 Na Zhao
Na Zhao Lei Jia3
Lei Jia3 Zhongdian Dong
Zhongdian Dong Bo Zhang
Bo Zhang- 1College of Fisheries, Guangdong Ocean University, Zhanjiang, China
- 2Southern Marine Science and Engineering Guangdong Laboratory-Zhanjiang, Zhanjiang, China
- 3Tianjin Fisheries Research Institute, Tianjin, China
- 4College of Life Science and Technology, Lingnan Normal University, Zhanjiang, China
- 5Guangdong Provincial Key Laboratory of Aquatic Animal Disease Control and Healthy culture, College of Fishery, Guangdong Ocean University, Zhanjiang, China
Proper DNA methylation in spermatozoa is essential for the normal development of fertilized embryos through gene expression regulation. Abnormal sperm DNA methylation is associated with male fertility impairment, offspring quality decline, and disease susceptibility. Compared with other epigenetic regulatory mechanisms (e.g., histone modification), DNA methylation is a stable regulator for the long-term transcriptional activity of genes. Sperm DNA methylation is crucial to offspring’s survival, development, and reproduction. However, it has not been well studied in teleost to a large extent, especially in some species with sex deviation or congenital sex abnormality in offspring. In the present study, DNA methylation profiles of pseudomale (ZW) and male (ZZ) tongue sole (Cynoglossus semilaevis) spermatozoa were characterized for differential methylation regions (DMRs) screening. The global methylation levels of the two sperm groups were highly methylated with no significant differences. For all kinds of genomic elements, the mean methylation level of the ZW group was higher than that of the ZZ group. The total numbers of Covered C annotated on the W chromosome of both groups were extremely small, suggesting that W-type sperm did not exist in pseudomales. A comparison of methylation levels on 20 sex-related genes between sperm and gonad showed that the heterogeneity between tissue resources was greater than that between sexes, and the methylation level of most genes in gonads was lower than that in sperm. For integrative analysis of DNA methylation and miRomic profiles, 11 sex-related DMRs associated with 15 differentials micro RNAs (miRNAs) in spermatozoa were identified to present targeting relationships and regulatory trends of the two distinct epigenetic patterns. This study provides valuable and potential targets of coordination between two epigenetic mechanisms in the process of C. semilaevis sex congenital bias.
Introduction
Gametes, as an important vector for the transmission of genetic materials, have a high level of methylation (Murphy et al., 2018). The methylation pattern of the embryo is reshaped after fertilization. The basic mode of intergenerational inheritance of epigenetic information is to transfer DNA methylation profiling in gametes to offspring in vertebrates (Woods et al., 2018; Iwanami et al., 2020). DNA methylation patterns in gametes of various species have different imprinting characteristics, especially in the process of zygotic genome activation. Within the same species, there are significant differences in DNA methylation patterns between male and female gametes (Potok et al., 2013). It was found in zebrafish (Danio rerio) that the DNA methylation pattern of sperm was more conservative than that of the egg. The paternal DNA methylation pattern was maintained throughout the early embryonic development, while the maternal methylation pattern was reprogrammed to match the paternal pattern (Jiang et al., 2013; Murphy et al., 2018), which was not applicable to medaka (Oryzias latipes). Current studies have shown that medaka embryos reset their DNA methylation patterns through active demethylation and gradual re-methylation, similar to mammals (Wang and Bhandari, 2020). The sperm genome of medaka was highly methylated, and the oocyte genome was hypo-methylated before fertilization.
As an important pattern of epigenetic regulation in gamete inheritance, DNA methylation is influenced by environmental factors (Yuan et al., 2019; Depince et al., 2020). Sperm DNA methylation profile altered when striped bass (Morone saxatilis) was reared for aquaculture rather than in wild since differential methylated related genes of wdr3/utp12 and GPCR families might be involved (Shao et al., 2018; Woods et al., 2018). Similar studies on Atlantic salmon (Salmo salar) and steelhead (Oncorhynchus mykiss) have revealed that one generation of captive breeding is enough to produce transgenerational epigenetic modification. Sperm methylation regions of wild and captive males are different, which is consistent with the epigenetic marks of domestication, phenotypic, and gene expression changes among fish offspring (Gavery et al., 2018; Rodriguez Barreto et al., 2019). Under different conditions of sperm cryopreservation, the changes in sperm DNA methylation profiling have been confirmed in a variety of fish, such as goldfish (Carassius auratus), zebrafish (Depincé et al., 2020) and tambaqui (Colossoma macropomum) (de Mello et al., 2017; Wang and Bhandari, 2020).
Half smooth tongue sole (Cynoglossus semilaevis), an economic mariculture species with sex size dimorphism, is popular in female-biased breeding in coastal areas of China (Zhang et al., 2020a; Zhang et al., 2020b; Zhao et al. 2021a). A high proportion of pseudomales without growth advantage occupy resources with no considerable return, which makes it difficult to break the circle of production capacity of C. semilaevis (Zhang et al., 2011; Zhang et al., 2019; Zhao et al., 2021b). C. semilaevis holds ZZ/ZW sex chromosome type, and the heredity of pseudomale is highly conservative (Chen et al., 2007; Chen et al., 2009; Zhang et al., 2019). Besides, the natural predisposition of pseudomales prompts epigenetics, an important breakthrough point to reveal the occurrence of pseudomales in C. semilaevis. DNA methylomes of gonads from different sexes’ C. semilaevis in a recent study have revealed that the methylation modification of pseudomales is inherited in pseudomale offspring, and sex-related genes have possessed lots of methylation inherited in the process of sex reversal of C. semilaevis (Shao et al., 2014). Furthermore, micro RNAs (miRNAs) in sperm of C. semilaevis showed obvious sex bias between male and pseudomale, and some of them were inherited in pseudomale offspring (Zhang et al., 2019; Zhao et al., 2022). However, our knowledge of the DNA methylome of gametes is limited in C. semilaevis, which is an important link between the gonad methylomic map and the sperm miRomic map.
In the present study, we characterized the global DNA methylation profiles in sperm of the male and pseudomale C. semilaevis. The DMRs were correlated with the differential miRNAs of sperm to reveal the potential interaction between two epigenetic regulation modes.
Materials and methods
Sampling
Tangshan Weizhuo aquaculture Ltd. (Hebei province, China) provided male and pseudomale C. semilaevis individuals as semen donors. A total of 30 living male fish obtained for semen production were artificially induced to sexual maturation. Semen (0.5 mL) was collected from each fish, and 15 mL (in total) of semen was mixed in ZZ males. The same procedure was applied to pseudomale fish (ZW group). Both males and pseudomales were kept in the same situation. Semen separated from both male and pseudomale sperm mixtures went through centrifugation at 5000 rpm for 10 min, and then the supernatant was removed. The sperm pellet was cryopreserved for whole genome bisulfite sequencing (WGBS). The mixed male sperm was labeled as the ZZ group, while the pseudomale sperm was labeled as the ZW group (Figure 1).
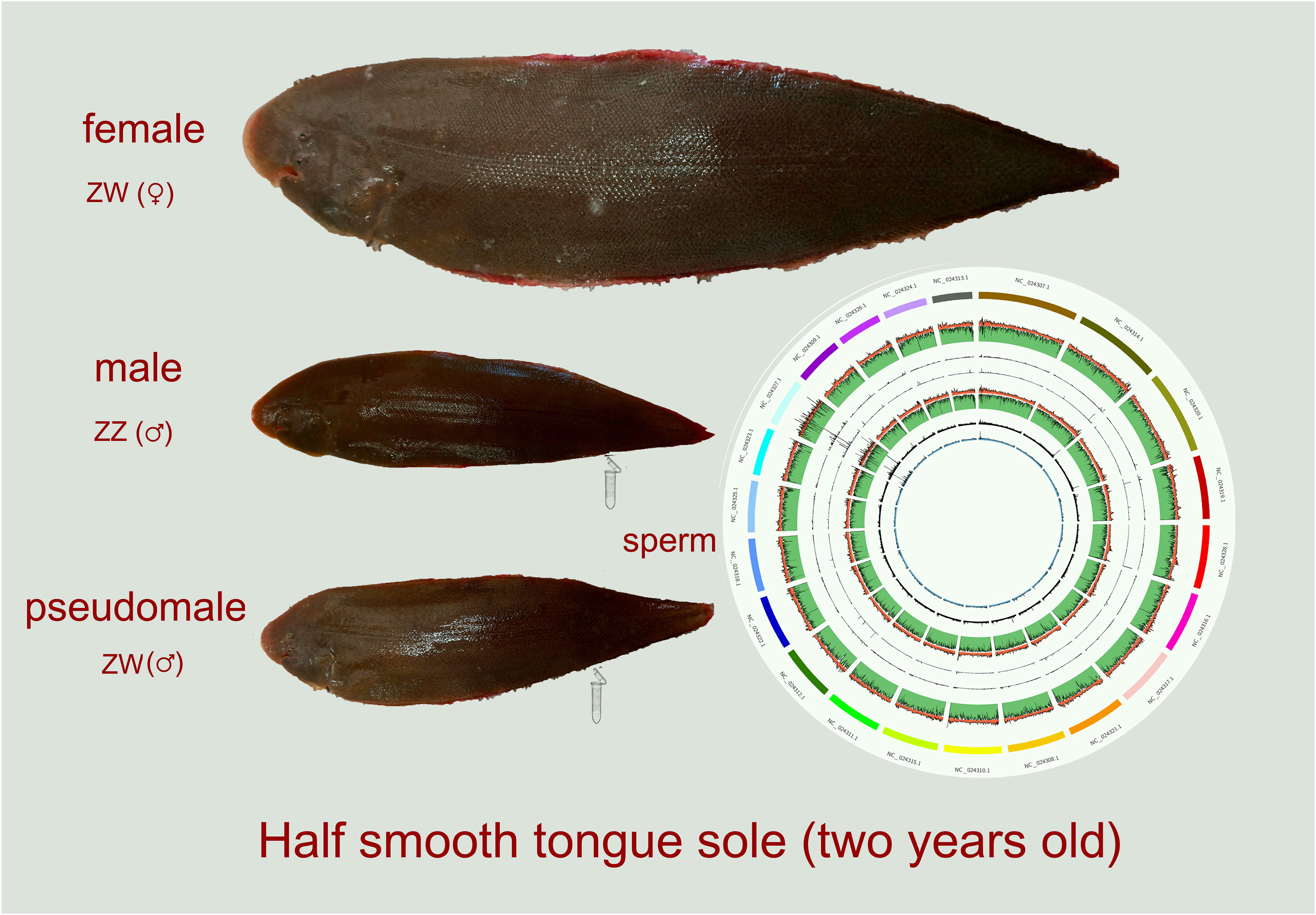
Figure 1 Schematic diagram of sex type grouping and sampling of Cynoglossus semilaevis. ZW (♀) represented females, ZZ (♂) represented normal males, and ZW (♂) represented pseudomales, all donors were about 2- year-old.
Construction of DNA library and Whole Genome Bisulfite Sequencing (WGBS) of sperm from ZW and ZZ C. semilaevi groups
Genomic DNA (gDNA) was extracted from the sperm samples, and the DNA libraries were established after electrophoresis. The gDNA was randomly interrupted to 200-300 bp, and DNA fragments were repaired with a poly-A tail and connected with sequencing splices in which all cytosines were methylated. Then, bisulfite treatment (EZ DNA methylation gold kit, Zymo Research) was performed. The unmethylated C became U (T after PCR amplification), while the methylated C (mC) remained unchanged. PCR amplification was performed to obtain the final DNA library. The raw sequencing data were filtered to remove the low-quality data, and clean reads were obtained. Then, clean reads were compared with the reference genome, and the methylation sites were detected and annotated. Bismark software (Krueger and Andrews, 2011) was used for comparative analysis of reference genomes of methylation data, which adopts in silico bisulfite conversion algorithm to solve the bisulfite transformation difficulties. Before comparing the reads with a reference genome, all C bases in the reads were converted into T bases. After converting all T bases, the sequence correspondence between the reads and the reference genome was equivalent to that before the bisulfite conversion so that the correct positioning of reads could be realized. The reference genome download link is as follows: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/523/025/GCF_000523025.1_Cse_v1.0.
Methylation site detection and DNA methylation profiling of sperm
To ensure the accuracy of the methylation site, the sites covering the number of reads > =5 were taken as the detected C sites (Covered C). In addition, methylated and unmethylated reads were detected at each site, and the probability of methylated reads obeyed binomial distribution. A binomial distribution test was conducted for the number of methylated and unmethylated C reads at each site. Assuming that the number of methylated C at a site is x, the coverage of the site is n, and the BS conversion rate is p, it is necessary to verify whether the occurrence of X mC is reliable. After False Discovery Rate (FDR) correction, the sites with a Q-value less than 0.05 were regarded as the methylation sites. Autosomes were windowed at 50 kb, and the number and methylation level of mC, mCG, mCHG, and mCHH in each window were counted. To explore the distribution of methylation levels in the gene region (genebody) and its upstream (2KB, promoter) and downstream (2KB, downstream) segments, a profiling analysis of methylation levels was conducted. Each segment was divided into 20 bins on average, and the methylation level in each bin reflected the trend of methylation level changes.
DMR and DMP screening
DMR is a specific region in the genome and can reflect the methylated regulation of transcription. DMP is a specific DMR located in the promoter region and may change the level of gene transcription expression, which is closely related to transcription regulation. DMR detection was analyzed using methylkit software. The Fisher’s exact test was done to analyze the DMR between groups. DMR was screened out according to the difference in mean methylation level in each window; methyl differs. =methyl case- methyl control (|methyl differs |. > 0.8 & p-value < 0.05) DMP, following similar rules: 2KB upstream of transcription start site (TSS) of the gene was used as promoter region, and the mean methylation level in this region was counted and compared between ZZ and ZW groups. DMP with a significant difference was screened out on | methyl diff | >0.6 and p-value<0.05 level. We drew volcano and heat maps for DMR and DMP, respectively. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted for DMR and DMP-related genes to determine biological functions and pathways, which were significantly associated with DMR/DMP-related genes. Top 30 GO terms and KEGG pathways were presented in DMR and DMP on sex chromosomes
DMP and DMR located on W and Z chromosomes of C. semilaevis were extracted and counted, respectively (methyl diff ± 0.1, P<0.05). Among the four groups, the top 8 DMR/DMP-related genes with the most significant methylation differences for each group were selected to draw a chord diagram (ranging from large to small according to the absolute value of methyl diff). The interval distribution of DMP/DMR on Z and W chromosomes was statistically analyzed, and a line diagram of DMPs/DMRs on sex chromosomes was drawn for counting the proportion of DMPs/DMRs in each interval among the total number of DMPs/DMRs.
Comparison of sex related DMR and DMP between sperm and gonads
The genome-wide methylation profiles of the gonads of C. semilaevis were well characterized in a previous study (Shao et al., 2014). We selected 20 sex-related genes with methylation data identified in that study and compared the mean methylation levels of these genes with our results. Datasets of gonads were obtained from the ovary and testis of female and male C. semilaevis, respectively. The degree of methylation was shown by a heatmap.
Integrative analysis of DNA methylation and miRomics of sperm
We obtained the miRomics profile datasets from sperm of the same samples in a recent study (Zhao et al., 2021b) for integrative analysis with global gDNA methylation profiling. Differential expressed miRNAs (DEM) and DMR/DMP were extracted and analyzed to reveal the interaction of these two epigenetic patterns as the following criteria: 1) Construction of the relationship between miRNAs and methylation-related target genes, such as DNA methyltransferases (Dnmts): maintenance methyltransferase (Dnmt1) and de novo methyltransferases (Dnmt3a, Dnmt3b, and Dnmt3L) as well as dnmt2 with weak DNA methyltransferase catalytic activity and demethylase related genes. 2) According to the distribution of miRNA in the genome, miRNA can be divided into two types: intergenic and intragenic miRNAs. For intergenic miRNA, the 2KB sequence upstream of the miRNA precursor was considered the promoter sequence; for intronic miRNA, the promoter sequence of the gene containing miRNA was considered the promoter of miRNA. The methylation level of the promoter region of both types of DEMs was analyzed. For the two types of DEMs, the DEMs with expression differences and the corresponding DEMs’ promotor methylation regions were identified and presented with heatmaps, respectively. Also, the linear graph of the methylation level of DEMs’ promoter and the expression level of DEMs were drawn to show the trend of coordination.
Results
Characteristics of genome-wide DNA methylation and mC site detection
Raw bases (16.61G and 21.79G) were obtained from original data files of sperm of ZW and ZZ groups, respectively, by Illumina Hiseq Xten. After quality control, we captured 15.80G and 21.01G clean bases, which occupied about 95.11% and 96.43% of the raw bases, respectively. The GC contents of the ZW and ZZ groups were 22.36% and 22.0%, respectively. The bisulfite conversion rates of ZW and ZZ samples were 99.39% and 99.60%, respectively. The proportion of mC in all detected C sites (Covered C) reached 87.78% and 84.73% in ZW and ZZ groups, respectively. In addition, there were three contexts (CG, CHH, and CHG) of the mC site. The three contexts together constituted mC. Of these, the CG context contributed the most of the mC methylation, occupying more than 98% of mC methylation in both ZW and ZZ groups (Figures 2A and 2a). The global methylation levels of ZW and ZZ were 92.92% and 92.95%, respectively, indicating no significant difference between ZW and ZZ groups on the genome-wide methylation level (Table 1). The total numbers of detected C sites (Covered C) counted on each chromosome of the ZW and ZZ groups showed that the numbers of Covered C in the ZZ group were higher than those in the ZW group. The numbers of Covered C annotated on the W chromosome of both groups were extremely small compared to other chromosomes, suggesting that the W chromosome did not exist (Figure 2B). For the mC site on chromosomes, the CG context in the mC site on each chromosome distributed relatively even, and there was no special enrichment or sparse distribution of CG context on any chromosome (Figures 2C and 2c).
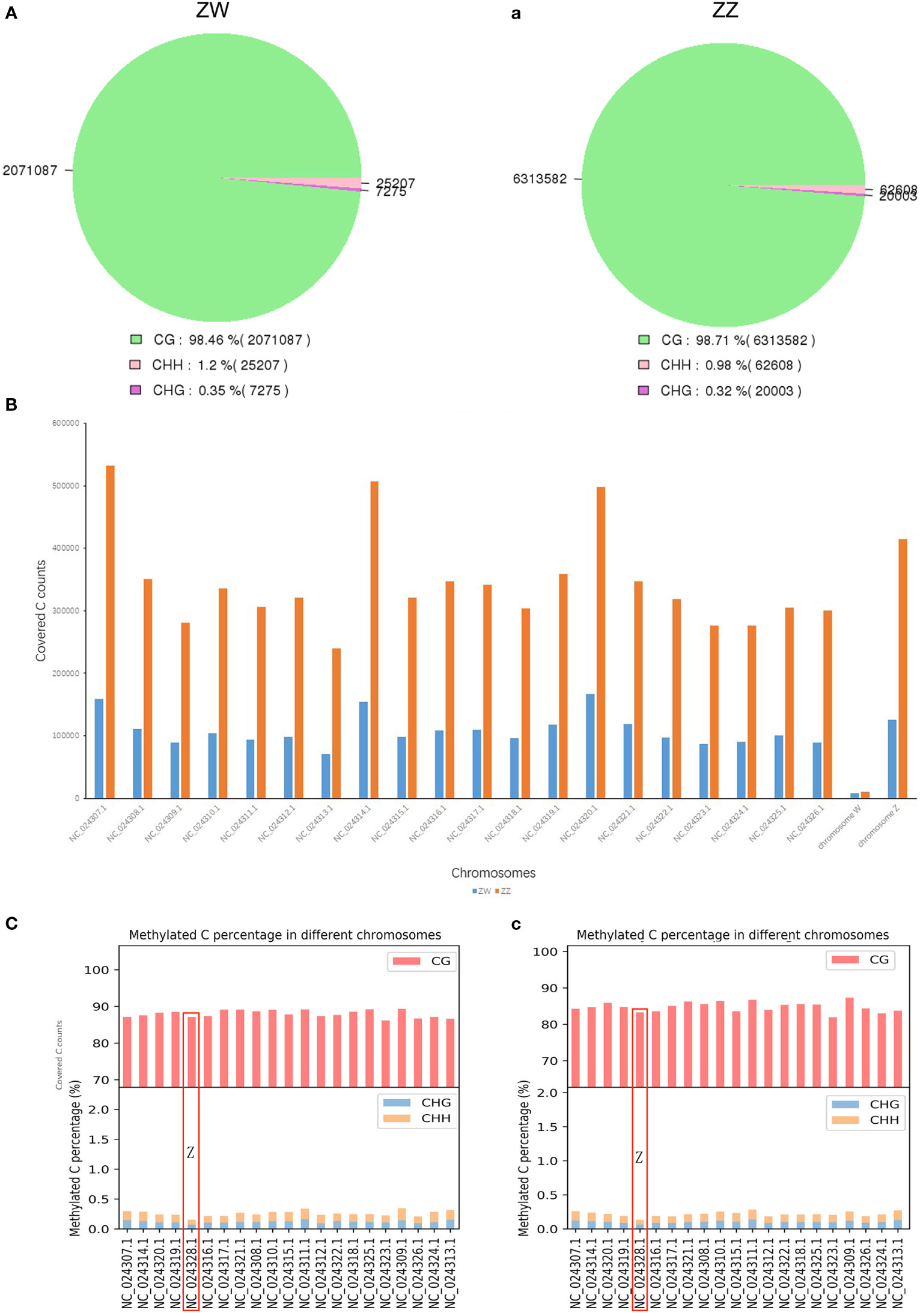
Figure 2 Genome-wide DNA methylation and methylated C site detection on chromosomes. (A) Contexts of methylation C site (CG, CHH, CHG) of DNA methylome in ZW group. (a) Contexts of methylation C site (CG, CHH, CHG) of DNA methylome in ZZ group. (B) Number of covered C annotated on W chromosome of both ZW and ZZ groups. (C) Percentage of Methylated C site distributed on each chromosome in ZW group. (c) Percentage of Methylated C site distributed on each chromosome in ZZ group.
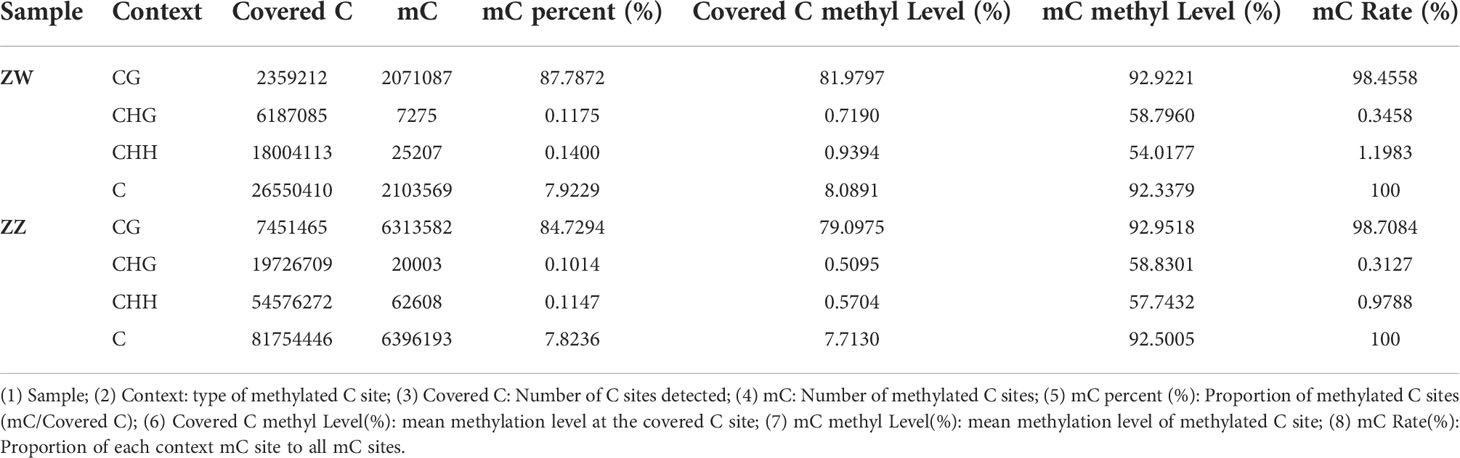
Table 1 Statistical results of methylation site test..
DNA methylation profiling of different genomic structural elements in ZW and ZZ sperm
Three contexts of mC sites (CG, CHG, and CHH) in the genomic elements, including gene, repeats, and TE, were presented as methylation profiling between the ZW and ZZ groups. No matter what type of mC site, or no matter where the elements were located in the genome, the mean methylated level in the ZW group was higher than that in the ZZ group (Figure 3). CG-type methylation sites accounted for the vast majority of C-site methylation types in each kind of genomic element. The mean methylation levels both exceeded 50% (Figure 3B), and the methylation level of TE exceeded 80% (Figure 3C). However, the methylation level of CG sites in the gene element changed greatly, especially in the promoter and downstream regions outside the genebody region. Promoter and downstream regions in both ZW and ZZ groups had sharp undulations. The promoter region showed a sharp downward trend, while the downstream region showed a sharp upward trend. Besides, the methylation level of ZW was always greater than that of ZZ for both DMRs and DMPs. In the promoter area, this difference decreased with the proximity to genebody, while this difference increased with the distance away from genebody in the downstream area (Figure 3A). For the 3 different genomic elements, the mean methylation levels of CHG and CHH sites were very low in both ZW and ZZ groups (less than 0.2%) (Figures 3D–F), but the difference of mean methylation levels between ZW and ZZ groups was highly significant (Figures 3G–I).
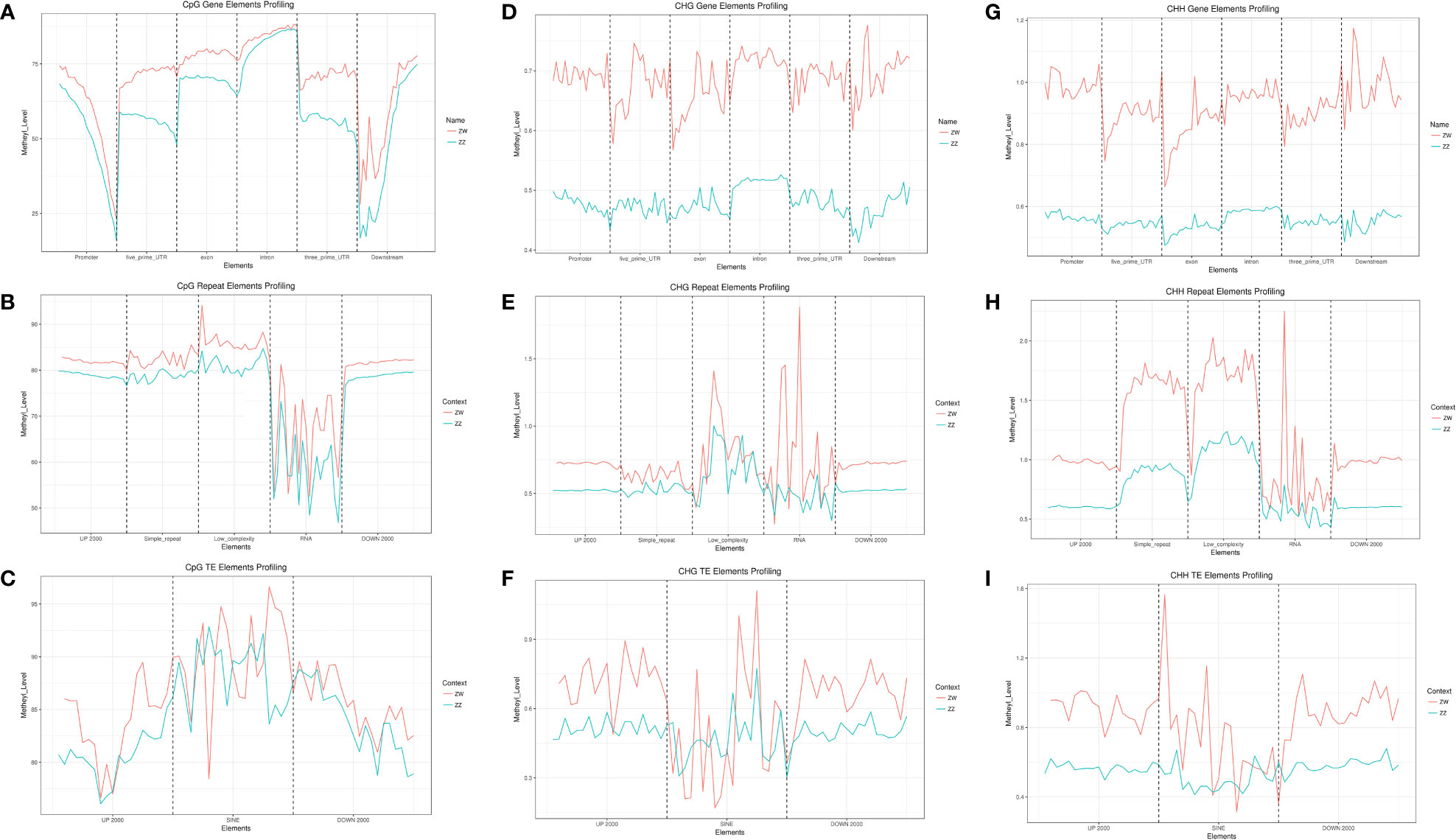
Figure 3 Genomic elements of three contexts of mC sites (CG, CHG, CHH) methylation profiling in ZW and ZZ groups. (A) CpG gene elements profiling of both ZW and ZZ groups. (B) CpG repeat elements profiling of both ZW and ZZ groups. (C) CpG TE elements profiling of both ZW and ZZ groups. (D) CHG gene elements profiling of both ZW and ZZ groups. (E) CHG repeat elements profiling of both ZW and ZZ groups. (F) CHG TE elements profiling of both ZW and ZZ groups. (G) CHH TE elements profiling of both ZW and ZZ groups. (H) CHH TE elements profiling of both ZW and ZZ groups. (I) CHH TE elements profiling of both ZW and ZZ groups. The red line represents ZW group and the blue line represents ZZ group.
Detection of DMRs and DMPs, related genes annotation, and enrichment analyses
There were 1045 DMRs detected between ZW and ZZ groups with 1037 hyper DMRs (methyl differs. > 0.8) and 398 hypo DMRs (methyl differs. <- 0.8) (Figures 4A, 4B). For DMPs screening, the methyl differs bar was set to 0.6, and there were still 505 DMPs remaining. Of these, methylated levels of 407 DMPs in ZW were higher than those in ZZ, while methylated levels of 98 DMPs were lower than those in ZZ (Figures 4E, 4F). DMRs and DMPs related genes were annotated for GO and KEGG pathway enrichments. Top 30 GO terms of DMRs between ZW and ZZ groups were most significantly enriched into the regulation of transmembrane ion transport and related receptor binding in biological process categories (Figure 4C). In KEGG pathway enrichment, P values of most enrichment pathways were greater than 0.05, indicating that the relevant enrichment was not significant. Only 5 pathways were significantly enriched, involving the metabolism and synthesis of amino acids and fatty acids (Figure 4D). The related terms and pathways were completely different from DMRs in GO and KEGG enrichment analyses. The top GO terms of DMPs were mainly involved in the nucleolus, RNA metabolic process, and Golgi network in cellular components. Molecular function categories, histone, and tRNA methyl transferase terms were significantly enriched (Figure 4G). Similar to DMRs, most of the top 30 KEGG pathways of DMPs enrichment in ZW and ZZ groups were not significant, and only the P values of three pathways (ribosome, protein export, and VEGF signaling pathway) were less than 0.05 (Figure 4H).
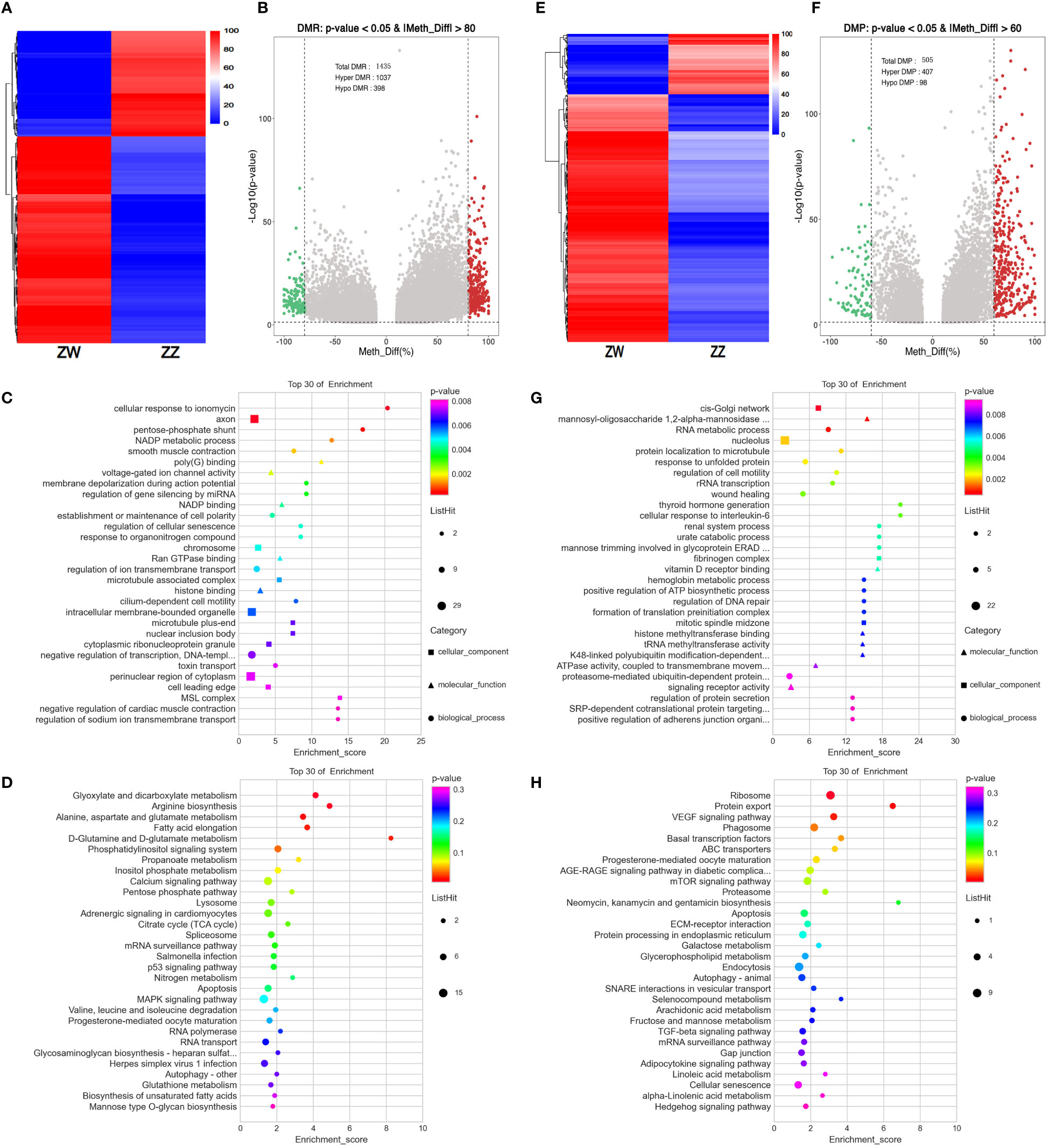
Figure 4 DMRs/DMPs feature and functional annotation analysis. (A) Heatmap of DMRs (on level of |methyl differs| > 0.8 & p value < 0.05) between ZW and ZZ groups showed the degree of differential methylation. (B) Volcano map of DMRs (on level of |methyl differs |. > 0.8 & p value < 0.05) between ZW and ZZ groups. Red dots represent hypermethylation and green dots represent hypomethylation (C) GO enrichment of Top 30 GO term of DMRs between ZW and ZZ groups (D) KEGG pathways enrichment of Top 30 KEGG pathways of DMRs between ZW and ZZ groups (E) Heatmap of DMPs (on level of |methyl differs |. > 0.6 & p value < 0.05) between ZW and ZZ groups showed the degree of differential methylation. (F) Volcano map of DMPs (on level of |methyl differs |. > 0.6 & p value < 0.05) between ZW and ZZ groups. Red dots represent hypermethylation and green dots represent hypomethylation (G) GO enrichment of Top 30 GO term of DMPs between ZW and ZZ groups (H) KEGG pathways enrichment of Top 30 KEGG pathways of DMPs between ZW and ZZ groups.
DMRs/DMPs in sex chromosomes of C. semilaevis sperm
We screened out DMPs and DMRs annotated on the Z and W chromosomes of C. semilaevis. The number of DMRs was greater than that of DMP. There was no exception on the sex chromosomes. Besides, the number of DMRs or DMPs on the Z chromosome was significantly greater than that on the W chromosome (Figure 5A). Top 8 DMPs and DMRs with the most significant methyl differences on W and Z chromosomes were screened out and presented by a Chord diagram. A total of 29 related genes were mapped into DMP-W, DMP-Z, DMR-W, and DMR-Z groups, with 21 hyper-methyl genes and eight hypo-methyl genes. Three of 29 related genes were shared by both DMPs and DMRs, where loc103396997 was mapped to both DMR-W and DMP-W. In contrast, golga3 and ccdc158 were mapped to both DMR-Z and DMP-Z (Figure 5B). The interval distribution line diagram of the DMPs and DMRs on sex chromosomes indicated that the intervals with the highest proportion of DMPs/DMRs were -0.1 to +0.3 in the 4 groups, which were mostly concentrated between interregional -0.1 and +0.3, which means that most DMPs and DMRs on Z and W chromosomes were concentrated in these three adjacent methylated level intervals. The line diagram was similar to a normal distribution-like curve and shifted to the right a little, although there was a deviated small sub-peak in DMP-W within the -0.3 interval. There were only 5 DMPs on W chromosome, and the proportion of DMPs in each interval was prone to statistical deviation (Figure 5C).
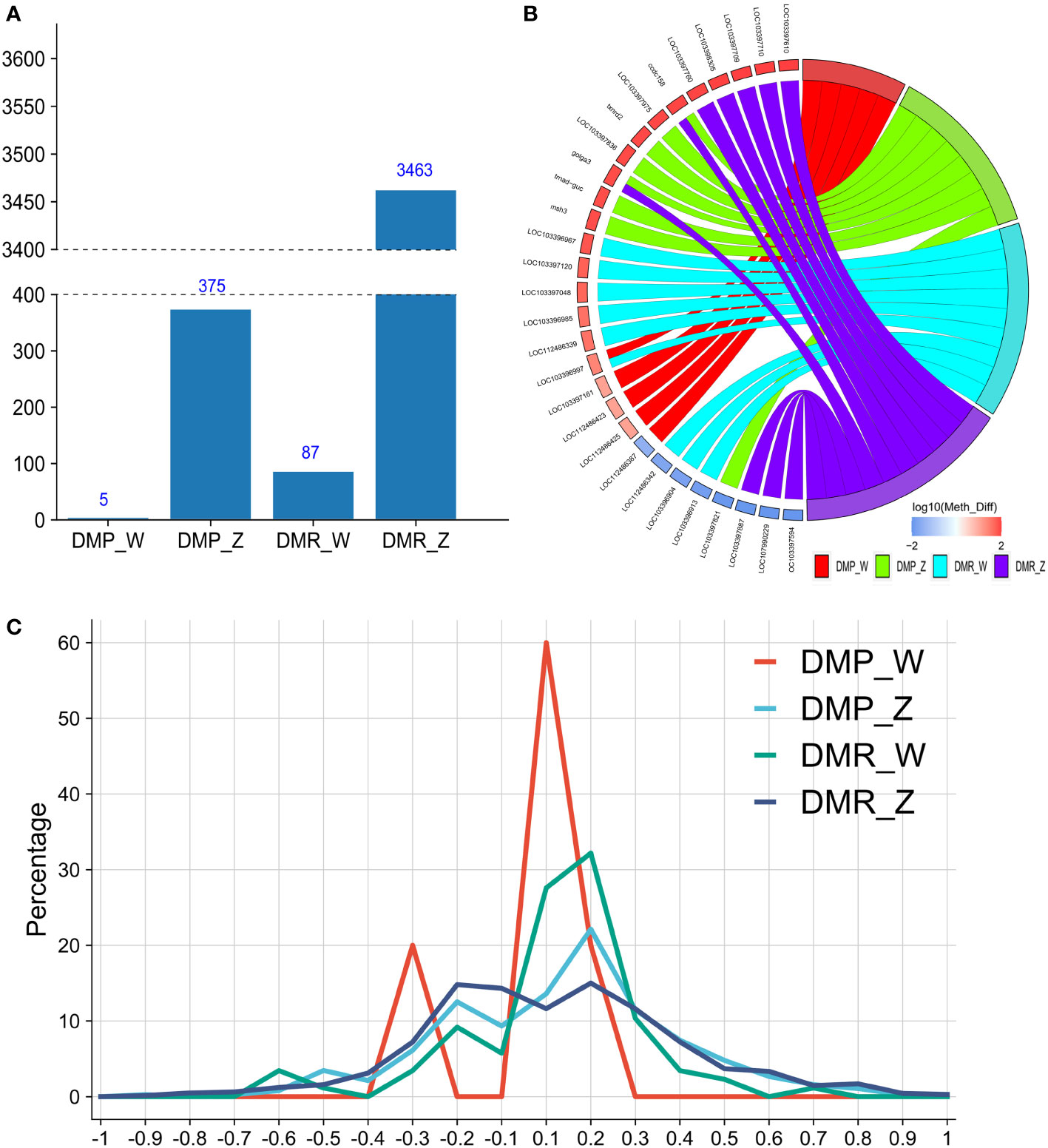
Figure 5 Distribution of DMRs/DMPs on sex chromosomes of ZW and ZZ groups. (A) Number of DMPs/DMRs annotated on W and Z chromosomes of C.semilaevis of both ZW and ZZ groups (methyl diff ± 0.1, P value less than 0.05). (B) Chord diagram of top eight DMR/DMP related genes with the most significant methylation differences of DMP-W, DMP-Z, DMR-W, and DMR-Z groups (Ranged from large to small according to absolute value of methyl diff). (C) Line diagram of the interval distribution of DMP/DMR on Z and W chromosomes. Red line represents DMP-W; Blue line represents DMP-Z; Green line represents DMR-W; Dark blue represents DMR-Z.
Comparison of methylation levels on sex-related genes between sperm and gonads
Twenty sex-related genes were used to compare the mean methylation level between sperm and gonads of C. semilaevis (Figure 6). The different methylation levels of sex-related genes from different sources (sperm vs. gonad) reflected significant heterogeneity between the two media. The heterogeneity between media was greater than that between sexes. Among the 20 genes, the methylation level of most genes in gonads was lower than that in sperm. Ctnnb1, fst, and lhx9 genes had high methylation levels in the testis of males, which was so different from their methylated performance of the ovary, but closer to sperm groups. Furthermore, amh and lgr8, only in testis, had higher methylation levels, while in ovary and sperm, they all had relatively lower methylation levels compared with testis (Figure 6).
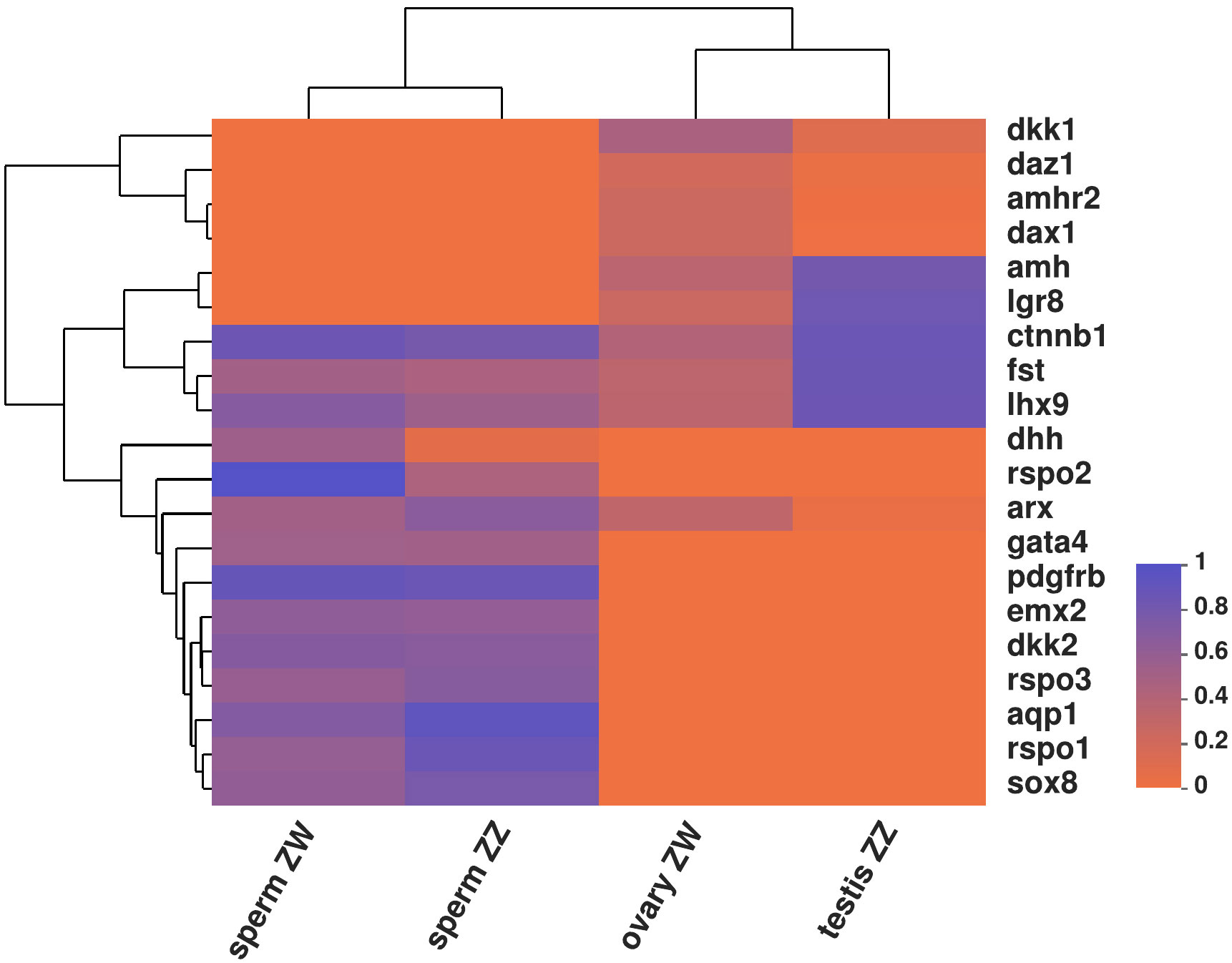
Figure 6 Heatmap of mean methylation levels on 20 sex related genes between sperm and gonad from C. semilaevis.
Interaction patterns of DMR/DMP and differential expressed miRNAs (DEMs)
We conducted integrative analysis between DMR/DMP and DEMs of sperm to uncover interaction patterns between DMP/DMR and DEMs. We found seven methyltransferases and demethylase-related target genes from DMPs corresponding to eight DEMs, as well as six methyltransferases and demethylase-related targets from DMRs corresponding to five DEMs (Figure 7A). Among 13 DEMs, only two DEMs belonging to the mir-140 family targeted both DMRs and DMPs, while the remaining 11 DEMs targeted either DMRs or DMPs, separately. We further investigated the methylation level of the promoter of DEMs and showed the expression level of DEMs and their promoter’ methylation level with a heat map. A total of 15 DEMs were presented in the heatmap. Except for four DEMs, the expression levels of the other 11 were relatively lower (Figure 7B). Corresponding to the 15 DEMs. There were 11 promoters of DMR/DMPs since some miRNAs had the same precursors or promoters in the same DMRs. The methylation level of the promoter of DEMs varied greatly from 2.27% to 97.14% (Figure 7C). The upward and downward trends of DEMs between ZW and ZZ groups were consistent with or negatively correlated with the hyper-methylated or hypo-methylated trend of DMR/DMPs, which was illustrated by the dot line diagram (Figures 7D and 7E). Compared with ZZ groups, most expressions of DEMs in ZW groups were up-regulated, and only two of 15 DEMs were downregulated. While considering the methylated levels of promoters of DMRs, 11 DMRs were hyper-methylated in the ZW group compared with the ZZ group. The expression trend of six DEMs, such as dre-miR-499-5p, ssa-miR-9a-1-3p, dre-miR-146a, dre-miR-182-3p, ssa-miR-210-3p, and hhi-miR-301, were consistent with the trend of their corresponding promoter methylation levels, while the remaining 10 DEMs showed an opposite trend.
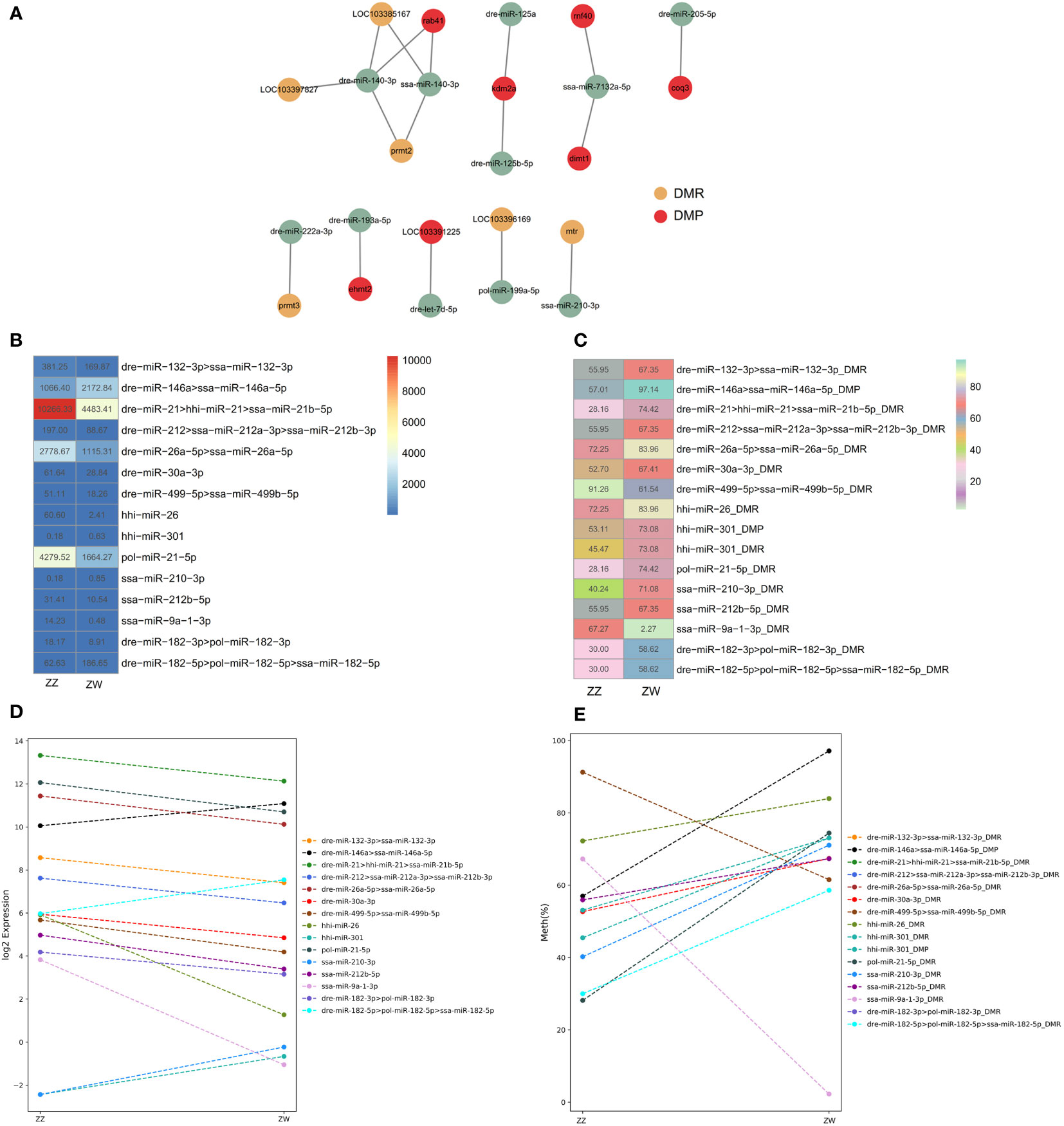
Figure 7 Interaction patterns of DMR/DMP and DEMs (A) Cytoscape of DEMs and their corresponding methyltransferases and demethylase related target DMR/DMP. Yellow represents DMR, red represents DMP, Green represents DEMs. (B) Heatmap of expression level of 15 DEMs between ZW and ZZ sperm groups. (C) Heatmap of the methylation level on promoters of a total 15 DEMs. (D) Dot line diagram illustrated trend of up-regulated and downregulated of 15 DEMs between ZW and ZZ groups. (E) Dot line diagram illustrated trend of hyper-methylated and hoper–methylated trend of DMR/DMPs of 15 DEMs between ZW and ZZ groups.
Discussion
Conservation of hypermethylation levels in sperm of teleost
DNAs of gametes are considered to be highly methylated in organisms from gametogenesis to fertilization. Until zygote genome activation, a new round of DNA methylation reprogramming leads the embryonic development to a totipotent state with an in-depth and complex process (Schulz and Harrison, 2019). Parental epigenome reset occurs throughout development, which may affect the balance between reprogramming and inheritance. For gametes carrying important genetic information, this orderly conservative design is a valuable accumulation deposited in the long evolution process of organisms (Dura et al., 2022). There were significant differences in DNA methylation levels between oocytes and spermatozoa in mice and humans (Smith and Meissner, 2013). The average methylation level of mouse spermatozoa was 80%, and that of oocytes was 50% (Ci and Liu, 2015). The average methylation level in human spermatozoa was about 54%, while that in oocytes was 48% (Guo et al., 2014). In teleost, the genome-wide methylation map of gametes in model fish bore the brunt of disclosure. Zebrafish oocytes showed a slightly lower level of methylation (75%-80% CpG methylation) than sperm hypermethylation (91%-95%), which is similar to mice (Jiang et al., 2013; Potok et al., 2013). For gametes of medaka, the paternal genome was highly methylated (83.03%), whereas the maternal genome possessed ultra-low methylation (26.59%) (Wang and Bhandari, 2020). This huge difference in the methylation level of male and female gametes is rare in fish. The spermatic mean methylation levels of both pseudomale and male C. semibreves reached 92% in this study. However, there were no significant differences between the two groups. As a non-model teleost, hypermethylation of sperm was also common. The level of methylated cytosine climbed to 89% in Atlantic salmon (Wellband et al., 2021), like in steelhead. The average methylation of a large part of CG sites exceeded 90%, which was consistent with the reports in other salmon species (Gavery et al., 2018). The total methylation rate of common carp (Cyprinus carpio) sperm was about 78.6% - 80.8% (Cheng et al., 2021). In contrast, the methylation rate of fresh semen in Gamitana (Colossoma macropomum) was 35.3%. Considering the high methylation level of other fish, the overall low methylation level in Gamitana was lower, which is mainly due to the limitation of the method with MspI and Hpall restriction enzymes digestion for methylome (de Mello et al., 2017). Based on the previous study, methylation difference among tissues of the same species was very small, at least less than that among species (Jabbari et al., 1997), It was found that the methylation level of tropical fish was lower than that of temperate fish species, and the methylation level of temperate fish was lower than that of polar fish (Varriale and Bernardi, 2006). In the same study, liver, testes, or muscle tissue represented the mean level of global methylation and reached the above conclusion, which is highly debatable. From the irregular changes in data of gamete methylation levels in the teleost, the hypothesis is too early to make.
Missing of W-type sperm in pseudomale C. semilaevis
Although we named the sperm group of pseudomale fish as the ZW group in this study, this did not mean that there were W-type sperm in the sperm of pseudomales. The nomenclature here was only derived from tracing the origin of the parent. W-type sperm was absent in semen contents of pseudomale C. semilaevis, resulting in the sex bias of offspring (Cui et al., 2018), whereas pseudomales can’t produce W-type sperm, which should be further explored. In our study, compared with other chromosomes, only a very small number of detected cover C sites of both ZW and ZZ sperms were aligned with the W chromosome of the reference genome, indicating that W-type sperm could hardly exist in the sperm of pseudomales. The few cover C sites in the alignment might come from homologous sequences on other chromosomes rather than the W chromosome. Few DMRs were annotated on the W chromosome, whereas major DMRs were more concentrated on other chromosomes. The difference between pseudomale sperm and male sperm was between the Z-type sperm of the two groups, and there was no W-type sperm involved.
Functional DMRs related genes potentially interacted with DEMs
Although no significant differences in spermatic methylation level were observed between ZW and ZZ groups, specific to differential methylation regions (e.g., various types of genomic structural elements), the hypermethylated ZW-biased DMRs were widespread in methylomes of the two groups. The number of up-methylated DMRs was more than three times that of down-methylated DMRs (ZW vs. ZZ). This trend also existed in DMRs on different chromosomes, especially on the sex chromosome. Corresponding to the distribution of DMRs on sex chromosomes in testes of tongue sole between pseudomale and male, ZW up-regulated methylation was also significantly higher than that in the ZZ group (Shao et al., 2014). DNA methylation can play a regulatory role in miRNA transcription by modification of the CPG region of miRNA promoter, like coding genes. Some miRNAs can target various enzymes related to DNA methylation modification, thereby affecting the methylation pattern of the whole genome (Chhabra, 2015). We captured methyltransferases and demethylase targets from DMPs corresponding to DEMs, and the expression trends (ZW vs. ZZ) of 6 miRNAs were consistent with those of their corresponding promoter methylation levels, while the expression trends of the remaining 10 DEMs were opposite.
Protein arginine methylation is catalyzed by members of the protein arginine methyltransferase (PRMT) family (Wang and Li, 2012). Prmt2, a type I arginine methyltransferase, attenuates tumor necrosis factor receptor-associated factor 6 (traf6)-mediated antiviral response. Prmt2 binds to the C terminus of traf6 to catalyze asymmetric arginine dimethylation of TRAF6 and exercise virus-related immune function (Zhu et al., 2021). Before establishing asymmetric dimethylarginine, PRMT3 can be used as an intermediate to form stable monomethyl arginine (Zhu et al., 2020). In the study, miR-140 and miR-222a as DEMs targeted the prmt2 and prmt3, respectively, to exert the function of arginine methylation. Another DMR, methionine synthase (mtr) gene, a DEM-miR-210’s target, with hypermethylated CpG sites in promoter, is associated with the preterm placenta in humans. It was demonstrated that the methylated level of the CpG site was negatively correlated with the vitamin B12 level in maternal plasma (Khot et al., 2017). Dimethyladenosine transferase 1 (dimt1), a conserved RNA methyltransferase (m26,6a), is crucial in ribosome biogenesis, cell viability, and protein synthesis. Dimt1 can also interact with miR-7132a, reflecting the possible connection and potential interaction between them (Shen et al., 2021). Besides, euchromatic histone lysine methyltransferase 2 (Ehmt2/G9a) (Ryu et al., 2022), and Lysine-specific demethylase 2A (KDM2A), (Xu et al., 2019), play important roles in a variety of cancers through regulation in multiple immune-related pathways.
Conclusion
We characterized and analyzed genome-wide DNA methylation profiles in sperm from male and pseudomale C. semilaevis in this study. The differential methylation genes were identified and correlated with the differential miRNAs to reveal the potential interaction between two epigenetic regulation modes. This study helps understand the potential role of sex-related genes modified with methylation and regulated by miRNAs. The future work will focus on the verification of the interaction between DEM and DMRs to reveal the targeting mechanism between them.
Data availability statement
The DNA methylation data in the study have been deposited to theNational Center for Biotechnology Information (NCBI)Sequence Read Archive (SRA) repository in BioProject: PRJNA871466, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA871466.
Ethics statement
The animal study was reviewed and approved by Academic Ethics Committee of Guangdong Ocean University.
Author contributions
BZ conceived and designed the project. NZ, ZD, and BZ carried out the computational analysis and expression profiling. NZ and LJ performed sampling and data analysis. NZ and BZ wrote the manuscript. LC and JYL edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from the Special Funding for Modern Agricultural Industrial Technology System (CARS-47-Z01), the Modern Industrial Technology System in Tianjin ITTFRS2017011, and the Nanhai Scholar Project of Guangdong Ocean University (QNXZ201903).
Acknowledgments
We thank a lot for the support from Fan Zhang, Shanghai OE Biomed Company (Shanghai, China).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Cheng Y., Vechtova P., Fussy Z., Sterba J., Linhartova Z., Rodina M., et al. (2021). Changes in phenotypes and DNA methylation of In vitro aging sperm in common carp cyprinus carpio. Int. J. Mol. Sci. 22 (11), 5925. doi: 10.3390/ijms22115925
Chen S. L., Li J., Deng S. P., Tian Y. S., Wang Q. Y., Zhuang Z. M., et al. (2007). Isolation of female-specific AFLP markers and molecular identification of genetic sex in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. (NY) 9 (2), 273–280. doi: 10.1007/s10126-006-6081-x
Chen S. L., Tian Y. S., Yang J. F., Shao C. W., Ji X. S., Zhai J. M., et al. (2009). Artificial gynogenesis and sex determination in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. (NY) 11 (2), 243–251. doi: 10.1007/s10126-008-9139-0
Chhabra R. (2015). miRNA and methylation: A multifaceted liaison. Chembiochem 16 (2), 195–203. doi: 10.1002/cbic.201402449
Ci W., Liu J. (2015). Programming and inheritance of parental DNA methylomes in vertebrates. Physiol. (Bethesda) 30 (1), 63–68. doi: 10.1152/physiol.00037.2014
Cui Y., Wang W., Ma L., Jie J., Zhang Y., Wang H., et al. (2018). New locus reveals the genetic architecture of sex reversal in the Chinese tongue sole (Cynoglossus semilaevis). Heredity (Edinb) 121 (4), 319–326. doi: 10.1038/s41437-018-0126-6
de Mello F., Garcia J. S., Godoy L. C., Depince A., Labbe C., Streit D. P. Jr. (2017). The effect of cryoprotectant agents on DNA methylation patterns and progeny development in the spermatozoa of colossoma macropomum. Gen. Comp. Endocrinol. 245, 94–101. doi: 10.1016/j.ygcen.2016.06.003
Depince A., Gabory A., Dziewulska K., Le Bail P. Y., Jammes H., Labbe C. (2020). DNA Methylation stability in fish spermatozoa upon external constraint: Impact of fish hormonal stimulation and sperm cryopreservation. Mol. Reprod. Dev. 87 (1), 124–134. doi: 10.1002/mrd.23297
Dura M., Teissandier A., Armand M., Barau J., Lapoujade C., Fouchet P., et al. (2022). DNMT3A-dependent DNA methylation is required for spermatogonial stem cells to commit to spermatogenesis. Nat. Genet. 54 (4), 469–480. doi: 10.1038/s41588-022-01040-z
Gavery M. R., Nichols K. M., Goetz G. W., Middleton M. A., Swanson P. (2018). Characterization of genetic and epigenetic variation in sperm and red blood cells from adult hatchery and natural-origin steelhead, oncorhynchus mykiss. G3 (Bethesda) 8 (11), 3723–3736. doi: 10.1534/g3.118.200458
Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., et al. (2014). The DNA methylation landscape of human early embryos. Nature 511 (7511), 606–610. doi: 10.1038/nature13544
Iwanami N., Lawir D. F., Sikora K., Meara C. O., Takeshita K., Schorpp M., et al. (2020). Transgenerational inheritance of impaired larval T cell development in zebrafish. Nat. Commun. 11 (1), 4505. doi: 10.1038/s41467-020-18289-9
Jabbari K., Caccio S., Pais de Barros J. P., Desgres J., Bernardi G. (1997). Evolutionary changes in CpG and methylation levels in the genome of vertebrates. Gene 205 (1-2), 109–118. doi: 10.1016/s0378-1119(97)00475-7
Jiang L., Zhang J., Wang J. J., Wang L., Zhang L., Li G., et al. (2013). Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 153 (4), 773–784. doi: 10.1016/j.cell.2013.04.041
Khot V. V., Yadav D. K., Shrestha S., Kaur L., Sundrani D. P., Chavan-Gautam P. M., et al. (2017). Hypermethylated CpG sites in the MTR gene promoter in preterm placenta. Epigenomics 9 (7), 985–996. doi: 10.2217/epi-2016-0173
Krueger F., Andrews S. R. (2011). Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27 (11), 1571–1572. doi: 10.1093/bioinformatics/btr167
Murphy P. J., Wu S. F., James C. R., Wike C. L., Cairns B. R. (2018). Placeholder nucleosomes underlie germline-to-Embryo DNA methylation reprogramming. Cell 172 (5), 993–1006.e1013. doi: 10.1016/j.cell.2018.01.022
Potok M. E., Nix D. A., Parnell T. J., Cairns B. R. (2013). Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 153 (4), 759–772. doi: 10.1016/j.cell.2013.04.030
Rodriguez Barreto D., Garcia de Leaniz C., Verspoor E., Sobolewska H., Coulson M., Consuegra S. (2019). DNA Methylation changes in the sperm of captive-reared fish: A route to epigenetic introgression in wild populations. Mol. Biol. Evol. 36 (10), 2205–2211. doi: 10.1093/molbev/msz135
Ryu T. Y., Kim K., Han T. S., Lee M. O., Lee J., Choi J., et al. (2022). Human gut-microbiome-derived propionate coordinates proteasomal degradation via HECTD2 upregulation to target EHMT2 in colorectal cancer. ISME J. 16 (5), 1205–1221. doi: 10.1038/s41396-021-01119-1
Schulz K. N., Harrison M. M. (2019). Mechanisms regulating zygotic genome activation. Nat. Rev. Genet. 20 (4), 221–234. doi: 10.1038/s41576-018-0087-x
Shao C., Li Q., Chen S., Zhang P., Lian J., Hu Q., et al. (2014). Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 24 (4), 604–615. doi: 10.1101/gr.162172.113
Shao G. M., Li X. Y., Wang Y., Wang Z. W., Li Z., Zhang X. J., et al. (2018). Whole genome incorporation and epigenetic stability in a newly synthetic allopolyploid of gynogenetic gibel carp. Genome Biol. Evol. 10 (9), 2394–2407. doi: 10.1093/gbe/evy165
Shen H., Gonskikh Y., Stoute J., Liu K. F. (2021). Human DIMT1 generates N2(6,6)A-dimethylation-containing small RNAs. J. Biol. Chem. 297 (4), 101146. doi: 10.1016/j.jbc.2021.101146
Smith Z. D., Meissner A. (2013). DNA Methylation: Roles in mammalian development. Nat. Rev. Genet. 14 (3), 204–220. doi: 10.1038/nrg3354
Varriale A., Bernardi G. (2006). DNA Methylation and body temperature in fishes. Gene 385, 111–121. doi: 10.1016/j.gene.2006.05.031
Wang X., Bhandari R. K. (2020). The dynamics of DNA methylation during epigenetic reprogramming of primordial germ cells in medaka (Oryzias latipes). Epigenetics 15 (5), 483–498. doi: 10.1080/15592294.2019.1695341
Wang Y. C., Li C. (2012). Evolutionarily conserved protein arginine methyltransferases in non-mammalian animal systems. FEBS J. 279 (6), 932–945. doi: 10.1111/j.1742-4658.2012.08490.x
Wellband K., Roth D., Linnansaari T., Curry R. A., Bernatchez L. (2021). Environment-driven reprogramming of gamete DNA methylation occurs during maturation and is transmitted intergenerationally in Atlantic salmon. G3 (Bethesda) 11 (12). doi: 10.1093/g3journal/jkab353
Woods L. C. III, Li Y., Ding Y., Liu J., Reading B. J., Fuller S. A., et al. (2018). DNA Methylation profiles correlated to striped bass sperm fertility. BMC Genomics 19 (1), 244. doi: 10.1186/s12864-018-4548-6
Xu W. H., Liang D. Y., Wang Q., Shen J., Liu Q. H., Peng Y. B. (2019). Knockdown of KDM2A inhibits proliferation associated with TGF-beta expression in HEK293T cell. Mol. Cell Biochem. 456 (1-2), 95–104. doi: 10.1007/s11010-018-03493-5
Yuan C., Wang L., Zhu L., Ran B., Xue X., Wang Z. (2019). N-acetylcysteine alleviated bisphenol a-induced testicular DNA hypermethylation of rare minnow (Gobiocypris rarus) by increasing cysteine contents. Ecotoxicol Environ. Saf. 173, 243–250. doi: 10.1016/j.ecoenv.2019.02.035
Zhang B., Wang X. L., Sha Z. X., Yang C. G., Liu S. S., Wang N., et al. (2011). Establishment and characterization of a testicular cell line from the half-smooth tongue sole, cynoglossus semilaevis. Int. J. Biol. Sci. 7 (4), 452–459. doi: 10.7150/ijbs.7.452
Zhang B., Zhao N., Jia L., Che J., He X., Liu K., et al. (2020a). Identification and application of piwi-interacting RNAs from seminal plasma exosomes in cynoglossus semilaevis. BMC Genomics 21 (1), 302. doi: 10.1186/s12864-020-6660-7
Zhang B., Zhao N., Jia L., Peng K., Che J., Li K., et al. (2019). Seminal plasma exosomes: Promising biomarkers for identification of Male and pseudo-males in cynoglossus semilaevis. Mar. Biotechnol. (NY) 21 (3), 310–319. doi: 10.1007/s10126-019-09881-2
Zhang B., Zhao N., Liu Y., Jia L., Fu Y., He X., et al. (2019). Novel molecular markers for high-throughput sex characterization of cynoglossus semilaevis. Aquaculture 513, 734331. doi: 10.1016/j.aquaculture.2019.734331
Zhang B., Zhao N., Peng K., He X., Chen C. X., Liu H., et al. (2020b). A combination of genome-wide association study screening and SNaPshot for detecting sex-related SNPs and genes in cynoglossus semilaevis. Comp. Biochem. Physiol. Part D Genomics Proteomics 35, 100711. doi: 10.1016/j.cbd.2020.100711
Zhao N., Jia L., Deng Q., Zhu C., Zhang B. (2022). Comparative piRNAs profiles give a clue to transgenerational inheritance of sex-biased piRNAs in cynoglossus semilaevis. Mar. Biotechnol. 24 (2), 335–344. doi: 10.1007/s10126-022-10109-z
Zhao N., Zhang B., Jia L., He X. X., Bao B. L. (2021a). Extracellular vesicles piwi-interacting RNAs from skin mucus for identification of infected Cynoglossus semilaevis with vibrio harveyi. Fish Shellfish Immunol. 111, 170–178. doi: 10.1016/j.fsi.2021.02.001
Zhao N., Jia L., Zhang B. (2021b). Sex bias miRNAs inCynoglossus semilaevis could play a role in transgenerationalinheritance. Comp Biochem Physiol PartGenomicsProteomics 39:100853. doi: 10.1016/j.cbd.2021.100853
Zhu J., Li X., Sun X., Zhou Z., Cai X., Liu X., et al. (2021). Zebrafish prmt2 attenuates antiviral innate immunity by targeting traf6. J. Immunol. 207 (10), 2570–2580. doi: 10.4049/jimmunol.2100627
Keywords: sperm, DNA methylation, microRNAs, sex, teleost
Citation: Zhao N, Jia L, Chen L, Lin J, Dong Z and Zhang B (2022) Integrative sperm DNA methylation and miRomics analysis highlights interaction of two epigenetic patterns of pseudomale inheritance in teleost. Front. Mar. Sci. 9:1022091. doi: 10.3389/fmars.2022.1022091
Received: 07 September 2022; Accepted: 27 October 2022;
Published: 11 November 2022.
Edited by:
Vikash Kumar, Central Inland Fisheries Research Institute (ICAR), IndiaReviewed by:
Jinxiang Liu, Ocean University of China, ChinaMartin Psenicka, University of South Bohemia, Czechia
Copyright © 2022 Zhao, Jia, Chen, Lin, Dong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Zhang, zb611273@163.com; Zhongdian Dong, zddong@gdou.edu.cn