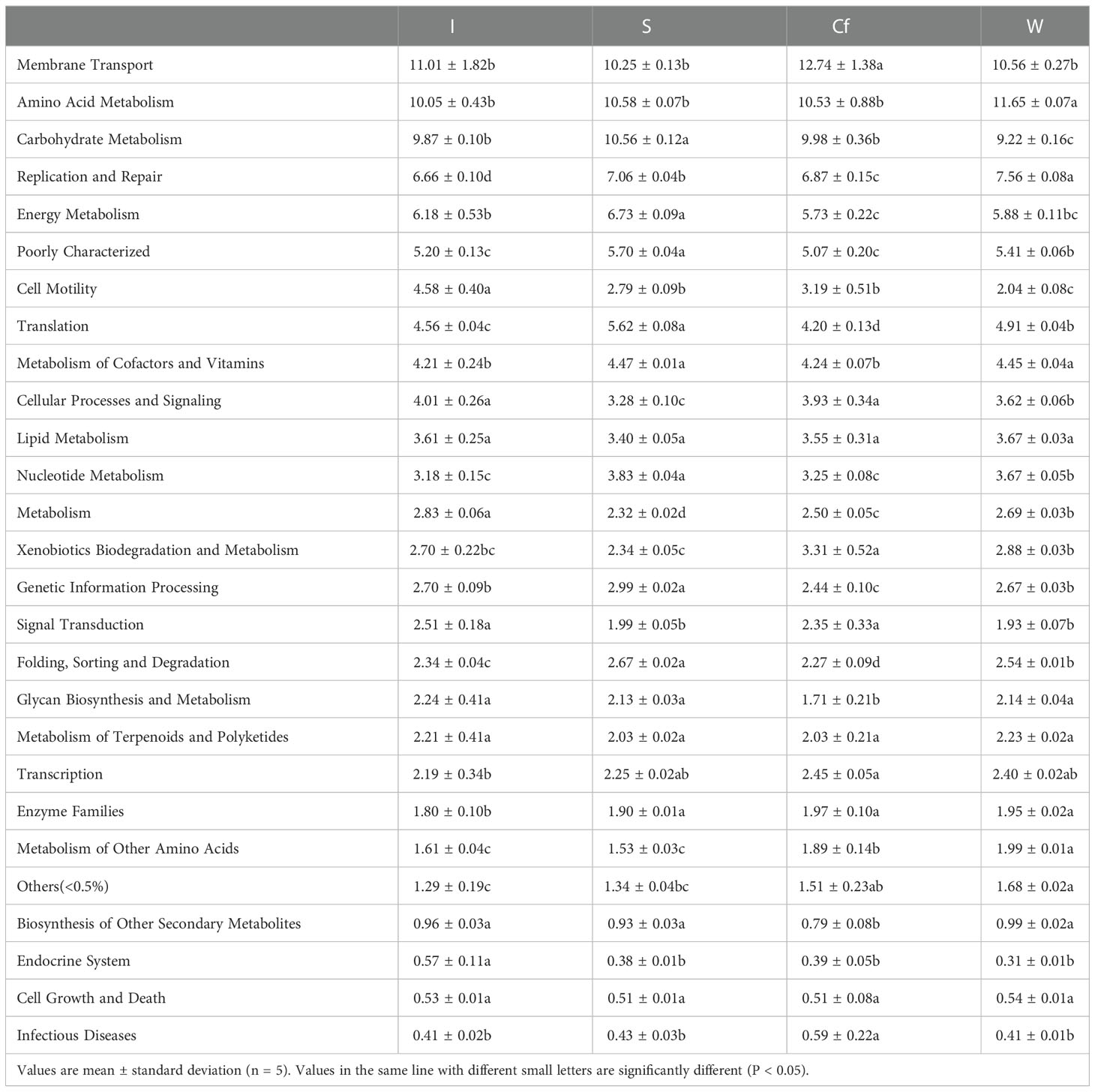
Table 3 The functional composition of bacteria community of different samples.
As an important component of farming ecosystems, bacterial communities play an important role in promoting material cycling and energy flow and are crucial in maintaining ecosystem balance (Xiong et al., 2016). The insurance hypothesis proposes that increasing biodiversity can help maintain ecosystem stability by preventing ecosystem function decline due to environmental fluctuations caused by the compensatory nature between species (Yachi and Loreau, 1999). Several studies have shown that bacterial communities in sediments have higher diversity compared with those in aquaculture water environments and the intestines of aquatic animals (Song et al., 2020; Sun et al., 2020; Zhou et al., 2021). Our study produced similar results, namely, the community richness and diversity of species in the sediments were higher than those in the other samples, suggesting that the sediments had higher bacterial diversity and a more stable bacterial community structure than water, the intestine of S. nudus, and the coelomic fluid of S. nudus in the marine area probably because sediments are rich in organic materials, such as aquatic debris and feces, which serve as a nutrient source for bacterial growth.
At the phylum level, Proteobacteria dominated in all the samples (48.82%–58.36%), and it mainly comprised gamma-, delta-, epsilon-, and alpha-proteobacteria. Similar results have been obtained for the benthic Apostichopus japonicus (Wang et al., 2021). Proteobacteria was the most abundant phylum in the water, sediments, and intestine samples in this study mainly because of the overwhelming dominance of Proteobacteria in the molecular biological classification or phenotypic classification among prokaryotes (Gupta, 2000). Previous studies have shown that Bacteroidetes is widely distributed in nature and an efficient degrader of organic matter in ecosystems, thus facilitating material cycling (Thomas et al., 2011). At the same time, members of Bacteroidetes can efficiently break down algal polysaccharides and participate in the metabolic conversion of nutrients, such as proteins and carbohydrates, thereby helping the host obtain energy from nutrients (Chen et al., 2018). The sub-dominant phylum in water and the intestine is Bacteroidetes, which may be abundant in algal nutrients in water and the intestine. Chloroflexi, as a sub-dominant phylum in sediments, can break down and convert nutrients, such as phytoplankton debris and feces, into humic acid, which can be used to improve the surrounding substrate environment (Björnsson et al., 2002; Gao et al., 2021). Studies have shown that Bacillus is a typical probiotic that can be used to prevent Vibrio infection, and its large reduction can lead to illness of the organism due to pathogenic infection (Sun et al., 2020). Notably, in this study, the relative abundance of Firmicutes (20.74%) in the coelomic fluid of S. nudus was second only to that of Proteobacteria (58.36%), but the relative abundance of Bacillus was higher than that of the two; hence, it can be identified and isolated as a candidate strain of probiotic bacteria for development, and it may be one of the reasons no large-scale disease has been reported in S. nudus. In summary, except for the dominance of Proteobacteria within S. nudus and its culture environment, considerable differences were observed in the other dominant phyla, indicating a certain independence between the environment and the composition of aquatic animal flora (Xiong et al., 2022).
S. nudus is benthic; it feeds mainly on organic debris in the surface layer of mudflat sediments and swims briefly sometimes in the water, but it spends most of its time submerged in sediments to avoid hostile organisms (Li JW, et al., 2017). Through these activities, S. nudus is closely linked to water and sediment. Therefore, understanding the relationship between aquatic animals and the bacterial community structure in the aquaculture environment is crucial in aquatic animal disease prevention and environmental regulation (Sun et al., 2020). Our cluster analysis and OTU Venn diagram analysis showed that the bacterial community compositions of the intestinal tract and coelomic fluid of S. nudus were highly similar, and 31.08% of the OTUs in the intestinal tract were consistent with those in the coelomic fluid. This result suggests the occurrence of frequent exchanges between the intestinal tract and coelomic fluid of S. nudus, but the means through which the exchanges occur are unknown and need to be studied comprehensively. The results of this study also showed that the bacterial community composition in the intestine was more similar to that in the sediments than to that in the culture water environment, with 37.37% of the OTUs in the intestine agreeing with those in the water compared with 48.20% for the sediments. The high similarity between the bacterial communities in the intestine and sediments may be related to the feeding habits of S. nudus. Large amounts of bacteria enter the intestine with the organic matter in the sediments, and some of these bacteria cannot be digested and colonize the intestine to maintain the bacterial metabolism in the intestine. A similar result has been obtained for other aquatic animals (Huang et al., 2018; Sheng et al., 2021; Zhou et al., 2021). In addition, the water and sediments in the culture environment clustered as one team, and 72.40% of the OTUs were consistent with those in the sediments; their bacterial communities were much more similar than those of water and intestine or intestine and sediments. This finding indicates that the interaction between water and sediments is more direct and closer than that between water and intestine or intestine and sediments (Zhao et al., 2018).
Bacterial communities are functionally related to ecological processes (Escalas et al., 2017). Our results showed that although bacterial communities in different surrounding environment differ greatly in their taxonomic composition, they are similar in their functional composition. This finding further suggests that bacteria do not exist in isolation but form complex networks of ecological interactions through multiple interactions, such as cooperation, competition, and predation (Faust and Raes, 2012). In this study, bacterial community functions related to metabolism were dominant in different surrounding environment (48.68%–50.47%) and mainly included amino acid metabolism, carbohydrate metabolism, and energy metabolism. This result may be related to the need of S. nudus to exhibit a high anabolic capacity to obtain sufficient energy for maintaining normal development of the organism (Yang et al., 2020; Huang et al., 2021).
We systematically characterized the bacterial communities in the intestine and coelomic fluid of S. nudus in relation to the structure of the bacterial community in the culture environment and revealed the relationships between them. Our results showed that the highest bacterial community richness and diversity were found in the sediments. The coelomic fluid of S. nudus contained high concentrations of Bacillus and could be a source of potential probiotic bacteria for isolation and culture. The bacterial community composition in the intestine was more similar to that in the sediments than in water. This study provides information that can help establish a sustainable microecological regulation management strategy for S. nudus culture and serves as a reference for the development and utilization of probiotics.
The data presented in the study are deposited in the NCBI Sequence Read Archive repository, accession number PRJNA896370.
QW, CY, and YD designed the research. RZ, YL, and JH conducted the research. RZ, JH, CY, and QW analyzed the data. JH, RZ, CY, QW, YL, and YD contributed to the final writing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Science and Technology Department, Guangdong Province (Grant number: 2021A05190, 2021A05171, 2020A04009 and 163-2019-XMZC-0009-02-0059).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adrianov A. V., Maiorova A. S. (2010). Reproduction and development of common species of peanut worms (Sipuncula) from the Sea of Japan. Russian J. Mar. Biol. 36 (1), 1–15. doi: 10.1134/S1063074010010013
Aires T., Muyzer G., Serrão E. A., Engelen A. H. (2019). Seaweed loads cause stronger bacterial community shifts in coastal lagoon sediments than nutrient loads. Front. Microbiol. 9, 3283. doi: 10.3389/fmicb.2018.03283
Bang C., Dagan T., Deines P., Dubilier N., Duschl W. J., Fraune S., et al. (2018). Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zoology 127, 1–19. doi: 10.1016/j.zool.2018.02.004
Björnsson L., Hugenholtz P., Tyson G. W., Blackall L. L. (2002). Filamentous chloroflexi (green non-sulfur bacteria) are abundant in wastewater treatment processes with biological nutrient removal. Microbiology 148 (8), 2309–2318. doi: 10.1099/00221287-148-8-2309
Blancheton J. P., Attramadal K. J. K., Michaud L., d’Orbcastel E. R., Vadstein O. (2013). Insight into bacterial population in aquaculture systems and its implication. Aquacultural Eng. 53, 30–39. doi: 10.1016/j.aquaeng.2012.11.009
Brooks A. W., Kohl K. D., Brucker R. M., van Opstal E. J., Bordenstein S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PloS Biol. 14 (11), e2000225. doi: 10.1371/journal.pbio.2000225
Cao F. J., Zhong R. Z., Yang C. Y., Hao R. J., Wang Q. H., Liao Y. S., et al. (2020). Transcriptomic analysis of differentially expressed genes in the larval settlement and metamorphosis of peanut worm Sipunculus nudus. Aquaculture Rep. 18, 100475. doi: 10.1016/j.aqrep.2020.100475
Carbone D., Faggio C. (2016). Importance of prebiotics in aquaculture as immunostimulants. effects on immune system of Sparus aurata and Dicentrarchus labrax. Fish Shellfish Immunol. 54, 172–178. doi: 10.1016/j.fsi.2016.04.011
Cárdenas C. A., Bell J. J., Davy S. K., Hoggard M., Taylor M. W. (2014). Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiol. Ecol. 88 (3), 516–527. doi: 10.1111/1574-6941.12317
Carrier T. J., Reitzel A. M. (2018). Convergent shifts in host-associated microbial communities across environmentally elicited phenotypes. Nat. Commun. 9 (1), 1–9. doi: 10.1038/s41467-018-03383-w
Chen J., Robb C. S., Unfried F., Kappelmann L., Markert S., Song T., et al. (2018). Alpha- and beta-mannan utilization by marine bacteroidetes. Environ. Microbiol. 20 (11), 4127–4140. doi: 10.1111/1462-2920.14414
Du X. D., Chen Z. A., Deng Y. W., Wang Q. H. (2009). Comparative analysis of genetic diversity and population structure of Sipunculus nudus as revealed by mitochondrial COI sequences. Biochem. Genet. 47 (11), 884–891. doi: 10.1007/s10528-009-9291-x
Du X. D., Chen Z. A., Deng Y. W., Wang Q. H., Huang R. L. (2008). Genetic diversity and population structure of the peanut worm (Sipunculus nudus) in southern China as inferred from mitochondrial 16S rRNA sequences. Israeli J. Aquaculture-Bamidgeh 60, 20495.
Escalas A., Troussellier M., Yuan T., Bouvier T., Bouvier C., Mouchet M. A., et al. (2017). Functional diversity and redundancy across fish gut, sediment and water bacterial communities. Environ. Microbiol. 19 (8), 3268–3282. doi: 10.1111/1462-2920.13822
Fan L. F., Li Q. X. (2019). Characteristics of intestinal microbiota in the pacific white shrimp Litopenaeus vannamei differing growth performances in the marine cultured environment. Aquaculture 505, 450–461. doi: 10.1016/j.aquaculture.2019.02.075
Fan L. F., Wang Z. L., Chen M. S., Qu Y. X., Li J. Y., Zhou A. G., et al. (2019). Microbiota comparison of pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 657, 1194–1204. doi: 10.1016/j.scitotenv.2018.12.069
Faust K., Raes J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10 (8), 538–550. doi: 10.1038/nrmicro2832
Foster K. R., Schluter J., Coyte K. Z., Rakoff Nahoum S. (2017). The evolution of the host microbiome as an ecosystem on a leash. Nature 548 (7665), 43–51. doi: 10.1038/nature23292
Gao Z. W., Liu F. H., Jia M. Q., Zhang G. G., Huang J., Wu G. H., et al. (2021). Illumina-based high-throughput sequencing analysis of bacterial community structure and diversity in beidagang wetland sediment, tianjin. J. Tianjin Normal Univ. (Natural Sci. Edition) 41 (4), 45–52. doi: 10.19638/j.issn1671-1114.20210407
Gupta R. S. (2000). The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol. Rev. 24 (4), 367–402. doi: 10.1111/j.1574-6976.2000.tb00547.x
Hou D., Huang Z., Zeng S., Liu J., Weng S., He J. (2018). Comparative analysis of the bacterial community compositions of the shrimp intestine, surrounding water and sediment. J. Appl. Microbiol. 125 (3), 792–799. doi: 10.1111/jam.13919
Huang F., Pan L. Q., Song M. S., Tian C. C., Gao S. (2018). Microbiota assemblages of water, sediment, and intestine and their associations with environmental factors and shrimp physiological health. Appl. Microbiol. Biotechnol. 102 (19), 8585–8598. doi: 10.1007/s00253-018-9229-5
Huang J. Q., Zhong R. Z., Yang C. Y., Wang Q. H., Liao Y. S., Deng Y. W. (2021). Dietary carbohydrate and protein levels affect the growth performance of juvenile peanut worm (Sipunculus nudus): an LC–MS-based metabolomics study. Front. Mar. Sci. 8, 913. doi: 10.3389/fmars.2021.702101
Kamada N., Chen G. Y., Inohara N., Núñez G. (2013). Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 14 (7), 685–690. doi: 10.1038/ni.2608
Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31 (9), 814–821. doi: 10.1038/nbt.2676
Li J. W., Hu R. P., Guo Y. J., Chen S. W., Xie X. Y., Qin J. G., et al. (2019). Bioturbation of peanut worms Sipunculus nudus on the composition of prokaryotic communities in a tidal flat as revealed by 16S rRNA gene sequences. Microbiol. Open 8 (8), e00802. doi: 10.1002/mbo3.802
Li T. T., Li H. A., Gatesoupe F. J., She R., Lin Q., Yan X. F., et al. (2017). Bacterial signatures of “Red-operculum” disease in the gut of crucian carp (Carassius auratus). Microbial Ecol. 74 (3), 510–521. doi: 10.1007/s00248-017-0967-1
Li J. W., Xie X. Y., Zhu C. B., Guo Y. J., Chen S. W. (2017). Edible peanut worm (Sipunculus nudus) in the beibu gulf: resource, aquaculture, ecological impact and counterplan. J. Ocean Univ. China 16 (5), 823–830. doi: 10.1007/s11802-017-3310-z
Li J. W., Zhu C. B., Guo Y. J., Xie X. Y., Huang G. Q., Chen S. W. (2015). Experimental study of bioturbation by Sipunculus nudus in a polyculture system. Aquaculture 437, 175–181. doi: 10.1016/j.aquaculture.2014.12.002
Moriarty D. J. W. (1997). The role of microorganisms in aquaculture ponds. Aquaculture 151 (1-4), 333–349. doi: 10.1016/S0044-8486(96)01487-1
Musella M., Wathsala R., Tavella T., Rampelli S., Barone M., Palladino G., et al. (2020). Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 717, 137209. doi: 10.1016/j.scitotenv.2020.137209
Sheng P., Chen Q., Han Q. X., Lu J. Q., Zhang B., Jiang M. W., et al. (2021). Bacterial community in gut, shell sediment, and surrounding water of Hyriopsis cumingii fed three different microalgal diets. Aquaculture 540, 736701. doi: 10.1016/j.aquaculture.2021.736701
Song M. S., Pan L. Q., Zhang M. Y., Huang F., Gao S., Tian C. C. (2020). Changes of water, sediment, and intestinal bacterial communities in Penaeus japonicus cultivation and their impacts on shrimp physiological health. Aquaculture Int. 28 (5), 1847–1865. doi: 10.1007/s10499-020-00562-9
Sun F. L., Wang C. Z., Chen L. J., Weng G. Z., Zheng Z. P. (2020). The intestinal bacterial community of healthy and diseased animals and its association with the aquaculture environment. Appl. Microbiol. Biotechnol. 104 (2), 775–783. doi: 10.1007/s00253-019-10236-z
Sun F. L., Wang Y. S., Wang C. Z., Zhang L., Tu K., Zheng Z. P. (2019). Insights into the intestinal microbiota of several aquatic organisms and association with the surrounding environment. Aquaculture 507, 196–202. doi: 10.1016/j.aquaculture.2019.04.026
Taylor M. W., Radax R., Steger D., Wagner M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71 (2), 295–347. doi: 10.1128/MMBR.00040-06
Thomas F., Hehemann J. H., Rebuffet E., Czjzek M., Michel G. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol. 2, 93. doi: 10.3389/fmicb.2011.00093
Wang Y. P., Shi T. T., Huang G. Q., Gong J. (2018). Molecular detection of eukaryotic diets and gut mycobiomes in two marine sediment-dwelling worms, Sipunculus nudus and Urechis unicinctus. Microbes environments 33(3), 290–300. doi: 10.1264/jsme2.ME18065
Wang L., Wei C., Chang Y. Q., Ding J. (2021). Response of bacterial community in sea cucumber Apostichopus japonicus intestine, surrounding water and sediment subjected to high-temperature stress. Aquaculture 535, 736353. doi: 10.1016/j.aquaculture.2021.736353
Wei D. D., Xing C. G., Hou D. W., Zeng S. Z., Zhou R. J., Yu L. F., et al. (2021). Distinct bacterial communities in the environmental water, sediment and intestine between two crayfish-plant coculture ecosystems. Appl. Microbiol. Biotechnol. 105 (12), 5087–5101. doi: 10.1007/s00253-021-11369-w
Xiong J. B., Dai W. F., Li C. H. (2016). Advances, challenges, and directions in shrimp disease control: the guidelines from an ecological perspective. Appl. Microbiol. Biotechnol. 100 (16), 6947–6954. doi: 10.1007/s00253-016-7679-1
Xiong X. Y., Zhao Y. F., Wang Z. C. (2022). Analysis of microbial community structure of channel catfish Ictalurus punctatus intestine and culture water. Aquaculture Sci. 41 (4), 589–596. doi: 10.16378/j.cnki.1003-1111.20175
Yachi S., Loreau M. (1999). Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis. Proc. Natl. Acad. Sci. 96 (4), 1463–1468. doi: 10.1073/pnas.96.4.1463
Yang C. Y., Zhang J. W., Zhong R. Z., Guo Z. C., Wang Q. H., Zheng Z. (2020). Characterization of metabolomic differences in peanut worm Sipunculus nudus between breeding and nonbreeding seasons. Aquaculture Rep. 16, 100271. doi: 10.1016/j.aqrep.2019.100271
Zhang C. X., Dai Z. R., Cai Q. X. (2011). Anti-inflammatory and anti-nociceptive activities of Sipunculus nudus l. extract. J. Ethnopharmacology 137 (3), 1177–1182. doi: 10.1016/j.jep.2011.07.039
Zhang J. W., Su Y. L., Zhong R. Z., Wang Q. H., Jiao Y., Yang C. Y. (2021). Transcriptome analysis reveals key molecular events of germinal vesicle breakdown in Sipunculus nudus. Aquaculture Res. 52 (4), 1757–1766. doi: 10.1111/are.15031
Zhao Y. T., Zhang X. X., Zhao Z. H., Duan C. L., Chen H. E., Wang M. M., et al. (2018). Metagenomic analysis revealed the prevalence of antibiotic resistance genes in the gut and living environment of freshwater shrimp. J. Hazardous Materials 350, 10–18. doi: 10.1016/j.jhazmat.2018.02.004
Zheng Z., Liao Y. S., Ye J. M., Yang C. Y., Adzigbli L., Wang Q. H., et al. (2021). Microbiota diversity in pearl oyster Pinctada fucata martensii intestine and its aquaculture environment. Front. Mar. Sci. 8, 655698. doi: 10.3389/fmars.2021.655698
Zhou L., Qu Y. Y., Qin J. G., Chen L. Q., Han F. L., Li E. C. (2021). Deep insight into bacterial community characterization and relationship in the pond water, sediment and the gut of shrimp (Penaeus japonicus). Aquaculture 539, 736658. doi: 10.1016/j.aquaculture.2021.736658
Keywords: Sipunculus nudus, bacterial community, intestine, coelomic fluid, water, sediment
Citation: Zhong R, Huang J, Liao Y, Yang C, Wang Q and Deng Y (2022) Insights into the bacterial community compositions of peanut worm (Sipunculus nudus) and their association with the surrounding environment. Front. Mar. Sci. 9:1076804. doi: 10.3389/fmars.2022.1076804
Received: 22 October 2022; Accepted: 12 December 2022;
Published: 23 December 2022.
Edited by:
Yafei Duan, South China Sea Fisheries Research Institute, ChinaReviewed by:
Quanwei Song, CNPC Research Institute of Safety and Environmental Technology, ChinaCopyright © 2022 Zhong, Huang, Liao, Yang, Wang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingheng Wang, d2FuZ3FpbmdoZW5nX2hhaWRhQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.