 Kang Ma
Kang Ma Ze Ren
Ze Ren Jiaming Ma
Jiaming Ma Nannan Chen
Nannan Chen Jingling Liu
Jingling Liu- 1State Key Laboratory of Water Environment Simulation, School of Environment, Beijing Normal University, Beijing, China
- 2Research and Development Center for Watershed Environmental Eco-Engineering, Advanced Institute of Natural Sciences, Beijing Normal University, Zhuhai, China
Estuarine ecosystems interconnect freshwater and marine environments, and comprise multiple highly dynamic and complex microhabitats. The resident microbiota in estuary is influenced by contrasting microenvironmental heterogeneity. However, the bacterial patterns and assembly processes in different microhabitats of estuarine ecosystem are not well studied. Here, we investigated the bacterial diversity, functions and community assembly mechanisms of mangrove soil, river sediment and overlying water in a subtropical estuary. Results showed that similar profiles of bacterial communities existed in the mangrove soil and river sediment and were dominated by Proteobacteria, Bacteroidetes and Acidobacteria. In terms of different microhabitats, the lowest alpha diversity of bacterial communities was found in overlying water and were dominated by Proteobacteria, Actinobacteria and Bacteroidetes. Meanwhile, the functional potential genes associated with carbon metabolisms were also substantially different in the three microhabitats. The relative abundance of genes connected to aerobic carbon respiration was significantly higher in overlying water than in the other two microhabitats. Bacterial communities in river sediments were enriched for genes associated with aerobic methane oxidation. The strong environmental heterogeneity of the three nearby microhabitats shaped the taxonomic and functional composition of the bacterial communities in estuarine ecosystem. Moreover, the plant rhizosphere effect increased the proportion of the dispersal limitation processes in mangrove soils compared to that in river sediments, while the overlying water was fluid and had less environmental selection processes compared to that in mangrove soil and river sediment. The bacterial communities in river sediment construct a more clustered network, while the overlying water network showed the highest complexity. Our findings reveal the differences of bacterial patterns and community assembly mechanisms in distinct microhabitats of estuarine ecosystems, and provide important insights for a comprehensive understanding of the mechanisms to maintain estuarine wetland conservation under environmental changes.
Introduction
The estuarine ecosystem represents the transition between freshwater and marine environments and is influenced by both aquatic realms (Bernhard and Bollmann, 2010). Due to strong terrestrial-oceanic interactions, estuaries have complex organism community biodiversity of both freshwater and marine origin, composed of microorganisms, aquatic animals, and riparian vegetations (Regnier et al., 2013; Weinke and Biddanda, 2018). Acting as a filter along the land-ocean aquatic continuum, estuaries comprise highly dynamic and complex habitats and play a vital role in biogeochemical processing (Bauer et al., 2013; Rosentreter et al., 2021). In recent years, estuaries have become more susceptible to increasing pressures from growing human activities related to eutrophication, urban construction, and general environmental degradation (Weinke and Biddanda, 2018; Yuan et al., 2019; Zhou et al., 2021). Microbes are the most prevalent organisms in natural ecosystems and are sensitive indicators of ecosystem stress. They constitute aquatic food webs, participate significantly in various biogeochemical cycles, and maintain ecosystem functions (Fuhrman, 2009). Thus, understanding the estuarine bacterial community is of great importance for the sustainability of estuarine ecosystem functions in natural environments (Vasar et al., 2022).
Lying at the interface between the river, marine, and terrestrial ecosystems, estuarine ecosystems comprise some of the most productive ecosystems in the world, and receive large amounts of organic materials and nutrient from ocean and rivers (Regnier et al., 2022). It has been reported that vegetated habitats in estuary sustain the highest rates of carbon sequestration, contributing to climate change mitigation and adaptation (Li et al., 2021). Due to the differences of vegetation types, hydrological conditions and land conversion, the environmental factors in different microenvironments of estuarine ecosystems have very strong heterogeneity (Rogers et al., 2019; Tee et al., 2021; Wang et al., 2021). The sediment microhabitats are kept submerged under overlying water, but the soil microhabitats were alternately exposed and submerged for the hydrological processes in estuaries (Liu et al., 2018b). Compared to nonfluidic sediment and soil, the fluidic overlying water is more susceptible to external environment variables with respect to atmospheric changes and terrestrial hydrologic processes (Liu et al., 2018a; Lei et al., 2020; Liao et al., 2020). The bacterial communities in planktonic and sedimentary microhabitats in estuary could be highly diverse and variable. The underlying sediments and riparian soil exhibit similar properties in some way, while vegetations can selectively enriched specific microbial populations by rhizosphere effects (Yu et al., 2020; Zhuang et al., 2020). Moreover, the confluence of freshwater and tidewater helps to change nutrients and salinity levels, which are main factors controlling the bacterial communities (Cloern et al., 2017; Zhang et al., 2021c). Previous studies have highlighted that microbial communities living in microenvironments differ significantly in community profiles (Jiao et al., 2020; Zhang et al., 2021b). As the central roles of the biological carbon cycles in natural ecosystems, bacteria in estuarine ecosystems carry out the dichotomous functions of carbon inputs and outputs, and the balance between these two processes might shift the ecosystems from carbon sinks to carbon sources (Jansson and Hofmockel, 2020). However, bacterial diversity patterns and the functional changes in nearby microhabitats of estuaries have not been systematically investigated, limiting our understanding of bacterial diversity patterns and functional changes, which are highly relevant to mitigating estuarine degradation and adaptive management of estuarine wetlands under intensifying global changes.
Microbial diversity patterns in natural ecosystems are also driven by community assembly mechanisms, including deterministic and stochastic processes (Ofiteru et al., 2010; Stegen et al., 2013). Revealing the underlying mechanisms that shape microbial community assembly is useful for understanding community stability and ecosystem function (Dini-Andreote et al., 2015; Zhou and Ning, 2017). Based on niche, neutral, and process theories, the ecological models were proposed to investigate the assembly mechanisms of the microbial community. The stochastic, deterministic and undominated processes operate the microbial assembly mechanism simultaneously (Chase, 2010). Furthermore, a conceptual framework was proposed and exhibited that community assembly mechanisms are five ecological processes (Vellend, 2010). Under this framework, the stochastic processes were categorized as homogeneous selection or heterogeneous selection, and the deterministic processes were categorized as homogenizing dispersal and dispersal limitation (Vellend, 2010). Many studies have reported ecological assembly processes in river, lake, marine, and soil habitats. For example, environmental preferences could result in habitat filtration and selection. It was found that heterogeneous selection was the most important assembly process in acid mine drainage lakes (She et al., 2021). Heterogeneous selection and dispersal limitation are two domain assembly processes in the plant rhizosphere soil of grassland (Zhong et al., 2022). In addition, co-occurrence network method was applied to uncover potential interactions of bacterial taxa, and elucidate the patterns ranging from pairs of taxa to complex communities (Barberán et al., 2014). Although many recent studies have been conducted on microbial assembly in many aquatic and terrestrial habitats, the different microhabitats of estuarine ecosystems are poorly studied. Such knowledge is essential for increasing the understanding of the assembly mechanism of microbial communities in estuarine ecosystems.
Based on the variation of oxygen and water fluxion along the transition zone from waterward to landward, the estuarine sedimentary compartments were divided into sediments and riparian soils (Wang et al., 2021). The flowing water not only acts as a carrier of sea and upper river substances, but also serves as the exchange medium for soil and sediment (Battin et al., 2009; Zhang et al., 2022). It is generally accepted that both microhabitat type and physicochemical parameters are key factors shaping the microbial community (Lozupone and Knight, 2007). Different microhabitats may impose different stresses (e.g., oxygen, rhizosphere effect, or fluidity of environmental media) that drive community assembly processes, thus altering the species co-occurrence patterns and carbon cycling. Therefore, we chose a typical subtropical estuarine ecosystem with the riparian zones wholly covered by mangroves as our study area. The purpose of this study was to explore (1) the bacterial community profiles and functional differences of mangrove soils, river sediments, and overlying water in estuarine ecosystems; (2) the dominant ecological processes governing the assembly mechanisms and species interactions of bacterial communities in different microhabitats. Given the contrasting environmental heterogeneity of the three microenvironments, we hypothesized that bacterial taxa exhibited distinct carbon cycling function and community assemblage processes in different habitats.
Methods
Study area and sampling strategy
Our study area was conducted at the Zhenhai Bay (21°44’00″ to 21°56’30″N, 112°24’00″ to 112°33’00″E) of Guangdong Province, China (Supplementary Figure 1), which located in subtropical zone. The annual precipitation of the Zhenhai Bay is 2683 mm and the annual air temperature is 23.7°C. In this study area, the tide is an irregular semidiurnal tide, and the riparian zones are covered with natural mangroves. We chose six sites in the river center and established sampling belts from river center to riparian zone (Supplementary Figure 1). The riverway distance between each sampling transect was about three kilometers. A more detailed description of the sampling sites is provided in Table S1. Paired overlying water and surface river sediment samples were collected synchronously from the river center. The mangrove soil samples were collected parallel to each sampling site from riparian zones, respectively. At each location, three replicates of the water samples were randomly collected at a depth of 0.5 m by Ruttner sampler (Hydro-Bios, Altenholz, Germany) and combined at each sampling location. Microbial samples of water samples were collected by filtering water through 0.22 μm Millipore polycarbonate membranes and immediately frozen in dry ice (-80°C) in the field. The remaining water samples were acid fixed and stored at -4°C for chemical analyses. For sediments and soils, three replicates of samples were randomly collected and combined at each site using a pre-cleaned grab sampler. Microbial samples of soil and sediment samples were connected in a 45-mL sterile centrifuge tube and immediately frozen in dry ice in the field. The remaining soil and sediment samples were air-dried and stored for the determination of chemical properties.
Physicochemical analyses
Environmental variables were measured according to the methods used in our previous studies (Ren et al., 2020). At the time of sampling, a multiparameter instrument (YSI ProPlus, Yellow Springs, Ohio, USA) was used to measure the pH and electrical conductivity (EC) of water samples in the field. Dissolved organic carbon (DOC) levels in the water samples were determined by a Shimadzu TOC analyzer (TOC-VCPH, Shimadzu Scientific Instruments, Columbia, MD, United States). Total nitrogen (TN) and total phosphorus (TP) in the water samples was analyzed by colorimetric method after oxidation according to EPA 300.0 and 365.3. For soil and sediment samples, pH and electric conductivity was measured in a 1:2.5 and 1:5 air-drying samples to distilled water ratio using pH and conductivity meter respectively (Yang et al., 2012). The soil total carbon (TC) and nitrogen (TN) contents were determined by air-drying samples and sieving them to 2 mm, before determination by combustion (Thermo Fisher Scientific Flash Smart Elemental Analyzer, Bremen, Germany). An ICP AES Optima 8000 instrument (PerkinElmer, Waltham, MA, USA) was used to measure the TP.
Molecular methods
The total genomic DNA of water samples was extracted from the filter using DNeasy PowerWater kit (QIGEN, Germany) according to the manufacturer’s instruction. For soil and sediment samples, the total genomic DNA was extracted from ~0.5 g samples using DNeasy PowerWater kit (QIGEN, Germany) according to the standard instruction. The forward primer (343 F 5’-TACGGRAGGCAGCAG-3’) and reverse primer (798 R5’-AGGGTATCTAATCCT-3’) were used amplify the V3–V4 variable regions of the 16S rRNA genes (Nossa et al., 2010). According to manufacturer’s instructions, the PCR amplifications for each DNA sample were performed using the following procedure as our previous study (Ma et al., 2022). Amplified DNA was purified using the Gel Extraction Kit (Qiagen, Hilden, Germany) and then validated by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States) and quantified by Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, United States). The amplicon libraries were sequenced on an Illumina MiSeq platform (Illumina, Inc., San Diego, CA, USA) according to standard instructions.
Raw sequence data were trimming firstly, to detect and cut off ambiguous bases and low-quality sequences (average quality score below 20) by using Trimmomatic (version 0.35) (Bolger et al., 2014). Then, the assembly of paired-end reads were generated by FLASH software (Magoc and Salzberg, 2011). Quality filtering of raw sequences was processed using the QIIME 1.9.1 (Caporaso et al., 2010; Bokulich et al., 2013). The obtained gene tags were compared with the Silva database (version 138.1) to remove the chimera sequences (Quast et al., 2013). Based on trimming sequences, operational taxonomic units (OTUs) were further clustered at 97% identity threshold against the Silva database (Schloss, 2009). At last, the sequences of all samples were homogenized to equal depth (48948 per sample), prior to further analysis. Raw sequence data can be accessed at the China National Center for Bioinformation (GSA: CRA007468).
Bioinformatic analysis
Based on the sequences data, the alpha diversity indices were calculated and the significance of each index among groups was tested and visualized using test line regression tools in Hiplot (https://hiplot.com.cn). The Beta-diversity (Bray-Curtis distance) was calculated and Adonis test was performed in the “vegan” package (version 2.5-7) in the R statistical software to illustrate the community dissimilarity and the principal coordinates analysis (PCoA). The biomarkers of bacterial communities (Wilcoxon test < 0.05, threshold > 4.0, Kruskal–Wallis test < 0.05) were identified by using least discriminant analysis (LDA) method (Segata et al., 2011). To assess the relationship between the environmental variables and bacterial communities, redundancy analysis (RDA) was performed in the “vegan” package (version 2.5-7) in the R statistical software.
Based on the PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states, version 1.0) analysis of the 16S rRNA gene sequence data, the functional profiles of the bacterial communities in this study were predicted (Langille et al., 2013). The nearest sequence taxon index (0.082 for bacteria) in this study was calculated, indicating a high accuracy of the PICRUSt prediction (Langille et al., 2013). By using the predicted metagenomes, the KEGG orthologs (KOs, https://www.genome.jp/kegg/) were clustered and different pathway levels (levels 1–3) were calculated. Then, the KOs associated with the carbon cycles in microhabitats were further extracted (Supplementary Table 2) and the relative abundance of each pathway was calculated (Lauro et al., 2011). The figure of carbon conversion step was modified from (Lauro et al., 2011) and (Llorens-Mares et al., 2020).
To analysis the community assembly processes in each microhabitat in this study, we firstly constructed the phylogenetic trees in the FastTree (Version 2.1.11) software based on 16S rRNA gene sequences (Price et al., 2009). Then, we divided the taxonomic groups based on phylogenetic relationships between OTUs (12-48 OTUs) to improve the accuracy of community assembly processes inferring in the ‘iCAMP’ package (Ning et al., 2020). The bin size (24) and threshold of phylogenetic distance (0.05) were determined by phylogenetic signal. Based on 1000-times randomization of the taxa across the tips of the phylogenetic tree, the beta net relatedness index (βNRI) and Raup–Crick metric (RC) was likewise calculated to obtain the relative importance of five community assembly processes: homogeneous selection, homogeneous dispersal, dispersal limitation, heterogeneous selection (variable selection), and undominated (including ecological drift, diversification, weak selection, and/or weak dispersal. This framework uses the threshold described to divide community assembly processes (Ning et al., 2020). Finally, the relative importance of ecological processes to community assembly at the subcommunity and bin levels was ascertained.
The bacterial co-occurrence network analysis was established in this study. The OTUs dataset was simplified firstly with the retaining only OTUs of relative abundance > 1% in all samples (Barberán et al., 2014). Secondly, based on the OTU levels, the possible pairwise Spearman’s rank correlations were calculated, and connections mean strong (Spearman R > 0.8 or R < -0.8) and significant (P- adjust < 0.05) correlation were extracted by using Benjamini and Hochberg (BH) methods (Benjamini and Hochberg, 1995). At last, the Gephi software (WebAtlas, Paris, France) was used to visual network patterns and calculate network topological parameters (Bastian et al., 2009). All statistical tests described were performed in the R environment (Version 4.1.1) (R Core Team, 2017).
Results
Alpha and beta diversity
A total of 881064 clean reads were obtained after quality filtering, and 24705 OTUs were divided according to the 97% similarity threshold in this study. Rarefication curves approached a plateau, confirming that adequate sequencing depth was achieved (Supplementary Figure 2). Among the OTUs identified, 5676, 4282 and 1573 (mean OUT number in each group) were specific to the mangrove soil, river sediment and overlying water, respectively, and 2837 were shared between the three microhabitats (Supplementary Figure 3). However, the most shared OTUs between each two matrices were found in the mangrove soil and river sediment, which was much higher than the shared OTUs between each with overlying water.
The alpha diversity of bacterial communities in coexisting microhabitats was calculated based on the OTUs (Figure 1). The results showed that, the community richness of the mangrove soil, river sediment and overlying water was between 6166 and 7105, 6079 and 6479, 1586 and 2593, respectively. The one-way ANOVA analysis was used to identify the significant differences in alpha diversity indices among groups. The observed OTUs, Shannon, Chao1 and phylogenetic diversity indexes were significantly (p < 0.05) lower in overlying water samples than in the other two microhabitat samples, and no significant difference was found between the mangrove soils and river sediments. In total, these findings indicated that the similarities of OTUs only existed in the mangrove soil and river sediment, and the overlying water had more unique community diversity.
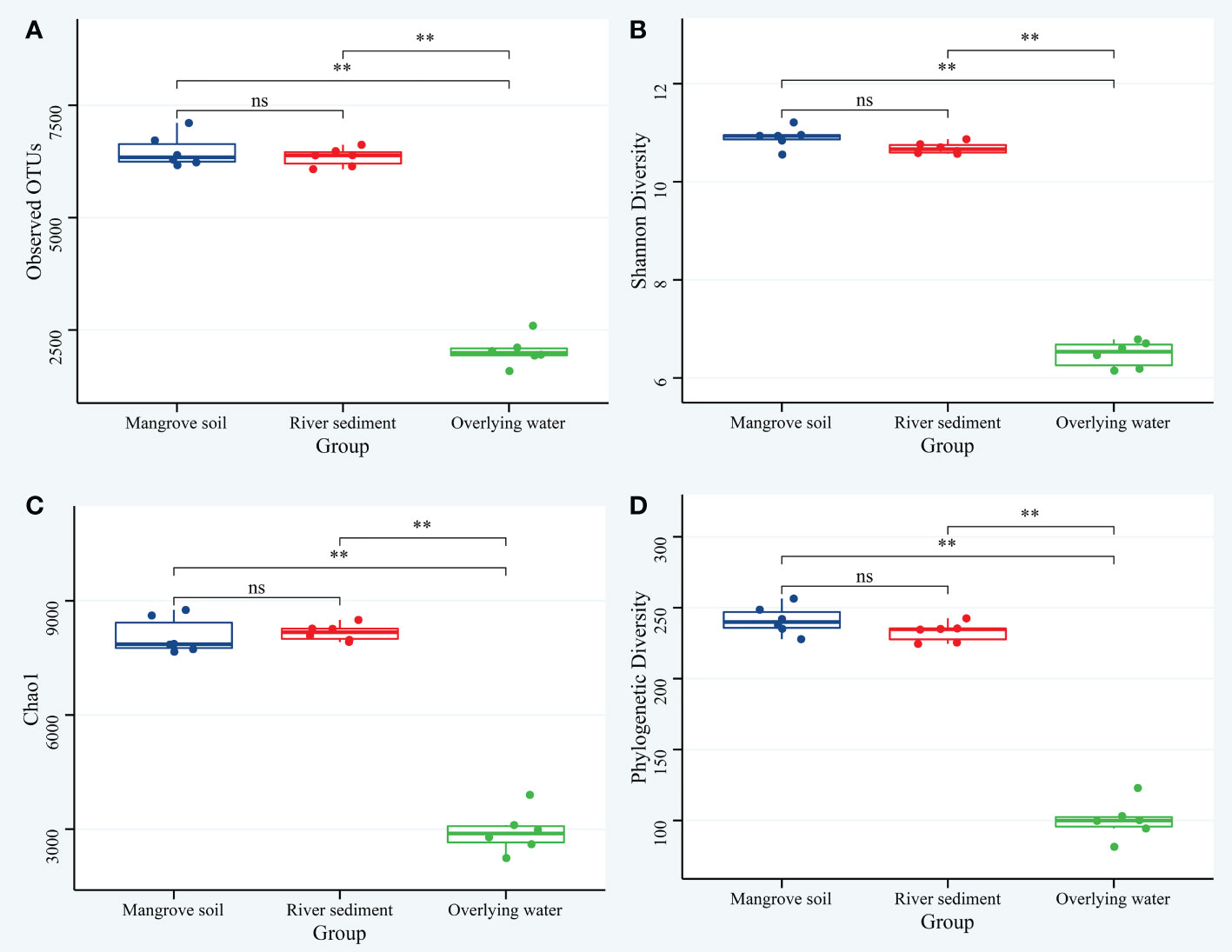
Figure 1 The alpha diversity indices of the bacterial communities in the mangrove soil, river sediment and overlying water. (A) observed OTUs; (B) Shannon diversity; (C) Chao1 diversity; (D) Phylogenetic Diversity. Significance of the one-way ANOVA tests: ns, not-significant; **p < 0.01.
Principal coordinates analysis (PCoA) was applied to investigate the heterogeneity of the samples (Figure 2). The first axis revealed that the planktonic bacterial communities differed significantly (p < 0.05) from the sedimentary bacterial communities. According to the Adonis test, the community dissimilarity of bacteria was affected by the differences of microhabitats (R = 0.61, p < 0.001). The results demonstrated that overlying water and river sediment samples were different, although they were collected from the same sites.
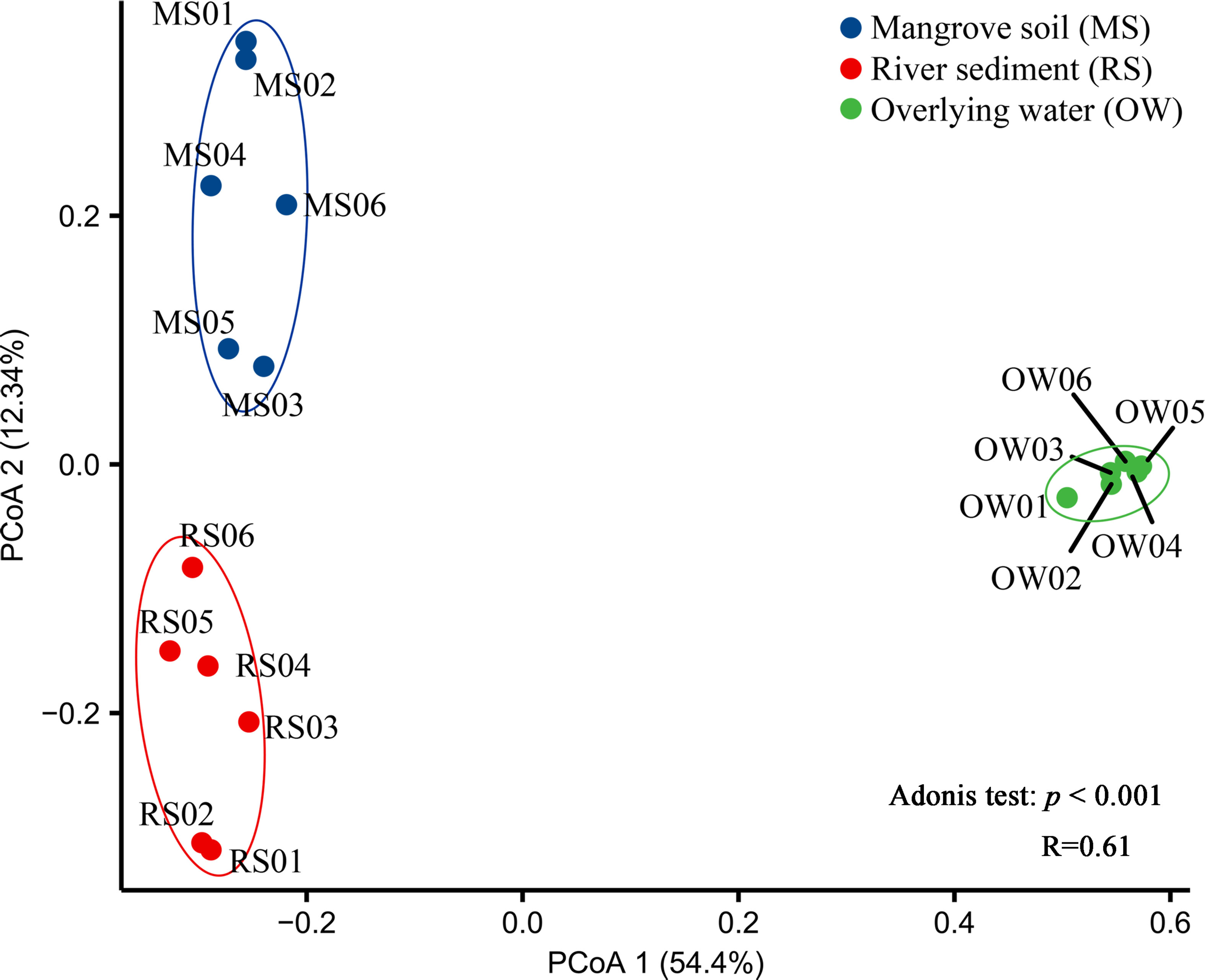
Figure 2 Principal Coordinate Analysis (PCoA) analysis of taxonomic composition of bacterial communities using Bray–Curtis dissimilarity based on the relative abundance of OTUs. Adonis test: p < 0.001, R =0.61.
Bacterial composition and functional prediction
At the phylum level, Proteobacteria was dominant (mean relative abundance, 67.84%) in all samples, followed by Bacteroidetes (10.85%), Actinobacteria (6.32%), Acidobacteria (3.48%), Gemmatimonadetes (2.89%), Firmicutes (2.02%), Nitrospirae (1.86%), Epsilonbacteraeota (0.85%), Spirochaetes (0.60%), and Chloroflexi (0.58%); these top 10 bacterial phyla constituted 97.29% of the average relative abundance (Supplementary Figure 3). Furthermore, the distinct bacterial community compositions of the three habitats (mangrove soil, river sediment and overlying water) are shown in Figure 3A. The Proteobacteria and Actinobacteria phyla had the highest relative abundances in overlying water, while other phyla were lowest compared to mangrove soils and river sediments. Proteobacteria was dominant, with mean relative abundances of 61.40% (mangrove soil), 65.13% (river sediment), and 76.98% (overlying water). However, Bacteroidetes was more abundant in mangrove soil and river sediment, with mean relative abundances of 14.56% and 10.19%, respectively, and Actinobacteria was more abundant in river sediment, with a mean relative abundance of 11.24%. The least discriminant analysis (LDA) method was applied to calculate the linear discriminant analysis effect size (LEfSe) of taxonomic differences between the three microhabitats (Figure 3B). Eleven, 21 and 20 biomarkers (LDA > 4.0, p < 0.05) were identified in the mangrove soil, river sediment and overlying water respectively and the discriminative taxa are shown in Figure 3C. Moreover, the phyla Deltaproteobacteria, Gammaproteobacteria and Alphaproteobacteria, were the main biomarkers in the mangrove soil, river sediment and overlying water microhabitats respectively.
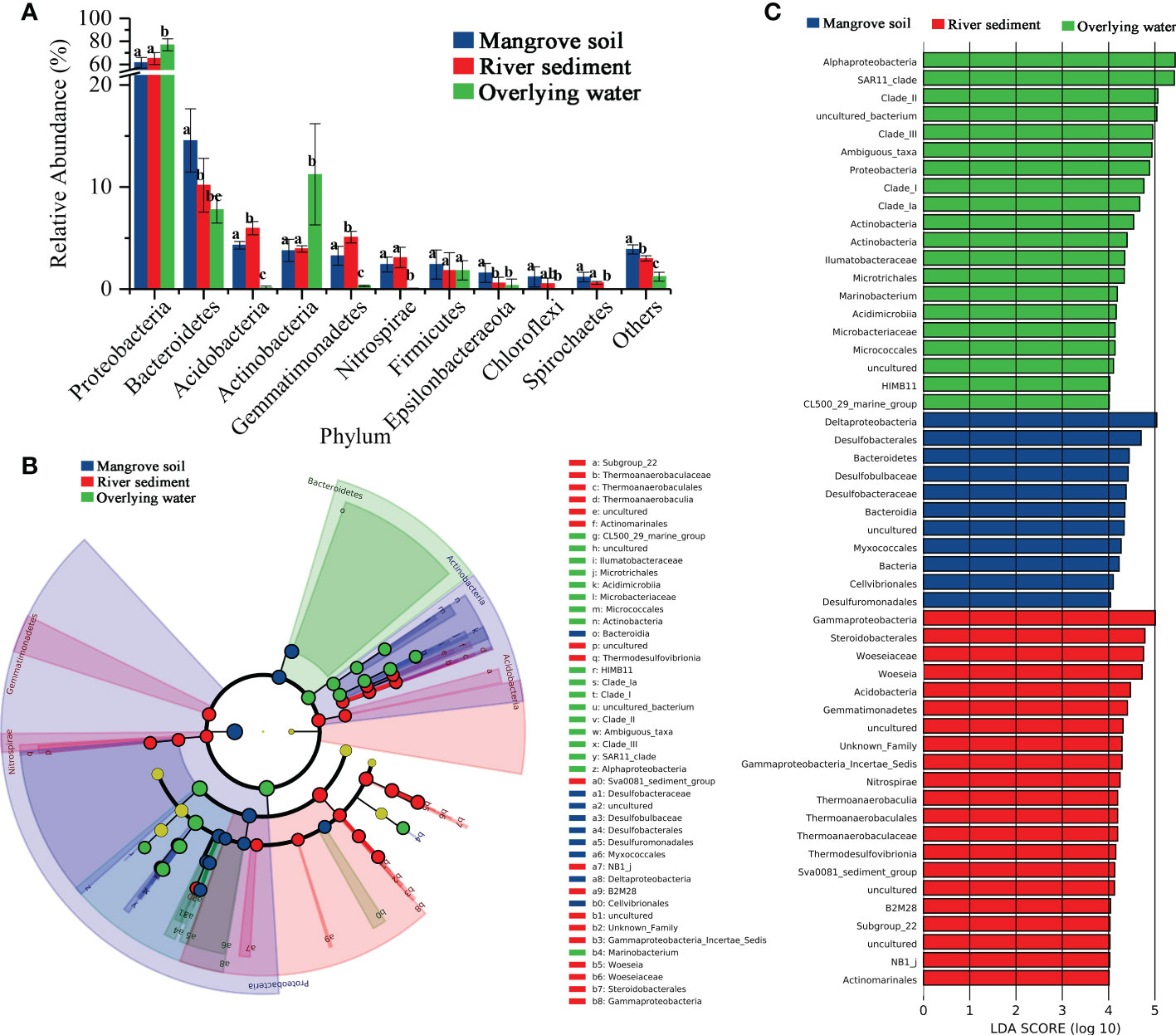
Figure 3 Relative abundances of bacterial phyla in distinct microhabitats (A). The cladogram of bacterial communities obtained using LEfSe analysis (B). The lineages with linear discriminant analysis (LDA) score of the abundance of taxa (C). Letters (a, b, and c) indicate significant difference of p < 0.01 between the sources (one-way ANOVA test). Only the taxa that LDA value above 4.0 are shown.
The biogeochemical cycling potential mediated by bacterial community in the three microhabitats were predicted using PICRUSt, and the genetic potential of the conversion steps associated with carbon cycle (Figure 4, Supplementary Table 2). The abundance of genes related to carbon cycling processes in mangrove soils was similar to that in river sediments. Compared to mangrove soil, only aerobic CH4 oxidation was significantly (p < 0.05) enriched in river sediments (Figure 4A). Moreover, all genes for the carbon cycle except for aerobic respiration in water were significantly (p < 0.05) lower than genes in mangrove soil and river sediment, indicating distinct patterns in overlying water (Figure 4B). The aerobic respiration of overlying water showed the highest abundance in the carbon cycles in the three microhabitats. In addition, the relative abundance of aerobic CH4 oxidation in river sediment was higher than that in mangrove soil, which might imply methane production potential in the two microhabitats (Figures 4C, D).
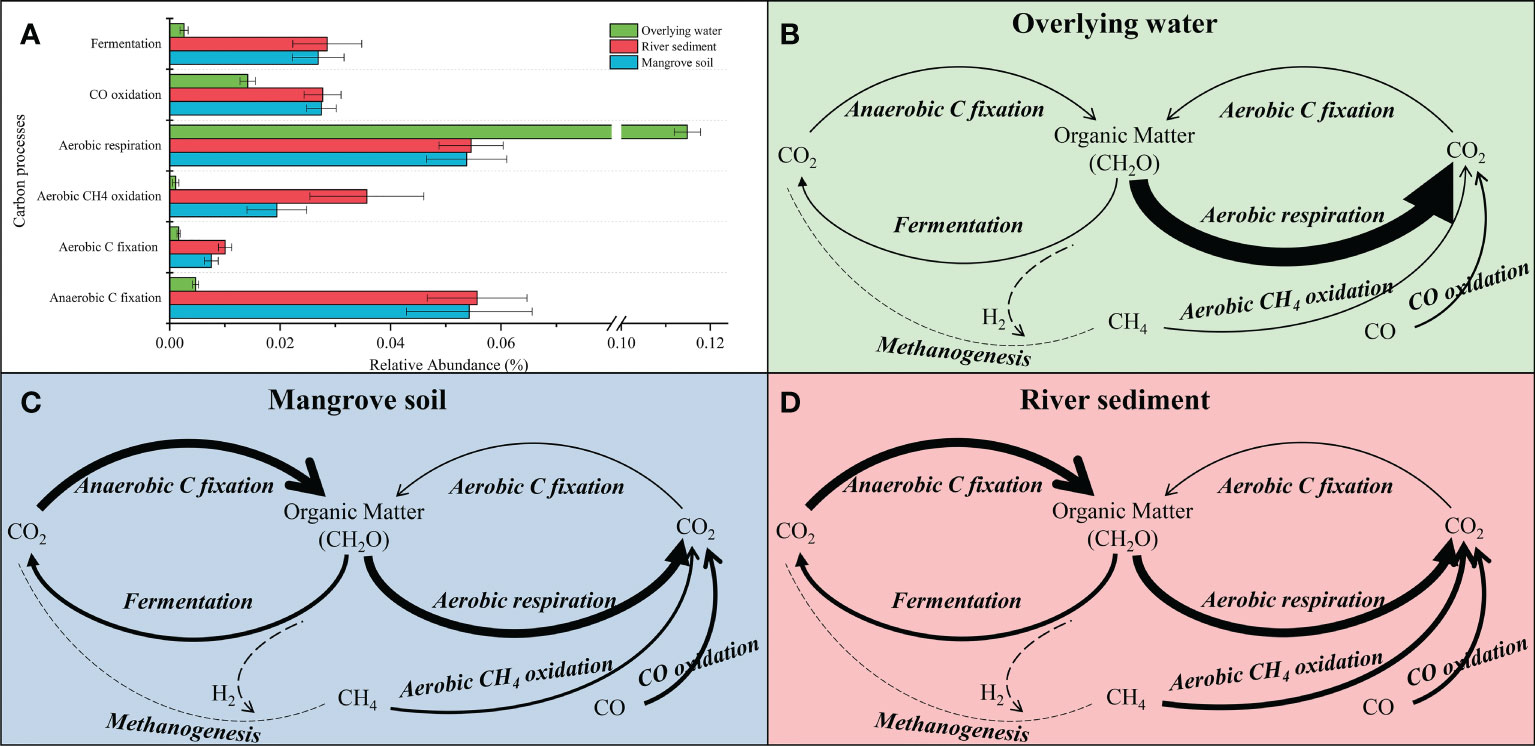
Figure 4 Relative abundance of genes response to carbon processes for bacterial communities (A). The genetic potential for each conversion step in the carbon cycles in overlying water (B), mangrove soil (C) and river sediment (D) microhabitats. The width of each arrow line is proportional to the relative abundance of the KEGG orthologies associated with the pathways. The dotted lines represent the conversion step without marker genes being detected.
Assembly processes and co-occurrence networks
The community assembly mechanism comprised of stochastic and deterministic processes was further revealed in the three microhabitats (Figure 5). The main ecological processes of overlying water were driving by the undominated and homogeneous selection processes (Figure 5). Homogeneous selection and dispersal limitation were the main processes for both river sediment and mangrove soil, revealing higher deterministic components for sedimentary substances rather than fluid water. Furthermore, the assembly mechanisms of the bacterial communities in mangrove soil and river sediment microhabitats were similar, but there were some differences. A higher proportion of the deterministic component (heterogeneous selection and homogenous selection) was found in river sediment than mangrove soil (Figure 5). Thus, the bacterial community assembly mechanism may be more complicated due to plant establishment.
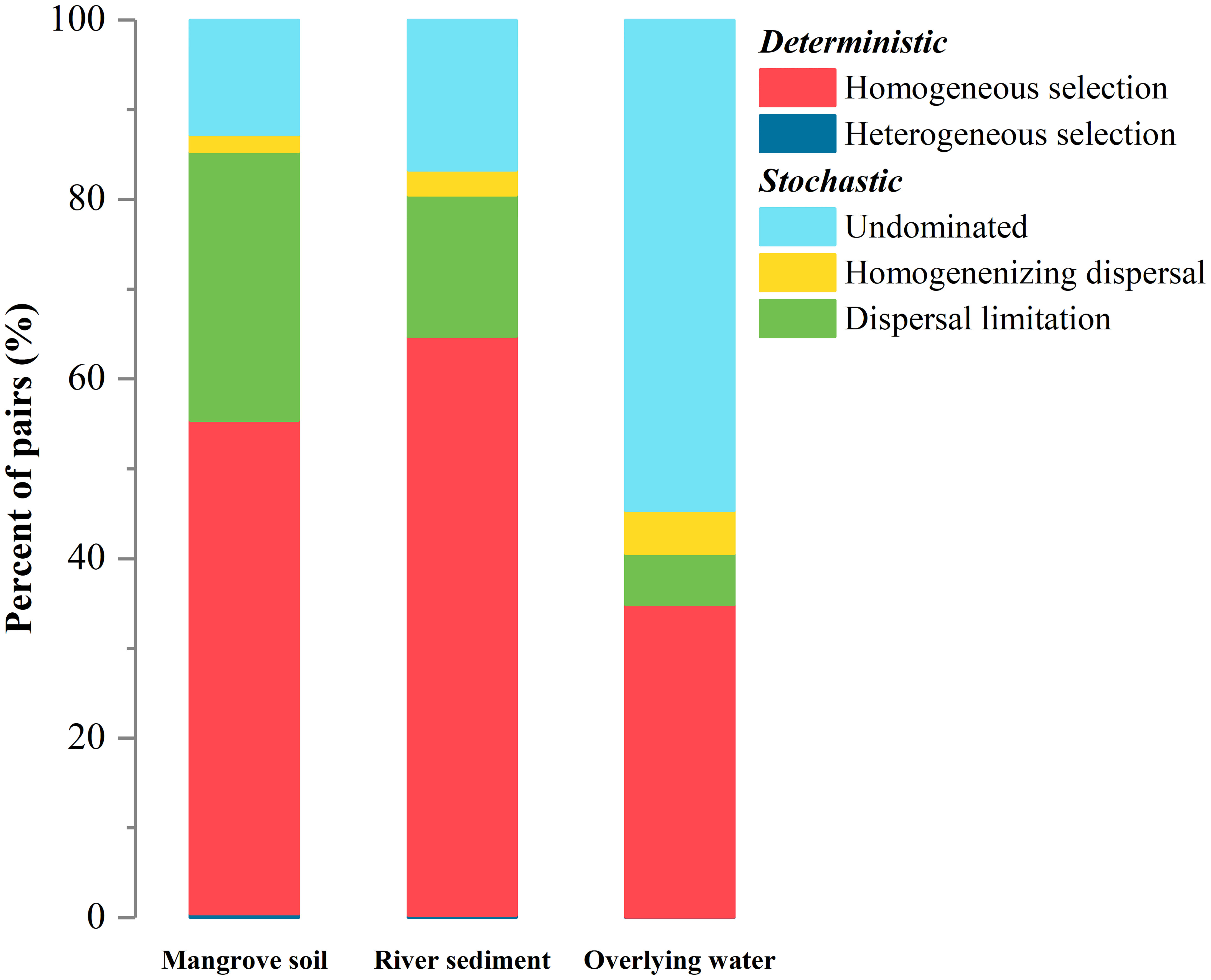
Figure 5 Relative importance of different ecological processes to bacterial community assemblies in different microhabitats.
To investigate the potential interactions between microbial communities of different microhabitats, three co-occurrence networks were constructed (Figure 6), and the topological characteristic parameters were calculated (Table 1). The network edge and node number of the three microhabitats gradually declined from the river sediment and mangrove soil to the overlying water, indicating that the complexity of community networks is different in the whole estuarine ecosystem. The overlying water network had the highest clustering coefficient and the lowest average path length, which suggests that the overlying water bacterial network was more complex with the OTUs more interconnected than mangrove soil and river sediment networks (Figure 6C). The network complexity (represented by the number of edges and nodes) was different between the mangrove soil and river sediment networks, with the river sediment network showing the highest complexity (Figure 6B). The average degree was higher in river sediment than in mangrove soil (Table 1). Meanwhile, the average path length and modularity were higher in mangrove soil than in river sediment, indicating that the mangrove soil bacterial network had a more clustered topology (Figure 6A).
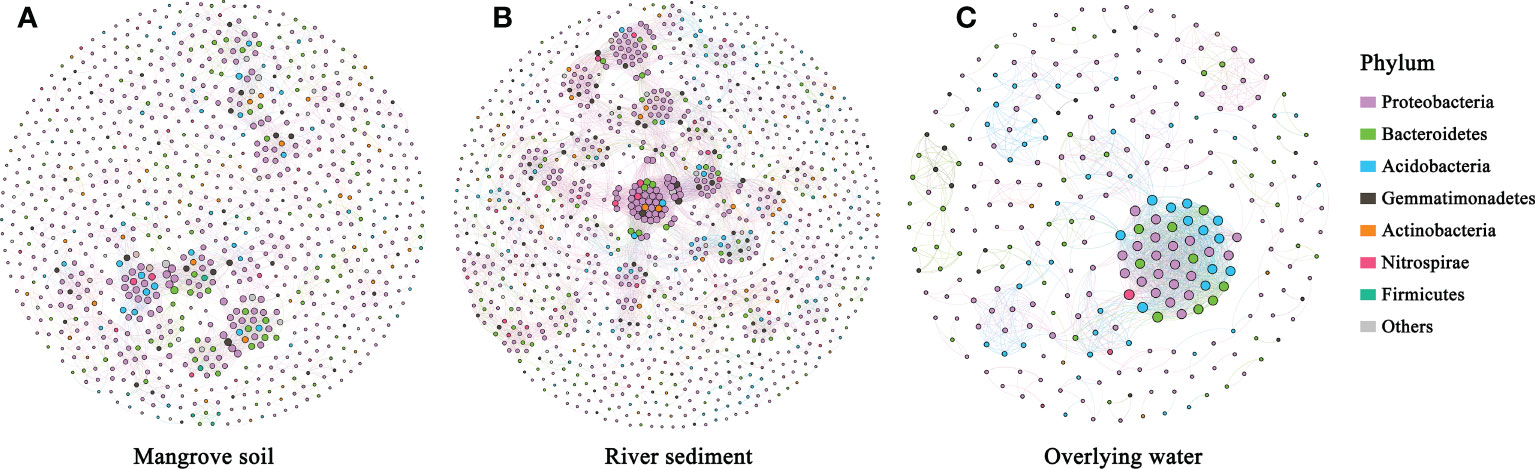
Figure 6 The co-occurrence networks of bacterial communities colored by phylum in mangrove soil (A), river sediment (B) and overlying water (C) microhabitats.
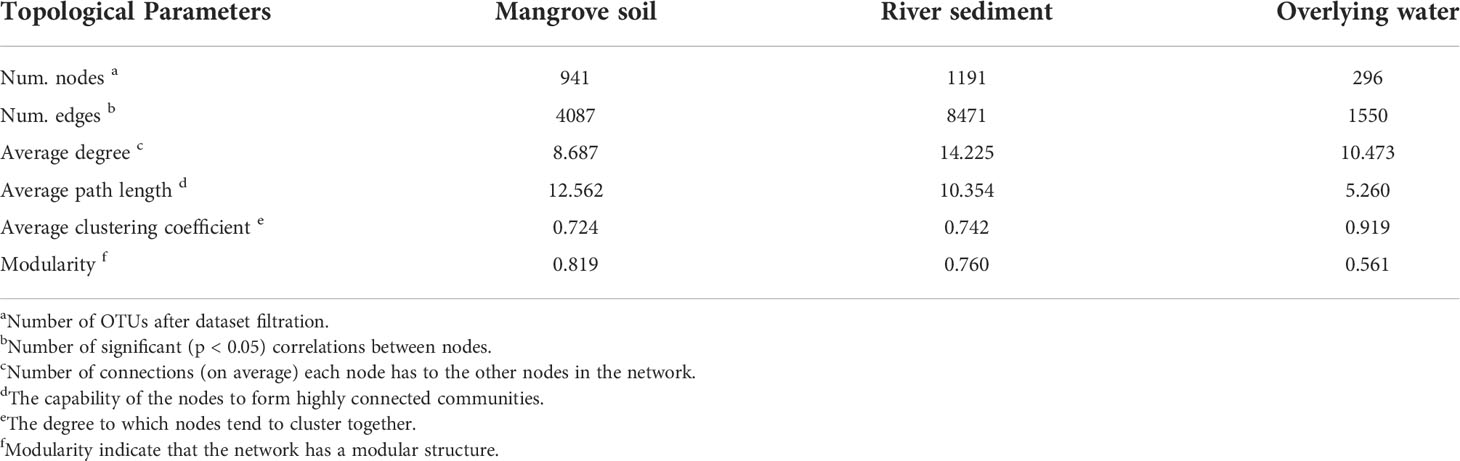
Table 1 Major topological properties of the ecological networks of bacterial communities three microhabitats.
Environmental drivers
The community diversity and composition revealed the discrepancy of bacterial communities in the three microhabitats, and the effects of physiochemical factors on bacterial communities in each microhabitat were further revealed by db-RDA (Figure 7). As a result (Supplementary Table 2), TC (p < 0.05) were identified as key drivers affecting the bacterial communities in mangrove soil and positively correlated with RDA1. Moreover, the EC (p < 0.05) and TN (p < 0.05) showed significant effects on bacterial communities in river sediment. For overlying water, EC (p < 0.05) and TP (p < 0.05) had significant effects on bacteria. The two axes explained 69.02%, 71.35% and 84.51% of the relationship between environmental parameters and the bacterial communities in the mangrove soil, river sediment and overlying water samples, respectively.
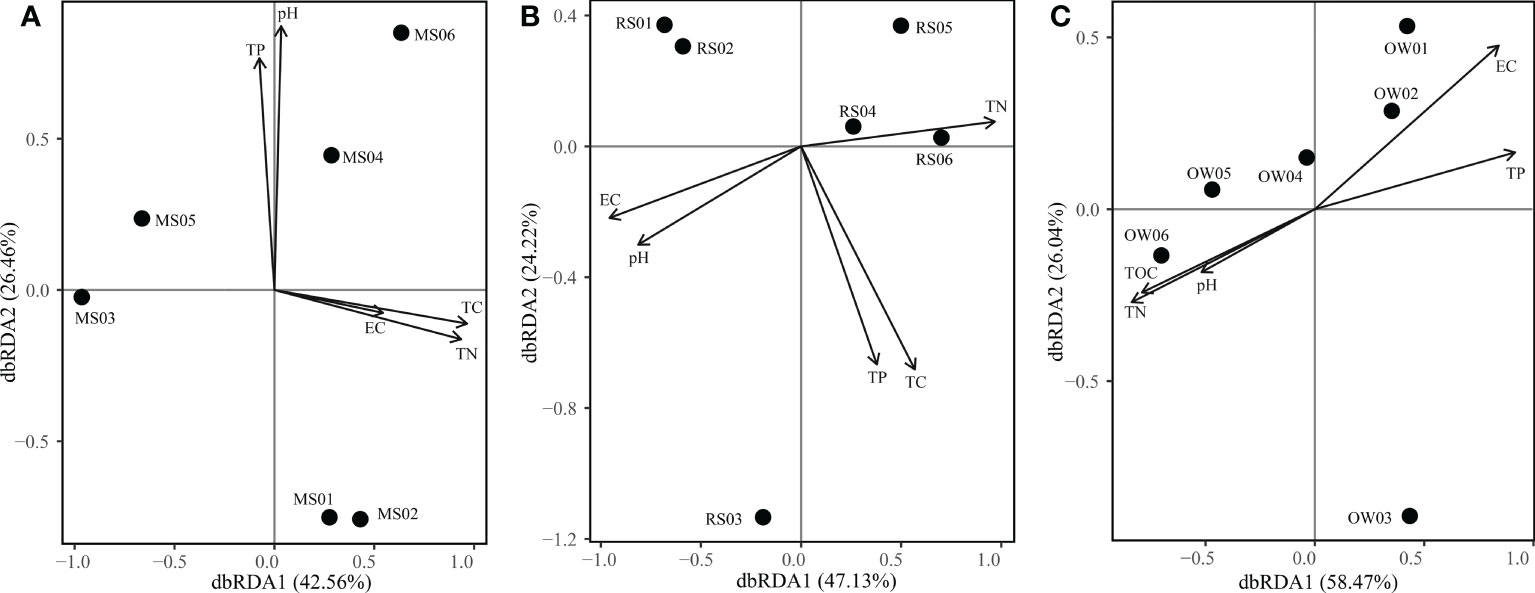
Figure 7 Distance-based redundancy analyses (RDA) of bacterial communities in mangrove soil (A), river sediment (B) and overlying water (C).
Discussion
The mangrove soil, river sediments, and overlying water microenvironments are nearby and highly interconnected microhabitats in the estuarine ecosystem. In these microhabitats, microorganisms play key roles in the function of biogeochemical and hydrological cycles. It is, therefore, important to understand how bacterial communities inhabiting different microhabitats, and how distinct environmental conditions drive assembly mechanisms in each microhabitat. In this study, we systematically investigated the diversity, community composition, assembly process and function of bacterial communities in mangrove soil, river sediment and overlying water in a subtropical estuary.
Variations in bacterial diversity, structure and function
As expected, distinctions in bacterial communities in the three microhabitats were found, indicating that the diversity and composition of microbial communities were substantially different in distinct microhabitats (mangrove soil, river sediment and overlying water). In our study, the mangrove soil and river sediment microhabitats shared the highest OTUs in the estuarine ecosystem (Supplementary Figure 2). The bacterial communities in these two microhabitats had significantly higher alpha-diversity than in those in overlying water samples, which was in agreement with previous studies (Liu et al., 2021; Zhang et al., 2021b; Dang et al., 2022; Zhang et al., 2022). Soils and sediments recruit and subsequently exchange microbes with surrounding areas, which are considered to be the cause of the main discrepancy between watery and sedimentary (soil and sediment) microhabitats in aquatic systems (Zhang et al., 2021b; Zhang et al., 2022). However, it was interesting that there was no significant difference in alpha diversity between mangrove soil and river sediment microhabitats in this study (Figure 1). Due to tidal processes, the sediment microhabitats are kept submerged but the soil microhabitats were alternately exposed and submerged in estuarine ecosystems (Liu et al., 2018b). In our study area, the sediment samples were collected in the river center, which is in perpetual anaerobic conditions. Consequently, the results suggested that the submerged condition had no significant effects on the bacterial alpha diversity but may have effects on the composition of bacterial community. Previous studies have demonstrated that nutrients and environmental variables significantly drive microbial community diversity and composition in aquatic environments (Kaestli et al., 2017; Jones et al., 2018). In our study, the major bacterial phyla of the mangrove soil, river sediment and overlying water microhabitats were similar, while their relative abundances differed in distinct microhabitats (Figure 3A). The Proteobacteria and Bacteroidetes phyla are known to be dominant surface colonizers in aquatic ecosystems, and are involved in the cycle of carbon rich substances in aquatic habitats (Chaudhry et al., 2012; Jiang et al., 2019; Ren et al., 2022). Furthermore, Actinobacteria was identified as a specific phylum in water, which was proven in other aquatic ecosystems (Hoshino et al., 2020; Chao et al., 2021). In addition, we observed that Bacteroidetes was significantly enriched in the mangrove soil samples, which may be due to the plant rhizosphere effect (Chaparro et al., 2014; Chen and Gu, 2017).
The diversity patterns of bacterial communities in different microhabitats also influence microbial carbon metabolism (Yu et al., 2022). In overlying water, the enrichment of aerobic respiration genes is likely a reflection of high cyanobacteria (Ren et al., 2022). Due to the turbulent flow of the estuary and the oxygen exchange of water-air, the genes related to the aerobic carbon respiration process are enriched in overlying water compared to the soil and sediment (Figure 4). In contrast, based on the absence of light and isolation from the atmosphere, the bacteria in river sediment microhabitats exhibited strong anaerobic carbon fixation and fermentation processes. The microorganisms of soil and sediment are usually supplied by heterotrophic and lithotrophic metabolisms in natural ecosystems (Bond-Lamberty et al., 2018; Liu et al., 2021). The chemolithotrophy might supplied for the anaerobic carbon fixation in river sediment microhabitats, and particulate organic carbon would be decomposed into smaller molecules by bacteria, ultimately to CO2 (Christner et al., 2014). Finally, the bacteria in river sediments showed higher the methane production potential than those in mangrove soils, which might decrease the carbon function of estuarine ecosystems.
Assembly processes and species interactions of the bacterial communities
Unraveling the community assembly of communities is a critical topic in microbial ecology, especially for different habitats (Ning et al., 2020). Microhabitat shifts could cause distinct community diversity and structure of bacterial communities as previously proven in our study, and change the balance between stochastic and deterministic community assembly processes (Zhong et al., 2022). The contribution of deterministic components to the assembly of overlying water, mangrove soil, and river sediment bacterial communities increased in turn (Figure 5). This might be mainly due to the fluidity differences of the three environmental media (Zhang et al., 2022). Estuarine ecosystems are highly connected with river and marine ecosystems, and the fluidity of microorganisms in water microhabitats leads to the highest contribution of homogeneous selection and the lowest dispersal limitation compared to mangrove soil and river sediment microhabitats (Wang et al., 2015; Isabwe et al., 2018). Furthermore, we found that the contribution of dispersal limitation to the assembly of the sediment microbial community was lower than that in the mangrove soil. This may be attributed to the moderate mobility of sediments due to the transporting effect of overlying waterflow (Wu et al., 2013; Liu et al., 2018a). Additionally, the rhizosphere effects in mangrove soil microhabitats play important roles in community assembly mechanisms. The bacterial communities in river sediments were structured more by heterogeneous selection processes than those in mangrove soil. This difference could be explained by the riparian vegetation; the higher heterogeneous selection processes in sediment microhabitats lead to increased dissimilarity between bacterial communities (Vieira et al., 2020; Ma et al., 2022; Zhong et al., 2022). Consequently, the fluidity of microorganisms and the vegetation rhizosphere effect play major roles in bacterial community assembly in different microhabitats in estuarine ecosystems.
Microbial networks are useful for exploring potential interactions between species (Freilich et al., 2018). In the present study, the co-occurrence patterns of microbial communities in mangrove soil, river sediment and overlying water microhabitats were explored (Figure 6). Significant differences were found in the compositions and structures of the three microbial co-occurrence networks, suggesting that microhabitats had significant influence on the co-occurrence networks of bacteria. Based on the analysis of the overall network topology, overlying water had the highest connectance property in the estuarine ecosystem, which indicated that the water microhabitat had more efficient biochemical cycles. The highest modularity of the mangrove soil microhabitat suggested more complexity and stability of vegetation ecosystems (Zhong et al., 2022). Thus, this implied that the rhizosphere effect of mangroves improved the stability of bacterial communities (Shi et al., 2016; Poosakkannu et al., 2017; Tian et al., 2018).
Impacts of physicochemical factors on estuarine bacteria
The environment of the estuarine system is affected by the interaction of the sea and land (Wang et al., 2019). Previous studies have shown that physicochemical factors regulate the community diversity, structure and function of microorganisms (Ren et al., 2019; Sun et al., 2020). In this study, different significant factors were found in distinct microhabitats (Figure 7). Along the river estuary, bacteria are highly controlled by the EC and nutrients, which is in agreement with other estuaries (Chi et al., 2021; Liu et al., 2021; Zhang et al., 2021a). Interestingly, TC and TN were important variables in shaping the bacterial communities of mangrove soil microhabitats, whereas EC and TP were the main factors of river sediment and overlying water microhabitats. There may be two reasons for this result. First, the plant growth of mangrove soil microhabitats requires more nutrient substances and further influences the microbes by the rhizosphere effect (Zhuang et al., 2020; Zhong et al., 2022). In addition, compared to mangrove soil, the sediment microhabitat is submerged continually, and more susceptible to the salinity of the tidal overlying water in the estuarine system (Xu et al., 2020; Zhang et al., 2021c). These findings suggest that the microhabitat discrepancy was the largest source of the variations in bacterial diversity and community composition.
Conclusion
Our study revealed that bacterial communities from adjacent microhabitats have differential patterns and assembly processes in a subtropical estuary. It was found that similar profiles of bacterial communities existed in the mangrove soil and river sediment and the overlying water had the lowest alpha diversity. Functional potentials suggested that the aerobic carbon respiration would be more enriched in water than in the other two microhabitats based on the oxygenated environment. Bacterial communities in river sediments were enriched for genes associated with aerobic methane oxidation. The results indicated distinct taxonomic and functional composition in mangrove soil, river sediment and overlying water. Moreover, the plant rhizosphere effect increased the proportion of the dispersal limitation processes in mangrove soils compared to that in river sediments, while the overlying water was fluid and had less environmental selection processes compared to that in mangrove soil and river sediment. The bacteria of the river sediment microhabitat construct a clustered network, while the overlying water network showed the highest complexity in the estuarine ecosystem. The contrasting bacterial diversity and structure reflected that the strong environmental heterogeneity of the three microhabitats shapes the function and community assembly of the bacterial community. Overall, our study reveals differential patterns and assembly mechanisms of bacterial communities in distinct microhabitats of estuarine ecosystems, and improves comprehensive understanding of maintaining estuarine wetland conservation under environmental changes.
Data availability statement
The data presented in the study are deposited in the China National Center for Bioinformation repository, accession number CRA007468.
Author contributions
KM, ZR, and JL conceived the ideas and designed the study. KM and ZR collected the samples and performed most laboratory analysis. KM, ZR, and JM analyzed the data.KM wrote the manuscript with contributions and comments from all co-authors. NC and JL significantly improved earlier version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Key Project of National Natural Science Foundation of China (U1901212).
Acknowledgments
The authors would like to thank Zhihui Wu, Sheng Dong and Cheng Zhang for their assistances in the laboratory work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1039387/full#supplementary-material
References
Barberán A., Bates S. T., Casamayor E. O., Fierer N. (2014). Erratum: Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 8 (4), 952–952. doi: 10.1038/ismej.2013.236
Bastian M., Heymann S., Jacomy M. (2009). Gephi: An open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media.
Battin T. J., Luyssaert S., Kaplan L. A., Aufdenkampe A. K., Richter A., Tranvik L. J. (2009). The boundless carbon cycle. Nat. Geosci. 2 (9), 598–600. doi: 10.1038/ngeo618
Bauer J. E., Cai W. J., Raymond P. A., Bianchi T. S., Hopkinson C. S., Regnier P. A. (2013). The changing carbon cycle of the coastal ocean. Nature 504 (7478), 61–70. doi: 10.1038/nature12857
Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B-Statistical Method. 57 (1), 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bernhard A. E., Bollmann A. (2010). Estuarine nitrifiers: New players, patterns and processes. Estuar. Coast. Shelf Sci. 88 (1), 1–11. doi: 10.1016/j.ecss.2010.01.023
Bokulich N. A., Subramanian S., Faith J. J., Gevers D., Gordon J. I., Knight R., et al. (2013). Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat. Methods 10 (1), 57–59. doi: 10.1038/nmeth.2276
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bond-Lamberty B., Bailey V. L., Chen M., Gough C. M., Vargas R. (2018). Globally rising soil heterotrophic respiration over recent decades. Nature 560 (7716), 80–83. doi: 10.1038/s41586-018-0358-x
Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7 (5), 335–336. doi: 10.1038/nmeth.f.303
Chao C., Wang L., Li Y., Yan Z., Liu H., Yu D., et al. (2021). Response of sediment and water microbial communities to submerged vegetations restoration in a shallow eutrophic lake. Sci. Total Environ. 801, 149701. doi: 10.1016/j.scitotenv.2021.149701
Chaparro J. M., Badri D. V., Vivanco J. M. (2014). Rhizosphere microbiome assemblage is affected by plant development. ISME J. 8 (4), 790–803. doi: 10.1038/ismej.2013.196
Chase J. M. (2010). Stochastic community assembly causes higher biodiversity in more productive environments. Science 328 (5984), 1388–1391. doi: 10.1126/science.1187820
Chaudhry V., Rehman A., Mishra A., Chauhan P. S., Nautiyal C. S. (2012). Changes in bacterial community structure of agricultural land due to long-term organic and chemical amendments. Microb. Ecol. 64 (2), 450–460. doi: 10.1007/s00248-012-0025-y
Chen J., Gu J. D. (2017). Faunal burrows alter the diversity, abundance, and structure of AOA, AOB, anammox and n-damo communities in coastal mangrove sediments. Microb. Ecol. 74 (1), 140–156. doi: 10.1007/s00248-017-0939-5
Chi Z., Zhu Y., Li H., Wu H., Yan B. (2021). Unraveling bacterial community structure and function and their links with natural salinity gradient in the yellow river delta. Sci. Total Environ. 773, 145673. doi: 10.1016/j.scitotenv.2021.145673
Christner B. C., Priscu J. C., Achberger A. M., Barbante C., Carter S. P., Christianson K., et al. (2014). A microbial ecosystem beneath the West Antarctic ice sheet. Nature 512 (7514), 310–313. doi: 10.1038/nature13667
Cloern J. E., Jassby A. D., Schraga T. S., Nejad E., Martin C. (2017). Ecosystem variability along the estuarine salinity gradient: Examples from long-term study of San Francisco bay. Limnology Oceanography 62, S272–S291. doi: 10.1002/lno.10537
Dang C., Wang J., He Y., Yang S., Chen Y., Liu T., et al. (2022). Rare biosphere regulates the planktonic and sedimentary bacteria by disparate ecological processes in a large source water reservoir. Water Res. 216, 118296. doi: 10.1016/j.watres.2022.118296
Dini-Andreote F., Stegen J. C., van Elsas J. D., Salles J. F. (2015). Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. U.S.A. 112 (11), E1326–E1332. doi: 10.1073/pnas.1414261112
Freilich M. A., Wieters E., Broitman B. R., Marquet P. A., Navarrete S. A. (2018). Species co-occurrence networks: Can they reveal trophic and non-trophic interactions in ecological communities? Ecology 99 (3), 690–699. doi: 10.1002/ecy.2142
Fuhrman J. A. (2009). Microbial community structure and its functional implications. Nature 459 (7244), 193–199. doi: 10.1038/nature08058
Hoshino T., Doi H., Uramoto G. I., Wormer L., Adhikari R. R., Xiao N., et al. (2020). Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. U.S.A. 117 (44), 27587–27597. doi: 10.1073/pnas.1919139117
Isabwe A., Yang J. R., Wang Y., Liu L., Chen H., Yang J. (2018). Community assembly processes underlying phytoplankton and bacterioplankton across a hydrologic change in a human-impacted river. Sci. Total Environ. 630, 658–667. doi: 10.1016/j.scitotenv.2018.02.210
Jansson J. K., Hofmockel K. S. (2020). Soil microbiomes and climate change. Nat. Rev. Microbiol. 18 (1), 35–46. doi: 10.1038/s41579-019-0265-7
Jiang W., Tian X., Li L., Dong S., Zhao K., Li H., et al. (2019). Temporal bacterial community succession during the start-up process of biofilters in a cold-freshwater recirculating aquaculture system. Bioresour Technol. 287, 121441. doi: 10.1016/j.biortech.2019.121441
Jiao C., Zhao D., Zeng J., Guo L., Yu Z. (2020). Disentangling the seasonal co-occurrence patterns and ecological stochasticity of planktonic and benthic bacterial communities within multiple lakes. Sci. Total Environ. 740, 140010. doi: 10.1016/j.scitotenv.2020.140010
Jones A. C., Hambright K. D., Caron D. A. (2018). Ecological patterns among bacteria and microbial eukaryotes derived from network analyses in a low-salinity lake. Microb. Ecol. 75 (4), 917–929. doi: 10.1007/s00248-017-1087-7
Kaestli M., Skillington A., Kennedy K., Majid M., Williams D., McGuinness K., et al. (2017). Spatial and temporal microbial patterns in a tropical macrotidal estuary subject to urbanization. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01313
Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31 (9), 814–821. doi: 10.1038/nbt.2676
Lauro F. M., DeMaere M. Z., Yau S., Brown M. V., Ng C., Wilkins D., et al. (2011). An integrative study of a meromictic lake ecosystem in Antarctica. ISME J. 5 (5), 879–895. doi: 10.1038/ismej.2010.185
Lei M., Li Y., Zhang W., Niu L., Wang L., Zhang H. (2020). Identifying ecological processes driving vertical and horizontal archaeal community assemblages in a contaminated urban river. Chemosphere 245, 125615. doi: 10.1016/j.chemosphere.2019.125615
Liao H., Yen J. Y., Guan Y., Ke D., Liu C. (2020). Differential responses of stream water and bed sediment microbial communities to watershed degradation. Environ. Int. 134, 105198. doi: 10.1016/j.envint.2019.105198
Li Y., Fu C., Zeng L., Zhou Q., Zhang H., Tu C., et al. (2021). Black carbon contributes substantially to allochthonous carbon storage in deltaic vegetated coastal habitats. Environ. Sci. Technol. 55 (9), 6495–6504. doi: 10.1021/acs.est.1c00636
Liu X., Pan J., Liu Y., Li M., Gu J. D. (2018b). Diversity and distribution of archaea in global estuarine ecosystems. Sci. Total Environ. 637-638, 349–358. doi: 10.1016/j.scitotenv.2018.05.016
Liu L., Sun F., Zhao H., Mi H., He S., Chen Y., et al. (2021). Compositional changes of sedimentary microbes in the Yangtze river estuary and their roles in the biochemical cycle. Sci. Total Environ. 760, 143383. doi: 10.1016/j.scitotenv.2020.143383
Liu T., Zhang A. N., Wang J., Liu S., Jiang X., Dang C., et al. (2018a). Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze river. Microbiome 6 (1), 16. doi: 10.1186/s40168-017-0388-x
Llorens-Mares T., Catalan J., Casamayor E. O. (2020). Taxonomy and functional interactions in upper and bottom waters of an oligotrophic high-mountain deep lake (Redon, pyrenees) unveiled by microbial metagenomics. Sci. Total Environ. 707, 135929. doi: 10.1016/j.scitotenv.2019.135929
Lozupone C. A., Knight R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U.S.A. 104 (27), 11436–11440. doi: 10.1073/pnas.0611525104
Magoc T., Salzberg S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27 (21), 2957–2963. doi: 10.1093/bioinformatics/btr507
Ma J., Ma K., Liu J., Chen N. (2022). Rhizosphere soil microbial community under ice in a high-latitude wetland: Different community assembly processes shape patterns of rare and abundant microbes. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.783371
Ning D., Yuan M., Wu L., Zhang Y., Guo X., Zhou X., et al. (2020). A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 11 (1), 4717. doi: 10.1038/s41467-020-18560-z
Nossa C. W., Oberdorf W. E., Yang L., Aas J. A., Paster B. J., Desantis T. Z., et al. (2010). Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J. Gastroenterol. 16 (33), 4135–4144. doi: 10.3748/wjg.v16.i33.4135
Ofiteru I. D., Lunn M., Curtis T. P., Wells G. F., Criddle C. S., Francis C. A., et al. (2010). Combined niche and neutral effects in a microbial wastewater treatment community. Proc. Natl. Acad. Sci. U.S.A. 107 (35), 15345–15350. doi: 10.1073/pnas.1000604107
Poosakkannu A., Nissinen R., Mannisto M., Kytoviita M. M. (2017). Microbial community composition but not diversity changes along succession in arctic sand dunes. Environ. Microbiol. 19 (2), 698–709. doi: 10.1111/1462-2920.13599
Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26 (7), 1641–1650. doi: 10.1093/molbev/msp077
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41 (Database issue), D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing
Regnier P., Friedlingstein P., Ciais P., Mackenzie F. T., Gruber N., Janssens I. A., et al. (2013). Anthropogenic perturbation of the carbon fluxes from land to ocean. Nat. Geosci. 6 (8), 597–607. doi: 10.1038/Ngeo1830
Regnier P., Resplandy L., Najjar R. G., Ciais P. (2022). The land-to-ocean loops of the global carbon cycle. Nature 603 (7901), 401–410. doi: 10.1038/s41586-021-04339-9
Ren Z., Gao H., Luo W., Elser J. J. (2022). Bacterial communities in surface and basal ice of a glacier terminus in the headwaters of Yangtze river on the qinghai-Tibet plateau. Environ. Microbiome 17 (1), 12. doi: 10.1186/s40793-022-00408-2
Ren Z., Niu D., Ma P., Wang Y., Wang Z., Fu H., et al. (2020). Bacterial communities in stream biofilms in a degrading grassland watershed on the qinghai-Tibet plateau. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01021
Ren Z., Qu X., Zhang M., Yu Y., Peng W. (2019). Distinct bacterial communities in wet and dry seasons during a seasonal water level fluctuation in the largest freshwater lake (Poyang lake) in China. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01167
Rogers K., Kelleway J. J., Saintilan N., Megonigal J. P., Adams J. B., Holmquist J. R., et al. (2019). Wetland carbon storage controlled by millennial-scale variation in relative sea-level rise. Nature 567 (7746), 91–95. doi: 10.1038/s41586-019-0951-7
Rosentreter J. A., Wells N. S., Ulseth A. J., Eyre B. D. (2021). Divergent gas transfer velocities of CO2, CH4, and N2O over spatial and temporal gradients in a subtropical estuary. J. Geophysical Research-Biogeosciences 126 (10), e2021JG006270. doi: 10.1029/2021JG006270
Schloss P. D. (2009). A high-throughput DNA sequence aligner for microbial ecology studies. PloS One 4 (12), e8230. doi: 10.1371/journal.pone.0008230
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12 (6), R60. doi: 10.1186/gb-2011-12-6-r60
She Z., Pan X., Wang J., Shao R., Wang G., Wang S., et al. (2021). Vertical environmental gradient drives prokaryotic microbial community assembly and species coexistence in a stratified acid mine drainage lake. Water Res. 206, 117739. doi: 10.1016/j.watres.2021.117739
Shi S., Nuccio E. E., Shi Z. J., He Z., Zhou J., Firestone M. K. (2016). The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 19 (8), 926–936. doi: 10.1111/ele.12630
Stegen J. C., Lin X., Fredrickson J. K., Chen X., Kennedy D. W., Murray C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7 (11), 2069–2079. doi: 10.1038/ismej.2013.93
Sun B., Liu J. L., Meng B., Bao K. (2020). Structural variability and Co-occurrence pattern differentiation in rhizosphere microbiomes of the native invasive plant echinochloa caudate in momoge national nature reserve, China. Wetlands 40 (3), 587–597. doi: 10.1007/s13157-019-01209-z
Tee H. S., Waite D., Lear G., Handley K. M. (2021). Microbial river-to-sea continuum: gradients in benthic and planktonic diversity, osmoregulation and nutrient cycling. Microbiome 9 (1), 190. doi: 10.1186/s40168-021-01145-3
Tian J., He N. P., Kong W. D., Deng Y., Feng K., Green S. M., et al. (2018). Deforestation decreases spatial turnover and alters the network interactions in soil bacterial communities. Soil Biol. Biochem. 123, 80–86. doi: 10.1016/j.soilbio.2018.05.007
Vasar M., Davison J., Sepp S. K., Mucina L., Oja J., Al-Quraishy S., et al. (2022). Global soil microbiomes: A new frontline of biome-ecology research. Global Ecol. Biogeography 31 (6), 1120–1132. doi: 10.1111/geb.13487
Vellend M. (2010). Conceptual synthesis in community ecology. Q Rev. Biol. 85 (2), 183–206. doi: 10.1086/652373
Vieira S., Sikorski J., Dietz S., Herz K., Schrumpf M., Bruelheide H., et al. (2020). Drivers of the composition of active rhizosphere bacterial communities in temperate grasslands. ISME J. 14 (2), 463–475. doi: 10.1038/s41396-019-0543-4
Wang Y., Liu L., Chen H., Yang J. (2015). Spatiotemporal dynamics and determinants of planktonic bacterial and microeukaryotic communities in a Chinese subtropical river. Appl. Microbiol. Biotechnol. 99 (21), 9255–9266. doi: 10.1007/s00253-015-6773-0
Wang S., Wang X., Jiang Y., Han C., Jetten M. S. M., Schwark L., et al. (2021). Abundance and functional importance of complete ammonia oxidizers and other nitrifiers in a riparian ecosystem. Environ. Sci. Technol. 55 (8), 4573–4584. doi: 10.1021/acs.est.0c00915
Wang F. F., Yan J. G., Ma X., Qiu D. D., Xie T., Cui B. S. (2019). Tidal regime influences the spatial variation in trait-based responses of suaeda salsa and edaphic conditions. Ecosphere 10 (3), e02642. doi: 10.1002/ecs2.2642
Weinke A. D., Biddanda B. A. (2018). From bacteria to fish: Ecological consequences of seasonal hypoxia in a great lakes estuary. Ecosystems 21 (3), 426–442. doi: 10.1007/s10021-017-0160-x
Wu B., Tian J., Bai C., Xiang M., Sun J., Liu X. (2013). The biogeography of fungal communities in wetland sediments along the changjiang river and other sites in China. ISME J. 7 (7), 1299–1309. doi: 10.1038/ismej.2013.29
Xu H., Zhang S., Ma G., Zhang Y., Li Y., Pei H. (2020). 18S rRNA gene sequencing reveals significant influence of anthropogenic effects on microeukaryote diversity and composition along a river-to-estuary gradient ecosystem. Sci. Total Environ. 705, 135910. doi: 10.1016/j.scitotenv.2019.135910
Yang Y., Ji C., Ma W., Wang S., Wang S., Han W., et al. (2012). Significant soil acidification across northern china's grasslands during 1980s-2000s. Global Change Biol. 18 (7), 2292–2300. doi: 10.1111/j.1365-2486.2012.02694.x
Yuan J. J., Xiang J., Liu D. Y., Kang H., He T. H., Kim S., et al. (2019). Rapid growth in greenhouse gas emissions from the adoption of industrial-scale aquaculture. Nat. Climate Change 9 (4), 318. doi: 10.1038/s41558-019-0425-9
Yu L., Bai J. H., Huang L. B., Zhang G. L., Wang W., Wang X., et al. (2022). Carbon-rich substrates altered microbial communities with indication of carbon metabolism functional shifting in a degraded salt marsh of the yellow river delta, China. J. Cleaner Production 331, 129898. doi: 10.1016/j.jclepro.2021.129898
Yu X. L., Yang X. Q., Wu Y. J., Peng Y. S., Yang T., Xiao F. S., et al. (2020). Sonneratia apetala introduction alters methane cycling microbial communities and increases methane emissions in mangrove ecosystems. Soil Biol. Biochem. 144, 107775. doi: 10.1016/j.soilbio.2020.107775
Zhang L., Delgado-Baquerizo M., Shi Y., Liu X., Yang Y., Chu H. (2021b). Co-Existing water and sediment bacteria are driven by contrasting environmental factors across glacier-fed aquatic systems. Water Res. 198, 117139. doi: 10.1016/j.watres.2021.117139
Zhang C., Du X. P., Zeng Y. H., Zhu J. M., Zhang S. J., Cai Z. H., et al. (2021a). The communities and functional profiles of virioplankton along a salinity gradient in a subtropical estuary. Sci. Total Environ. 759, 143499. doi: 10.1016/j.scitotenv.2020.143499
Zhang S., Li K. X., Hu J. M., Wang F., Chen D. H., Zhang Z. J., et al. (2022). Distinct assembly mechanisms of microbial sub-communities with different rarity along the nu river. J. Soils Sediments 22 (5), 1530–1545. doi: 10.1007/s11368-022-03149-4
Zhang X., Qi L., Li W., Hu B. X., Dai Z. (2021c). Bacterial community variations with salinity in the saltwater-intruded estuarine aquifer. Sci. Total Environ. 755 (Pt 1), 142423. doi: 10.1016/j.scitotenv.2020.142423
Zhong Y., Sorensen P. O., Zhu G., Jia X., Liu J., Shangguan Z., et al. (2022). Differential microbial assembly processes and co-occurrence networks in the soil-root continuum along an environmental gradient. iMeta 1 (2), e18. doi: 10.1002/imt2.18
Zhou J., Ning D. (2017). Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 81 (4), e00002–17. doi: 10.1128/MMBR.00002-17
Zhou L., Wang P., Huang S., Li Z., Gong H., Huang W., et al. (2021). Environmental filtering dominates bacterioplankton community assembly in a highly urbanized estuarine ecosystem. Environ. Res. 196, 110934. doi: 10.1016/j.envres.2021.110934
Keywords: estuarine ecosystem, bacterial diversity, carbon cycle, assembly processes, co-occurrence network
Citation: Ma K, Ren Z, Ma J, Chen N and Liu J (2022) Differential patterns and assembly processes of bacterial communities from distinct microhabitats in a subtropical estuary. Front. Mar. Sci. 9:1039387. doi: 10.3389/fmars.2022.1039387
Received: 08 September 2022; Accepted: 27 October 2022;
Published: 11 November 2022.
Edited by:
Milko Alberto Jorquera, University of La Frontera, ChileReviewed by:
Jia Jia, Yellow River Institute of Hydraulic Research, ChinaShanze Li, China Institute of Water Resources and Hydropower Research, China
Copyright © 2022 Ma, Ren, Ma, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingling Liu, amluZ2xpbmdAYm51LmVkdS5jbg==