 Sebastian G. Gornik1*†
Sebastian G. Gornik1*† Ira Maegele1
Ira Maegele1 Elizabeth A. Hambleton1†Philipp A. Voss1
Elizabeth A. Hambleton1†Philipp A. Voss1 Ross F. Waller2Annika Guse1*†
Ross F. Waller2Annika Guse1*†- 1Centre for Organismal Studies, Heidelberg University, Heidelberg, Germany
- 2Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom
Dinoflagellates are a diverse and ecologically important group of single-celled eukaryotes. Many are photosynthetic autotrophs while others are predatory, parasitic, or symbiotic. One major group — the Symbiodiniaceae — is well known for its role as coral symbionts that provide the coral host with vital nutrients. While genetic transformation protocols have been published for some non-symbiotic dinoflagellate species, robust methods for genetic manipulation of coral symbionts are lacking, hindering a detailed molecular understanding of this critical symbiotic interaction. Here, we describe the successful transformation of coral symbiont Breviolum minutum (strain SSB01). Using Golden Gate modular plasmid assembly and electroporation, we drove transient NLS-GFP expression from an endogenous dinoflagellate virus nuclear protein (DVNP) promoter and successfully targeted GFP to the dinoflagellate nucleus. We further determined that puromycin can efficiently select transformed cells using the puromycin N-acetyltransferase (pac) resistance gene. Transformed cells could be maintained under antibiotic selection for at least 12 months without losing resistance, albeit with slowly attenuating fluorescence signal. We thus tested the expression of hybrid GFP-2A-PAC polypeptides under the control of a single promoter sequence to overcome loss of fluorescence, but lack of efficient 2A cleavage seemingly hindered antibiotic selection interfering GFP function. Despite this, our transformation approach now allows unanswered questions of dinoflagellate biology to be addressed, as well as fundamental aspects of dinoflagellate-coral symbiosis.
Introduction
Dinoflagellates are highly successful single-celled eukaryotes that represent one of the most abundant and species-rich groups of single-celled phytoplankton in the sunlit layers of all oceans on planet Earth (de Vargas et al., 2015). The group evolved 245-208 million years ago, and today, comprises at least 2,400 extant species (Hackett et al., 2004; Janouškovec et al., 2017) of which approximately fifty percent are free-living and photosynthetic, while the remaining can be symbiotic, parasitic, or predatory (Hackett et al., 2004; Janouškovec et al., 2017).
Despite the diversity seen across the group, all dinoflagellates share a number of specialized cellular features that set them apart from other organisms. For example, they often possess gargantuan genomes with up to 185 Gb of DNA (Wisecaver and Hackett, 2011) and, fascinatingly, can manage these massive genomes without the classical use of histones. Instead, dinoflagellates utilize a non-nucleosomal system for nuclear DNA packaging comprising novel proteins that are of viral and bacterial origin called dinoflagellate viral nucleoproteins (DVNPs) and histone-like proteins (HLPs), respectively (Wisecaver and Hackett, 2011; Gornik et al., 2019). Furthermore, in comparison to other eukaryotes, higher-order analysis of the genetic and spatial organization of dinoflagellate genomes reveals excess non-coding DNA (Wisecaver and Hackett, 2011; Aranda et al., 2016; Gornik et al., 2019) coupled with an unusual enrichment of genes near the ends of chromosomes. The genes themselves are organized in alternating, unidirectional blocks with a marked correlation between gene orientation, transcription, and chromosome folding (Marinov et al., 2021; Nand et al., 2021). Additionally, dinoflagellates exhibit trans-splicing of a conserved spliced leader into mRNAs. This includes polycistronic mRNAs that are presumably processed into individual transcripts following transcription (Zhang et al., 2007; Shoguchi et al., 2013; Roy et al., 2018). Dinoflagellate genomes show limited regulation at a transcriptional level suggesting that much of the control of protein expression levels occurs post-transcriptionally (Akbar et al., 2018). The mitochondrial and plastid genomes of dinoflagellates are unusually reduced and often fragmented with complex transcript modifications (Lin, 2011; Wisecaver and Hackett, 2011; Jackson et al., 2012a; Jackson et al., 2012b). Dinoflagellate plastids are derived through secondary, tertiary and, in some cases, quaternary endosymbiosis (Archibald, 2009; Keeling, 2010; Waller and Koreny, 2017) and their photosynthesis relies on unique light-harvesting complexes (Carbonera et al., 2014).
Despite the abundance of interesting features and ecological relevance of dinoflagellates, we currently lack a detailed understanding of most major aspects of the cell biology of these important organisms. This is largely due to the fact that the vast majority of dinoflagellates are understudied and experimental molecular tools, including genetic transformation, are rudimentary, not yet robust and exist only for a few select species (Fernando Ortiz-Matamoros et al., 2015; Ortiz-Matamoros et al., 2015; Nimmo et al., 2019; Sakamoto et al., 2019; Faktorová et al., 2020; Sprecher et al., 2020; Einarsson et al., 2021). Owing to their ecological importance as the drivers of both the trophic and structural foundation of coral reef ecosystems, efforts to establish genetic transformation methods for a representative dinoflagellate symbiont of corals from the family Symbiodiniaceae started 25 years ago. Apparent success was reported in 1997 by Lohuis and Miller for S. microadriaticum (strain CS-153) using silicon carbide whiskers for transgene delivery. Expression of the reporter gene β-glucuronidase (GUS) and genes to infer resistance to hygromycin and geneticin, used as selectable markers, were driven by the heterologous promoters from Cauliflower Mosaic Virus (35S) and Agrobacterium tumefaciens nopaline synthase (nos), as well as by the bi-directional p1’2’ promoter. Subsequently, Ortiz-Matamoros et al. (2015) described the transformation of Fugacium kawagutii (formerly: Symbiodinium kawagutii), Symbiodinium sp. strain Mf11.5b.1, and Symbiodinium microadriaticum (strain MAC-CassKB8) using similar approaches. Specifically, they used glass bead agitation in the presence or absence of Agrobacterium using the heterologous ‘nos’ promoter from A. tumefaciens to drive the expression of BAR (bialaphos resistance), which confers resistance to glufosinate (also known as phosphinothricin), the active ingredient in the herbicide Basta® (BASF). However, both methods have not been able to be repeated by others (Chen et al., 2019) and for Ortiz-Matamoros et al. GFP expression was short-lived and cells did not grow or divide afterwards. Chen et al. (2019) and Faktorová et al. (2020) systematically tested various standard methods including the previously published methods comprising electroporation, biolistics (gene gun), and agitation with glass beads to transform S. microadriaticum (strain CCMP2467) and Breviolum minutum (strain NIES-4271). They tried the previously used S35 promoter, as well as a number of putative endogenous promoters (actin, tubulin A, tubulin B, Hsp90, psbJ, psbA) and terminator regions (Hsp90, actin) identified from the corresponding S. microadriaticum genome in attempts to express the chloramphenicol resistance gene. Despite all efforts, drug-resistent Symbiodiniaceae dinoflagellates were not obtained (Chen et al., 2019). Thus, to date, a robust and reproducible coral symbiont transformation method that could ultimately be utilized to analyze the molecular mechanisms of the dinoflagellate-coral symbiosis is still lacking.
Here, we report the stable genetic transformation of a coral symbiont that belongs to the Symbiodiniaceae family within the genus Breviolum (strain SSB01) (Xiang et al., 2013). SSB01 originates from the sea anemone Aiptasia (Exaiptasia diaphana, line H2), a model organism to investigate dinoflagellate-coral symbiosis (Baumgarten et al., 2015; Grawunder et al., 2015; Rädecker et al., 2018); however, it also readily establishes symbiosis with other anemone strains and reef-building corals (Xiang et al., 2013; Hambleton et al., 2014; Wolfowicz et al., 2016). It grows quickly in isolation and serves as a representative endosymbiont in many experimental studies (Xiang et al., 2013; Hambleton et al., 2014; Xiang et al., 2015; Hambleton et al., 2019). Thus, SSB01 is well-suited for experimental genetic studies. Using an adapted modular Golden Gate vector system (Engler et al., 2014; Einarsson et al., 2021) and electroporation we introduced several plasmid DNAs into SSB01 cells and successfully expressed GFP which we targeted to the cell nucleus using an intrinsic DVNP-derived nuclear localization signal (Gornik et al., 2012). We achieved stable transformation with constructs designed to confer puromycin resistance via PAC expression, with cultures surviving at least 12 months under antibiotic selection. With long-term maintenance of cultures, we found diminishing GFP fluorescence, so we also explored the application of 2A peptides to express an NLS-eGFP-PAC hybrid polyprotein. This is the first report of successful genomic transformation in a member of the Breviolum genus of dinoflagellates and a symbiont of corals. Ultimately, this work has established reliable genetic tools that can be applied to the investigation of the cellular and molecular mechanisms underpinning the dinoflagellate-coral symbiosis.
Material and methods
Algal cell culture
Breviolum sp. (strain SSB01 (Xiang et al., 2013)), Symbiodinium sp. (strains SSA01 (Xiang et al., 2013), SSA02 and SSA03 (Xiang et al., 2013)), Effrenium voratum (strain SSE01 (Xiang et al., 2013); CCMP421), Cladocopium sp. (Clade C; CCMP2466) and Durusdinium sp. (Clade D; CCMP2556) cells were grown in cell culture flasks in 0.22 μm filter-sterilized 1X Diago’s IMK medium (#392-01331, Wako Pure Chemical Corporation) at 26°C on a 12h light/12h dark cycle under 20–25 μmol m−2 s−1 of photosynthetically active radiation (PAR), as measured with an Apogee PAR Quantum meter (#MQ-200; Apogee).
Algal staining and microscopy
Approximately 1.0 x 106 alga cells were fixed in 4% paraformaldehyde for 15 minutes, pelleted at 10,000 x g, resuspended in PBS-T (0.2% (v/v) Triton X-100) and incubated for 2 hours. Following this, cells were washed in PBS twice and resuspended in 500 µl PBS before addition of 1 µl of 10% KOH in H2O and 50 µl of Calcofluor White stain (1mg ml-1 Calcofluor White, 0.5 mg ml-1 Evans blue; #18909, Sigma Aldrich). Cells were incubated at room temperature (RT) for 15 minutes and washed three times using 500 µl PBS per wash. To stain cellular DNA the second wash contained an additional 10 µl of 0.1 mg ml-1 DAPI (4’,6-Diamidin-2-phenylindol) and was incubated for 20 minutes. Cells were mounted in 10 µl of molten (45°C) glycerol jelly (2.5 g gelatin, 15 ml H2O, 17 ml glycerol and 10 µl phenol) and stored at RT prior to microscopy using a Leica TCS SP8 confocal microscope with a 63x glycerol immersion objective (numerical aperture = 1.30). Images were recorded using Leica LAS X software and processed using imageJ/Fiji. DAPI/calcofluor and plastid autofluorescence were excited with 405- and 633-nm laser lines, respectively. Fluorescence emission was detected at 410-499 nm for DAPI/calcofluor and 645-741 nm for plastid autofluorescence.
Genomic DNA extractions
Genomic DNA was extracted from approximately 1.0x105 algal cells using a DNeasy blood and tissue spin-column kit (#69504, Qiagen) following manufacturer instructions.
Phylogenies
To generate LSU rDNA phylogenies, we extracted genomic DNA from SSA01, SSA02, SSA03, SSB01, SSE01, Clade C, and Clade D cells as described above and amplified their LSU rDNA sequences by PCR (for primers see: Supplementary File 1) using Q5® high-fidelity DNA polymerase (#M0491S, NEB). Blunt-end amplicons were then gel extracted using a GeneJET PCR purification kit (#K0701, Thermo Fisher), ligated into pJET1.2 using a CloneJet PCR cloning kit (#K1231, Thermo Fisher), and, following CloneJet plasmid Miniprep extraction (#K0502, Thermo Fisher), sequenced by Sanger method using universal primers such a T7-forward supplied by the sequencing vendor (Eurofins Genomics). Per strain/species we sequenced at least 5 replicates and generated a consensus sequence per strain/species. We then re-aligned these consensus sequences with published alignments (LaJeunesse et al., 2018) using MUSCLE (Edgar, 2004) to then calculate phylogenies using IQ-TREE 2 (Minh et al., 2020) at standard settings. The resulting tree was finalized using FigTree 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and Adobe Illustrator CS6. Original tree files with accession numbers are provided (Supplementary File 2).
SSB01 transcriptome assembly and assessment of completeness
Using publicly available reads for Breviolum sp. SSB01 (BioProject PRJNA591730) we de-novo assembled a full transcriptome. To this end the available paired-end reads were first QC filtered, adapter-trimmed and deduplicated using fastq-mcf (https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md; last accessed 13/09/2021) prior to trinity assembly (Grabherr et al., 2011) using the following settings: Trinity –no_normalize_reads –trimmomatic –seqType fq –max_memory 50G –CPU 64. Using cegma (Parra et al., 2007) the resulting transcriptome was compared for completeness to an existing transcriptome assembly that was generated from the same raw read data (Xiang et al., 2015).
Sample collection, library preparation, sequencing, transcriptome assembly and quantitation of expression
At 6-7 days post-fertilization approximately 300 ml−1 larvae were infected for 24-48h with 1.0x105 ml−1 symbionts or left uninfected. Per replicate, three to five larvae were transferred to 5ml calcium- and magnesium-free artificial seawater (CMF-ASW; https://doi.org/10.1101/pdb.rec12053) and incubated for 5 minutes as described (Jacobovitz et al., 2021). Following this, the larvae were dissociated in 70μl Pronase (0.5% in CMF-ASW; #10165921001, Sigma Aldrich) and sodium thioglycolate (1% in CMF-ASW; #T0632, Sigma Aldrich) for approximately 2 minutes to remove ectodermal cells after pipetting up and down larvae three to five times. The endoderm was then transferred to 70μl FASW and residual ectodermal cells were mechanically removed. Endodermal cells were separated using tweezers, and pools of 7-20 cells (either symbiotic or aposymbiotic cells from symbiotic animals) were picked using microcapillary needles (#GB100T-8P, Science Products) with an opening diameter of 8-12 μm (Micropipette Puller P-97, Sutter Instrument). Capillaries were prefilled with 4.3 μl lysis buffer (0.2% Triton X-100, 1 U μl−1 Protector RNase Inhibitor (#333539001; Sigma Aldrich), 1.25 μM oligo-dT30VN and 2.5 mM dNTP mix). Cells were flushed out of the capillary with additional lysis buffer and flash frozen. Sequencing libraries were prepared by RNA reverse transcription and pre-amplification of complementary DNA of 21 PCR cycles, as described (Picelli et al., 2014); and prepared and sequenced using a NextSeq 500 (Illumina) generating 75-base pair paired-end reads. Resulting reads were deposited at NCBI’s SRA with the following accession: SRP229372. Paired-end reads were QC filtered, adapter-trimmed and deduplicated using fastq-mcf and mapped to the newly generated SSB01 transcriptome (see above) using HISAT2 version 2.1.0 with default settings (except –X 2000 –no-discordant –no-unal –no-mixed). Transcript expression was quantified in Trinity version 2.5.1 using salmon version 0.10.2 with default settings for read data from in-hospite and cultured symbionts generated in this study and for read data from cultured symbionts from Xiang et al. (2015). Principal component analysis was conducted using a perl script supplied with Trinity for all samples. Differential expression was analysed using DESeq2 (log2 [fold change] ≥ 1; adjusted P ≤ 0.05). A custom KNIME workflow was generated to process and analyse the data. The R package ComplexHeatmap was used to generate expression heatmaps of HLP and DVNP transcripts.
Promoter identification and RNA read mapping
The DVNP.17 promoter sequence was identified in the Breviolum minutum genome using blastn with the DVNP.17 CDS as query against an in-house database in Geneious 8 (Biomatters). RNA reads were mapped using bowtie2 at standard settings. Alignments and coverage data of uniquely mapped reads were extracted using samtools and visualized in Geneious 8. The TATA- and TTTT-boxes were annotated manually. Annotation of polyA-sites and a spliced leader transplicing site was based on mapping data.
Antibiotics growth assays
Glufosinate-ammonium (BASTA/PESTANAL®; 45520, Sigma-Aldrich) and puromycin (#0240, Carl Roth) growth assays were carried out in IMK medium. Initially 50 and 100 µg ml-1 concentrations of both antibiotics were tested. Here, SSB01 cells in exponential growth were split to approximately 1.2 -1.5 x 105 cells in 15 ml cultures and cell numbers were measured at the start of the experiment, on day 1, 4, 9 and 14 using an automated cell counter (TC20, BioRad) with careful mixing before pipetting. All cultures were measured in duplicate. For long-term exposure experiments puromycin was added to a concentration of 100 µg/ml. Control cultures without puromycin were prepared accordingly without antibiotics. Two puromycin-treated and two control cultures were kept in humid chambers to avoid evaporation and were grown under standard conditions. Cell growth was measured once a week using an automated cell counter (TC20, BioRad) with careful mixing before pipetting. All cultures were measured in duplicate. Cultures were kept for 8 weeks without splitting.
Golden Gate cloning
Golden Gate cloning and domestication (removal of internal BsaI and BpiI sites) was performed as previously described (Engler et al., 2014; Einarsson et al., 2021). For site directed mutagenesis of BsaI and BpiI sites a Q5® Site-Directed Mutagenesis kit (#E0554S, NEB) was used. All PCR and mutagenesis primers are listed in Supplementary File 1 and resulting plasmids for SSB01 transformation are provided as fasta file in Supplementary File 3. Genomic DNA from SSB01 was extracted as described above and used as template for PCRs of promoter, terminator and NLS sequences. Templates for H2B, 2A-pac and eGFP were synthesized as gblocks™ (IDT, Belgium; Supplementary File 1). Level 0 plasmids were generated by CloneJet PCR cloning kit as described above. All plasmids were Sanger sequenced following CloneJet plasmid Miniprep extraction (#K0502, Thermo Fisher). Where necessary, for different constructs, overhangs were adjusted using specific primers via PCR (Supplementary File 1). Golden Gate restriction-ligation reactions for level M construction were performed in 0.2 mL tubes with 200 ng of acceptor vector, 400 ng of each insert DNA component, 400 U T4 DNA ligase (#M0202, NEB), 2 ml 10 mM ATP, 10 U BsaI (#ER0292, Thermo Fisher), 2 µl of Buffer G (#BG5, Thermo Fisher) and sterile H2O in 20 µl. The reactions were performed in a BioRad S1000 thermocycler as follows: 3 cycles of 10 minutes at 37°C and 10 minutes at 16°C, followed by final incubation at 37°C for 10 minutes and then 20 minutes at 65°C. For assembly of level 1 constructs the reaction contained 20 U BpiI, (#ER1011, Thermo Fisher) and 2 µl CutSmart Buffer (#7204S, NEB) instead of 10 U BsaI and 2 µl of Buffer G. The incubation cycles were: 3 cycles of 10 minutes at 40°C and 10 minutes at 16°C followed by final incubation at 50°C for 10 minutes and then 20 minutes at 80°C. The reaction mixture was added to DH5a competent cells (#C2987H, NEB) and heat shock transformation performed according to the manufacturer’s recommendation. Transformed cells were selected on LB plates containing the appropriate antibiotic (ampicillin for level 0 (pJET2.1), kanamycin for level 1 and ampicillin for level M), and IPTG and X-gal for blue-white screening. All Golden Gate modules and final constructs used in this study are available upon request.
Transformation
SSB01 cells in exponential growth were split (1:1) three days before transformation. Per transfection reaction, approximately 1.0 x 106 cells were spun down at 5,000 rpm for 10 minutes at 26°C. The supernatant was carefully removed and cells were resuspended in 100 µl of transfection solution (Nucleofector™ Basic Solution 1 for parasites with supplement 1), which also contained the transfection vectors (10 µg per vector; at approximately 4-5 µg µl-1) and 15 µg of the bacterial cloning vector pUC19 (at approximately 5 µg µl-1) as carrier plasmid that was added to assist transformation as described (Faktorová et al., 2020). Cells were transferred to electroporation cuvettes and electroporated using program D-023 on a LONZA Nucleofector™ 2b device. Following electroporation cells were immediately transferred evenly across 4 x 1 ml fresh IMK on a 6-well plate and incubated at 26°C in the dark. After 1 hour, 1 µl of ampicillin (1 mg ml-1) was added per well. Cells were then grown at 26°C in a humid chamber under standard conditions. When using antibiotic selection, puromycin was added to a final concentration of 100 µg ml-1 using 1 ml of 200 µg ml-1 puromycin per 1 ml of existing cell culture 36 hours post transformation. After a 1-week incubation cells were transferred from the 6-well plates to cell culture flasks and the culture volume increased to 10 ml. Cell cultures were split (1:1) regularly every 14-21 days thereafter in either IMK or IMK containing 100 µg ml-1 of puromycin when grown under selection.
Live microscopy of transformed cells
For live imaging transformed cells were incubated with 10 µl of 0.1 mg ml-1 DAPI for 10 minutes to stain for cellular DNA. The cells were then spun down at 5,000 rpm and, following removal of the supernatant, mounted in 10 µl of molten (45°C) glycerol jelly. Cells were immediately imaged as described above for DAPI and plastid auto-fluorescence. Additionally, the GFP signal was excited with a 488-nm laser line and fluorescence emission was detected at 501-550 nm.
RNA extraction, cDNA synthesis, PCR amplification and sequencing for transgenesis confirmation
RNA was extracted from 1.0x105 transformed SSB01 cells using an RNeasy mini spin-column kit (#74004, Qiagen) following manufacturer instructions. A total of 1 µg of total RNA was used to generate cDNA using a ReadyScript® cDNA-synthesis kit (#RDRT, Sigma Aldrich). Resulting cDNA was used as template for PCRs using pac- and/or GFP-specific primer pairs (Supplementary File 1). Blunt-end amplicons were gel extracted, cloned and sequenced by Sanger method as described above.
Protein extraction, SDS-PAGE, Coomassie-staining and anti-PAC Western blots
Approximately 0.5x106 SSB01 cells were pelleted at 16.000 x g for 10 minutes and resuspended in 50 µl of 1X Laemli buffer (50 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 0.005% bromophenol blue) by careful pipetting up and down several times. Re-suspended samples were then incubated at 100°C for 10 minutes in an Eppendorf thermo block under constant shaking at 1,000 rpm. Per sample 15 µl of extract were loaded and run on a 10% Bis-Tris SDS-polyacrylamide gel at 80 V for 15 minutes and then 120 V until about 1 cm before the loading front ran out of the gel. Gels were either stained using Coomassie or subjected to Western blotting. Coomassie staining was performed as described (Yan, 2011). Western blot transfer onto nitrocellulose was performed at 120 V for 90 minutes in Towbin buffer (25 mM Tris, 192 mM glycine, pH 8.3, 20% methanol (v/v)). Successful transfer was confirmed using Ponceau S staining (0.1% Ponceau S in H2O (v/v)). The membrane was then blocked overnight in 5% milk in PBS-T (0.1% Triton-X100). A commercial recombinant polyclonal rabbit anti-PAC antibody (#1HCLC, Invitrogen) was used at a 1:500 dilution in 1% milk in PBS-T and incubated at RT for 2 hours or at 4°C overnight. The membrane was washed 3 x 10 minutes in PBS-T. Peroxidase-coupled AffiniPure goat anti-rabbit IgG antibody (#111-035-144, Jackson ImmunoResearch) was used at a 1:5000 dilution in 1% milk in PBS-T and incubated for 30-60 minutes at RT. Following a 3 x 10 minutes wash in PBS-T the membrane was transferred to a plastic sheet, subjected to 150 µl of SuperSignal™ West Pico Chemiluminescent Substrate (#34579, Thermo Fisher) and visualized on an ECL Chemocam Imager (INTAS).
Results & discussion
Identification in the phylogenetic position of the dinoflagellate symbiont model strain SSB01 and selection of strong and functional promoter and terminator sequences
Several Symbiodiniaceae species, including members of the genera Symbiodinium, Durusdinium, Gerakladium, and Breviolum, can establish symbiosis with a number of protists, corals, and sea anemones in nature (LaJeunesse et al., 2018). Common clonal strains cultivated for use in the laboratory are SSA01, SSA02, SSA03, SSB01, SSE01, as well as Clade C and Clade D (Xiang et al., 2013; Hambleton et al., 2014; Wolfowicz et al., 2016). As the basis for genomic analyses and promoter identification, we aimed to match those strains to available, sequenced genomes by using existing LSU rDNA-based phylogenetic analyses. Specifically, we determined the LSU rDNA sequences for the SSA01, SSA02, SSA03, SSB01, SSE01, Clade C, and Clade D strains and added their sequences to existing alignments (LaJeunesse et al., 2018). We then re-calculated the corresponding phylogenetic tree. This revealed that the closest available genomic resource for SSB01 is represented by Breviolum minutum CCMP2460 (Figure 1A). Our subsequent analyses focused on SSB01 as it is a commonly used endosymbiont model for dinoflagellate-coral symbiosis that grows relatively quickly in isolation from its host.
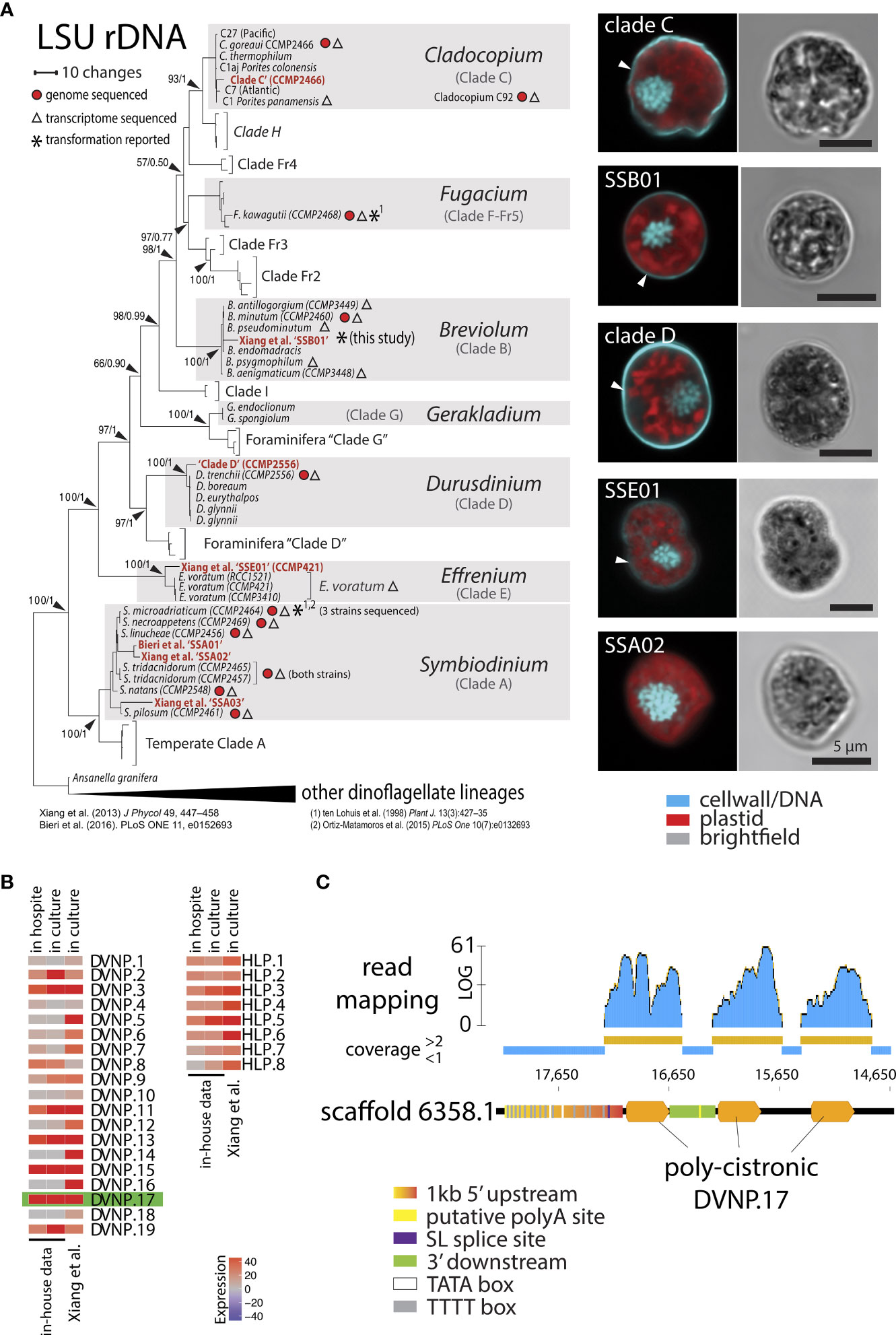
Figure 1 (A) Phylogeny and morphological presentation of laboratory strains used in this study. Cell nuclei were stained with DAPI and cell walls with Calcofluor White. (B) Heat map showing the TPM (Transcripts per million) expression of chromatin proteins DVNP.1-19 and HLP-I.1-8 from Breviolum sp. ‘SSB01’ in culture and in hospite from in-house and public RNA-seq data (Xiang et al, 2015). (C) Overview of the genomic locus of DVNP.17 depicting regulatory elements and mapped RNA-reads seemingly showing the poly-cistronic nature of the gene body.
The selection of promoter sequences plays a significant role in transgene expression, a task that is complex in dinoflagellates. Due to the occurrence of trans-splicing in dinoflagellates, the identification of pre-mRNA UTRs and transcription start sites is not straightforward. Moreover, the promoter regions of multi-exonic genes of dinoflagellates are difficult to recognize as dinoflagellate genes exist in both multiple gene copy arrays and tandem gene arrays (Lin et al., 2015; Mendez et al., 2015; Nand et al., 2021). Thus, we aimed to identify functionally-important, highly-abundant, small, and preferably single exon genes that are highly expressed in SSB01, rather than relying on commonly-used heterologous viral promoters or putative promoters of endogenous housekeeping genes as were previously used for Symbiodiniaceae (Fernando Ortiz-Matamoros et al., 2015; Ortiz-Matamoros et al., 2015; Chen et al., 2019).
Based on existing transcriptomic data from multiple dinoflagellate species, it is known that genes for the DVNP and HLP families of nuclear proteins are among the most highly-expressed non-organelle, non-ribosomal, small, single exon genes (Gornik et al., 2012; Marinov and Lynch, 2016). We therefore used publicly available RNA sequencing reads for SSB01 to newly generate a de novo transcriptome assembly (Supplementary File 4). This resulted in a more complete data set (89.92% versus 86.69% completeness) than the previously published transcriptome (Xiang et al., 2015) as indicated by CEGMA assessment (Supplementary File 5). Following read-mapping and quantification of expression, we identified several DVNP and HLP candidates among the top 500 expressed genes in SSB01 (data not shown). In total, we identified 8 HLP and 19 DVNP genes in the SSB01 transcriptome (Supplementary File 6) with the number of DVNPs being consistent with the 24 DVNPs reported for B. minutum (Marinov and Lynch, 2016). Utilizing both published and in-house single-cell RNA sequencing data, which allowed us to compare two lifestyle states of symbionts: free-living (i.e., in culture) and in hospite, we found that DVNP.17 was the highest expressed DVNP in both cultured and in hospite symbionts (Figure 1B). Next, we queried the publicly available genome of Breviolum minutum (BioProject PRJDB732) for the genomic locus of DVNP.17. We reasoned that its 5’ upstream sequence was likely to represent a strong promoter because it only exists at a single genomic locus rather than as multiple copies across the genome. Indeed, we found only a single locus for DVNP.17 in the available genomic resource for B. minutum. Interestingly, the genomic locus for DVNP.17 seems to consist of a poly-cistronic array, where a poly-cistronic pre-mRNA is presumably split by trans-splicing into three mono-cistronic, mature mRNAs after transcription (Figure 1C). This observation is consistent with the proposed mechanism for the function of the dinoflagellate spliced leader (Zhang et al., 2007). We therefore presumed the DVNP promoter region to be 1 kb upstream of the start codon of the first DVNP.17 open reading frame (ORF). The 3’ terminator region was defined as the region downstream of the stop codon of the first DVNP.17 and upstream of the start codon of the next DVNP ORF, resulting in a 3’ regulatory sequence with a length of 268 bp. By comparing transcript sequences and the putative promoter, we were able to identify the primary 5’ spliced leader (SL) splice site (Figure 1C). Upstream of this site, we also identified two classic, eukaryotic TATA-boxes. Interestingly, the putative promoter region also contains 14 TTTT-motifs upstream of the SL splice site. According to functional data from Crypthecodinium cohnii and analysis of the Fugacium kawagutii genome content in dinoflagellates, such TTTT motifs may act as strong, alternative TATA-binding protein binding sites, allowing transcription initiation in a non-TATA-box manner (Guillebault et al., 2002; Lin et al., 2015). Thus, irrespective of whether TATA or TTTT motifs are preferred transcription initiation sites in Breviolum ‘SSB01,’ the promoter we identified has putative regulatory sequences that would satisfy both conditions. Lastly, transcript-to-genome comparison showed that the 3’ terminator region contained a putative poly-A signal and poly-A site (Figure 1C).
Golden Gate modular plasmid assembly for generating SSB01 transformation vectors
We utilized Golden Gate modular plasmid assembly to develop a highly versatile genetic modification method for SSB01 transformation (Figure 2A). Such a modular assembly method allows multiple genes to be expressed simultaneously from a single expression vector. To start, ‘level 0’ plasmids carrying inserts of interest flanked by BsaI (a Type IIS restriction enzyme) sites with adjacent specific overhangs are cloned to direct the order of module assembly (Figure 2A). Following this, modules of interest can be assembled as ‘scarless’ (lacking the original restriction enzyme recognition site) expression units into ‘level 1’ acceptor plasmids using the type IIS restriction enzyme BsaI. Level 1 plasmids are designed in such a way that adjacent type IIS BplI restriction enzyme sites flank the newly assembled expression unit. Specific level 1 plasmids exist that define the future position of the expression unit in a multi-gene ‘level M’ plasmid, where up to seven of these units can be assembled, simultaneously. Alternatively, when less than seven expression units are desired, the assemblies are capped with an ‘end linker’ (Engler et al., 2014; Einarsson et al., 2021). To allow modular assembly, all modules (inserts of interest) need to be ‘domesticated’ using site directed mutagenesis (SDM) to eliminate any endogenous non-flanking type IIS restriction enzyme sites for both BsaI and BpII.
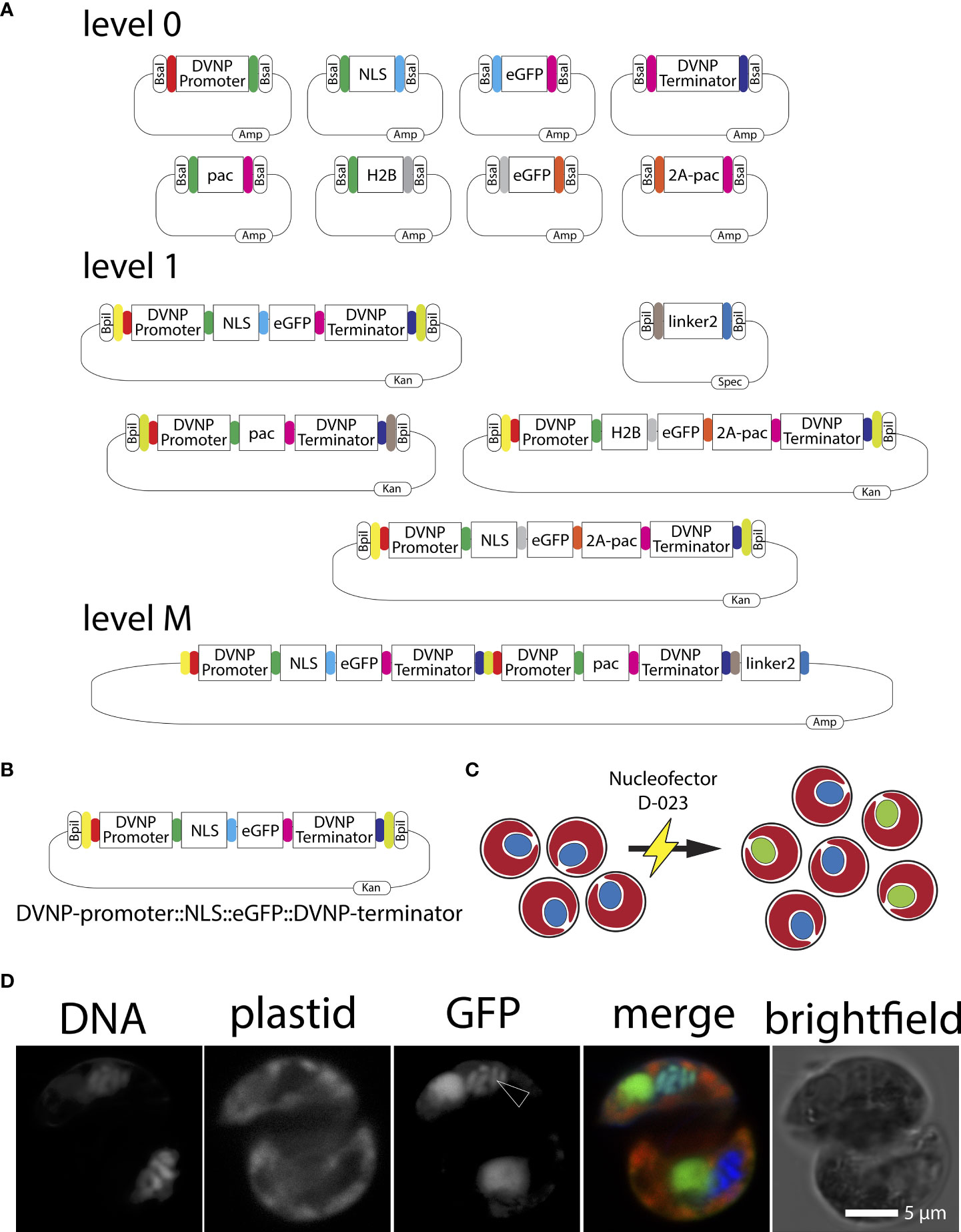
Figure 2 (A) Graphical representation of level 0, level 1, end-linker, and level M Golden Gate modules used in this study. Kan=kanamycin, Amp=ampicillin, Spec=spectinomycin. (B) Graphical representation of the DVNP-promoter::NLS::eGFP::DVNP-terminator level 1 position 1 module. (C) Cartoon depicting the Nucleofector electroporation of Breviolum SSB01 cells (red plastid auto-fluorescence with blue nuclei) and resulting localization of the nuclear targeted NLS-eGFP (red plastid auto-fluorescence-containing cells with green nuclei) following transformation. (D) Confocal image of a transformed SSB01 cell showing expression and translocation of GFP to the nucleus. Note: all SSB01 cells contain a structure that resembles an accumulation body as described by Wakefield et al. (2000). This accumulation body very often sits near the nucleus and exhibits strong autofluorescence at 501-550 nm when excited with a 488-nm laser line.
SSB01 transformation: proof-of-principle
We cloned and ‘domesticated’ several level 0 plasmids containing our identified promoter and terminator sequences of SSB01 DVNP.17, and eGFP with the addition of overlaps to allow ordered assembly into level 1 or level M vectors (Figure 2A). Primers designed using B. minutum sequences yielded both the DVNP.17 promoter and terminator sequences supporting the close relation between SSB01 and B. minutum as revealed by above phylogenies (Figure 1A). When designing the proof-of-principle vector, we reasoned that visual confirmation of cytosolic GFP expression may prove challenging as all members of the Symbiodiniaceae, including SSB01, are photosynthetic and therefore, exhibit a broad (400 to 700 nm) and strong plastid-derived auto-fluorescent signal when examined by fluorescent microscopy (Figure 1A). The cell’s nucleus is not auto-fluorescent and is not visible at wavelengths necessary for GFP detection (see for example the control in Figure 3C). Thus, we sought to direct eGFP towards the nucleus of SSB01 cells to localize DNA to provide as location clearly distinct from the auto-fluorescence of the plastids, as graphically depicted in Figure 2C.
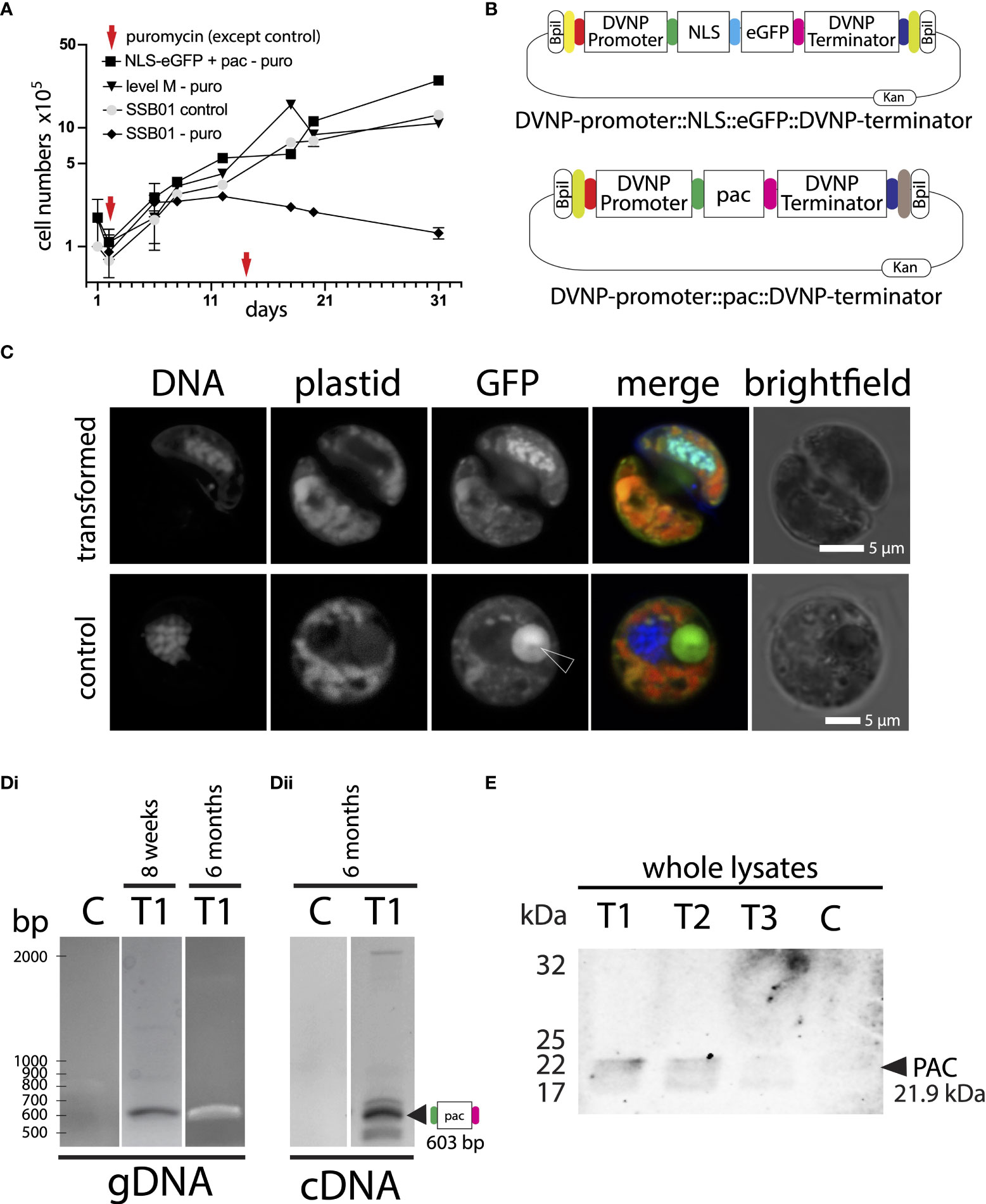
Figure 3 (A) Addition of puromycin 100 µg ml-1 at day 1 and day 14 shows clear growth inhibition in untransformed SSB01 cells (SSB01-puro). Transfected cells (NLS-eGFP+pac-puro and level M-puro) and cells without addition of puromycin (SSB01-control) grow steadily and at a similar rate. (B) Graphical representation of the DVNP-promoter::NLS-eGFP::DVNP-terminator level 1 position 1 and DVNP-promoter::pac::DVNP-terminator level 1 position 2 modules. (C) Confocal image of transformed and untransformed SSB01 cells clearly showing expression and translocation of GFP to the nucleus in the presence of the expression vector. Note: all SSB01 cells contain a structure that resembles an accumulation body as described by Wakefield et al. (2000). This accumulation body very often sits near the nucleus and exhibits strong autofluorescence at 501-550 nm when excited with a 488-nm laser line. (Di) PCR and RT-PCR for pac in gDNA and cDNA from double transformed NLS-eGFP/PAC transformant T1 indicating apparent good expression of pac at 8 weeks post-transformation. (Dii) RT-PCR for pac in cDNA from double transformed NLS-eGFP/PAC transformant T1 indicating that good expression of pac persists for at least 6 months post-transformation. (E) Anti-PAC Western blot showing that cells from transformants T1-T3 express PAC at 8 weeks post transformation, but with variable intensities.
Gornik et al. (2012) showed that the lysine-rich N-terminal region of DVNPs acts as strong nuclear localization sequence (NLS). Thus, we hypothesized that the lysine-rich sequence in SSB01 DVNP.17 would have a similar effect. We defined a 30 amino acid long, lysine-rich N-terminal region of SSB01 DVNP.17 as the NLS and cloned this into an additional level 0 plasmid. This plasmid contained an adjacent type IIS restriction enzyme BsaI and necessary overlaps to allow the following order of level 1 position 1 module assembly: DVNP-promoter::NLS::eGFP::DVNP-terminator (Figure 2B). This module was then used to test our strategy for SSB01 transformation using the Golden Gate modular system, with the putative endogenous promoter sequences and targeting signals.
To introduce the plasmid DNA carrying the DVNP-promoter::NLS::eGFP::DVNP-terminator construct into SSB01 cells, we used electroporation as described for P. marinus, O. marina and K. brevis with modifications ( (Fernández Robledo et al., 2008; Faktorová et al., 2020; Einarsson et al., 2021); and methods). Although electroporation yielded low efficacy and efficiency (approx. 0.1%; we detected 1-2 GFP-positive cells per 1,000 cells observed), we were able to successfully identify GFP positive cells at 10 days following transformation with a strong nuclear GFP signal (Figure 2D). Interestingly, the GFP signal overlapped considerably with the DNA signal, which may indicate that the high lysine content of the DVNP-NLS may also be capable of binding DNA, consistent with observations in Toxoplasma gondii nuclei (Gornik et al., 2012). We further observed that all SSB01 cells contain a structure that resembles an accumulation body described by Wakefield et al. (2000). This accumulation body most often sits near the nucleus and exhibits a strong autofluorescent signal at 501-550 nm when excited with a 488-nm laser line (Figure 2D). This structure also occurs in untransformed cells when excited at 488-nm.
We also tested the DVNP-promoter::NLS::eGFP::DVNP-terminator construct in SSE01, SSA02, and Clade D symbionts; however, while we saw some GFP-positive cells in SSE01 (Supplementary Figure 7), GFP expression was absent for SSA02 and Clade D cells (data not shown). Both SSA02 (Symbiodinium), Clade D (Durusdinium) and SSE01 (Effrenium) belong to Symbiodniiaceae genera that are genetically distinct from SSB01 (Breviolum); see also Figure 1A and it has yet to be determined how distinct the DVNP promoters of these genera are on a genetic level. We would have expected them to be reasonable conserved and thus, lack of expression in SSA02 and Clade D is somewhat surprising. We would have expected that the DVNP promoter works well across all members of the Symbiodiniaceae; this, however, seems to not be the case. It seems to be only functional in SSB01 and SSE01. It remains to be determined however, if this is indeed caused genetic distance or if in fact some other biological parameters such as the thickness and composition of their cell wall, their cell cycle momentum or general fitness under the culture conditions applied hinders efficient transformation in some of the genera tested in this study. Moreover, it may be that the transformation solution used in this study is toxic to SSA02 and Clade D cells, but not to SSB01 and SSE01; so testing other solutions may overcome the lack of transformation success for SSA02 and Clade D cells. Because transformation was successful in SSB01 cells using the current protocol no further attempts were made to test different solutions, light intensities, day- or night times or to optimize transformation for SSA02 and clade D cells. And, since SSE01 represents an entirely free-living non-symbiotic member of the Symbiodiniaceae (LaJeunesse et al., 2018) subsequent analyses only focused on SSB01.
SSB01 transformation: Puromycin selection
After demonstrating that transformation is achievable, we set out to optimize transformation efficiency in SSB01 cells utilizing an antibiotic or herbicide as putative selectable markers. Numerous antibiotics and herbicides including hygromycin, Geneticin/G418, chloramphenicol, Basta® (glufosinate), blasticidin, and atrazine were shown to inhibit the growth of dinoflagellate species including S. microadriaticum, Oxyrrhis marina, and the dinoflagellate-related, non-photosynthetic, parasitic P. marinus (Chen et al., 2019; Sprecher et al., 2020; Einarsson et al., 2021). However, with the exception of P. marinus, only a few of these resulted in successful selection allowing propagation of transformed cells. Although the inhibitory effect of puromycin on dinoflagellate growth was not previously reported, its efficacy in P. marinus has been shown (Sakamoto et al., 2019). We thus tested puromycin for their ability to inhibit SSB01 growth under culturing conditions with the known herbicide glufosinate (as a positive control). In doing so, we determined that puromycin concentrations >100 µg ml-1 and glufosinate >10 µg ml-1 were effective in impeding SSB01 growth in culture (Supplementary Figure 8A). It should be noted that we observed a marked reduction of cell growth using puromycin at concentrations between 10-50 µg ml-1, but full inhibition of growth consistently occurred only at concentrations of 100 µg ml-1 and above (Supplementary Figure 8B).
The glufosinate/phosphinothricin in Basta® is a structural analog of glutamate that binds to glutamine synthetase irreversibly, which inhibits the synthesis of glutamine from ammonia into glutamate, ultimately impairing amino acid metabolism and leading to death (Tian et al., 2015). Mechanistically, BAR (a phosphinothricin acetyltransferase), inactivates the herbicide by transferring an acetyl group from acetyl coenzyme A (acetyl-CoA) to the α-NH2 group of glufosinate/phosphinothricin, resulting in herbicide inactivation (Christ et al., 2017). However, the expression of BAR has off-target effects on cellular amino acid metabolism arising from enzyme promiscuity. We wanted to avoid long-term exposure to glufosinate/phosphinothricin and therefore decided we would continue working only with puromycin, which, in contrast to the Basta® herbicide, inhibits protein synthesis at the ribosome with no known off-target effects (Aviner, 2020). Next, we tested whether a single dose of puromycin is effective in inhibiting growth for an extended period of time. We found that addition of 100 µg ml-1 puromycin to SSB01 at a density of 1.0 x 105 cells per milliliter successfully inhibits cell growth for at least 31 days with an additional dose of 100 µg ml-1 puromycin at day 14 (Figure 3A “SSB01–puro”). In fact, in a separate experiment we found that a single application of 100 µg ml-1 puromycin inhibits growth of SSB01 for as much as 2 months (Supplementary Figure 8C). We thus conclude that using the selection marker pac encoding puromycin N-acetyltransferase, which confers puromycin resistance in P. marinus, yeast, and other cell culture systems, in combination with a single application of pyromycin at 100 µg ml-1 should also serve to effectively select transformants following transformation in SSB01. In P. marinus, an IC50 of approximately 5 µg ml-1 was determined and effective selection of transformants was conferred at a concentration of 100 µg ml-1, which aligns with the working concentrations we propose here (Sakamoto et al., 2019; Einarsson et al., 2021).
We generated a plasmid carrying a DVNP-promoter::pac::DVNP-terminator construct (Figure 3B). The pac sequence was domesticated by synthesis, PCR amplified, and then cloned as a level 0 plasmid with overlaps to allow ordered assembly into a level 1 position 2 vector that further allows level M vector assembly with the level 1 position 1 NLS-eGFP vector from above (see also Figure 2A). The plasmid was then co-transfected with the NLS-eGFP level 1 position 1 plasmid we generated and used earlier, acting as a visual marker for successful transfection. Following electroporation of the two plasmids, cells were allowed to grow without selection for 24 hours. Then, puromycin was added to a final concentration of 100 µg ml-1. Fresh puromycin was added every 14 days or whenever splitting cultures.
We performed 4 independent transformations (T1-T4). Transformations were successful in all 4 cases since cells grew in culture upon puromycin selection (for an example growth curve see Figure 3A “NLS-eGFP+pac”); however, in we only observed GFP positive cells within 10 days post transformation in 3 out of 4 transformant lines (T1-T3) with similar patterns as observed previously with the DVNP-promoter::NLS-eGFP::DVNP-terminator vector only (for an example see: Figure 3C; we detected approx. 10 GFP-positive cells per 1,000 cells observed). No GFP-positive cells were identifiable in transformant line T4 or control transformations (not shown). As expected, control and untransformed cultures did not grow in the presence of puromycin (not shown). In an attempt to overcome the sparse GFP expression, we manually picked clusters of positive cells to enrich for them (not shown); however, we observed that GFP expression eventually weakened from 4 weeks onwards and ceased completely within 6 months in these cultures, presumably since the selective marker and GFP were provided on separate plasmids lending to loss of GFP without loss of resistance, despite the ongoing selection pressure. Cells only needed to maintain the DVNP-promoter::pac::DVNP-terminator sequence for survival, and the loss of the DVNP-promoter::NLS-eGFP::DVNP-terminator expression unit had no impact on cell survival. This was also evident from the original transformant lines T1-T3, where many cells completely lacked GFP expression, but still grew under selection; presumably having eliminated the DVNP-promoter::NLS-eGFP::DVNP-terminator plasmid or transgene NLS-eGFP expression in most cells. Likewise, in T4, no GFP-positive cells were ever observed suggesting low or lack of DVNP-promoter::NLS-eGFP::DVNP-terminator plasmid uptake or complete elimination of transgene expression in these cells.
At 8 weeks and 6 months post-transformation, we used PCR to confirm both presence and expression of the pac and gfp genes in genomic DNA from transformant lines T1 - T4 (Figure 3Di for transformant T1 and Supplementary File 9). At 6 months, we also determined whether the pac and gfp genes were actively expressed using cDNA from RT-PCRs from fresh RNA extractions (Figure 3Dii for transformant T1 and Supplementary File 9). For all PCRs, the sizes of resulting amplicons corresponded with expected sizes. Amplicon sequences were confirmed by Sanger sequencing (not shown). We noted that the pac signal was strong in all clones at both 8 weeks and 6 months in genomic DNA and was also evident in cDNA after 6 months; however, gfp detection was less consistent, especially after 6 months, where genomic presence and expression of gfp was not reliably detected anymore; consistent with either elimination of the vector and/or substantial loss of transgene expression (Supplementary File 9). This reduction is consistent with weakening GFP fluorescence; and this may indicate that gfp plasmids and expression are progressively lost following electroporation. Loss of gfp has no impact on selection; moreover, gfp expression may even negatively affect cell growth taking up energy otherwise available for cell growth. Western blots against lysates of the 6-month sample for T1-T3 with an anti-pyromycin N-acetyltransferase (PAC) antibody also confirmed expression of PAC (Figure 3E). Transformant line T4 was not probed by Western blot, as this culture grew very slowly and at low densities under permanent puromycin selection and lacked fluorescence throughout; presumably reflecting low level expression of the pac gene and absence of gfp as seen by semi-quantitative RT-PCR (Supplementary File 9).
SSB01 transformation: Multi expression units
Attempting to overcome the progressive elimination of GFP expression that we observed in our transformations, we generated a level M plasmid carrying the expression unit for both NLS-eGFP and PAC by Golden Gate cloning: DVNP-promoter::NLS-eGFP::DVNP-terminator::DVNP-promoter::pac::DVNP-terminator (Figure 4A); allowing the co-expression of NLS-eGFP and PAC from the same plasmid under control of identical, but separate, DVNP promoter regions. As before, the cells grew well in the presence of puromycin (Figure 3A “level M-puro”) and we detected GFP 10 days after transformation (Figure 4B) in approximately 1% of cells under selection (we detected 10 GFP-positive cells per 1,000 cells observed.) However, we still observed the progressive loss of GFP expression over the course of several weeks in sub-cultures that were manually enriched for positive clones by pipetting GFP-positive cells into fresh culture dishes. This is also consistent with data after 4 weeks where we found gDNA for egfp and pac via PCR but only cDNA for pac (Figure 4C). Expression of PAC was confirmed by Western blot (Figure 4D). We suspect that over time the promoter and/or ORF region of egfp degraded, as its expression was not directly essential for retention of puromycin resistance, albeit on the same plasmid, it was under control of a second, independent DVNP promoter. In an effort to overcome this problem, we attempted to use a self-cleaving P2A peptide to generate polyproteins from a single promoter expression unit, however, we found that cleavage was very unreliable and erratic and often did not work at all (see Supplementary Results and Supplementary File 10).
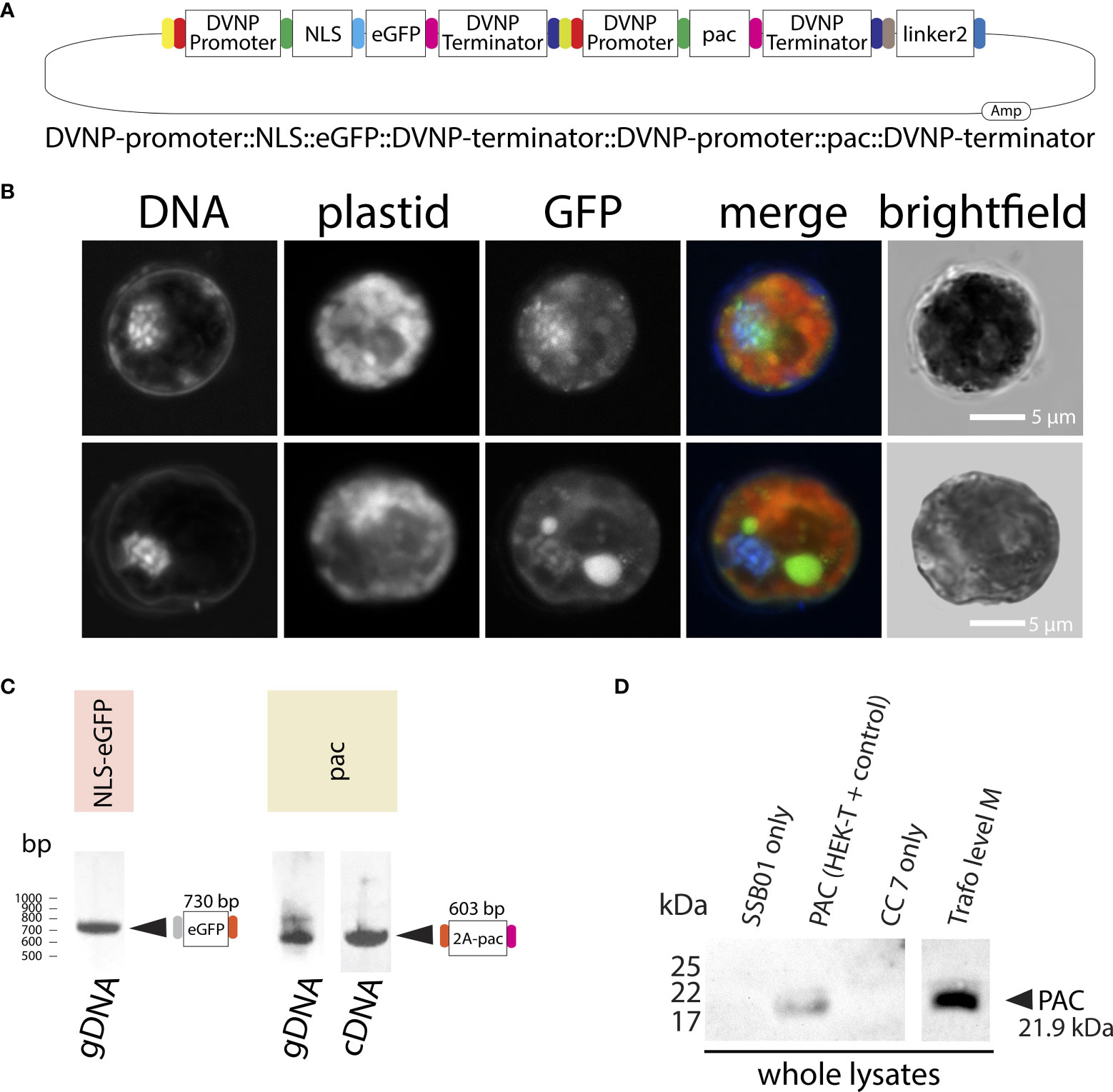
Figure 4 (A) Graphical representation of the level M transformation vector comprising DVNP-promoter::NLS-eGFP::DVNP-terminator level 1 position 1, DVNP-promoter::pac::DVNP-terminator level 1 position 2 and end linker 2 modules. (B) Confocal images of transformed SSB01 cells showing expression and translocation of GFP to the nucleus in the presence of the expression vector. Note: the large GFP-positive structure in one of the cells is the result of strong autofluorescence of an accumulation body. (C) PCR results for eGFP and (RT-)PCR results for pac in transformed cells. (D) Anti-PAC Western blot showing that transformed cells express PAC post transformation. For a whole blot see: Supplementary File 10.
Conclusion
Using Golden Gate modular plasmid assembly, we generated several ‘level 0’ entry vectors using both SSB01-derived and generic or synthesized sequences. From these we effectively generated a number of ‘level 1’ expression units and one ‘level M’ vector containing two independent expression units. We successfully transformed cells of the coral symbiont SSB01 belonging to the dinoflagellate genus Breviolum using several constructs, and we successfully targeted GFP to the nucleus of these cells using a DVNP-derived NLS under control of an endogenous promoter for DVNP. We further determined that puromycin at 100 µg ml-1 can be used to efficiently select transformed cells when provided with a puromycin N-acetyltransferase gene (pac). Efficiency of transformation was low, but well within the expected range of 0.0005 - 0.1 for stably transformed marine protists (Faktorová et al., 2020; Einarsson et al., 2021). While beyond the scope of this study efficiency of this method will likely improve over time should other promoters, fluorophores and electroporation buffers be tested and used in future.
We found that transformed cells can be maintained for at least 6 months without loss of antibiotic resistance, albeit with slow loss of fluorescence. We also tested the utility of a 2A peptide to generate hybrid peptides under the control of a single promoter sequence; however, we found that cleavage was very unreliable. Nevertheless, in our work we identified a good promoter sequence that consistently allowed us to drive at least two different genes (GFP and pac) on a single plasmid. We built a GoldenGate-like modular transformation method that will in future allow addressing many of the unsolved questions of dinoflagellate biology, with a particular emphasis on the biology of the dinoflagellate-coral symbiosis. For example, to understand whether certain proteins at the surface of the symbionts promote or inhibit initiation of symbiosis could be investigated by expressing GFP-tagged candidate proteins under the control of the DVNP promoter. Furthermore, using Breviolum SSB01 cells that express NLS-eGFP and other, untagged Symbiodiniaceae strains, the dynamics of symbiont uptake and competition during recovery following a bleaching event can now be studied in great detail. On the cellular level, organelle-targeted proteins and the role of N-terminal targeting signals can now be investigated using putative targeting signals such as the N-terminal bipartite leader, presumed to play a role in dinoflagellate plastid targeting. With a successful transformation method for SSB01, it will now be important to focus on further developing and optimizing this system by stabilizing fluorescence and quantifying and characterizing new promoters. Lastly, extensions of this method to other dinoflagellates species may also have economic impact. It may allow, for example, for the genetic optimization and modification of Crypthecodinium cohnii cells, which have been used for the commercial production of oils rich in omega-3 polyunsaturated fatty acid and docosahexaenoic acid since the mid 1990s. In this context Diao et al. (2018) showed that deletion of a RuBisCO gene promoted cell growth and increased the lipid content of C. cohnii under heterotrophic conditions compared with those of the wild-type. Such modifications may allow for more reliable harvests by overexpressing target genes involved in the associated biosynthetic pathways (Diao et al., 2018).
Data availability statement
The Breviolum sp. SSB01 RNA-seq dataset analyzed in this study is accessible via NCBI (BioProject PRJNA591730). Sequence accessions IDs and sources are given at the relevant sections throughout the text or are found in the supplementary material. Alignments and resulting phylogenies are provided in the supplementary data. All Golden Gate modules and final constructs used in this study are available upon request.
Author contributions
SG and AG conceived the study. SG developed the methodology, performed the formal analyses, performed major parts of the investigation, curated and interpreted the data and wrote the original draft of the manuscript. PV generated samples for RNA-seq. IM helped with dinoflagellate culturing and carried out and conceptualized the SSB01 antibiotics resistance experiments. EH and IM generated Symbiondinaceae rRNA sequences. RW generated the Golden Gate level 1, linker 2 and level M plasmids. AG provided resources and acquired major funding. SG, AG and RW reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the Deutsche Forschungsgemeinschaft (DFG) (EmmyNoether Program Grant GU 1128/3-1), the Government of Canada ( New Frontiers in Research Fund - Exploration Grant NFRFE-2019-00189) and H2020 European Research Council (ERC Consolidator Grant 724715) to AG, by the Gordon and Betty Foundation (grants 4977, 4977.01) to RW. The Baden-Württemberg Landesgraduiertenförderung Program (Evolutionary Novelty and Adaptation) provided a PhD scholarship to PV within the graduate school.
Acknowledgments
The authors want to thank members of the Waller lab at the Department of Biochemistry at the University of Cambridge for discussion and feedback. We also wish to thank the Katrin Gundel for carrying out the initial antibiotics growth assays. We also thank the Frischknecht laboratory at the parasitology unit at the center for infectious diseases at Heidelberg University for providing access to a Nucleofector IIb device (Lonza). We further thank D. Pavlinic and V. Benes (GeneCore Facility, EMBL Heidelberg) for assistance with the Smart-Seq2 protocol and sequencing library preparation; D. Ibberson (Deepseqlab, Heidelberg University) for assistance with the Smart-Seq2 protocol.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor CV declared a past co-authorship with the author AG.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1035413/full#supplementary-material
References
Akbar M., Ahmad A., Usup G., Bunawan H. (2018). Current knowledge and recent advances in marine dinoflagellate transcriptomic research. J. Mar. Sci. Eng. 6, 13. doi: 10.3390/jmse6010013
Aranda M., Li Y., Liew Y. J., Baumgarten S., Simakov O., Wilson M. C., et al. (2016). Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 6, 39734. doi: 10.1038/srep39734
Archibald J. M. (2009). The puzzle of plastid evolution review. Curr. Biol. 19, R81–R88. doi: 10.1016/j.cub.2008.11.067
Aviner R. (2020). The science of puromycin: From studies of ribosome function to applications in biotechnology. Comput. Struct. Biotechnol. J. 18, 1074–1083. doi: 10.1016/j.csbj.2020.04.014
Baumgarten S., Simakov O., Esherick L. Y., Liew Y. J., Lehnert E. M., Michell C. T., et al. (2015). The genome of aiptasia, a sea anemone model for coral symbiosis. Proc. Natl. Acad. Sci. U.S.A. 112, 11893–11898. doi: 10.1073/pnas.1513318112
Carbonera D., Di Valentin M., Spezia R., Mezzetti A. (2014). The unique photophysical properties of the peridinin-Chlorophyll-α-Protein. Curr. Protein Pept. Sci. 15, 332–350. doi: 10.2174/1389203715666140327111139
Chen J. E., Barbrook A. C., Cui G., Howe C. J., Aranda M. (2019). The genetic intractability of Symbiodinium microadriaticum to standard algal transformation methods. PloS One 14, e0211936. doi: 10.1371/journal.pone.0211936
Christ B., Hochstrasser R., Guyer L., Francisco R., Aubry S., Hörtensteiner S., et al. (2017). Non-specific activities of the major herbicide-resistance gene BAR. Nat. Plants 3, 937–945. doi: 10.1038/s41477-017-0061-1
de Vargas C., Audic S., Henry N., Decelle J., Mahé F., Logares R., et al. (2015). Ocean plankton. eukaryotic plankton diversity in the sunlit ocean. Science 348, 1261605. doi: 10.1126/science.1261605
Diao J., Song X., Zhang X., Chen L., Zhang W. (2018). Genetic engineering of Crypthecodinium cohnii to increase growth and lipid accumulation. Front. Microbiol. 9, 492. doi: 10.3389/fmicb.2018.00492
Edgar R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5, 1–19. doi: 10.1186/1471-2105-5-113
Einarsson E., Lassadi I., Zielinski J., Guan Q., Wyler T., Pain A., et al. (2021). Development of the myzozoan aquatic parasite Perkinsus marinus as a versatile experimental genetic model organism. Protist 172, 125830. doi: 10.1016/j.protis.2021.125830
Engler C., Youles M., Gruetzner R., Ehnert T.-M., Werner S., Jones J. D. G., et al. (2014). A golden gate modular cloning toolbox for plants. ACS synthetic Biol. 3, 839–843. doi: 10.1021/sb4001504
Faktorová D., Nisbet R. E. R., Fernández Robledo J. A., Casacuberta E., Sudek L., Allen A. E., et al. (2020). Genetic tool development in marine protists: emerging model organisms for experimental cell biology. Nat. Methods 17, 481–494. doi: 10.1038/s41592-020-0796-x
Fernández Robledo J. A., Lin Z., Vasta G. R. (2008). Transfection of the protozoan parasite Perkinsus marinus. Mol. Biochem. Parasitol. 157, 44–53. doi: 10.1016/j.molbiopara.2007.09.007
Fernando Ortiz-Matamoros M., Villanueva M. A., Islas-Flores T. (2015). Transient transformation of cultured photosynthetic dinoflagellates (Symbiodinium spp.) with plant-targeted vectors. Cienc. Marinas 41, 21–32. doi: 10.7773/cm.v41i1.2449
Gornik S. G., Ford K. L., Mulhern T. D., Bacic A., McFadden G. I., Waller R. F. (2012). Loss of nucleosomal DNA condensation coincides with appearance of a novel nuclear protein in dinoflagellates. Curr. Biol. 22, 2303–2312. doi: 10.1016/j.cub.2012.10.036
Gornik S. G., Hu I., Lassadi I., Waller R. F. (2019). The biochemistry and evolution of the dinoflagellate nucleus. Microorganisms 7, 245. doi: 10.3390/microorganisms7080245
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grawunder D., Hambleton E. A., Bucher M., Wolfowicz I., Bechtoldt N., Guse A. (2015). Induction of gametogenesis in the cnidarian endosymbiosis model Aiptasia sp. Sci. Rep. 5, 15677. doi: 10.1038/srep15677
Guillebault D., Sasorith S., Derelle E., Wurtz J.-M., Lozano J.-C., Bingham S., et al. (2002). A new class of transcription initiation factors, intermediate between TATA box-binding proteins (TBPs) and TBP-like factors (TLFs), is present in the marine unicellular organism, the dinoflagellate crypthecodinium cohnii. J. Biol. Chem. 277, 40881–40886. doi: 10.1074/jbc.M205624200
Hackett J. D., Anderson D. M., Erdner D. L., Bhattacharya D. (2004). Dinoflagellates: a remarkable evolutionary experiment. Am. J. Bot. 91, 1523–1534. doi: 10.3732/ajb.91.10.1523
Hambleton E. A., Guse A., Pringle J. R. (2014). Similar specificities of symbiont uptake by adults and larvae in an anemone model system for coral biology. J. Exp. Biol. 217, 1613–1619. doi: 10.1242/jeb.095679
Hambleton E. A., Jones V. A. S., Maegele I., Kvaskoff D., Sachsenheimer T., Guse A. (2019). Sterol transfer by atypical cholesterol-binding NPC2 proteins in coral-algal symbiosis. Elife 8, e43923. doi: 10.7554/eLife.43923.025
Jackson C. J., Gornik S. G., Waller R. F. (2012a). A tertiary plastid gains RNA editing in its new host. Mol. Biol. Evol. 30, 788–792. doi: 10.1093/molbev/mss270
Jackson C. J., Gornik S. G., Waller R. F. (2012b). The mitochondrial genome and transcriptome of the basal dinoflagellate hematodinium sp.: character evolution within the highly derived mitochondrial genomes of dinoflagellates. Genome Biol. Evol. 4, 59–72. doi: 10.1093/gbe/evr122
Jacobovitz M. R., Rupp S., Voss P. A., Maegele I., Gornik S. G., Guse A. (2021). Dinoflagellate symbionts escape vomocytosis by host cell immune suppression. Nat. Microbiol. 6, 769–782. doi: 10.1038/s41564-021-00897-w
Janouškovec J., Gavelis G. S., Burki F., Dinh D., Bachvaroff T. R., Gornik S. G., et al. (2017). Major transitions in dinoflagellate evolution unveiled by phylotranscriptomics. Proc. Natl. Acad. Sci. U.S.A. 114, E171–E180. doi: 10.1073/pnas.1614842114
Keeling P. J. (2010). The endosymbiotic origin, diversification and fate of plastids. Philos. Trans. R. Soc. B: Biol. Sci. 365, 729–748. doi: 10.1098/rstb.2009.0103
LaJeunesse T. C., Parkinson J. E., Gabrielson P. W., Jeong H. J., Reimer J. D., Voolstra C. R., et al. (2018). Systematic revision of symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 28, 2570–2580.e6. doi: 10.1016/j.cub.2018.07.008
Lin S. (2011). Genomic understanding of dinoflagellates. Res. Microbiol. 162, 551–569. doi: 10.1016/j.resmic.2011.04.006
Lin S., Cheng S., Song B., Zhong X., Lin X., Li W., et al. (2015). The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350, 691–694. doi: 10.1126/science.aad0408
Marinov G. K., Lynch M. (2016). Diversity and divergence of dinoflagellate histone proteins. G3 6, 397–422. doi: 10.1534/g3.115.023275
Marinov G. K., Trevino A. E., Xiang T., Kundaje A., Grossman A. R., Greenleaf W. J. (2021). Transcription-dependent domain-scale three-dimensional genome organization in the dinoflagellate Breviolum minutum. Nat. Genet. 53, 613–617. doi: 10.1038/s41588-021-00848-5
Mendez G. S., Delwiche C. F., Apt K. E., Lippmeier J. C. (2015). Dinoflagellate gene structure and intron splice sites in a genomic tandem array. J. Eukaryotic Microbiol. 62, 679–687. doi: 10.1111/jeu.12230
Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., Haeseler, von A., et al. (2020). IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Nand A., Zhan Y., Salazar O. R., Aranda M., Voolstra C. R., Dekker J. (2021). Genetic and spatial organization of the unusual chromosomes of the dinoflagellate Symbiodinium microadriaticum. Nat. Genet. 53, 618–629. doi: 10.1038/s41588-021-00841-y
Nimmo I. C., Barbrook A. C., Lassadi I., Chen J. E., Geisler K., Smith A. G., et al. (2019). Genetic transformation of the dinoflagellate chloroplast. eLife 8, e45292. doi: 10.7554/eLife.45292.014
Ortiz-Matamoros M. F., Islas-Flores T., Voigt B., Menzel D., Baluška F., and Villanueva M. A. (2015). Heterologous DNA uptake in cultured Symbiodinium spp. aided by agrobacterium tumefaciens. PloS One 10, e0132693. doi: 10.1371/journal.pone.0132693
Parra G., Bradnam K., Korf I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Picelli S., Faridani O. R., Björklund Å.K., Winberg G., Sagasser S., Sandberg R. (2014). Full-length RNA-seq from single cells using smart-seq2. Nat. Protoc. 9, 171–181. doi: 10.1038/nprot.2014.006
Rädecker N., Raina J.-B., Pernice M., Perna G., Guagliardo P., Kilburn M. R., et al. (2018). Using aiptasia as a model to study metabolic interactions in cnidarian-symbiodinium symbioses. Front. Physiol. 9,214. doi: 10.3389/fphys.2018.00214
Roy S., Jagus R., Morse D. (2018). Translation and translational control in dinoflagellates. Microorganisms 6, 30. doi: 10.3390/microorganisms6020030
Sakamoto H., Hirakawa Y., Ishida K.-I., Keeling P. J., Kita K., Matsuzaki M. (2019). Puromycin selection for stable transfectants of the oyster-infecting parasite Perkinsus marinus. Parasitol. Int. 69, 13–16. doi: 10.1016/j.parint.2018.10.011
Shoguchi E., Shinzato C., Kawashima T., Gyoja F., Mungpakdee S., Koyanagi R., et al. (2013). Draft assembly of the symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408. doi: 10.1016/j.cub.2013.05.062
Sprecher B. N., Zhang H., Lin S. (2020). Nuclear gene transformation in the dinoflagellate oxyrrhis marina. Microorganisms 8, 126. doi: 10.3390/microorganisms8010126
Tian Y.-S., Xu J., Zhao W., Xing X.-J., Fu X.-Y., Peng R.-H., et al. (2015). Identification of a phosphinothricin-resistant mutant of rice glutamine synthetase using DNA shuffling. Sci. Rep. 5, 15495. doi: 10.1038/srep15495
Wakefield T. S., Farmer M. A., Kempf S. C. (2000). Revised description of the fine structure of in situ “zooxanthellae” genus symbiodinium. Biol. Bull. 199, 76–84. doi: 10.2307/1542709
Waller R. F., Koreny L. (2017). Plastid complexity in dinoflagellates: A picture of gains, losses, replacements and revisions. Secondary Endosymbio. 84, 105–143. doi: 10.1016/bs.abr.2017.06.004
Wisecaver J. H., Hackett J. D. (2011). Dinoflagellate genome evolution. Annu. Rev. Microbiol. 65, 369–387. doi: 10.1146/annurev-micro-090110-102841
Wolfowicz I., Baumgarten S., Voss P. A., Hambleton E. A., Voolstra C. R., Hatta M., et al. (2016). Aiptasia sp. larvae as a model to reveal mechanisms of symbiont selection in cnidarians. Sci. Rep. 6, 32366. doi: 10.1038/srep
Xiang T., Hambleton E. A., DeNofrio J. C., Pringle J. R., Grossman A. R. (2013). Isolation of clonal axenic strains of the symbiotic dinoflagellate Symbiodinium and their growth and host specificity. J. Phycol. 49, 447–458. doi: 10.1111/jpy.12055
Xiang T., Nelson W., Rodriguez J., Tolleter D., Grossman A. R. (2015). Symbiodinium transcriptome and global responses of cells to immediate changes in light intensity when grown under autotrophic or mixotrophic conditions. Plant J. 82, 67–80. doi: 10.1111/tpj.12789
Yan Q. (2011). Rapid coomassie protein SDS-gel staining. Bio-protocol. 1, e83–e83. doi: 10.21769/BioProtoc.83
Keywords: Symbiodiniaceae, dinoflagellate transformation, Aiptasia, Breviolum minutum, SSB01, coral symbiosis, coral biology, Alveolata
Citation: Gornik SG, Maegele I, Hambleton EA, Voss PA, Waller RF and Guse A (2022) Nuclear transformation of a dinoflagellate symbiont of corals. Front. Mar. Sci. 9:1035413. doi: 10.3389/fmars.2022.1035413
Received: 02 September 2022; Accepted: 13 October 2022;
Published: 09 November 2022.
Edited by:
Christian Robert Voolstra, University of Konstanz, GermanyReviewed by:
Guoxin Cui, King Abdullah University of Science and Technology, Saudi ArabiaJean-Baptiste Raina, University of Technology Sydney, Australia
Copyright © 2022 Gornik, Maegele, Hambleton, Voss, Waller and Guse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annika Guse, YW5uaWthLmd1c2VAYmlvbG9naWUudW5pLW11ZW5jaGVuLmRl; Sebastian G. Gornik, c2ViYXN0aWFuLmdvcm5pa0Bjb3MudW5pLWhlaWRlbGJlcmcuZGU=
†Present address: Elizabeth A. Hambleton, Division of Microbial Ecology, Department for Microbiology and Ecosystem Science, Centre for Microbiology and Environmental Systems Science, University of Vienna, Vienna, Austria
Annika Guse, Faculty of Biology, Ludwig-Maximilians-Universität Munich, Munich, Germany
Sebastian G. Gornik, Faculty of Biology, Ludwig- Maximilians-Universität Munich, Munich, Germany