 Yuhang Ren†
Yuhang Ren† Yuan Tian
Yuan Tian Haishen Wen
Haishen Wen Xin Qi
Xin Qi Yun Li
Yun Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 03 October 2022
Sec. Global Change and the Future Ocean
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1024218
This article is part of the Research Topic The Adaptation and Response of Aquatic Animals in the Context of Global Climate Change View all 18 articles
Hypoxia is one of the most important environmental stressors in aquatic ecosystems. To deal with the hypoxia environment, fishes exhibit a series of physiological and molecular responses to maintain homeostasis and organism functions. In the present study, hypoxia-induced changes in gene expression profiles and alternative splicing (AS) events in spotted sea bass (Lateolabrax maculatus), a promising marine-culture fish species in China, were thoroughly investigated by RNA-Seq analysis. A total of 1,242, 1,487 and 1,762 differentially expressed genes (DEGs) were identified at 3 h, 6 h and 12 h in gills after hypoxia stress. Functional enrichment analysis by KEGG and GSEA demonstrated that HIF signal network system was significantly activated and cell cycle process was remarkably suppressed in response to hypoxia. According to the temporal gene expression profiles, six clusters were generated and protein-protein interaction (PPI) networks were constructed for the two clusters that enriched with hypoxia-induced (cluster 2) or -suppressed genes (cluster 5), respectively. Results showed that HIF signaling related genes including vegfa, igf1, edn1, cox2b, cxcr4b, ctnnb1, and slc2a1a, were recognized as hubs in cluster 2, while mcm2, chek1, pole, mcm5, pola1, and rfc4, that tightly related to cell cycle, were down-regulated and considered as hubs in cluster 5. Furthermore, a total of 410 differential alternative splicing (DAS) genes were identified after hypoxia, which were closely associated with spliceosome. Of them, 63 DAS genes also showed differentially expressed levels after hypoxia, suggesting that their expression changes might be regulated by AS mechanism. This study revealed the key biological pathways and AS events affected by hypoxia, which would help us to better understand the molecular mechanisms of hypoxia response in spotted sea bass and other fish species.
Fish are frequently exposed to variations in oxygen levels due to the rapid fluctuations in patterns of oxygen production and consumption in aquatic environment (Nikinmaa and Rees, 2005). As one of the most important environmental stressors in aquatic ecosystems, hypoxia is usually defined as the dissolved oxygen (DO) levels of less than 2.0 mg/L, which is low enough to negatively influence the normal physiological conditions of fishes (Hrycik et al., 2017). If severe hypoxia persists, fish will eventually die and result in serious yield losses (Abdel-Tawwab et al., 2019). Hypoxia occurres with temperature, seasonal and compositional changes in water (Xiao, 2015). In aquaculture system, hypoxia can be accelerated by several factors such as high stocking density, water pollution, excessive nutrient enrichment and human activities (Ng and Chiu, 2020). To deal with the hypoxia environment, fishes exhibit a number of behavioral, morphological, physiological and molecular responses to maintain homeostasis and organism functions. For example, in order to get more oxygen, they try to breathe directly by emerging at the water surface (Xiao, 2015), or enlarge the respiratory surface area by remodeling their gills (Sollid et al., 2005). Some fishes make a rapid reorganization of cellular metabolism to suppress ATP and O2 consumption (Richards, 2011), and increase the number of red blood cells to improve oxygen-carrying capacity (Claireaux et al., 1988). Moreover, hypoxia response is always correlated with the activation of anaerobic metabolism and the increase of anaerobic glycolysis, in order to meet the high energy demand during hypoxia (Abdel-Tawwab et al., 2019).
At the molecular level, several proteins and signaling pathways have been reported to involve in hypoxia response. Among them, hypoxia-inducible factor 1 (HIF-1) is recognized as master regulator of the cellular responses to hypoxia by promoting target genes transcription under hypoxia (Déry et al., 2005). HIF-1 is a heterodimeric DNA-binding complex consisting of an oxygen-labile α-subunit (HIF-1α) and a stably expressed β-subunit (HIF-1β). Under normoxia (normal oxygen condition), HIF-1α is hydroxylated by prolyl hydroxylase domain-containing enzymes (PHDs) whose activities are closely related with oxygen levels, and eventually degraded by proteasome. In contrast, when exposed to hypoxia conditions, HIF-1α is not hydroxylated because of the catalytic activity of PHD is inhibited by the lack of oxygen. Meantime, HIF-1 heterodimers rapidly accumulate and bind to hypoxia response elements (HREs) contained in the promoter or enhancer regions of the hypoxia-inducible genes (Bartoszewski et al., 2019). These HIF-1 target genes were demonstrated to involve in numbers of signaling pathways such as redox status (Semenza, 2001), AMPK (Bohensky et al., 2010), MAPK (Zhang et al., 2013), PI3K-Akt (Zhang et al., 2018), mTOR (Wei et al., 2019), and VEGF (Kajimura et al., 2006), which affect multiple aspects of the physiological responses including proliferation, apoptosis, and differentiation (Zhu et al., 2013). Moreover, it has recently become clear that HIF-1 pathway is not a linear cascade of signal transduction but a multi-level regulatory network representing a high degree of complexity (Bárdos and Ashcroft, 2005; Yee Koh et al., 2008; Zheng et al., 2008; Agani and Jiang, 2013; Fábián et al., 2016).
Investigation into the hypoxia responses of fish will not only help us to understand the strategies and mechanisms for environmental stress adaptation in fish, but will also guide us in improving fish hypoxia tolerance capabilities and preventing economic losses in aquaculture, and artificial breeding of hypoxia-tolerant fish strain/species in the future. In recent years, with the rapid development of high-throughput sequencing technology, RNA-Seq tool has been successfully used in studying the molecular responses to hypoxia in various fish species, and several known and novel hypoxia-related genes and pathways have been identified in different tissues. For examples, in large yellow croaker (Larimichthys crocea), genes involved in innate immunity, ion transport and glycolytic pathways were significantly differentially expressed in the gill and/or heart under hypoxia, indicating they may contribute to maintain cellular energy balance during hypoxia (Mu et al., 2020). In bighead carp (Hypophthalmichthys nobilis), the cardiac transcriptomic analysis identified that MAPK signaling pathway played a key role in cardiac tolerance after acute hypoxia treatment (Zhou et al., 2020). In the gill of Nile tilapia (Oreochromis niloticus), the hypoxia responsive genes were significantly enriched in energy and immune-related pathways including glycolysis, metabolic process and antigen processing and presentation (Li et al., 2017). For channel catfish (Ictalurus punctatus), RNA-Seq analysis with swimbladder samples revealed that many genes affected by hypoxia were enriched in the HIF, MAPK, PI3K/Akt/mTOR, VEGF and Ras signaling pathways (Yang et al., 2018). Moreover, liver transcriptomes under hypoxia have been widely studied in aquaculture fishes such as large yellow croaker (Ding et al., 2020), silver sillago (Sillago sihama) (Tian et al., 2020a), largemouth bass (Micropterus salmoides) (Sun et al., 2020), golden pompano (Trachinotus blochii) (Sun et al., 2021b), rainbow trout (Oncorhynchus mykiss) (Hou et al., 2020) and Nile tilapia (Oreochromis niloticus) (Ma et al., 2021).
Alternative splicing (AS) mechanism are widely existed in eukaryotes that could generate multiple transcripts and enables different proteins to be synthesized from one single gene (Nilsen and Graveley, 2010). This regulatory mechanism significantly expands transcript variability and proteome diversity, servicing as an enhancing ability to cope with biotic or abiotic stresses in eukaryotes, including fishes (Mastrangelo et al., 2012; Lee and Rio, 2015). It has been documented that there are changes in splicing patterns for a set of functional genes that play central roles in cold-acclimation responses in Atlantic killifish (Fundulus heteroclitus), threespine stickleback (Gasterosteus aculeatus) and zebrafish (Danio rerio) (Healy and Schulte, 2019). AS events of several RNA-binding proteins are significantly induced by different salinity environments in turbot (Scophthalmus maximus), tongue sole (Cynoglossus semilaevis) and steelhead trout (Oncorhynchus mykiss), which may affect the splicing decisions of many downstream target genes in response to osmoregulation (Tian et al., 2022). However, the AS events related to hypoxia adaptation are only reported in Nile tilapia until now. A total of 103 genes undergo differential usage of exons and splice junction events after acute hypoxia stress in the heart of Nile tilapia, the biological function of which are tightly associated with structural constituent of ribosome, structural molecule activity and ribosomal protein (Xia et al., 2017).
Spotted sea bass (Lateolabrax maculatus), also known as Chinese sea bass, is a euryhaline and eurythermic fish species which broadly distributed along the coasts of China and Indo-China Peninsula (Chen et al., 2019). Because of its fast growth, high nutritive value and pleasant taste, the industry of spotted sea bass is considered as one of the leading aquaculture marine fish in China (Wen et al., 2016; Liu et al., 2020), with the production capacity of 195,000 tons last year (Yearbook, 2021). However, spotted sea bass is threatened by hypoxia in natural watersheds resulted from ongoing global warming and environmental pollution, as well as in cultured conditions due to the sustained high temperature in summer and high-density farming (Dong et al., 2020). To date, studies about hypoxia responses of spotted sea bass remains largely unknown. The fish gill is the dominant organ responsible for physiological exchanges with the surrounding environment, playing a primary role in gas exchange and is the first target under hypoxia (Evans et al., 2005), but the underlying molecular basis has not been explored in spotted sea bass. Therefore, in this study, hypoxia-induced changes in gene expression profiles and AS events in gills were investigated by RNA-Seq analysis in spotted sea bass. Our results revealed the key biological pathways and AS events affected by hypoxia, and candidate regulatory hub genes involving in hypoxia response, which will provide new insights into the elucidation of the responses and adaption mechanism to environmental stress in fish.
This study was carried out in accordance with the rules and approval of the respective Animal Research and Ethics Committees of Ocean University of China (Permit Number: 20141201), and was not involved in any endangered or protected species.
A total of 60 healthy spotted sea bass (body length: 48.76 ± 4.26 cm, body weight: 178.25 ± 18.56 g) were obtained from Shuangying Aquatic Seeding Co., Ltd. (Lijin, Shandong, China). Before hypoxia experiment, the individuals were acclimated in the square tank (5 m × 5 m × 1 m, L × W × H) for two weeks. All the water was obtained from the natural sea area in Bohai, China (38.04°N, 118.58°E), followed by plain sedimentation, sand filtration and UV sterilization. During the periods of acclimation, the water quality conditions were kept relatively constant and suitable for the culture of spotted sea bass, including temperature (17.0 ± 1.0°C), dissolved oxygen saturation (7.0 ± 0.5 mg/L), salinity (27 - 30) and pH (6.5 - 7.5).
After acclimation, fish individuals were transferred to three tanks (0.5 m × 0.5 m × 1 m, L × W × H) for hypoxia challenge experiment and the density was set as 20 individuals per tank. The oxygen level in each tank was quickly reduced to 1.1 ± 0.14 mg/L within ~ 1h by bubbling nitrogen gas. Then, the hypoxia experiment started and lasted for 12 h. During the periods of experiment, dissolved oxygen levels were maintained relatively constant by pumping the mixture of air and nitrogen gas and continuously monitored using YSI DO200 oxygen meter (YSI EcoSense, OH, USA) every ten minutes. 3 individuals per tank (a total of 9 individuals for each time point) were anesthetized and euthanized with MS-222, and collected for gill tissues at 0 h (before hypoxia experiment), 3 h, 6 h and 12 h after hypoxia (named as T0, T3, T6 and T12 groups, respectively). Then gill tissues were quickly frozen in liquid nitrogen until RNA extraction.
Total RNA of gill tissues was extracted using TRIzol reagents (Invitrogen, USA). The concentration of total RNA was determined by NanoDrop 2000 (Thermo Scientific, Waltham, MA), and RNA integrity was checked by the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Equal quantity of high-quality RNA from the 3 individuals each tank was pooled as one sample, and 3 replicated samples were generated for each time point to minimize the individual differences. A total of 12 sequencing libraries (3 replicated samples × 4 time points), named T0 (T0-1, T0-2, T0-3), T3 (T3-1, T3-2, T3-3), T6 (T6-1, T6-2, T6-3), and T12 (T12-1, T12-2, T12-3), were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA), respectively. Sequencing was conducted on the Illumina Novaseq platform, generating 150 bp paired-end reads.
Raw reads were treated to discard adapter sequences and ambiguous nucleotides using fastp v0.20.1 software to generate the clean reads. The reference genome file of spotted sea bass (Assembly ID: GCA_004028665.1) was used for the following bioinformatics analysis. Index of the reference genome was built using “hisat2-build” function of Hisat2 v2.1.0 software (Kim et al., 2015) and paired-end clean reads were mapped to the reference genome using Hisat2 with the default parameters. The count matrixes were constructed using featureCounts software from Subread v1.6.4 program, which were then transformed into fragments per kilobase of transcript per million fragments mapped (FPKM) for the normalization of gene expression levels. DEGs among the three comparisons (T3 vs. T0, T6 vs. T0 and T12 vs. T0) were calculated using the scripts run_DE_analysis.pl in Trinity program (Grabherr et al., 2011), and the calculation method was performed using the DESeq2 package. The threshold of DEGs were set as false discovery rate (FDR)-p < 0.05 and |log2 (fold change)| ≥ 1. Expression cluster analysis of time-series data was performed based on fuzzy c-means using Mfuzz v2.48.0 R package (Kumar and Futschik, 2007) and the number of centers was set as 6.
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DEGs were implemented using clusterProfiler v4.2.2 R package (Yu et al., 2012) and FDR-p < 0.05 was considered as statistically significant. Additionally, Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was performed to interpret expression matrix from RNA-Seq data. The FPKMs of all genes in gill transcriptome were used for the calculation of normalized enrichment score (NES) in GSEA. |NES|>1, FDR-p < 0.25 and Nominal (NOM) p-value < 0.05 were regarded as the significant threshold.
The PPI network was constructed via STRING v11.5 (Szklarczyk et al., 2016). Minimum required interaction score was settled as 0.4. The predicted network was analyzed and visualized by Cytoscape v3.8.0 software. The highly connected protein nodes were determined by Cyto-Hubba v0.1, a plug-in of Cytoscape software. Significant modules in PPI network were identified by MCODE (Bader and Hogue, 2003) v1.6.1 of Cytoscape, which meet the following parameters: MCODE score ≥ 5, degree cut-off = 2, node score cut-off = 0.2, max depth = 100, and k-score = 2.
rMATS v4.0.1 (Shen et al., 2014) was utilized to determine DAS events by computing the inclusion level from two-group RNA-Seq data with replicates. Five classical types of DAS events were identified including exon skipping (ES), intron retention (IR), alternative 3’ splice sites (A3SS), alternative 5’ splice sites (A5SS), and mutually exclusive exon (ME). In details, rMATS applied a hierarchical framework to simultaneously model the variability among replicates, and estimated uncertainty of inclusion and exclusion isoform proportions in individual replicates. Mature mRNAs including or excluding additional sequences were regarded as inclusion or exclusion isoforms, respectively (Ding et al., 2017). A likelihood-ratio test was used to calculate the p-value and FDR-p of the inclusion levels between two sets of RNA-Seq datasets. FDR-p < 0.05 was set as the threshold for significant DAS events. DAS genes were determined and their KEGG functional enrichment analysis was performed by the same procedures as for DEGs.
To validate the gene expression results of DEGs generated from RNA-Seq analysis, qPCR was performed to detect the relative expression levels of several DEGs among different pairwise comparisons. In details, total RNA of gills collected from hypoxia experiment was reverse-transcribed to cDNA using PrimeScript RT reagent kit (Takara, Shiga, Japan) following the manufacturer’s instructions. 10 × diluted cDNA (~100ng/μl) was served as the template for PCR amplification, which was conducted using FastPfu reagent kit (TransGen, Beijing, China). The gene specific primers used in qPCR were designed using Primer 5 software and listed in Supplementary Table 1. qPCR was performed on the StepOne Plus Real-Time PCR system (Applied Biosystems, CA, USA) using the SYBR Premix Ex Taq™ Kit (Takara, Shiga, Japan). qPCR reaction system consisted of 2 μl of cDNA, 10 μl SYBR premix Ex Taq, 0.4 μl forward primers, 0.4 μl reverse primers, 0.4 μl ROX reference dye, and 6.8 μl ddH2O in a final volume of 20 μl, which was repeated in triplicate and was run in accordance with the following procedure: 95°C for 30 s, then 40 cycles of 95°C for 10 s and 55°C for 30 s, and finally 72°C for 30 s. The 18S ribosomal RNA was considered as the internal reference gene (Strepparava et al., 2014). The expression levels were calculated using the 2−ΔΔCt method.
The cDNA samples were subsequently used as templates for following RT-PCR experiment, which was performed to validate the identified DAS events. Transcript-specific primers were designed to span the predicted splicing events using Primer 5 software and Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Supplementary Table 1). PCR condition was set as follows: 94°C for 5 min, following by 35 cycles of 94°C for 30 s, melting temperature for 30 s, 72°C for a time period that depends on the product sizes, and finally extended at 72°C for 10 min. PCR products were monitored on 1% agarose gel stained by GelStain (TransGen, Beijing, China).
In the present study, all the data are shown as the mean ± standard deviation (SD). Correlation coefficient between the results of DEGs in RNA-Seq and qPCR were determined by SPSS 22.0 software (SPSS Inc., Chicago, USA). One-way ANOVA was conducted followed by Duncan’s multiple tests to identify significance differences when P < 0.05.
In the present study, RNA-Seq analysis was performed on the gill tissues of spotted sea bass at 0 h, 3 h, 6 h and 12 h after hypoxia. As a result, a total of 655,653,966 raw reads were generated respectively, on average, 99.12% of which were considered as clean reads after quality control. Then, all clean reads were aligned to the reference genome of spotted sea bass with mapping ratio ranging from 83.14% to 92.50%, and the unique mapping ratios were different between 80.91% and 89.91% (Supplementary Table 2).
As results showed, the total number of DEGs increased as the hypoxia-treating time was prolonged (Figure 1). In details, a total of 1,242, 1,487 and 1,762 DEGs were identified through the comparisons of T3 vs. T0, T6 vs. T0 and T12 vs. T0, respectively. In those comparison groups, 914 DEGs were significantly up-regulated and 328 DEGs were down-regulated in T3 vs. T0 group (Figure 1A), and 853 up-regulated DEGs and 634 down-regulated DEGs were identified in T6 vs. T0 group (Figure 1B), as well as 1,254 up-regulated and 508 down-regulated DEGs were discovered in T12 vs. T0 group (Figure 1C). The number of DEGs were greatly increased with the prolongation of hypoxia stress from 3 h to 12 h. Notably, 607 DEGs were shared among all three comparison groups, and 181 (overlapped between T3 vs. T0 and T6 vs. T0), 205 (overlapped between T6 vs. T0 and T3 vs. T0), and 247 (overlapped between T6 vs. T0 and T12 vs. T0) DEGs were coexisted in two comparison groups, respectively. Meantime, 249 (T3 vs. T0), 452 (T6 vs. T0) and 703 (T12 vs. T0) DEGs was only appeared in unique comparison group (Supplementary Figure 1).
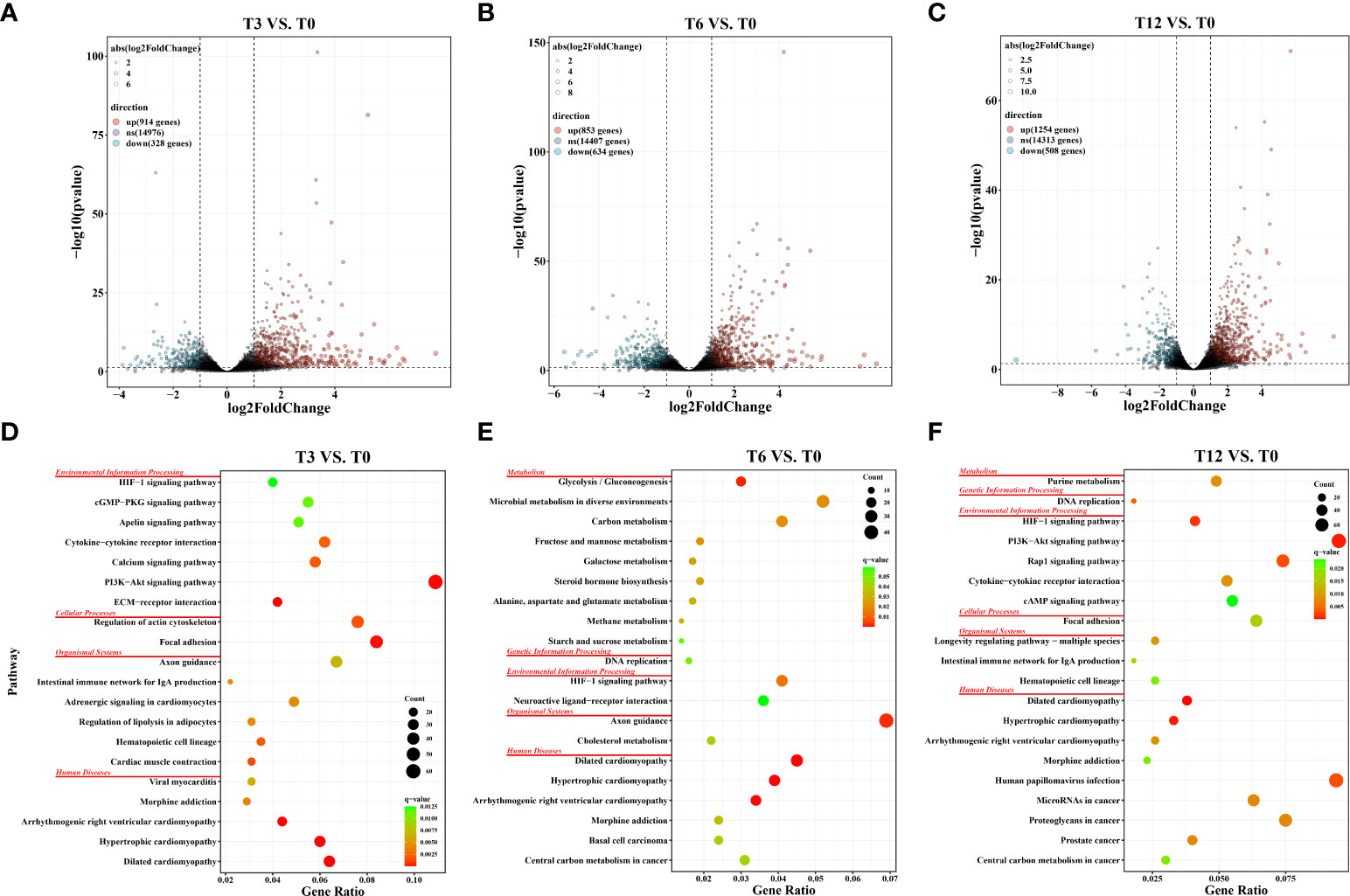
Figure 1 Statistics of differentially expressed genes (DEGs) and KEGG pathway enrichment analysis in gills of spotted sea bass at different time points after hypoxia. (A) Volcano plot of DEGs in the comparison group of T3 vs. T0; (B) Volcano plot of DEGs in the comparison group of T6 vs. T0; (C) Volcano plot of DEGs in the comparison group of T12 vs. T0; (D) KEGG pathway enrichment analysis of DEGs in the comparison group of T3 vs. T0; (E) KEGG pathway enrichment analysis of DEGs in the comparison group of T6 vs. T0; (F) KEGG pathway enrichment analysis of DEGs in the comparison group of T12 vs. T0;.
To confirm the accuracy of our bioinformatic analysis results for gene expression pattern of RNA-Seq data, the expression levels of 10 randomly selected genes were detected by qPCR experiment at 0 h, 3 h, 6 h and 12 h after hypoxia, including hif-1α, L-lactate dehydrogenase A chain (ldha), hexokinase-1 (hk1), glyceraldehyde-3-phosphate dehydrogenase (gapdh), pyruvate kinase M1/2 (pkm), aldolase, fructose-bisphosphate A (aldoa), insulin like growth factor binding protein 1 (igfbp1), insulin like growth factor binding protein 4 (igfbp4), ephrin A2 (epha2) and transforming growth factor beta 3 (tgfβ−3). As shown in Supplementary Figure 2, expression patterns determined by qPCR were consistent with the results of RNA-Seq analysis with the R2 = 0.87, 0.93 and 0.89 in the pairwise comparisons of T3 vs. T0, T6 vs. T0 and T12 vs. T12, respectively. The results indicated the accuracy and reliability of our bioinformatic analysis results.
KEGG enrichment analysis was conducted to investigate the potential biology function of DEGs. Results revealed that a total of 31 (T3 vs. T0), 17 (T6 vs. T0) and 26 (T12 vs. T0) pathways were significantly enriched (FDR-p < 0.05) in the three comparison groups, respectively. The top 20 KEGG pathways (ranked by FDR-p) in each comparison group were displayed in Figures 1D–F, which were categorized into distinct KEGG orthology classes including environmental information processing, organismal systems, cellular processes, metabolism, genetic information processing and human disease. Among them, the two functional classes, environmental information processing and metabolism, which harbored a large number of significantly enriched pathways in our study, were recognized playing essential roles in sensing stress associated with changes in environmental dissolved oxygen and intracellular transduction. For example, at 3 h after hypoxia, the significantly enriched pathways were concentrated in signaling transduction and signaling molecules and interactions, such as ECM-receptor interaction, PI3K-Akt signaling pathway, Calcium signaling pathway, Cytokine-cytokine receptor interaction, Apelin signaling pathway, cGMP-PKG signaling pathway, and HIF-1 signaling pathway (Figure 1D). At 6 h, DEGs involved in four carbohydrate metabolism processes including Glycolysis/Gluconeogenesis, Fructose and mannose metabolism, Galactose metabolism, Starch and sucrose metabolism, as well as two lipid metabolism processes including Steroid hormone biosynthesis and Alanine, aspartate and glutamate metabolism, were significantly enriched (Figure 1E). At 12 h, the DEGs were also significantly enriched in signaling pathways like PI3K-Akt signaling pathway, HIF-1 signaling pathway, Rap1 signaling pathway, Cytokine-cytokine receptor interaction, and cAMP signaling pathway. Notably, the HIF-1 signaling pathway was enriched in all three tested time points after hypoxia (Figure 1F), confirming the key roles of HIF pathway in the response to hypoxia.
In addition to determine the KEGG enrichment information of these DEGs, we performed GSEA analysis for interpreting gene expression data of the gills under hypoxia at the level of gene sets, by using canonical pathways (CP) collection in the Molecular Signatures Database (MSigDB). We identified the significant gene sets within each comparison group (T3 vs. T0, T6 vs. T0, T12 vs. T0), and the top 20 most significant enrichments (ranked in descending order of the |NES-value|) were listed in Supplementary Table 3. Overall, the most significantly enriched gene sets positively correlated with all the hypoxia-treatment groups (gene sets that up-regulated in T3, T6 and T12) included HIF-1-alpha transcription factor network, HIF-2-alpha transcription factor network, Glycolysis and Gluconeogenesis, and Cori cycle (Figure 2 and Supplementary Table 3). Additionally, signal transduction associated pathways such as signaling by Type 1 Insulin-like Growth Factor 1 Receptor (IGF1R), Insulin receptor, VEGF, as well as metabolism pathways including Oxidative phosphorylation and Respiratory electron transport, were also enriched in the gill after hypoxia (Supplementary Table 3). Alternatively, the most significant enriched gene sets positively correlated with the control group (gene sets that up-regulated in T0) were mainly concentrated in cell cycle and DNA replication related processes such as Cell Cycle Mitotic, Cell Cycle Checkpoints, Mitotic G1 phase and G1/S transition, G2/M Checkpoints, DNA Replication Pre-Initiation, Activation of the pre-replicative complex, and other associated pathways (Figure 2 and Supplementary Table 3).
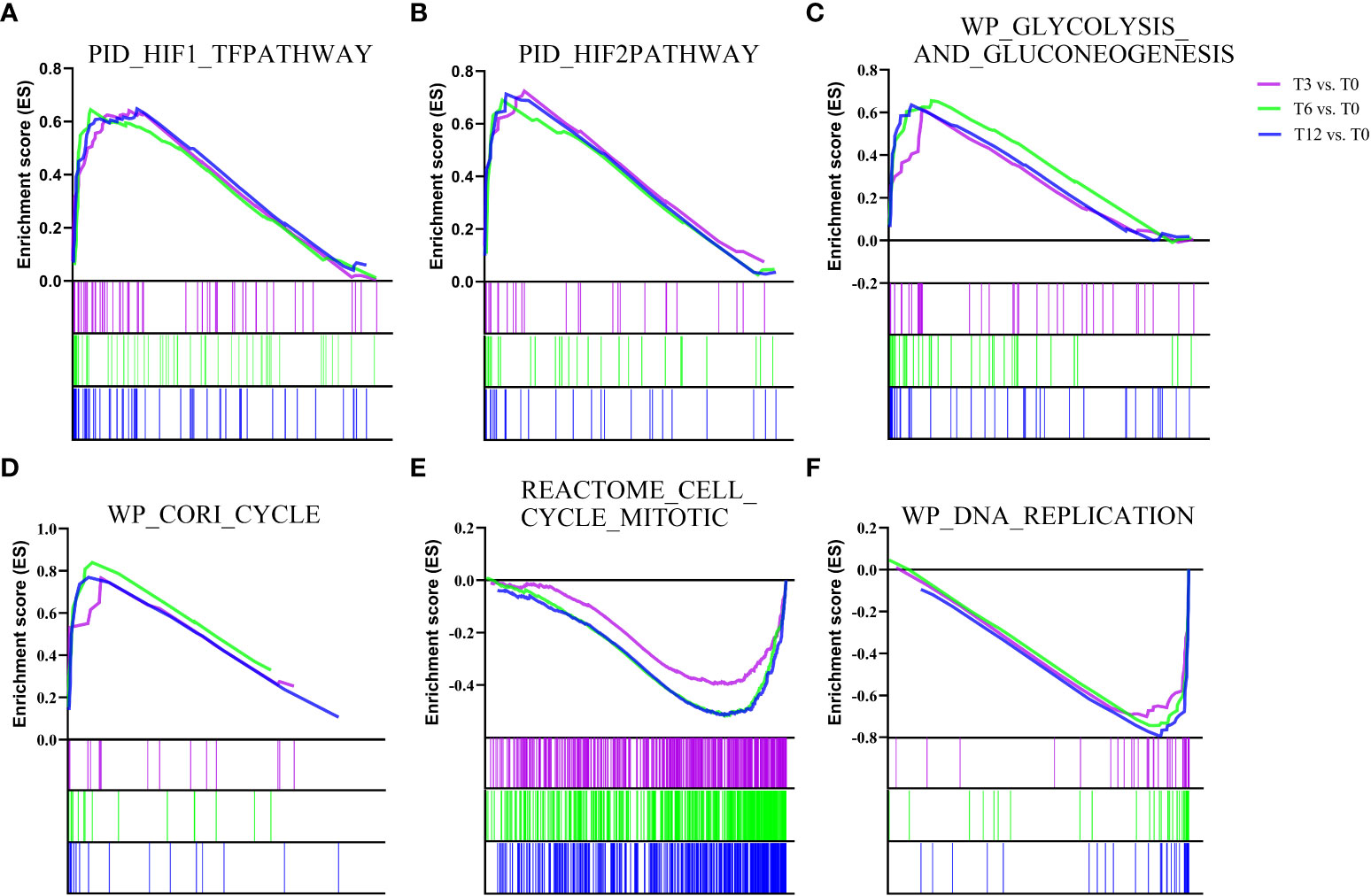
Figure 2 Gene Set Enrichment analysis (GSEA) plots showing the most enriched gene sets in hypoxia treatment groups (T3, T6 and T12) and control group (T0). The most significant enriched gene sets positively correlated with hypoxia treatment groups (T3, T6 and T12) are (A) PID_HIF1_TFPATHWAY, (B) PID_HIF2PATHWAY, (C) WP_GLYCOLYSIS_AND_GLUCONEOGENESIS, and (D) WP_CORI_CYCLE. The most significant enriched gene sets positively correlated with control group (T0) are (E) REACTOME_CELL_CYCLE_MITOTIC and (F) WP_DNA_REPLICATION. The enrichment score of each comparison is showed with different color lines.
A total of 2,796 DEGs were assigned to six clusters, the gene number of which varied from 422 to 541 (Figure 3). The six clusters represented six distinct patterns of expression trajectories in the time course, and the detected clusters of co-expressed genes indicated co-regulation. As we mainly focus on the most relevant regulation networks (genes) interacted with hypoxia environment, the two clusters including cluster 2 and cluster 5, which enriched with hypoxia-responsive genes and pathways, were further analyzed. As shown in Figure 3A, with the prolonging of hypoxia time, the expression pattern of DEGs in cluster 2 showed a general upward trend, while expression of DEGs in cluster 5 exhibited the overall downward trend. KEGG enrichment analysis showed that DEGs in cluster 2 were significantly enriched in signal transduction pathways including HIF-1 signaling pathway, MAPK signaling pathway, AMPK signaling pathway, PI3K-Akt signaling pathway and Insulin signaling pathway, as well as carbohydrate metabolism pathways such as Glycolysis/Gluconeogenesis, Fructose and mannose metabolism, and Galactose metabolism (Figure 3B). The DEGs in cluster 5 were enriched in 1) replication and repair related pathways containing DNA replication, Base excision repair, Mismatch repair, Homologous recombination, and Fanconi anemia pathway; 2) Cell cycle; and 3) several nucleotide metabolism and lipid metabolism pathways (Figure 3C).
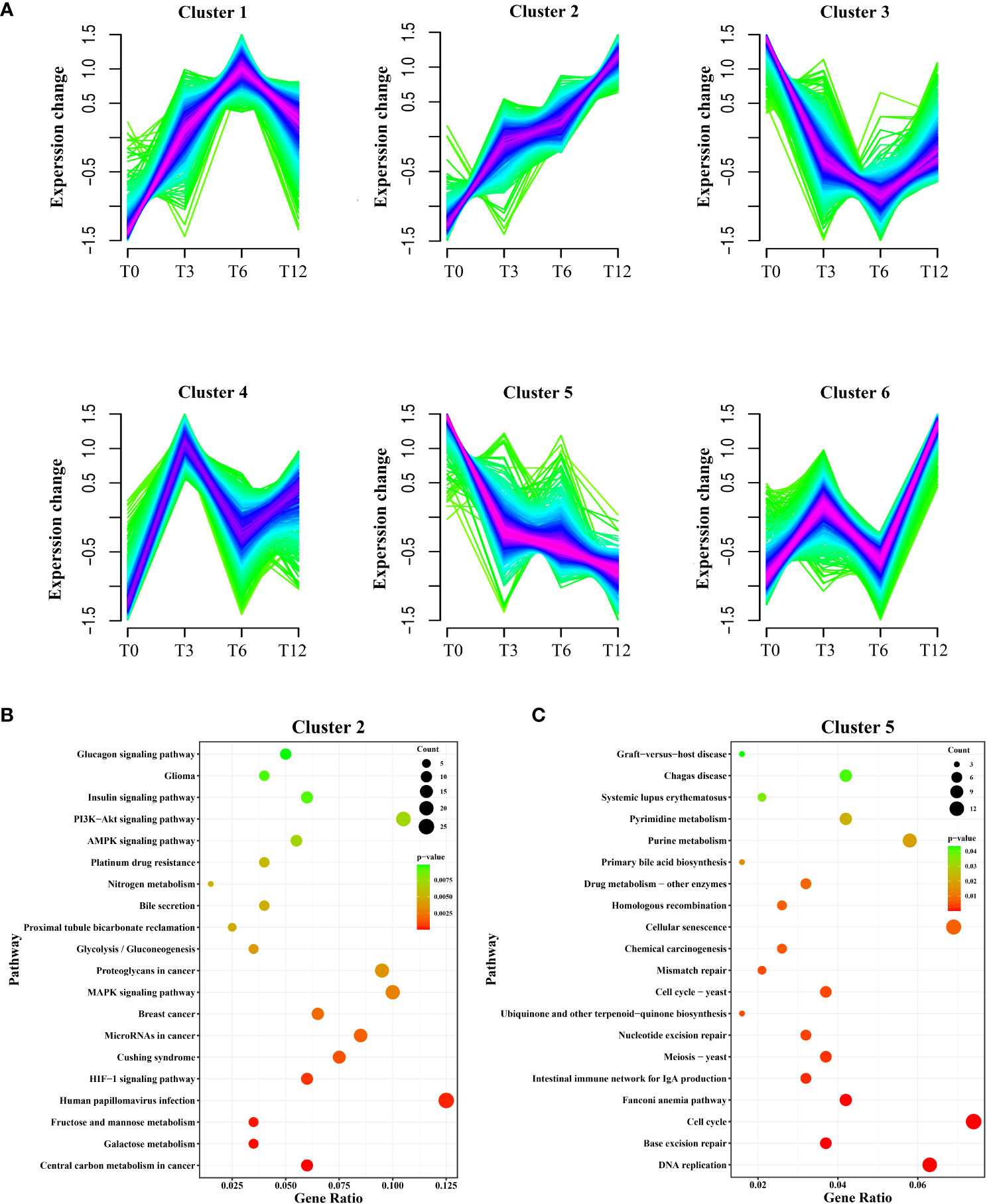
Figure 3 Soft clustering profiles of DEGs after hypoxia. (A) The dynamic expression patterns of the DEGs were analyzed by Mfuzz R package. Six clusters (cluster 1-6) were identified based on the similarity in expression patterns. Different colors indicate the match degrees between changes of genes and the major changes of the clusters. Fuchsia, blue and green represent high, moderate and low match degrees, respectively. (B) KEGG enrichment analysis of DEGs in cluster 2. (C) KEGG enrichment analysis of DEGs in cluster 5.
PPI networks were constructed and hub genes were identified for co-expression genes of cluster 2 and cluster 5. STRING analysis identified 206 nodes and 240 edges for cluster 2, as well as 246 nodes and 817 edges for cluster 5. Their networks were visualized using Cytoscape software, showing in Figures 4A, B. Cyto-Hubba of Cytoscape was used for determining highly connected protein nodes (hubs) by four algorithms (MCC, MNC, Degree and EPC). The ranks and names for top 10 hub genes of each algorithm were shown in Supplementary Table 4. The results showed that for cluster 2, all four methods identified vascular endothelial growth factor A (vegfa), insulin-like growth factor 1 (igf1), endothelin 1 (edn1), cyclooxygenase 2b (cox2b), chemokine (C-X-C motif) receptor 4b (cxcr4b), catenin beta 1 (ctnnb1), and solute carrier family 2 member 1a (slc2a1a) as hub genes, and hk1 was considered as hub gene by three of the four algorithms (Supplementary Table 4). For cluster 5, six hub genes, that were minichromosome maintenance 2 (mcm2), replication factor C subunit 4 (rfc4), minichromosome maintenance 5 (mcm5), DNA polymerase alpha 1 catalytic subunit (pola1), DNA polymerase epsilon catalytic subunit A (pole), and HK1 checkpoint homolog (chek1), were identified by all four algorithms. Additionally, cell division cycle 20 (cdc20), recombinase rad51 (rad51), replication protein A1 (rpa1) and gins complex subunit 2 (gins2) were suggested as hub genes by three algorithms (Supplementary Table 4).
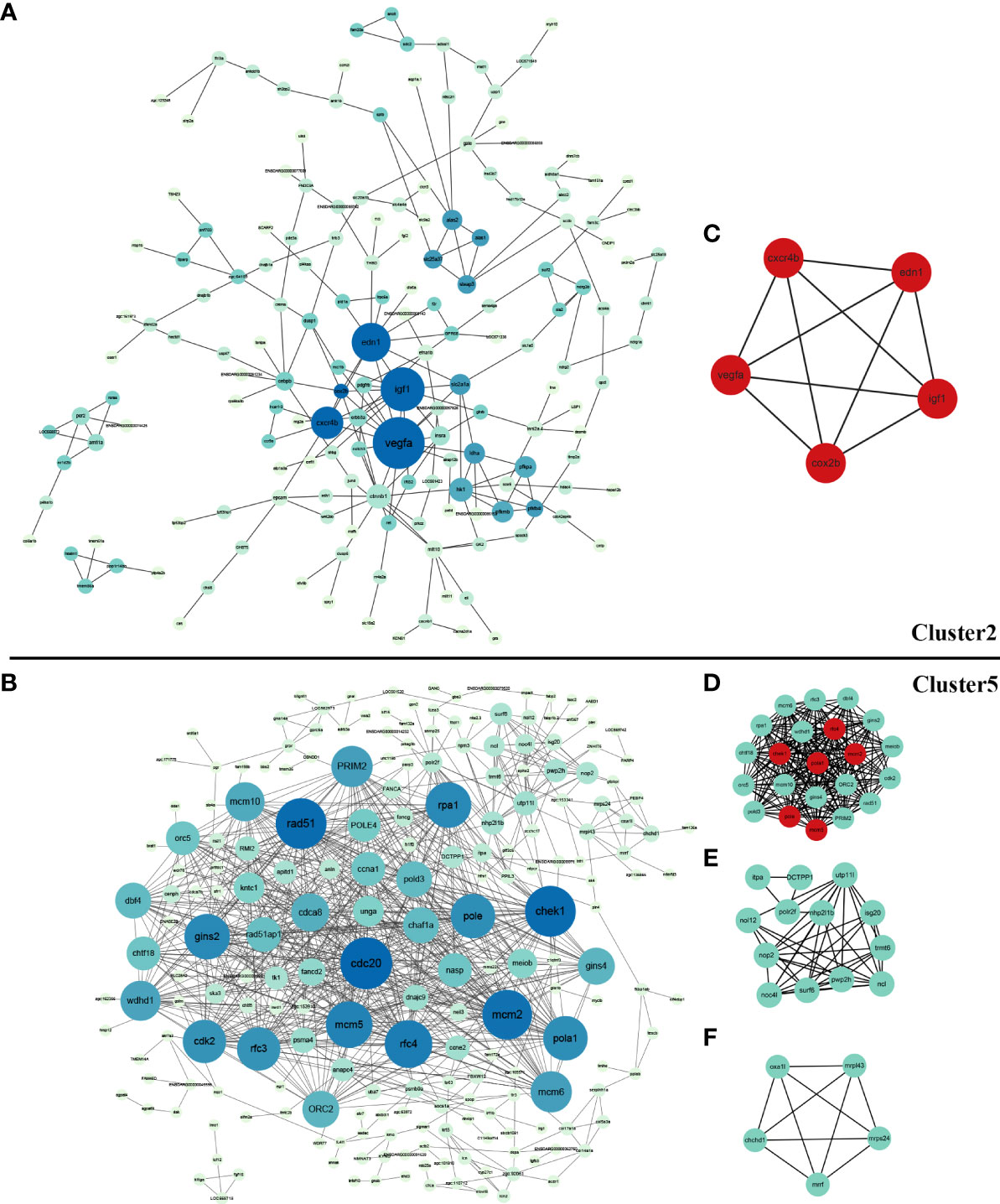
Figure 4 Protein-protein interaction (PPI) network and significant module of DEGs in cluster 2 and cluter 5. (A) PPI network of DEGs in cluster 2 created by STRING and visualized using Cytoscape software; (B) PPI network of DEGs in cluster 5 created by STRING and visualized using Cytoscape software; (C) The significant module of cluster 2 identified by MCODE (score = 5.000). The hub genes identified by Cyto-Hubba analysis were shown in red; (D) The most significant module of cluster 5 identified by MCODE (score =20.286); (E) The second significant module of Cluster 5 identified by MCODE (score =7.167); (F) The third significant module of Cluster 5 identified by MCODE (score =5.000). The hub genes identified by Cyto-Hubba analysis are shown in red.
MCODE defined one and three significant modules from the PPI networks of cluster 2 and cluster 5, respectively. For cluster 2, the five genes in the significant module (vegfa, igf1, edn1, cox2b, cxcr4b) all belonged to hub genes with top-ranking obtained from the Cyto-Hubba analysis (Figure 4C). For cluster 5, the identified hub genes including mcm2, mcm5, pola, pole, chek1, rfc4 were also included in the most significant module (Figures 4D–F).
Taken together, from the enrichment analysis results generated by above methods, the genes and pathways involved in HIF signal network system were induced most significantly in response to hypoxia, meanwhile the cell cycle related genes and pathways were dramatically suppressed by hypoxia. Putative pathways and genes involved in low oxygen response in spotted sea bass gills were illustrated in Figure 5, and their gene expression levels were shown in Supplementary Tables 5, 6. In addition, their potential biological functions were discussed in detail in the Discussion section.
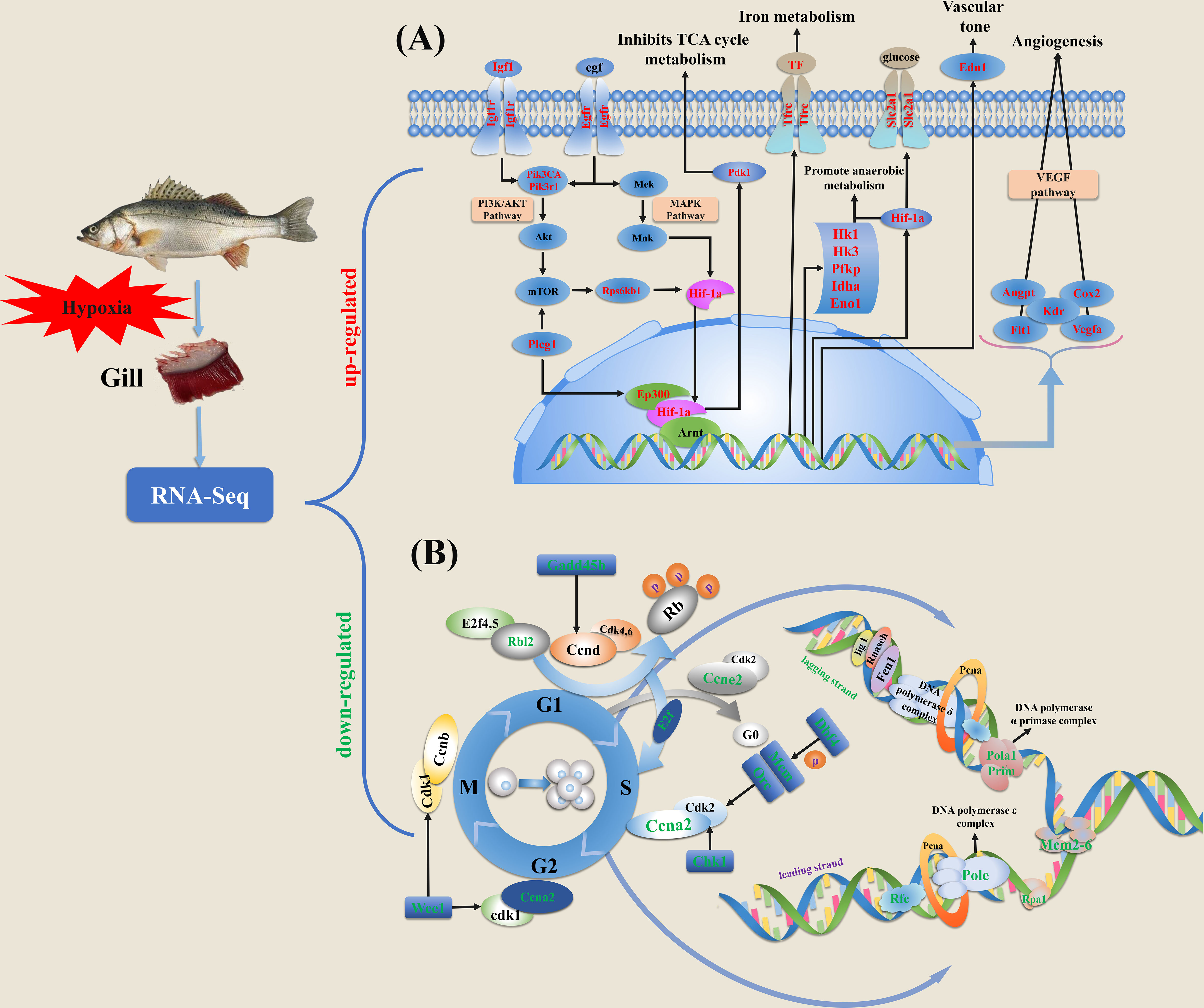
Figure 5 Schematic diagram of key genes and pathways in response to hypoxia based on the expression profiles in gills of spotted sea bass. (A) The genes involved in HIF-1 signal network system were significantly induced by hypoxia. (B) The genes involved in cell cycle progression were significantly suppressed by hypoxia stress. Cell cycle is generally consisted of four different phases, including M, S, G1 and G2. DEGs in HIF-1 signal network system and cell cycle progression were labeled with red and green texts, respectively. The detailed information of these genes involved in HIF-1 signaling pathway and cell cycle are given in Supplementary Tables 3, 4, respectively.
In order to survey AS events in genes associated with hypoxia response in gills, DAS events were determined between two sets of RNA-Seq samples within each comparison group using rMATS. As a result, a total of 400 DAS events were identified after hypoxia including 166, 176 and 295 DASs in comparison group of T3 vs. T0, T6 vs. T0 and T12 vs. T0, which were derived from 153, 158 and 262 genes, respectively (Table 1). Among the five types of AS events (ES, IR, A3SS, A5SS, and ME), ES was the most abundant type, accounting for more than 80% of the total DAS events (Table 1). To validate the reliability of the bioinformatic analysis results for AS, four predicted DAS genes with ES type (slc6a13, zmym2, tnip1, msrb3) were selected, and specific primers of the inclusion isoforms were designed and the sizes were validated by RT-PCR. The results demonstrated that the amplified product sizes were consistent with predicted target fragments (Supplementary Figure 3).

Table 1 Statistics of DAS events detected in gills after hypoxia.
The KEGG functional enrichment analysis result for the DAS genes indicated the most significant pathway was spliceosome (Figure 6A), which is directly related to the occurrence of DAS events. Notably, 63 DAS genes were also determined as DEGs after hypoxia (Figure 6B and Supplementary Table 7), suggesting that the hypoxia-response expression of these genes might be regulated by the alternative splicing mechanism.
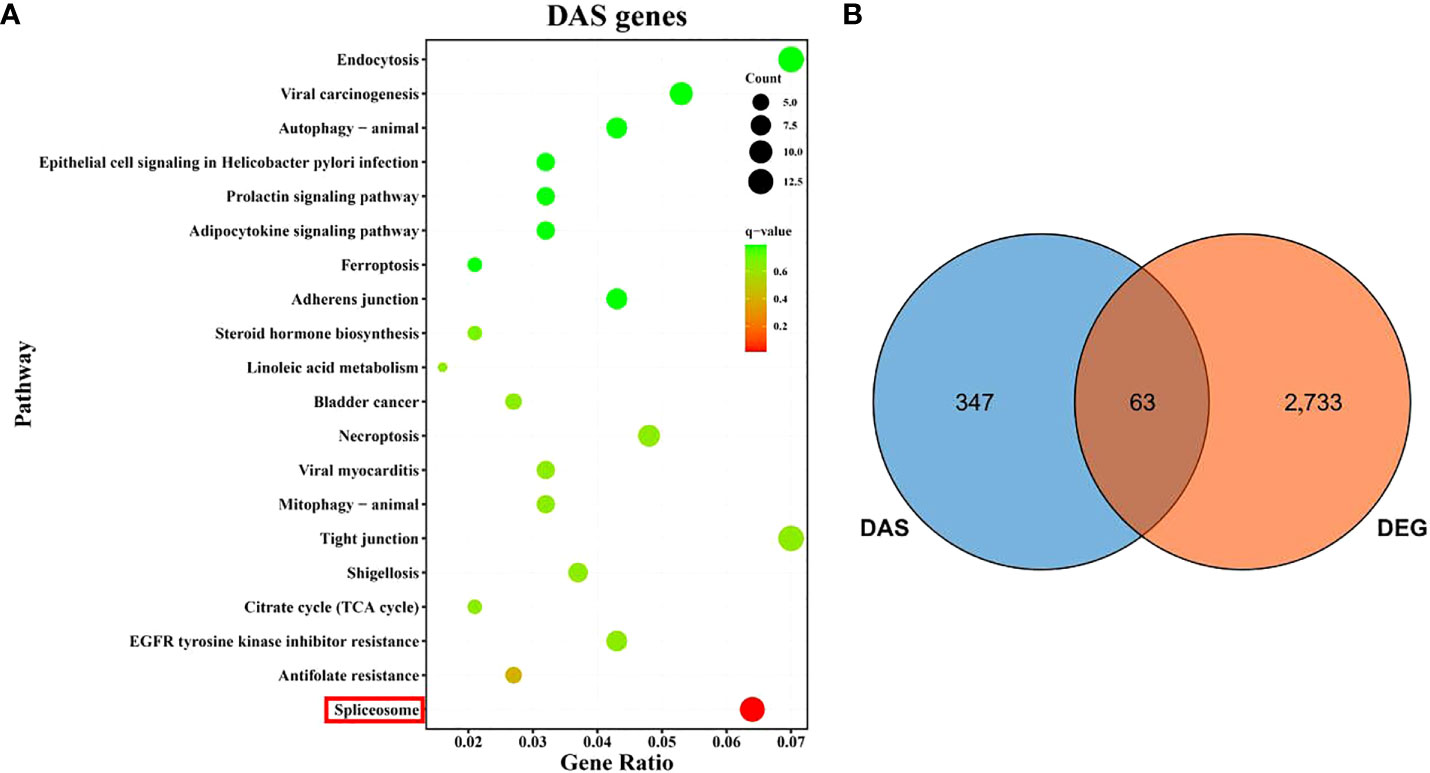
Figure 6 An overview of differential alternative splicing (DAS) events in gills of spotted sea bass after hypoxia. (A) KEGG enrichment analysis of DAS genes. (B) Venn diagram showing the overlap of DEGs and DAS genes.
Fish are often challenged to survive in hypoxia environments caused by various factors such as high temperature, low atmospheric pressure and eutrophication in water (Abdel-Tawwab et al., 2019; Sun et al., 2020). Although fishes are capable to make quickly reactions to hypoxia to maintain normal physical activity, a lack of oxygen will have detrimental effects on growth, reproduction and survival (Wang et al., 2017). Therefore, to prevent yield loss, increasing attention should be taken into account on DO levels and elucidating the mechanisms of fish reaction under hypoxia. Over the past decade, the profound changes in gene expression profiles were generated based on transcriptomic datasets, which have been widely employed in investigating the molecular basis for hypoxia-responsive and -adaptive mechanisms in a number of fish species. Fish gills play dominant roles in aquatic gas exchange and are responsible for making behavioral, morphological and physiological adaptions to hypoxia conditions (Wu et al., 2017; Abdel-Tawwab et al., 2019). Previous studies have characterized the plastic hypoxia-induced changes in gill morphology and cellular ultrastructure in several fish species (Sollid et al., 2003; Sollid and Nilsson, 2006; Matey et al., 2008; Wu et al., 2017), but the underlying molecular basis in gills was still lacking. In this study, we performed the in-depth analysis of transcriptomic datasets of spotted sea bass gills to better understand the molecular changes affected by hypoxia and the underlying adaptive mechanisms. It was noted that the number of DEGs were greatly increased with the prolongation of hypoxia stress from 3 h to 12 h. It suggested that a series of adaptive mechanisms may be gradually activated to deal with hypoxia stress and maintain homeostasis in gills of spotted sea bass.
Several proteins and signaling pathways have been reported to function in hypoxia adaptation in fishes, and the changes of their expressions can trigger a great number of biological processes (Zhu et al., 2013). In our study, through function enrichment annotation analysis by DEGs-based KEGG, or GSEA analysis based on all the gene expression data under hypoxia in spotted sea bass, the transcriptional inducible genes and pathways in gills were significantly enriched in HIF signal network (Figures 1D–F and 2). This evolutionarily conserved signal network included HIF homologs and their targets, and hypoxia adaptation signaling pathways such as PI3K-Akt, MAPK, AMPK, VEGF and calcium signaling pathway, which have been extensively reported in vertebrate species (Xiao, 2015; Xie et al., 2019).
HIF-α is recognized as a key modulator for the regulation of the hypoxia signaling pathway. Among the three isoforms of HIF-α (HIF-1α, HIF-2α and HIF-3α) that have been identified in vertebrates, HIF-1α and HIF-2α are widely studied and considered particular critical for hypoxia response. They share similar domain structures, heterodimerize with the stable HIF-1β and bind to hypoxia responsive element (HRE), but their regulations on the expression of genes may be different (Loboda et al., 2010). In mammals, HIF-1α is expressed ubiquitously in all cells, whereas HIF-2α are selectively expressed in certain tissues (Majmundar et al., 2010). For the central adaptation to hypoxia that shift towards non-oxidative forms of carbon metabolism and ATP production, both HIF-1α and HIF-2α play essential functions in modulating cellular metabolisms and controlling redox homeostasis, but through distinct transcriptional programs (Majmundar et al., 2010). For example, glucose consumption and glycolysis were reported to be promoted primarily by HIF-1α, while fatty acid storage was promoted by HIF-2α (Loboda et al., 2010; Majmundar et al., 2010). In our results, both HIF-1α and HIF-2α were identified to be expressed in gills of spotted sea bass, and their mRNA expressions were regulated by hypoxia (Supplementary Table 5). In addition, several KEGG pathways related to carbohydrate and lipid metabolism processes were significantly induced by hypoxia exposure (Figures 1D–F). The related up-regulated DEGs potentially to be targeted by HIF-α included genes encoding pdk1 that represses the flux of pyruvate into acetyl-CoA and suppresses O2 consumption (Simon, 2006), edn1 used for vasomotor control (Koltsova et al., 2014), slc2a1 that facilitate cellular glucose uptake, cox2b that promotes angiogenesis, hk1, hk3, ldha, pfkp, gapdh, and eno1 as metabolic enzymes that reduce oxygen consumption and promote anaerobic glycolysis (Rye and LaMarr, 2015) (Figure 5A and Supplementary Table 5). However, the specific targets regulated by HIF-1α and HIF-2α in fish gills couldn’t be distinguished based on expression patterns, which need to be further investigated. In addition, we performed GSEA analysis as it could provide valuable information based on expression levels of all the genes in an experiment instead of only considering those DEGs (Subramanian et al., 2005). Our results also indicated that hypoxia-treatment groups (T3, T6 and T12) were most positively correlated with enriched gene sets, like HIF-1-alpha transcription factor network, HIF-2-alpha transcription factor network, Glycolysis and Gluconeogenesis and Cori cycle (Figure 2).
As the master regulator of the transcriptional response to hypoxia, HIF is not merely a transducer but an integrator of multiple signaling pathways (Fábián et al., 2016). Numerous signaling pathways have been reported to function in hypoxia adaptation. VEGF has been demonstrated as the common target gene shared by HIF-1α and HIF-2α (Loboda et al., 2010), and the HIF-α/VEGF signaling pathway showed to play a crucial role in angiogenesis which stimulates the proliferation of blood vessels to increase oxygen supply (Zhu et al., 2013). In our study, the mRNA expression levels of numerous genes involved in signaling by VEGF was significantly induced by hypoxia, such as vegfa, fms-related tyrosine kinase 1 (flt1), kinase insert domain receptor (kdr), angiopoietin-like 4 (angpt) and cox2b (Figure 5A and Supplementary Table 5). Although not detected by KEGG enrichment analysis based on DEGs, we noted that the gene sets of VEGF and angiogenesis were positively correlated with hypoxia treatment groups (T3 and T12) by GSEA analysis (Supplementary Table 3). In addition, PI3K/Akt, MAPK and AMPK pathways have been reported to play important roles in hypoxia response in both higher vertebrates and fish species (Zhu et al., 2013). The PI3K/Akt signaling pathway is activated by IGF1, which increase synthesis and transcriptional activity of HIF-1α by preventing its ubiquitination and degradation (Yang et al., 2018). The MAPK signaling pathway is essential for controlling the production of reactive oxygen species (ROS) and oxygen homeostasis (Seifried et al., 2007), and has been proved to be implicated in regulating the activity of HIF (Bracken et al., 2003). For AMPK signaling pathway, hypoxia can lead to the activation of AMPK, which initiates various adaptive responses to decreased ATP levels or reduced oxygen levels (Zhu et al., 2013). Moreover, in brain of mammals, PI3K and MAPK pathways have been reported to be involved in stimulating HIF in angiogenesis and could be activated by the insulin signaling pathway, which suggested that PI3K and MAPK pathways might act as cross-talk between the insulin signaling pathway and the angiogenesis pathway (Zeng et al., 2016). Correspondingly, in our study, we found that insulin, PI3K/AKt, MAPK and AMPK signaling pathways were all enriched based on the expression patterns of genes in cluster 2, which showed a general upward trend towards hypoxia (Figure 3) The identified DEGs involved in these pathways included insulin receptor (insr), hk1 in the insulin signaling pathway, platelet-derived growth factor receptor beta-like (pdgfrb), CREB binding protein (creb), growth hormone receptor 2 (ghr) in the PI3K/AKt signaling pathway, transcription factor jun-D (jund), igf1 in the MAPK signaling pathway and ATP-dependent 6-phosphofructokinase, platelet type (pfkp), cyclin, C-terminal domain (cycd) in the AMPK signaling pathway (Supplementary Table 5). This result indicated the potential critical involvement and conservative functions of these genes and signaling pathways in gills of spotted sea bass after hypoxia stress, and future studies are required to elucidate how the hypoxia signaling cascade can integrate information from many other signaling pathways.
Furthermore, we performed PPI analysis and identified hub genes in cluster 2, which showed continuously induced expression patterns by hypoxia in our study. Notably, the identified hub genes in cluster 2 (vegfa, igf1, edn1, cox2b, cxcr4b, slc2a1) were all related to HIF signal network system and supposed to be targeted by HIF-α (Figure 4 and Supplementary Table 4). The significant module identified by MCODE included five hub genes (vegfa, igf1, edn1, cox2b and slc2a1), and their potential roles in response to hypoxia have been mentioned above. In addition, hypoxia enhances cxcr4 expression by activating HIF-1α in several types of tumor cells (Korbecki et al., 2021). Moreover, there are extensive interactions among those hub genes (Figure 4). For example, cox2 was reported to play a critical role in VEGF induction and stimulation of angiogenesis (Kaidi et al., 2006), meanwhile VEGF was proved to be one of the principal factors produced by hypoxia myocytes that is responsible for the induction of endothelial cell cox2 expression (Wu et al., 2003). Overall, in line with expectations, the HIF signal network system plays the most important roles in response to hypoxia in gills of spotted sea bass, and the functions of HIF target genes and related pathways may highly conserved from teleost to mammals.
In mammals, it have been demonstrated that hypoxia served as a stimulus for cell cycle arrest, which might be mediated through the non-transcriptional role as an inhibitor of MCM helicase activity of HIF-1α (Hubbi et al., 2013). MCM helicase is composed of six protein subunits termed MCM2-7 and is a principal component of the prereplicative complex, which assemble at origins of replication during G1 before the onset of DNA replication. Under hypoxia, the HIF-1α protein may bind to the MCM proteins, maintaining the complex in a loaded but inactive state (Hubbi and Semenza, 2015). Moreover, evidence showed that MCM mRNA expression was inhibited in a HIF-1α-dependent manner (Semenza, 2011). Conversely, individual MCM subunit such as MCM2, MCM3, MCM5 or MCM7 can inhibit the activity of HIF-1α, which functions as negative regulators of HIF activity (Hubbi et al., 2011). Thus HIF and MCM proteins act in a mutually antagonistic manner, however, the ability of HIF-1α to inhibit cell cycle progression under severe hypoxia may be dominant over the ability of MCMs to promote cell cycle progression by inhibiting HIF-1α (Semenza, 2011).
Although the relationship of hypoxia and cell cycle arrest has not revealed in teleosts, in our study, it can be obviously observed that numerous cell cycle related genes and pathways were significantly suppressed in gills of spotted sea bass under hypoxia (Figures 1D–F and 2). Consistent with the findings in mammals, genes encoding MCM proteins (mcm2-6 and mcm10) were dramatically down-regulated in the hypoxia treatment groups than that in the control group (Supplementary Table 6). Besides MCM helicase, several other types of DEGs which are functioning for DNA replication at the S phase showed down-regulation, such as genes encoding: 1) the different catalytic subunits of DNA polymerase complex (pola1, DNA polymerase delta 3 subunit 3 (pold3), pole, DNA polymerase epsilon 4 subunit (pole4)); 2) the replication factor C (replication factor C subunit 3 (rfc3) and replication factor C subunit 4 (rfc4)) which act as an activator of DNA polymerases; 3) the replication protein A complex (rpa1) that binds to single-stranded DNA; 4) the subunits of the origin recognition complex (origin recognition complex subunit 1-2, 4-5 (orc1-2, orc4-5)) that form a core complex and bind specifically to origins of replication; 5) the subunit of DNA primase (DNA primase large subunit 2 (prim2)) which forms a heterodimer to function as RNA polymerase to synthesize small RNA primers that are used to create Okazaki fragments on the lagging strand of the DNA. In addition, the expression levels of genes function in other cell cycle stages, including G1 phase and G1/S transition (ccne2, rbl2, anapc4-5), cell cycle checkpoints (cell division cycle associated 8 (cdca8), chek1, protein DBF4 homolog A (dbf4), wee1-like protein kinase (wee1), DNA excision repair protein ERCC-6-like (ercc6l)), and chromosome maintenance (centromere protein H (cenph), centromere protein K (cenpk), telomeric repeat-binding factor 1 (terf1)) exhibited remarkably downregulation under hypoxia (Figure 5B and Supplementary Table 6). Overall, a large numbers of cell cycle related genes were suppressed significantly by hypoxia in our study, indicating hypoxia may mediate severely inhibition of cell cycle progression in fish gills.
AS plays critical roles at the post-transcriptional level of gene regulation by producing structurally and functionally distinct mRNA and protein variants (Blencowe, 2006). In recent years, accumulating studies have showed that AS in teleost species served as the tightly regulated processes to cope with environmental stresses. For example, through transcriptome analysis, AS has been demonstrated as important regulator in channel catfish and rainbow trout in response to heat stress (Tan et al., 2019; Sun et al., 2021a), in common carp (Cyprinus carpio) and Atlantic killifish during cold acclimation (Healy and Schulte, 2019; Long et al., 2020), in spotted sea bass during salinity adaptation (Tian et al., 2020b), as well as in Nile tilapia after hypoxia stress (Li et al., 2017; Xia et al., 2017). In our study, for the first time, the hypoxia stress induced AS changes in gills of spotted sea bass has been investigated by transcriptome analysis. We found that among the typical patterns of AS (ES, IR, A3SS, A5SS, and ME), ES was the most abundant type, accounting for more than 80% of the total DAS events in gills under hypoxia (Table 1). Moreover, in our previous study, ES was proved to be the most enriched type of AS event in spotted sea bass based on the RNA-Seq data generated by pooling tissues (Tian et al., 2020b). The similar result was also reported in rainbow trout, catfish and common carp (Tan et al., 2018; Long et al., 2020; Ali et al., 2021). It has been demonstrated that animals have higher rates of ES than other eukaryotes (Patthy, 2019). Besides, ES is reported to be the most common AS event leading to a variety of human diseases, due to the loss of functional domains/sites or shifting of the open reading frame (ORF) (Kim et al., 2020). Since the correct processing for the production of a mature mRNA requires specialized interaction with the splicing machinery, we hypothesized that hypoxia stress may induce alteration of the communication between the transcriptional and splicing machinery, which may result in widespread ES and play important role in response to hypoxia (Dutertre et al., 2010).
Notably, KEGG functional analyses revealed that the genes undergoing hypoxia-induced DAS in gills of spotted sea bass were mainly involved in spliceosome (Figure 6). In consistence with our findings, DAS involved in spliceosome were significantly enriched in the brain of common carp during cold adaption (Long et al., 2020), in both gill and liver of spotted sea bass during salinity acclimation (Tian et al., 2020b), and in the liver of catfish after heat stress (Tan et al., 2019). Spliceosome is known as a large and ribonucleoprotein complex, which are differently assembled at distinct introns and accomplishes the feat of intron removal from the messenger RNA precursors (pre-mRNA) of eukaryotic cells (Jenkins and Kielkopf, 2017; Yan et al., 2019). It suggested that some important elements in spliceosome, RNA splicing regulators, underwent different splicing events between normoxia and hypoxia, which may alter their function in pre-mRNA splicing and affect the splicing decisions of numerous downstream target genes to deal with hypoxia stress and maintain homeostasis in gills of spotted sea bass. The results were largely consistent with the previous observation that splicing factors themselves often undergo auto- or cross-regulation by AS in response to environmental and developmental cues (Staiger and Brown, 2013; Tian et al., 2022). However, the underlying mechanism need to be investigated further in the future.
In the current study, the in-depth analysis of transcriptomic datasets of spotted sea bass gills have been performed to study the molecular changes including the gene expression patterns and AS profiles affected by hypoxia, which provided further insights into the underlying adaptive mechanisms. As results, a total of 1,242, 1,487 and 1,762 DEGs were identified through the comparisons of T3 vs. T0, T6 vs. T0 and T12 vs. T0, respectively. KEGG enrichment analysis based on these DEGs and GSEA analysis performed with the whole gene expression datasets indicated that HIF signal network system was significantly activated and cell cycle related genes and pathways were suppressed in response to hypoxia in gills of spotted sea bass. Six clusters were generated based on dynamic gene expression patterns, and PPI networks were constructed and hub genes were recognized for the cluster 2 and cluster 5, which enriched mostly with hypoxia-responsive genes and pathways. Vegfa, igf1, edn1, cox2b, cxcr4b, ctnnb1, and slc2a1a were identified as hubs for hypoxia-induced genes (cluster 2), and mcm2, chek1, pole, mcm5, pola1, rfc4 were considered as hubs for down-regulated genes (cluster 5). Besides, a total of 153, 158 and 262 DAS genes were identified in comparison group of T3 vs. T0, T6 vs. T0 and T12 vs. T0, which were mainly enriched in spliceosome. Of them, 63 DAS genes also showed differentially expressed levels after hypoxia, suggesting that the expression changes of these genes might be regulated by the AS mechanism. Our results will lay the valuable basis for elucidated the molecular mechanisms that contribute to short-term hypoxia acclimation in spotted sea bass and other fish species.
The data presented in the study are deposited in the NCBI Sequence Read Archive (SRA) repository, accession number SRR17822288-SRR17822299.
The animal study was reviewed and approved by Animal Research and Ethics Committees of Ocean University of China (Permit Number: 20141201).
YR: Methodology, Software, Visualization, Validation and Original draft. YT: Conceptualization and Formal analysis. XM: Data curation. HW: Funding acquisition, Resources and Investigation. XQ: Resources and Investigation. JL: Methodology and Validation. YL: Project administration, Supervision and Writing - review & editing. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China [grant numbers 2018YFD0900101]; the National Natural Science Foundation of China [grant numbers 32072947]; and the China Agriculture Research System of MOF and MARA [grant numbers CARS-47].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1024218/full#supplementary-material
Abdel-Tawwab M., Monier M. N., Hoseinifar S. H., Faggio C. J. (2019). Fish response to hypoxia stress: Growth, physiological, and immunological biomarkers. Fish Physiol. Biochem. 45, 997–1013. doi: 10.1007/s10695-019-00614-9
Agani F., Jiang B. H. (2013). Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 245, 51. doi: 10.2174/1568009611313030003
Ali A., Thorgaard G. H., Salem M. (2021). PacBio iso-seq improves the rainbow trout genome annotation and identifies alternative splicing associated with economically important phenotypes. Front. Genet. 12. doi: 10.3389/fgene.2021.683408
Bader G. D., Hogue C. (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 4, 1–27. doi: 10.1186/1471-2105-4-2
Bárdos J. I., Ashcroft M. (2005). Negative and positive regulation of HIF-1: A complex network. Biochim. Biophys. Acta 1755, 107–120. doi: 10.1016/j.bbcan.2005.05.001
Bartoszewski R., Moszyńska A., Serocki M., Cabaj A., Polten A., Ochocka R., et al. (2019). Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 33, 7929–7941. doi: 10.1096/fj.201802650RR
Blencowe B. (2006). Alternative splicing: New insights from global analyses. Cell 126, 37–47. doi: 10.1016/j.cell.2006.06.023
Bohensky J., Leshinsky S., Srinivas V., Shapiro I. (2010). Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 25, 633–642. doi: 10.1007/s00467-009-1310-y
Bracken C. P., Whitelaw M. L., Peet D. J. (2003). The hypoxia-inducible factors: Key transcriptional regulators of hypoxic responses. Cell. Mol. Life Sci. 60, 1376–1393. doi: 10.1007/s00018-003-2370-y
Chen B., Li Y., Peng W., Zhou Z., Shi Y., Pu F., et al. (2019). Chromosome-level assembly of the Chinese seabass (Lateolabrax maculatus) genome. Front. Genet. 10. doi: 10.3389/fgene.2019.00275
Claireaux G., Thomas S., Fievet B., Motais R. (1988). Adaptive respiratory responses of trout to acute hypoxia. II. blood oxygen carrying properties during hypoxia. Respir. Physiol. 74, 91–98. doi: 10.1016/0034-5687(88)90143-0
Déry M. A., Michaud M. D., Richard D. E. (2005). Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 37, 535–540. doi: 10.1016/j.biocel.2004.08.012
Ding J., Liu C., Luo S., Zhang Y., Gao X., Wu X., et al. (2020). Transcriptome and physiology analysis identify key metabolic changes in the liver of the large yellow croaker (Larimichthys crocea) in response to acute hypoxia. Ecotoxicol. Environ. Saf. 189, 109957. doi: 10.1016/j.ecoenv.2019.109957
Ding L., Rath E., Bai Y. (2017). Comparison of alternative splicing junction detection tools using RNA-seq data. Curr. Genomics 18, 268–277. doi: 10.2174/1389202918666170215125048
Dong H., Sun Y., Duan Y., Li H., Li Y., Liu Q., et al. (2020). The effect of teprenone on the intestinal morphology and microbial community of Chinese sea bass (Lateolabrax maculatus) under intermittent hypoxic stress. Fish Physiol. Biochem. 46, 1873–1882. doi: 10.1007/s10695-020-00838-0
Dutertre M., Sanchez G., De Cian M. C., Barbier J., Dardenne E., Gratadou L., et al. (2010). Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct. Mol. Biol. 17, 1358–1366. doi: 10.1038/nsmb.1912
Evans D. H., Piermarini P. M., Choe K. (2005). The multifunctional fish gill: Dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol. Rev. 85, 97–177. doi: 10.1152/physrev.00050.2003
Fábián Z., Taylor C. T., Nguyen L. (2016). Understanding complexity in the HIF signaling pathway using systems biology and mathematical modeling. J. Cell. Mol. Med. 94, 377–390. doi: 10.1007/s00109-016-1383-6
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Healy T. M., Schulte P. S. (2019). Patterns of alternative splicing in response to cold acclimation in fish. J. Exp. Biol. 222, jeb193516. doi: 10.1242/jeb.193516
Hou Z., Wen H., Li J., He F., Li Y., Qi X. (2020). Environmental hypoxia causes growth retardation, osteoclast differentiation and calcium dyshomeostasis in juvenile rainbow trout (Oncorhynchus mykiss). Sci. Total Environ. 705, 135272. doi: 10.1016/j.scitotenv.2019.135272
Hrycik A. R., Almeida L. Z., Höök T. O. (2017). Sub-Lethal effects on fish provide insight into a biologically-relevant threshold of hypoxia. Oikos 126, 307–317. doi: 10.1111/oik.03678
Hubbi M. E., Kshitiz Gilkes D. M., Rey S., Wong C. C., Luo W., Kim D., et al. (2013). A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci. Signal. 6, ra10–ra10. doi: 10.1126/scisignal.2003417
Hubbi M. E., Luo W., Baek J. H., Semenza G. L. (2011). MCM proteins are negative regulators of hypoxia-inducible factor 1. Mol. Cell. 42, 700–712. doi: 10.1016/j.molcel.2011.03.029
Hubbi M. E., Semenza G. L. (2015). An essential role for chaperone-mediated autophagy in cell cycle progression. Autophagy 11, 850–851. doi: 10.1080/15548627.2015.1037063
Jenkins J. L., Kielkopf C. L. (2017). Splicing factor mutations in myelodysplasias: Insights from spliceosome structures. Trends Genet. 33, 336–348. doi: 10.1016/j.tig.2017.03.001
Kaidi A., Qualtrough D., Williams A. C., Paraskeva C. (2006). Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 66, 6683–6691. doi: 10.1158/0008-5472.CAN-06-0425
Kajimura S., Aida K., Duan C. (2006). Understanding hypoxia-induced gene expression in early development: In vitro and in vivo analysis of hypoxia-inducible factor 1-regulated zebra fish insulin-like growth factor binding protein 1 gene expression. Mol. Cell Biol. 26, 1142–1155. doi: 10.1128/MCB.26.3.1142-1155.2006
Kim D., Langmead B., Salzberg S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kim P., Yang M., Yiya K., Zhao W., Zhou X. (2020). ExonSkipDB: functional annotation of exon skipping event in human. Nucleic Acids Res. 48, D896–D907. doi: 10.1093/nar/gkz917
Koltsova S. V., Shilov B., Birulina J. G., Akimova O. A., Haloui M., Kapilevich L. V., et al. (2014). Transcriptomic changes triggered by hypoxia: Evidence for HIF-1α-independent,[Na+] i/[K+] i-mediated, excitation-transcription coupling. PloS One 9, e110597. doi: 10.1371/journal.pone.0110597
Korbecki J., Kojder K., Kapczuk P., Kupnicka P., Gawrońska-Szklarz B., Gutowska I., et al. (2021). The effect of hypoxia on the expression of CXC chemokines and CXC chemokine receptors-a review of literature. Int. J. Mol. Sci. 22, 843. doi: 10.3390/ijms22020843
Kumar L., Futschik M. E. (2007). Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7. doi: 10.6026/97320630002005
Lee Y., Rio D. C. (2015). Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 84, 291–323. doi: 10.1146/annurev-biochem-060614-034316
Li H. L., Lin H. R., Xia J. (2017). Differential gene expression profiles and alternative isoform regulations in gill of Nile tilapia in response to acute hypoxia. Mar. Biotechnol. 19, 551–562. doi: 10.1007/s10126-017-9774-4
Liu Y., Wang H., Wen H., Shi Y., Zhang M., Qi X., et al. (2020). First high-density linkage map and QTL fine mapping for growth-related traits of spotted sea bass (Lateolabrax maculatus). Mar. Biotechnol. 22, 526–538. doi: 10.1007/s10126-020-09973-4
Loboda A., Jozkowicz A., Dulak J. (2010). HIF-1 and HIF-2 transcription factors-similar but not identical. Mol. Cells 29, 435–442. doi: 10.1007/s10059-010-0067-2
Long Y., Li X., Li F., Ge G., Liu R., Song G., et al. (2020). Transcriptional programs underlying cold acclimation of common carp (Cyprinus carpio l.). Front. Genet. 111122. doi: 10.3389/fgene.2020.556418
Majmundar A. J., Wong W. J., Simon M. C. (2010). Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 40, 294–309. doi: 10.1016/j.molcel.2010.09.022
Ma J., Qiang J., Tao Y., Bao J., Zhu H., Li L., et al. (2021). Multi-omics analysis reveals the glycolipid metabolism response mechanism in the liver of genetically improved farmed tilapia (GIFT, Oreochromis niloticus) under hypoxia stress. BMC Genomics 22, 1–16. doi: 10.1186/s12864-021-07410-x
Mastrangelo A. M., Marone D., Laidò G., De Leonardis A. M., De Vita P. (2012). Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 186, 40–49. doi: 10.1016/j.plantsci.2011.09.006
Matey V., Richards J. G., Wang Y., Wood C. M., Rogers J., Davies R., et al. (2008). The effect of hypoxia on gill morphology and ionoregulatory status in the lake qinghai scaleless carp, Gymnocypris przewalskii. J. Exp. Biol. 211, 1063–1074. doi: 10.1242/jeb.010181
Mu Y., Li W., Wei Z., He L., Zhang W., Chen X. (2020). Transcriptome analysis reveals molecular strategies in gills and heart of large yellow croaker (Larimichthys crocea) under hypoxia stress. Fish Shellfish Immunol. 104, 304–313. doi: 10.1016/j.fsi.2020.06.028
Nikinmaa M., Rees B. B. (2005). Oxygen-dependent gene expression in fishes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1079–R1090. doi: 10.1152/ajpregu.00626.2004
Ng J. C., Chiu J. M. (2020). Changes in biofilm bacterial communities in response to combined effects of hypoxia, ocean acidification and nutrients from aquaculture activity in three fathoms cove. Mar. Pollut. Bull. 156, 111256. doi: 10.1016/j.marpolbul.2020.111256
Nilsen T. W., Graveley B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463. doi: 10.1038/nature08909
Patthy L. (2019). Exon skipping-rich transcriptomes of animals reflect the significance of exon-shuffling in metazoan proteome evolution. Biol. Direct. 14, 1–4. doi: 10.1186/s13062-019-0231-3
Richards J. G. (2011). Physiological, behavioral and biochemical adaptations of intertidal fishes to hypoxia. J. Exp. Biol. 214, 191–199. doi: 10.1242/jeb.047951
Rye P. T., LaMarr W. A. (2015). Measurement of glycolysis reactants by high-throughput solid phase extraction with tandem mass spectrometry: Characterization of pyrophosphate-dependent phosphofructokinase as a case study. Anal. Biochem. 482, 40–47. doi: 10.1016/j.ab.2015.03.029
Seifried H. E., Anderson D. E., Fisher E. I., Milner J. A. (2007). A review of the interaction among dietary antioxidants and reactive oxygen species. J. Nutr. Biochem. 18, 567–579. doi: 10.1016/j.jnutbio.2006.10.007
Semenza G. L. (2001). HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 13, 167–171. doi: 10.1016/s0955-0674(00)00194-0
Semenza G. L. (2011). Hypoxia. cross talk between oxygen sensing and the cell cycle machinery. Am. J. Physiol. Cell Physiol. 301, C550–C552. doi: 10.1152/ajpcell.00176.2011
Shen S., Park J. W., Lu Z., Lin L., Henry M. D., Wu Y. N., et al. (2014). rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl. Acad. Sci. U. S. A. 111, E5593–E5601. doi: 10.1073/pnas.1419161111
Simon M. C. (2006). Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab. 3, 150–151. doi: 10.1016/j.cmet.2006.02.007
Sollid J., De Angelis P., Gundersen K., Nilsson G. E. (2003). Hypoxia induces adaptive and reversible gross morphological changes in crucian carp gills. J. Exp. Biol. 206, 3667–3673. doi: 10.1242/jeb.00594
Sollid J., Nilsson G. E. (2006). Plasticity of respiratory structures-adaptive remodeling of fish gills induced by ambient oxygen and temperature. Respir. Physiol. Neurobiol. 154, 241–251. doi: 10.1016/j.resp.2006.02.006
Sollid J., Weber R. E., Nilsson G. E. (2005). Temperature alters the respiratory surface area of crucian carp Carassius carassius and goldfish Carassius auratus. J. Exp. Biol. 208, 1109–1116. doi: 10.1242/jeb.01505
Staiger D., Brown J. W. (2013). Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell. 25, 3640–3656. doi: 10.1105/tpc.113.113803
Strepparava N., Wahli T., Segner H., Petrini O. J. (2014). Detection and quantification of Flavobacterium psychrophilum in water and fish tissue samples by quantitative real time PCR. BMC Microbiol. 14, 1–10. doi: 10.1186/1471-2180-14-105
Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. doi: 10.1073/pnas.0506580102
Sun J., Liu Y., Jiang T., Li Y., Song F., Wen X., et al. (2021b). Golden pompano (Trachinotus blochii) adapts to acute hypoxic stress by altering the preferred mode of energy metabolism. Aquaculture 542, 736842. doi: 10.1016/j.aquaculture.2021.736842
Sun J., Liu Z., Quan J., Li L., Zhao G., Lu J. (2021a). RNA-Seq analysis reveals alternative splicing under heat stress in rainbow trout (Oncorhynchus mykiss). Mar. Biotechnol. 17, 1–13. doi: 10.1007/s10126-021-10082-z
Sun J., Zhao L., Wu H., Liu Q., Liao L., Luo J., et al. (2020). Acute hypoxia changes the mode of glucose and lipid utilization in the liver of the largemouth bass (Micropterus salmoides). Sci. Total Environ. 713, 135157. doi: 10.1016/j.scitotenv.2019.135157
Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., et al. (2016). The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, 362–368. doi: 10.1093/nar/gkw937
Tan S., Wang W., Tian C., Niu D., Zhou T., Jin Y., et al. (2019). Heat stress induced alternative splicing in catfish as determined by transcriptome analysis. Comp. Biochem. Physiol. Part D Genomics Proteomics. 29, 166–172. doi: 10.1016/j.cbd.2018.11.008
Tan S., Wang W., Zhong X., Tian C., Niu D., Bao L., et al. (2018). Increased alternative splicing as a host response to Edwardsiella ictaluri infection in catfish. Mar. Biotechnol. 20, 729–738. doi: 10.1007/s10126-018-9844-2
Tian Y., Gao Q. F., Dong S. L., Zhou Y. G., Yu H., Liu D., et al. (2022). Genome-wide analysis of alternative splicing (AS) mechanism provides insights into salinity adaptation in the livers of three euryhaline teleosts, including Scophthalmus maximus, Cynoglossus semilaevis and Oncorhynchus mykiss. Biology 11, 222. doi: 10.3390/biology11020222
Tian C., Lin X., Saetan W., Huang Y., Shi H., Jiang D., et al. (2020a). Transcriptome analysis of liver provides insight into metabolic and translation changes under hypoxia and reoxygenation stress in silver sillago (Sillago sihama). Comp. Biochem. Physiol. Part D Genomics Proteomics. 36, 100715. doi: 10.1016/j.cbd.2020.100715
Tian Y., Wen H., Qi X., Zhang X., Sun Y., Li J., et al. (2020b). Alternative splicing (AS) mechanism plays important roles in response to different salinity environments in spotted sea bass. Int. J. Biol. Macromol. 155, 50–60. doi: 10.1016/j.ijbiomac.2020.03.178
Wang Q., Shen W., Hou C., Liu C., Wu X., Zhu J. (2017). Physiological responses and changes in gene expression in the large yellow croaker Larimichthys crocea following exposure to hypoxia. Chemosphere 169, 418–427. doi: 10.1016/j.chemosphere.2016.11.099
Wei X., Ai K., Li H., Zhang Y., Li K., Yang J. (2019). Ancestral T cells in fish require mTORC1-coupled immune signals and metabolic programming for proper activation and function. J. Immunol. 203, 1172–1188. doi: 10.4049/jimmunol.1900008
Wen H. S., Zhang M., He F., Li J. (2016). Research progress of aquaculture industry and its seed engineering in spotted sea bass (Lateolabrax maculatus) of China. Fish Inf. Strategy 31, 105–111. doi: 10.13233/j.cnki.fishis.2016.02.005
Wu C., Liu Z., Li F., Chen J., Jiang X., Zou S. (2017). Gill remodeling in response to hypoxia and temperature occurs in the hypoxia sensitive blunt snout bream (Megalobrama amblycephala). Sci. Total Environ. 479, 479–486. doi: 10.1016/j.scitotenv.2019.135157
Wu G., Mannam A. P., Wu J., Kirbis S., Shie J., Chen C., et al. (2003). Hypoxia induces myocyte-dependent COX-2 regulation in endothelial cells: role of VEGF. Am. J. Physiol. Heart Circ. Physiol. 285, H2420–H2429. doi: 10.1152/ajpheart.00187.2003
Xia J., Li H., Li B., Gu X., Lin H. (2017). Acute hypoxia stress induced abundant differential expression genes and alternative splicing events in heart of tilapia. Gene 639, 52–61. doi: 10.1016/j.gene.2017.10.002
Xiao W. (2015). The hypoxia signaling pathway and hypoxic adaptation in fishes. Sci. China Life Sci. 58, 148–155. doi: 10.1007/s11427-015-4801-z
Xie Y., Shi X., Sheng K., Han G., Li W., Zhao Q., et al. (2019). PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia. Mol. Med. Rep. 19, 783–791. doi: 10.3892/mmr.2018.9713
Yang Y., Fu Q., Wang X., Liu Y., Zeng Q., Li Y., et al. (2018). Comparative transcriptome analysis of the swimbladder reveals expression signatures in response to low oxygen stress in channel catfish, Ictalurus punctatus. Physiol. Genomics 50, 636–647. doi: 10.1152/physiolgenomics.00125.2017
Yan C., Wan R., Shi Y. (2019). Molecular mechanisms of pre-mRNA splicing through structural biology of the spliceosome. Cold Spring Harb. Perspect. Biol. 11, a032409. doi: 10.1101/cshperspect.a032409
Yee Koh M., Spivak-Kroizman T. R., Powis G. (2008). HIF-1 regulation: Not so easy come, easy go. Trends Biochem. Sci. 33, 526–534. doi: 10.1016/j.tibs.2008.08.002
Yu G., Wang L., Han Y., He Q. (2012). clusterProfiler: an r package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Zeng Y., Zhang L., Hu Z. (2016). Cerebral insulin, insulin signaling pathway, and brain angiogenesis. Neurol. Sci. 37, 9–16. doi: 10.1007/s10072-015-2386-8
Zhang Q., Cui B., Li H., Li P., Hong L., Liu L., et al. (2013). MAPK and PI3K pathways regulate hypoxia-induced atrial natriuretic peptide secretion by controlling HIF-1 alpha expression in beating rabbit atria. Biochem. Biophys. Res. Commun. 438, 507–512. doi: 10.1016/j.bbrc.2013.07.106
Zhang Z., Yao L., Yang J., Wang Z., Du G. (2018). PI3K/Akt and HIF−1 signaling pathway in hypoxia−ischemia. Mol. Med. Rep. 18, 3547–3554. doi: 10.3892/mmr.2018.9375
Zheng X., Linke S., Dias J. M., Zheng X., Gradin K., Wallis T. P., et al. (2008). Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. U. S. A. 105, 3368–3373. doi: 10.1073/pnas.0711591105
Zhou Y., Luo W., Yu X., Wang J., Feng Y., Tong J. (2020). Cardiac transcriptomics reveals that MAPK pathway plays an important role in hypoxia tolerance in bighead carp (Hypophthalmichthys nobilis). Animals 101483. doi: 10.3390/ani10091483
Keywords: RNA-Seq, hypoxia-inducible factor, cell cycle, alternative splicing, hypoxia
Citation: Ren Y, Tian Y, Mao X, Wen H, Qi X, Li J, Li J and Li Y (2022) Acute hypoxia changes the gene expression profiles and alternative splicing landscape in gills of spotted sea bass (Lateolabrax maculatus). Front. Mar. Sci. 9:1024218. doi: 10.3389/fmars.2022.1024218
Received: 21 August 2022; Accepted: 20 September 2022;
Published: 03 October 2022.
Edited by:
Hongsheng Yang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Changxu Tian, Guangdong Ocean University, ChinaCopyright © 2022 Ren, Tian, Mao, Wen, Qi, Li, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Li, eXVubGkwMTE2QG91Yy5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.