 Qingfei Li1†
Qingfei Li1† Jinghua Chen
Jinghua Chen- 1School of Marine Science and Engineering, Qingdao Agricultural University, Qingdao, China
- 2Healthy Aquaculture Laboratory of Sichuan Province, Tongwei Agricultural Development Co., Ltd., Chengdu, China
Introducton
The high-throughput sequencing technologies serve as a promising tool to facilitate the molecular biological research of aquaculture and fishery species. Especially, the revolutionary next-generation sequencing (NGS) technology has largely enhanced the capacity to decode DNA in a large-scale and cost-effective manner, and a considerable amount of genetic information has been widely applied to address a variety of biological questions related to physiology, growth, nutrition, breeding, and genetics and genomics of major aquaculture species (Abdelrahman et al., 2017). Nevertheless, NGS poses great challenges including the requirement of sophisticated bioinformatics skills and the availability of sufficient data-processing resources. Moreover, the characterization of the transcriptome profile constructed by NGS sequencing are insufficiently accurate and complete since the precise annotation requires up-to-date databases and automatic analysis procedures. Additionally, the short-read length output hindered the discovery of novel transcripts and characterization of gene functions, thus preventing further understanding of specific biological processes (Metzker, 2010).
To address those limitations inherent in NGS sequencing, the cutting-edge third-generation sequencing, also referred to as single-molecule real-time (SMRT) sequencing was developed by the PacBio company (Munroe and Harris, 2010), which functions by anchoring a DNA polymerase to the bottom surface of Zero-Mode Waveguide (ZMW) nanostructure (Levene et al., 2003). The SMRT technology has four major advantages compared to the NGS platforms: Firstly, the most prominent advantage is its long, continuous reads from 5’ untranslated region (UTR) to 3’ polyadenylation without assembly required, providing an average read length of up to 20,000 bases, 100 times longer than that generated by the Illumina RNA sequencing (only between 100 and 200 bases). Secondly, SMRT sequencing features the “sequencing-by-synthesis” process based on a real-time imaging of fluorescently tagged dNTPs which are incorporated into complementary DNA strands mediated by DNA polymerases, conferring it much more efficient and faster sequencing than others. Besides, this real-time sequencing technology could perform sequencing for individual template molecule without PCR amplification, resulting in low degree of bias when dealing with those hard-to-sequence regions such as high-GC content genomes and large structural variations (Nakano et al., 2017). Thirdly, observation of each nucleotide in a single DNA molecule allows for the detection of base modifications, such as methylation, through measuring changes in the kinetics of the fluorescence pulses (Roberts et al., 2013). Finally, SMRT sequencing has a relatively high error rate (10%-15%), but unlike the NGS, those errors are random without bias, therefore, those deviations could be effectively eliminated by multiple sequencing, ensuring a high sequencing accuracy (99.999%) (Roberts et al., 2013). Since the advent of this sequencing technology, it has become powerful in a variety of applications such as sequencing of the full transcriptome (Bashir et al., 2012), elucidation of mutations in target regions (English et al., 2012), de novo assembly of unknown genomes (Pendleton et al., 2015), sequencing of giant short tandem repeats (Ummat and Bashir, 2014), and characterization of alternative mRNA splicing isoforms (Harvey et al., 2021).
The greenfin horse-faced filefish, also known as filefish, features its laterally compressed body, blue-green fins, rough skin and a spine-like first dorsal fin. This fish is a temperate demersal species inhabiting in the coastal regions of South Korea, China, and Japan (Liu et al., 2017). As a fish species with great commercial value, the annual catch of filefish in the East China Sea once reached 330 thousand tons, if no less than. Unfortunately, overfishing, environment deterioration and lacking fishery regulations lead to the drastic decline of the wild resources. In recent years, a considerable number of measures such as artificial propagation and intensive aquaculture have been taken to repopulate this fish species (Mizuno et al., 2012). Thanks to the rapid technological advances that offer vast opportunities to apply genomic and molecular techniques, the production efficiency and profitability of aquaculture practice has been largely improved (Bian et al., 2020). Using nanopore sequencing and Hi-C technology, the first chromosome-level of Thamnaconus modestus was constructed in 2021, containing 474.31 Mb of the genome, and 20.923 annotated genes (Bian et al., 2020). In spite of this major progress, most existing gene information was obtained based on the in silico prediction and the annotation of alternative isoforms and analysis of untranslated region was scarce. To fill this gap, SMRT technology was used to construct a high-quality full-length reference transcriptome database for functional gene annotation, diversified splicing isoforms characterization, coding sequence (CDS) identification, lncRNA prediction, transcriptional factor analysis, and landscapes of SSR motifs, etc. Those results will provide valuable genomic resources for further studies on the transcriptional regulation and annotation of functional genes that play important roles in the biological processes of Thamnaconus modestus
Data description
Sample collection and RNA extraction
Six Thamnaconus modestus biological samples at different developmental stages, namely fertilized eggs, 1 day-after-hatch (DAH) larvae, 7 DAH larvae, 21 DAH juveniles, 42 DAH juveniles and tissue samples (heart, gills, brain, liver, spleen, intestine, muscle, gonads) from individual male and female broodstock (~425 g) were collected from Yinze Fishery Co., Ltd., Wendeng, Shandong. The embryos, larvae, juveniles, and broodstocks were reared in indoor flow-through cement tanks with ample supply of filtered seawater at 22-26°C. All samples were washed and quickly frozen in liquid nitrogen before being stored in -80°C refrigerator for further experiments. RNA was extracted from the biological samples prepared before using the TRIzol reagent method according to the published literature (Li et al., 2020). RNA integrity and purity were examined by 1% agarose gel electrophoresis and NanoDrop UV analysis, respectively. And the concentration of extracted RNA was then quantified using a spectrophotometer. Only the high-quality RNA with an OD260/280 value of 1.8 ~ 2.0 would be considered for subsequent usage. Equal amounts of RNA extraction from each of six sampling stages were pooled to generate one mixed sample (~5 ug) for SMRT library preparation.
Library construction and PacBio SMRT sequencing
For the preparation of SMRT sequencing library, the RNA extraction from the samples aforementioned were used to generate the full-length cDNA using the NEBNext Single Cell/Low Input cDNA Synthesis & Amplification Module (New England Biolabs, Inc.). Subsequently, PCR amplification was performed using the synthesized cDNA template, and the resultant products were subject to BluePippin™ Size Selection System (Sage Science, Beverly, MA, USA) for length selection. After repairing DNA damage and ends of fragmented DNA, hairpin adapters were ligated to both ends of target double-stranded DNA molecules to form a closed single-stranded circular DNA template called SMRT bell. Each SMRT bell template was then combined to polymerases using the DNA/Polymerase Binding Kit (PacBio, USA) (Chin et al., 2013). Sequencing with 1-6k cDNA was carried out on the PACBIO SEQUEL I platform by Biomarker Technologies Corporation. Benchmarking Universal Single-Copy Orthologs (BUSCO) tool suite (3.0.2) provided a quantitative evaluation of the completeness of non-redundant full-length transcriptome within the scope of eukaryote lineage_odb10 (Simão et al., 2015). The results indicated that 41.7% (1,078 genes) were complete and single-copy BUSCOs, 38.9% (1006 genes) were complete and duplicated BUSCOs, 8.0% (1006 genes) were fragmented, and 11.4% (295 genes) were missing BUSCOs. Herein, we detected a high level of duplication, which may be due to the fact that the present transcriptome is not filtered for isoforms (Manni et al., 2021).
Quality filtering and long reads processing
Raw reads were processed to obtain circular consensus sequencing (CCS) reads using Iso-seq v3.4.0 pipeline in SMRT Link V10.1 following the criteria of Full Passes >=3 and Predicted Accuracy>=0.9. Next, full-length, non-chimeric (FLNC) transcripts were determined by scanning the poly(A) tail signal and the 5’/3’ cDNA primers. High quality FL transcripts were classified with the criteria post-correction accuracy above 99%. Then Iterative Clustering for Error Correction (ICE) algorithm was used to obtain consensus isoforms and FL consensus sequences from ICE was polished using Quiver software. Next, the reduction of redundancy was carried out by using cDNA-Cupcake 28.0.0. In brief, FL consensus sequences were mapped to the reference genome using the minimap2 2.20-r1061 (-ax splice -uf –secondary=no -C5), the resultant reads were further collapsed by cDNA-Cupcake 28.0.0 (min-coverage=85% and min-identity =90%). As presented in Figure 1A, a total of 70.02 Gb of subreads were generated with 649,867 polished CCSs including 527,444 FLNC sequences. 118,452 non-redundant transcripts were produced from 210,435 high-confidence consensus sequences, from which 19,299 novel gene loci and 95,583 new transcripts were identified after differential splicing analysis.
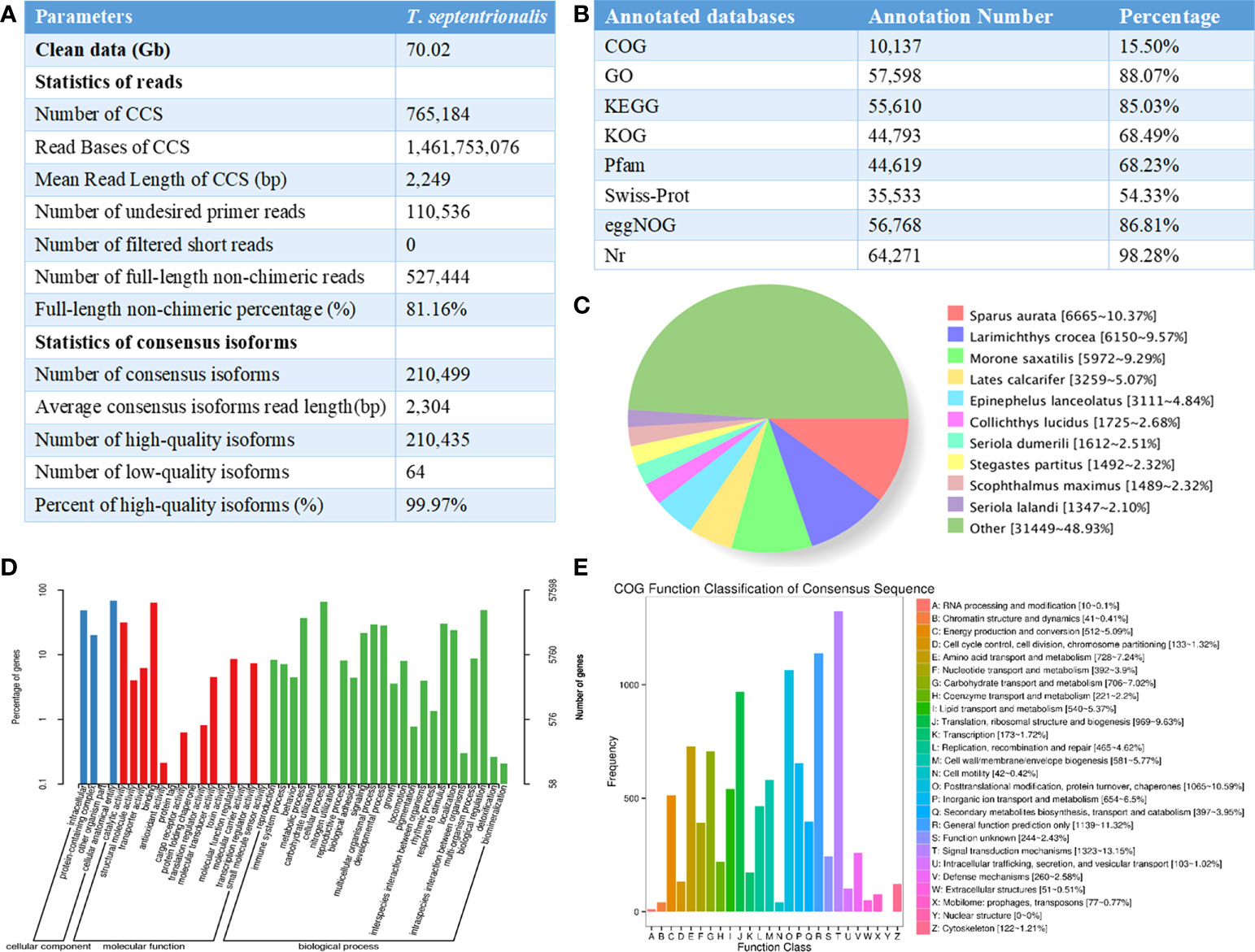
Figure 1 (A) Summary for the full-length transcriptome of Thamnaconus modestus; (B) Statistics of the annotation results; (C) The distribution diagram of species with homologous sequences in NR. (D) Go functional annotation of the Thamnaconus modestus transcriptome. The x-axis represents different gene categories, the right and left y-axis indicate the number of transcripts and percentage of genes. (E) Functional annotation of the T. transcriptome full-length transcripts using COG database, letters on the x-axis represents different COG categories shown detail on the right legend, the y-axis represents the number of the transcripts.
Gene functional annotation
All the non-redundant transcript sequences were functionally annotated using DIAMOND 2.0.4 software based on the following databases: NCBI non-redundant protein sequences (NR), Protein family (Pfam), KOG/COG/eggNOG (Clusters of Orthologous Groups of proteins), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) (Yuan et al., 2020). The E-value threshold for functional assignment was set at “1e-5”, and the parameter were “-k 100 -e -evalue 1e-5 -f 5”. In total, 65,397 new transcripts were functionally annotated by aligning to the databases, and the annotation rates were shown in Figure 1B.
To validate the full-length transcripts of Thamnaconus modestus, the non-redundant transcripts generated by SMRT sequencing were served as queries against other fishes. Results in NR database showed the species that share the highest gene sequence similarity with Thamnaconus modestus. As shown in Figure 1C, the top four species were Sparus aurata (6665, 10.37%), Larimichthys crocea (6150, 9.57%), Morone saxatlis (5972, 9.29%), and Lates calcarifer (3259, 5.07%), respectively. GO enrichment analysis was undertaken to perform the functional annotation of the Thamnaconus modestus full-length transcripts. All GO annotations were classified into three main categories, namely “cellular components”, “biological processes”, and “molecular functions”. Cellular process is the most enriched group in the category of “biological process”, binding is the most abundant term in “molecular function” category, and cellular anatomical entity is the most highly represented in “cellular component” (Figure 1D). The COG analysis indicated that 10,057 transcripts were grouped into 26 categories. The dominant three subgroups were signal transduction mechanisms (1,323, 13.15%), general function prediction only (1,139, 11.32%), and posttranslational modification, protein turnover and chaperones (1,065, 10.59%), respectively (Figure 1E).
Gene structure analysis
Prediction of candidate coding regions and transcription factors
TransDecoder v5.0.0 identifies coding regions and untranslated regions within transcripts generated by full-length transcript assembly using the PacBio sequencing. A total of 72,918 predicted CDS, including 48,939 complete opening reading frames (ORFs), and the length distributions of the deduced proteins are shown in Figures 2A, B. TFs are considered as the central part of the transcriptional regulatory system. In this present study, TFs were identified by the animal TFDB 2.0 database (Zhang et al., 2014), and hmmscan 3.1b2 search was used to identify TFs according to the Pfam files of the transcription factor family (Finn et al., 2011). A total of 3,427 putative TFs were identified in the full-length transcriptome, and the top five TF families are Zinc finger C2H2 (Zf-C2H2), Homeobox, ZBTB, bHLH, and High Mobility Group (HMG) (Figure 2C), indicating their important roles of those identified TFs in the regulatory mechanism of transcription.
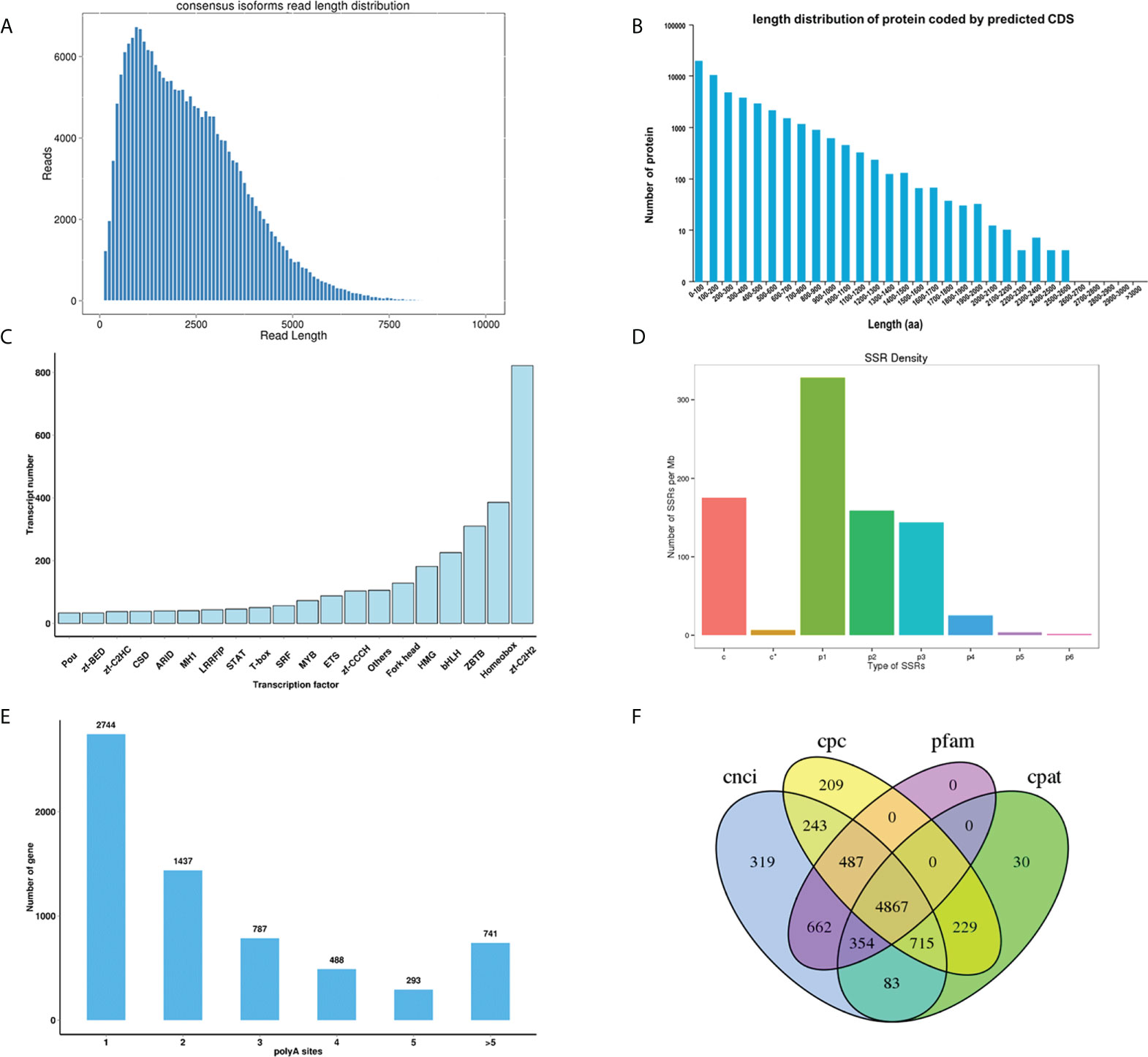
Figure 2 (A) Consensus isoforms read length distribution of Thamnaconus modestus; (B) The coding protein length distribution of the predicted CDS; (C) Transcriptional factor prediction; (D) Distribution of different types of SSRs. (E) APA polyA sites distribution; (F) Venn diagram of the number of lncRNAs identified by CNCI, CPC, Pfam and CPAT.
Alternative splicing analysis
SMRT sequencing is able to identify alternative splicing (AS) events by directly comparing isoforms of the same gene. Transcripts were validated based on known reference genomic annotations. Using Astalavista v3.2 tool (Wang et al., 2015), a total of 18,936 AS events were detected among the transcripts, which were then clustered into five AS types, namely intron retention (IR), exon skipping (ES), alternative donor sites (AD), alternative acceptor sites (AA) and Mutually Exclusive Exons (MEE), with IR (48.82%) being the most abundant type, followed by ES (21.08%), AA (16.49%), AD (11.68%), and MEE (1.87%).
Simple sequence repeats analysis
SSRs (also known as microsatellites) are a group of short tandem DNA repeats that have been used as valuable genetic markers for applied molecular breeding studies and basic genetic diversity analyses (Bhattarai et al., 2021). SSRs of the transcriptome were located and identified from transcripts with a length of over 500 bp using MISA-web 1.0 (http://pgrc.ipk-gatersleben.de/misa/misa.html). Results indicated that seven types, totaling 339,514 SSRs were identified in the present transcriptome, among which the Mono-nucleotide motif (142,815) was the most abundant type, followed by di nucleotide (109,157), compound SSR (84,118), Tri nucleotide (71,244), Tetra nucleotide (13,929), Penta nucleotide (1,606), Hexa nucleotide motifs (763) (Figure 2D and Supplementary Table 1).
Polyadenylation and alternative poly analysis
APA analysis was conducted with Transcriptome Analysis Pipeline for Isoform Sequencing (TAPIS) 1.1.3 program (https://bitbucket.org/comp_bio/tapis/overview). Results showed that 6,490 genes have at least one poly(A) site, and 741 genes have at least five poly(A) sites (Figure 2E)
Long-chain noncoding RNA prediction
LncRNA, as the name implies, is a type of RNA molecule with a length of more than 200 nt, and they do not encode proteins. LncRNA plays important roles in the regulation of gene transcription by controlling chromatin organization and telomere replication (Batista and Chang, 2013). Moreover, it has been well elucidated that lncRNA could modulate mRNA stability and regulate translation in the cytoplasm (Kung et al., 2013). In this study, four computational approaches include Coding Potential Calculator 2 (CPC2) 0.1 (Kong et al., 2007), Coding-Non-Coding Index (CNCI) V2 (Sun et al., 2013), Coding Potential Assessment Tool (CPAT) 1.2.2 (Wang et al., 2013), Pfam protein structure domain analysis (PfamScan) 1.60 (Finn et al., 2016) were combined to sort non-protein coding RNA candidates from putative protein-coding RNAs that were filtered out using a minimum length and exon number threshold, identifying a shared dataset of 4867 lncRNAs from the non-redundant full-length transcripts (Figure 2F). According to their locus information on the reference genome, those identified lncRNAs were further classified into four categories (sense, antisense, intronic, and normal) (Supplementary Figure 1). Further efforts need to be made to investigate the functions of those novel lncRNAs.
In the present study, we used PacBio Iso-seq sequencing platform to obtain a comprehensive full-length transcriptome of Thamnaconus modestus of different developmental stages. The publication of full-length transcript isoforms improves the accuracy and reliability of gene annotation, development of molecular markers, and lncRNA prediction. Thus, this comprehensive full-length transcriptome could serve as a valuable resource for ongoing research of functional genes, molecular markers, molecular events, and signaling pathways, providing support for the exploration of interrelationship exist between genomic basis and mechanism of biological processes of Thamnaconus modestus.
Data availability statement
The datasets presented in this study can be found in online Short Read Archive (SRA) of NCBI repositories via accession number: PRJNA862466.
Ethics statement
The animal study was reviewed and approved by the Institutional Animal care and Use committee of Qingdao Agricultural University, Qingdao, China.
Author contributions
QL conceptualized the experiment. NW, CS, and HM collected tissue samples. QL and NW performed the bioinformatics analysis. QL drafted the manuscript and revised the manuscript. LZ and JC provided the funding to conduct the project. All authors contributed to the article and approved the submitted version.
Funding
This project was financially supported by Shandong Technical System of Fish Industry (SDAIT-12-05), the National Natural Science Foundation of China (32102807).
Conflict of interest
LZ is employed by Tongwei Agricultural Development Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1028231/full#supplementary-material
References
Abdelrahman H., ElHady M., Alcivar-Warren A., Allen S., Al-Tobasei R., Bao L., et al. (2017). Aquaculture genomics, genetics and breeding in the united states: current status, challenges, and priorities for future research. BMC Genomics 18 (1), 191. doi: 10.1186/s12864-017-3557-1
Bashir A., Klammer A. A., Robins W. P., Chin C.-S., Webster D., Paxinos E., et al. (2012). A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 30 (7), 701–707. doi: 10.1038/nbt.2288
Batista P. J., Chang H. Y. (2013). Long noncoding RNAs: cellular address codes in development and disease. Cell 152 (6), 1298–1307. doi: 10.1016/j.cell.2013.02.012
Bhattarai G., Shi A., Kandel D. R., Solís-Gracia N., da Silva J. A., Avila C. A. (2021). Genome-wide simple sequence repeats (SSR) markers discovered from whole-genome sequence comparisons of multiple spinach accessions. Sci. Rep. 11 (1), 9999. doi: 10.1038/s41598-021-89473-0
Bian L., Li F., Ge J., Wang P., Chang Q., Zhang S., et al. (2020). Chromosome-level genome assembly of the greenfin horse-faced filefish (Thamnaconus septentrionalis) using Oxford nanopore PromethION sequencing and Hi-c technology. Mol. Ecol. Resour 20 (4), 1069–1079. doi: 10.1111/1755-0998.13183
Chin C.-S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10 (6), 563–569. doi: 10.1038/nmeth.2474
English A. C., Richards S., Han Y., Wang M., Vee V., Qu J., et al. (2012). Mind the gap: Upgrading genomes with pacific biosciences RS long-read sequencing technology. PloS One 7 (11), e47768. doi: 10.1371/journal.pone.0047768
Finn R. D., Clements J., Eddy S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39 (Web Server issue), W29–W37. doi: 10.1093/nar/gkr367
Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., et al. (2016). The pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44 (D1), D279–D285. doi: 10.1093/nar/gkv1344
Harvey S. E., Lyu J., Cheng C. (2021). Methods for characterization of alternative RNA splicing. Methods Mol. Biol. 2372, 209–222. doi: 10.1007/978-1-0716-1697-0_19
Kong L., Zhang Y., Ye Z. Q., Liu X. Q., Zhao S. Q., Wei L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35 (Web Server issue), W345–W349. doi: 10.1093/nar/gkm391
Kung J. T., Colognori D., Lee J. T. (2013). Long noncoding RNAs: past, present, and future. Genetics 193 (3), 651–669. doi: 10.1534/genetics.112.146704
Levene M. J., Korlach J., Turner S. W., Foquet M., Craighead H. G., Webb W. W. (2003). Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299 (5607), 682–686. doi: 10.1126/science.1079700
Li Q., Cui K., Wu M., Xu D., Mai K., Ai Q. (2020). Polyunsaturated fatty acids influence LPS-induced inflammation of fish macrophages through differential modulation of pathogen recognition and p38 MAPK/NF-κB signaling. Front. Immunol. 11 (2238). doi: 10.3389/fimmu.2020.559332
Liu K., Zhang L., Zhang Q., Chen S., Liu C., Bian L. (2017). Study on thamnaconus septentrionalis under industrial aquaculture condition. J. Aquaculture 44 (3), 35–40 (in Chinese). doi: 10.3969/j.issn.1007-9580.2017.03.006
Manni M., Berkeley M. R., Seppey M., Zdobnov E. M. (2021). BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 1 (12), e323. doi: 10.1002/cpz1.323
Metzker M. L. (2010). Sequencing technologies — the next generation. Nat. Rev. Genet. 11 (1), 31–46. doi: 10.1038/nrg2626
Mizuno K., Shimizu-Yamaguchi S., Miura C., Miura T. (2012). Method for efficiently obtaining fertilized eggs from the black scraper Thamnaconus modestus by natural spawning in captivity. Fisheries Sci. 78 (5), 1059–1064. doi: 10.1007/s12562-012-0527-z
Munroe D. J., Harris T. J. R. (2010). Third-generation sequencing fireworks at Marco island. Nat. Biotechnol. 28 (5), 426–428. doi: 10.1038/nbt0510-426
Nakano K., Shiroma A., Shimoji M., Tamotsu H., Ashimine N., Ohki S., et al. (2017). Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 30 (3), 149–161. doi: 10.1007/s13577-017-0168-8
Pendleton M., Sebra R., Pang A. W., Ummat A., Franzen O., Rausch T., et al. (2015). Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12 (8), 780–786. doi: 10.1038/nmeth.3454
Roberts R. J., Carneiro M. O., Schatz M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14 (6), 405. doi: 10.1186/gb-2013-14-6-405
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351%JBioinformatics
Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41 (17), e166. doi: 10.1093/nar/gkt646
Ummat A., Bashir A. (2014). Resolving complex tandem repeats with long reads. Bioinformatics 30 (24), 3491–3498. doi: 10.1093/bioinformatics/btu437
Wang L., Park H. J., Dasari S., Wang S., Kocher J. P., Li W. (2013). CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 41 (6), e74. doi: 10.1093/nar/gkt006
Wang J., Ye Z., Huang T. H.-M., Shi H., Jin V. (2015). A survey of computational methods in transcriptome-wide alternative splicing analysis. Biomol Concepts 6 (1), 59–66. doi: 10.1515/bmc-2014-0040
Yuan H., Zhang X., Zhao L., Chang H., Yang C., Qiu Z., et al. (2020). Characterization and analysis of full-length transcriptomes from two grasshoppers, gomphocerus licenti and mongolotettix japonicus. Sci. Rep. 10 (1), 14228. doi: 10.1038/s41598-020-71178-5
Keywords: single-molecule real-time sequencing, alternative splicing, greenfin horse-faced filefish (Thamnaconus modestus), full-length transcript, simple sequence repeat, long non-coding RNA
Citation: Li Q, Wang N, Sui C, Mao H, Zhang L and Chen J (2022) PacBio single molecule real-time sequencing of a full-length transcriptome of the greenfin horse-faced filefish Thamnaconus modestus. Front. Mar. Sci. 9:1028231. doi: 10.3389/fmars.2022.1028231
Received: 25 August 2022; Accepted: 09 September 2022;
Published: 27 September 2022.
Edited by:
Rodrigo Vidal, University of Santiago, ChileReviewed by:
Nyok Sean Lau, Universiti Sains Malaysia (USM), MalaysiaPing Liu, Yellow Sea Fisheries Research Institute (CAFS), China
Copyright © 2022 Li, Wang, Sui, Mao, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinghua Chen, Y2hlbmpoQHFhdS5lZHUuY24=
†These authors have contributed equally to this work