
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 25 October 2022
Sec. Marine Biology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1014336
 Bing Xie1†Chuan-yue Chen2†
Bing Xie1†Chuan-yue Chen2† Xiaolin Zhang1*Si-yuan Li1Zhong-qi Gu3Ji Huang3Jian-yu He1,4
Xiaolin Zhang1*Si-yuan Li1Zhong-qi Gu3Ji Huang3Jian-yu He1,4 Peng-zhi Qi1
Peng-zhi Qi1 Bao-ying Guo1
Bao-ying Guo1 Zhi Liao1
Zhi Liao1 Xiaojun Yan1*
Xiaojun Yan1*Being an industrially valuable species of bivalve, Mytilus is widespread in the area around the eastern coast of China. Starvation is an environmental stress that mussels often encounter in their growth. However, few report carried out on the physiological and biochemical response and molecular regulation and adaptation mechanism of mussels under starvation. To illustrate the molecular mechanism of hosts of Mytilus to starvation, the gill transcriptome was used for analysis before and after 9 days of starvation in Mytilus using the Illumina/HiSeq-2000 deep sequencing platform, accompanied by the study of differentially expressed genes (DEGs). In total, 42.137 GB of clean data were obtained from six sample libraries, the average amount of clean data of each sample is 7.023 GB, and 58,540 unigenes that average 780 bp in length were assembled. Unigenes were illustrated by commenting them against the NR, Swiss-Prot, Pfam, String, GO and KEGG databases. After 9 days of starvation, 2,188 and 2,672 genes were determined to be significantly up- or down-regulated expression genes, separately. Amongst, 4,860 genes were associated with 260 pathways, contains vital enrichment pathways, such as “Metabolic”, “PI3K-Akt signaling pathway”, “Phagosome”, “Apoptosis” and “Lysosome”. In inclusion, autophagy cells were investigated by an electron microscopy, alongside further observations of the expression of autophagy-related genes in gill tissue before and after starvation. The data indicates that the amount of autophagosomes increased and found that atg2, atg6 and atg13 were significantly up-regulated. These results indicated that Mytilus may use autophagy to cope with their damage after starvation. With our research results, we have contributed to a deeper understanding of the role of the molecular mechanisms of immune defence in Mytilus under environmental stress, which has given insights into mussel breeding and the understanding of the molecular mechanisms of Mytilus immune system.
Mytilus is commonly known to be commercially and ecologically vital and is widely distributed throughout the world (Wang, 2008; Liu, 2021). Mussel genera are strongly endeared to a variety of environmental changes. However, with the increasing density of mussel farming and the particularity of environmental changes, farmed mussels are suffering from various environmental stresses, including temperature, acidification, oxidation, and starvation (Zhang et al., 2014; Dong and Zhang, 2016; Falfushynska and Piontkivska, 2020; Kong et al., 2021). It has been reported that heat stress affects cardiac function and cell metabolism (Zhang et al., 2014; Dong and Zhang, 2016), acidification stress has a negative effect on growth and calcification (Zhang et al., 2014), with oxidative stress can produce reactive oxygen species and dangerously compromise the host integrity (Kong et al., 2021), and starvation stress has also been shown to be one of the major threats to the mussel farming industry (Orban et al., 2002). Nevertheless, the molecular mechanisms connected with starvation stress in Mytilus remain scarce and fragmented up to now.
Mytilus mainly feed on natural algae and organic debris in the ocean, while starvation is a major problem often encountered in the growth of mussels due to improper feeding, insufficient abundance, uneven spatial distribution, seasonal change or drastic environmental change in natural sea areas (Li et al., 2010). Starvation will not only lead to inadequate nutrition of shellfish, but also affect the weight, metabolism and energy reserves of other aquatic organisms to a certain extent (Dai et al., 2018; Kankuan et al., 2019; Xu et al., 2021). Moreover, imbalance in nutrient supply will also lead to metabolic disorders in aquatic organisms, and eventually shorten the life span of many different organisms (Dai et al., 2018; Kankuan et al., 2019; Xu et al., 2021). At the same time, starvation stress will affect the adaptability of marine organisms to the aquatic environment, may prolong or even fail to adapt, and may further affect the growth, defense system and reproduction of marine organisms (Mahapatra et al., 2017; Haider et al., 2020). Therefore, exploring the consequences of starvation stress-induced nutritional restriction is very important to further understanding the potential defense and molecular adaptation mechanisms of Mytilus.
In aquatic animals, starvation and nutritional deficiency will cause significant physiological changes and destroys a variety of organelles, contains mitochondria, of which are accountable for the synthesis of ATP and free radicals, and activates cellular autophagy (Kamogashira et al., 2015; Włodarczyk et al., 2017; Bialik and Dasari, 2018; Lőrincz and Juhász, 2020). Autophagy is a commonly used self-feeding phenomenon observed in eukaryotic cells. By degrading long-lived proteins and impaired organelles, it is a major pathway for cellular repair, so that cells can recycle nutrients and continue to survive under stress conditions (Ham and Raju, 2017; Gao, 2019). Under certain circumstances, autophagy will selectively degrade certain macromolecules and organelles, including cytoplast to vacuole transport (Cvt) pathway, lysosomal autophagy (pexophagy) and mitochondrial autophagy (mitophagy), which is activated in organism’s responses to stress environments (Sciarretta et al., 2011; Ham and Raju, 2017; Gao, 2019). Autophagy may be divided with six instances: the induction, the vesicle nucleation and the expansion, the substrate recognition, the autophagosome formation, the autophagosome lysosome fusion and the substrate degradation (Sciarretta et al., 2011; Ham and Raju, 2017; Gao, 2019). After the cells are induced by autophagy signals, a “lipid like” membrane structure is formed in the cytoplasm, which is called phagophore. The autophagosome elongates, surrounds the target product to be degraded, develops a closed bilayer of membrane autophagosomes and converges with the lysosome to formed an autolysosome, so as to degrade the wrapped materials, and the generated fatty acids, amino acids and other substances can be transported to the cytoplasm for recycling. In this procedure, there are over 30 autophagy-related genes (atgs) whose primary function is to mediate the formation of autophagy via four of the protein complexes (Tsukada and Ohsumi, 1993; Sciarretta et al., 2011; Ham and Raju, 2017). Unc-51-like kinase 1 (Ulk1) complex (ulk1-atg101-fip200-atg13) to participate in the autophagy induction phase, type III of phosphatidylinositol 3-kinase (PI3K) enzyme complex (beclin1-vps34-atg14) to participate in the nucleation phase of autophagy vesicles, atg12-atg5-atg16 ubiquitination complex to participate in the extension phase of autophagy vesicles, lc3-II-pe ubiquitination complex to participate in the extension of autophagy vesicles and the formation of autophagosomes (Tsukada and Ohsumi, 1993; Mizushima et al., 2002; Sciarretta et al., 2011; Ham and Raju, 2017; Gao, 2019). Besides, starvation represents a potentially life-threatening issue for all organisms, so the host will evolve a set of mechanisms to cope with the pressure of starvation (Tsukada and Ohsumi, 1993; Mizushima et al., 2002; Hara et al., 2006; Komatsu et al., 2007; Sciarretta et al., 2011). Autophagy is an evolutionary well-conserved process of intracellular metabolic catabolism that becomes activated under starvation as a result. The organisms lacking the process of autophagy die when starved, which emphasizes the crucial importance of autophagy to the survival of organisms (Sciarretta et al., 2011; Ham and Raju, 2017; Bialik and Dasari, 2018; Gao, 2019). Therefore, this study focused on the role of starvation in inducing autophagy in mussels, and analyzed autophagy coding genes from the autophagy mechanism level, so as to understand the biological effects of autophagy on mussels.
The dramatic evolution of next-generation sequencing technologies has led to a transcriptomics and genomic signature revolution in recent decades. The most frequent use of the high throughput sequencing technology is to enable functional genomics investigations, which includes worldwide gene expression, novel gene discovery, full-length gene assortments, single nucleotide polymorphism (SNP) and simple sequence repeat (SSR) detection (Zhan et al., 2014; Zhou et al., 2017). The digital gene expression on which the sequencing platform is based has been increasingly applied to a larger number of aquatic molluscs, such as Crassostrea gigas Thunberg, 1793 (Blanca et al., 2011; Huang et al., 2011; Gavery and Roberts, 2012), Pecten maximus Linnaeus, 1758 (Li et al., 2020), Haliotis midae Linnaeus, 1758 (Zhao et al., 2012b), Pinctada martensii Dunker, 1880 (Van Der Merwe et al., 2011; Pauletto et al., 2014), Chlamys farreri K. H. Jones & Preston, 1904 (Zhao et al., 2012a), Mytilus galloprovincialis Lamarck, 1819 (Shi et al., 2013; Cai et al., 2014), Mytilus edulis Linnaeus, 1758 (Costa et al., 2009; Fu et al., 2014; Rosani and Domeneghetti, 2014), Mytilus chilensis Hupé, 1854 (Philipp et al., 2012; Tanguy et al., 2013a) for investigating their behavior in response to environmental stress. In the presented research, the transcriptome of Mytilus gill tissue was examined after 9 days of starvation by Illumina RNA-seq technology. The transcriptome profiles of the control group and the starvation group were measured and contrasted to determine the autophagy-related genes and biological processes related to the innate defense mechanism response of Mytilus under short-term starvation. This finding will enhance understanding of the molecular mechanisms underlying the adaptive response of the Mytilus to environmental stress.
The mature thick-shelled Mytilus coruscus (shell length 60-70 mm) were collected from Aquatic Farm located at Shengsi Island, East China Sea. The mussels accelerated in laboratory conditions for one week in a 300 L aquarium at 20 ± 2°C and a salinity of 28 ± 1‰ before treatments were carried out. Daily feeding of Mytilus with commercial spirulina pollen and filtered seawater replacement for half of the aquarium was performed daily. Following acclimation, the mussels were randomly distributed into control (Con) and starved (Sta) groups. Both Con and Sta groups have triplicate tanks, with 6 individuals per tank. The mussels in Con group were fed normally and the mussels in Sta groups were deprived of feeding for 9 days. At the end of the treatment, three mussels per replicate from Con and Sta groups were dissected by cutting off the adductor muscle. The gill tissue acquired was preserved at -80°C until RNA was isolated for transcriptome sequencing analysis. Meanwhile, three mussels were sampled for autophagosome electron microscope observation.
Following the manufacturer’s instructions, total RNA was isolated from tissues using TRIzol® reagent (Invitrogen) and genomic DNA was extracted using DNase I (TaKaRa). the Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit) as well as agarose gel electrophoresis were used to evaluate the isolated RNA for integrity and purity of the extracted RNA. Alternatively, RNA was quantified using a Nano-Drop 2000 spectrophotometer (NanoDrop® Technologies Inc). For the construction of the sequencing libraries, factoring was performed only on samples of high quality RNA (OD260/280 = 1.8- 2.2, OD260/230≥ 2.0, RIN≥ 6.5, 28 S: 18S≥ 1.0, > 1 μg). RNA-seq transcriptome libraries were assembled under the Illumina (San Diego, CA) TruSeqTM RNA samples preparation kit, using 1 μg of total RNA. The mRNA was separated from the total RNA and segmented as approximately 200 nt. Double-stranded cDNA was integrated using the SuperScript double-stranded cDNA synthesis kit (Invitrogen, CA) and random hexamer primers (Illumina). Next, the refined cDNA was end-repaired, attached to an adapter, and were subjected to enrichment. Following quantification and validation, the cDNA library was assembled on a cBot (Truseq PE Cluster Kit v3- cBot- HS, Illumina) to yield clusters on stream cells, and the assembled stream cells were sequenced with matched ends on a Nova-seq 6000 system (Illumina). The tailoring and qualitative control of raw paired end reads with default settings of SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.Com/najoshi/sickle). The elementary sequencing profiles were subsequently performed with quality control (QC) using RSeQC (v2. 3. 6) to confirm they were fit for analysis. Mapped reads for of each sample were compiled from String Tie (https://ccb.jhu.edu/software/stringtie) in a reference-based assortment process (Tanguy et al., 2013b; Núñez-Acuña and Gallardo-Escárate, 2013; Pertea et al., 2015). Remaining clean reads were generated using Trinity software (v2012_10_05) with default settings after removal of aptamers, elicited sequences and low-quality sequences. The transcripts were assorted and clustered by the Chrysalis cluster tool, where the lengthiest sequence in the cluster was reserved and specified as a “unigenes”. Unigenes were annotated by BLASTX alignment against NR (NCBI Non-Redundant Protein Sequences), GO (Gene Ontology, http://www.geneontology.org), String (Functional Enrichment Analysis of Protein Interaction Networks, http://cn.string-db.org/), KEGG (Kyoto encyclopedia of genes and genomes), Swiss-Prot (Protein sequence database, https://www.sib.swiss/swiss-prot) and Pfam (Protein family, http://pfam.xfam.org) databases with E values less than 1.0e-5 and HMMER (v3.1) with default settings. The alignment of results for highest homology was applied to ascertain the sequence identity orientation of single genes. High quality clean reads from each RNA-seq library were plotted against the assigned transcripts using the Bowtie2 (v2.4.4) routine. In order for the DEGs to be identified between different samples, the expression level of for instance each transcript was determined based on fragments per kilobase of exon model per million mapped fragments (FPKM) approach (http://deweylab.biostat.wisc.edu/rsem), which was used to determine gene enrichment (Philipp et al., 2012). Basically, differential expression analysis was conducted using DESeq2 (Tanguy et al., 2013a), with FDR ≤ 0.05. DEGs with |log2FC| (Fold change)> 1 and FDR ≤ 0.05 were identified as genes that were markedly differentially expressed. In inclusion, functional enrichment analyses encompassing GO and KEGG were also implemented to determine which DEGs were markedly enriched in GO terms and metabolic pathways at Bonferroni-corrected P values ≤ 0.05 by comparison with the whole transcriptome background. GO functional enrichment and KEGG pathways performed with KOBAS (http://kobas.cbi.pku.edu.cn/home.do) (Kim et al., 2015).
RNA was extracted and measured by UV spectrophotometer and agarose electrophoresis. Based on the manufacturer’s instructions, transcripts ®RT Kit (perfect real time) (TaKaRa, Dalian, China) was used to convert transcribe the total RNA of each sample is subjected to a test to yield the first cDNA strand. The expression of autophagy-related coding genes was investigated by performing quantitative real-time PCR (qRT-PCR). premier 5.0 was used to design the primers. Primer information for each gene has been listed in Table 1. The total volume of qRT-PCR is 20 µL, which contains 10 µL 2 × SYBR premix Ex Taq (TaKaRa, Dalian, China), 0.40 µmol/L and 0.1 ± 0.02 µg cDNA template for each primer. The three stage qRT-PCR response was conducted as follows: pre-denaturation at 95°C for 30 seconds, followed by denaturation at 95°C for 5 seconds, annealing at 58-60°C for 20 seconds and extension at 72°C for 20 seconds for 40 cycles, and the transcript levels of the genes were determined by the 2-ΔΔCt method and standardized with the reference gene (β-actin) (He et al., 2021). Three samples were tested for each group, with three technical replicates of each test. In a further step, the same RNA samples were used to perform validation of the RNA seq analysis in the transcriptome analysis of the gills. The primers that were used for qRT-PCR to be verified are shown in Supplementary Table 1.

Table 1 Primer table for qRT-PCR detection of autophagy gene.
The gill tissue samples were taken out from the 2.5% glutaraldehyde solution, washed with PBS buffer (pH 7.4) for 3 times, then added with 2% osmic acid, immobilise at 4°C over 60 minutes in the light of darkness, followed by washing with PBS buffer (pH 7.4). Then dehydrate with gradient acetone (30%, 50%, 70%, 80%, 90%, 95%, 100%) and transfer to anhydrous acetone for 15 min, and the samples were sealed and packaged by spurr resin embedding agent, then polymerized in bake oven at 37-60°C. After the temperature naturally drops to room temperature, take out the embedded plate from the oven. Slice with ultra-thin microtome. The sections were observed and photographed under the transmission electron microscope (Miao et al., 2019).
The statistical analysis of all figures was carried out by one-way analysis of variance (ANOVA) with SPSS software package. All results are shown as mean ± standard deviation (SD) and significant differences are shown as P < 0.05.
For a better understanding of the molecular variations in the metabolic and immune reaction of starvation-treated Mytilus, two cDNA libraries were generated from the gills of starved and normal feeding mussels. Following replicate studies and assured quality control, the Illumina HiSeqTM 2000 was utilised to sequence cDNA libraries. And further PCA testing of gill tissue samples from the mussels showed that the majority of samples were tightly clustered with a clear segregation between treated and control samples (Supplementary Figure 1), which illustrates the stability and reproducibility of the sequencing platform Illumina used in this study. 47, 180, 816 and 55, 215, 552 raw reads were obtained from the gill fraction of the Sta and Con groups respectively. Having filtered out low quality reads, short sequences and low complexity sequences, 47, 126, 196 and 55, 089, 960 clean reads were remained for de novo assembly (Table 2). The values for percentage Q30 and percentage GC among the clean reads in the two DGE libraries were 92.92% and 93.13%, and 36.89% and 36.31%, respectively. Upon comparing all clean reads to the reference database, 30,893,705 (69.340%) and 41,209,086 (74.803%) were revealed as unique matches. Ultimately, the average amount of clean data of each sample is 7.023 GB, 58,540 unigenes with an average length of 780 bp were then assigned. The transcriptome databases were submitted to the NCBI SRA database under submission number SUB10509738. The BLAST software was utilized to search all unigenes against the NR, Swiss-Prot, Pfam, String, GO and KEGG databases. Of these, 40, 817 (69.7%) were found within NR, 14, 826 (24.7%) within Swiss-port, 10, 143 (17.3%) within KEGG, 12, 304 (45.0%) within Swiss-Prot, 30,423 (52. 0%) within Pfam, 13, 870 (23. 7%) were traced in GO and 9,466 (16.7%) in the String database (Supplementary Table 2). For identification of digenomes, the gill transcriptome database of Mytilus after 9 days of starvation and normal feeding was used to analyze FPKM. Expression changes of more than two-fold and an FDR (false discovery rate) < 0.05 were criteria selected to determine genes that were significantly up or down during starvation. A total of 4,860 DEGs were identified using this criterion, comprised of 2,188 up-regulated and 2,672 down-regulated expressed genes (Figure 1).
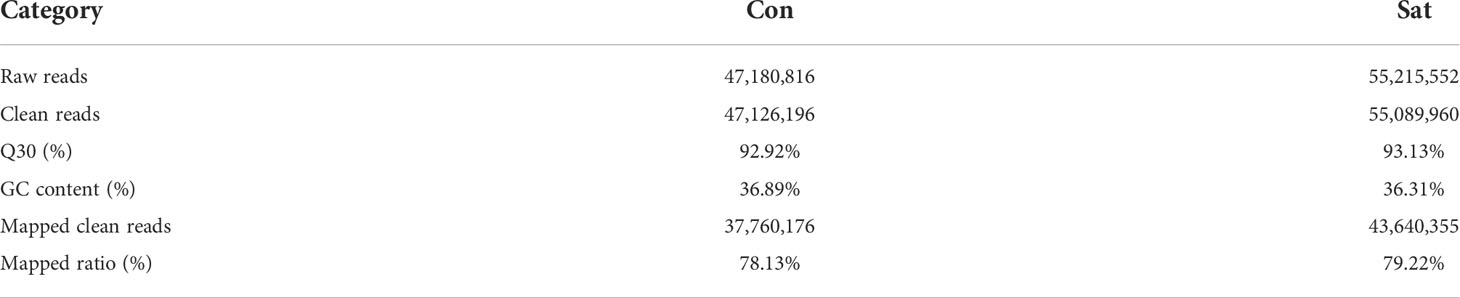
Table 2 Summary of sequencing results.
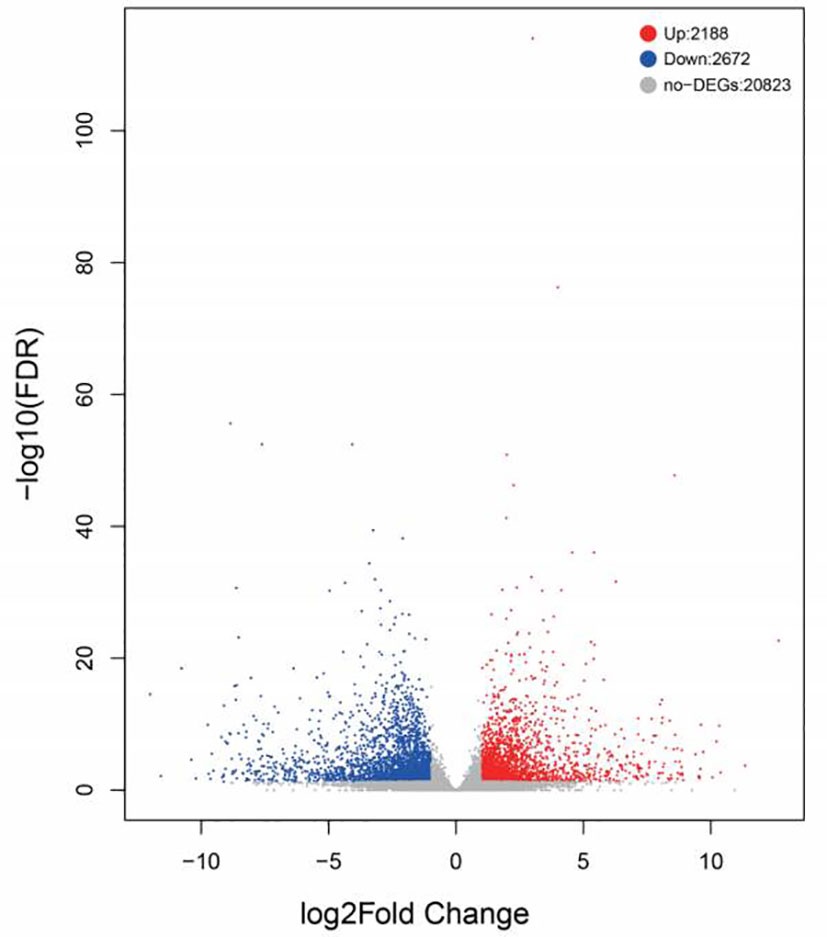
Figure 1 Volcano plot of differentially expressed genes identified between the control and starvation groups. The blue dots represent the down-regulation of gene expression, the red dots represent the up-regulation of gene expression, and the black dots represent the gene with no significant difference in expression.
Subsequently the annotated DEGs were inspected and GO terms were assigned as a reference for enrichment analysis. The significant enrichment GO pathways were determined by Fisher’s exact tests (P value < 0.05). DEGs in biological processes have been identified as being primarily related to protein metabolism, lipid metabolism, glucose metabolism and nucleic acid metabolism. The dominant subclass of cellular components is the cytoplasmic fraction, and within the molecular functional categories, the most prominent subclass is oxidoreductase activity (Figure 2). According to the GO enrichment results of DEGs in gill, the most abundant pathways in the Sta group were cytoplasm, cilium and microtubule compared with the control group (Figure 3). In order to determine the biological pathways active in Mytilus during the 9-day starvation state, DEG was annotated to typical signalling pathways in the KEGG database. Ultimately, a total of 2,791 genes were mapped to 260 statistically significant categories (P < 0.05) (Supplementary Table 3). The most prominent pathways including metabolic pathways, biosynthesis of secondary metabolites, neuroactive ligand-receptor interactions and focal adhesions. The most abundant group of these is the lysosome (Figure 4). In addition, it is worth noting that a large number of KEGG pathways are associated with autophagy were found in starvation Mytilus, such as metabolic, phagosome, pI3K-akt and TNF pathways (Table 3), and among these KEGG pathways, some well-known autophagy-related genes were also identified respectively (Supplementary Figure 2). At the same time, we used the KEGG database to integrate these three pathways, and 15 DEGs were selected as the nodes to create a network diagram related to, protein systhesis, glycometabolism, and oxidative stress (Supplementary Figure 3). Therefore, we carried out some further analysis to study the role of autophagy in starvation.
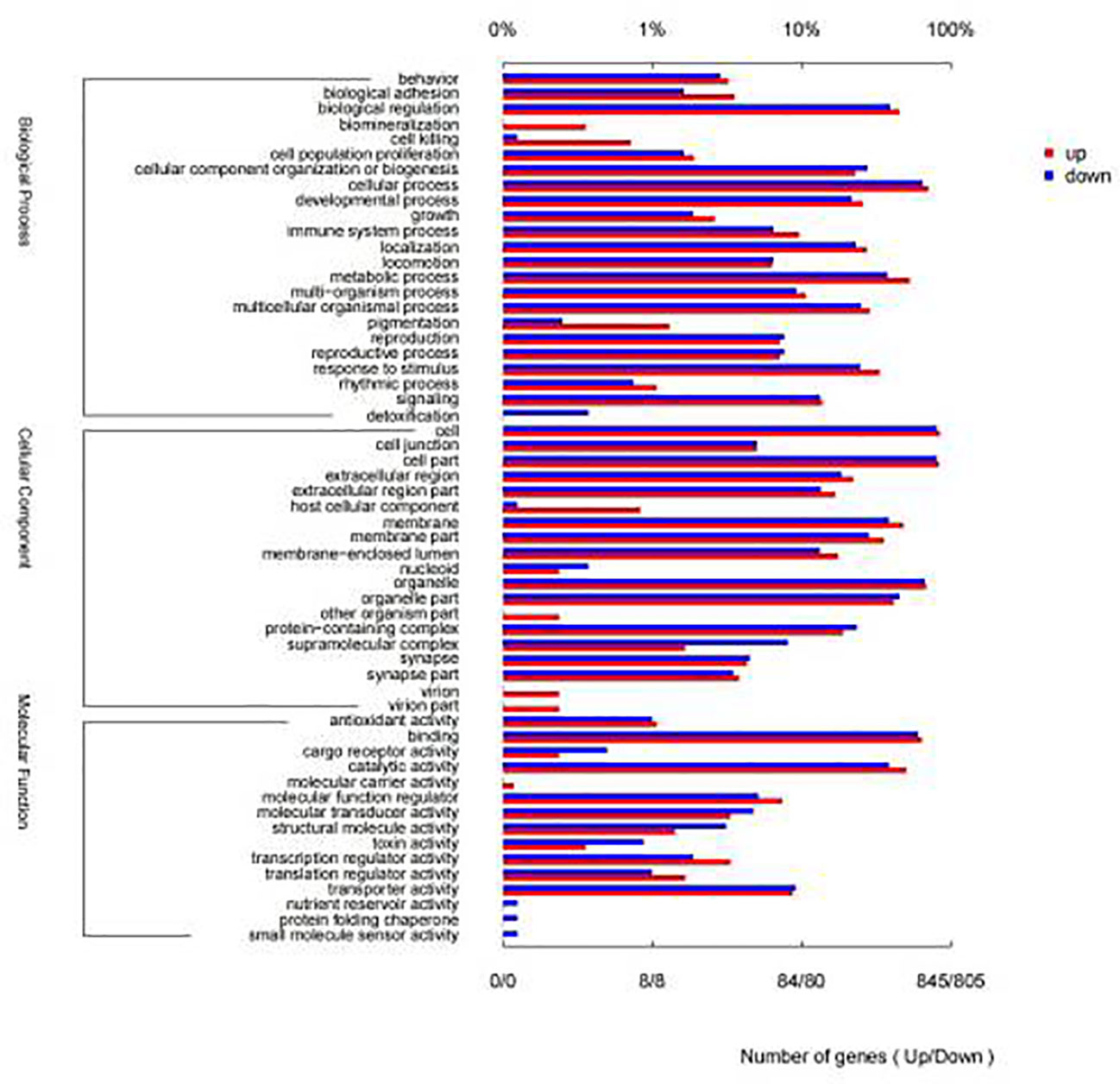
Figure 2 Gene ontology classifications of assembled unigenes. A total of 845 unigenes were categorized into 3 functional categories: biological process, cellular component and molecular function.
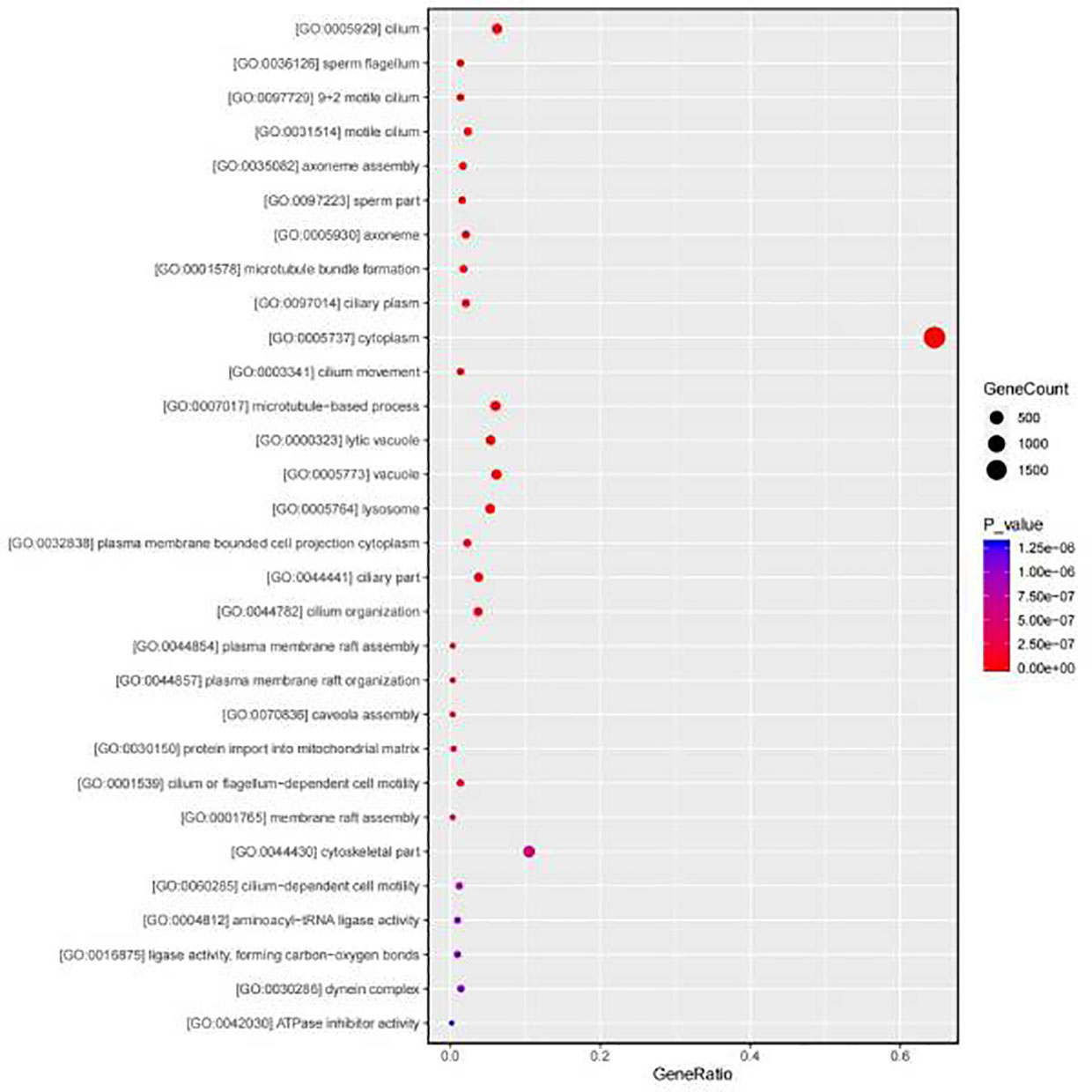
Figure 3 GO classifications of DEGs. The most abundant pathways are cilium and microtubule.
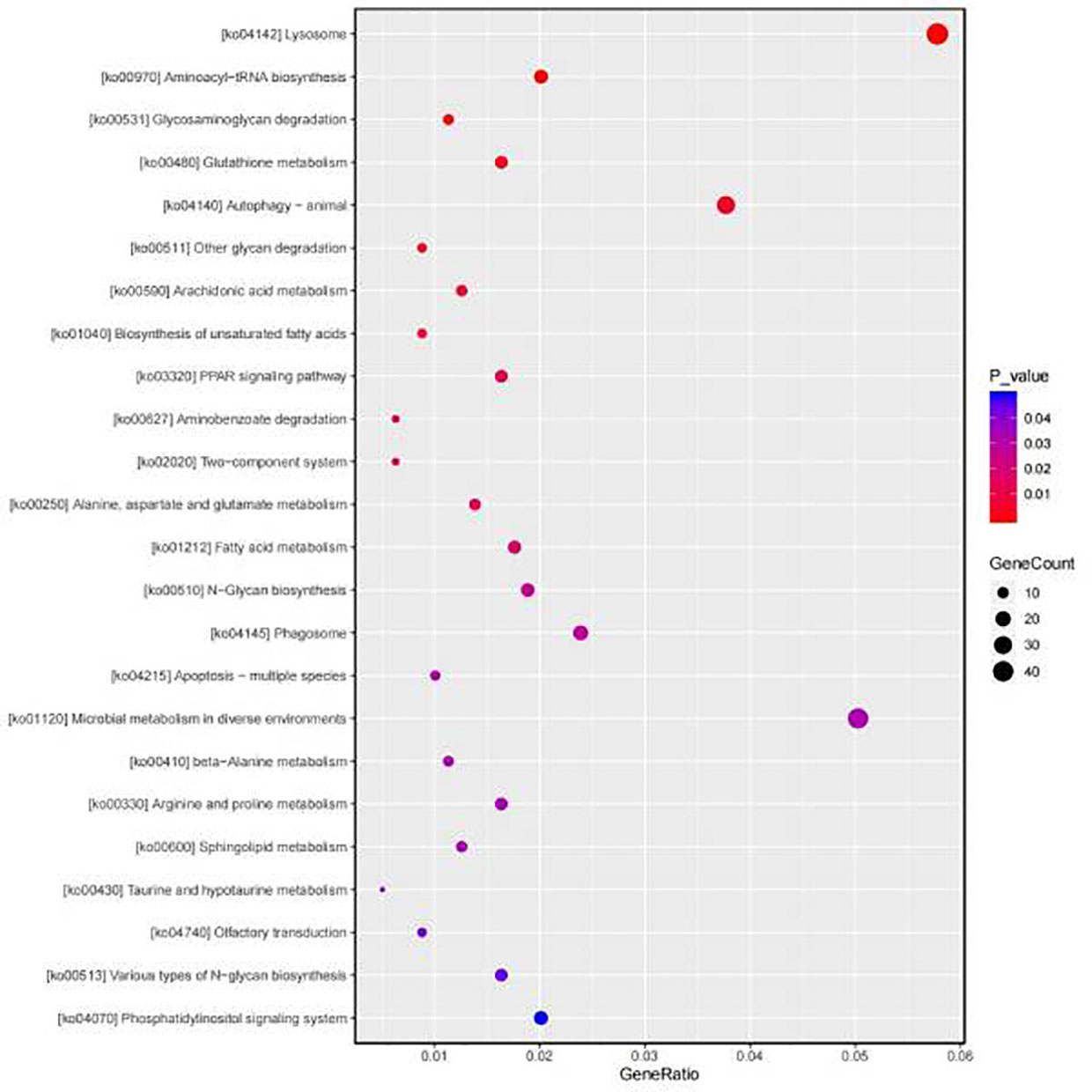
Figure 4 KEGG classifications of DEGs. The most abundant pathway is lysosome.

Table 3 Number of top 20 KEGG pathways and DEGs associated with autophagy.
Through the electron microscope analysis of the gill tissue, we found that the number of autophagosomes in the gill tissue of the starvation treatment group changed significantly compared with the control group (Figure 5A). The number of autophagosomes in the gill tissue of mussels increased under starvation conditions. These can be attributed to the fact that mussels respond to their upheaval by decomposing their contents through more autophagosomes under starvation. In order to further study the molecular changes of autophagy, we screened autophagy related coding genes for qRT-PCR (Figure 5B), and found that atg2, atg6 and atg13 were significantly up-regulated. This result confirmed that the autophagy pathway has important biological effects during starvation.
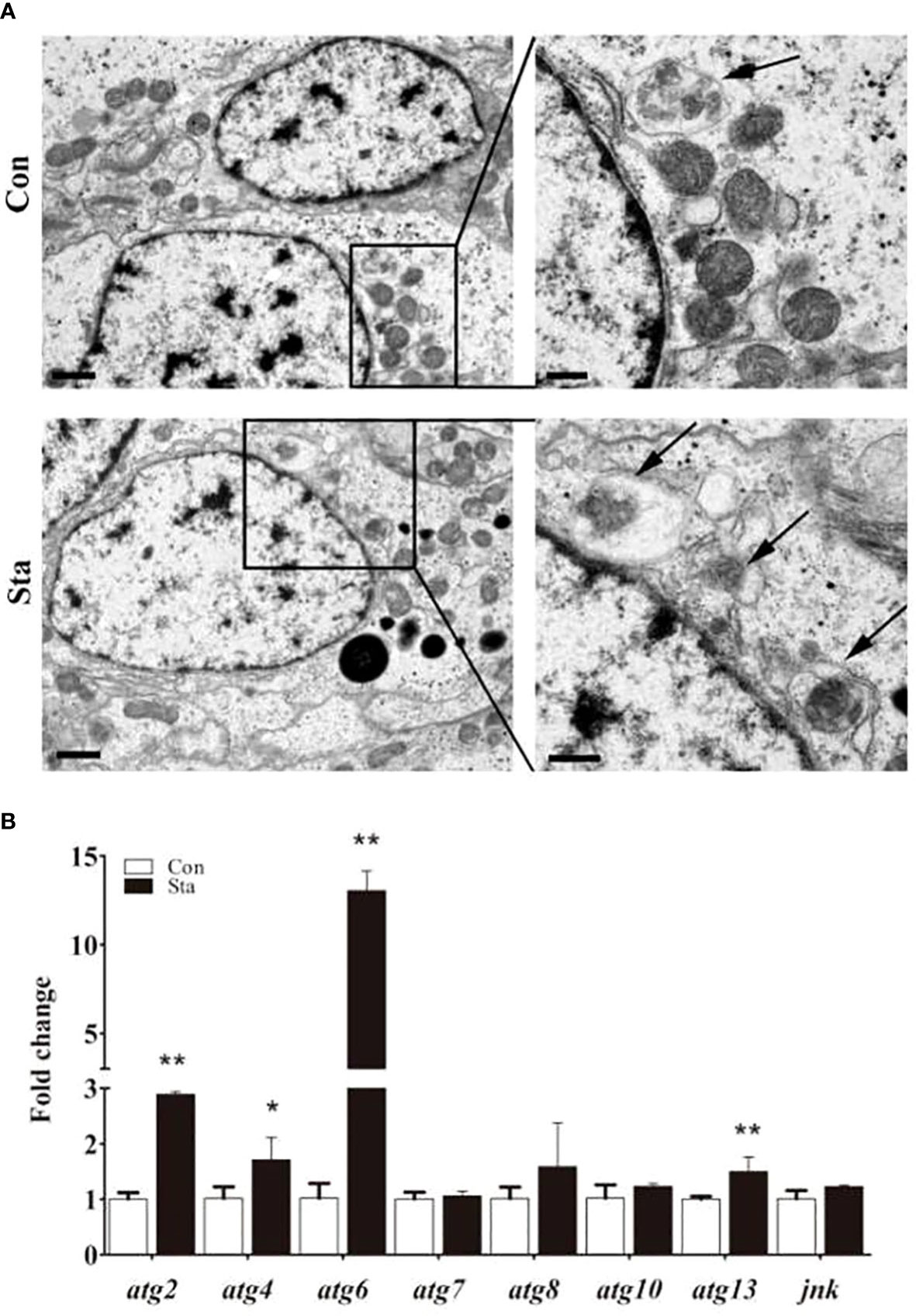
Figure 5 Autophagosome transmission electron microscope (A) and autophagy gene expression (B). ATG mRNA expression levels in mussel gill were detected by qRT-PCR. Values are shown as means ± SD (n=3), *P<0.05 or **P<0.01 versus Con group.
The relevant mRNA expression levels of 12 genes in the transcriptome was randomly examined using qRT-PCR for the transcriptome sequencing library. qRT-PCR profiling of the melting curves showed that all genes had a product. Relevant mRNA expression levels were plotted against the transcriptome analysis results. As illustrated in Supplementary Figure 4, there were 12 genes which showed a consistent orientation in the transcriptome library. The reliability of the RNA seq results was demonstrated
Molecular studies of metabolism during starvation in Mytilus are limited, and there is currently no report that describes the transcriptome profile of the gill during this particular condition. To increase our knowledge on the effect of starvation on gene expression in Mytilus, we employed RNA-seq to explore the effect of starvation stress on the gene expression patterns of Mytilus gill. In this study, A combined total with 163,961 transcripts with an average length of 820 bp were forecast from the clean reads. After the removal of redundancy, 58,540 unigenes were assembled with an average length of 780 bp. A totally of 4,860 DEGs were identified, comprising 2,188 up-regulated genes and 2,672 down-regulated genes, indicating that starvation stress affects the whole genome transcription of Mytilus gill.
There are a number of DEGs implicated in glycolysis, lipid and protein metabolism, like those encoding genes for key enzymes engaged in fatty acid metabolism, amino degradation and among other biological functions. The energy supply and demand of mussel under starvation stress can be met by accurately adjusting the amount of glucose. Glycolysis and gluconeogenesis are the opposite metabolic pathways related to carbohydrate decomposition and synthesis, which are crucial to the survival of shellfish. Especially during starvation, the energy provided by glycolysis and gluconeogenesis is more important (Jitrapakdee, 2012; Wang et al., 2021b). In this study, DEGs related to glucose metabolism were annotated in KEGG pathway, indicating that the expression level of most enzymes encoding genes involved in glycolysis/gluconeogenesis pathway has changed significantly. For example, the important metabolic enzymes involved in gluconeogenesis, fructose-1,6-diphosphate esterase (FBP) and phosphoenolpyruvate carboxylation kinase (PEPCK) (Jitrapakdee, 2012), were also down-regulated expression in the transcriptome of gill tissue after 9 days of starvation. This finding was also observed by the large yellow croaker, in which the expression of pyruvate kinase (PK) and glucose-6-phosphate dehydrogenase (FBP) gene were significantly decreased after 21 days of starvation (Qian et al., 2016). On the other hand, previous studies by Metón et al. (2003), the enzyme activities of PK and G6P-DH were significantly decreased after 18 days of starvation, and this phenomenon was also reported in rainbow trout muscle (Johansen and Overturf, 2006), similarly, we also found the down regulation of these metabolic genes in the results of Mytilus transcriptome analysis.
Lipids are the main energy source for mussels. In the state of hunger, the body provides energy through fat catabolism to maintain its own steady state, and it is also an indispensable part of body tissue and bioactive substances (Kolb, 1982). Through transcriptome analysis, we discovered that starvation stress resulted in significant up-regulation of genes encoding proteins related to fatty acid metabolism in Mytilus (Figure 4 and Supplementary Table 3), such as fatty acid-binding protein coding gene (fabp), fatty acid metabolism coding gene(fabd) and fatty acid-binding protein coding gene (fabf). Apolipoproteins, which are a complex of plasma lipoproteins, are up-regulated in the liver of different aquatic organisms, which bind to lipids and are subsequently transported to different tissues via the blood (Kolb, 1982; Liu et al., 2003). FABP (fatty acid-binding protein) plays an important role in promoting fatty acid solubilization, transportation and metabolism (Liu et al., 2003). Furthermore, other up-regulated or down-regulated genes involved in fat digestion and absorption pathways were found by transcriptome analysis that fatty acid metabolism was active after starvation (Supplementary Table 3). The consequences of these transcriptomic studies may demonstrate the beginning of a redistribution of lipid reserves and cholesterol metabolism in Mytilus and justify attempts to increase lipid metabolism in Mytilus during starvation. Therefore, we conclude that Mytilus may respond to early starvation by adjusting sugar and fat metabolism during starvation.
Gill tissue is an important organ of shellfish that conducted filter feeding and immune response (Wang et al., 2021a). In present study, gill RNA-seq and TEM data have shown that the most abundant pathways in the Sta group were cytoplasm, cilium and microtubule in the GO enrichment results. And cilium is an important filter feeding organs in mussel gill tissues (Dong et al., 2017; Tassanakajon et al., 2018), thus, the cilium function of the starved mussels was impaired, and autophagy bodies began to increase in the gill cilia of the mussels by transmission electron microscopy. And transcriptome analysis showed that there were great changes in the genes of autophagy related regulatory pathways in Mytilus in mussels under starvation, and the genes encoded by proteins involved in autophagy and apoptosis pathways were significantly up-regulated, such as ras enzyme coding gene (rab5), vacuolar Protein Sorting (vps4) and vacuolar Protein Sorting (vps36). Additionally, among all DEGs, some well-known autophagy-related genes, including, patamycin protein (mTOR), tumor suppressor protein (p53), etc, were also identified respectively. Autophagy is a homeostasis process, when organs are challenged by nutritional stress, autophagy breaks down cytoplasmic components to maintain organ homeostasis (Byrne et al., 2016; Nakamura and Yoshimori, 2017). Although several ATG proteins have been reported in mollusks, there is still little information about the components of the molecular mechanism of autophagy pathway in this phylum. We observed the increase in the number of autophagosomes in the gill tissues of mussels under starvation, which may indicate that during the stress environment, mussels maintain the stability of their internal environment by increasing the number of autophagosomes in their tissues, facilitating the fusion of more lysosomes, and finally degrading the contents of cells. This finding is similar to the response of lamprey autophagy when stimulated (Luo et al., 2018). Furthermore, the mRNA expression levels of atg2, atg4, atg6 and atg13 in the gills of starved mussels were significantly up-regulated, which undoubtedly further confirmed that autophagy has a great biological effect under starvation stress, and may be an important way for mussels to regulate their homeostasis under stress. This finding is consistent with earlier reports on oysters, which show that autophagy activates energy supply under environmental stress, and autophagy is considered to have a protective effect during environmental stress (Creagh et al., 2003; Samokhvalov et al., 2008), indicating the large increase of autophagy bodies is a possible sign of mussels’ self-protection and mussels resist starvation by activating autophagy through a series of pathway changes.
Macroautophagy that occurs in all eukaryotic cells is a conserved catabolic process during which specific cellular components are transported into lysosomes and broken down (Creagh et al., 2003; Mizushima et al., 2008; Nakamura and Yoshimori, 2017), The procedure begins with the capture of unnecessary material into autophagosomes, followed by autophagosome-lysosome fusion to yield autolysosomes that degrade cargo (Creagh et al., 2003; Mizushima et al., 2008). From previous reports (Levine and Kroemer, 2008; Cullup et al., 2013; Byrne et al., 2016), the deletion of autophagosome lysosome related coding genes is related to various diseases, and affecting the related coding genes may lead to immune deficiency. For example, Rab coding genes may lead to neuropathy, and the interaction between ATG and other genes may also lead to neuropathy (Verhoeven et al., 2003; Levine and Kroemer, 2008; Cullup et al., 2013; Byrne et al., 2016; Nakamura and Yoshimori, 2017). In our study, through the transcriptome analysis and the qRT-PCR of gill tissues after starvation, we found that the most abundant KEGG pathway is lysosome, and atg2, atg4, atg6 and atg13 are significantly up-regulated under starvation, which indicates that these atg genes may be molecular markers of mussel response to autophagy pathway under starvation. It may indicate that the products of autophagy related genes are involved in endosomal lysosomal processes other than autophagy. Therefore, the combined effects of autophagy and lysosomes are likely to play a great role in the adaptation of Mytilus to starvation stress.
In conclusion, the gill tissues of Mytilus starved for 9 days were sequenced by RNA-seq, and a large number of differentially expressed autophagy and metabolic related genes and signal pathways were detected. The significant changes in glucose metabolism and autophagy transcription caused by starvation stress indicate that mussels may first respond to starvation by regulating their stored glucose, and then activate autophagy pathway, so as to increase the recycling of material capacity and better balance and stability. These findings deepen our understanding of the molecular adaptation mechanism of Mytilus to starvation, which will help to develop new aquaculture strategies to meet various external challenges in the aquaculture industry.
The data presented in the study are deposited in the SRA repository, accession number PRJNA770460.
X-JY, X-LZ, and ZL designed the experiments and approved the final version of the manuscript. BX, C-YC, Z-QG, S-YL and JH carried out the experiments. J-YH, P-ZQ and B-YG completed the data analysis. BX and X-LZ wrote this manuscript. X-JY, Z-QG and X-LZ directed the manuscript revision. All authors contributed to the article and approved the submitted version.
This work was supported by grant from the National Natural Science Foundation of China (42020104009, 41976111, 42176099), the Natural Science Foundation for Distinguished Young Scholars of Zhejiang province (LR22D060002), the Project of Zhoushan Science and Technology Bureau (2019F12004) and the Science Foundation of Donghai Laboratory (Grant No. DH-2022KF0219).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1014336/full#supplementary-material
Bialik S., Dasari S. K., Kimchi A. (2018). Autophagy-dependent cell death - where, how and why a cell eats itself to death J. Cell. Sci. 131(18), 1–12. doi: 10.1242/jcs.215152
Blanca J. M., Cañizares J., Ziarsolo P., Esteras C., Mir G., Nuez F., et al. (2011). Melon transcriptome characterization: Simple sequence repeats and single nucleotide polymorphisms discovery for high throughput genotyping across the species. Plant Genome 4, 118–131. doi: 10.3835/plantgenome2011.01.0003
Byrne S., Jansen L., Jm U. K.-I., Siddiqui A., Lidov H. G., Bodi I., et al. (2016). EPG5-related syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain 139(3), 765–781. doi: 10.1093/brain/awv393
Cai Y., Pan L., Hu F., Jin Q., Liu T. (2014). Deep sequencing-based transcriptome profiling analysis of Chlamys farreri exposed to benzo [a] pyrene. Gene 551, 261–270. doi: 10.1016/j.gene.2014.09.003
Costa M. M., Prado-Alvarez M., Gestal C., Li H., Roch P., Novoa B., et al. (2009). Functional and molecular immune response of mediterranean mussel (Mytilus galloprovincialis) haemocytes against pathogen-associated molecular patterns and bacteria. Fish Shellfish Immunol. 26, 515–523. doi: 10.1016/j.fsi.2009.02.001
Creagh E. M., Conroy H., Martin S. J. (2003). Caspase activation pathways in apoptosis and immunity. Immunol. Rev. 193, 10–21. doi: 10.1034/j.1600-065x.2003.00048.x
Cullup T., Kho A. L., Dionisi-Vici C., Brandmeier B., Smith F., Urry Z., et al. (2013). Recessive mutations in EPG5 cause syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 45, 83–87. doi: 10.1038/ng.2497
Dai W. F., Zhang J. J., Qiu Q. F., Chen J., Yang W., Ni S., et al. (2018). Starvation stress affects the interplay among shrimp gut microbiota, digestion and immune activities. Fish Shellfish Immunol. 80, 191–199. doi: 10.1016/j.fsi.2018.05.040
Dong W., Chen Y., Lu W., Wu B., Qi P. (2017). Transcriptome analysis of Mytilus coruscus hemocytes in response to Vibrio alginnolyficus infection. Fish Shellfish Immunol. 70, 560–567. doi: 10.1016/j.fsi.2017.08.034
Dong Y., Zhang S. (2016). Ecological relevance of energy metabolism: transcriptional responses in energy sensing and expenditure to thermal and osmotic stresses in an intertidal limpet. Funct. Ecol. 30, 1539–1548. doi: 10.1111/1365-2435.12625
Falfushynska H., Piontkivska H. (2020). Effects of intermittent hypoxia on cell survival and inflammatory responses in the intertidal marine bivalves Mytilus edulis and. Crassostrea gigas. 223, 1–13. doi: 10.1242/jeb.217026
Fu X., Sun Y., Wang J., Xing Q., Zou J., Li R., et al. (2014). Sequencing based gene network analysis provides a core set of gene resource for understanding thermal adaptation in zhikong scallop Chlamys farreri. Mol. Ecol. Resour 14, 184–198. doi: 10.1111/1755-0998.12169
Gao Q. (2019). Oxidative stress and autophagy. Adv. Exp. Med. Biol. 1206, 179–198. doi: 10.1007/978-981-15-0602-49
Gavery M. R., Roberts S. B. (2012). Characterizing short read sequencing for gene discovery and RNA-seq analysis in Crassostrea gigas. Comp. Biochem. Physiol. Part D Genomics Proteomics 7, 94–99. doi: 10.1016/j.cbd.2011.12.003
Haider F., Timm S., Bruhns T., Noor M. N., Sokolova I. M. (2020). Effects of prolonged food limitation on energy metabolism and burrowing activity of an infaunal marine bivalve. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 250, 110780. doi: 10.1016/j.cbpa.2020.110780
Ham P. B., Raju R. (2017). Mitochondrial function in hypoxic is chemic injury and influence of aging. Prog. Neurobiol. 157, 92–116. doi: 10.1016/j.pneurobio.2016.06.006
Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
He Z., He J., Wang J., Zhang X., Liao Z. (2021). Comparative transcriptomic analysis of gill and gonad from Mytilus under antibiotics treatment followed by different bacteria challenge. Aquaculture 547, 737457. doi: 10.1016/j.aquaculture.2021.737457
Huang Y., Huang X., Yan Y., Cai J., Ouyang Z., Cui H., et al. (2011). Transcriptome analysis of orange-spotted grouper (Epinephelus coioides) spleen in response to Singapore grouper iridovirus. BMC Genomics 12, 556. doi: 10.1186/1471-2164-12-556
Jitrapakdee S. (2012). Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 44, 33–45. doi: 10.1016/j.biocel.2011.10.001
Johansen K. A., Overturf K. (2006). Alterations in expression of genes associated with muscle metabolism and growth during nutritional restriction and refeeding in rainbow trout. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 144, 119–127. doi: 10.1016/j.cbpb.2006.02.001
Kamogashira T., Fujimoto C., Yamasoba T. (2015). Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed. Res. Int. 2015, 617207. doi: 10.1155/2015/617207
Kankuan W., Wanichanon C., Morani F., Thongrod S., Titone R., Siangcham T., et al. (2019). Starvation promotes autophagy associated maturation of the testis in the Giant freshwater prawn, Macrobrachium rosenbergii. Front. Physiol. 10. doi: 10.3389/fphys.2019.01219
Kim D., Langmead B., Salzberg S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163. doi: 10.1016/j.2007.10.035
Kong N., Han S., Fu Q., Yu Z., Song L. (2021). Impact of ocean acidification on the intestinal microflora of the pacific oyster crassostrea gigas. Aquaculture 546, 737365. doi: 10.1016/j.Aquaculture.2021.737365
Lőrincz P., Juhász G. (2020). Autophagosome-lysosome fusion. J. Mol. Biol. 432, 2462–2482. doi: 10.1016/j.jmb.2019.10.028
Levine B., Kroemer G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li G., Li J., Li D. (2010). Seasonal variation in nutrient composition of Mytilus coruscus from China. J. Agric. Food Chem. 58, 7831–7837. doi: 10.1021/jf101526c
Liu R. Z., Denovan-Wright E. M., Wright J. M. (2003). Structure, linkage mapping and expression of the heart-type fatty acid binding protein gene (fabp3) from zebrafish (Danio rerio). Eur. J. Biochem. 270, 3223–3234. doi: 10.1046/j.1432-1033.2003.03705.x
Li R., Zhang W., Lu J., Zhang Z., Mu C., Song W., et al. (2020). The whole- genome sequencing and hybrid assembly of Mytilus coruscus. Front. Genet. 11. doi: 10.3389/fgene.2020.00440
Luo L., Lu J., Wang Q., Chen S., Xu A., Yuan S. (2018). Autophagy participates in innate immune defense in lamprey. Fish Shellfish Immunol. 83, 416–424. doi: 10.1016/j.fsi.2018.09.016
Mahapatra E., Dasgupta D., Bhattacharya N., Mitra S., Banerjee D., Goswami S., et al. (2017). Sustaining immunity during starvation in bivalve mollusc: A costly affair. Tissue Cell 49, 239–248. doi: 10.1016/j.tice.2017.02.005
Metón I., Fernández F., Baanante I. V. (2003). Short and long-term effects of refeeding on key enzyme activities in glycolysis gluconeogenesis in the liver of gilthead seabream (Sparus aurata). Aquaculture 225, 99–107. doi: 10.1016/S0044-8486(03)00281-3
Miao F., Wu Z. D., Wu J. L., Shi S. J., Zeng X., Wang J., et al. (2019). Transmission electron microscopic observation on gonad of Oncomelania hupensis off spring bred in wei shan lake areas. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 32, 195–197. doi: 10.16250/j.32.1374.2019088
Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Mizushima N., Ohsumi Y., Yoshimori T. (2002). Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429. doi: 10.1247/csf.27.421
Nakamura S., Yoshimori T. (2017). New insights into autophagosome lysosome fusion. J. Cell. Sci. 130, 1209–1216. doi: 10.1242/jcs.196352
Núñez-Acuña G., Gallardo-Escárate C. (2013). Identification of immune related SNPs in the transcriptome of Mytilus chilensis through high-throughput sequencing. Fish Shellfish Immunol. 35, 1899–1905. doi: 10.1016/j.fsi.2013.09.028
Orban E., Di Lena G., Nevigato T., Casini I., Marzetti A., Caproni R. (2002). Seasonal changes in meat content, condition index and chemical composition of mussels (Mytilus galloprovincialis) cultured in two different Italian sites. Food Chem. 77 (1), 57–65. doi: 10.1016/S0308-8146(01)00322-3
Pauletto M., Milan M., Moreira R., Novoa B., Figueras A., Babbucci M., et al. (2014). Deep transcriptome sequencing of pecten maximus hemocytes: a genomic resource for bivalve immunology. Fish Shellfish Immunol. 37, 154–165. doi: 10.1016/j.fsi.2014.01.017
Pertea M., Pertea G. M., Antonescu C. M., Chang T. C., Mendell J. T., Salzberg S. L. (2015). Stringtie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Philipp E. E., Kraemer L., Melzner F., Poustka A. J., Thieme S., Findeisen U., et al. (2012). Massively parallel RNA-sequencing identifies a complex immune gene repertoire in the lophotrochozoan Mytilus edulis. PloS One 7, e33091. doi: 10.1371/journal.pone.0033091
Qian B., Xue L., Huang H. (2016). Liver transcriptome analysis of the Large yellow croaker (Larimichthys crocea) during fasting by using RNA-seq. PloS One 11, e0150240. doi: 10.1371/journal.pone.0150240
Rosani U., Domeneghetti S. (2014). Target capture and massive sequencing of genes transcribed in Mytilus galloprovincialis. BioMed. Res. Int., 538–549. doi: 10.1155/2014/538549
Samokhvalov V., Scott B. A., Crowder C. M. (2008). Autophagy protects against hypoxic injury in elegans. Autophagy 4, 1034–1041. doi: 10.4161/auto.6994
Sciarretta S., Hariharan N., Monden Y., Zablocki D., Sadoshima J. (2011). Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr. Cardiol. 32, 275–281. doi: 10.1007/s00246-010-9855-x
Shi Y., Yu C., Gu Z., Zhan X., Wang Y., Wang A. (2013). Characterization of the pearl oyster (Pinctada martensii) mantle transcriptome unravels biomineralization genes. Mar. Biotechnol. (NY) 15, 175–187. doi: 10.1007/s10126-012-9476-x
Tanguy M., Mckenna P., Gauthier-Clerc S., Pellerin J., Danger J. M., Siah A. (2013a). Functional and molecular responses in Mytilus edulis hemocytes exposed to bacteria, Vibrio splendidus. Dev. Comp. Immunol. 39, 419–429. doi: 10.1016/j.dci.2012.10.015
Tanguy M., Mckenna P., Gauthier-Clerc S., Pellerin J., Danger J. M., Siah A. (2013b). Sequence analysis of a normalized DNA library of Mytilus edulis hemocytes exposed to Vibrio splendidus LGP32 strain. Results Immunol. 3, 40–50. doi: 10.1016/j.rinim.2013.04.001
Tassanakajon A., Rimphanitchayakit V., Visetnan S., Amparyup P., Somboonwiwat K., Charoensapsri W., et al. (2018). Shrimp humoral responses against pathogens: antimicrobial peptides and melanization. Dev. Comp. Immunol. 80, 81–93. doi: 10.1016/j.dci.2017.05.009
Tsukada M., Ohsumi Y. (1993). Isolation and characterization of autophagy defective mutants of saccharomyces cerevisiae. FEBS Lett. 333, 169–174. doi: 10.1016/0014-5793(93)80398-e
Van Der Merwe M., Franchini P., Roodt-Wilding R. (2011). Differential growth-related gene expression in abalone (Haliotis midae). Mar. Biotechnol. (NY) 13, 1125–1139. doi: 10.1007/s10126-011-9376-5
Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J. M., et al. (2003). Mutations in the small GTPase late endosomal protein RAB7 cause charcot marie-tooth type 2b neuropathy. Am. J. Hum. Genet. 72, 722–727. doi: 10.1086/367847
Wang M., Li B., Wang J., Xie S., Zhang L. (2021b). Skin transcriptome and physiological analyses reveal the metabolic and immune responses of yellow catfish (Pelteobagrus fulvidraco) to acute hypoxia. Aquaculture 546, 737277. doi: 10.1016/j.aquaculture.2021.737277
Wang H., Zhang H., Zhong Z., Sun Y., Wang M., Chen H., et al. (2021a). Molecular analyses of the gill symbiosis of the bathymodiolin mussel gigantidas platifrons. iScience 24, 101894. doi: 10.1016/j.isci.2020.101894
Włodarczyk A., Sonakowska L., Kamińska K., Marchewka A., Wilczek G., Wilczek P., et al. (2017). The effect of starvation and refeeding on mitochondrial potential in the midgut of neocaridina davidi (Crustacea, malacostraca). PloS One 12, e0173563. doi: 10.1371/journal.pone.0173563
Xu H., Jiang Y., Miao X. M., Tao Y. X., Xie L. (2021). A model construction of starvation induces hepatic steatosis and transcriptome analysis in zebra fish larvae. Biology (Basel) 10. doi: 10.3390/biology10020092
Zhang S., Han G. D., Dong Y. W. (2014). Temporal patterns of cardiac performance and genes encoding heat shock proteins and metabolic sensors of an intertidal limpet cellana toreuma during sublethal heat stress. J. Therm Biol. 41, 31–37. doi: 10.1016/j.jtherbio.2014.02.003
Zhan Z., Xie X., Cao H., Zhou X., Zhang X. D., Fan H., et al. (2014). Autophagy facilitates TLR4 and TLR3 triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy 10, 257–268. doi: 10.4161/auto.27162
Zhao X., Wang Q., Jiao Y., Huang R., Deng Y., Wang H., et al. (2012a). Identification of genes potentially related to biomineralization and immunity by transcriptome analysis of pearl sac in pearl oyster pinctada martensii. Mar. Biotechnol. (NY) 14, 730–739. doi: 10.1007/s10126-012-9438-3
Zhao X., Yu H., Kong L., Li Q. (2012b). Transcriptomic responses to salinity stress in the pacific oyster Crassostrea gigas. PloS One 7, e46244. doi: 10.1371/journal.pone.0046244
Keywords: Mytilus, starvation, transcriptomes, autophagy, pathway
Citation: Xie B, Chen C-y, Zhang X, Li S-y, Gu Z-q, Huang J, He J-y, Qi P-z, Guo B-y, Liao Z and Yan X (2022) Response of the metabolic and autophagy pathways in Mytilus under starvation. Front. Mar. Sci. 9:1014336. doi: 10.3389/fmars.2022.1014336
Received: 08 August 2022; Accepted: 06 October 2022;
Published: 25 October 2022.
Edited by:
Yunyan Deng, Institute of Oceanology (CAS), ChinaReviewed by:
Mohammad Vatanparast, Johannes Gutenberg University Mainz, GermanyCopyright © 2022 Xie, Chen, Zhang, Li, Gu, Huang, He, Qi, Guo, Liao and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolin Zhang, emhhbmd4aWFvbGluQHpqb3UuZWR1LmNu; Xiaojun Yan, eWFueGpAempvdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.