
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 01 November 2022
Sec. Microbial Symbioses
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1024117
This article is part of the Research Topic Diversity, Dynamics and Function of Host-Associated Microbiomes in Marine Organisms View all 11 articles
 Xiaoling Wan1
Xiaoling Wan1 Jia Li1
Jia Li1 Renmao Tian2
Renmao Tian2 Richard William McLaughlin3
Richard William McLaughlin3 Yujiang Hao1,4Junyi Wu5Zhengyang Wang5Fei Fan1,4
Yujiang Hao1,4Junyi Wu5Zhengyang Wang5Fei Fan1,4 Ding Wang1,4
Ding Wang1,4 Jinsong Zheng1,4*
Jinsong Zheng1,4*Understanding the effects of human care on the dynamics of the host-associated microbiota is critical for the health management of dolphins living in an aquarium. Yet this aspect remains relatively unexplored. Here, by utilizing 16S rRNA gene sequencing, we profiled the blowhole and gut bacterial communities of two bottlenose dolphins (Tursiops truncatus) and a Chinese white dolphin (Sousa chinensis) reared in the same indoor pool, based on year-round surveillance. In addition, we compared these dolphin microbiotas with those previously published datasets from wild dolphins. Our results showed that both the blowhole and the gut of the two dolphin species under human care shared a more similar microbiome than members of the same dolphin species across different habitats (human care vs wild). However, the effects of human care on the dolphin microbiome from the two body sites varied. In the aquarium, bacterial alpha diversities differed significantly between the two body sites, and the seasonal stability of the bacterial community was more evident in the gut than in the blowhole. Additionally, the blowhole bacterial composition and the predicted functional capacity from the two dolphin species showed differences and were less convergent than their gut microbiota over a decade-long cohabitation. Further analyses showed that heterogeneous and homogeneous selections (i.e., deterministic processes) contributed more to the blowhole than to the gut bacterial communities, while a dispersal limitation (i.e., a stochastic process) was more important for the gut microbiota. The present study provides the first comparative evidence that the gut microbiota may be more plastic in response to the human care environment than the blowhole microbiota. This improves our understanding of dolphin health management under human care and helps to predict their microbial responses to environmental changes.
As top predators in marine ecosystems, cetaceans are critical sentinel and indicator species for ecosystem health and integrity, which has caught global attention (Parsons et al., 2015). However, due to the pervasiveness of anthropogenic pressures, at least a quarter of the world’s cetaceans have been confirmed as endangered or even extinct (Turvey et al., 2007; Simmonds and Eliott, 2009). Human care has been put forward as one of the complementary approaches to in situ and natural ex situ conservation practices for some endangered small cetacean species (Wang et al., 2005; Curry et al., 2013; Muka and Zarpentine, 2021). Although the care of Dolphinarium animal husbandry specialists can facilitate the diagnosis and treatment of injured or sick individuals and preserve genetic diversity (Muka and Zarpentine, 2021), it also changes the animals’ diet and restricts their behaviors, which may lead to physiological changes, disease susceptibility (e.g., gastrointestinal and respiratory diseases) and early mortality (Lott and Williamson, 2017; Nabi et al., 2020; Segawa et al., 2020). As a result, there is an urgent need to develop effective assessment tools that can improve cetaceans’ long-term survival and welfare under human care.
In recent decades there has been a growing appreciation of the ability to alter the microbiome to positively affect the health of the animal which could aid in the conservation of threatened species (Jin et al., 2019; West et al., 2019). Microbes, especially those which colonize the gut and respiratory tract, are extremely important in maintaining the health of the host by affecting the immune defenses, digestion and nutrient absorption, behavior, reproduction, etc., (Vuong et al., 2017; Yadav et al., 2018; Invernizzi et al., 2020; Mallott et al., 2020; Bosco and Noti, 2021). There is strong support for systemic microbial communication between the microbiota found in the gastrointestinal and respiratory tracts (Enaud et al., 2020). Multiple lines of evidence show that the host-associated microbiota co-diversifies with their host lineages and can be further shaped by the external environment (e.g., dietary and habitat conditions) (Bik et al., 2016; Bai et al., 2021; Jin et al., 2021; Mallott and Amato, 2021). Compared to wild animals, animals under human care may undergo a series of changes that influence their microbiome, e.g., dietary restrictions, antibiotic interventions, and increased exposure to human-associated microbes (Hyde et al., 2016). The disturbance of a healthy microbiome may disrupt immune homeostasis and promote opportunistic bacterial pathogens (Toor et al., 2019; Kaul et al., 2020). Therefore, as recently proposed, integrating human care along with host microbial research is becoming more important in going forward for conservation programs (Dallas and Warne, 2022). Despite recent advancements, studies on the microbiome of cetaceans under human care remain limited.
Several recent studies have reported the influences of the human care environment on the cetacean microbiota. For example, Suzuki et al. (2019) found that the gut microbial alpha diversity and the dominant phyla of common bottlenose dolphins (Tursiops truncatus) differed in three aquaria. Suzuki et al. (2021) found that Indo-Pacific bottlenose dolphins (T. aduncus) under human care and wild environments harbored habitat-specific microbial groups and that several pathogenic bacterial genera were only detected in the dolphins under human care, which highlighted the importance of monitoring pathogenic bacteria in cetaceans under human care. Another study provided evidence that the blowhole microbiota of common bottlenose dolphins was individual-specific and stable over eight months (Vendl et al., 2021). Regardless of the potential importance of human care for the health of captive cetaceans, the seasonal stability, and the degree to which human care humanizes the microbiota of different cetacean taxa have not been explored.
For these reasons, by using 16S rRNA gene-based high-throughput sequencing, we characterized the taxonomic and potential functional profiles of the blowhole and fecal microbiota of two common bottlenose dolphins and a Chinese white dolphin (Sousa chinensis) reared in the same indoor pool, based on year-round surveillance. Firstly, we uncovered the seasonal dynamics of the blowhole and gut microbiota of both dolphin species. Then we examined whether and how the host microbiota (blowhole and gut) converged between these two cohabiting dolphin species. Lastly, we explored the effects of human care on the dolphins’ microbiota through a comparison with their wild counterparts from the previously published datasets. In this study, we aim to enhance our knowledge about how human care influences the dolphin microbiota to facilitate their health monitoring in the future.
Two bottlenose dolphins (ID: BD1 and BD2) and one Chinese white dolphin (ID: CWD) housed in the Dolphinarium of Nanning Zoo, China, were included in this study (Table S1). These three dolphins were rescued from the wild and have lived in the Dolphinarium since 2007 under the same housing conditions (i.e., the same diet and aquarium parameters). There were no relations between these animals. They were able to breathe out and defecate promptly on command, allowing for a biological sample (exhaled blow or feces) to be gathered in no more than 10 min in a non-invasive way. Sample collection was generally performed at the end of the month around 4 pm during the whole course of the study. To eliminate potential contamination by pool water and skin, the skin regions near the blowhole and the anus were wiped gently using alcohol pads before sampling. Specifically, feces were collected using a sterilized plastic spoon immediately after defecating and were then transferred to a 5-mL sterile Eppendorf tube. At least 5 g of feces was collected for each dolphin at each sampling time point. For blow samples, a 50-mL sterile Eppendorf tube was placed 2-3 cm above the blowhole, and 2-4 exhalations from the same individual were collected per tube. In total two tubes were used to collect the blow from one individual. After fecal and blowhole sampling each month, a total of the 5-L pool water was gathered and filtered through a mixed cellulose esters filter paper (47-um pore diameter and 0.45-um pore size). The filters were stored with 95% ethanol until DNA extraction. Feces, blow, and water samples were collected monthly over a period of 12 months from Dec. 2015 to Nov. 2016, except for the Mar. 2016 sample. Sample details are shown in Table S2. Monthly mean water temperatures were recorded by the dolphin trainers (Figure S1). All samples were preserved at -80 °C until DNA extraction.
One week before each sampling event, the blood biochemical and blood routine tests from three dolphins were evaluated by veterinarians. The test results were within the normal range and all dolphins were considered healthy during all sampling events. No antibiotics were given to dolphins two weeks before each sampling event. The sampling procedures used in this study were approved by the Regulations of the People’s Republic of China for the Implementation of Wild Aquatic Animal Protection (promulgated in 1993), adhering to all ethical guidelines and legal requirements in China.
The ZR Faecal DNA Kit (Zymo Research Incorporated, CA, USA) was applied to extract the metagenomic DNA from frozen samples. Universal primers 341F-806R were then used to amplify the V3-V4 regions of bacterial 16S rRNA genes for library constructions (Roggenbuck et al., 2014). PCR products of different samples were pooled and run on the Illumina HiSeq 2500 platform (2 × 250 bp paired ends).
The raw sequencing reads were analyzed via an internal pipeline Amplicon Sequencing Analysis Pipeline (Wan et al., 2018). In brief, after a quality check of raw sequences using FastQC, sequences with high quality were then trimmed to remove primer sequences (Brown et al., 2017). Clean reads were then processed to generate operational taxonomic units (OTUs) by UPARSE at the 97% cutoff (Edgar, 2013). The representative sequence of each OTU was classified into taxa with a confidence threshold of 0.8 on the RDP classifier using the SILVA database (Cole et al., 2014). FastTree was applied to generate phylogenetic trees from all representative sequences (Price et al., 2009). To enable comparisons with previously-published datasets of the bottlenose dolphin and the Chinese white dolphin living in other habitats, we downloaded the respiratory (PRJNA279427) and rectal sequences (PRJNA174530) of bottlenose dolphins inhabiting San Diego Bay and Sarasota Bay, USA (Bik et al., 2016) and intestinal sequences (PRJNA610950) of the wild Chinese white dolphin stranding in Guangdong Province, China (Wan et al., 2021). The above steps were repeated for sequencing processing for all downloaded 16S rRNA gene sequences.
PICRUST version 2.0 (Douglas et al., 2020) was applied to predict functions based on the taxonomy affiliations from the 16S rRNA gene sequences. To obtain the potential pathogens from 16S rRNA gene sequences, the Functional Annotation of PROkaryotic TAXa (FAPROTAX) database was further used to sort out potentially pathogenic groups, including ‘animal parasites or symbionts’, ‘intracellular parasites’, ‘invertebrate parasites’, ‘human pathogens all’, ‘human pathogens diarrhea’, ‘human pathogens gastroenteritis’, ‘human pathogens meningitis’, ‘human pathogens nosocomia’, ‘human pathogens pneumonia’, ‘human pathogens septicemia’, ‘plant pathogen’ and ‘fish parasites’, which has been experimentally verified to threaten the welfare of humans, aquatic animals or plants (Louca et al., 2016; Zhou et al., 2020).
To eliminate potential contamination originating from the water samples, we identified contaminated OTUs using the R package {decontam} following the same parameters outlined in the study by Vendl et al., 2021. In total, we discarded 339 OTUs from the dolphin microbiota which likely originated from the pool water (Figure S2). We then removed seven blowhole samples and two fecal samples from the dataset, as the number of reads was extremely low (< 6,000) (Table S2). The resultant OTU abundance tables from these remaining samples were rarefied to the same number (at the minimum sequence number of 7,551) of sequences per sample to ensure equal sampling depth using the rrarefy function (R package {vegan}) (Weiss et al., 2017).
Microbial α-diversity indices, including taxonomic (richness) and phylogenetic (Faith’s PD) diversities, were computed (R package {Picante}) (Kembel et al., 2010). To evaluate whether both α-diversity indices were correlated with the sampling month for each dolphin, we fitted linear mixed effects models (Lindstrom and Bates, 1990) with a fixed effect of ‘sampling month’ and random effects of ‘dolphin ID’. The α-diversity indices between groups were further tested by applying the Wilcoxon rank sum test. To assess the difference in the microbial communities between the groups, non-parametric permutation tests, including the multiple response permutation procedure (MRPP), analysis of similarity (ANOSIM), and permutational multivariate analysis of variance (ADONIS), were computed, based on Bray-Curtis and weighted UniFrac distances. Non-metric multidimensional scaling (NMDS) analyses were further calculated to visualize patterns of community dissimilarity across samples. The LEfSe (Linear discriminant analysis Effect Size) was applied to identify biomarker functional potentials between groups (P < 0.05, LDA score > 3.0) via the website http://huttenhower.sph.harvard.edu/galaxy/.
The microbial community assembly processes were initially quantified using the null model approach (Stegen et al., 2013). Bray-Curtis-based Raup-Crick (RCBray) and β-mean nearest taxon distance (β-NTI) values of each pairwise comparison between two samples were calculated using the iCAMP (Ning et al., 2020). A β-NTI value < -2 or > 2 indicates that homogeneous selection or variable selection (deterministic processes) is predominant in shaping the community composition, whereas |β-NTI | < 2 indicates that neutral assembly processes account for the communities. Further, RCBray was used to partition the stochastic ecological processes into dispersal limitation, undominated and homogenizing dispersal at the cutoff of ± 0.95 (Stegen et al., 2013).
For all 80 samples, a total of 2,831,917 sequences were obtained, with an average of 35,398 (ranging from 19,415 to 44,181) sequences per sample, resulting in 2,714 OTUs. After removing 339 contaminated OTUs that were likely from the pool water (Figure S2), as well as nine dolphin samples with low reads number of less than 6,000 (Table S2), 2,539 OTUs were obtained from 71 samples in total. The rarefaction curves of the observed OTUs suggested that the diversity of blow, fecal, and pool water microbiota sampled in this study were well represented after resampling at the minimum sequence number of 7,551 (Figure S3).
Based on the taxonomic assignment, we found that the blow, feces, and pool water samples exhibited unique microbial profiles, respectively. Proteobacteria (~51.17%) and Bacteroidetes (~32.20%) constitute the main component (~83.37%) of the dolphin blow microbiota, whereas the dolphin fecal microbiota was dominated by Firmicutes (~73.99%) and a lesser degree Proteobacteria (~16.57%) (Figure 1A). Pool water samples were overwhelmingly dominated by Proteobacteria (~96.21%) (Figure 1A). The blow microbiota was mainly composed of Tenacibaculum (~6.35%) and Arcobacter (~4.35%) (Figure 1B). The fecal bacteria were dominated by Romboutsia (~19.28%), Clostridium XI (~16.22%), Clostridium sensu stricto (~12.82%), and unclassified Peptostreptococcaceae (~10.03%) (Figure 1B). In the pool water, unclassified Alphaproteobacteria (~50.05%), Halomonas (~23.94%), and unclassified Hyphomonadaceae (~18.32%) were the most prevalent (Figure 1B). Non-metric multidimensional scaling (NMDS) analyses further demonstrated that, for each dolphin species, the blow and feces contained disparate bacterial communities (Adonis: P < 0.001), which were well separated from the pool water community (Adonis: P < 0.001) (Figures 1C, D). As for α-diversity parameters (richness and PD), even though there was no significant inter-individual differentiation (Figures S4A, B), both α-diversity indices of the blow bacteria were significantly higher than either fecal or pool water bacteria (Figures S4C, D, P < 0.05). Nevertheless, fecal and pool water bacterial diversities were comparable (Figures S4C, D, P > 0.05).
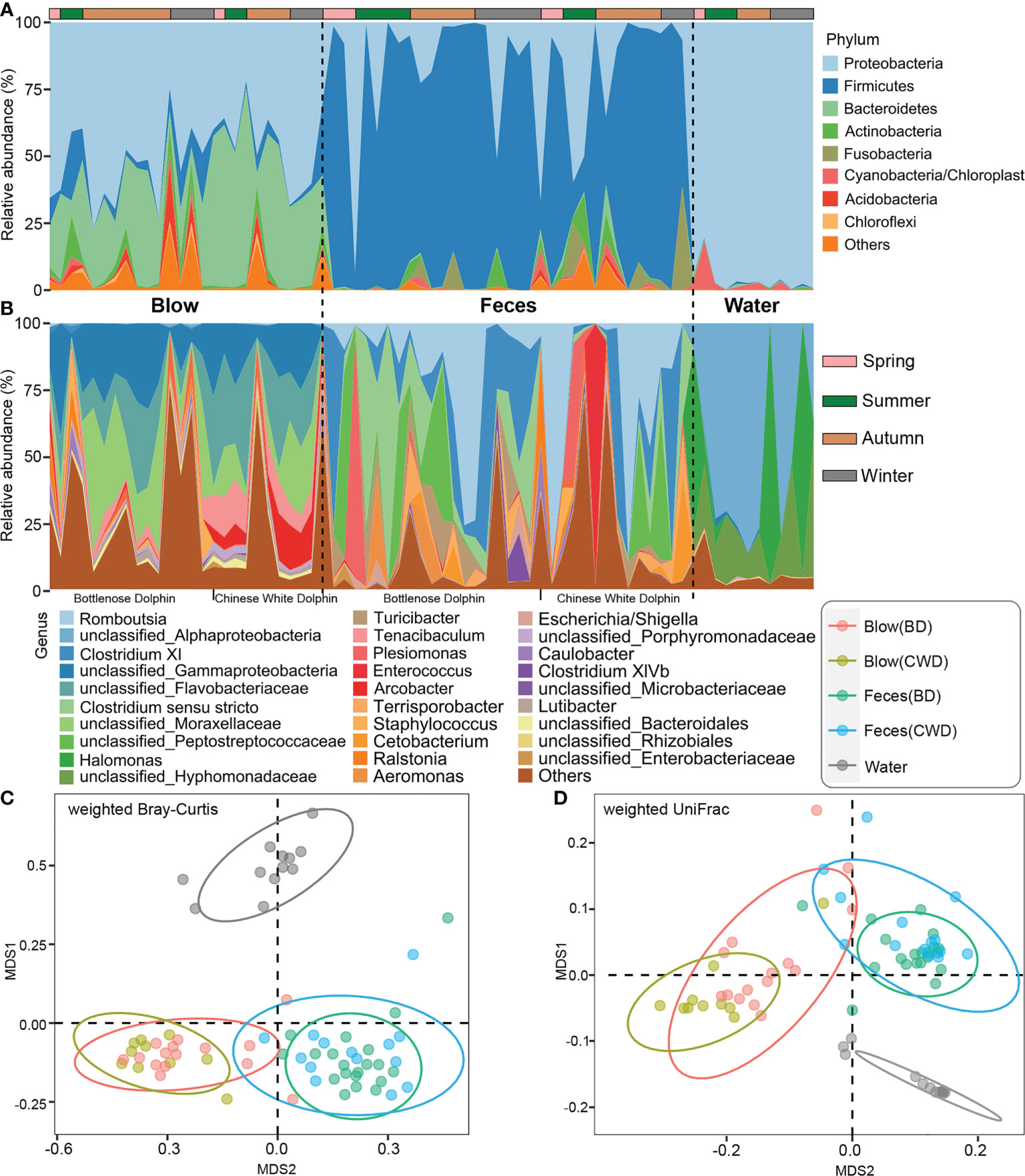
Figure 1 Composition and comparison of microbiota from the dolphin blow, feces, and pool water. (A), the relative abundance of each phylum. A total of 28 phyla were detected. Only the top 8 most abundant phyla are shown here; the remaining 20 phyla as well as unclassifiable sequences were grouped as ‘Others’. (B), the relative abundance of each genus. A total of 571 genera, including unclassified genera, were detected. Only the top 29 most abundant genera are shown here; the remaining genera including unclassifiable sequences were grouped as ‘Others’. The horizontal lines at the top indicate the collecting season of each specimen. (C), the non-metric multidimensional scaling (NMDS) analysis of different sampling groups at the OTU level based on weighted Bray-Curtis distances. (D), the NMDS analysis of different sampling groups at the OTU level based on weighted UniFrac distances. BD indicates the bottlenose dolphin, and CWD indicates the Chinese white dolphin.
Neither blow nor fecal α-diversity indices (richness and PD) differed significantly over time (Table S3, P > 0.05). In general, for each dolphin, the microbial diversity indices of fecal samples over one year were more variable than the blow samples, as revealed by CV coefficient of variation (CV) values (Table 1). Only the blow PD of dolphin BD2 showed a higher CV value than that in the feces (Table 1). No significant seasonal variation in the blow or fecal microbial composition at the OTU level was detected among the four seasonal groups (winter, spring, summer, and autumn) (Adonis, P > 0.05; Table S4) or two seasonal groups (winter-spring and summer-autumn) (Adonis, P > 0.05; Table S5). Interestingly, when samples were divided into two temperature groups (high-temperature and low-temperature groups) based on the monthly mean pool water temperature, feces rather than blow samples, exhibited significant microbial composition shifts (Adonis, P < 0.05; Table S6), signifying that fecal microbial composition is more sensitive to water temperature compared to the blow microbiota.
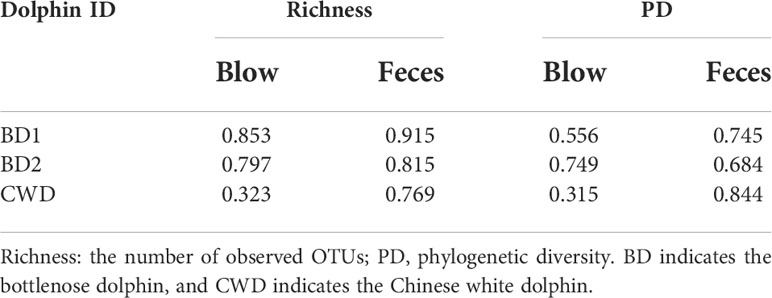
Table 1 The coefficient of variation of microbial diversity indices over one year in each dolphin.
We further named the OTUs that were detected in each dolphin across 80% of all sampling events as the “intra-core” (i.e., temporal core). A minimal part (0.07-13.17%) of the total OTUs were detected to constitute a dolphin’s temporal core, which accounted for a relatively high proportion (54.35-86.55%) of the total reads of each dolphin, except for the blow dataset of the bottlenose dolphin BD1 (0.84%) (Table S7). In total, 70 intra-core OTUs were shared by both blow and fecal samples, accounting for on average 59.10% of all the dolphin microbial reads. The blow-associated intra-core of the Chinese white dolphin harbored a higher proportion of Bacteroidetes (43.91%) compared to bottlenose dolphins (21.17%) (Figure 2A). As for the fecal intra-cores, Firmicutes (63.02%) was most prevalent, followed by Proteobacteria (6.46%) (Figure 2A). A higher proportion of intra-core bacteria in the blow (59.79%) compared to the feces (10.97%) were novel and could therefore not be assigned a genus (Figure 2B). Among the classifiable genera, Tenacibaculum (7.05%), Arcobacter (5.15%), and Staphylococcus (1.36%) were abundant in the blow samples (Figure 2B). Whereas, Romboutsia (20.20%), Clostridium XI (15.72%), Clostridium sensu stricto (5.48%), Terrisporobacter (2.76%), Plesiomonas (4.63%), Turicibacter (4.29%) and Escherichia/Shigella (1.23%) were prevalent in the feces (Figure 2B).
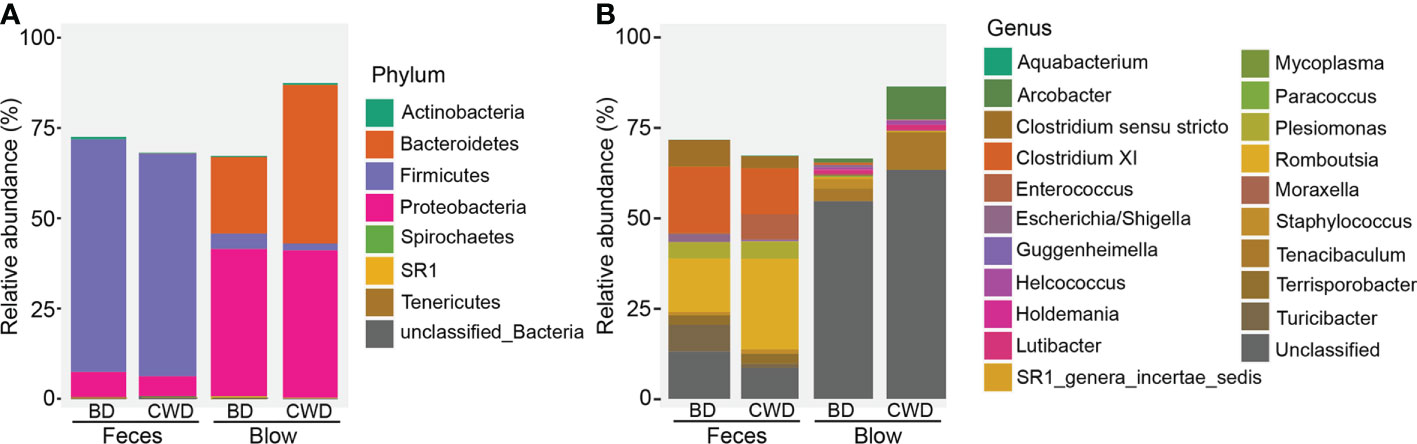
Figure 2 Taxonomic composition of 70 intra-cores detected from each dolphin. (A), Phylum level. (B), Genus level. BD indicates the bottlenose dolphin, and CWD indicates the Chinese white dolphin.
The hierarchical clustering analysis showed that the fecal microbiota from both dolphin species clustered closer to each other, whereas the blow microbiota differed substantially between species (Figure 3). This pattern is further supported by three different non-parametric multivariate statistical tests (Table 2). Then we identified OTUs of blow microbiota which was able to discriminate dolphin species classification, with an overall accuracy of 96% (random forest models). Interestingly, the top 30 most discriminatory OTUs (1.72% of the overall blow OTUs) accounted for an average of 30.06% of the relative read abundance of the blow microbiota (Figure S5 and Table S8). Among them, nine OTUs were assigned at the genus level, with Tenacibaculum (4.35%), Staphylococcus (1.29%), and Ralstonia (1.24%) at a higher proportion (Table S8).
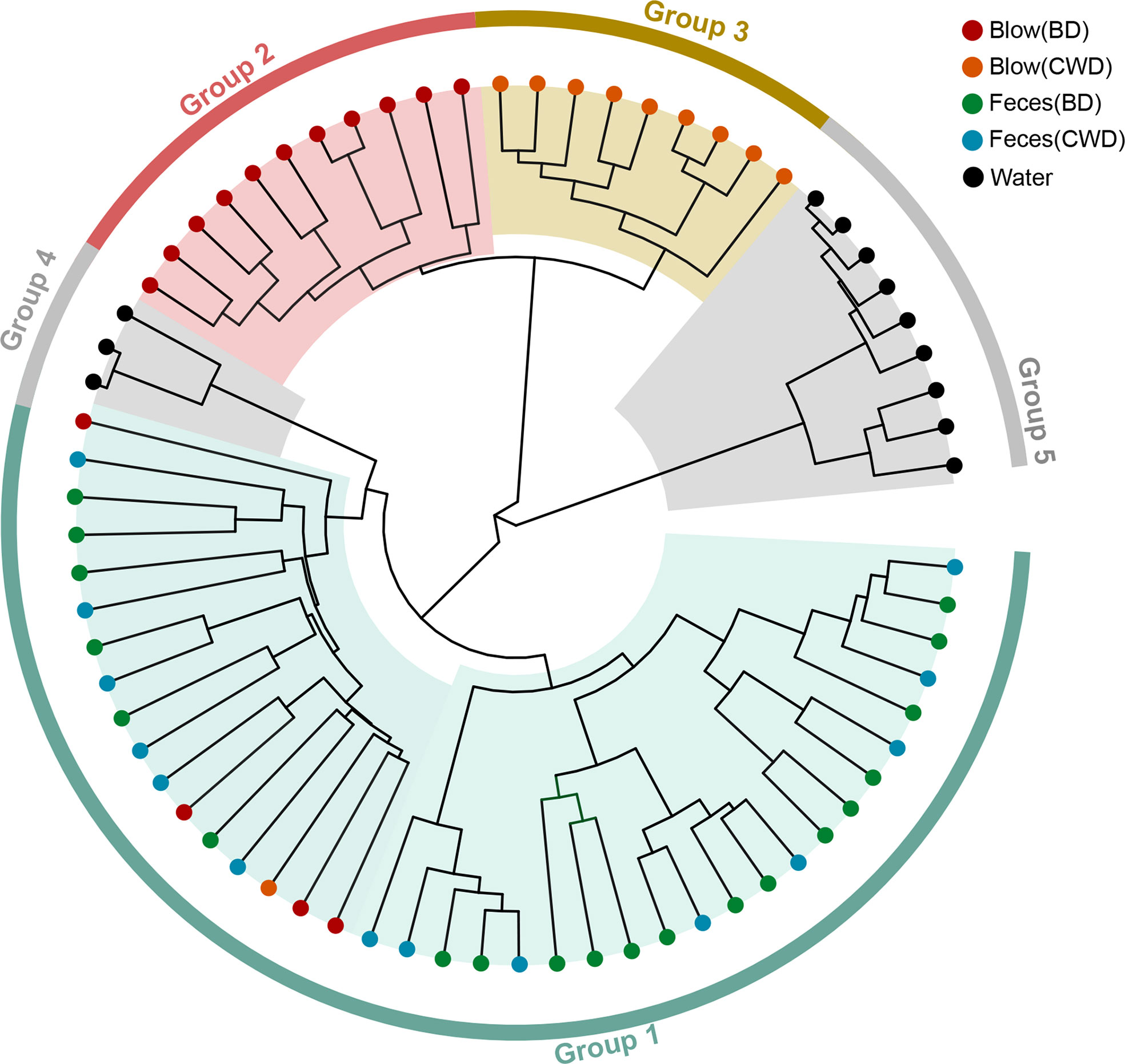
Figure 3 The hierarchical clustering analysis of all samples. Different colors of the outside ring represent that all samples were grouped into five clusters (group 1, group 2, group 3, group 4 and group 5) using unweighted pair group method with arithmetic mean (UPGMA) based on the microbial taxonomic composition at the OTU level. The terminal node of each branch corresponded to the individual sample. Colors of the nodes reflected different sampling groups in the legend. BD indicates the bottlenose dolphin, and CWD indicates the Chinese white dolphin.
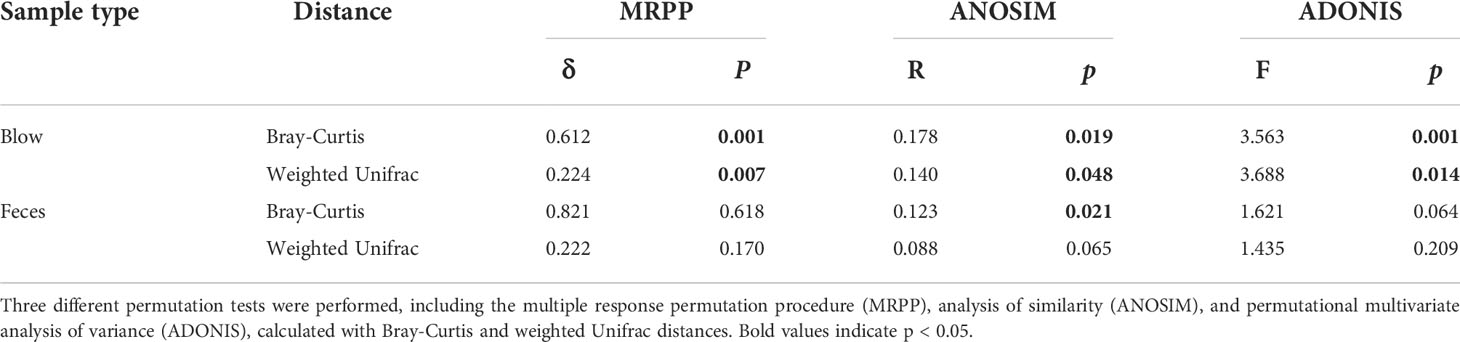
Table 2 Significance tests on the effects of species on the respiratory and intestinal microbial community composition.
By quantifying deviations in the microbial phylogenetic turnover, we found that both blow and fecal bacterial communities were primarily governed by neutral processes (dispersal limitation and drift) (Figure 4). However, feces (-0.49) exhibited significantly lower β-NTI than the blow (0.056) (Figure 4A). Specifically, deterministic assembly (heterogeneous and homogeneous selections, 2.54% and 22.87%, respectively) showed greater importance to the blow (0.46% and 8.36%, respectively), while the stochastic process was higher in the fecal bacterial community (dispersal limitation: 38.70%) than the blow bacterial community (dispersal limitation: 30.55%) (Figure 4B).
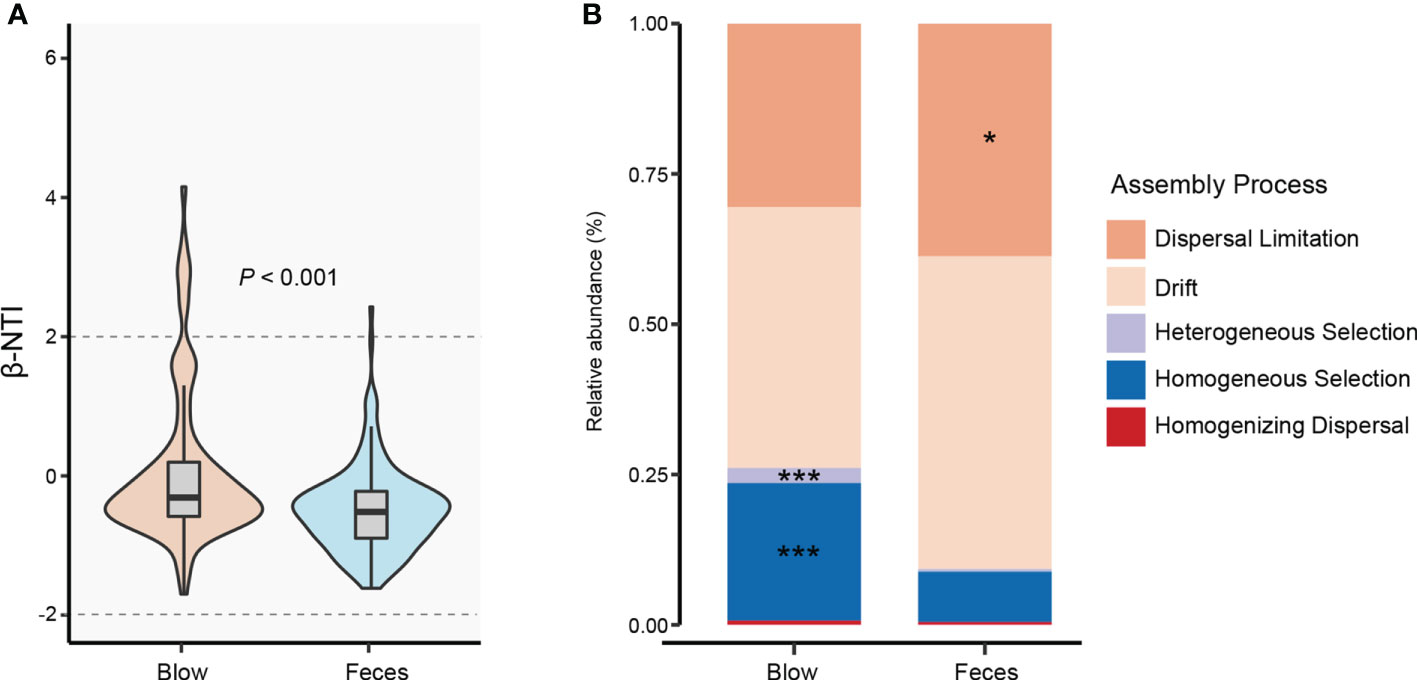
Figure 4 Bacterial community assembly processes for blow and feces in both dolphin species. (A), the distribution of the bacterial weighted beta nearest taxon index (β-NTI) in the blow and feces. The P value is from Wilcox rank sum test. (B), the relative abundance of different assembly processes in the blow and feces. Asterisks represent statistically greater proportions at the following significance levels: * P < 0.05, *** P < 0.001.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that a total of 297 pathways were detected across all dolphin samples. Based on functional profiles, the blow microbiota was distinct from the feces (Adonis: P < 0.001), having a significantly higher relative abundance of Vibrio cholerae infection, biofilm formation-Vibrio cholerae, epithelial cell signaling in Helicobacter pylori infection, Salmonella infection, Influenza A, lipid metabolism, etc. (Figures 5A, B). Whereas, pathways such as bacterial invasion of epithelial cells, Staphylococcus aureus infection, amino acid metabolism, and energy metabolism were enriched in the feces (Figure 5A).
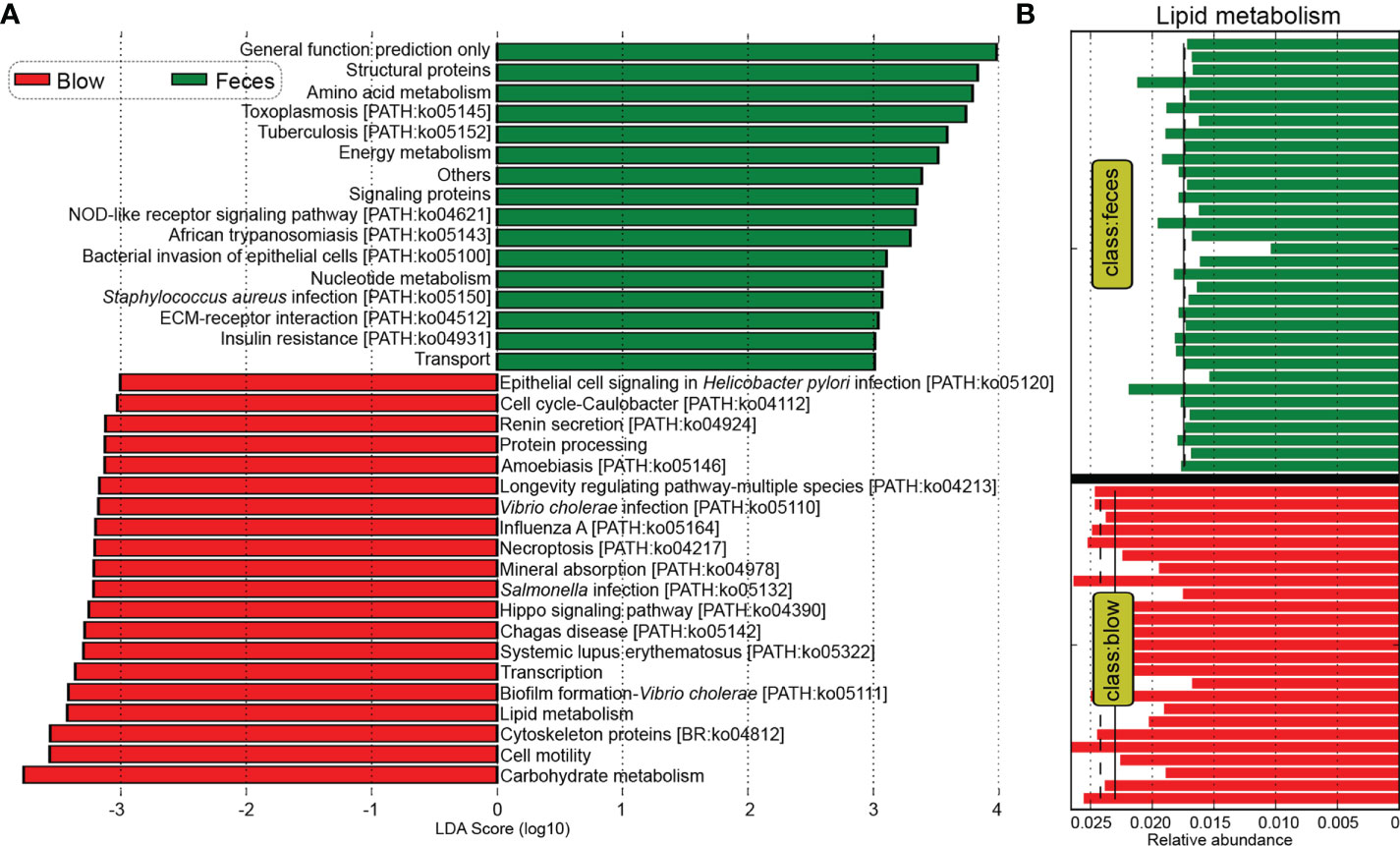
Figure 5 Comparative analyses of potential functions between the blow and fecal microbiomes. (A), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched in the blowhole and fecal microbiomes by the Linear discriminant analysis Effect Size (LEfSe) analysis. (B), the lipid metabolism pathway was significantly higher in the blow than in the fecal microbiome. The mean and median relative abundance are indicated with solid and dashed lines, respectively.
Consistent with the taxonomic profiles, the predicted functional composition of fecal microbiomes from both dolphin species was similar, while the predicted functional composition of the blow microbiomes from the two dolphin species was discriminated against each other significantly (Figures 6A, B; Table S9, P < 0.05). A LEfSe analysis was used to further characterize the significant pathways in the blow samples between the two dolphin species. The results showed that quorum sensing, secretion system, two-component system, and ABC transporters were abundant in the bottlenose dolphin, while ribosome was enriched in the Chinese white dolphin (Figure 6C).
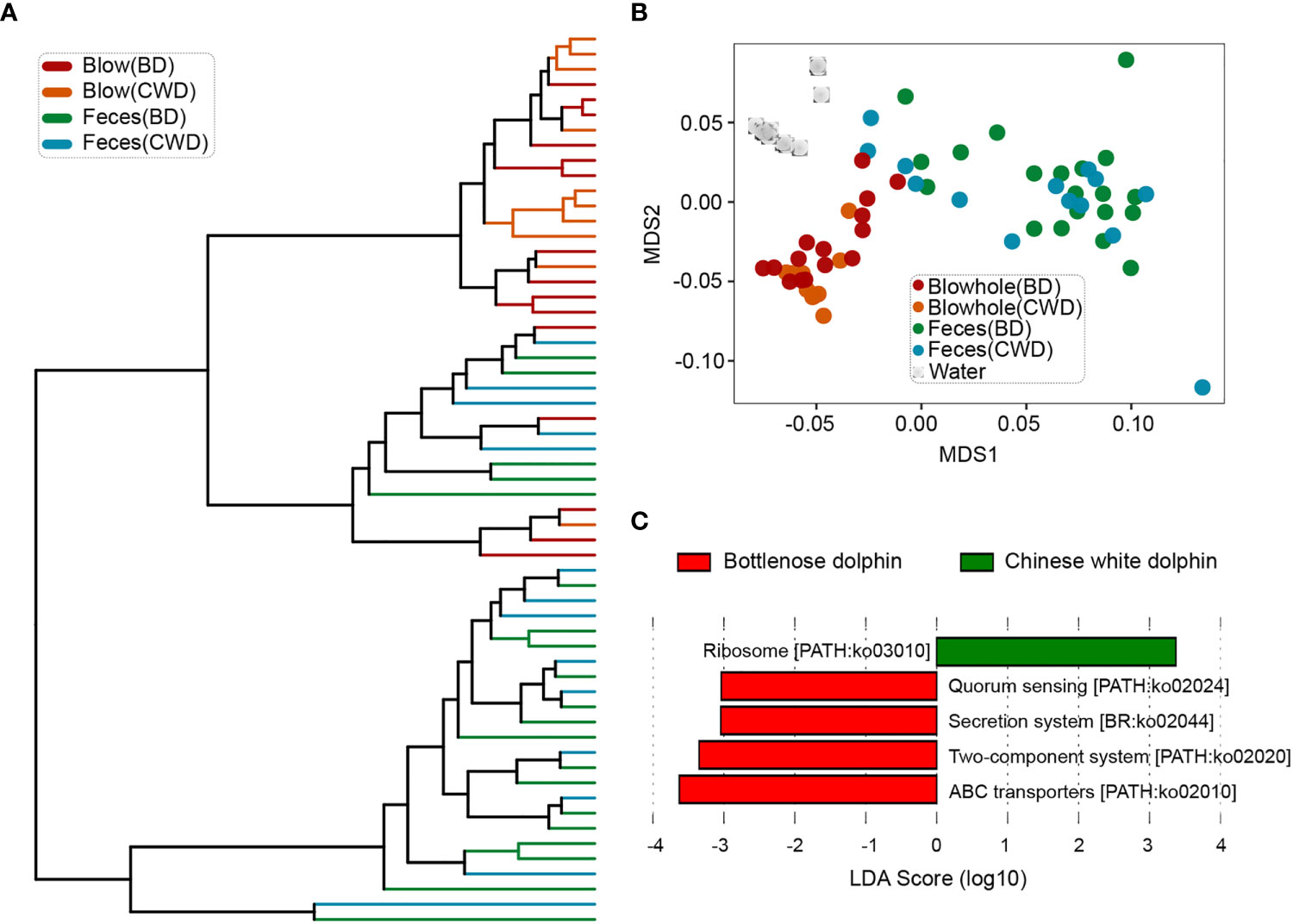
Figure 6 Convergence of the predicted functional composition of the dolphin microbiomes. (A), the hierarchy clustering analysis of the dolphin microbial functional composition. (B), the non-metric dimensional scaling (NMDS) analysis of microbial functional compositions based on the Bray-Curtis distances. (C), pathways that were over-represented for both dolphin species identified by the Linear discriminant analysis Effect Size (LEfSe) analysis. BD indicates the bottlenose dolphin, and CWD indicates the Chinese white dolphin.
To further evaluate the effects of the human care environment on the dolphin microbiota, blow and fecal microbiota data derived from this present study were compared with published datasets found in the studies (Bik et al., 2016; Wan et al., 2021). Our analyses revealed that the microbial composition of dolphin species under human care tended to be separate from their wild counterparts as determined by NMDS clustering analysis (Figure S6). Further statistical tests corroborated the NMDS results. Specifically, for both the blow and feces, the community dissimilarity, as revealed by four different distance matrices, between both captive dolphin species was significantly lower than that between dolphins under human care and their wild conspecifics, except the blow samples as calculated by the weighted UniFrac distance matrix (Figure 7).
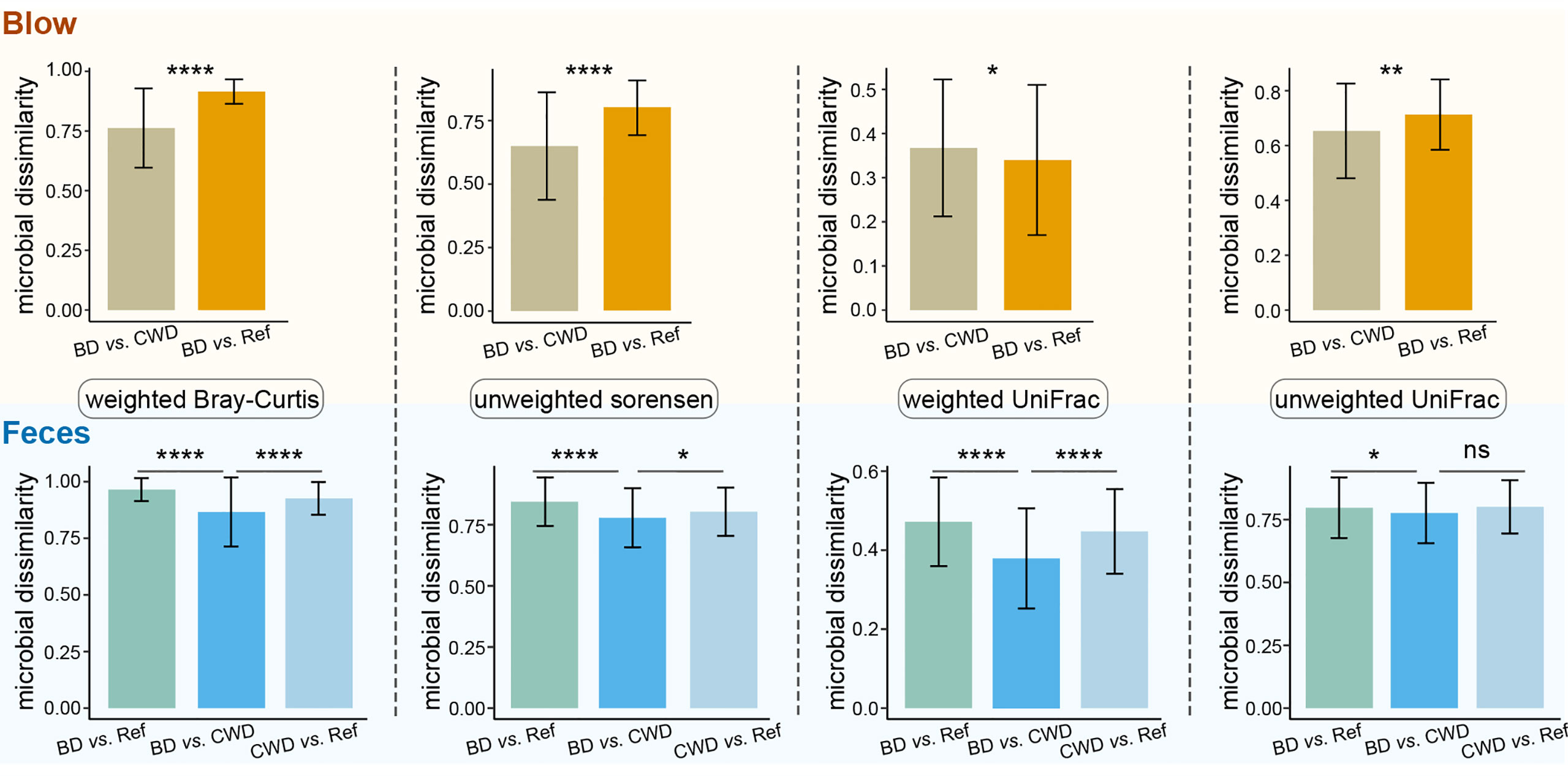
Figure 7 Information on within-group community dissimilarities for blow and fecal microbiotas. In these panels, higher microbial dissimilarity values indicate that these microbial samples are more dissimilar in composition. BD vs. CWD, the dissimilarity between bottlenose dolphins (BD) and the Chinese white dolphin (CWD). BD vs. Ref, the dissimilarity between bottlenose dolphins (BD) and the referred published datasets of their conspecifics. CWD vs. Ref, the dissimilarity between the Chinese white dolphin (CWD)and the referred published datasets of their conspecifics. Statistical significance is based on Wilcoxon rank sum test; **** P < 0.0001, ** P < 0.01, * P < 0.05. ns, not significant.
Among all the OTUs obtained from our current study, 57 OTUs (2,579 sequences) were assigned to three potential pathogenic groups (animal parasites or symbionts, intracellular parasites, and human pathogens all) based on the FAPROTAX database (Figure 8). This only accounted for an average of 0.48% of the total abundance. These potential pathogenic OTUs included species from Prevotella, Chlamydiales, Roseburia, Roseomonas, Campylobacter, Stenotrophomonas, Moraxella, Legionella, Edwardsiella, Proteus, Aquicella, Coxiellaceae and Pasteurellaceae (Figure 8). Prevotella and Stenotrophomonas were widespread in the blow, feces, and pool water samples, with a higher relative abundance in the blow. Moraxella was only detected in the blow, and Coxiella was only detected in the feces (Figure 8).
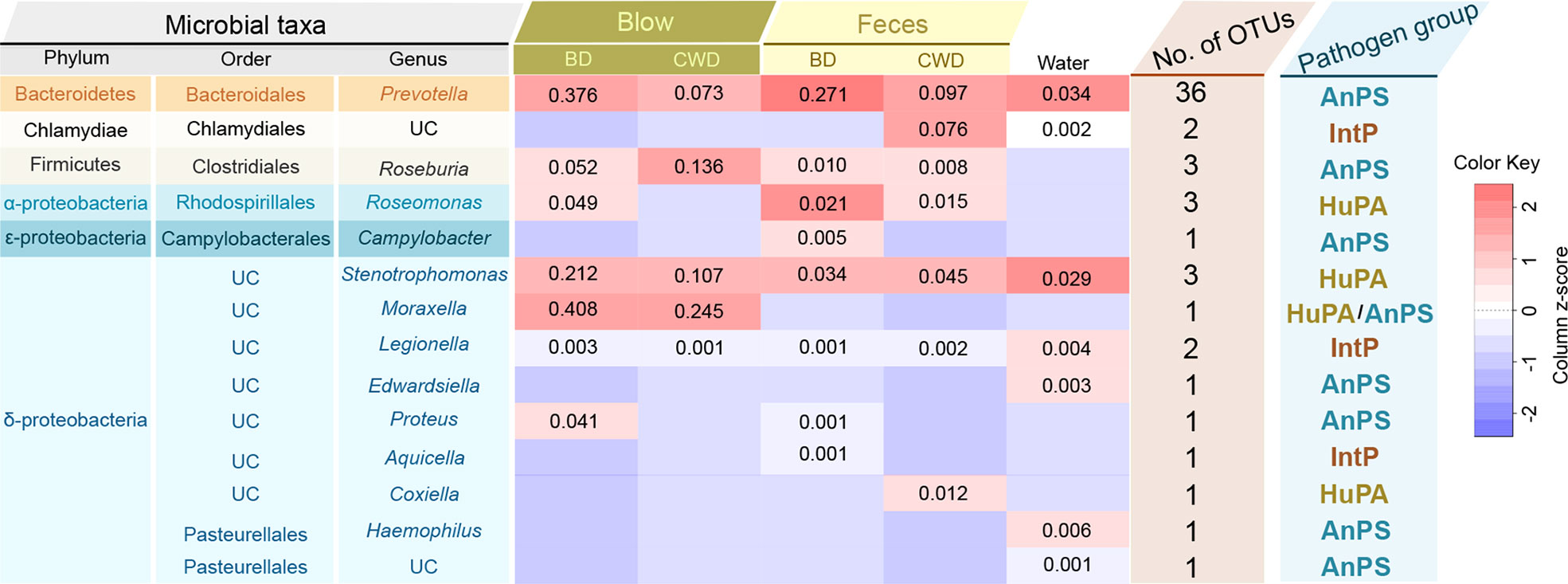
Figure 8 Taxonomic and functional information of potential pathogenic taxa. ‘Microbial taxa’ indicates the classification details of each potential pathogenic taxa. UC denotes ‘unclassified’. The heatmap in the middle indicates the log-transformed and column-scaled abundance data of potential pathogenic taxa present in five different sampling groups. BD, bottlenose dolphins; CWD, the Chinese white dolphin. Decimals indicate the relative abundance (%) of each pathogenic taxa, and the empty value indicates zero. ‘No. of OTUs’ indicates the number of OTUs detected from each potential pathogenic taxon. ‘Pathogen group’ indicates potentially pathogenic group of the pathogenic taxa predicted based on the FAPROTAX database. AnPS, animal parasites or symbionts; IntP, intracellular parasites; HuPA, human pathogens all.
Although keeping cetaceans in human care remains controversial, it cannot be denied that populations under human care are ideal study subjects that can be used to address some biological questions which will ultimately contribute to species conservation efforts. Recent research has revealed the influences of the human care environment on the mammalian gut microbiome (McKenzie et al., 2017; Frankel et al., 2019). However, how the human care environment impacts the microbial communities of different dolphin species is yet to be assessed. In this study, utilizing a one-year time series of 16S rRNA gene sequencing data, we compared the blow and fecal microbial composition and functional potentials of two cohabiting dolphin species (two bottlenose dolphins and one Chinese white dolphin) in an indoor pool. We found that although their blow and fecal bacterial communities stayed stable over one year, the blow diversity was less variable and its microbial composition was less sensitive to pool water temperature compared to the feces. Moreover, the blow microbial composition and functional potentials from two dolphin species under human care were species-specific and less convergent than their fecal microbiota over a decade-long cohabitation. This result indicates that bacteria from the interior respiratory tract are likely to be less alterable by human care, whereas bacteria from the intestinal tract are prone to be more plastic in response to the human care environment.
The bacterial community composition in the blow and fecal samples were distinct (Figure 1). For example, Romboutsia, which mainly has an intestinal origin (Gerritsen et al., 2019), was detected abundantly (~19.28%) in the feces, rather than in the blow, from all sampled dolphins (Figure 1B). This abundant genus may play essential roles in dolphins’ intestinal nutrient absorptions as it covers a variety of metabolic capabilities regarding carbohydrate utilization, fermentation of single amino acids, anaerobic respiration, and metabolic end products (Gerritsen et al., 2019). Similar to the previous findings of the blow from free-ranging bottlenose dolphins (Robles-Malagamba et al., 2020), Tenacibaculum (~6.35%) and Arcobacter (~4.35%) (Figure 1B) also dominated in the blow specimens in the present study. Thus, both genera may be commensal residents of the respiratory tract of dolphins. However, these genera also contained several pathogenic species that widely infect both fish and invertebrates (Habib et al., 2014; Ramees et al., 2017). The specific roles that these species play in the respiratory tract need further experimental validation.
The respiratory tract and gut represent two major protective barriers against the external environment in mammals and could get infected easily. Despite the speculated significance of dysbacteriosis in respiratory diseases, there are relatively few studies on the cetacean respiratory microbiome and it has been less explored than their gut microbiome. The respiratory tract, historically considered sterile in healthy humans, harbors diverse communities of microbes (Dickson et al., 2016). As cetaceans surface from the water through a blowhole, fresh DNA, lipids, proteins, bacteria, and viruses are forcefully exhaled out from their lungs with their secretions (Foot et al., 2006). Therefore, unlike gut microbes which are constantly receiving exogenous dietary ingredients, microbiota associated with the interior respiratory tract are prone to receive less disturbance from the external environment and may remain relatively unchanged once colonized. This may at least partly explain why the blow microbiotas of the three dolphins under human care showed relatively higher seasonal constancy, during the one year, and less convergence between dolphin species than the fecal microbiome in this study. However, when compared with their wild conspecifics, both the blow and fecal microbiome of the two dolphin species shared a more similar microbiome than members of the same dolphin species across different habitats (Figures 7 and S6), signifying that the human care environment may exert strong effects on structuring dolphin microbiotas distinct from the wild.
Shared bacterial communities between individuals often occur from vertical and/or horizontal transmission (Inoue and Ushida, 2003). In the present study, horizontal transmission resulting from shared environments and socialization between individuals may account for some of the shared bacteria observed between the two dolphin species under human care. To be specific, they were fed with the same thawed fish that had been washed with saline and they swam near one another. It’s likely that in the process they transferred microbes. The prevalent influence of the human care environment suggests that environmental microorganisms or potential pathogens might colonize a dolphin easily. In this study, a considerable number (4/7) of potentially pathogenic members detected in the pool water also were found in the feces (Figure 8). It’s likely that the potentially fecal pathogens derived from the adjacent pool water, or that the dolphins defecated potential pathogens into the pool water. Therefore, ensuring water quality and reducing or eliminating potential pathogens in the aquarium should aid in maintaining the gut health of dolphins in the human care environment. The dolphins in this study lived in a relatively constant indoor condition, where animals are not exposed to a diurnal cycle in water temperature, nor is there the same level of habitat complexity that is found in the natural environment. For example, the annual water temperature fluctuation amplitude in the aquarium was artificially regulated to approximately 6.50°C, with the minimum monthly mean water temperature at 21.79°C in February and the maximum at 28.29°C in July (Figure S1). The pool water was periodically disinfected with chlorine. Therefore, as revealed in our study, the seasonal dynamics of the fecal bacterial composition from dolphins under human care were likely driven by pool water temperature (Table S6). It is well known that temperature is a crucial abiotic factor structuring the composition and function of animal gut microbiomes (Sepulveda and Moeller, 2020). Our results are likely to confirm previous findings that gut microbiota may assist hosts in coping with seasonal thermoregulatory demands (Baniel et al., 2021). Since gut microbiota is critical in regulating many hosts’ physiological processes, such as metabolic and immune system functions (Kau et al., 2011; Frame et al., 2020), as well as their adaptive evolution (Moeller et al., 2019), gut microbial dysbiosis may provide a mechanism for temperature variation influences on animal fitness in endangered populations (Sepulveda and Moeller, 2020). Since fecal bacteria are sensitive to low-level water temperature changes, even in controlled indoor conditions, it is speculated that dolphin gut microbiome could be disrupted rapidly by acute temperature changes. On the other hand, hosts can reshape their gut microbial communities to compensate for the environmental effects (e.g., temperature), resulting in gut microbial resilience to environmental change (Kokou et al., 2018). For example, in COVID-19 patients, the gut microbiota restored more rapidly with increased bacterial diversity and enrichment of beneficial commensal than the respiratory microbiome, and the former then promoted the restoration of the latter via bacterial cross-talk and resulted in a synchronous restoration of both organs (Xu et al., 2021). Thus, maintaining intestinal flora homeostasis is highly significant for both intestinal and respiratory homeostatic balance and health.
Respiratory illnesses, especially those caused by bacteria, are considered a threat to cetaceans in both human care and wild populations (Acevedo-Whitehouse et al., 2010; Waltzek et al., 2012). If non-invasive blow sampling can aid in the respiratory health monitoring of cetaceans has been one of the major concerns for conservation biologists. In agreement with previous observations (Nelson et al., 2019; Vendl et al., 2021), our findings supported that blow sampling of captive dolphins can be used for their health monitoring in conservation practices. Firstly, the dolphin blow displayed more highly diverse bacterial communities compared to the feces and pool water samples, even if the potentially contaminated reads originating from the pool water were removed (Figure S1). Furthermore, the blow bacterial composition and functional potentials were distinct from the feces and pool water (Figure 1). Therefore, dolphin blow samples may mirror the pulmonary microbial communities. However, a handful of studies showed potential water-borne microbes contaminated in the blow (Nelson et al., 2019). This may be overcome by first wiping the blowhole, using alcohol pads, before sampling the dolphins living under human care or collecting biological replicates. Secondly, the blow bacterial community stayed relatively stable over the sampling period of one year. Species-specific bacteria were detected in each dolphin species even with an over decade-long cohabitation in which potential horizontal transmission could have occurred. In addition, the dolphin blows maintained highly abundant intra-cores (Table S7). This longitudinal observation provides additional evidence that the cetacean blow microbiota may be persistent and relatively independent of environmental influences (i.e., seasonal fluctuations and host phylogeny) in the human care environment. Lastly, we observed a small portion (0.94%) of potential bacterial pathogens in the blow, including members of the genera Prevotella, Roseburia, Roseomonas, Stenotrophomonas, Moraxella, Legionella, and Proteus (Figure 8). All of these are known potential pathogens of both animals and humans (Louca et al., 2016). Since we find no obvious signs of illness during the sampling, the presence of potential pathogens in the blow may be commensals to the dolphin lungs. However, this result showed that blow samples could be a method used to detect putative pathogenic bacteria in the respiratory tract of dolphins.
This study casts new light on explaining the differences between the blow and fecal bacterial communities by calculating their community assembly processes using the null model framework, which is widely acknowledged in microbial ecology (Stegen et al., 2013; Xie et al., 2022). The importance of homogeneous selection in structuring blow bacterial communities over one year is consistent with the strong relationships between bacterial communities and host species and individuality (e.g., host species-specificity and individual-specificity) (Lima et al., 2012; Vendl et al., 2021). In contrast, we observed that stochastic processes contributed more to the fecal community structure, primarily due to the drift and dispersal limitation processes (Figure 4), which may indicate that fecal bacteria exchanged between communities and fluctuated due to chance events in the dolphins under human care. These differences in community assembly processes likely reflect different transmission routes of the blow and fecal bacteria. For example, as observed in the present study, compared to the blow bacteria, the fecal bacteria tended to be more horizontally transferred between animals in the human care facility. It’s important to notice that the differences between reared dolphins and their wild counterparts from previous studies may be due to different experimental methods (including 16S amplicon region, PCR process, and sequencing platform), which may not be as accurate as sampling and sequencing both reared and wild dolphins under the same experiment condition.
In conclusion, this study demonstrated that human care exerted different impacts on the blow and gut bacterial communities of two dolphin species over a decade-long cohabitation. Typically, compared to the blow bacteria, the fecal bacteria between the two-dolphin species were more variable over one year, more convergent between dolphin species, and more sensitive to the pool water temperature. These results improve our understanding of how the human care environment alters the dolphin microbiome from different body sites, which provides an empirical baseline for diagnosing abnormal shifts in symbiont communities of dolphins under human care. In the future blow and fecal monitoring should be considered as an important part of cetacean health management.
The data presented in the study are deposited in the NCBI repository, accession number PRJNA865874.
The animal study was reviewed and approved by People’s Republic of China for the Implementation of Wild Aquatic Animal Protection.
XW and JZ designed this research. XW and JL collected the samples and performed the lab experiments. XW and RT completed the data analysis. XW and JZ wrote the manuscript. RM, YH, JW, ZW, FF, and DW assisted with experiments and advice on the manuscript. All authors contributed to discussions, and revisions, and approved the final manuscript.
This study was partly supported by grants from the National Natural Science Foundation of China (No. 31870372), China Postdoctoral Science Foundation (No. 2020M682530), and the Bureau of Science & Technology for Development, Chinese Academy of Sciences (No. ZSSD-004).
We thank Dr. Xianyuan Zeng, Junhua Ji, and the dolphin trainers in Nanning Zoo for their assistance in collecting specimens. We also appreciate Dr. Youchuan Wei in Guangxi University for helping with sample preprocessing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1024117/full#supplementary-material
Acevedo-Whitehouse K., Rocha-Gosselin A., Gendron D. (2010). A novel non-invasive tool for disease surveillance of free-ranging whales and its relevance to conservation programs. Anim. Conserv. 13 (2), 217–225. doi: 10.1111/j.1469-1795.2009.00326.x
Bai S., Zhang P., Zhang C., Du J., Du X., Zhu C., et al. (2021). Comparative study of the gut microbiota among four different marine mammals in an aquarium. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.769012
Baniel A., Amato K. R., Beehner J. C., Bergman T. J., Mercer A., Perlman R. F., et al. (2021). Seasonal shifts in the gut microbiome indicate plastic responses to diet in wild geladas. Microbiome 9 (1), 1–20. doi: 10.1186/s40168-020-00977-9
Bik E. M., Costello E. K., Switzer A. D., Callahan B. J., Holmes S. P., Wells R. S., et al. (2016). Marine mammals harbor unique microbiotas shaped by and yet distinct from the sea. Nat. Commun. 7 (1), 1–13. doi: 10.1038/ncomms10516
Bosco N., Noti M. (2021). The aging gut microbiome and its impact on host immunity. Genes Immun. 22 (5), 289–303. doi: 10.1038/s41435-021-00126-8
Brown J., Pirrung M., McCue L. A. (2017). FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33 (19), 3137–3139. doi: 10.1093/bioinformatics/btx373
Cole J. R., Wang Q., Fish J. A., Chai B., McGarrell D. M., Sun Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42 (D1), D633–D642. doi: 10.1093/nar/gkt1244
Curry B. E., Ralls K., Brownell R. L. Jr. (2013). Prospects for captive breeding of poorly known small cetacean species. Endanger. Species Res. 19 (3), 223–243. doi: 10.3354/esr00461
Dallas J. W., Warne R. W. (2022). Captivity and animal microbiomes: potential roles of microbiota for influencing animal conservation. Microb. Ecol., 1–19. doi: 10.1007/s00248-022-01991-0
Dickson R. P., Erb-Downward J. R., Martinez F. J., Huffnagle G. B. (2016). The microbiome and the respiratory tract. Annu. Rev. Physiol. 78, 481. doi: 10.1146/annurev-physiol-021115-105238
Douglas G. M., Maffei V. J., Zaneveld J. R., Yurgel S. N., Brown J. R., Taylor C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38 (6), 685–688. doi: 10.1038/s41587-020-0548-6
Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 (10), 996–998. doi: 10.1038/nmeth.2604
Enaud R., Prevel R., Ciarlo E., Beaufils F., Wieërs G., Guery B., et al. (2020). The gut-lung axis in health and respiratory diseases: A place for inter-organ and inter-kingdom crosstalks. Front. Cell. Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.00009
Foot N. J., Orgeig S., Daniels C. B. (2006). The evolution of a physiological system: the pulmonary surfactant system in diving mammals. Resp. Physiol. Neurobi. 154 (1-2), 118–138. doi: 10.1016/j.resp.2006.04.012
Frame L. A., Costa E., Jackson S. A. (2020). Current explorations of nutrition and the gut microbiome: a comprehensive evaluation of the review literature. Nutr. Rev. 78 (10), 798–812. doi: 10.1093/nutrit/nuz106
Frankel J. S., Mallott E. K., Hopper L. M., Ross S. R., Amato K. R. (2019). The effect of captivity on the primate gut microbiome varies with host dietary niche. Am. J. Primatol. 81 (12), e23061. doi: 10.1002/ajp.23061
Gerritsen J., Hornung B., Ritari J., Paulin L., Rijkers G. T., Schaap P. J., et al. (2019). A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. BioRxiv 845511. doi: 10.1101/845511
Habib C., Houel A., Lunazzi A., Bernardet J.-F., Olsen A. B., Nilsen H., et al. (2014). Multilocus sequence analysis of the marine bacterial genus Tenacibaculum suggests parallel evolution of fish pathogenicity and endemic colonization of aquaculture systems. Appl. Environ. Microbiol. 80, 5503–5514. doi: 10.1128/AEM.01177-14
Hyde E. R., Navas-Molina J. A., Song S. J., Kueneman J. G., Ackermann G., Cardona C., et al. (2016). The oral and skin microbiomes of captive komodo dragons are significantly shared with their habitat. mSystems 1 (4), e00046–e00016. doi: 10.1128/mSystems.00046-16
Inoue R., Ushida K. (2003). Vertical and horizontal transmission of intestinal commensal bacteria in the rat model. FEMS Microbiol. Ecol. 46 (2), 213–219. doi: 10.1016/S0168-6496(03)00215-0
Invernizzi R., Lloyd C. M., Molyneaux P. L. (2020). Respiratory microbiome and epithelial interactions shape immunity in the lungs. Immunology 160 (2), 171–182. doi: 10.1111/imm.13195
Jin L., Huang Y., Yang S., Wu D., Li C., Deng W., et al. (2021). Diet, habitat environment and lifestyle conversion affect the gut microbiomes of giant pandas. Sci. Total Environ. 770, 145316. doi: 10.1016/j.scitotenv.2021.145316
Jin Song S., Woodhams D. C., Martino C., Allaband C., Mu A., Javorschi-Miller-Montgomery S., et al. (2019). Engineering the microbiome for animal health and conservation. Exp. Biol. Med. 244 (6), 494–504. doi: 10.1177/1535370219830075
Kau A. L., Ahern P. P., Griffin N. W., Goodman A. L., Gordon J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature 474 (7351), 327–336. doi: 10.1038/nature10213
Kaul D., Rathnasinghe R., Ferres M., Tan G. S., Barrera A., Pickett B. E., et al. (2020). Microbiome disturbance and resilience dynamics of the upper respiratory tract during influenza a virus infection. Nat. Commun. 11 (1), 1–12. doi: 10.1038/s41467-020-16429-9
Kembel S. W., Cowan P. D., Helmus M. R., Cornwell W. K., Morlon H., Ackerly D. D., et al. (2010). Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26 (11), 1463–1464. doi: 10.1093/bioinformatics/btq166
Kokou F., Sasson G., Nitzan T., Doron-Faigenboim A., Harpaz S., Cnaani A., et al. (2018). Host genetic selection for cold tolerance shapes microbiome composition and modulates its response to temperature. eLlife 7, e36398. doi: 10.7554/eLife.36398
Lima N., Rogers T., Acevedo-Whitehouse K., Brown M. V. (2012). Temporal stability and species specificity in bacteria associated with the bottlenose dolphins respiratory system. Env. Microbiol. Rep. 4 (1), 89–96. doi: 10.1111/j.1758-2229.2011.00306.x
Lindstrom M. J., Bates D. M. (1990). Nonlinear mixed effects models for repeated measures data. Biometrics 46, 673–687. doi: 10.2307/2532087
Lott R., Williamson C. (2017). “Cetaceans in captivity,” in Marine mammal welfare. animal welfare, vol. 17 . Ed. Butterworth A. (Cham: Springer). doi: 10.1007/978-3-319-46994-2_11
Louca S., Parfrey L. W., Doebeli M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353 (6305), 1272–1277. doi: 10.1126/science.aaf4507
Mallott E. K., Amato K. R. (2021). Host specificity of the gut microbiome. Nat. Rev. Microbiol. 19 (10), 639–653. doi: 10.1038/s41579-021-00562-3
Mallott E. K., Borries C., Koenig A., Amato K. R., Lu A. (2020). Reproductive hormones mediate changes in the gut microbiome during pregnancy and lactation in phayre’s leaf monkeys. Sci. Rep. 10 (1), 1–9. doi: 10.1038/s41598-020-66865-2
McKenzie V. J., Song S. J., Delsuc F., Prest T. L., Oliverio A. M., Korpita T. M., et al. (2017). The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 57 (4), 690–704. doi: 10.1093/icb/icx090
Moeller A. H., Gomes-Neto J. C., Mantz S., Kittana H., Segura Munoz R. R., Schmaltz R. J., et al. (2019). Experimental evidence for adaptation to species-specific gut microbiota in house mice. mSphere 4 (4), e00387–e00319. doi: 10.1128/mSphere.00387-19
Muka S., Zarpentine C. (2021). Cetacean conservation and the ethics of captivity. Biol. Conserv. 262, 109303. doi: 10.1016/j.biocon.2021.109303
Nabi G., McLaughlin R. W., Khan S., Hao Y., Chang M. (2020). Pneumonia in endangered aquatic mammals and the need for developing low-coverage vaccination for their management and conservation. Anim. Health Res. Rev. 21 (2), 122–130. doi: 10.1017/S1466252320000158
Nelson T. M., Wallen M. M., Bunce M., Oskam C. L., Lima N., Clayton L., et al. (2019). Detecting respiratory bacterial communities of wild dolphins: implications for animal health. Mar. Ecol. Prog. Ser. 622, 203–217. doi: 10.3354/meps13055
Ning D., Yuan M., Wu L., Zhang Y., Guo X., Zhou X., et al. (2020). A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 11 (1), 1–12. doi: 10.1038/s41467-020-18560-z
Parsons E. C. M., Baulch S., Bechshoft T., Bellazzi G., Bouchet P., Cosentino A. M., et al. (2015). Key research questions of global importance for cetacean conservation. Endanger. Species Res. 27 (2), 113–118. doi: 10.3354/esr00655
Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26 (7), 1641–1650. doi: 10.1093/molbev/msp077
Ramees T. P., Dhama K., Karthik K., Rathore R. S., Kumar A., Saminathan M., et al. (2017). Arcobacter: an emerging food-borne zoonotic pathogen, its public health concerns and advances in diagnosis and control-a comprehensive review. Vet. Quart. 37 (1), 136–161. doi: 10.1080/01652176.2017.1323355
Robles-Malagamba M. J., Walsh M. T., Ahasan M. S., Thompson P., Wells R. S., Jobin C., et al. (2020). Characterization of the bacterial microbiome among free-ranging bottlenose dolphins (Tursiops truncatus). Heliyon 6 (6), e03944. doi: 10.1016/j.heliyon.2020.e03944
Roggenbuck M., Bærholm Schnell I., Blom N., Bælum J., Bertelsen M. F., Sicheritz-Pontén T., et al. (2014). The microbiome of new world vultures. Nat. Commun. 5, 5498. doi: 10.1038/ncomms6498
Segawa T., Ohno Y., Tsuchida S., Ushida K., Yoshioka M. (2020). Helicobacter delphinicola sp. nov., isolated from common bottlenose dolphins Tursiops truncatus with gastric diseases. Dis. Aquat. Organ. 141, 157–169. doi: 10.3354/dao03511
Sepulveda J., Moeller A. H. (2020). The effects of temperature on animal gut microbiomes. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00384
Simmonds M. P., Eliott W. J. (2009). Climate change and cetaceans: concerns and recent developments. J. Mar. Biol. Assoc. 89 (1), 203–210. doi: 10.1017/S0025315408003196
Stegen J. C., Lin X., Fredrickson J. K., Chen X., Kennedy D. W., Murray C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7 (11), 2069–2079. doi: 10.1038/ismej.2013.93
Suzuki A., Akuzawa K., Kogi K., Ueda K., Suzuki M. (2021). Captive environment influences the composition and diversity of fecal microbiota in indo-pacific bottlenose dolphins, Tursiops aduncus. Mar. Mammal Sci. 37 (1), 207–219. doi: 10.1111/mms.12736
Suzuki A., Segawa T., Sawa S., Nishitani C., Ueda K., Itou T., et al. (2019). Comparison of the gut microbiota of captive common bottlenose dolphins Tursiops truncatus in three aquaria. J. Appl. Microbiol. 126 (1), 31–39. doi: 10.1111/jam.14109
Toor D., Wasson M. K., Kumar P., Karthikeyan G., Kaushik N. K., Goel C., et al. (2019). Dysbiosis disrupts gut immune homeostasis and promotes gastric diseases. Int. J. Mol. Sci. 20 (10), 2432. doi: 10.3390/ijms20102432
Turvey S. T., Pitman R. L., Taylor B. L., Barlow J., Akamatsu T., Barrett L. A., et al. (2007). First human-caused extinction of a cetacean species? Biol. Lett. 3 (5), 537–540. doi: 10.1098/rsbl.2007.0292
Vendl C., Nelson T., Ferrari B., Thomas T., Rogers T. (2021). Highly abundant core taxa in the blow within and across captive bottlenose dolphins provide evidence for a temporally stable airway microbiota. BMC Microbiol. 21 (1), 20. doi: 10.1186/s12866-020-02076-z
Vuong H. E., Yano J. M., Fung T. C., Hsiao E. Y. (2017). The microbiome and host behavior. Annu. Rev. Neurosci. 40 (1), 21–49. doi: 10.1146/annurev-neuro-072116-031347
Waltzek T. B., Cortés-Hinojosa G., Wellehan J. F. X. Jr., Gray G. C. (2012). Marine mammal zoonoses: a review of disease manifestations. Zoonoses Public Helth. 59 (8), 521–535. doi: 10.1111/j.1863-2378.2012.01492.x
Wang D., Hao Y., Wang K., Zhao Q., Chen D., Wei Z., et al. (2005). Aquatic resource conservation. the first Yangtze finless porpoise successfully born in captivity. Environ. Sci. Pollut. R. 12 (5), 247–250. doi: 10.1065/espr2005.08.284
Wan X., Li J., Cheng Z., Ao M., Tian R., Mclaughlin R. W., et al. (2021). The intestinal microbiome of an indo-pacific humpback dolphin (Sousa chinensis) stranded near the pearl river estuary, China. Integr. Zool. 16 (3), 287–299. doi: 10.1111/1749-4877.12477
Wan X., McLaughlin R. W., Zheng J., Hao Y., Fan F., Tian R., et al. (2018). Microbial communities in different regions of the gastrointestinal tract in East Asian finless porpoises (Neophocaena asiaeorientalis sunameri). Sci. Rep. 8 (1), 14142. doi: 10.1038/s41598-018-32512-0
Weiss S., Xu Z., Peddada S., Amir A., Bittinger K., Gonzalez A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5 (1), 1–18. doi: 10.1186/s40168-017-0237-y
West A. G., Waite D. W., Deines P., Bourne D. G., Digby A., McKenzie V. J., et al. (2019). The microbiome in threatened species conservation. Biol. Conserv. 229, 85–98. doi: 10.1016/j.biocon.2018.11.016
Xie Y., Ouyang Y., Han S., Se J., Tang S., Yang Y., et al. (2022). Crop rotation stage has a greater effect than fertilisation on soil microbiome assembly and enzymatic stoichiometry. Sci. Total Environ. 815, 152956. doi: 10.1016/j.scitotenv.2022.152956
Xu R., Lu R., Zhang T., Wu Q., Cai W., Han X., et al. (2021). Temporal dynamics of human respiratory and gut microbiomes during the course of COVID-19 in adults. Commun. Biol. 4 (1), 1–11. doi: 10.1101/2020.07.21.20158758
Yadav M., Verma M. K., Chauhan N. S. (2018). A review of metabolic potential of human gut microbiome in human nutrition. Arch. Microbiol. 200 (2), 203–217. doi: 10.1007/s00203-017-1459-x
Keywords: dolphin, gut microbiota, blowhole microbiota, convergence, human care
Citation: Wan X, Li J, Tian R, McLaughlin RW, Hao Y, Wu J, Wang Z, Fan F, Wang D and Zheng J (2022) The effects of human care on the blowhole and gut microbiotas of two cohabiting dolphin species based on a year-round surveillance. Front. Mar. Sci. 9:1024117. doi: 10.3389/fmars.2022.1024117
Received: 21 August 2022; Accepted: 17 October 2022;
Published: 01 November 2022.
Edited by:
Yunyun Zhuang, Ocean University of China, ChinaReviewed by:
Susan Smith, Mystic Aquarium, United StatesCopyright © 2022 Wan, Li, Tian, McLaughlin, Hao, Wu, Wang, Fan, Wang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinsong Zheng, emhlbmdqaW5zb25nQGloYi5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.