
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 30 November 2022
Sec. Aquatic Microbiology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1046808
This article is part of the Research TopicMicrobes from Marine Distinctive EnvironmentsView all 19 articles
 Tianying Chen1
Tianying Chen1 Yunyun Zhuang1,2*Chang Chen1
Yunyun Zhuang1,2*Chang Chen1 Xuewei Mao1Ruping Ge1
Xuewei Mao1Ruping Ge1 Hongju Chen1,2
Hongju Chen1,2 Jianwei Chen3,4Liang Fu5
Jianwei Chen3,4Liang Fu5 Zuosheng Yang6
Zuosheng Yang6 Guangxing Liu1,2
Guangxing Liu1,2The Sansha Yongle blue hole (SYBH) is the world’s deepest blue hole known so far and its unique hydrochemical characteristics make it a valuable site for studying biodiversity and ecological processes. Here, we used metabarcoding approach to investigate the diversity, assembly mechanism and co-occurrence pattern of planktonic microbiome in SYBH. Our results revealed the distinct separation of communities from upper oxic, middle oxic and suboxic-anoxic layer and significant day-night difference was detected in the upper-layer community of microeukaryotes, indicating potential diel migration. Stochastic processes played a significant role in the community assembly of microeukaryotes, while deterministic processes dominated in prokaryotes, confirming the stronger environmental filtering on prokaryotes as also suggested by the correlation with environmental variables. Microeukaryotes were less sensitive to environments but significantly affected by cross-domain biointeraction. When comparing the subcommunities of different abundance, we found that abundant taxa were widespread while rare taxa were habitat-specific. In co-occurrence network, over 87% of the interactions and 19 out of 20 keystone OTUs (Operational Taxonomic Units) were affiliated to moderate or rare taxa, suggesting the importance of non-abundant taxa in maintaining community stability. The predominant positive edges in the network pointed to that interspecies cooperation may be one of the ecological strategies in SYBH. Overall, we highlight the divergent assembly pattern and different driving forces in shaping plankton microbiome in SYBH, which may advance current understanding on diversity and dynamics of marine life in blue hole ecosystems.
Marine blue holes are underwater sinkholes open to the marine environment, which formed in carbonate bank during glacial periods when sea level was below current sea level and were submerged during interglacial periods (Mylroie et al., 1995; Wyrwoll et al., 2006). They have been found on island, coral reef and peninsular coastlines around world’s oceans. The geomorphology of blue holes contributes to the special hydrochemical characteristics including low photosynthetic oxygen production, tidally-influenced upper water, stratified water column with limited vertical mixing. Typically, the permanent thermo-halocline and the hypoxic/anoxic layer below are found in blue holes with high level of hydrogen sulfide formed in deep waters (Gonzalez, 2010). Being relatively isolated ecosystems, blue holes are described as time capsules providing evidence of Earth’s history regarding past climate change, karst processes, and life evolution (Mylroie et al., 1995; Swart et al., 2010). They are also considered to be extreme environments for life with poor supply of light, nutrient and oxygen (Northup and Lavoie, 2001; Krstulović et al., 2013). In spite of this, highly diverse organisms, new and blue hole-specific species ranging from Arthropoda, Mollusca, Ciliophora, Proteobacteria to Woesearchaeota have been documented (Iliffe and Kornicker, 2009; Ricci et al., 2015; Patin et al., 2021; Kajan et al., 2022). Biodiversity of microbes and benthic fauna in blue holes have been found to be significantly higher than that in ambient waters (Garman et al., 2011). Spatial heterogeneity is one of the reasons that produce major biodiversity pattern, and potentially making blue hole a hotspot of microbial diversity. How the community is structured, how the organisms adapt and thrive, and how these biological evidences reflect the ecosystem stability and functions in blue holes have become hot topics of research. For example, photoheterotroph utilizing a range of rhodopsins has been proposed to be an important lifestyle of microbes in Shark Bay Blue Bole (Kindler et al., 2021).
The Sansha Yongle Blue Hole (SYBH, N16°31.5′, E111°46.1′, Figure 1) located at the eastern end of the Yongle Atoll of the Xisha Islands, South China Sea is the deepest blue hole ever discovered in the world, with the observed deepest portion at 301.19 m (Li et al., 2018). The 3D topography of the SYBH and underwater photography revealed the sinuous underwater structure with two large transitions at 76-78 m and 158 m, and no water or material exchange with the outside open sea (Li et al., 2018). Several investigations detailed the hydrochemical properties in the SYBH (Bi et al., 2018; Yao et al., 2018; Xie et al., 2019), which were different from others with fresh water injection or subsurface connections to the sea (Gonzalez, 2010). Strong vertical stratification in the hole is controlled by temperature gradients and three distinct layers including an oxic layer, a chemocline and an anoxic layer of deep water have been characterized (Xie et al., 2019). Unique hydrochemical characteristics of SYBH, particularly the strong vertical heterogeneity, make it a valuable and challenging site for uncovering the fundamental ecological processes.
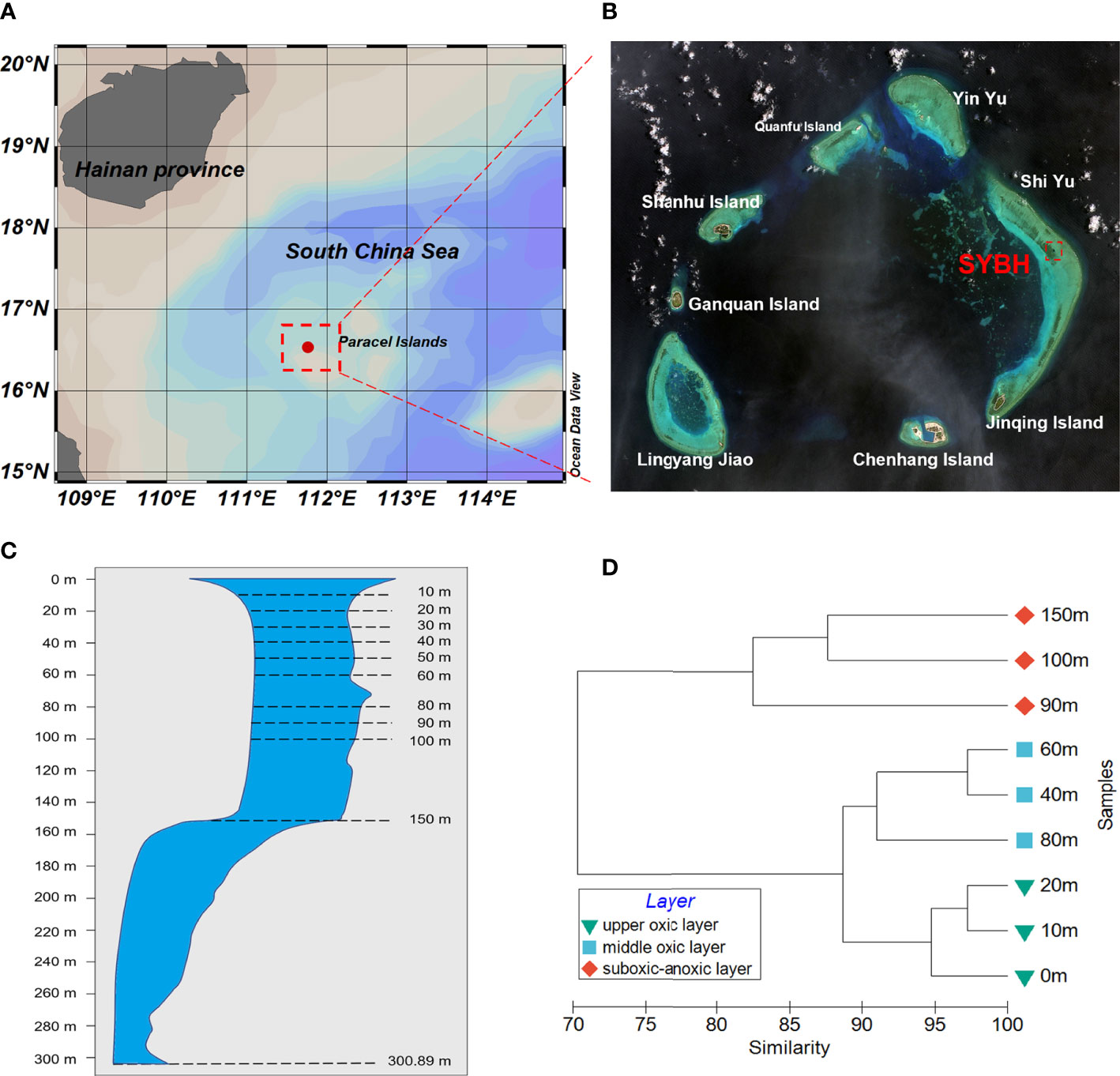
Figure 1 (A, B) Location of the Sansha Yongle Blue Hole (SYBH). (C) Vertical section through the SYBH and dash lines indicate the sampling depths in this study and the deepest depth of 300.89 m (modified from Li et al.,2018). (D) Vertical heterogeneity shown by the dendrogram of cluster analysis based on environmental variables at each depth in the SYBH.
In aquatic ecosystems, plankton serve as one of the sensitive indicators of biodiversity and environmental changes, and underpin important ecosystem functions (e.g., Ibarbalz et al., 2019; Sunagawa et al., 2020), thus scientists have started to explore the community composition of plankton and its relationship with environmental variables in SYBH. Prokaryotic plankton including bacteria and archaea community (He et al., 2019; Zhang et al., 2021), microeukaryotic plankton (Liu et al., 2019), microphytoplankton (Ge et al., 2020) and mesozooplankton (Chen et al., 2018) in SYBH have been investigated separately, with some lineages such as a genus of gram-negative bacteria Vibrio (Li et al., 2020a) and foraminifera (Li et al., 2020b) being addressed. However, the assembly mechanism of plankton microbiome, the role of subcommunities of different abundance and the potential microbial biointeraction remain elusive, which is crucial for understanding the ecological processes in SYBH.
Metabarcoding approaches have been widely used to study the diversity, assembly mechanism and biological interaction of planktonic microbiomes (e.g., de Vargas et al., 2015; Sunagawa et al., 2020; Burki et al., 2021), but in most studies, prokaryotes and eukaryotes are analyzed separately. Although amplicon libraries for metabarcoding of prokaryotes and eukaryotes have to be prepared separately due to the distinct nucleotide characters, they form microbial assemblage in natural waters and play roles together in ecological processes such a microbial food web. Recently, increasing attention has been paid to joint analysis of microbial prokaryotic and eukaryotic plankton in aquatic ecosystems (Jiang et al., 2018; Wu et al., 2020; Lu et al., 2022; Sun et al., 2022), revealing that the distribution patterns and assembly mechanisms are distinct between domains, and cross-domain interaction plays an important role in shaping the plankton community, even overriding abiotic factor in some cases. Which process contributes more to the community assembly? The answer needs the quantification of the relative influence of ‘habitat heterogeneity and environment-induced species sorting’ (niche theory) versus ‘random demographic fluctuation and disperse’ (neutral theory) (Stegen et al., 2013), which may depend on geographic scale and strength of environmental gradients (Hanson et al., 2012; Morrison-Whittle and Goddard, 2015). Moreover, it has been widely accepted that rare biosphere plays important roles in community turnover, driving functions and maintaining stability of ecosystem (Debroas et al., 2015; Lynch and Neufeld et al., 2015; Jia et al., 2018), particularly in extreme environment like deep sea (Sogin et al., 2006). Also, taxa with moderate abundance (also called intermediate subcommunity) have been found to be highly metabolic active and determine the plankton community transition among seasons (Sun et al., 2017). Previous studies on microbial plankton pointed out that the composition, diversity, assembly processes and response to environment of non-abundant subcommunity are different from those of abundant one (Logares et al., 2014; Xue et al., 2018). Removing rare taxa from dataset may neglect some keystone species and hinder the understanding of community dynamics (Gobet et al., 2012).
In this study, based on high-throughput sequencing of 18S rRNA and 16S rRNA genes, we investigated the community composition, diversity, assembly mechanism and co-occurrence network of microbial plankton communities in the SYBH. We hypothesized that (1) microbial plankton community are structured by vertical environmental gradient and day-night variation in the SYBH; (2) drivers of microeukaryotic and prokayrotic plankton community and their sensitivity to environmental filtering are different; (3) non-dominant subcommunities play a central role in diversity and biological interaction. As the first attempt, we jointly analyzed eukaryotic and prokaryotic microbe, and compared the subcommunities of different abundance in the SYBH. It is hoped that this study will contribute to a deeper understanding on community assembly and drivers of microbial plankton in the SYBH, which may have implication in conservation strategies.
Samples were collected from the SYBH (16°31 ‘30 “N, 111°46 ‘05 “E) at the depths of 0-150 m (Figure 1C) on the night of March 14 and the day of March 17, 2017. Temperature, salinity, dissolved oxygen (DO) and depth of all water samples were measured in situ using a CTD profiler. Seawater was collected by Niskin bottles mounted on a rosette, equipped with CTD (SeaBird SBE 19 Plus), and then was prefiltered through 200-µm mesh to remove large zooplankton. A subsample (4~6 L) of the prefiltered water was passed successively through 5-µm and 0.45-µm polycarbonate membranes (Millipore, USA), which was then stored in liquid nitrogen until DNA extraction. Another 500 mL subsample was filtered by the manner for the analysis of chlorophyll a. The filter membrane was extracted with 10 mL of 90% v/v acetone aqueous solution for 24 h at 4°C and in darkness, centrifuged at 1500 g for 10 min, and the supernatant was measured with a fluorescence spectrophotometer (Hitachi F-4500, Japan). H2S was determined in site with a spectrophotometer at 670 nm after color development, and pH was determined with a Mettler-Toledo Delta 320 pH meter. The seawater for nutrient determination was filtered through 0.45-μm acetate filter membranes that were soaked and washed in dilute hydrochloric acid before use, and the filtrates were immediately frozen at -20°C. Nutrients were measured using colorimetric method by AA3 continuous flow analyzer (Seal Analytical Company, UK). The detection limits of NO3−, NO2−, NH4+, PO43− and SiO32− were 0.02, 0.01, 0.04, 0.02 and 0.01 μmol mL-1, respectively, and the uncertainty of repeated determination was less than 5%-10%. Vertical profiles of multiple hydrochemical parameters in this study was adopted from Bi et al. (2018) and Yao et al. (2018).
Membrane filters of different pore sizes from the same depth were pooled together. A total of 18 samples (10 from nighttime and 8 from daytime) were subjected to DNA extraction following the method described by Yuan et al. (2015). Briefly, each membrane filter was cut with sterilizing scissors and immersed in 1 mL DNA extraction buffer (100 mM EDTA, 1% SDS and 10 μg mL-1 proteinase K) for 24-h incubation at 56°C. After incubation, DNA was extracted using the CTAB method, further purified using the DNA Clean & Concentrator kit (Zymo Research, USA), and eluted in 25 μL 10mM Tris-HCl. The concentrations and purities of DNA were measured on a NanoDrop 2000 Spectrophotometer (Thermo Scientific, USA). DNA integrity was assessed by gel electrophoresis.
The V4 region of the eukaryotic 18S rDNA gene was amplified with the primers MEG_F (5’- GGCAAGTCTGGTGCCAG -3’) and MEG_R (5’- GACTACGACGGTATCTRATCRTCTTCG -3’) (Bråte et al., 2010). The PCR was performed as follows: 95°C for 5 min, followed by 25 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 40 s, with a final extension at 72°C for 10 min. The V4 hypervariable region of prokaryotic 16S rDNA was amplified with 515F (5’- GTGYCAGCMGCCGCGGTAA -3’) and 806R (5’- GGACTACNVGGGTWTCTAAT -3’) (Caporaso et al., 2011). The PCR was performed as follows: 95°C for 5 min, followed by 25 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 40 s, with a final extension at 72°C for 10 min. PCR reactions were performed in a total volume of 25 µL using Ex Taq (Takara, China) with 20-300 ng template gDNA. All amplicons were paired-end sequenced (2×300 bp) using Illumina MiSeq platform (GENEWIZ, SuZhou, China).
Raw reads were subjected to quality filtration by trimming sequencing adaptor, primers, low quality bases and too short reads (<200 bp) with Cutadapt (v1.9.1), Vsearch (v1.9.6) and Qiime (v1.9.1) (Martin, 2011; Rognes et al., 2016; Bolyen et al., 2019). The remaining high-quality paired-end reads were assembled into metabarcodes with fastq mergepairs module of Vsearch (v1.9.6) and non-singleton ones were clustered into OTUs (Operational Taxonomic Units) with a 97% identity. Then the rest of metabarcodes were mapped to the OTU representative sequences to generate the OTU abundance profile. For taxonomic annotations, the representative OTU sequences of 18S rRNA genes were taxonomically classified by sintax algorithm (Edgar, 2016) compared with PR2 database (Guillou et al., 2012), and the confidence threshold was set to 0.8. The representative OTU sequences of 16S rRNA gene were taxonomically classified by Bayesian algorithm of the RDP classifier (Ribosomal Database Program, v2.2; Wang et al., 2007) using Silva128 Database (Quast et al., 2012). Taxonomic annotation was further manually checked particularly for highly abundant and ambiguous metabarcodes. The ecological functions of bacterial communities were further annotated using Functional Annotation of Prokaryotic Taxa (FAPROTAX) (Louca et al., 2016).
In addition to the total community, abundant, moderate, and rare subcommunities were analyzed separately. We defined ‘abundant’ OTUs as those having relative abundances above 0.1% of total sequences, ‘rare’ OTUs as those having relative abundances below 0.01% and ‘moderate’ OTUs as those having relative abundances between 0.01% and 0.1% (Campbell et al., 2011).
For alpha-diversity, richness and Shannon diversity index were calculated for each sample using the ‘vegan’ package in R (v4.0.3). Significance of the difference in alpha-diversity between groups was tested using the Wilcoxon rank sum test. Non-Metric Multidimensional Scaling (NMDS) ordination of community structure was performed using Bray-Curtis dissimilarity matrices in PRIMER 6 (Clarke and Gorley, 2009). Global and pairwise differences among groupings of samples were further tested by ANOSIM within PRIMER 6. Mantel tests with 9999 permutations were performed using the ‘vegan’ package in R, to examine the Spearman’s correlation between abiotic factors (temperature, salinity, pH, DO, nutrient and etc.), cross-domain biotic factors (PCoA 1 and 2 value of community) and the Bray-Curtis distances of communities. The circos plot are visualized using the ‘circlize’ package in R.
To quantify the relative importance of deterministic and stochastic processes in microbial communities and subcommunities with different abundance, the phylogenetic null model of Stegen et al. (2013) was applied. Phylogenetic turnover between communities was first used to determine the effects of selection, followed by OTU turnover to determine the effects of dispersal and drift. Values of βNTI greater than 2 or less than −2 indicate the impact of environmental pressures on heterogeneous and homogeneous selection, respectively. If the |βNTI| is < 2 but with an |RCbray| >0.95, the community assembly is controlled by homogenizing dispersal or dispersal limitation, respectively. In addition, the |βNTI| < 2 and a |RCbray| < 0.95 suggest that the community assembly is not dominated by any single process, consisting of weak selection, weak dispersal, and diversification or drift. (Stegen et al., 2013).
Spearman’s correlations between OTU relative abundance were calculated using the ‘rcorr’ function in the ‘Hmisc’ package, based on which co-occurrence network analysis was conducted. To reduce false-positive prediction, OTUs present in ≥6 samples with robust correlation (spearman rho > |0.8| and P<0.01) were considered for network analysis. Networks were visualized using Gephi (v0.9.2). Topological properties of nodes, including degree, betweenness centrality and modularity, were also calculated in Gephi. A random network with the same number of nodes and edges as a real network is generated in the ‘igraph’ R package. The topological characteristics of real and random networks, including modularity, clustering coefficient network diameter and average path length, are calculated and compared. Nodes with high degree (>50) and low betweenness centrality (<5000) values were considered to be keystone species in the co-occurrence network.
The vertical profiles of several environmental parameters in SYBH showed depth-dependent pattern (Supplementary Figure 1). Temperature, pH and DO decreased with depth. Oxycline appeared around 80-90 m and the water became anoxic below 100 m. Contrastingly, salinity and the concentrations of silicate, phosphate increased with depth, which was more evident at depths below 80 m. Turbidity, the concentration of nitrate and nitrite showed a single-peak pattern with the maxima detected at 100 m, and 90 m, 40 m, respectively. The concentration of chlorophyll a also showed a single-peak pattern, with the maximum found at 40 m and 20 m at daytime and nighttime, respectively. The maximum of H2S concentration was detected at 150 m, which was the deepest sampling depth of this study, mirroring the anoxic condition. Considering all the measured environmental variables, the vertically heterogeneous environments above 150 m in the SYBH could be categorized into two major clusters: the oxic layer in the top 80 m and the suboxic-anoxic layer below 90 m. The oxic layer could be further divided to upper oxic layer (0-20 m) and middle oxic layer (40-80 m) (Figure 1D).
Microeukaryotic and prokaryotic plankton communities were examined separately in 18 samples collected from nine layers of the SYBH. Rarefaction curves and Shannon-Wiener curves depicted a leveling off with increased sequences for all samples, indicating that sequencing depth was sufficient to identify the majority of OTU richness and community diversity (Supplementary Figure 2). A total of 1,817,031 high-quality 18S rDNA metabarcodes and 1,083,134 high-quality 16S rDNA metabarcodes were obtained, and yielded 6,121 and 2,883 OTUs, respectively (Supplementary Table 1). Among these, 948 (relative abundance 4.29%) eukaryotic OTUs and 160 (relative abundance 1.61%) prokaryotic OTUs did not match any sequence in the reference databases (the ‘unclassified’ category in Figure 2), pointing to the undocumented genetic diversity in SYBH.
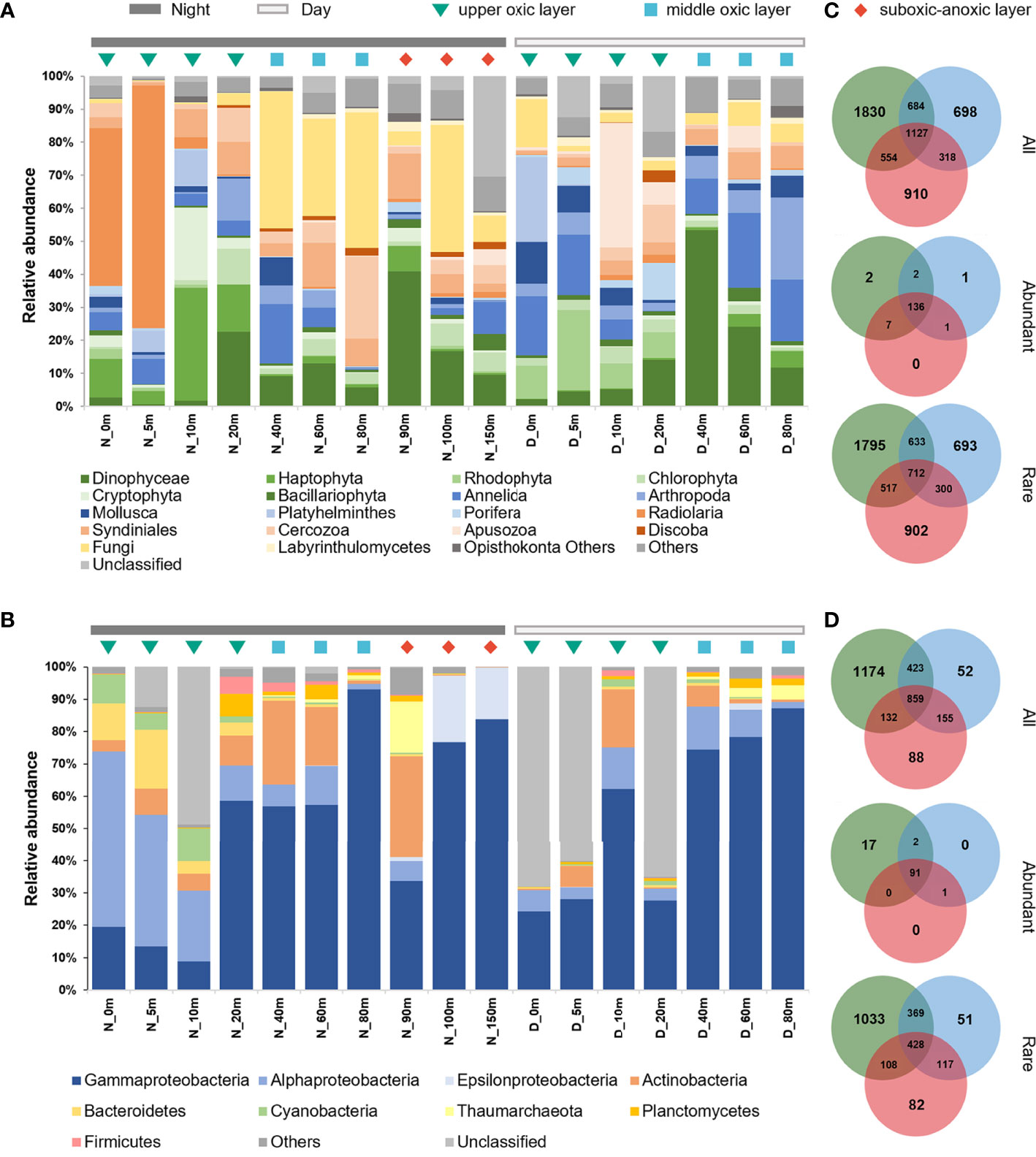
Figure 2 Community composition of plankton microbiome in the SYBH. The relative abundances of microeukaryotic (A) and prokaryotic (B) plankton community across depths and time of the day. D, Day; N, Night. Venn diagrams showing the numbers of unique and shared OTUs of microeukaryotes (C) and prokaryotes (D) among three layers. All, whole community; Abundant, abundant plankton subcommunities; Rare, rare plankton subcommunities.
The taxonomic composition of planktonic microbiome in the SYBH was uneven across depths and the sampling times of the day (Figure 2). Microeukaryotic plankton community was dominated by Dinophyceae, Annelida and Fungi (Figure 2A). Dinophyceae and Annelida accounted for 13.55% and 14.02% of the reads within microeukaryotes, respectively, but there was no significant difference between day and night communities, nor among the three layers. Rhodophyta, Radiolaria and Fungi showed the most vertically heterogenous distribution (P<0.05) with Rhodophyta and Radiolaria mainly distributed in the upper oxic layer, while Fungi in the middle oxic layer. Such depth partitioning was more pronounced at nighttime (Supplementary Figure 3), leading to the significant day-night difference (P<0.05) in the upper oxic layer community of Haptophyta, Rhodophyta, Radiolaria, Apusozoa and Labyrinthulomycetes.
The prokaryotic plankton community was dominated by Proteobacteria, Actinobacteria, Bacteroidetes, Cyanobacteria, Planctomycetes and Thaumarchaeota, which accounted for over 90% of the total sequences (Figure 2B). Within Proteobacteria, Gammaproteobacteria (average 52.60%) and Alphaproteobacteria (11.72%) dominated across all samples except for those from 100 m and 150 m at nighttime, where Epsilonproteobacteria and Gammaproteobacteria were abundant class (Figure 2B). Although no significant day-night difference was detected for prokaryotic plankton community, Cyanobacteria, Gammaproteobacteria, Epsilonproteobacteria, and Thaumarchaeota showed depth-dependent distribution (P<0.05). Cyanobacteria were mainly distributed in the upper oxic layer, Gammaproteobacteria in the middle oxic layer, while Epsilonproteobacteria and Thaumarchaeota in the suboxic-anoxic layer. Despite the low abundance, the relative abundance of nitrite-oxidizing bacteria (NOB) affiliated with the phylum Nitrospinae peaked at 90 m, where that of ammonium-oxidizing archaea (AOA) of the phylum Thaumarchaeota also peaked (Supplementary Figure 4). Such vertical niche partition was more remarkable when considering the ecological functions of prokaryotes (Supplementary Figure 5). Most of chemoheterotrophs were mainly distributed in the upper and middle oxic layers. Nitrifier and aerobic ammonium oxidizer were enriched at the middle oxic layer and peaked at oxycline of 80-90 m. Contrastingly, lineages responsible for dark oxidation of sulfur and sulfur-containing compounds were mainly found below 100 m.
After categorizing OTUs by relative abundance, we found that both eukaryotic and prokaryotic plankton communities in the SYBH were dominated by few abundant taxa (Supplementary Table 2). Among the total 6121 eukaryotic OTUs, 2.4% of OTUs (149) were classified as abundant taxa and accounted for 82.0% of total sequences. In contrast, 90.7% of OTUs (5552) were classified as rare taxa but only accounted for 5.4% of total sequences. Similar pattern was also observed in prokaryotes, as abundant taxa accounted for 3.9% of OTUs (111) but 77.1% of total sequences, while rare taxa accounted for 75.9% of OTUs (2188) but 5.8% of total sequences. Moreover, 91.28% of abundant 18S rDNA OTUs and 81.98% of abundant 16S rDNA OTUs were commonly found in three layers, accounting for 79.37% and 70.87% of total reads, respectively (Figures 2C, D). Nevertheless, the majority of layer-specific OTUs belonged to rare taxa. Plankton community composition also differed between abundant and rare taxa (Supplementary Figure 6). Abundant subcommunity of microeukaryotic plankton was dominated by Annelida, Dinophyceae and Fungi, while 14 lineages including Phaeophyceae, Hemichordata, Centroheliozoa and Perkinsea were exclusively present in rare subcommunity. Both abundant and rare subcommunities of prokaryotic plankton were dominated by Gammaproteobacteria, but Nitrospinae, Betaproteobacteria, Deltaproteobacteria and Acidobacteria was exclusively found in rare subcommunity.
NMDS ordinations and ANOSIM analysis revealed the distinct separation of microplankton communities from the upper oxic, middle oxic and suboxic-anoxic layer, suggesting that the microbiome assembly was mainly shaped by vertical heterogeneity of environment for both eukaryotic (R=0.592, P=0.001) and prokaryotic communities (R=0.552, P=0.001) (Supplementary Table 3). This was also the case when analyzing abundant, moderate and rare subcommunities separately (Supplementary Figure 7, Supplementary Table 3). The alpha-diversity were also compared between different layers and subcommunities (Figures 3A, B). Rare subcommunities of both microeukaryotic and prokaryotic plankton contained a significantly greater diversity of OTUs than abundant and moderate counterparts (Supplementary Figure 8). The alpha diversity of prokaryotic plankton gradually decreased with depths which was significant in rare subcommunity (P<0.05), while no significant change was detected in that of microeukaryotes across depths (Figures 3A, B). Moreover, significant day-night difference was only detected in the upper layer community of microeukaryotic plankton (R=0.948, P<0.05) (Supplementary Table 3), of which the rare subcommunity at daytime had significantly higher richness and diversity than that at nighttime (Supplementary Figure 9). All these results indicated that rare subcommunities contributed significantly to the biodiversity in the SYBH, and potentially the sensitivity of microeukaryotic and prokaryotic plankton to environment differed.
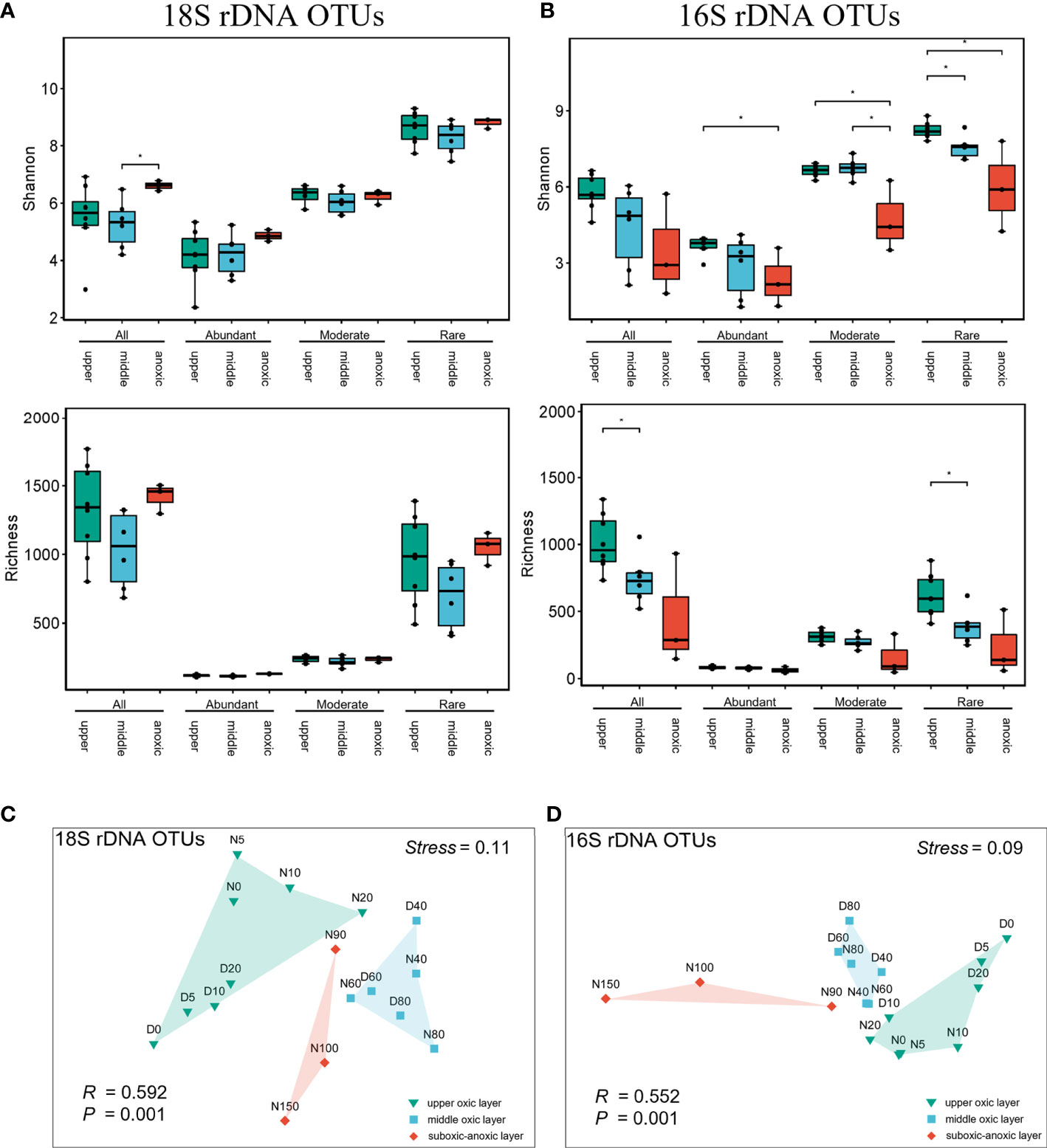
Figure 3 Community structuring of microeukaryotic and prokaryotic plankton in the SYBH. The richness and Shannon Index of microeukaryotic (A) and prokaryotic (B) plankton communities. The error bars indicate SD, and asterisks (*) indicate significant differences at the level of P<0.05. (C, D) Non-metric multidimensional scaling (NMDS) ordination of plankton communities based on Bray–Curtis distances. upper, upper oxic layer; middle, middle oxic layer; anoxic, suboxic-anoxic layer. All, whole community; Abundant, Moderate and Rare indicate the abundant, moderate and rare plankton subcommunities, respectively.
Mantel test further suggested that prokaryotic plankton had stronger correlation with environmental variables than microeukaryotic plankton did (Figure 4; Supplementary Table 4). Except for the concentrations of inorganic nitrogen and chlorophyll a, all the other physicochemical properties showed a significant positive correlation with overall prokaryotic plankton community and three subcommunities (R=0.251~0.728, P<0.05). Contrastingly, the influence of environmental factors on microeukaryotic community was much weaker (R=0.210~0.474, P<0.05) and varied among subcommunities, of which rare subcommunity was most sensitive to environmental variables with salinity (R=0.657, P<0.001) and temperature (R=0.728, P<0.001) showed the strongest correlation. We also assessed the influence of cross-domain biotic factor on planktonic microbiome (Table 1). Mantel tests revealed that the rare subcommunities of microeukaryotic plankton were more sensitive to the cross-domain biotic factors, and the moderate and rare subcommunity of prokaryotic plankton played significant roles in microeukayotic community variation (R=0.344~0.524, P<0.01).
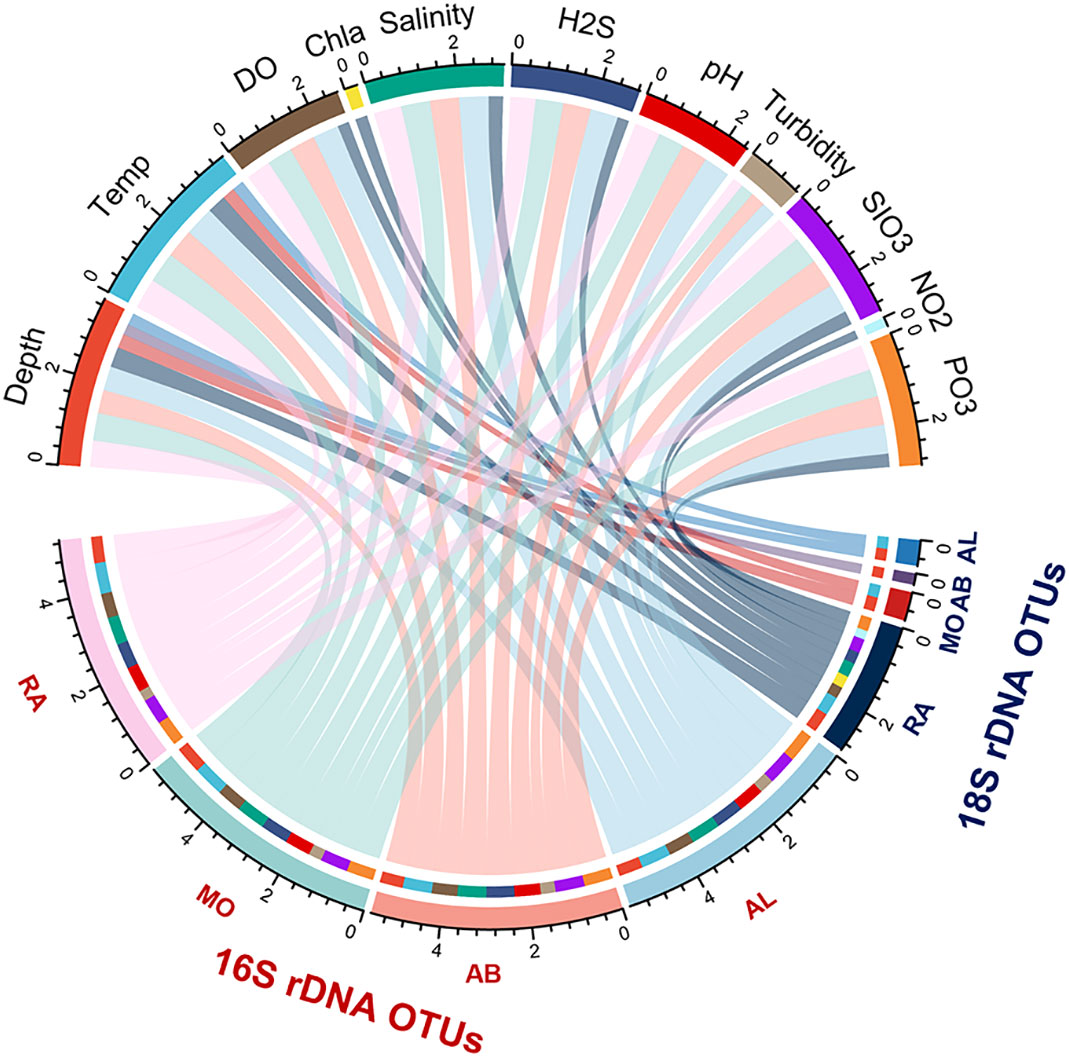
Figure 4 The circos plot depicts the effects of environmental variables on the beta diversity of microeukaryotes and prokaryotes plankton communities revealed by Mantel tests. Each environmental variable and subcommunity are depicted by the arcs. The correlation between the relative abundance of community and environmental variable are depicted by the arcs with specific color. The area of ribbons indicate the contribution of each variables with significant positive correlation. Detailed results of Mantel test are shown in Supplementary Table 4. RA, Rare subcommunity; MO, Moderate subcommunity; AB, Abundant subcommunity; AL, whole community.

Table 1 Mantel tests showing the effects of cross-domain biotic factors on microeukaryotic and prokaryotic plankton communities.
Given the contribution of rare taxa in the biodiversity of SYBH and potential effect of cross-domain biotic factor on planktonic community, we performed co-occurrence network analysis (Figure 5). All obtained network exhibited a scale-free and non-random distribution with power-law R2 = 0.824 (Supplementary Figure 10, Supplementary Table 5). The network was composed of 9238 positive and 416 negative edges, among which 1,343, 2,360 and 5,951 edges were from microeukaryote-prokaryote, microeukaryote-microeukaryote, and prokaryote-prokaryote connections with 62.74% of the negative edges were found between microeukaryotes and prokaryotes. Nodes linked by cross-domain edges were mainly composed of Gammaproteobacteria, Alphaproteobacteria and Syndiniales (Supplementary Table 7). Among 1629 nodes, the majority (49.54%) was rare OTUs followed by moderate (37.57%) and abundant (12.83%) OTUs (Figure 5A). The interactions with rare taxa were found to be most frequent in the network (Supplementary Table 6). Nodes were mainly represented by Gammaproteobacteria (18.97%), Alphaproteobacteria (13.20%), Syndiniales (6.02%). Eighteen prokaryotic OTUs and 2 eukaryotic OTUs were identified as keystone species with high degree (>50) and low betweenness centrality (<5000), which consisted of 6 rare OTUs, 13 moderate OTUs and 1 abundant OTU belong to Gammaproteobacteria, Alphaproteobacteria, Planctomycetacia, Actinobacteria, Verrucomicrobia, Radiolaria (Figure 5C; Supplementary Table 8). Among these keystone species, the top 2 OTUs with highest degree were affiliated to rare OTUs of Planctomycetes and Alphaproteobacteria.
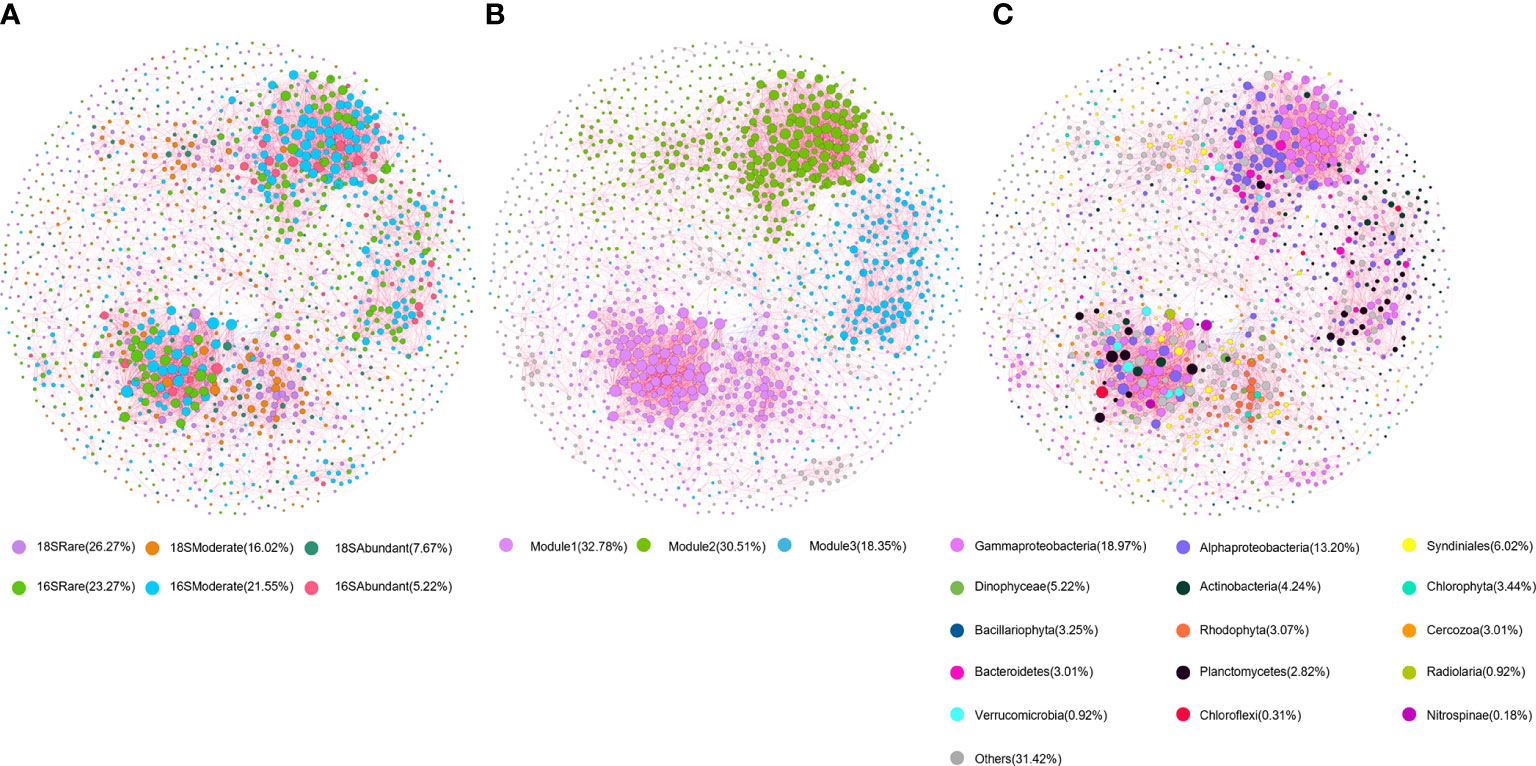
Figure 5 Co-occurrence network of planktonic microbiome in SYBH. The network was established by calculating correlations among OTUs. The nodes in the network are colored according to (A) subcommunities of different abundance; (B) modules and (C) taxa. The red and blue lines represent positive and negative correlations, respectively. The sizes of the nodes are scaled to the degree of connection.
The co-occurrence network was clustered into three major modules accounting for 81.64% of the total nodes (Figure 5B). All three modules were dominated by rare OTUs affiliated to Alphaproteobacteria and Gammaproteobacteria (Figure 5C). Three modules of prokaryotic OTUs exhibited distinct distribution pattern along the vertical gradient which was not observed for microeukaryotes (Supplementary Figure 11). Most of the prokaryotic OTUs from module I had higher relative abundance in the middle oxic layer, while the relative abundance of OTUs in module II and III was higher in the upper oxic layer (Supplementary Figure 11).
The relative contribution of each ecological process underpinning the assembly of microbial community was quantified by the phylogenetic null model analysis (Figure 6). It was found that stochastic processes, mainly dispersal limitation (83.09%), made crucial contributions to microeukaryotic plankton assembly, while prokaryotic assembly was dominated by deterministic processes consisting of heterogenous (42.65%) and homogenous selection (47.79%). Nevertheless, the effect of ecological drift was minor (<0.74%). The role of different ecological processes varied among subcommunities (Figure 6). Dispersal limitation made a significant contribution to microeukaryotic subcommunities assembly, accounting for 97.06%, 91.91%, 66.18% in abundant, moderate and rare subcommunities, respectively. Homogenous selection was not detected in abundant subcommunity but more diverse processes played a role in rare subcommunity. For prokaryotes, abundant taxa were driven mainly by dispersal limitation (54.41%) and heterogeneous selection (41.18%). The moderate taxa were mainly driven by deterministic processes (70.59%) with heterogeneous and homogeneous selection accounting for 42.65% and 27.94%, respectively, and dispersal limitation also made a significant contribution (27.21%). The effect of deterministic processes was overwhelming (100%) in rare subcommunity with homogenous and heterogenous selection contributed 57.35% and 42.65%, respectively. Generally, the contribution of deterministic processes to the microbial assembly increased with the taxa abundance, while that of stochastic processes showed the opposite trend.

Figure 6 The relative contributions of the ecological processes in shaping plankton microbiome. Abundant, Moderate and Rare indicate the abundant, moderate and rare plankton subcommunities, respectively.
Environmental heterogeneity is regarded as a universal driver of biodiversity (Stein et al., 2014), thus the strong vertically heterogeneous environment in the SYBH explained its higher plankton diversity than that in outer reef slope and other Chinese marginal seas (He et al., 2019; Liu et al., 2019; Li et al., 2020b). In this study, our joint analysis of microeukaryotic and prokaryotic plankton found that planktonic microbiome above 150 m in the SYBH was vertically structured along the abiotic gradients and grouped into three clusters as upper oxic layer (0-20 m), middle oxic layer (40-80 m) and suboxic-anoxic layer (90-150 m) (Figures 1D, 3). This finding is consistent with previous studies on Vibrio community (Li et al., 2020a) and overall bacterial communities (Zhang et al., 2021) in the SYBH, and such depth partitioning of microbial community was also found in the Hospital Hole (Davis and Garey, 2018). The decrease of richness and diversity with depths was detected in prokaryotic plankton community in the SYBH (Figure 3B) which is also in concert with previous findings of overall bacterial community (Zhang et al., 2021). Contrastingly, no significant variation of diversity was detected in microeukaryotes across depths (Figure 3A), indicating the different sensitivity of eukaryotic and prokaryotic community to the strong vertical stratification.
The vertical variation of relative abundance was particularly pronounced in some lineages. As expected, the abundance of oxygenic photosynthetic phytoplankton decreases has light decays at the depth of 90 m in the hole (Bi et al., 2018), which explained the greater abundance of Haptophyta, Rhodophyta and Cyanobacteria in the upper oxic layer (P<0.05, Supplementary Figure 3). However, no significant vertical variation among three layers was detected in the most dominant phytoplankton Dinophyceae, of which the versatile nutrient utilization and mixotrophy might make Dinophyceae less sensitive to the light availability and nutrient input (Edwards, 2019), and consequently thrive in the bule hole. Fungi dominated by the phylum Ascomycota was enriched (average 21.36%) in the middle oxic layer of the SYBH. The depth-dependent distribution of pelagic Fungi has been characterized to be universal in the global oceans (Morales et al., 2019) and integrated omics analysis pointed to that depth is also a significant factor in structuring the function of Fungi, particularly in carbonhydrate and lipid degradation (Morales et al., 2019; Chrismas and Cunliffe, 2020; Baltar et al., 2021). Thus, given the important roles of pelagic Fungi in marine biogeochemical cycles, the vertical distribution of Fungi in the SYBH suggests a potential variation in organic carbon speciation with depth.
With the increase of depth, the relative abundance of ammonium oxidizer Thaumarchaeota and nitrite oxidizer Nitrospinae increased and peaked at 90 m (Supplementary Figure 4) with a sharp reduction downward. Such vertical profile of nitrifying consortium well matches that of inorganic nitrogen concentrations, indicating that the main biological process below euphotic zone is nitrification and shifts to denitrification and anaerobic ammonium oxidation (annammox) below 90 m as speculated in previous studies (He et al., 2019; Xie et al., 2019). Moreover, increasing evidence has shown that the reciprocal feeding on diverse dissolved organic nitrogen (DON) such as urea and cyanate between Thaumarchaeota and Nitrospinae may fuel the nitrification in deep ocean (Palatinszky et al., 2015; Pachiadaki et al., 2017; Kitzinger et al., 2019), which might be overlooked in the past. In upper sunlit but N-deplete ocean waters, DON may also play a role in ‘regenerated’ primary production (Kamennaya et al., 2008). The depth-dependent ecological roles of DON are also mirrored by the distribution and expression of genes related to DON metabolism which differed significantly among surface and mesopelagic zones in the global oceans (Mao et al., 2022). Such vertical pattern could be more pronounced in the strongly stratified SYBH and as the reduced form of nitrogen, DON could be particularly important in structuring the microbial community in suboxic-oxic layer. Although no DON measurement has been so far attempted for SYBH, in the future omic approaches could be utilized to characterizing DON pool as it has been done in other ocean waters (Palatinszky et al., 2015; Pachiadaki et al., 2017; Mao et al., 2022).
The relative abundance of Epsilonproteobacteria sharply increased at the depth of 100 m and retained high (15.99%) down to 150 m. Most members of Epsilonproteobacteria are chemoautotrophs and play crucial roles in biogeochemical cycle of carbon, nitrogen and sulfur in extreme environments such as deep-sea hydrothermal vents (Akerman et al., 2013; Meyer and Huber, 2014). It has been reported to be more abundant in the deep layer of SYBH than those in other caves and other areas of South China Sea (He et al., 2019). The dominancy of Epsilonproteobacteria at suboxic-oxic layer can be attributed to the sulfidic environment below 100 m in SYBH which is totally disconnected from adjacent ocean (Bi et al., 2018; Xie et al., 2019).
The diel vertical distribution of mesozooplankton, micro- and nanophytoplankton in the SYBH have been characterized based on morphological analysis (Chen et al., 2018; Ge et al., 2020). Results in these studies showed that diel vertical variation was mainly detected in the oxic layer (<90 m) and the strong thermohalocline around 80-90 m is considered to be the barrier of vertical migration for plankton. In this study, metabarcoding analysis also revealed the significant daytime-nighttime difference of community composition, which was restricted in the upper oxic layer and was mainly detected in microeukaryotic community (Supplementary Figure 3).
Radiolarian was most abundant in the upper oxic layer of nighttime, reaching the highest relative abundance at 5 m (Figure 2A), suggesting their diel periodicity. Hu et al. (2018) reported the significant diel rhythmicity in the field community of Rhizaria including radiolarians and their acantharians relatives in the North Pacific Subtropical Gyre, which had higher metabolic activities at nighttime compared to that at daytime. Diel vertical migration (DVM) was also observed in acantharians in the Atlantic Ocean, but the pattern could vary among different waters (Massera Bottazzi and Andreoli, 1982). Members of Rhizaria are commonly found in the euphotic zone, host algal endosymbionts and prey on a variety of microorganisms (Decelle et al., 2012; Ohtsuka et al., 2015). Their diel rhythmicity could be related to the availability of light and picocyanobacterial prey (Massera Bottazzi and Andreoli, 1982; Hu et al., 2018), which may also explain why the different daily distribution of plankton only found in the upper layer of SYBH. Given the importance of Rhizaria in global plankton biomass and contribution to carbon export (Biard et al., 2016; Caron, 2016; Guidi et al., 2016), further investigation is desired to fully portrait their diversity and distribution in the SYBH.
Besides DVM, we also postulated that diel horizontal migration (DHM) could be a potential factor shaping the distribution of eukaryotic plankton in SYBH: plankton inhabit the walls of SYBH at daytime and horizontally migrate into water column at nighttime. Nocturnal horizontal migration has been observed in both freshwater and marine ecosystems (Webb and Wooldridge, 1990; White, 1998) and is hypothesized to help organisms find prey and avoid predator, like DVM does. It has been suggested that abiotic conditions are less likely to influence DHM than DVM when horizontal gradients of abiotic conditions are weaker relative to the vertical gradients (Burks et al., 2002), which is also the case in the SYBH. This could partly explain that relative to prokaryotes, the day-night difference of community composition was more evident in microbial eukaryotes, who were less sensitive to abiotic environmental factors in the SYBH (Figure 4). Moreover, viable sampling techniques are desired in the future to quantify the DHM organisms and assess the factors influencing DHM.
Analysis of diversity, abiotic and biotic factors, co-occurrence network and assembly mechanism consistently pointed to the divergent assembly pattern and driving forces for prokaryotic and microeukaryotic plankton community in SYBH. Vertical heterogeneity significantly shaped the community composition of microeukaryotic plankton without affecting overall diversity (Figures 3A, B). Abiotic factors played a minor role in shaping microeukaryotic plankton community in SYBH, which is often found to be more influential in other aquatic ecosystems such as the northwestern Pacific Ocean (Wu et al., 2020). Moreover, compared to prokayrotes, microeukaryotic plankton community had less compact network topology with lower average degree and number of intradomain edges. The modularity of microeukaryotes OTUs in network showed less depth partitioning. Network complexity and connectivity are considered to be associated with sensitivity of microbes to environmental disturbances (Zhou et al., 2010). Weaker network connectivity of microeukaryotic community than bacterial community has been found in previous study, indicating the community assembly is less determined by environmental selection processes (Jiang et al., 2018), which is consistent with our results in assembly mechanism.
Quantifying the relative importance of each ecological process in community assembly highlighted the different roles in shaping the composition of microeukaryotic and prokaryotic plankton community in SYBH. We observed that stochastic processes, mainly dispersal limitation played a significant role in the community assembly of microeukaryotic plankton, which is consistent with previous findings in reservoir, intertidal zone, and costal upwelling system (Xue et al., 2018; Kong et al., 2019; Sun et al., 2022). In comparison to prokaryotic plankton, microeukaryotes were more dispersal limited which has hypothesized to increase with body size in planktonic communities (Finlay, 2002; Soininen et al., 2013; Villarino et al., 2018). Very limited vertical mixing and exchange with adjacent waters in SYBH forms a strong barrier to dispersal for larger planktonic organisms. Such barriers include persistent oceanographic features like thermhalocline and other sharp environmental gradients and the effect of barriers varies across taxa depending on both body size and abundance (Finlay, 2002; Martiny et al., 2006; Jenkins et al., 2007). Furthermore, although the role of ecological drift was minor for both planktonic community assembly, the contribution of it was slightly higher in microeukaryotic plankton. This still could be explained by the generally smaller size of prokaryotic plankton as smaller organisms are expected to have lower demographic stochasticity and ecological drift (Rosindell et al., 2011), thus resulting in higher population density than larger organisms. Ecological drift is also expected to be more significant with decreasing population size and community size (Liu et al., 2018). However, using community snapshots by amplicon sequencing may not fully quantify the random demographic events (Stegen et al., 2015) and effect of size and abundance on ecological drift of natural assemblage remain uncertain with scarce of field data (Liu et al., 2018).
Contrastingly, deterministic processes dominated over the stochastic processes in prokaryotic community but contributed much less to the assembly of microeukaryotic communities, confirming the stronger environmental filtering on prokaryotes in SYBH as shown by mantel test (Figure 4; Supplementary Table 4). The difference of assembly mechanism between prokaryotic and microeukaryotic community in SYBH is consistent with previous research on protist and bacteria in global surface waters (Logares et al., 2020) and upwelling system (Sun et al., 2022), but contradict with that in East China Sea’s euphotic zone where protist community is mainly structured by selection processes (Wu et al., 2018). Such inconsistency has been discussed in several studies on microbiomes across different ecosystems (Xue et al., 2018; Xiong et al., 2021; Lu et al., 2022; Sun et al., 2022), supporting that the combined effect of species, straits, habitat type, spatiotemporal scale and environmental gradient determine the relative contribution of determinism and stochasticity in shaping the microbial landscape (Chase and Myers, 2011; Hanson et al., 2012; Morrison-Whittle and Goddard, 2015).
Collectively, our results showed that microeukaryotic plankton community in SYBH was less sensitive to environmental filtering compared with prokaryotes. Their different physiological characteristics, particularly body size, fundamentally determine the community turn over, community size, physiological plasticity, niche breaths and eventually environmental sensitivity (Shade et al., 2012). Despite of weaker environmental sensitivity, the effect of cross-domain biotic factor, i.e. effect of prokaryotic community on microeukaryotic community, was significant, pointing to the importance of bio-interaction in shaping microeukaryotic community in SYBH. In present study of SYBH, there were more positive interactions than negative ones in co-occurrence network, suggesting that interspecies cooperation such as symbiosis (Supplementary Table 5) may play a critical role in supporting ecosystem function and stability in SYBH. Mutual interactions in the form of symbiosis can increase the fitness of hosts and allow them to exploit new ecological niches, which is prevalence in aquatic ecosystem and particularly important in extreme habitats such as hydrothermal vent and anoxic environment (Zehr and Caron, 2022). Obligate symbiosis with the chemoautotrophs including methanogens and sulfur oxidizers might be restricted in anoxic or microoxic environment, and movement of host like ciliate between oxic and anoxic zones may favor the symbiont growth (Dziallas et al., 2012). Denitrification in anoxic environments could be also performed by eukaryotic microbes like foraminifera harboring denitrifying symbionts (Bernhard et al., 2012). Thus, we also expect diverse symbiotic relationships would favor the organisms in blue hole to cope with extreme conditions like anoxia, low light and poor supply of labile organic matter (Northup and Lavoie, 2001; Krstulović et al., 2013).
About 14% of edges in co-occurrence network were between microeukaryotes and prokaryotes, indicating the nonnegligible cross-domain interaction in SYBH planktonic ecosystem. Among these, microeukayotes were most associated with Proteobacteria, and bacteria were most associated with Syndiniales (Supplementary Table 7), which was also found in the euphotic zone of Northwest Pacific Ocean (Wu et al., 2020) and permanently anoxic Cariaco Basin (Suter et al., 2022). Microbial interaction, especially parasitism involving Syndiniales is a very significant factor in structuring the global plankton community (Lima-Mendez et al., 2015) and particularly important in maintaining microbial food web productivity in resources-limited waters (Suter et al., 2022). Interestingly, although the network possessed a higher proportion of positive correlations than negative ones, 62.74% of negative correlations existed between microeukaryotes and prokaryotes, suggesting their predation or other competition relationship may play an important role in the pelagic food web of SYBH where the supply of nutrient and organic matter is limited (Boenigk and Arndt, 2002; Melanie and Sara, 2016). In comparison to prokaryotic plankton, we expect more complex biotic factors shaping the microeukaryotic assembly in SYBH, including microbe-microbe symbiosis, predation by mesoplankton or micronekton and diel migration at both vertical and horizontal directions. Although it has been pointed out biological interaction based on rDNA-derived OTU correlation may be overestimated due to variable rDNA copy number among some lineage such alveolates, next generation sequencing enables us to discover a vast array of potential biotic interactions in batch (Lima-Mendez et al., 2015), when morphological observation and physiological experiment are impractical.
As normally found in most habitats (Pedros-Alio, 2012; Lynch and Neufeld, 2015), a skewed abundance distribution of plankton was also observed in SYBH, as the community across depths was dominated by a few abundant taxa. The richness of rare subcommunities was 7-fold and 6-fold greater than that of abundant ones for microeukaryotes and prokaryotes, respectively (Supplementary Figure 8), corroborating the well-recognized idea that rare biosphere contribute significantly to the microbial diversity (Debroas et al., 2015; Lynch and Neufeld et al., 2015). The sensitivity to environments also differed between subcommunities as supported by multiple evidence. Firstly, abundant taxa were commonly found in three layers in SYBH while rare taxa were habitat-specific (Figure 2C, D). Secondly, although deterministic processes represented played a minor role in shaping the assembly of microeukaryotic plankton community, the selection effect increased from abundant to rare subcommuniteis and the enhancement was more pronounced in prokaryotic plankton (Figure 6). This indicates that in comparison with abundant taxa, rare taxa were more sensitive to local environmental filtering, which in concert with previous studies on planktonic microbiomes in freshwater ecosystems (Liu et al., 2015; Xue et al., 2018). The high abundance resulted from active growth mirrors that abundant taxa are more competitive in resource acquisition and better adapted to habitats. However, high diversity and functional redundancy of rare taxa make them important in ecosystem resilience (Jousset et al., 2017).
Furthermore, non-abundant taxa are indispensable in maintaining the structure of microbial community as denoted in co-occurrence network with over 87% of significant biological interactions involved non-abundant taxa. Moreover, except for one abundant OTU, all keystone species in co-occurrence network were belonged to moderate and rare taxa. Compared to eukaryotic plankton of reservoir (Xue et al., 2018), the role of moderate taxa was more prominent in SYBH, which has been also found in estuarine ciliate community (Sun et al., 2017). Rare taxa are more likely to be ignored through morphological analysis and low-resolution sampling due to habitat specificity. Although deep sequencing of metabarcodes unveiled the rare biosphere in diverse habitats (Pedros-Alio, 2012), the role of rare taxa in maintaining the microbiome stability has not been addressed in SYBH. Moreover, much less attention has been paid to moderate taxa, which could contribute up to 30% of abundance (Logares et al., 2014) and critical to the dynamics of community (Mangot et al., 2013; Sun et al., 2017). Out of 20 keystone OTUs, 13 were affiliated with moderate taxa which is considered as initiating components in network with longer evolutionary history (Barabási, 2009). Future study on temporal variation of microbial community in SYBH could provide precise assessment on the role of non-abundant taxa on microbiome stability and have important implication in conservation strategy.
Metabarcoding approaches come with some limitations. The most common one is the variation in rDNA gene copy number among different taxa, especially in microbial eukaryotes, hinders the directly translation of the rDNA read counts into absolute abundance of organisms (Burki et al., 2021). The rDNA copy number per genome positively correlates to cell size and biovolume (de Vargas et al., 2015). Thus, cautions should be taken when interpreting amplicon sequencing data, especially for organisms of large size or unique genome architecture (e.g., ciliate). Although metabarcoding analysis only provides relative or semiquantitative information, one can assume that such molecular biases would be relatively constant across samples and will allow the comparison between different ecological conditions (Pawlowski et al., 2016). Therefore, rDNA OTUs and reads can still be rough proxies for assessing, respectively, the genetic biodiversity and biovolume of microbial eukaryotic communities with great ecological relevance (de Vargas et al., 2015; Pawlowski et al., 2016).
Since the SYBH has been discovered for only six years, we are still at the stage of preliminary exploration and have to admit the sampling difficulty and technical limitation. SYBH sits on the coral reef and it is impractical to set up a large observation platform equipped with comprehensive and powerful facilities. Meanwhile, sinuous underwater structure and the slope at the depth about 150 m increase the difficulty of vertically sampling. Although several professional-grade facilities such as underwater remotely operated vehicle (ROV) have been exploited to obtain geomorphological profile of SYBH, more flexible sampler with less disturbance to organism is desired. In the future, portable and autonomous facility or unmanned aerial vehicles (UAV)-based system could provide a promising approach for remote sampling in the SYBH (Horricks et al., 2022).
Although the vertical profile of hydrochemical properties and microbial plankton community structure in the SYBH showed generally similar pattern among multiple investigations (He et al., 2019; Xie et al., 2019; Li et al., 2020a), variation in the depth of Deep Chlorophyll Maximum and community composition are still observed. Whether these inconsistencies are resulted from different sampling time of the day and season, or occasional observation is still unclear. Thus, replicate, high-resolution and time-series samples are needed to fully portrait the spatiotemporal distribution of life in SYBH. Moreover, currently available information about plankton diversity in SYBH are based on morphological identification (Chen et al., 2018; Ge et al., 2020; Fu et al., 2020) and DNA metabarcoding (He et al., 2019; Liu et al., 2019; Li et al., 2020a; Li et al., 2020b) which are impossible to differentiate the dead and live organisms. Thus, profiling the viability, metabolic activity and machinery of organism, particularly for those survive in anoxic conditions, is crucial for unveiling the ecological strategy and evolutionary adaptation of marine life in the SYBH, which could be achieved by viability assessment (Eva-Maria and Meysnan, 2012) and RNA-based approaches. Finally, integrated investigation on organisms from broad size spectrum and multiple habitats (plankton, bentho and neckton) are desired, which is essential for improve our understanding on the nature of bio-interaction and ecological functions in SYBH.
The data presented in the study are deposited in China National GeneBank DataBase (https://db.cngb.org/), accession number CNP0003459.
YZ and HC conceived and coordinated the project with help from LF, ZY and GL. CC and RG carried out the sampling with help from LF. YZ, CC and XM processed the samples by extracting DNA and preparing the amplicon libraries. TC, YZ and JC analyzed the data. TC and YZ wrote the manuscript with input from all authors. ZY and GL supervised the work. All authors contributed to the article and approved the submitted version.
This work was supported by the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (No.LSKJ202202804), the National Natural Science Foundation of China (No. 41876156, No. 42076146), the Fundamental Research Funds for the Central Universities (No. 201762038, No. 201812019) and Research and Development Program for the South China Sea of the Sansha Track Ocean Coral Reef Conservation Research Institute (No. HJLD17-01).
We are grateful to the Sansha Track Ocean Coral Reef Conservation Research Institute expedition vessel for their support. We also thank Libao Yang, Honglin Ma and Lin Chen for their assistance in sampling. We also thank Peng Yao and Naishuang Bi for their advice on the manuscript.
Author JC is employed by BGI, Qingdao, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1046808/full#supplementary-material
Akerman N. H., Butterfield D. A., Huber J. A. (2013). Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor Epsilonproteobacteria in diffuse hydrothermal vent fluids. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00185
Baltar F., Zhao Z., Herndl G. J. (2021). Potential and expression of carbohydrate utilization by marine fungi in the global ocean. . Microbiome 9, 106. doi: 10.1186/s40168-021-01063-4
Barabási A. L. (2009). Scale-free networks: A decade and beyond. Science 325, 412–413. doi: 10.1126/science.1173299
Bernhard J. M., Edgcomb V. P., Casciotti K. L., McIlvin M. R., Beaudoin D. J. (2012). Denitrification likely catalyzed by endobionts in an allogromiid foraminifer. ISME J. 6, 951–960. doi: 10.1038/ismej.2011.171
Biard T., Stemmann L., Picheral M., Mayot N., Vandromme P., Hauss H., et al. (2016). In situ imaging reveals the biomass of giant protists in the global ocean. Nature 532, 504–507. doi: 10.1038/nature17652
Bi N., Liang F., Chen H., Liu R., Lin C., Liu Q., et al. (2018). Hydrographic features of the yongle blue hole in the south China sea and their influential factors. Chin. Sci. Bulletin 63, 2184–2186. doi: 10.1360/N972017-01329
Boenigk J., Arndt H. (2002). Bacterivory by heterotrophic flagellates: community structure and feeding strategies. Antonie van Leeuwenhoek. 81, 465–480. doi: 10.1023/a:1020509305868
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bråte J., Logares R., Berney C., Ree D. K., Klaveness D., Jakobsen K. S., et al. (2010). Freshwater perkinsea and marine-freshwater colonizations revealed by pyrosequencing and phylogeny of environmental rDNA. ISME J. 4, 1144–1153. doi: 10.1038/ismej.2010.39
Burki F., Sandin M. M., Jamy M. (2021). Diversity and ecology of protists revealed by metabarcoding. Curr. Biol. 31, R1267–R1280. doi: 10.1016/j.cub.2021.07.066
Burks R. L., Lodge D. M., Jeppesen E., Lauridsen T. L. (2002). Diel horizontal migration of zooplankton: Costs and benefits of inhabiting the littoral. Freshw. Biol. 47, 343–365. doi: 10.1046/j.1365-2427.2002.00824.x
Campbell B. J., Yu L., Heidelberg J. F., Kirchman D. L. (2011). Activity of abundant and rare bacteria in a coastal ocean. Proc. Natl. Acad. Sci. United States America 108, 12776–12781. doi: 10.1073/pnas.1101405108
Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Lozupone C. A., Turnbaugh P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Caron D. A. (2016). Ocean science: the rise of rhizaria. Nature 532, 444–445. doi: 10.1038/nature17892
Chase J. M., Myers J. A. (2011). Disentangling the importance of ecological niches from stochastic processes across scales. Philos. Trans. R. Soc B 366, 2351–2363. doi: 10.1098/rstb.2011.0063
Chen C., Fu L., Bi N., Ge R., Liu G., Zhuang Y., et al. (2018). Zooplankton community composition and diel vertical distribution in the yongle blue hole, xisha islands, south China sea. Oceanologia Limnologia Sinica 49, 10. doi: 10.11693/hyhz20171200328
Chrismas N., Cunliffe M. (2020). Depth-dependent mycoplankton glycoside hydrolase gene activity in the open ocean–evidence from the Tara oceans eukaryote metatranscriptomes. ISME J. 14, 2361–2365. doi: 10.1038/s41396-020-0687-2
Davis M. C., Garey J. R. (2018). Microbial function and hydrochemistry within a stratified anchialine sinkhole: A window into coastal aquifer interactions. Water 10, 972–972. doi: 10.3390/w10080972
Debroas D., Hugoni M., Domaizon I. (2015). Evidence for an active rare biosphere within freshwater protists community. Mol. Ecol. 24, 1236–1247. doi: 10.1111/mec.13116
Decelle J., Siano R., Probert I., Poirier C., Not F. (2012). Multiple microalgal partners in symbiosis with the acantharia Acanthochiasma sp. (Radiolaria). Symbiosis 58, 233–244. doi: 10.1007/s13199-012-0195-x
de Vargas C., Audic S., Henry N., Decelle J., Mahé F., Logares R., et al. (2015). Eukaryotic plankton diversity in the sunlit ocean. Science 348, 29223618–29223625. doi: 10.1126/science.1261605
Dziallas C., Allgaier M., Monaghan M. T., Grossart H.-P. (2012). Act together-implications of symbioses in aquatic ciliates. Front. Microbio 3. doi: 10.3389/fmicb.2012.00288
Edgar R. (2016). SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv. doi: 10.1101/074161
Edwards K. F. (2019). Mixotrophy in nanoflagellates across environmental gradients in the ocean. Proc. Natl. Acad. Sci. 116, 6211–6220. doi: 10.1073/pnas.1814860116
Eva-Maria Z., Meysman F. J.R. (2012). Dead or alive? viability assessment of micro- and mesoplankton. J. Plankton Res. 34, 493–509. doi: 10.1093/plankt/fbs018
Finlay B. J. (2002). Global dispersal of free-living microbial eukaryote species. Science 296, 1061–1063. doi: 10.1126/science.1070710
Fu M., Sun P., Li Y., Pu X., Yuan C., Zhang X. (2020). Phytoplankton community structure in sansha yongle blue hole and its adjacent area of the south China Sea. Adv. Mar. Sci. 38, 304–316. doi: 10.3969/j.issn.1671-6647.2020.02.010
Garman K. M., Rubelmann H., Karlen D. J., Wu T., Garey J. R. (2011). Comparison of an inactive submarine spring with an active nearshore anchialine spring in Florida. Hydrobiologia 677, 65–87. doi: 10.1007/s10750-011-0740-2
Ge R., Fu L., Bi N., Chen C., Liu G., Zhuang Y., et al. (2020). Diel vertical distribution of phytoplanktonin yongle blue hole, xisha islands in spring. Periodical Ocean Univ. China 50, 65–73. doi: 10.16441/j.cnki.hdxb.20180428
Gobet A., Böer S., Huse S., Beusekom J. E. E., Quince C., Sogin M. L., et al. (2012). Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 6, 542–553. doi: 10.1038/ismej.2011.132
Gonzalez B. C. (2010). Novel bacterial diversity in an anchialine blue hole on abaco island, bahamas. master’s thesis (College Station, TX, USA: Texas A&M University).
Guidi L., Chaffron S., Bittner L., Eveillard D., Larhlimi A., Roux S., et al. (2016). Plankton networks driving carbon export in the oligotrophic ocean. Nature 532, 465–470. doi: 10.1038/nature16942
Guillou L., Bachar D., Audic S., Bass D., Berney C., Bittner L., et al. (2012). Protist ribosomal reference database (PR2): A catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/nar/gks1160
Hanson C. A., Fuhrman J. A., Horner-Devine M. C., Martiny J. B. (2012). Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
He H., Fu L., Liu Q., Fu L., Bi N., Yang Z., et al. (2019). Community structure, abundance and potential functions of bacteria and archaea in the sansha yongle blue hole, xisha, south China Sea. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02404
Horricks R. A., Bannister C., Lewis-McCrea L. M., Hicks J., Watson K., Reid G. K. (2022). Comparison of drone and vessel-based collection of microbiological water samples in marine environments. Environ. Monit Assess. 194, 439. doi: 10.1007/s10661-022-10095-8
Hu S. K., Connell P. E., Mesrop L. Y., Caron D. A. (2018). A hard day's night: diel shifts in microbial eukaryotic activity in the north pacific subtropical gyre. Front. Mar. Sci. 5. doi: 10.3389/fmars.2018.00351
Ibarbalz F. M., Henry N., Brando M. C., Martini S., Busseni G., Byrne H., et al. (2019). Global trends in marine plankton diversity across kingdoms of life. Cell 179, 1084–1097.e21. doi: 10.1016/j.cell.2019.10.008
Iliffe T. M., Kornicker L. S. (2009). Worldwide diving discoveries of living fossil animals from the depths of anchialine and marine caves. Smithson Contrib Smithsonian Contributions to Mar. Sci. 38, 269–280.
Jenkins D. G., Brescacin C. R., Duxbury C. V., Elliott G. A., Evans J. A., Grablow K. R., et al. (2007). Does size matter for dispersal distance? Glob. Ecol. Biogeogr. 16, 415–425. doi: 10.1111/j.1466-8238.2007.00312.x
Jia X., Dini-Andreote F., Salles J. F. (2018). Community assembly processes of the microbial rare biosphere. Trends Microbiol. 26, 738–747. doi: 10.1016/j.tim.2018.02.011
Jiang Y., Lei Y., Yang Y., Korpelainen H., Niinemets U., Li C. (2018). Divergent assemblage patterns and driving forces for bacterial and fungal communities along a glacier forefield chronosequence. Soil Biol. Biochem. 118, 207–216. doi: 10.1016/j.soilbio.2017.12.019
Jousset A., Bienhold C., Chatzinotas A., Gallien L., Gobet A., Kurm V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.174
Kajan K., Cukrov N., Cukrov N., Bishop-Pierce R., Orlić S. (2022). Microeukaryotic and prokaryotic diversity of anchialine caves from Eastern Adriatic Sea islands. Microb. Ecol. 83, 257–270. doi: 10.1007/s00248-021-01760-5
Kamennaya N. A., Chernihovsky. M. P., Anton. F. (2008). The cyanate utilization capacity of marine unicellular cyanobacteria. Limnology Oceanography 53, 2485–2494. doi: 10.4319/lo.2008.53.6.2485
Kindler G. S., Wong H. L., Larkum A. W. D., Johnson M., Macleod F. I., Burns B. P. (2021). Genome-resolved metagenomics provides insights into the functional complexity of microbial mats in blue holes, shark bay. FEMS Microbiol. Ecol. 98, fiab158. doi: 10.1093/femsec/fiab158
Kitzinger K., Padilla C. C., Marchant H. K., Hach P. F., Herbold C. W., Kidane A. T., et al. (2019). Cyanate and urea are substrates for nitrification by thaumarchaeota in the marine environment. Nat. Microbiol. 4, 234–243. doi: 10.1038/s41564-018-0316-2
Kong J., Wang Y., Warren A., Huang B., Sun P. (2019). Diversity distribution and assembly mechanisms of planktonic and benthic microeukaryote communities in intertidal zones of southeast fujian, China. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02640
Krstulović N., Šolić M., Šantić D., Maršić-Lučić J., Ordulj M., Šestanović S. (2013). Microbial community structure in two anchialine caves on mljet island (Adriatic Sea). Acta Adriat 54, 183–198.
Li T., Feng A., Liu Y., Li Z., Guo K., Jiang W., et al. (2018). Three-dimensional (3D) morphology of sansha yongle blue hole in the south China Sea revealed by underwater remotely operated vehicle. Sci. Rep. 8, 17122. doi: 10.1038/s41598-018-35220-x
Li Q., Lei Y., Morard R., Li T., Wang B. (2020b). Diversity hotspot and unique community structure of foraminifera in the world’s deepest marine blue hole – sansha yongle blue hole. Sci. Rep. 10, 10257. doi: 10.1038/s41598-020-67221-0
Li B., Liu J., Zhou S., Fu L., Yao P., Chen L., et al. (2020a). Vertical variation in Vibrio community composition in sansha yongle blue hole and its ability to degrade macromolecules. Mar. Life Sci. Technol. 2, 60–72. doi: 10.1007/s42995-019-00003-4
Lima-Mendez G., Faust K., Henry N., Decelle J., Colin S., Carcillo F., et al. (2015). Determinants of community structure in the global plankton interactome. Science 348, 1262073. doi: 10.1126/science.1262073
Liu Y., He H., Fu L., Liu Q., Yang Z., Zhen Y. (2019). Environmental DNA sequencing reveals a highly complex eukaryote community in sansha yongle blue hole, xisha, south China Sea. Microorganisms 7, 624. doi: 10.3390/microorganisms7120624
Liu J., Vellend M., Wang Z., Yu M. (2018). High beta diversity among small islands is due to environmental heterogeneity rather than ecological drift. J. Biogeography 45, 2252–2261. doi: 10.1111/jbi.13404
Liu L., Yang J., Yu Z., Wilkinson D. M. (2015). The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 9, 2068–2077. doi: 10.1038/ismej.2015.29
Logares R., Audic S., Bass D., Bittner L., Boutte C., Christen R., et al. (2014). Patterns of rare and abundant marine microbial eukaryotes. Curr. Biol. 24, 813–821. doi: 10.1016/j.cub.2014.02.050
Logares R., Deutschmann I. M., Junger P. C., Giner C. R., Krabberød A. K., Schmidt T. S. B., et al. (2020). Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome 8, 55. doi: 10.1186/s40168-020-00827-8
Louca S., Parfrey L. W., Doebeli M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Lu L., Tang Q., Li H., Li Z. (2022). Damming river shapes distinct patterns and processes of planktonic bacterial and microeukaryotic communities. Environ. Microbiol. 24, 1760–1774. doi: 10.1111/1462-2920.15872
Lynch M. D. J., Neufeld J. D. (2015). Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 13, 217–229. doi: 10.1038/nrmicro3400
Mangot J. F., Domaizon I., Taib N., Marouni N., Duffaud E., Bronner G., et al. (2013). Short-term dynamics of diversity patterns: evidence of continual reassembly within lacustrine small eukaryotes. Environ. Microbiol. 15, 1745–1758. doi: 10.1111/1462-2920.12065
Mao X., Chen J., van Oosterhout C., Zhang H., Liu G., Zhuang Y., et al. (2022). Diversity, prevalence, and expression of cyanase genes (cynS) in planktonic marine microorganisms. ISME J. 16, 602–605. doi: 10.1038/s41396-021-01081-y
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBNet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martiny J. B. H., Bohannan B. J. M., Brown J. H., Colwell R. K., Fuhrman J. A., Green J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Micro 4, 102–112. doi: 10.1038/nrmicro1341
Massera Bottazzi E., Andreoli M. (1982). Distribution of adult and juvenile acantharia (Protozoa sarcodina) in the Atlantic ocean. J. Plankton Res. 4, 757–777. doi: 10.1093/plankt/4.4.757
Melanie G., Sara M. (2016). The ecology and evolution of microbial competition. Trends Microbiol. 24, 833–845. doi: 10.1016/j.tim.2016.06.011
Meyer J., Huber J. (2014). Strain-level genomic variation in natural populations of Lebetimonas from an erupting deep-sea volcano. ISME J. 8, 867–880. doi: 10.1038/ismej.2013.206
Morales S. E., Biswas A., Herndl G. J., Baltar F. (2019). Global structuring of phylogenetic and functional diversity of pelagic fungi by depth and temperature. Front. Mar. Sci. 6. doi: 10.3389/fmars.2019.00131
Morrison-Whittle P., Goddard M. R. (2015). Quantifying the relative roles of selective and neutral processes in defining eukaryotic microbial communities. ISME J. 9, 2003–2011. doi: 10.1038/ismej.2015.18
Mylroie J. E., Carew J. L., Moore A. I. (1995). Blue holes: definition and genesis. Carbonates Evaporites 10, 225–233. doi: 10.1007/BF03175407
Northup D. E., Lavoie K. H. (2001). Geomicrobiology of caves: a review. Geomicrobiol J. 18, 199–222. doi: 10.1080/01490450152467750
Ohtsuka S., Suzaki T., Horiguchi T., Suzuki N., Not F. (2015). Marine protists: diversity and dynamics (Tokyo: Springer Japan).
Pachiadaki M. G., Sintes. E., Bergauer K., Brown J. M., Record R., Swan B. K., et al. (2017). Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation. Science 358, 1046–1051. doi: 10.1126/science.aan8260
Palatinszky M., Herbold C., Jehmlich N., Pogoda M., Han P., Bergen M., et al. (2015). Cyanate as an energy source for nitrifiers. Nature 524, 105–108. doi: 10.1038/nature14856
Patin N. V., Dietrich Z. A., Stancil A., Quinan M., Beckler J. S., Hall E. R., et al. (2021). Gulf of Mexico blue hole harbors high levels of novel microbial lineages. ISME J. 15, 2206–2232. doi: 10.1038/s41396-021-00917-x
Pawlowski J., Lejzerowicz F., Apotheloz-Perret-Gentil L., Visco J., Esling P. (2016). Protist metabarcoding and environmental biomonitoring: time for change. Eur. J. Protisto 55, 12–25. doi: 10.1016/j.ejop.2016.02.003
Pedros-Alio C. (2012). The rare bacterial biosphere. Annu. Rev. Mar. Sci. 4, 449–466. doi: 10.1146/annurev-marine-120710-100948
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ricci S., Sacco Perasso C., Antonelli F., Davidde Petriaggi B. (2015). Marine bivalves colonizing Roman artefacts recovered in the gulf of pozzuoli and in the blue grotto in Capri (Naples, italy): boring and nestling species. Int. Biodeterioration Biodegradation 98, 89–100. doi: 10.1016/j.ibiod.2014.12.001
Rognes T., Flouri T., Nichols B., Quince C., Mahé F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. doi: 10.7717/peerj.2584
Rosindell J., Hubbell S. P., Etienne R. S. (2011). The unified neutral theory of biodiversity and biogeography (MPB-32) (Princeton: Princeton University Press).
Shade A., Peter H., Allison S. D., Baho D. L., Berga M., Burgmann H., et al. (2012). Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00417
Sogin M., Morrison H., Huber J., Welch D., Huse S., Neal P., et al. (2006). Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc. Natl. Acad. Sci. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Soininen J., Korhonen J. J., Luoto M. (2013). Stochastic species distributions are driven by organism size. Ecology 94, 660–670. doi: 10.1890/12-0777.1
Stegen J. C., Lin X., Fredrickson J. K., Chen X., Kennedy D. W., Murray C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069–2079. doi: 10.1038/ismej.2013.93
Stegen J. C., Lin X., Fredrickson J. K., Konopka A. E. (2015). Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00370
Stein A., Gerstner K., Kreft H. (2014). Environmental heterogeneity as a universal driver of species richness across taxa, biomes and spatial scales. Ecol. Lett. 17, 866–880. doi: 10.1111/ele.12277
Sunagawa S., Acinas S. G., Bork P., Bowler C., Eveillard D., Gorsky G., et al. (2020). Tara oceans: towards global ocean ecosystems biology. Nat. Rev. Microbiol. 18, 428–445. doi: 10.1038/s41579-020-0364-5
Sun P., Huang L., Xu D., Huang B., Chen N., Warren A. (2017). Marked seasonality and high spatial variation in estuarine ciliates are driven by exchanges between the a’bundant’ and ‘intermediate’ biospheres. Sci. Rep. 7, 9494. doi: 10.1038/s41598-017-10308-y
Sun P., Wang Y., Huang X., Huang B., Wang L. (2022). Water masses and their associated temperature and cross-domain biotic factors co-shape upwelling microbial communities. Water Res. 215, 118274. doi: 10.1016/j.watres.2022.118274
Suter E. A., Pachiadaki M., Taylor G. T., Edgcomb V. P. (2022). Eukaryotic parasites are integral to a productive microbial food web in oxygen-depleted waters. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.764605
Swart P. K., Arienzo M., Broad K., Clement A., Kakuk B. (2010). Blue holes in Bahamas: repositories of climate, anthropogenic, and archaeological changes over the past 300 000 years. J. Earth Sci. 21, 265. doi: 10.1007/s12583-010-0231-9
Villarino E., Watson J. R., Jönsson B., Gasol J. M., Salazar G., Acinas S. G., et al. (2018). Large-Scale ocean connectivity and planktonic body size. Nat. Commun. 9, 142. doi: 10.1038/s41467-017-02535-8
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Webb P., Wooldridge T. H. (1990). Diel horizontal migration of Mesopodopsis slabberi (Crustacea: Mysidacea) in algoa bay, southern Africa. . Mar. Ecol. Progress 62, 73–77. doi: 10.3354/meps062073
White M. G. (1998). Horizontal distribution of pelagic zooplankton in relation to predation gradients. Ecography 21, 44–62. doi: 10.1111/j.1600-0587.1998.tb00393.x
Wu P., Li D., Kong L., Li Y., Zhang H., Xie Z., et al. (2020). The diversity and biogeography of microeukaryotes in the euphotic zone of the northwestern pacific ocean. Sci. Total Environ. 698, 134289. doi: 10.1016/j.scitotenv.2019.134289
Wu W., Lu H. P., Sastri A., Yeh Y., Gong G., Chou W., et al. (2018). Contrasting the relative importance of species sorting and dispersal limitation in shaping marine bacterial versus protist communities. ISME J. 12, 485–494. doi: 10.1038/ismej.2017.183
Wyrwoll K., Zhu Z., Collins L. B., Hatcher B. G. (2006). Origin of blue hole structures in coral reefs: Houtman abrolhos, Western Australia. J. Coast. Res. 22, 202–208. doi: 10.2112/05A-0015.1
Xie L., Wang B., Pu X., Xin M., He P., Li C., et al. (2019). Hydrochemical properties and chemocline of the sansha yongle blue hole in the south China Sea. Sci. Total Environ. 649, 1281–1292. doi: 10.1016/j.scitotenv.2018.08.333
Xiong C., He J., Singh B. K., Zhu Y., Wang J., Li P., et al. (2021). Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ. Microbiol. 23, 1907–1924. doi: 10.1111/1462-2920.15262
Xue Y., Chen H., Yang J., Liu M., Huang B., Yang J. (2018). Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 12, 2263–2277. doi: 10.1038/s41396-018-0159-0
Yao P., Chen L., Fu L., Zhang Y., Bi N., Wang L., et al. (2018). Controls on vertical nutrient distributions in the sansha yongle blue hole, south China sea. Chin. Sci. Bulletin 63, 2393–2402. doi: 10.1360/N972018-00155
Yuan J., Li M., Lin S. (2015). An improved DNA extraction method for efficient and quantitative recovery of phytoplankton diversity in natural assemblages. PloS One 10, e0133060–e0133060. doi: 10.1371/journal.pone.0133060
Zehr J. P., Caron D. A. (2022). “Symbiosis in the ocean microbiome,” in The marine microbiome. the microbiomes of humans, animals, plants, and the environment, vol. 3 . Eds. Stal L. J., Cretoiu M. S. (Springer, Cham), 535–577. doi: 10.1007/978-3-030-90383-1_13
Zhang Y., Xu H., Wang L., Liu R., Fu L., Lin K. (2021). Unique bacterial communities and potential function along the vertical gradient in the deepest marine blue hole. Environ. Microbiol. Rep. 13, 911–927. doi: 10.1111/1758-2229.13001
Keywords: Sansha Yongle blue hole, planktonic microbiome, microbial diversity, assembly mechanism, rare taxa, microbial interaction
Citation: Chen T, Zhuang Y, Chen C, Mao X, Ge R, Chen H, Chen J, Fu L, Yang Z and Liu G (2022) Metabarcoding reveals the differential sensitivity of planktonic microbiome to environmental filtering and biointeraction in Sansha Yongle blue hole. Front. Mar. Sci. 9:1046808. doi: 10.3389/fmars.2022.1046808
Received: 17 September 2022; Accepted: 14 November 2022;
Published: 30 November 2022.
Edited by:
Shan He, Ningbo University, ChinaReviewed by:
Pengbin Wang, Ministry of Natural Resources, ChinaCopyright © 2022 Chen, Zhuang, Chen, Mao, Ge, Chen, Chen, Fu, Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunyun Zhuang, eXVueXVuLnpodWFuZ0BvdWMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.