 Jing Huang1,2†
Jing Huang1,2† Lei Song
Lei Song Xiqiu Han
Xiqiu Han Ying Huang
Ying Huang- 1State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 3School of Biology and Biological Engineering, South China University of Technology, Guangzhou, Guangdong, China
- 4China General Microbiological Culture Collection Center, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 5Key Laboratory of Submarine Geosciences and Second Institute of Oceanography, Ministry of Natural Resources, Hangzhou, China
A consensus is emerging on marine microbial biogeography. However, knowledge of the biodiversity and biogeographic patterns of deep-sea microbes and the drivers is far from adequate. In this study, we investigated the diversity and biogeography of prokaryotes at the Indian Ocean Ridges (IOR) using 16S rRNA gene and dsrB sequencing combined with bioinformatics analyses. A total of 39 hydrothermal field sediment (HFS) and non-hydrothermal field sediment (NHFS) samples were collected from the Carlsberg Ridge (CR) and Southwest Indian Ridge (SWIR). Seventy-four prokaryotic phyla were detected in the samples, of which 13 phyla were distributed across all samples. The composition of prokaryotic communities diverged between HFSs and NHFSs, but not between the CR and SWIR. Sixteen phyla were differentially enriched between HFSs and NHFSs. The community structure was significantly correlated to calcium, sulfur, total phosphorus, and total nitrogen, the first three of which were the main distinguishing factors of HFSs and NHFSs. Moreover, a weak distance–decay pattern was observed among the NHFS communities. PICRUSt predictions revealed functional discrepancies in carbon and nitrogen metabolism between the HFS and NHFS communities, and dsrB sequencing analysis indicated higher diversity of sulfate reduction genes and taxa in HFSs than in NHFSs. The diversity and relative abundance of corresponding functional taxa and genes suggested that the nitrogen cycle might be centralized in an energy-generating manner to support the microbial community in NHFSs while the sulfur cycle is more important for the HFS community. These findings provide new insights into microbial biogeography and metabolic differentiation along the IOR that are influenced by hydrothermal activity.
Introduction
Mid-ocean ridges (MOR) are the divergent boundary of the oceanic plates above the ocean seafloor, forming by rapid upwelling of mantle material and characterized by high seismicity and heat flow (Iyer et al., 2003). Hydrothermal vents often occur at MOR, acting as mass and heat exchange between the oceans and Earth’s interior and exhibiting unique physical and geochemical characteristics that are distinct from other ocean habitats. Therefore, compared to non-hydrothermal field sediments (NHFSs), hydrothermal field sediments (HFSs) are enriched for some specific chemical substances (Jannasch and Mottl, 1985; Baker et al., 1995; Simoneit and Fetzer, 1996). These characteristics may have a significant effect on the microbial communities in HFSs (Orcutt et al., 2011). Studies have shown that in the prokaryotic communities in Atlantic and Pacific HFSs, Actinobacteria, Deltaproteobacteria, Gammaproteobacteria, Epsilonproteobacteria, Nitrospirae, and Thaumarchaeota are dominant and Planctomycetes is minor; and Alphaproteobacteria, Betaproteobacteria, Bacteroidetes, Chlorobi, and Dependentiae are dominant in Atlantic but not Pacific HFSs (Teske et al., 2002; López-García et al., 2003; Nercessian et al., 2005; Nunoura et al., 2010; Cerqueira et al., 2017; Storesund et al., 2018). For the prokaryotic communities in NHFSs of Atlantic, Pacific, as well as Arctic Oceans, Gammaproteobacteria, Planctomycetes, Chloroflexi, and Actinobacteria have been reported to be dominant (Newberry et al., 2004; Schauer et al., 2010; Jorgensen et al., 2012; Zhang et al., 2015; Zeng et al., 2017; Varliero et al., 2019). Some lineages can be found in both HFSs and NHFSs (Supplementary Figure S1) and in different oceans, suggesting a continuous biosphere and a cosmopolitan distribution pattern. However, knowledge gaps still exist regarding the biodiversity and biogeography of microbes along MOR.
The biogeographic distribution of marine microbes has been reported to correlate with both environmental and geographic factors. Several studies have reported that water depth, hydrography, total organic carbon (TOC), total phosphorus (TP), iron, manganese, calcium, sulfate, and significantly affect marine microbial community structures (German et al., 1998; Holmfeldt et al., 2009; Schauer et al., 2010; Qian et al., 2011; Jorgensen et al., 2012). As the environmental variables between HFSs and NHFs diverge markedly, it is worth exploring whether they play a dominant role in prokaryotic community assembly in the deep sea. In addition, the view of distance-decay pattern describes an increase in community dissimilarities as spatial distance increases (Martiny et al., 2011). When the spatial scale is larger than 3000 km, geographic factors may outweigh environmental factors, but the reliable division criterion for spatial scale is dictated by specific habitats (He et al., 2007). To better understand the biogeography of marine microbes originated from distinct habitats across large distances, the influence of different ecological factors on structuring HFS and NHFS prokaryotic communities needs to be explored.
On the other hand, marine microbes also play important roles in overall oceanic processes and global nutrient cycling (Orcutt et al., 2011). Marine photosynthetic bacteria, chemoautotrophic bacteria and deep sea archaea mediate many important metabolic processes of the carbon cycle, such as carbon fixation (Berg, 2011; Hugler and Sievert, 2011) and methane metabolism (Kirchman, 2012; Carini et al., 2014; McGlynn et al., 2018). The full nitrogen biogeochemical cycle has been found in marine ecosystems, including N2-fixation, nitrification, assimilatory nitrate reduction, denitrification, anaerobic ammonium oxidation (anammox) and dissimilatory nitrate reduction (Teske et al., 1994; Song et al., 2014; Hutchins and Fu, 2017). A large fraction of bacteria and archaea in marine sediments participate in the sulfur cycle through a variety of processes that result in the oxidation or the production of sulfide compounds (Sievert et al., 2007; Anantharaman et al., 2018; Meier et al., 2019). Sulfate-reducing prokaryotes (SRPs) use sulfate as electron acceptors to mineralize organic carbon or oxidize hydrogen for energy (Nakagawa et al., 2004), which would occur in deep-sea hydrothermal vents where seawater-derived sulfate is abundant but oxygen is absent (Frank et al., 2013). Different functional microbes can play different roles in the processes of nutrient cycles, while some microbes can take part in more than one cycle. For instance, Betaproteobacteria participates in both sulfur oxidation and ammonia oxidation, and Thaumarchaeota can couple carbon and nitrogen cycles (Pester et al., 2011). The nutrient cycling has been extensively explored by surveying the corresponding microbial functional genes. The dsr genes encode dissimilative sulfite reductase (DSR), which is a key enzyme in the process of sulfate reduction, and the gene of DSR beta subunit (dsrB) is the best studied molecular marker of sulfate-reducing microbes in environment (Müller et al., 2015; Pelikan et al., 2016).
Compared to the Atlantic and Pacific MOR, the Indian Ocean Ridges (IOR) including their hydrothermal ecosystems are unique, with contrasting spreading rates (ranging from <16 to ~80 mm/year), distinct mineral composition, and lower organic carbon fluxes, but have been less explored (Perez et al., 2021). Furthermore, compared to the faunal communities on the IOR, much less attention has been paid to the microbiotas although they are recognized as the keystone to ecosystem function (Perez et al., 2021). The Southwest Indian Ridge (SWIR) is an ultraslow (<16 mm/year) spreading ocean ridge and the only known modern migration route of deep-sea vent fauna between the Atlantic and Indian Oceans (German et al., 1998; Tao et al., 2012). The sulfide deposits and/or metalliferous sediments resource areas are usually enriched in the hydrothermal fields on the SWIR (Tao et al., 2012; Tao et al., 2014). Recently, a handful of studies on prokaryotic communities of SWIR sediments revealed that Acidobacteria, Bacteroidetes, Proteobacteria, Planctomycetes, and Actinobacteria were the major phyla in both HFSs and NHFSs; while Chloroflexi, Gemmatimonadetes, Nitrospirae, Thaumarchaeota, and Schekmanbacteria were dominant in HFSs but not in NHFSs (Li et al., 2013; Li et al., 2014; Yang et al., 2020a; Lecoeuvre et al., 2021). The Carlsberg Ridge (CR) is a slow (24–26 mm/year) spreading ocean ridge which was kept almost unknown until the cruise of 2003 (Murton and Taylor, 2003). As a distal portion of the IOR, the CR has significant implications for the colonization and dispersal of hydrothermal ecosystems (Tao et al., 2013). The basalt-hosted Wocan hydrothermal fields are located on the Northwest slope of an axial volcanic ridge on the CR, and sediments in Wocan are rich in chalcopyrite, pyrite, and Fe-oxyhydroxides (Wang et al., 2017; Popoola et al., 2019). The bacterial communities in the water column of a Wocan hydrothermal plume-influenced zone were found to be dominated by Proteobacteria, Bacteroidota, Firmicutes, and Desulfobacterota (Xie et al., 2022). However, the microbial diversity and their ecological functions in the CR sediments remain largely unknown.
In the present study, the diversity and community structure of prokaryotes in HFSs and NHFSs of the CR and SWIR were explored using 16S rRNA gene Miseq-sequencing. The variations in prokaryotic communities between HFSs and NHFS and between the CR and SWIR were determined, and the cosmopolitan lineages present in each sample were identified. The environmental and geographic factors correlated to the beta diversity of the prokaryotes were analyzed at different scales. Moreover, the metagenomic functional pathways of the deep-sea prokaryotes were predicted by PICRUSt and the potential functional groups that might play significant roles in the S cycle were revealed by dsrB amplicon sequencing. The results contribute to the understanding of microbial distribution patterns in the deep ocean.
Materials and methods
Sample collection and physicochemical analysis
Thirty-four NHFS (N1, N2, N7-N29, MS1, MS2, and M1-M7) and five HFS samples (N3-N6 from Wocan and M8 from Longqi) were collected along the CR (N1-N29) and SWIR (MS1, MS2, and M1-M8) (Figure 1) during the DY115-21, DY115-22, DY125-26, and DY-28I cruises of the DaYang YiHao and Haiyang 20 research vessels (RVs) using TV-guided grabs or multi-core samplers (Φ10 × 60 cm). The samples were transferred into sterile 200-ml plastic boxes and frozen at -80°C until further processing in the laboratory. The longitude, latitude, water depth, and temperature of the sampling sites were recorded by the RVs.
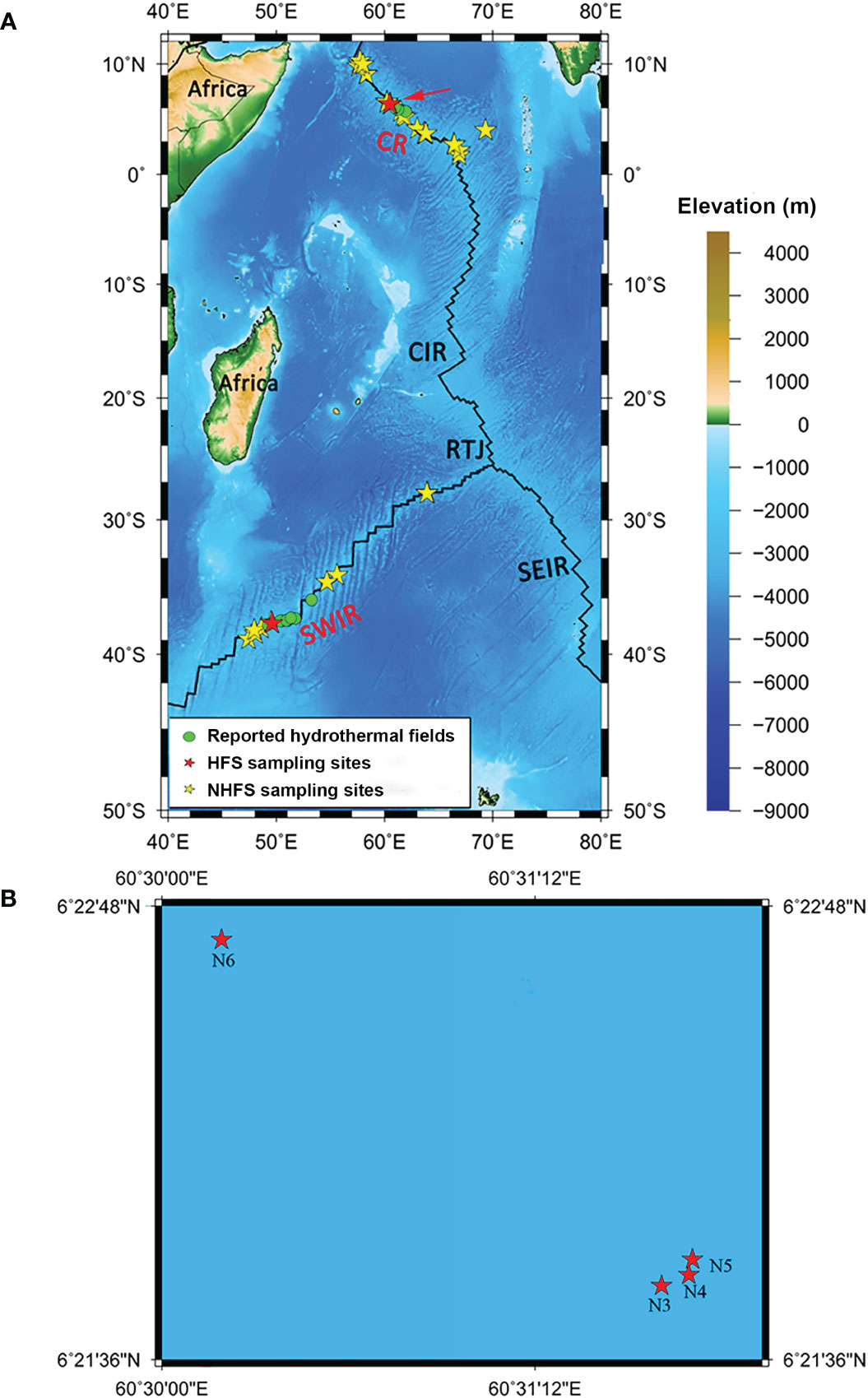
Figure 1 The map of sampling locations in this study. (A) The whole 39 sampling sites on the Southwest Indian Ridge (SWIR) and the Carlsberg Ridge (CR). (B) Magnified view of the sampling area of CR marked by a red arrow in A, where N3-N5 were from the hydrothermal field Wocan-1 and N6 from Wocan-2. Green dots indicate the hydrothermal fields from previous studies (Tao et al., 2012; Tao et al., 2013; Tao et al., 2014). Red and yellow stars indicate the sampling sites of hydrothermal field sediments (HFSs) and non-hydrothermal field sediments (NHFSs), respectively. SEIR, Southeast Indian Ridge; CIR, Central Indian Ridge; RTJ, Rodrigues Triple Junction (25.5°S, 70°E).
The physicochemical analysis of the samples was conducted at the Analysis Center of Tsinghua University using the methods as previously described (Chen et al., 2016). Detailed information of the samples was provided in Table S1. Significant differences of environmental factors between the HFS and NHFS samples were determined using the Wilcoxon test. The map of sampling sites was drawn using the Generic Mapping Tools (GMT) (Wessel and Smith, 1998) (http://gmt.soest.hawaii.edu/) and plotted using the Mercator projection and NUVEL-1A plate boundaries (http://jules.unavco.org/GMT/).
DNA extraction, PCR amplification of the 16S rRNA and dsrB genes, and Miseq sequencing
The total DNA was extracted from 5 g (wet weight) of each of the 39 sediment samples which was carefully homogenized before subsampling. The procedure was as previously described (Chen et al., 2016). Amplifications were conducted using the primer set F515 (5’-GTGCCAGCMGCCGCGGTAA-3’) and R806 (5’-GGACTACHVGGGTWTCTAAT-3’) that targets V4 region of the prokaryotic 16S rRNA genes. The primers were labeled with a unique 6-nt barcode for each sample. The PCR of each sample was performed in triplicate with a 25 μL reaction mixture containing 10 ng purified DNA template under following thermal cycles: initial denaturation at 98°C for 1 min; 35 cycles at 98°C for 10 sec, 50°C for 30 sec, and 72°C for 30 sec; and a final extension at 72°C for 5 min. The triplicate PCR products were mixed together and purified using the Takara PCR fragment purification kit (Takara, Japan), followed by the ligation of ‘A’ sequencing adaptor. The purified PCR amplicons were combined in equimolar amounts and submitted to Novogene Co. Beijing for Illumina 250 bp paired-end sequencing on a MiSeq machine.
Fragments (~350 bp) of bacterial dsrB gene related to the sulfur cycle was amplified using primers DSRp2060F (5’-CAACATCGTYCAYACCCAGGG-3’) and DSR4R (5’-GTGTAGCAGTTACCGCA-3’) (Wagner et al., 1998; Geets et al., 2006). The PCR was performed in triplicate with a 50 μL reaction mixture containing 50 ng purified DNA template under following thermal cycles: initial denaturation at 94°C for 2 min; 15 cycles at 94°C for 10 sec, 62°C for 30 sec, and 72°C for 20 sec; 20 cycles at 94°C for 10 sec, 48°C for 15 sec, and 72°C for 20 sec; and a final extension at 72°C for 2 min. The PCR products were purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA), and then submitted to Beijing Genomics Institute (BGI) Tech., where the library was constructed and sequenced using Illumina Miseq PE 300.
16S rRNA gene sequencing data processing and bioinformatics analyses
Raw fastq sequences were first demultiplexed according to the unique barcode by the software QIIME v 1.9.1 (Quantitative Insights Into Microbial Ecology) (Caporaso et al., 2010b), and then pairs of reads were merged with FLASH (Fast Length Adjustment of Short Reads) (Magoč and Salzberg, 2011) under default settings. The merged reads were quality filtered by QIIME. Sequences that contained ambiguous bases (N) were discarded, and sequences were truncated at any sites containing more than three consecutive bases with a Phred quality score (Q) < 20; only those with > 75% (as percentage of total read length) consecutive high-quality base calls were included in further analyses (Bokulich et al., 2013). Chimeric sequences were detected and excluded using the QIIME implementation of the UCHIME algorithm (Edgar et al., 2011) based on the Greengenes database version 13.8 (DeSantis et al., 2006).
Statistics analyses of the high-quality 16S rRNA gene sequences were performed with QIIME and USEARCH as described previously (Chen et al., 2016). Briefly, the 16S rRNA gene sequences were assigned to operational taxonomic units (OTUs) at a similarity cutoff of 97% by UPARSE (Edgar, 2013). After discarding all singletons, representative sequences of the OTUs were aligned using PyNAST (Caporaso et al., 2010a). The phylogenetic tree of the representative sequences was constructed using FastTree (Price et al., 2010). The RDP classifier (Cole et al., 2014) was used to annotate taxa based on the SILVA database version 132 (Yilmaz et al., 2014) with a 50% bootstrap cutoff. To avoid sampling errors, subsamples of the minimal number of sequences (9,231 sequences) per sample were randomly selected to compare the alpha and beta diversity between samples. The estimators of Shannon diversity, Chao1 richness, and Faith’s phylogenetic diversity (PD) were calculated in QIIME. The Wilcoxon test was used to measure the difference of environmental factors between the HFS and NHFS samples.
Bray-Curtis distance and Euclidean distance matrices were used to measure the dissimilarities of prokaryotic communities and geochemical characteristics, respectively, and the non-metric multidimensional scaling (NMDS) analysis was used to visualize the dissimilarities between the samples. Nonparametric analysis of similarities (ANOSIM) was performed on Bray-Curtis community dissimilarities. Standardization was performed to make all geographic and physicochemical parameters comparable between the different units of different variables using the scale function in R v4.0.2 (https://www.r-project.org/). The “Geography” library was used to calculate the paired geographic distances between samples based on latitude and longitude coordinates in R. A Spearman’s rank correlation between the communities and geochemical factors was calculated. Mantel test, Redundancy analysis (RDA), and Bioenv analysis were conducted to assess the relationships between prokaryotic communities and environmental variables with 999 permutations. These analyses were carried out with the vegan package v2.5-7 in R (Oksanen et al., 2020). The differentially abundant taxa were determined by the linear discriminant analysis (LDA) effect size (LEfSe) online software (http://huttenhower.sph.harvard.edu/lefse/) (Segata et al., 2011). The threshold on the logarithmic LDA score for distinguishing features was set to 3.0. The software PICRUSt2 (Douglas et al., 2020) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database were used to infer functional potentials of the microbial communities based on the 16S rRNA gene amplicon sequencing profile. STAMP v2.0.0 (Parks et al., 2014) was used to determine significant putative KEGG pathways with two-side Welch’s t-test (P value < 0.05) and the Benjamini-Hochberg FDR (Benjamini and Hochberg, 1995) correction for multiple tests.
dsrB sequencing data analyses
The sequence processing was the same as above. The USEARCH software (Edgar, 2010) was used to divide the dsrB sequences into different OTUs based on 90% sequence similarity, removing the chimera by the denovo method. All OTUs were annotated using the NCBI blastx method (Altschul et al., 1997) and Non-redundant protein sequence (NR) database (Pruitt et al., 2007). After discarding the sequences with E-value > e-10, coverage < 50%, or bit-score < 50, and filtering out the false positive sequences annotated as non-dsrB genes, the high-quality sequences were obtained for subsequent analysis. Alpha diversity of the dsrB sequences was analyzed as above. The sequences were compared to those of the DsrAB database constructed by Müller (Müller et al., 2015) through local blast analysis to retrieve the most similar reference sequences. The phylogenetic tree of the representative dsrB sequences and related reference sequences was constructed using MEGA7 (Kumar et al., 2016).
Data accessibility
The metadata of the 16S rRNA gene and dsrB amplicon sequences from this study have been deposited in the NCBI Sequence Read Archive (SRA) under the accession numbers SRR17439176-SRR17439214 and SRR17440762-SRR17440769, respectively, and in the China National Microbiology Data Center (NMDC) under the accession numbers NMDC40016400-NMDC40016438 and NMDC40016793-NMDC40016800, respectively.
Results and discussion
Environmental characteristics of the samples
Three geographic and 12 physicochemical parameters of the samples were obtained and are summarized in Table S1. Five samples were from HFSs at water depths of 2800-3104 m and with pairwise geographic distances ranging from 0.1 to 5049 km; 34 samples were from NHFSs at water depths of 1608-4213 m, and the pairwise geographic distances between the sampling sites ranged from 1 to 5582 km (Figure 1). NMDS analysis based on the 12 physicochemical factors and water depth was used to evaluate the dissimilarities between the samples. As shown in Supplementary Figure S2, HFSs were obviously different from NHFSs while most SWIR samples were similar to CR samples, indicating that the environmental variables were significantly affected by habitat rather than by geographic distance. Compared to NHFSs, HFSs contained significantly lower content of Ca (Wilcoxon test, P < 0.0001) and lower pH (P < 0.001) while higher contents of TP (P < 0.001), Fe, S and TOC (all P < 0.05) (Figure 2).
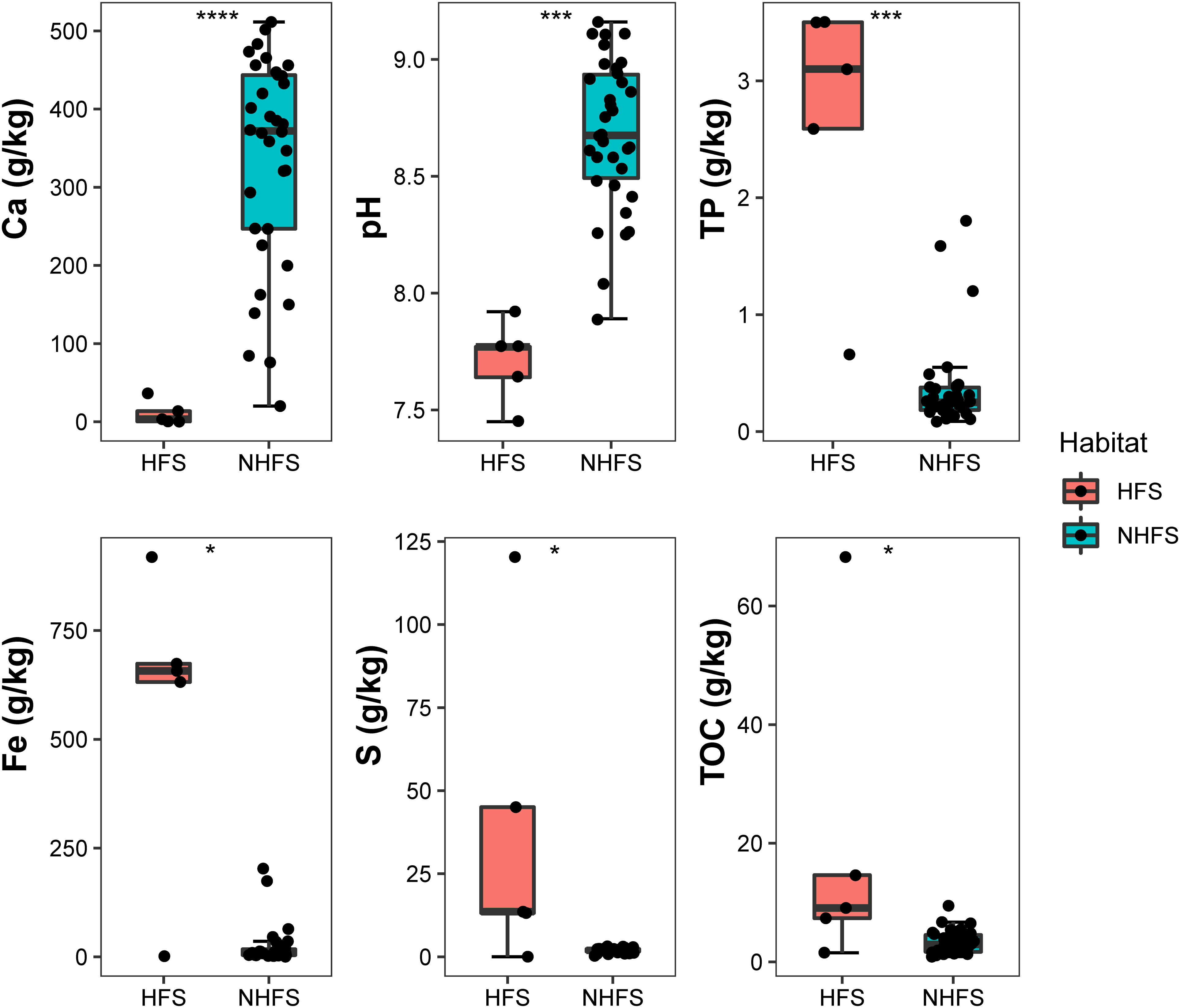
Figure 2 Environmental factors that are significantly different between the HFS and NHFS samples. Wilcoxon test, *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Diversity and distribution of prokaryotic communities
After quality control and chimera removal, a total of 1,534,540 16S rRNA gene sequences were obtained from the 39 samples, including 1,141,817 bacterial and 392,723 archaeal sequences. These sequences were binned into 14,978 non-singleton OTUs (1,663-6,260 OTUs per sample) at a 97% sequence similarity cutoff, with a coverage of 84.75-92.31% across the samples (Table S2). The OTUs belonged to 74 prokaryotic phyla (Supplementary Figure S3), 235 classes and 2,059 genera (892 classified genera and 1,167 unclassified genera). Proteobacteria (relative abundance 36.98%), Thaumarchaeota (23.31%), Planctomycetes (8.53%), Actinobacteria (5.44%), Acidobacteria (5.33%), Chloroflexi, Bacteroidetes, Gemmatimonadetes, and Nitrospirae were the dominant phyla (average relative abundance >1% in all samples) in the communities. The dominant classes (relative abundance >1%) were Nitrososphaeria, Gammaproteobacteria, Alphaproteobacteria, Deltaproteobacteria, Acidimicrobiia, Phycisphaerae, Planctomycetacia, Bacteroidia, Dehalococcoidia, Gemmatimonadetes, Nitrospira, and Anaerolineae, constituting 78.75% of the total sequences. At the genus level, Nitrosopumilus of the phylum Thaumarchaeota was the most abundant (7.31%), followed by unclassified_Nitrosopumilaceae (5.67%), Woeseia (3.95%), Escherichia-Shigella (2.85%), Urania-1B-19 marine sediment group (2.68%), unclassified_Actinomarinales (2.17%), unclassified_Kiloniellaceae (1.69%), Halomonas (1.63%), Sphingomonas (1.61%), Nitrospira (1.56%), uncultured_Gemmatimonadaceae (1.55%), and Pir4 lineage (1.31%).
There was no significant difference in alpha diversity of the community between the HFS and NHFS habitats or between the CR and SWIR locations (P > 0.05; Supplementary Figure S4). However, NMDS analysis at 97% OTU similarity of 39 subsamples of even reads showed a biogeographic distribution of the microbial community between HFSs and NHFSs but not between the CR and SWIR (Figure 3A). This result accords with the distribution of environmental variables (Supplementary Figure S2).
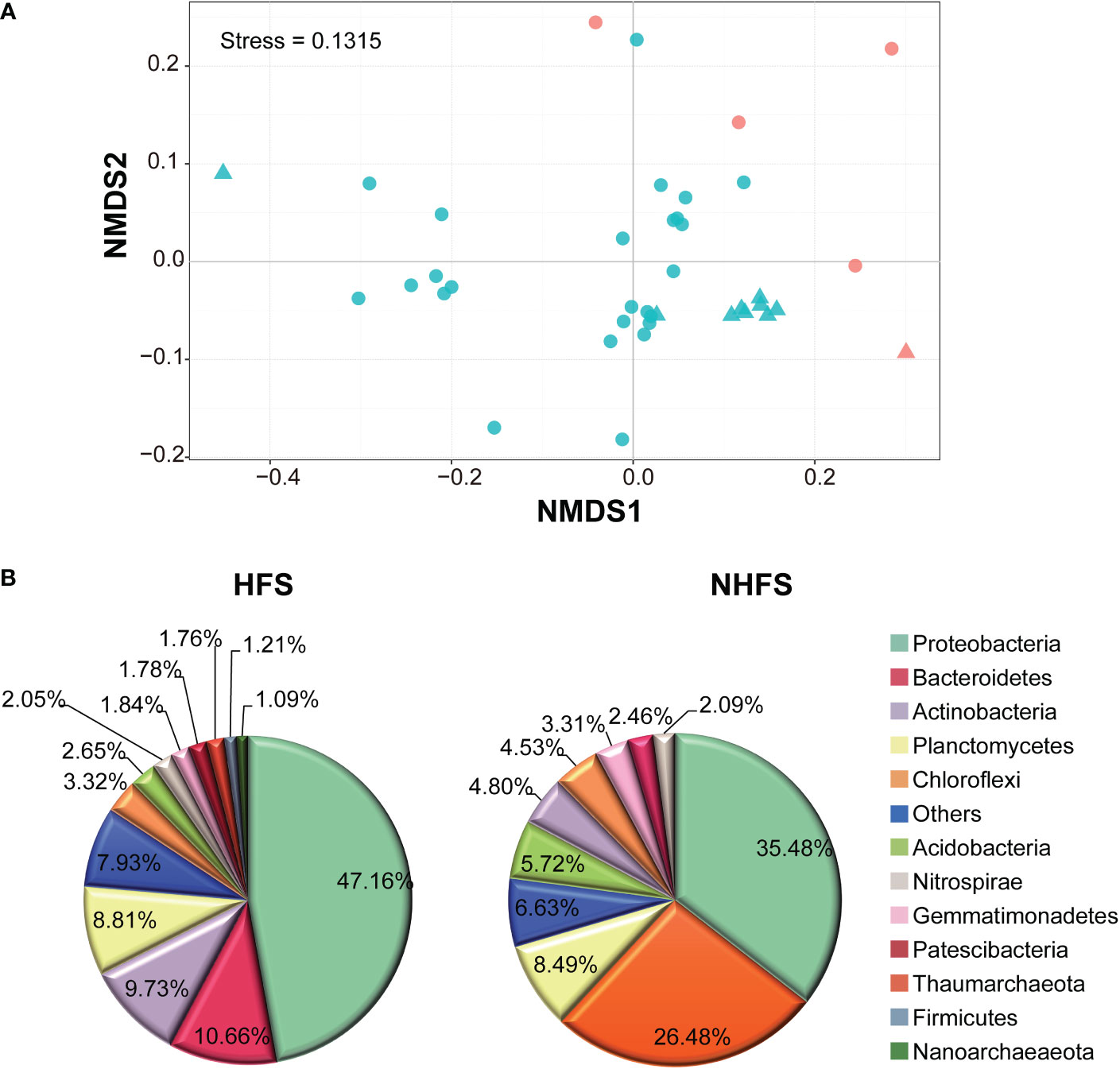
Figure 3 Differences between the HFS and NHFS samples in prokaryotic community structure. (A) NMDS of all 39 subsamples based on Bray-Curtis dissimilarities of 16S rRNA gene, representing the dissimilarity of the prokaryotic community composition in the samples at 97% OTU similarity. The red and blue colors indicate HFS and NHFS samples, respectively; the dots and triangles indicate CR and SWIR samples, respectively. (B) Pie graphs showing the dominant phyla (relative abundance >1%) in the HFS and NHFS samples.
Most of the above nine dominant phyla showed remarkably different relative abundances between HFSs and NHFSs (Figure 3B). Besides, Firmicutes, Nanoarchaeaeota, and Patescibacteria were also dominant in the HFS samples (Figure 3B). Using LEfSe at the phylum level, 16 of the 74 phyla were determined to be differentially abundant between HFSs and NHFSs (LDA > 3.0, P < 0.05; Figure 4). Nine phyla were enriched in HFSs, of which Proteobacteria, Aerophobetes, Bacteroidete, and Deferribacteres were the most enriched (LDA > 4.0, P < 0.05; Figure 4); seven phyla were enriched in NHFSs, with Thaumarchaeota the most enriched (LDA > 4.0, P < 0.05; Figure 4). It should not be neglected that Epsilonbacteraeota and Hydrothermarchaeota, previously reported to be enriched in HFSs (Kato et al., 2010; Zhou et al., 2020), were also enriched in our HFS samples (LDA > 3.0, P < 0.05; Figure 4). Epsilonproteobacteria are important chemolithotrophic primary producers in deep-sea hydrothermal vent systems (Kato et al., 2010; Waite et al., 2017), and Hydrothermarchaeota are widely distributed in HFSs (Zhou et al., 2020). Among the 2,059 genera, 54 (16 classified and 38 unclassified genera, accounting for 54.36% of all reads) were detected to be differentially abundant between HFSs and NHFSs by LEfSe (LDA > 3.0, P < 0.05; Supplementary Figure S5). HFSs and NHFSs were enriched for 28 and 26 genera, respectively. Achromobacter and Sphingomonas of Proteobacteria, uncultured_Bacteroidetes and Ambiguous_Actinobacteria were the most enriched in HFSs; while Nitrosopumilus and three uncultured genera of Thaumarchaeota and the uncultured Urania-1B-19 group of Planctomycetes were the most enriched in NHFSs (LDA > 4.0, P < 0.05, Supplementary Figure S5). Members of the genera Achromobacter and Sphingomonas have previously been found in deep-sea sediments (Zhang et al., 2014; Yang et al., 2020b; Kikukawa et al., 2021), and this is the first study to demonstrate that they are enriched in HFSs. In the previous studies of microbial diversity in deep-sea hydrothermal vents, Nitrosopumilus was found to adapt to inactive vents or cooler ecological niches of the vents (Zhang et al., 2016; Cerqueira et al., 2017). Our study further suggests that Nitrosopumilus might prefer NHFSs. As Nitrosopumilus consists of marine ammonia-oxidizing archaea, this genus may play a significant role in the carbon and nitrogen cycling in non-hydrothermal environments of the global ocean (Walker et al., 2010; Qin et al., 2017).
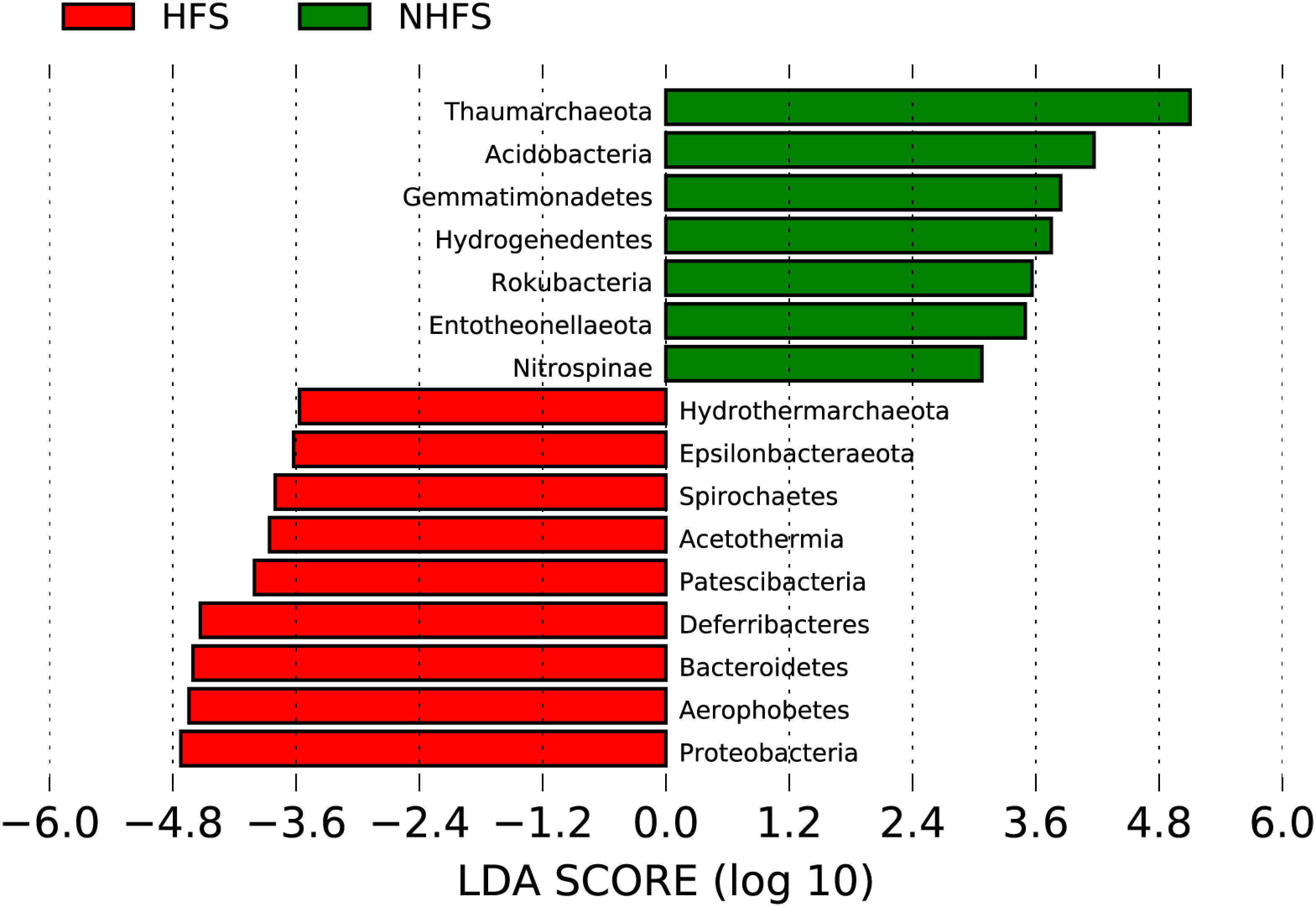
Figure 4 Taxonomic representation of the statistical differences at the phylum level between the prokaryotic communities in HFSs and NHFSs. The differential phyla in the histogram were ranked by effect size with an LDA threshold of 3.0.
Although the microbial community compositions were obviously different between HFSs and NHFSs, 190 OTUs were commonly present in all samples. These common OTUs accounted for only 1.27% of the total OTUs but 50.81% of the total reads, with the relative abundance of each OTU ranging from 0.02 to 4.93%. These OTUs belonged to 13 phyla (Supplementary Figure S6A), most of which have been reported to be common phyla in marine. Only 11 of these OTUs had a relative abundance of >1% each, and seven of them belonged to Nitrosopumilaceae of Thaumarchaeota. The phyla Proteobacteria (13.56%) and Thaumarchaeota (21.02%) were the most dominant in the common OTUs of HFSs and NHFSs, respectively (Supplementary Figure S6B). At the genus level, these common OTUs belonged to 106 genera, and 78 of them were unclassified, indicating that most of them represent novel taxa that have been little studied.
Our data on the SWIR samples contained all the phyla that were detected in a previous clone library study on semi-consolidated carbonate sediments of the SWIR (Li et al., 2014) (Supplementary Figure S1). The relative abundances of most of these phyla were largely consistent between the studies, but Planctomycetes, Chloroflexi, Nitrospirae, and Thaumarchaeota were more abundant while Acidobacteria and Verrucomicrobia were less abundant in our study. In addition, one dominant phylum detected here, Gemmatimonadetes, was absent in the previous study. The dominant prokaryotic taxa showed in HFSs of our study were contrast to those reported in a recent study on the newly discovered serpentinite-hosted Old City hydrothermal field of the SWIR (Lecoeuvre et al., 2021). The Old City hydrothermal chimneys had low relative abundances of Actinobacteria, Bacteroidetes, Proteobacteria, and Nanoarchaeaeota while more abundant Schekmanbacteria and Thaumarchaeota. The varied microbial communities in different samples of the SWIR may be attributed to different local geochemistry factors.
Factors influencing the community structure
The concordant NMDS biplots of prokaryotic communities and physicochemical factors suggested a linkage between the community structure and environmental factors (Figure 3A and Supplementary Figure S2). This relationship was supported by the significant Spearman correlation between the community compositions and the environmental attributes of all 39 samples (Figure 5A). RDA analysis revealed that all of the environmental factors explained as much as 44.85% of the overall variance of the communities (P = 0.001), with Ca, S, TP, and TN showing significant effects (Figure 5B; Table S3). Bioenv analysis indicated that water depth, Ca, S, nitrate nitrogen, Fe, and TN were the main environmental variables affecting the beta diversity (dissimilarity) among communities, explaining as much as 36.04% of the overall variance. As the content of Ca, S, TP, and Fe differed significantly between HFSs and NHFSs (Figure 2), these results sustain that the environmental differences between the two habitats shape the prokaryotic community structure. The results also align with our previous finding that Ca and TP, as essential nutrients, significantly correlate to prokaryotic communities along the SWIR (Chen et al., 2016). Moreover, S and Fe have been reported to be enriched in deep-sea hydrothermal vents (Jannasch and Mottl, 1985) and play an important role in fueling the microbes present in the vent habitats on the SWIR (Cao et al., 2014).
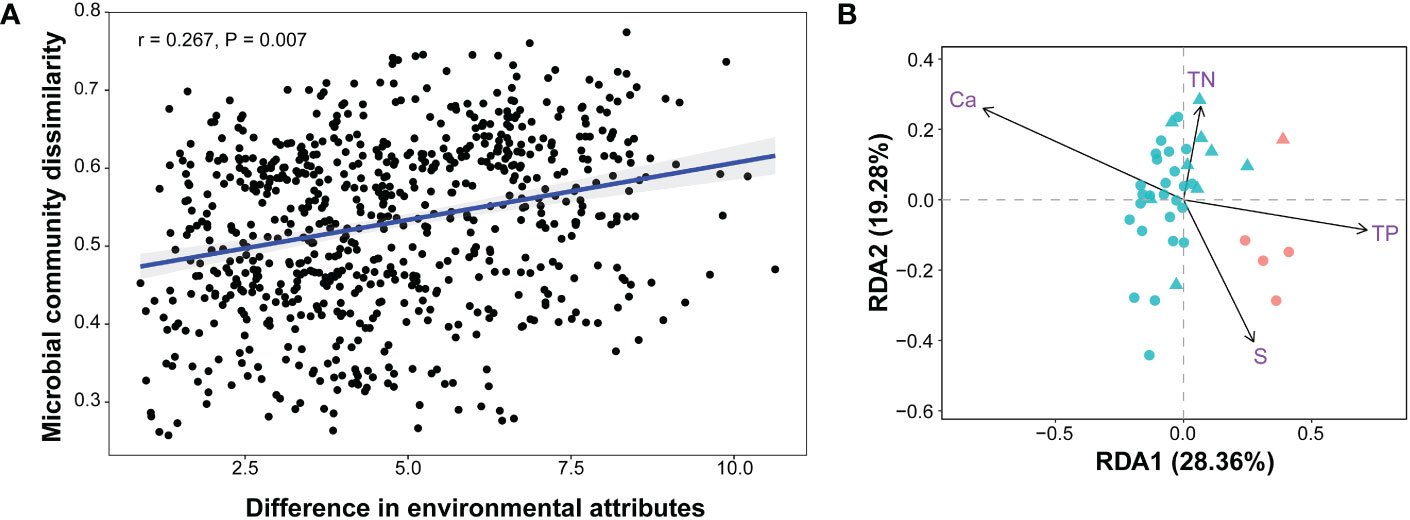
Figure 5 Correlations between the prokaryotic communities and environmental attributes (12 physicochemical factors and depth) of all 39 samples. Bray-Curtis distances of prokaryotic communities and Euclidean distances of environmental attributes among samples were calculated. (A) Spearman correlation. (B) Redundancy analysis (RDA). The arrows indicate significant factors; the red and blue colors indicate HFS and NHFS samples, respectively; the dots and triangles indicate CR and SWIR samples, respectively.
To quantify the contribution of spatial distance to prokaryotic community variability, Spearman correlations between Bray-Curtis community dissimilarities and geographic distances were calculated. There was no significant correlation across all samples. However, when focusing on the NHFS samples only, a weak significant correlation was observed (r = 0.195, P = 0.022; Supplementary Figure S7). The NHFS samples had relatively similar environmental variables but geographic distance up to 5582 km, it was therefore expected that dispersal limitation would become important at such a spatial scale. This is consistent with the commonly observed distance–decay pattern (Martiny et al., 2011). For the whole dataset containing both the HFS and NHFS samples, the remarkable difference in environmental variables probably disrupts the overall distance–decay pattern, supporting the Baas-Becking hypothesis that ‘the environment selects’ (Martiny et al., 2006). Together, the prokaryotic biogeographic pattern along the IOR is readily shaped by environmental selection between HFSs and NHFSs while also affected by large spatial distance (>5000 km).
Potential functional diversity and distribution patterns of functional microbes
To explore the functional capabilities of the prokaryotic communities in the sediments of the IOR, we used PICRUSt2 (Douglas et al., 2020) to predict the metabolic functions from the 16S rRNA gene sequences. A total of 437 metabolic pathways (level 3) belonging to 49 level 2 and seven level 1 KEGG pathway groups were predicted in all the samples. As shown in Supplementary Figure S8, nine level 2 KEGG pathway groups were significantly different in abundance between the HFS and NHF communities (P < 0.05). The HFS communities were mainly enriched for pathways related to lipid metabolism, unclassified: signaling and cellular processes, and cell growth and death (Supplementary Figure S8), reflecting the ability of microbes to adapt to dynamic changes in hydrothermal habitats. On the other hand, pathways of energy metabolism, metabolism of cofactors and vitamins, and transcription were mainly enriched in the NHFS communities. The microbes in NHFS may be committed to obtaining more energy to maintain physiological metabolic activities.
Energy metabolism pathways related to the cycles of carbon, nitrogen, and sulfur elements showed some differences between the HFS and NHFS samples (Figure 6). Compared to HFSs, NHFSs contained more abundant genes involved in carbon fixation pathways (P = 0.021; Figure 6A, Supplementary Figure S9). The genes required for the 3-hydroxypropionate/4-hydroxybutyrate (3-HP/4-HB) pathway for CO2 fixation, i.e., accA encoding the acetyl-coenzyme A (CoA) carboxylase and abfD encoding the 4-HB-CoA dehydratase, were predicted. Although accA was abundant in HFSs, abfD was lacked in the samples; whereas both genes were abundant in NHFSs (Figure 6B). The abundant Thaumarchaeota and some Crenarchaeota in NHFSs likely use the 3-HP/4-HB pathway for autotrophic carbon fixation (Pester et al., 2011). Meanwhile, the three key genes of the reductive tricarboxylic acid (rTCA) cycle, namely aclB, oorA, and por/nifJ, were also predicted with a higher abundance in NHFSs. Microbes utilizing the rTCA cycle to fix CO2 are ubiquitous in microaerophilic to anaerobic and sulfuric-rich environments, such as deep-sea hydrothermal vents (Campbell and Cary, 2004; Nakagawa and Takai, 2008). As for methane metabolism pathways, the genes encoding copper-dependent particulate methane monooxygenase (pMMO) for methane oxidation (pmoA, pmoB, and pmoC) were enriched in NHFSs, probably contributed by methane-oxidizing bacteria of Alphaproteobacteria, Gammaproteobacteria, and NC10 (Kirchman, 2012; Offre et al., 2013). Moreover, methanotrophs seemed to overwhelm methanogens in both HFSs and NHFSs, as the average abundance of methane-oxidizing genes such as pmo were more than 10 times that of methanogenic genes such as mcr. This may be attribute to the fact that methane is abundant in deep sea sediments (Baker et al., 1995).
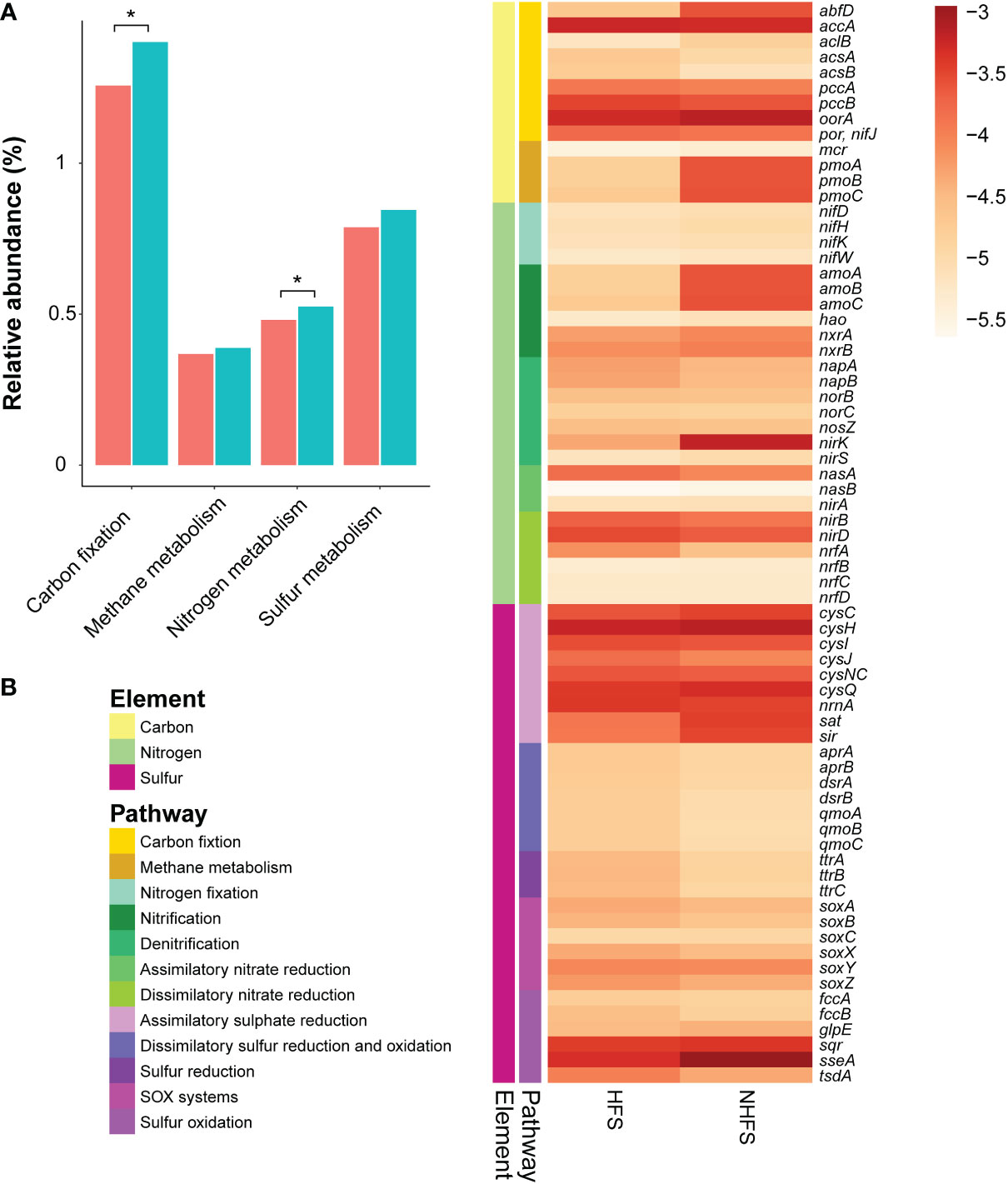
Figure 6 The relative abundance of element metabolism pathways and related genes involved in energy metabolism in HFSs and NHFSs based on KEGG orthologous gene identification by PICRUSt2 analysis. (A) Comparison of the relative abundance of carbon, nitrogen, and sulfur metabolism pathways between the two habitats. The red and blue colors indicate HFSs and NHFSs, respectively.*, P < 0.05 by Welch’s t-test. (B) Heat map showing the distribution of representative genes involved in energy metabolism in the two habitats. The color bar indicates the logarithm of relative abundance of the genes. SOX systems, sulfur oxidizing protein systems.
A number of gene families related to nitrogen metabolism were predicted (Figure 6), forming a complete nitrogen cycle. In general, nitrogen metabolism pathways were enriched in NHFSs (P = 0.048) (Figure 6A). The gene families involved in N2-fixation, including nifD, nifH, nifK, and nifW, showed little difference in relative abundance between HFSs and NHFSs (Figure 6B). It was noticeable that the genes for ammonia oxidation (amoA, amoB, and amoC) were much more abundant in NHFSs than in HFSs (Figure 6B). Congruently, prokaryotes related to ammonia oxidation, especially members of Thaumarchaeota, had a relative abundance in NHFSs more than 10 times that in HFSs (Figure 4 and Supplementary Figure S5). These results support that N cycle is more important in NHFSs than in HFSs, because nitrification, containing ammonia oxidation and nitrite oxidation, is the important process in N cycle and ammonia oxidation is the rate-limiting step of nitrification. Furthermore, gene families for each process of denitrification were predicted, including napAB, nirKS, norBC, and nosZ, where nirK (encoding Cu-containing nitrite reductase, Cu-NIR) was remarkably more abundant in NHFSs than in HFSs. Meanwhile, some assimilatory and dissimilatory nitrate reduction genes (nasA, nirBD, and nrfA) were enriched in HFSs.
Collectively, the gene families involved in carbon fixation and ammonia oxidation pathways were more abundant in NHFSs than in HFSs. Both of the two pathways seem to be related to Thaumarchaeota, which was significantly enriched in NHFs according to the result of LEfSe (Figure 4). It has been illustrated that some members of Thaumarchaeota support their chemolithoautotrophic lifestyle in the process of ammonia oxidation through the 3-HP/4-HB pathway (Pester et al., 2011). Given the high relative abundances of both the genes and taxa participating in the N and C cycles in NHFSs, the N cycle might be centralized in an energy-generating manner to support the microbial community in NHFSs.
On top of that, diverse genes involved in the S cycle were also predicted (Figure 6B). The assimilatory sulfate reduction pathway was abundant compared to the other pathways in both HFSs and NHFSs. Assimilatory sulfate reduction is the predominant way for microbes to integrate sulfur from environment into cells, and plays a vital role in microbial sulfur metabolism. On one hand, the sulfate assimilation process is the key source of cysteine in microorganisms; on the other hand, this pathway also involves in the detoxification of heavy metals in the organisms (Salazar et al., 2013; Wheaton et al., 2016), which may help deep-sea microbes to withstand high concentrations of metal ions. Sulfate represents one of the most common electron acceptors in the ocean. Genes related to the dissimilatory sulfate reduction pathway were mainly found from Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria in the samples and assigned to the apr, dsr, and qmo families. These gene families had a slightly higher relative abundance in HFS than in NHFS (Figure 6B). The dsr genes might be involved in both dissimilatory sulfur reduction and oxidation. Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria were also the major classes related to sulfur oxidation, contributing gene families such as soxABCXYZ, fccAB, sqr and sseA. To sum up, the S cycle-related pathways were relatively complete and enriched in both the HFS and NHFS samples with no significant difference between the two habitats.
Diversity and distribution of functional genes involved in S cycling
Although the overall relative abundance of sulfur metabolism pathways was not significantly different between HFSs and NHFSs (Figure 6), prokaryotes with sulfate-reducing ability exist widely in deep-sea hydrothermal vents (Frank et al., 2013; Meier et al., 2019). To further explore the difference of sulfate-reducing prokaryotes between HFSs and NHFSs, we used four HFSs (N3, N4, N5, and N6) and four NHFSs (N8, N12, N15, and N18) from CR as representative samples to investigate the diversity and distribution of dsrB genes, the well-known molecular marker of sulfate reducers. NMDS analyses of the physicochemical factors and 16S rRNA gene-based microbial communities both showed that the eight samples also clustered according to habitat (Supplementary Figure S10). A total of 427,499 high-quality dsrB sequences were obtained, which were divided into 129 OTUs according to 90% nucleotide similarity. The two habitats shared 25 dsrB OTUs, and the HFS samples showed a significant higher alpha diversity of dsrB than the NHFS samples (P < 0.05; Table S4). A comparison to the DsrAB reference database (Müller et al., 2015) and subsequent phylogenetic analysis (Figure 7) showed that the OTUs were assigned into 13 class-level taxa, mainly uncultured and unclassified clades. The vast majority of dsrB OTUs (92/129) were clustered with sequences from marine sources. Only a small proportion of the OTUs were clustered with sequences of known sulfate-reducing strains, most of which belong to Deltaproteobacteria, dominated by Desulfobulbaceae and followed by Desulfobacteraceae and Syntrophobacteraceae. Very few OTUs were affiliated to Clostridia of the phylum Firmicutes. Deltaproteobacteria and Firmicutes are already known as common sulfate-reducing bacteria (Kaneko et al., 2007; Robador et al., 2014). In addition, dsrB sequences related to Thermodesulfobacteria (OTU35) were detected only in the HFS samples, in line with the thermophilic characteristic of the taxon. A previous study found that Thermodesulfobacteria existed in high abundance as sulfate-reducing microorganisms in deep-sea hydrothermal chimneys in the Okinawa trough and Izu-Bonin arc (Nakagawa et al., 2004). Forty-two OTUs (61.7% of the total sequence) had a similarity of < 90% with the known sequences in the reference (Müller et al., 2015) and NR database, and thus could be considered as putative novel dsrB genes. Among them, OTU86 was the most dominant OTU (26.3% of the total sequence), existing in all samples and showing a low similarity (< 77%) to the known dsrB sequences. This OTU may represent a potential novel bacterial phylogroup widely distributed in CR sediments.
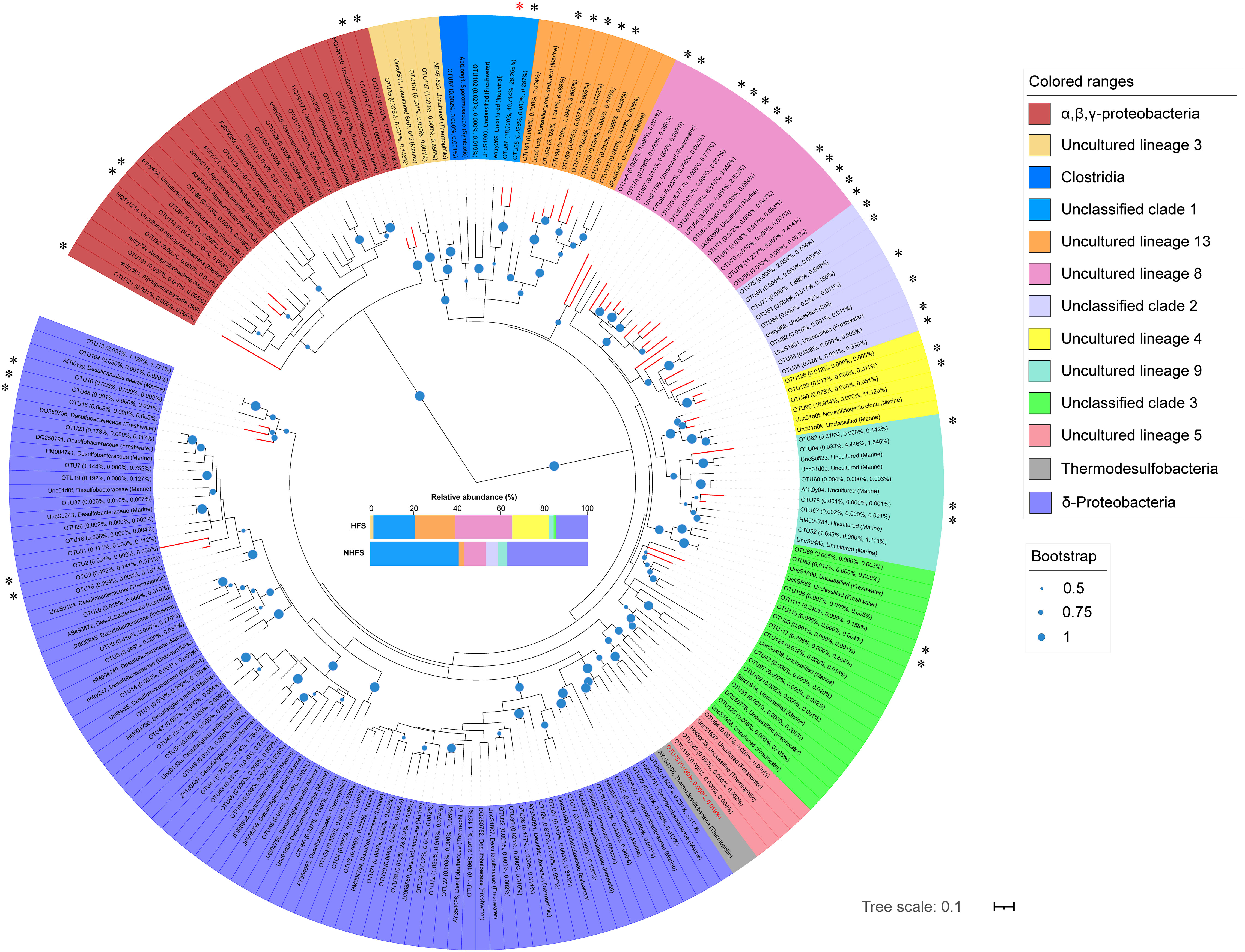
Figure 7 Maximum-likelihood phylogenetic tree of the representative bacterial dsrB sequences in this study and the relevant sequences retrieved from the reference (Müller et al., 2015). The average relative abundances of OTUs in the HFS, NHFS, and total samples are shown at the first, second, and third positions in the parentheses, respectively. The source habitats of the reference sequences are also given in the parentheses. Colors represent different microbial classes. The compositions of these classes in HFSs and NHFSs are shown in the middle of the figure. Bootstrap values higher than 50% of 1,000 resamples are shown by dots of different sizes at branch points. Scale bar represents 0.1 nucleotides substitution per site. Branches of potential novel dsrB genes are shown in red, and corresponding OTUs are marked with “*”, with the most abundant “OTU86” marked in red. The sequence name of “OTU35” is in red.
In short, dsrB sequencing analysis further confirms the existence of diverse and novel sulfate/sulfite-reducing microbes in the IOR sediments. The HFS samples contain higher diversity of sulfate reduction genes and taxa than the NHFS samples, inferring that the S cycle is more important for the HFS microbial community.
Conclusions
Our results demonstrate that the sediments along the SWIR and CR contain high microbial diversity. Both the total prokaryotes and functional groups have a biogeographic distribution, which is significantly shaped by environmental selection while also affected by large-scale geographic distance. Microbes in HFSs and NHFSs differed in the enrichment of genes related to C, N, and S metabolism pathways. NHFSs contain more abundant functional groups involved in the N and C cycles while HFSs contain a higher diversity of sulfate-reducing bacteria, reflecting the microbial adaptability to different habitats. This study provides further insights into the assembly patterns and metabolic differentiation of microbial communities at the IOR under the influence of hydrothermal activity. In future studies, enhanced sampling at a global geographic scale with more hydrothermal fields is needed to elucidate the microbial diversity, biogeography, and distribution mechanism along MOR. Also, culture and simulation studies are required to understand the ecological functions of deep-sea microbial communities.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
JH: Validation, Formal analysis, Investigation, Data Curation, Writing - Original Draft, Writing - Review and Editing, Visualization. PC: Methodology, Validation, Investigation, Data Curation, Writing - Original Draft, Visualization. YZ: Methodology, Investigation. JW: Resources. LS: Resources. XH: Resources, Writing - Review and Editing, Funding acquisition. YH: Conceptualization, Validation, Resources, Writing - Review and Editing, Visualization, Supervision, Project administration, Funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key R&D Program of China (2018YFC0310703 and 2021YFF0501304), the program of China Ocean Mineral Resources R&D Association (DY135-B2-02), and the National Natural Science Foundation of China (91751118).
Acknowledgments
We are grateful to all the scientists and crewmembers on the RVs during the expedition for their assistance in sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1072569/full#supplementary-material
Abbreviations
HFS, hydrothermal field sediment; NHFS, non-hydrothermal field sediment; CR, Carlsberg Ridge; SWIR, Southwest Indian Ridge; IOR, Indian Ocean Ridges; MOR, mid-ocean ridges.
References
Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi: 10.1093/nar/25.17.3389
Anantharaman K., Hausmann B., Jungbluth S. P., Kantor R. S., Lavy A., Warren L. A., et al. (2018). Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 12 (7), 1715–1728. doi: 10.1038/s41396-018-0078-0
Baker E. T., German C. R., Elderfield H. (1995). “Hydrothermal Plumes Over Spreading-Center Axes: Global Distributions and Geological Inferences.” in Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions eds Humphris S. E., Zierenberg R. A., Mullineaux L. S., Thomson R. E. (Washington: American Geophysical Union) 47–71.
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc B. 57 (1), 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Berg I. A. (2011). Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77 (6), 1925–1936. doi: 10.1128/Aem.02473-10
Bokulich N. A., Subramanian S., Faith J. J., Gevers D., Gordon J. I., Knight R., et al. (2013). Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 10 (1), 57–59. doi: 10.1038/nmeth.2276
Campbell B. J., Cary S. C. (2004). Abundance of reverse tricarboxylic acid cycle genes in free-living microorganisms at deep-sea hydrothermal vents. Appl. Environ. Microbiol. 70 (10), 6282–6289. doi: 10.1128/AEM.70.10.6282-6289.2004
Cao H., Wang Y., Lee O. O., Zeng X., Shao Z., Qian P.-Y. (2014). Microbial sulfur cycle in two hydrothermal chimneys on the southwest Indian ridge. mBio 5 (1), e00980–e00913. doi: 10.1128/mBio.00980-13
Caporaso J. G., Bittinger K., Bushman F. D., DeSantis T. Z., Andersen G. L., Knight R. (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26 (2), 266–267. doi: 10.1093/bioinformatics/btp636
Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 (5), 335–336. doi: 10.1038/nmeth.f.303
Carini P., White A. E., Campbell E. O., Giovannoni S. J. (2014). Methane production by phosphate-starved SAR11 chemoheterotrophic marine bacteria. Nat. Commun. 7 (5), 4346. doi: 10.1038/ncomms5346
Cerqueira T., Pinho D., Froufe H., Santos R. S., Bettencourt R., Egas C. (2017). Sediment microbial diversity of three deep-sea hydrothermal vents southwest of the Azores. Microb. Ecol. 74 (2), 332–349. doi: 10.1007/s00248-017-0943-9
Chen P., Zhang L., Guo X., Dai X., Liu L., Xi L., et al. (2016). Diversity, biogeography, and biodegradation potential of Actinobacteria in the deep-sea sediments along the southwest Indian ridge. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01340
Cole J. R., Wang Q., Fish J. A., Chai B., McGarrell D. M., Sun Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
DeSantis T. Z., Hugenholtz P., Larsen N., Rojas M., Brodie E. L., Keller K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72 (7), 5069–5072. doi: 10.1128/AEM.03006-05
Douglas G. M., Maffei V. J., Zaneveld J. R., Yurgel S. N., Brown J. R., Taylor C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38 (6), 685–688. doi: 10.1038/s41587-020-0548-6
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 (19), 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 (10), 996–998. doi: 10.1038/nmeth.2604
Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27 (16), 2194–2200. doi: 10.1093/bioinformatics/btr381
Frank K. L., Rogers D. R., Olins H. C., Vidoudez C., Girguis P. R. (2013). Characterizing the distribution and rates of microbial sulfate reduction at middle valley hydrothermal vents. ISME J. 7 (7), 1391–1401. doi: 10.1038/ismej.2013.17
Geets J., Borremans B., Diels L., Springael D., Vangronsveld J., van der Lelie D., et al. (2006). DsrB gene-based DGGE for community and diversity surveys of sulfate-reducing bacteria. J. Microbiol. Methods 66 (2), 194–205. doi: 10.1016/j.mimet.2005.11.002
German C., Baker E., Mevel C., Tamaki K. (1998). Hydrothermal activity along the southwest Indian ridge. Nature 395 (6701), 490–493. doi: 10.1038/26730
He J.-Z., Shen J.-P., Zhang L.-M., Zhu Y.-G., Zheng Y.-M., Xu M.-G., et al. (2007). Quantitative analyses of the abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea of a Chinese upland red soil under long-term fertilization practices. Environ. Microbiol. 9 (9), 2364–2374. doi: 10.1111/j.1462-2920.2007.01481.x
Holmfeldt K., Dziallas C., Titelman J., Pohlmann K., Grossart H. P., Riemann L. (2009). Diversity and abundance of freshwater Actinobacteria along environmental gradients in the brackish northern Baltic Sea. Environ. Microbiol. 11 (8), 2042–2054. doi: 10.1111/j.1462-2920.2009.01925.x
Hugler M., Sievert S. M. (2011). Beyond the Calvin cycle: Autotrophic carbon fixation in the ocean. Annu. Rev. Mar. Sci. 3, 261–289. doi: 10.1146/annurev-marine-120709-142712
Hutchins D. A., Fu F. (2017). Microorganisms and ocean global change. Nat. Microbiol. 2, 17058. doi: 10.1038/nmicrobiol.2017.58
Iyer S. D., Mukhopadhyay R., Drolia R. K., Ray D. (2003). Mid-ocean ridges, InRidge and the future. Curr. Sci. 85 (3), 272–276.
Jannasch H. W., Mottl M. J. (1985). Geomicrobiology of deep-sea hydrothermal vents. Science 229 (4715), 717–725. doi: 10.1126/science.229.4715.717
Jorgensen S. L., Hannisdal B., Lanzén A., Baumberger T., Flesland K., Fonseca R., et al. (2012). Correlating microbial community profiles with geochemical data in highly stratified sediments from the Arctic mid-ocean ridge. Proc. Natl. Acad. Sci. U. S. A. 109 (42), E2846–E2855. doi: 10.1073/pnas.1207574109
Kaneko R., Hayashi T., Tanahashi M., Naganuma T. (2007). Phylogenetic diversity and distribution of dissimilatory sulfite reductase genes from deep-sea sediment cores. Mar. Biotechnol. (NY). 9 (4), 429–436. doi: 10.1007/s10126-007-9003-7
Kato S., Takano Y., Kakegawa T., Oba H., Inoue K., Kobayashi C., et al. (2010). Biogeography and biodiversity in sulfide structures of active and inactive vents at deep-sea hydrothermal fields of the southern Mariana trough. Appl. Environ. Microbiol. 76 (9), 2968–2979. doi: 10.1128/AEM.00478-10
Kikukawa H., Okaya T., Maoka T., Miyazaki M., Murofushi K., Kato T., et al. (2021). Carotenoid nostoxanthin production by sphingomonas sp. SG73 isolated from deep sea sediment. Mar. Drugs 19 (5), 274. doi: 10.3390/md19050274
Kumar S., Stecher G., Tamura K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lecoeuvre A., Menez B., Cannat M., Chavagnac V., Gerard E. (2021). Microbial ecology of the newly discovered serpentinite-hosted old city hydrothermal field (southwest Indian ridge). ISME J. 15 (3), 818–832. doi: 10.1038/s41396-020-00816-7
Li J., Peng X., Zhou H., Li J., Sun Z. (2013). Molecular evidence for microorganisms participating in fe, Mn, and s biogeochemical cycling in two low-temperature hydrothermal fields at the southwest Indian ridge. J. Geophysical Research: Biogeosciences. 118 (2), 665–679. doi: 10.1002/jgrg.20057
Li J., Peng X., Zhou H., Li J., Sun Z., Chen S. (2014). Microbial communities in semi-consolidated carbonate sediments of the southwest Indian ridge. J. Microbiol. 52 (2), 111–119. doi: 10.1007/s12275-014-3133-1
López-García P., Duperron S., Philippot P., Foriel J., Susini J., Moreira D. (2003). Bacterial diversity in hydrothermal sediment and epsilonproteobacterial dominance in experimental microcolonizers at the mid-Atlantic ridge. Environ. Microbiol. 5 (10), 961–976. doi: 10.1046/j.1462-2920.2003.00495.x
Magoč T., Salzberg S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27 (21), 2957–2963. doi: 10.1093/bioinformatics/btr507
Martiny J. B. H., Bohannan B. J., Brown J. H., Colwell R. K., Fuhrman J. A., Green J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4 (2), 102–112. doi: 10.1038/nrmicro1341
Martiny J. B., Eisen J. A., Penn K., Allison S. D., Horner-Devine M. C. (2011). Drivers of bacterial beta-diversity depend on spatial scale. Proc. Natl. Acad. Sci. U. S. A. 108 (19), 7850–7854. doi: 10.1073/pnas.1016308108
McGlynn S. E., Chadwick G. L., O’Neill A., Mackey M., Thor A., Deerinck T. J., et al. (2018). Subgroup characteristics of marine methane-oxidizing ANME-2 archaea and their syntrophic partners as revealed by integrated multimodal analytical microscopy. Appl. Environ. Microbiol. 84 (11), e00399–e00318. doi: 10.1128/aem.00399-18
Meier D. V., Pjevac P., Bach W., Markert S., Schweder T., Jamieson J., et al. (2019). Microbial metal-sulfide oxidation in inactive hydrothermal vent chimneys suggested by metagenomic and metaproteomic analyses. Environ. Microbiol. 21 (2), 682–701. doi: 10.1111/1462-2920.14514
Müller A. L., Kjeldsen K. U., Rattei T., Pester M., Loy A. (2015). Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi) sulfite reductases. ISME J. 9 (9), 1152–1165. doi: 10.1038/ismej.2014.208
Murton B., Taylor R. (2003). Cruise report: CD149–RRS Charles Darwin 18th July to 6th August 2003. spreading-ridge geometry, hydrothermal activity, and the influence of modern and ancient hotspots on the Carlsberg Ridge-northwestern Indian ocean. (Southampton: Southampton Oceangraphy center).
Nakagawa T., Nakagawa S., Inagaki F., Takai K., Horikoshi K. (2004). Phylogenetic diversity of sulfate-reducing prokaryotes in active deep-sea hydrothermal vent chimney structures. FEMS. Microbiol. Lett. 232 (2), 145–152. doi: 10.1016/S0378-1097(04)00044-8
Nakagawa S., Takai K. (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS. Microbiol. Ecol. 65 (1), 1–14. doi: 10.1111/j.1574-6941.2008.00502.x
Nercessian O., Fouquet Y., Pierre C., Prieur D., Jeanthon C. (2005). Diversity of bacteria and archaea associated with a carbonate-rich metalliferous sediment sample from the rainbow vent field on the mid-Atlantic ridge. Environ. Microbiol. 7 (5), 698–714. doi: 10.1111/j.1462-2920.2005.00744.x
Newberry C. J., Webster G., Cragg B. A., Parkes R. J., Weightman A. J., Fry J. C. (2004). Diversity of prokaryotes and methanogenesis in deep subsurface sediments from the nankai trough, ocean drilling program leg 190. Environ. Microbiol. 6 (3), 274–287. doi: 10.1111/j.1462-2920.2004.00568.x
Nunoura T., Oida H., Nakaseama M., Kosaka A., Ohkubo S. B., Kikuchi T., et al. (2010). Archaeal diversity and distribution along thermal and geochemical gradients in hydrothermal sediments at the yonaguni knoll IV hydrothermal field in the southern Okinawa trough. Appl. Environ. Microbiol. 76 (4), 1198–1211. doi: 10.1128/AEM.00924-09
Offre P., Spang A., Schleper C. (2013). Archaea in biogeochemical cycles. Annu. Rev. Microbiol. 67, 437–457. doi: 10.1146/annurev-micro-092412-155614
Oksanen J., Blanchet F. G., Friendly M., Kindt R., Legendre P., McGlinn D., et al. (2020). Vegan: Community Ecology Package. R package version 2.5-7. Available at: https://CRAN.R-project.org/package=vegan.
Orcutt B. N., Sylvan J. B., Knab N. J., Edwards K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75 (2), 361–422. doi: 10.1128/MMBR.00039-10
Parks D. H., Tyson G. W., Hugenholtz P., Beiko R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30 (21), 3123–3124. doi: 10.1093/bioinformatics/btu494
Pelikan C., Herbold C. W., Hausmann B., Muller A. L., Pester M., Loy A. (2016). Diversity analysis of sulfite- and sulfate-reducing microorganisms by multiplex dsrA and dsrB amplicon sequencing using new primers and mock community-optimized bioinformatics. Environ. Microbiol. 18 (9), 2994–3009. doi: 10.1111/1462-2920.13139
Perez M., Sun J., Xu Q. Z., Qian P. Y. (2021). Structure and connectivity of hydrothermal vent communities along the mid-ocean ridges in the West Indian ocean: a review. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.744874
Pester M., Schleper C., Wagner M. (2011). The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 14 (3), 300–306. doi: 10.1016/j.mib.2011.04.007
Popoola S. O., Han X. Q., Wang Y. J., Qiu Z. Y., Ye Y., Cai Y. Y. (2019). Mineralogical and geochemical signatures of metalliferous sediments in wocan-1 and wocan-2 hydrothermal sites on the carlsberg ridge, Indian ocean. Minerals 9 (1), 26. doi: 10.3390/min9010026
Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5 (3), e9490. doi: 10.1371/journal.pone.0009490
Pruitt K. D., Tatusova T., Maglott D. R. (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65. doi: 10.1093/nar/gkl842
Qian P.-Y., Wang Y., Lee O. O., Lau S. C., Yang J., Lafi F. F., et al. (2011). Vertical stratification of microbial communities in the red Sea revealed by 16S rDNA pyrosequencing. ISME J. 5 (3), 507–518. doi: 10.1038/ismej.2010.112
Qin W., Heal K. R., Ramdasi R., Kobelt J. N., Martens-Habbena W., Bertagnolli A. D., et al. (2017). Nitrosopumilus maritimus gen. nov., sp nov., Nitrosopumilus cobalaminigenes sp nov., Nitrosopumilus oxyclinae sp nov., and Nitrosopumilus ureiphilus sp nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int. J. Syst. Evol. Microbiol. 67 (12), 5067–5079. doi: 10.1099/ijsem.0.002416
Robador A., Jungbluth S. P., LaRowe D. E., Bowers R. M., Rappé M. S., Amend J. P., et al. (2014). Activity and phylogenetic diversity of sulfate-reducing microorganisms in low-temperature subsurface fluids within the upper oceanic crust. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00748
Salazar C. N., Acosta M., Galleguillos P. A., Shmaryahu A., Quatrini R., Holmes D. S., et al. (2013). Analysis of gene expression in response to copper stress in Acidithiobacillus ferrooxidans strain D2, isolated from a copper bioleaching operation. Adv. Mat Res. 825, 157–161. doi: 10.4028/www.scientific.net/AMR.825.157
Schauer R., Bienhold C., Ramette A., Harder J. (2010). Bacterial diversity and biogeography in deep-sea surface sediments of the south Atlantic ocean. ISME J. 4 (2), 159–170. doi: 10.1038/ismej.2009.106
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12 (6), R60. doi: 10.1186/gb-2011-12-6-r60
Sievert S., Kiene R., Schulz-Vogt H. (2007). The sulfur cycle. Oceanography 20 (2), 117–123. doi: 10.5670/oceanog.2007.55
Simoneit B. R., Fetzer J. C. (1996). High molecular weight polycyclic aromatic hydrocarbons in hydrothermal petroleums from the gulf of California and northeast pacific ocean. Org. Geochem. 24 (10-11), 1065–1077. doi: 10.1016/S0146-6380(96)00081-2
Song B., Lisa J. A., Tobias C. R. (2014). Linking DNRA community structure and activity in a shallow lagoonal estuarine system. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00460
Storesund J. E., Lanzèn A., García-Moyano A., Reysenbach A. L., Øvreås L. (2018). Diversity patterns and isolation of Planctomycetes associated with metalliferous deposits from hydrothermal vent fields along the valu fa ridge (SW pacific). Antonie van Leeuwenhoek. 111 (6), 841–858. doi: 10.1007/s10482-018-1026-8
Tao C., Li H., Jin X., Zhou J., Wu T., He Y., et al. (2014). Seafloor hydrothermal activity and polymetallic sulfide exploration on the southwest Indian ridge. Chin. Sci. Bulletin. 59, 2266–2276. doi: 10.1007/s11434-014-0182-0
Tao C., Lin J., Guo S., Chen Y. J., Wu G., Han X., et al. (2012). First active hydrothermal vents on an ultraslow-spreading center: Southwest Indian ridge. Geology 40 (1), 47–50. doi: 10.1130/G32389.1
Tao C., Wu G., Deng X., Qiu Z., Han C., Long Y. (2013). New discovery of seafloor hydrothermal activity on the Indian ocean carlsberg ridge and southern north Atlantic ridge-progress during the 26th Chinese COMRA cruise. Acta Oceanologica Sinica. 32 (8), 85–88. doi: 10.1007/s13131-013-0345-x
Teske A., Alm E., Regan J., Toze S., Rittmann B., Stahl D. (1994). Evolutionary relationships among ammonia-and nitrite-oxidizing bacteria. J. Bacteriol. 176 (21), 6623–6630. doi: 10.1128/jb.176.21.6623-6630.1994
Teske A., Hinrichs K.-U., Edgcomb V., de Vera Gomez A., Kysela D., Sylva S. P., et al. (2002). Microbial diversity of hydrothermal sediments in the guaymas basin: evidence for anaerobic methanotrophic communities. Appl. Environ. Microbiol. 68 (4), 1994–2007. doi: 10.1128/AEM.68.4.1994-2007.2002
Varliero G., Bienhold C., Schmid F., Boetius A., Molari M. (2019). Microbial diversity and connectivity in deep-sea sediments of the south Atlantic polar front. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00665
Wagner M., Roger A. J., Flax J. L., Brusseau G. A., Stahl D. A. (1998). Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J. Bacteriol. 180 (11), 2975–2982. doi: 10.1128/JB.180.11.2975-2982.1998
Waite D. W., Vanwonterghem I., Rinke C., Parks D. H., Zhang Y., Takai K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00682
Walker C. B., de la Torre J. R., Klotz M. G., Urakawa H., Pinel N., Arp D. J., et al. (2010). Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine Crenarchaea. Proc. Natl. Acad. Sci. U. S. A. 107 (19), 8818–8823. doi: 10.1073/pnas.0913533107
Wang Y. J., Han X. Q., Petersen S., Frische M., Qiu Z. Y., Li H. M., et al. (2017). Mineralogy and trace element geochemistry of sulfide minerals from the wocan hydrothermal field on the slow-spreading carlsberg ridge, Indian ocean. Ore Geol. Rev. 84, 1–19. doi: 10.1016/j.oregeorev.2016.12.020
Wessel P., Smith W. H. (1998). New, improved version of generic mapping tools released. Eos Trans. Am. Geophysical Union. 79, 579–579. doi: 10.1029/98EO00426
Wheaton G. H., Mukherjee A., Kelly R. M. (2016). Transcriptomes of the extremely thermoacidophilic archaeon Metallosphaera sedula exposed to metal “shock” reveal generic specific metal responsesAppl. Environ. Microbiol. 82 (15), 4613–4627. doi: 10.1128/AEM.01176-16
Xie Q., Han X., Wei M., Qiu Z., Dong C., Wu Y., et al. (2022). Characteristics and evolution of bacterial communities in the wocan hydrothermal plume-influenced zone, carlsberg ridge, northwestern Indian ocean. Acta Microbiologica Sinica. 62 (6), 1974–1985. doi: 10.13343/j.cnki.wsxb.20220147
Yang S., Li X., Xiao X., Zhuang G., Zhang Y. (2020a). Sphingomonas profundi sp. nov., isolated from deep-sea sediment of the Mariana trench. Int. J. Syst. Evol. Microbiol. 70 (6), 3809–3815. doi: 10.1099/ijsem.0.004235
Yang Z., Xiao X., Zhang Y. (2020b). Microbial diversity of sediments from an inactive hydrothermal vent field, southwest Indian ridge. Mar. Life Sci. Technol. 2 (1), 73–86. doi: 10.1007/s42995-019-00007-0
Yilmaz P., Parfrey L. W., Yarza P., Gerken J., Pruesse E., Quast C., et al. (2014). The SILVA and “All-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Zeng Y.-X., Yu Y., Li H.-R., Luo W. (2017). Prokaryotic community composition in arctic kongsfjorden and sub-arctic northern Bering Sea sediments as revealed by 454 pyrosequencing. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02498
Zhang Z., Fan X., Gao X., Zhang X.-H. (2014). Achromobacter sediminum sp nov., isolated from deep subseafloor sediment of south pacific gyre. Int. J. Syst. Evol. Microbiol. 64 (Pt 7), 2244–2249. doi: 10.1099/ijs.0.062265-0
Zhang L., Kang M., Xu J., Xu J., Shuai Y., Zhou X., et al. (2016). Bacterial and archaeal communities in the deep-sea sediments of inactive hydrothermal vents in the southwest India ridge. Sci. Rep. 6, 25982. doi: 10.1038/srep25982
Zhang J., Sun Q.-L., Zeng Z.-G., Chen S., Sun L. (2015). Microbial diversity in the deep-sea sediments of iheya north and iheya ridge, Okinawa trough. Microbiol. Res. 177, 43–52. doi: 10.1016/j.micres.2015.05.006
Keywords: prokaryotes, community structure, 16S rRNA gene, dsrB, Carlsberg Ridge, Southwest Indian Ridge, element cycling
Citation: Huang J, Chen P, Zhu Y, Wang J, Song L, Han X and Huang Y (2023) Biogeography and potential ecological functions of prokaryotes in the hydrothermal and non-hydrothermal field sediments of the Indian Ocean Ridges. Front. Mar. Sci. 9:1072569. doi: 10.3389/fmars.2022.1072569
Received: 17 October 2022; Accepted: 13 December 2022;
Published: 12 January 2023.
Edited by:
Paraskevi Polymenakou, Hellenic Centre for Marine Research (HCMR), GreeceReviewed by:
Wei Xie, Sun Yat-sen University, Zhuhai Campus, ChinaHanzhi Lin, Science Systems and Applications, Inc., United States
Copyright © 2023 Huang, Chen, Zhu, Wang, Song, Han and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Huang, aHVhbmd5QGltLmFjLmNu
†These authors have contributed equally to this work