
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 02 December 2022
Sec. Marine Molecular Biology and Ecology
Volume 9 - 2022 | https://doi.org/10.3389/fmars.2022.1010319
This article is part of the Research TopicMarine Omics in a Changing Ocean: Modelling Molecular Pathways and Networks to Understand Species Acclimation and AdaptationView all 10 articles
 Christopher Powers1
Christopher Powers1 Fatma Gomaa2,3
Fatma Gomaa2,3 Elizabeth B. Billings1
Elizabeth B. Billings1 Daniel R. Utter3
Daniel R. Utter3 David J. Beaudoin2
David J. Beaudoin2 Virginia P. Edgcomb2
Virginia P. Edgcomb2 Colleen M. Hansel4
Colleen M. Hansel4 Scott D. Wankel4
Scott D. Wankel4 Helena L. Filipsson5
Helena L. Filipsson5 Ying Zhang1*
Ying Zhang1* Joan M. Bernhard2*
Joan M. Bernhard2*Certain benthic foraminifera thrive in marine sediments with low or undetectable oxygen. Potential survival avenues used by these supposedly aerobic protists include fermentation and anaerobic respiration, although details on their adaptive mechanisms remain elusive. To better understand the metabolic versatility of foraminifera, we studied two benthic species that thrive in oxygen-depleted marine sediments. Here we detail, via transcriptomics and metatranscriptomics, differential gene expression of Nonionella stella and Bolivina argentea, collected from Santa Barbara Basin, California, USA, in response to varied oxygenation and chemical amendments. Organelle-specific metabolic reconstructions revealed these two species utilize adaptable mitochondrial and peroxisomal metabolism. N. stella, most abundant in anoxia and characterized by lack of food vacuoles and abundance of intracellular lipid droplets, was predicted to couple the putative peroxisomal beta-oxidation and glyoxylate cycle with a versatile electron transport system and a partial TCA cycle. In contrast, B. argentea, most abundant in hypoxia and contains food vacuoles, was predicted to utilize the putative peroxisomal gluconeogenesis and a full TCA cycle but lacks the expression of key beta-oxidation and glyoxylate cycle genes. These metabolic adaptations likely confer ecological success while encountering deoxygenation and expand our understanding of metabolic modifications and interactions between mitochondria and peroxisomes in protists.
Foraminifera are widely distributed across the marine realm, where they can constitute large proportions of the benthos (Bernhard et al., 2000; Gooday et al., 2000). This diverse protistan group is broadly implicated in biogeochemical cycling, where some foraminifera and/or their symbionts perform approximately two-thirds of total denitrification (Risgaard-Petersen et al., 2006; Piña-Ochoa et al., 2010; Bernhard et al., 2012a), with some studies noting all (Choquel et al., 2021). Others are involved in sulfur cycling (Nomaki et al., 2016) and exhibit physiological responses, such as increased adenosine triphosphate (ATP) concentrations, to reactive oxygen species (ROS) (Bernhard and Bowser, 2008). Certain foraminiferan species, while exhibiting lower abundance in oxygenated habitats, thrive in hypoxic or anoxic sediments that are characterized by low oxygen or absence of dissolved oxygen, respectively (Bernhard et al., 1997; Bernhard et al., 2003; Levin et al., 2009).
Robust foraminiferan populations occur across the oxygen gradient within chemoclines. Chemoclines are often coupled with complex gradients of reduction-oxidation chemistry (Reimers et al., 1996) and considerable concentrations of hydrogen sulfide (Bernhard et al., 2003). Reactive oxygen species, such as oxygen radicals and hydrogen peroxide, can be produced in chemoclines via abiotic means such as the interaction of O2 with reduced metals, sulfide, or dissolved organic carbon, or through biotic means mediated by transmembrane NADPH oxidases (Hansel and Diaz, 2021). Certain chemocline-associated foraminifera contain masses of peroxisomes, often correlated with the presence of endoplasmic reticulum (Bernhard and Bowser, 2008). It has been hypothesized that peroxisomes can mediate foraminiferal aerobic respiration through catalase-induced O2 production (Bernhard and Bowser, 2008), but this hypothesis requires further investigation.
Protists inhabiting hypoxic or anoxic environments have been shown to modify the functions of their mitochondria and peroxisomes (Müller et al., 2012; Burki et al., 2013; Stairs et al., 2015; Gawryluk et al., 2016; Verner et al., 2021; Záhonová et al., 2022). The mitochondria related organelles (MROs), reduced forms of mitochondria characterized by the loss of key enzymes involved in oxygen-dependent metabolism, are often specialized for anaerobic energy generation through pyruvate fermentation, hydrogen production, substrate-level phosphorylation, and malate dismutation (Müller et al., 2012; Stairs et al., 2015; Leger et al., 2017), or known to abandon their roles in energy metabolism entirely (Jedelský et al., 2011; Burki et al., 2013; Leger et al., 2017). The metabolism of MROs can be evolutionarily linked to metabolic modifications of peroxisomes. In some obligate anaerobic protists, the peroxisomes are often absent, as in the cases of Giardia and Trichomonas (Gabaldón et al., 2016), or highly modified, as observed in Entamoeba histolytica, Pelomyxa schiedti, and Mastigameoba balamuthi (Le et al., 2020; Verner et al., 2021; Záhonová et al., 2022). Modifications to peroxisomal metabolism in anaerobes often entail loss of the O2-involving catalase and beta-oxidation functions (Le et al., 2020; Verner et al., 2021; Záhonová et al., 2022).
The known metabolic capacity in benthic foraminifera may include denitrification (Risgaard-Petersen et al., 2006; Woehle et al., 2018; Gomaa et al., 2021), ammonium assimilation (LeKieffre et al., 2018b; Gomaa et al., 2021), fermentation (Orsi et al., 2020; Gomaa et al., 2021), and utilization of dissolved organic matter (DeLaca et al., 1981; Orsi et al., 2020). Our recent findings, based on transcriptomic and metatranscriptomic analysis of two foraminiferan species, Nonionella stella and Bolivina argentea, collected from the oxygen-depleted Santa Barbara Basin (SBB), USA, provided insights into metabolic functions related to the survival of these two species in anoxia, such as anaerobic pyruvate metabolism and denitrification (Gomaa et al., 2021). Here, we performed additional analysis on the (meta)transcriptomes of these two species by computationally reconstructing metabolic responses of intracellular organelles to chemical amendments, which has not been elucidated in the two foraminifera.
N. stella and B. argentea inhabit distinct niches within the SBB (Table 1). N. stella is most abundant in severely hypoxic to euxinic (anoxic plus sulfidic) conditions, and is the sole calcareous foraminifer documented to survive long-term anoxia within the basin (Bernhard et al., 1997; Bernhard et al., 2006). N. stella sequesters chloroplasts (Bernhard and Bowser, 1999; Grzymski et al., 2002; Gomaa et al., 2021), likely from prey, proliferates peroxisome-endoplasmic reticulum (P-ER) complexes (Bernhard and Bowser, 2008), and apparently lacks food vacuoles (Gomaa et al., 2021). In contrast, B. argentea is abundant at the basin’s flanks, rarely occurring in the most oxygen-depleted conditions (Bernhard et al., 1997), contains solitary to small groups of peroxisomes, and exhibits evidence of digestion of chloroplasts and other organics (Bernhard et al., 2012a). Given their contrasting physiological features, we hypothesize these two species would demonstrate distinct metabolic responses to variable biogeochemistry, which is known to occur episodically within the SBB (Reimers et al., 1996; Kuwabara et al., 1999; Bernhard et al., 2003). Based on the identification of potential metabolic connectivity between peroxisomes and mitochondria, we illustrate the organelle-specific metabolic responses of N. stella and B. argentea to the addition of hydrogen peroxide (H2O2), nitrate (), and/or hydrogen sulfide (H2S) amendments under anoxia or hypoxia, thereby providing insights into the metabolic adaptations of foraminifera to chemocline biogeochemistry.
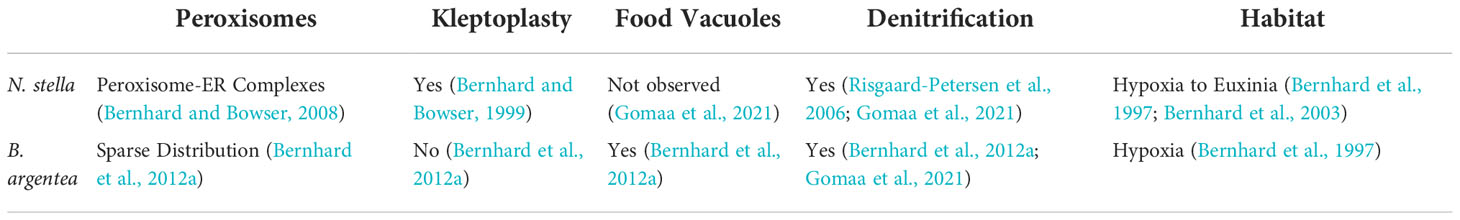
Table 1 Distinctive cytology and ecophysiology of N. stella and B. argentea.
Genes related to the function and biogenesis of peroxisomes were identified in the (meta)transcriptomes of N. stella and B. argentea (Figure 1). Specifically, transcripts were found encoding a transmembrane protein (Pex11) linked to remodeling of the peroxisome membrane for proliferation of peroxisomes (Koch et al., 2010; Williams et al., 2015), transport of fatty acids for peroxisomal beta-oxidation (Mindthoff et al., 2016), and initiation of contact between mitochondria and peroxisomes (Mattiazzi Ušaj et al., 2015). Transcripts of three receptor proteins (i.e. Pex5, Pex7, Pex19) that recognize signals for peroxisomal protein import were also identified (Brown and Baker, 2003). Further, expression of genes encoding AAA ATPases (i.e. Pex1, Pex6) and ubiquitination proteins (i.e. Pex4, Pex2, Pex10) demonstrated activities in the release of peroxisomal imported proteins from signal receptors and the recycling of receptor proteins, respectively (Brown and Baker, 2003). It is notable that transcription of all the Pex genes was observed in N. stella, while transcription of the Pex11 and Pex19 genes was not identified in B. argentea under any treatment (Figure 1; SI Table S1).
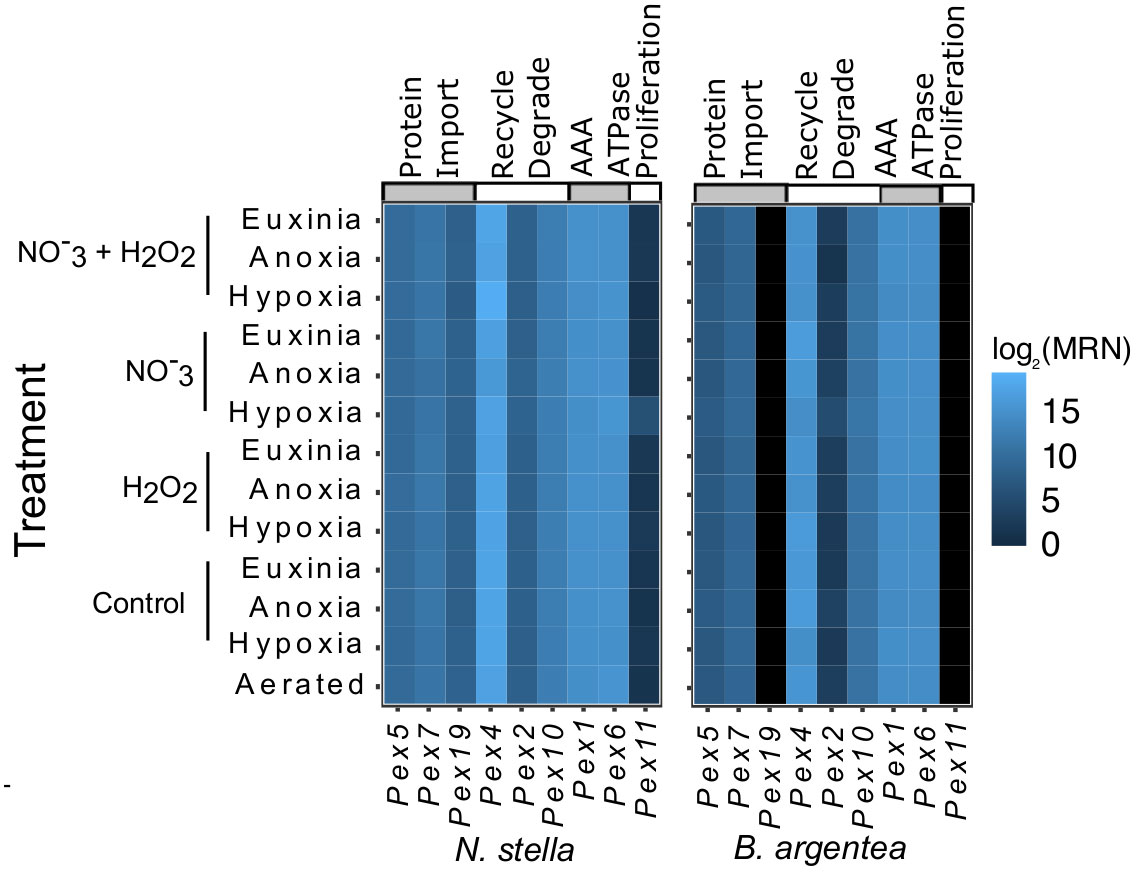
Figure 1 Heatmap presented by species showing the log2 transformed median ratio normalization (MRN) values for all the identified peroxisome biogenesis genes. Black indicates no expression.
A reconstruction of metabolic pathways, based on transcripts identified in the (meta)transcriptomes of N. stella and B. argentea, revealed an overall distinction in the metabolism of the two species (Figure 2). Protein localizations to the peroxisome were predicted based on a consensus of four motif identification procedures for each of the three peroxisome targeting signals, peroxisome targeting signal 1 (PTS1), peroxisome targeting signal 2 (PTS2), and the peroxisomal membrane protein import signal (PMP) (SI Table S2). Protein localizations for mitochondrial functions were predicted based on the consensus of multiple probabilities for mitochondrial localization signals (SI Table S3; Materials and Methods). The resulting localization predictions represent hypothesized final cellular locations based on our bioinformatics approaches and are summarized below.
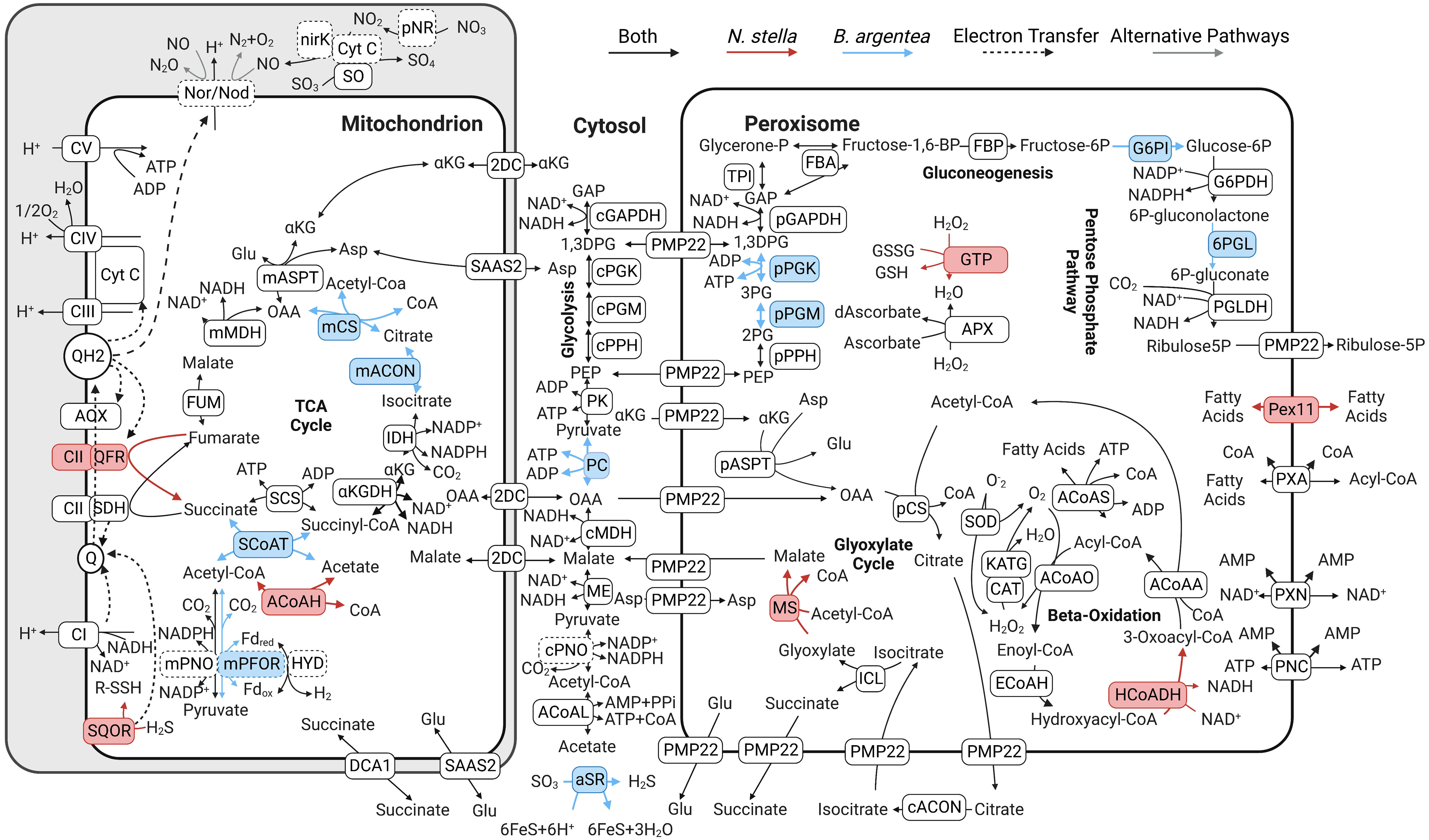
Figure 2 Pathway map of the predicted peroxisomal and mitochondrial metabolic processes in N. stella and B. argentea. Unique genes expressed in N. stella are shown with pink shade with the corresponding metabolic reactions indicated as red arrows. Unique genes expressed in B. argentea are shown with blue shade with the corresponding metabolic reactions indicated as blue arrows. Genes expressed in both species are shown in black boxes with the corresponding metabolic reactions indicated as black arrows. Biochemical conversions between metabolites are annotated with the abbreviation of corresponding genes, as defined in SI Table S1. Boxes with a dotted outline indicate genes that have been described previously (Gomaa et al., 2021). Gray arrows represent alternative pathways for the same genes, specifically with respect to the currently uncertain role of Nor/Nod (Gomaa et al., 2021). Q represents quinones/rhodoquinones and QH2 represents quinols/rhodoquinols. Cyt C represents Cytochrome (C) CI, CII, CIII, CIV, and CV, represent Complex I, II, III, IV, and V, respectively, of the electron transport chain. Dashed arrows indicate the flow of electrons.
Proteins involved in beta-oxidation and the glyoxylate cycle were predicted to localize in the peroxisome. Beta-oxidation, which has not been previously described in foraminifera, is linked to the detoxification of H2O2 or superoxide radicals by catalase/peroxidase (e.g. CAT, KATG) or superoxide dismutase (SOD), respectively. Beta-oxidation breaks down fatty acids for the production of acetyl-CoA, which in turn serves as a precursor metabolite for the glyoxylate cycle, leading to the production of central carbon metabolites, such as malate and oxaloacetate (OAA) (Figure 2). All genes associated with the above-mentioned peroxisomal pathways were expressed in N. stella, while transcripts that encode 3-hydroxyacyl-CoA dehydrogenase (HCoADH) of the beta-oxidation pathway and malate synthase (MS) of the glyoxylate cycle were not identified in B. argentea across all conditions examined (Figure 3). Similarly, genes encoding Pex11, which supports the transport of fatty acids for enabling beta-oxidation in the peroxisomes (Mindthoff et al., 2016), were expressed in N. stella but not in B. argentea (Figure 1).
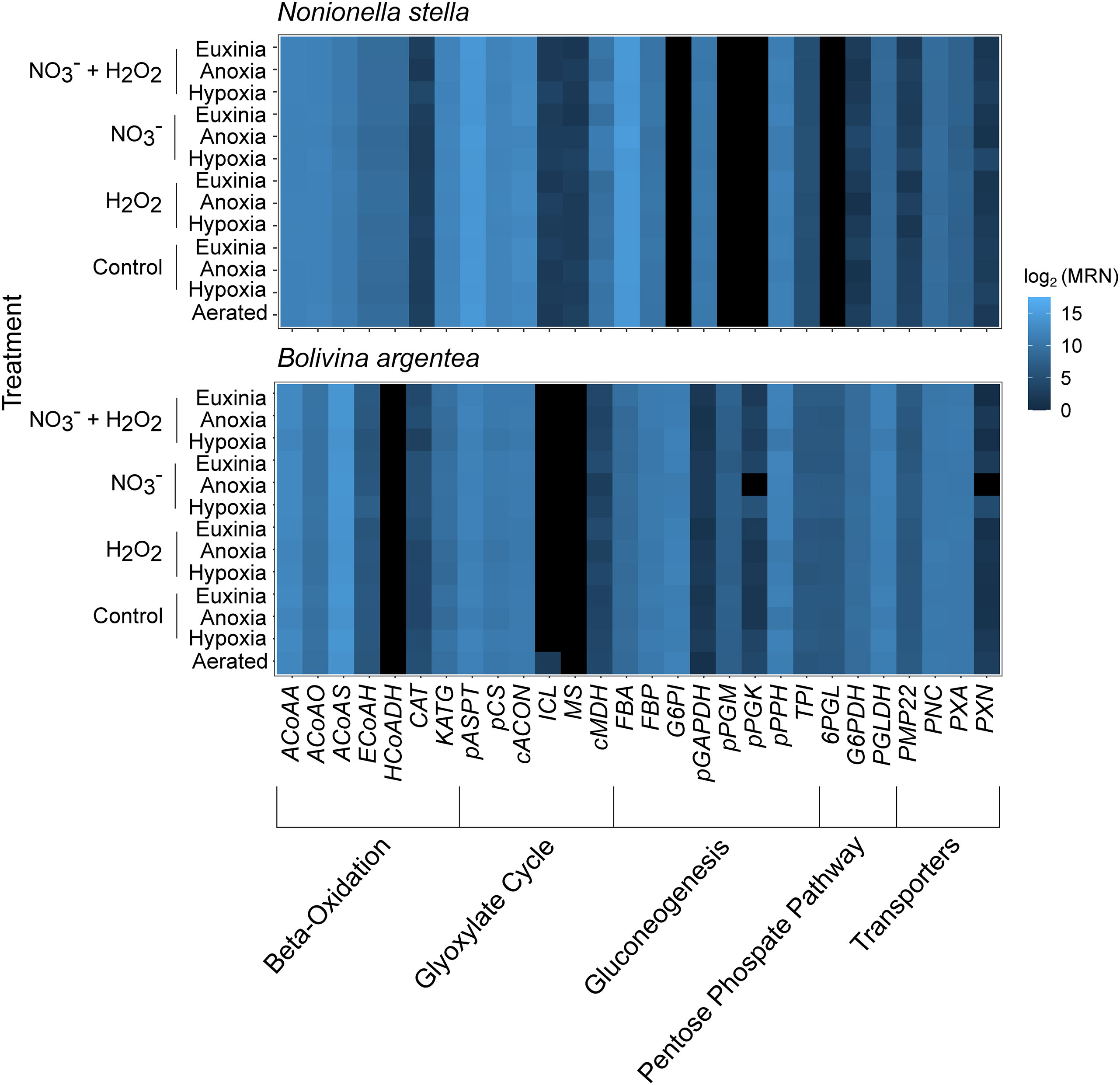
Figure 3 Heatmap showing the log2 transformed median ratio normalization (MRN) values for all genes involved in the peroxisomal pathways of each species. Black indicates no expression. Abbreviations align with those in SI Table S1.
Transcripts encoding fructose bisphosphatase (FBP), a committed step of gluconeogenesis, were identified and predicted to localize in the peroxisome of both N. stella and B. argentea. However, the predicted peroxisomal gluconeogenic pathways in the two species had distinct entry points using metabolites from the cytosol: 1,3-bisphospho-D-glycerate (1,3DPG) for N. stella; phosphoenolpyruvate (PEP) for B. argentea (Figure 2). Similarly, the oxidative pentose phosphate pathway was predicted to localize in the peroxisome of both species, but it appeared to be active in B. argentea but not in N. stella because N. stella lacked expression of genes encoding the peroxisome-localized 6-phosphogluconolactonase (6PGL) or glucose-6-phosphate isomerase (G6PI) (Figures 2, 3).
The prediction of protein localization in mitochondria also revealed distinct transcription of metabolic pathways in N. stella and B. argentea. Expression of genes for mitochondrial substrate-level phosphorylation was observed in both species, with transcripts encoding acetyl-CoA hydrolase (ACoAH) identified in N. stella but not B. argentea, and transcripts encoding succinate:acetate CoA transferase (SCoAT) found in B. argentea but not N. stella. Related to the utilization of acetyl-CoA, genes encoding pyruvate:NADP oxidoreductase (mPNO) were expressed in both species and genes encoding pyruvate:ferredoxin oxidoreductase (mPFOR) were expressed in B. argentea. Further, transcripts encoding the sulfide-oxidizing sulfide:quinone oxidoreductase (SQOR) were detected in N. stella, but not in B. argentea (SI Figure S1). Finally, a complete mitochondrial tricarboxylic acid (TCA) cycle was identified from the (meta)transcriptomes of B. argentea, while only a truncated TCA cycle, from oxaloacetate (OAA) to succinyl-CoA, was detected in the N. stella (meta)transcriptomes (Figure 2).
Core subunits of the electron transport chain (ETC) Complexes I-V were identified in both species. However, variations were observed in the subunit compositions of Complex II (CII) (SI Figure S1). Specifically, characterization of succinate dehydrogenase (SDH) was based on the overall sequence similarity to experimentally verified reference proteins and the presence of an isoleucine and other common variants in the quinone-binding site of the large cytochrome subunit (CII-SDHC) (Zhou et al., 2011). The characterization of quinol:fumarate oxidoreductase (QFR) was based on the presence of a glycine in a key position of the quinone-binding pocket, which indicated an essential modification in the QFR large cytochrome subunit (CII-QFRC) for the binding of rhodoquinone, an electron carrier required for anaerobic energy metabolism through fumarate reduction (Zhou et al., 2011; Del Borrello et al., 2019) (Figure 4A). While transcripts encoding the CII-SDHC were detected in both N. stella and B. argentea, putative transcripts of the CII-QFRC were identified solely in N. stella (Figure 4B). The putative CII-SDH and CII-QFR formed a monophyletic group with other foraminifera (e.g. Rosalina, Reticulomyxa, Sorites, Elphidium, Ammonia). Presence of the same QFR-conserved glycine mutation was observed among other taxa known to exhibit anaerobic metabolism (i.e. Mastigamoeba balamuthi, Blastocystis hominis, and the metazoans Ascaris suum, Caenorhabditis elegans, Ancylostoma ceylanicum) (Figure 4A). The biosynthesis of rhodoquinone was putatively identified in both N. stella and B. argentea based on the detection of transcripts encoding rhodoquinone biosynthesis protein A (RQUA) (Figure 4B), which catalyzes the conversion of ubiquinone to rhodoquinone (Bernert et al., 2019). Multiple sequence alignment revealed a conservation of quinone binding site residues between the RQUA proteins of foraminifera and other eukaryotes that perform fumarate reduction during exposure to anoxia (Figure 4C) (Bernert et al., 2019).
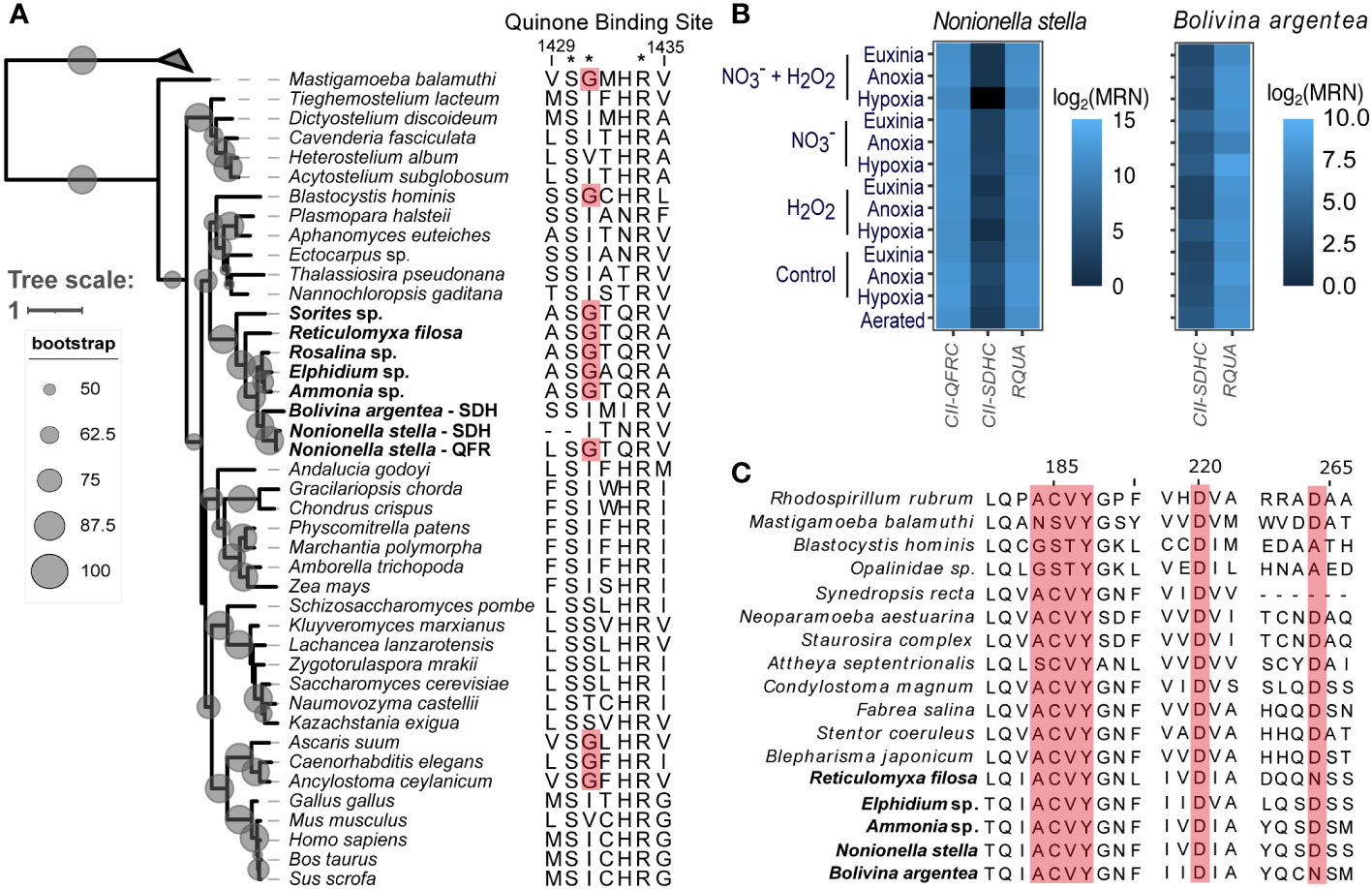
Figure 4 (A) Phylogenetic reconstruction of concatenated protein sequences from Complex II showing phylogenetic placement of the putative CII-QFR and CII-SDH from N. stella and B. argentea, with bootstrapping support values represented as scaled, gray-shaded circles. This phylogeny is rooted using bacterial fumarate reductase sequences. Leaves belonging to foraminifera are highlighted with bold font. The amino acid sequences to the right of the tree shows an alignment of the quinone-binding site in the CII large cytochrome C subunit. Asterisks indicate amino acids implicated in quinone binding. Sites highlighted in red background are QFR-specific mutations associated with the binding of rhodoquinone. (B) Expression of the CII-QFRC, CII-SDHC, and RQUA genes of N. stella and/or B. argentea represented as a heatmap for each species based on the log2 transformed median ratio normalization (MRN) values. (C) Multiple sequence alignment of the RQUA protein from foraminifera and other eukaryotes. Amino acid residues highlighted in red background indicate functional sites in the quinone-binding pocket. Sequences belonging to foraminifera are labeled with bold font.
The mitochondrial electron transport system of both N. stella and B. argentea was complemented by alternative oxidase (AOX), which is known to mediate redox state of the quinone pool (Millenaar et al., 1998), and denitrification proteins (composed of pNR, NirK, and Nor/Nod) (Woehle et al., 2018; Gomaa et al., 2021). The AOX and denitrification genes were constitutively expressed in both foraminifers (SI Table S1).
Proteins that were not predicted to locate in any specific organelles were classified as cytosolic proteins. Transcripts encoding cytosolic proteins, such as malate dehydrogenase (cMDH), malic enzyme (ME), and acetyl-CoA ligase (ACoAL), were identified from the (meta)transcriptomes of both N. stella and B. argentea (Figure 2). Finally, transcripts encoding the cytosolic pyruvate carboxylase (PC) and assimilatory sulfite reductase (aSR) were identified in B. argentea but not in N. stella (Figure 2; SI Table S1).
N. stella responses to the addition of chemical amendments were revealed by the identification of differentially expressed metabolic genes under exposure to H2O2 and , particularly within the anoxic condition (SI Table S4; Figure 5). The H2O2 amendment produced no significant differential expression of genes compared to the anoxia control (without either H2O2 or amendment) or the amendment of both H2O2 and . However, significant differential gene expression was observed between the H2O2 and the only amendment (Figure 5A). Specifically, genes associated with denitrification (pNR, NirK, and Nor/Nod), sulfite oxidation (SO), and Complex V ATP synthase (CV) were up-regulated in the H2O2 amendment. In contrast, genes associated with subunits of Complex II (CII) were down-regulated in H2O2 compared to the amendment. Genes related to several peroxisomal processes, such as the acyl-CoA importer (PXA), beta-oxidation of fatty acids (enoyl-CoA hydratase - ECoAH and 3-hydroxyacyl-CoA dehydrogenase - HCoADH), the glyoxylate cycle (cACON - cytosolic aconitase), gluconeogenesis (FBP), and Pex7, which encodes a receptor for peroxisomal matrix proteins, were also upregulated in the H2O2 amendment. Additional up-regulation of genes was observed among the cytosolic Enolase (cPPH) and glyceraldehyde-3-phosphate dehydrogenase (cGAPDH).
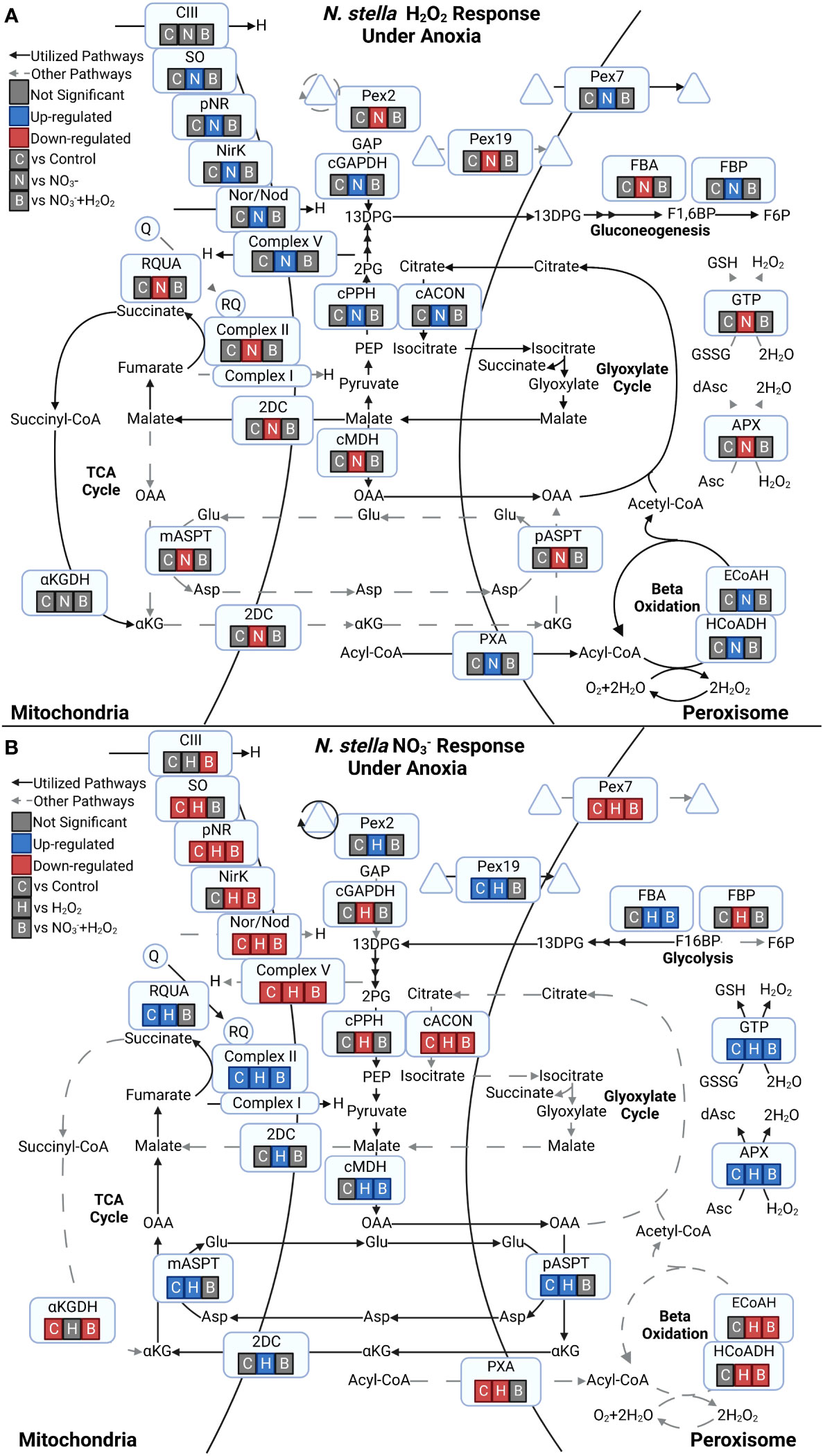
Figure 5 Responses to chemical amendments under anoxia for proteins predicted to the peroxisome and mitochondria in N. stella. (A) Differential expression of genes under the H2O2 amendment in contrast to the control (C), (N), and combined (B) amendments. (B) Differential expression of genes under the amendment in contrast to the control (C), H2O2 (H), and combined (B) amendments. Triangles in both panels represent transported and recycled proteins. Significant differential gene expressions between contrasts (adjusted p-value< 0.05) are highlighted in green (Up-regulated) or red (Down-regulated). Contrasting conditions with no significant differential gene expression are indicated in gray (Not Significant).
The transcriptional responses of N. stella to the addition of under anoxia included the up-regulation of genes related to fumarate reduction (CII and RQUA) and the down-regulation of genes associated with denitrification (pNR, NirK, Nor/Nod) and Complex III (CIII) (Figure 5B). Several genes related to carbon metabolism, including the cytosolic malate dehydrogenase (cMDH), mitochondrial and peroxisomal aspartate transaminase (mASPT and pASPT), mitochondrial 2-oxodicarboxylate carrier (2DC) and peroxisomal glycolysis (fructose-bisphosphate aldolase - FBA), were also up-regulated in the amendment. Lastly, the amendment also stimulated up-regulation of genes associated with peroxisome biogenesis (Pex19 and Pex2) and ROS response (ascorbate peroxidase - APX and glutathione peroxidase - GTP).
N. stella exhibited similar responses to the H2O2 and amendments under hypoxia as compared to anoxia, but with modifications. The amendment in hypoxia induced the up-regulation of CII, CIV, and genes encoding the peroxisome transmembrane protein Pex11 (SI Figure S3A). The combined amendment of both and H2O2 induced the up-regulation of denitrification gene NirK and carbon metabolism genes, such as 2-oxoglutarate dehydrogenase (aKGDH), cGAPDH, and cPPH (SI Figure S3B).
Besides the H2O2 and amendments, differential expression of metabolic genes was observed in N. stella in the presence or absence of oxygen and with or without the addition of H2S. For example, significant up-regulation of Complex IV (CIV) was identified in the aerated treatment compared to the hypoxic, anoxic, and euxinic treatments (SI Figure S2). Similarly, genes related to ROS responses, such as APX, GTP, and catalase-peroxidase (KATG), and the peroxisome biogenesis (Pex19), were up-regulated in the aerated condition (SI Table S4). Interestingly, the addition of H2S to the amendment of anoxia (SI Figure S4) induced a similar response as the H2O2 amendment of anoxia (Figure 5A), with the up-regulation of genes for denitrification (pNR, NirK, Nor/Nod), beta-oxidation (ECoAH and HCoADH), the glyoxylate cycle (cACON), gluconeogenesis (FBP), and peroxisome receptor gene Pex7 in the amendment under euxinia.
In contrast to the diverse transcriptional responses of N. stella to deoxygenation and chemical amendments, B. argentea exhibited up-regulation of genes only under the amendment in hypoxia. Specifically, genes related to the assimilatory sulfite reductase (aSR), glutathione reductase (GTR), and Complex I (CI) were up-regulated in the amendment compared to the hypoxia control and the H2O2 amendment (SI Figure S5). Several carbon metabolism genes, including the mitochondrial mMDH, cytosolic PC, and peroxisomal pPGK, were also upregulated in the amendment compared to the H2O2 amendment, but not in comparison to the hypoxia control (SI Figure S5). Unlike the active regulation of Pex genes in N. stella, no differential expression of the Pex genes was detected in B. argentea under the examined conditions.
Microbial eukaryotes have evolved variable combinations of carbon and energy metabolism in their organelles consistent with their ecological specialization, such as modifications observed in mitochondria (Shiflett and Johnson, 2010; Müller et al., 2012) and peroxisomes (Müller et al., 1968; Michels et al., 2005). Our (meta)transcriptomic analysis of two cytologically and ecophysiologically distinct foraminifera from the SBB, N. stella and B. argentea (Table 1), revealed contrasting utilization of these organelles. As expected from the peroxisome-ER complexes observed in previous cytological analyses (Bernhard and Bowser, 2008), our analysis showed N. stella actively expressed a wide range of genes regulating the function and proliferation of peroxisomes (Figure 1). However, B. argentea lacked the expression of key genes related to peroxisome protein localization (Pex19) and metabolite transport and peroxisome proliferation (Pex11), indicating reduced peroxisomal activities consistent with the sparse distribution of peroxisomes in B. argentea (Table 1).
The identification of transcripts encoding beta-oxidation and the glyoxylate cycle revealed that N. stella may use intracellular lipid droplets as a primary carbon source. This is consistent with the dearth of observed food vacuoles and the abundance of lipid droplets in N. stella (Gomaa et al., 2021) and in closely related species, such as Nonionellina labradorica (LeKieffre et al., 2018a). The predicted integration of beta-oxidation and the glyoxylate cycle in N. stella peroxisomes is similar to the glyoxysomes in vascular plants, which are specialized peroxisomes that break down fatty acids to support central carbon metabolism and are linked to the metabolism of mitochondria in germinating seeds (Eastmond and Graham, 2001; Ma et al., 2016). In contrast, B. argentea lacked the expression of key genes in beta-oxidation and the glyoxylate cycle (Figures 2, 3).
B. argentea instead were predicted to employ gluconeogenesis and the oxidative pentose phosphate pathway in the peroxisome (Figures 2, 3), indicating an active central carbon metabolism likely driven by digestion of phagocytosed organic materials. The predicted peroxisomal localization of gluconeogenesis in B. argentea could allow the simultaneous carbon metabolism in both the glycolytic and the gluconeogenic directions. This may serve as an alternative to allosteric regulation that prevents toxic accumulation of metabolic intermediates (Haanstra et al., 2008; Gualdrón-López et al., 2012), similar to that which may occur in the glycosomes described in diplonemids (Morales et al., 2016).
Based on predictions of protein localizations, the categorization of N. stella and B. argentea peroxisomes into a glyoxysome-like and a glycosome-like metabolism, respectively, is consistent with their different cytological traits and ecological distributions (Table 1). The predicted localization of the glyoxylate cycle to the peroxisomes of N. stella may represent an adaptation of protistan peroxisomes to use H2O2 for lipid metabolism in the SBB chemocline. The predicted compartmentalization of glycolysis and gluconeogenesis in B. argentea suggests this adaptation could be widespread in microeukaryotes and may reflect convergent evolution associated with hypoxia tolerance (Morales et al., 2016; Škodová-Sveráková et al., 2021).
The electron transport system of B. argentea and N. stella appeared to be an integration of oxygen-dependent and nitrate-dependent metabolisms, likely reflecting adaptations to hypoxic and/or anoxic environments. Diverse electron transport machinery in the mitochondria, including denitrification (Gomaa et al., 2021), alternative oxidase (AOX), and sulfide:quinone oxidoreductase (SQOR), was identified among the gene transcripts in either or both foraminifera (Figure 2; SI Figure S1). Genes encoding SQOR have been reported before in other foraminifers, such as Reticulomyxa filosa, Ammonia sp., and Elphidium margaritaceum (Stairs et al., 2018). The presence of enzymes associated with denitrification has been discussed in depth for both N. stella and B. argentea (Gomaa et al., 2021), as well as in other foraminifers (Woehle et al., 2018; Orsi et al., 2020). Additionally, the AOX contributes to redox balance by serving as an alternative path to the proton-pumping machinery (Complex III/IV) of the ETC (Millenaar et al., 1998), alleviating potential ROS or reactive nitrogen species (RNS) stress caused by the intracellular production of H2O2, oxygen radicals, and/or nitric oxide from electron leakage of the ETC (Zhao et al., 2019).
Despite oxygen being a common terminal electron acceptor for AOX, genes encoding this protein were expressed across all treatments, including both the anoxic and euxinic conditions (Orsi et al., 2020). Constitutive expression of this gene could have resulted from adaptations to rapid fluctuations of oxygen (Chaudhuri et al., 2006), endogenously sourced oxygen hypothetically produced by catalases in the anoxic treatment (Bernhard and Bowser, 2008) or oxygenic denitrification by Nor/Nod (Gomaa et al., 2021). Interestingly, recent evidence has shown that nitrite can serve as an alternative electron acceptor for AOX under hypoxia and anoxia in some plants (Vishwakarma et al., 2018), suggesting a potential mechanism for the activation of foraminiferan AOX under anoxia that deserves dedicated biochemical investigation. The observation of SQOR transcripts solely in N. stella indicates its capacity to use H2S as an electron donor to fuel respiration. The SQOR contributes to a reduced quinol pool that can be used by Nor/Nod to drive denitrification. This is corroborated by the upregulation of denitrification genes in N. stella with amendment under euxinia compared to anoxia (SI Figure S4). The expansion of N. stella’s electron transport system to utilize H2S as an alternative electron donor reflects an adaptive feature for the conservation of carbon sources in N. stella, which may be carbon limited as this SBB population of N. stella lacks evidence of digestion (Gomaa et al., 2021) and utilizes a truncated TCA cycle (Figure 2).
Quinol:fumarate oxidoreductase (QFR) is commonly considered an adaptive mechanism for anaerobic respiration in eukaryotic organisms under anoxic conditions (Del Borrello et al., 2019). We found constitutive expression of genes encoding QFR solely in N. stella (Figure 4B), suggesting potential adaptations of this species to an anaerobic lifestyle. The conservation of QFR based on the presence of a glycine residue in the large cytochrome subunit of foraminifers (Figure 4A) indicates an ancestral state of this taxon that may have evolved in an anoxic milieu. In N. stella, transcripts encoding RQUA of the rhodoquinone biosynthesis pathway further supports the potential presence of fumarate reduction (Stairs et al., 2018; Salinas et al., 2020). Interestingly, transcripts of succinate dehydrogenase (SDH) were observed in both N. stella and B. argentea. The SDH formed a monophyletic group with the QFR, suggesting that both the QFR and SDH may have originated from a common foraminiferal ancestor capable of fumarate reduction. Given the prevalence of QFR in other foraminifera and the expression of the RQUA gene in B. argentea, a QFR could be encoded in the B. argentea genome, although it was not detected in the (meta)transcriptomes represented in this study.
B. argentea additionally demonstrated potential capacities for substrate-level phosphorylation through the coupling of succinate:acetate CoA transferase (SCoAT) and succinyl-CoA synthetase (SCS). Genes encoding SCS and SCoAT have been previously identified in related species (Orsi et al., 2020), but this is the first documentation of the expression of the SCoAT gene in foraminifera. Carbon conservation was inferred from the predicted mitochondrial PFOR, the cytosolic PNO, and the cytosolic acetyl-CoA ligase (ACoAL), another gene previously not documented in foraminifera (Figure 2). The utilization of substrate-level phosphorylation and the constitutive expression of denitrification pathways by B. argentea under hypoxia indicated the potential lifestyle of a facultative anaerobe. The up-regulation of aSR and GTR by B. argentea in hypoxia amendment (SI Figure S5) was consistent with an increased tolerance of ROS stress for maintaining active carbon metabolism (Gill et al., 2013; Wang et al., 2016).
In contrast, N. stella demonstrated a wider range of metabolic responses to the H2O2, , and H2S amendments under both hypoxia and anoxia by using a combination of peroxisomal and mitochondrial pathways. The H2O2 amendment induced the up-regulation of genes associated with beta-oxidation and the glyoxylate cycle in N. stella, likely linked to the utilization of intracellular lipid droplets that are abundant in this species (Gomaa et al., 2021). The release of carbon substrates by beta-oxidation and the glyoxylate cycle appears to be coupled with carbon storage via gluconeogenesis (i.e. up-regulation of FBP) and a more active electron transport system, based on the up-regulation of the denitrification and SO genes (Figure 5A). The H2O2 amendment also stimulated up-regulation of ATP-producing Complex V genes, which was consistent with the experimentally measured increases in intracellular ATP production of N. stella with the addition of H2O2 (Bernhard and Bowser, 2008). The metabolism of N. stella under the H2O2 amendment may reflect its native metabolic state in the SBB sediments, as no differential gene expression was identified between the H2O2 amendments under anoxia or hypoxia versus their corresponding control (without amendment) conditions (Figure 5A; SI Table S4).
The amendment of N. stella induced differential gene expression compared to both the control and the H2O2 amendment. The down-regulation of FBP and the up-regulation of FBA in anoxia amendment may indicate the activation of carbon metabolism in the glycolytic direction, which can be an adaptive mechanism in response to the lack of carbon substrates due to down-regulation of the beta-oxidation (Figure 5B). The up-regulation of both RQUA and Complex II genes indicates the potential activation of fumarate reduction. This is coordinated with the down-regulation of denitrification genes and likely reflects a metabolism limited by electron donors, as denitrification competes with fumarate reduction for the quinone pool. Further evidence for limited availability of electron donors was observed under the anoxic amendment in comparison to the euxinic amendment. With H2S as an alternative electron donor via SQOR, N. stella showed an up-regulation of genes involved in denitrification, beta-oxidation, the glyoxylate cycle, and gluconeogenesis (SI Figure S4). This reflects an adaptive feature for the conservation of carbon sources in N. stella, which may be carbon limited as this species lacks evidence for digestion through the presence of food vacuoles (Gomaa et al., 2021). Lastly, the up-regulation of ROS response genes (e.g. APX, GTP) in the amendment of N. stella likely indicates cellular mitigation of the effects of electron leakage during fumarate reduction (Gawryluk and Stairs, 2021).
The evolution of mitochondria and peroxisomes are often coupled in eukaryotes (Le et al., 2020; Verner et al., 2021; Záhonová et al., 2022) and represents a spectrum of metabolic phenotypes (Müller et al., 2012; Stairs et al., 2015). Combinations of metabolic pathways known in MROs were predicted to localize in the mitochondria of N. stella and B. argentea (Figure 2); however, mitochondria of these foraminifers still maintain the oxygen-dependent ETC (CIII, CIV, AOX) (Figure 2) and may perform anaerobic respiration through denitrification (Gomaa et al., 2021). These unique metabolic capabilities have not been identified in any other protists and may allow our two benthic foraminifera to survive in both oxygen depleted and aerated conditions. The organelle-specific metabolic flexibility documented in this study offers a contrast to the mitochondrial metabolism of conventional MROs found in anaerobic protists. These differences suggest that the mitochondria of these two benthic foraminifera have evolved specialized metabolic tools that support their ability to respond to varied oxygen concentrations and milieu chemistry. The plasticity observed in the mitochondria was enhanced by the peroxisomes in both species, which are metabolically distinct not only from each other, but also from known anaerobic peroxisomes (Le et al., 2020; Verner et al., 2021; Záhonová et al., 2022). The selective pressures associated with survival in highly variable habitats have conferred our two foraminifera with diverse metabolic toolkits of mitochondrial and peroxisomal metabolism that enables survival in SBB chemoclines.
Overall, the two foraminifers demonstrated distinct metabolic adaptations to hypoxia and anoxia, and had different capacities to use compounds often abundant in SBB pore water (Risgaard-Petersen et al., 2006; Gomaa et al., 2021). This indicates benthic foraminiferal metabolism is not uniform across species. While the ability to perform denitrification and anaerobic energy metabolism are established for N. stella (Risgaard-Petersen et al., 2006; Gomaa et al., 2021), we show here that N. stella of the SBB are adept at surviving anoxia, having metabolically flexible mitochondria that function in coordination with the predicted metabolism of its peroxisomes. The metabolism predicted for B. argentea suggested the capacity to digest food vacuole contents as a facultative anaerobe. These species represent only two examples of the unique cytologies that occur along the SBB chemocline. Additional cytological diversity is known among other foraminifera, such as Bulminella tenuata, an endobiont-bearing species that also exhibits close associations between mitochondria and peroxisomes (Bernhard and Bowser, 2008), or an as-yet undescribed agglutinated foraminifer with coccoid endosymbionts that lacks peroxisome-ER complexes in favor of few peroxisomes (Bernhard et al., 2006; Bernhard et al., 2012b). The highly variable cytology observed among foraminifera of the SBB chemocline and elsewhere, such as hydrocarbon seeps (Bernhard et al., 2010), implies potential distinctions in their metabolic capacity, which may influence biogeochemical cycling and calcite biomineralization, as the presence of symbionts likely shifts δ13Ccalcite values in one foraminifera species (Bernhard, 2003). Further understanding the highly variable metabolic adaptations that can occur across foraminifera will allow for more informed reconstructions of paleoceanographic conditions and strengthen biogeochemical and ecological predictions of shifts in organismal and metabolic niches under future ocean deoxygenation. Lastly, these findings provide additional insights into the biogeochemical roles and the evolution of this ecologically significant protistan taxon that diversified in the Neoproterozoic.
Transcriptome and metatranscriptome data were obtained from the NCBI BioProject PRJNA714124. Details of the experimental settings were described in (Gomaa et al., 2021). Briefly, surface sediments containing either N. stella or B. argentea of the Santa Barbara Basin were subjected to laboratory treatments under four oxygen conditions (i.e. aerated, hypoxic, anoxic, and euxinic) and four chemical amendments: the no amendment (Control), nitrate (), hydrogen peroxide (H2O2), nitrate and hydrogen peroxide (), with 2-3 replicates in ~75 cm2 polypropylene containers for each species given each combination of the oxygen and amendment conditions. Following a treatment period of three days, N. stella or B. argentea were picked into pools of 25 (cleaned) conspecifics, and each pool was counted as a biological replicate for transcriptomic or metatranscriptomic sequencing using the PolyA- or Total RNA-based library preparation, respectively (Gomaa et al., 2021). Quality and integrity of each RNA sample were assessed with Nanodrop and Bioanalyzer (Agilent 2100), only samples with nanodrop values of 260:280 and 260:230 higher than 1.8 and 2.0, respectively, and Bioanalyzer RNA integrity numbers above 5 were selected for library preparation. Bar-coded library samples were quantified with Bioanalyzer (Agilent 2100) and TapeStation (Agilent 2200), and each sample was amplified with 11 to 15 rounds of PCR cycles in preparation for sequencing. Paired-end sequencing was performed using two sequencing runs on an Illumina NovaSeq platform for the polyA selected mRNA library and two sequencing runs on an Illumina NextSeq platform for the total RNA library, resulting in approximately 8 billion and 1.9 billion reads, respectively, from the two platforms. Overall, between 3-7 biological replicates per amendment/treatment were analyzed, with each biological replicate represented by pooling all sequences from its two sequencing runs. Detailed information of all samples referenced in this study and their corresponding data in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database are summarized in SI Table S5.
The N. stella and B. argentea transcriptomes and metatranscriptomes were quality checked and processed as described previously (Gomaa et al., 2021). Briefly, reads were quality checked with TrimGalore (Krueger, 2015), a wrapper script built around Cutadapt (Martin, 2011) and FastQC (Andrews, 2010). Each (meta)transcriptome was assembled individually using the de novo transcriptome assembly functions in rnaSPAdes v3.14.1 with default parameters (Bushmanova et al., 2019).
Due to the large size of the nucleotide dataset (greater than fifty million contigs), the lin-clust clustering algorithm from MMSEQS2 was used to cluster nucleotide sequences with a minimum sequence identity of 97% using default settings (Steinegger and Söding, 2018). Coding sequences (CDS) within a representative contig from each cluster were identified using geneMarkS-T with default settings (Tang et al., 2015) and the protein sequences encoded by the resulting CDS were further clustered using the Cluster Database at High Identity with Tolerance (CD-HIT) (Fu et al., 2012) with a minimum sequence identity of 99% and 95% to identify representative sequences for further annotations and taxonomic assignments, respectively. Representative protein sequences from the 99% CD-HIT clustering were annotated with EggNOG V.2 (Huerta-Cepas et al., 2019) to assign enzyme commission (EC) numbers using default settings. The EggNOG annotation did not capture all subunits of mitochondrial electron transport chain complexes. Therefore, targeted identification of these subunits was performed by examining the presence and organization of protein domains based on matches to hidden markov models (HMM) from the Pfam 27.0 database (Finn et al., 2014) using HMMER 3.1 (Eddy, 2011) with an independent e-value under 1E-5. Genes identified as missing or as expressed in only one species were further searched for using a homology-based approach by mapping reference proteins against the assembled contigs using tblastn (Zhang et al., 2000). Three sets of protein sequences were used as the reference database for this search: foraminifera proteins identified from the automated prediction of coding sequences for genes that were missing in one species, sequences of the ETC complexes from the Protein Data Bank (PDB IDs: SRFR, 3VRB, 1NTZ, and 5B3S for CII, CII, CIII, and CIV respectively), and the RQUA sequence from Reticulomyxa filosa (Accession: ETO28810.1). Sequences that shared 60% identity and 70% coverage to the reference proteins and were not already identified in the CDS predictions were added to the list of annotated genes. Two RQUA transcripts found through this search were included in the analysis despite sharing a sequence identity of 47.6% and 45.5% to the RQUA sequence from Reticulomyxa filosa based on the identification of binding sites known for RQUA (Figure 4C).
Representative protein sequences from the 95% CD-HIT clustering were mapped against the NCBI non-redundant protein database using the DIAMOND BLASTP (Buchfink et al., 2021). If a sequence had an alignment to foraminifera out of the top 25 best hits, it was taxonomically assigned to the foraminifera group. If a representative protein sequence had a majority of the top 25 hits assigned to diatoms, other eukaryotes, bacteria, or archaea, the sequence was taxonomically assigned based on the majority mapping.
Peroxisome localization was predicted using a consensus approach that combined motif-based identification of targeting signals with homology-based search of well-curated peroxisomal proteins (SI Figure S6). The Pex5, Pex7, and Pex19 genes were identified in the foraminiferal (meta)transcriptomes and the Pex5, Pex7, and Pex19 proteins recognize peroxisome targeting signal 1 (PTS1), peroxisome targeting signal 2 (PTS2), and peroxisomal membrane protein import signal (PMP), respectively. Correspondingly, these three signals were used to predict peroxisomal localization. The predictions were informed by a consensus of four strategies (SI Figure S6) using tools from the MEME Suite, version 5.1.1 (Bailey et al., 2009). All four strategies used the FIMO software (Grant et al., 2011) to scan translated proteins from the foraminifera transcriptome, using custom MEME motifs derived from different sets of putative peroxisomal proteins (SI Table S6; SI Figure S6). Strategy one used the MEME motif constructed from whole sequences that showed exact matches to the canonical PTS1 (Rucktäschel et al., 2011), PTS2 (Petriv et al., 2004), or PMP (Rottensteiner et al., 2004) signals, Strategy two was similarly initiated based on the exact matches of canonical motifs, but made site-specific constructions using Sites2Meme (Bailey et al., 2009). Strategy three used reference motif sequences from the peroxisomeDB to construct the MEME motif profiles using Sites2Meme. Strategy four aligned the peroxisomeDB reference sequences using MAFFT version 7.453 (Katoh et al., 2002) and converted them to the MEME motif using Sites2Meme. Each protein sequence identified from this pipeline was assigned a consensus score from one to four for each of the peroxisomal import signals by counting the number of strategies that resulted in a motif match to the target sequence. Confidence levels of peroxisome localization prediction were evaluated based on the number of strategies that support the motif-based mapping and a homology search to the peroxisomeDB reference database, a database of peroxisomal proteins from diverse organisms, including protists (Schlüter et al., 2010), using DIAMOND BLASTP (Buchfink et al., 2021) (SI Table S7).
Mitochondrion localization was predicted based on homology and a consensus of three predictors: MitoFates version 1.2, TargetP version 2.0, and PredSL (Petsalaki et al., 2006; Fukasawa et al., 2015; Armenteros et al., 2019), combinations of which have been utilized with success in putatively anaerobic protists previously (Eme et al., 2017; Stairs et al., 2021). Combined probabilities of mitochondrion localization were calculated by multiplying the probabilities from all three predictors to provide an aggregated estimation. In parallel, DIAMOND BLASTP (Buchfink et al., 2021) was used to identify homology from our assembly to a reference database consisting of mitochondrial proteins from OrganelleDB (Wiwatwattana et al., 2007), Mitocarta (Calvo et al., 2016), the Acanthomoeba castellanii mitochondrial proteome (Gawryluk et al., 2014), the Andalucia godoyi proteome (Gray et al., 2020), and mitochondrial sequences from the plant proteome database (Sun et al., 2009). Confidence levels were assigned to the putative mitochondrial proteins by integrating the combined probability of localization and the homology identifications to the reference mitochondrial protein database (SI Table S7).
Taxonomic assignments were additionally used for the filtering of peroxisomal and mitochondrial pathways. Only proteins taxonomically assigned to foraminifera or other eukaryotes were included in the metabolic reconstruction. A number of metabolic processes were placed to mitochondria because they represent known or proposed genes in mitochondria. These included subunits of the electron transport chain complexes and mitochondrial transporters (SI Table S1) identified through homology to the above-mentioned mitochondrial protein reference database and the transporter classification database (TCDB) (Saier et al., 2021) using DIAMOND BLASTP with a sequence identity cutoff of at least 30% and a reference coverage cutoff of at least 50%.
Similarly, some proteins were assigned to the peroxisome because they represent peroxisomal-relevant processes and/or were shown to co-localize with other peroxisomal proteins, including Pex proteins, transmembrane transporters, catalase, peroxidase, and isocitrate lyase (SI Table S1). Pex proteins were identified based on the EggNOG annotations using default settings and/or homology searches to the peroxisomeDB using DIAMOND BLASTP (Buchfink et al., 2021) with a sequence identity of at least 30% and a reference coverage of at least 50%. The presence and organization of protein domains in the Pex proteins were analyzed based on matches to HMMs from the Pfam 27.0 database (Finn et al., 2014) using HMMER 3.1 (Eddy, 2011) with an e-value under 1E-5. Transport of small soluble metabolites such as malate and succinate across the peroxisomal membrane was included in our prediction, as the transport of small metabolites is commonly mediated by a variety of non-selective channels through which these compounds can freely diffuse (Antonenkov and Hiltunen, 2012). Specific transporters for cofactors such as NAD, CoA, ATP, acyl-CoA, and fatty acids were identified through homology searches to known peroxisomal transporters from the Swiss-Prot database, release-2021_01 (Boeckmann et al., 2003), and the peroxisomeDB using DIAMOND BLASTP (Buchfink et al., 2021) with a sequence identity cutoff of at least 30% and a reference coverage cutoff of at least 50%. The catalase and peroxidase proteins are commonly known to localize in the peroxisome. Further, the peroxisomal localization of catalase in N. stella is seen through the presence of the crystalline cores formed by the catalase aggregates in its peroxisomes (Bernhard and Bowser, 2008). Isocitrate lyase was included because previous research showed that this protein can be localized without import signals to the peroxisome by co-importing as oligomeric proteins (Lee et al., 1997). The isocitrate lyases in our data are also homologous to the isocitrate lyases in the peroxisomeDB. Catalases are known to have a modified peroxisomal import signal, which may explain why they were not identified in our analysis (Oshima et al., 2008). While little support was found for the localization of the denitrification proteins (pNR, NirK, and Nor/Nod) to the mitochondria, its involvement in respiration may require an association with the mitochondria. This was proposed in previous studies by our group and others (Woehle et al., 2018; Gomaa et al., 2021), so these proteins were included in our reconstruction of mitochondrial metabolism.
Proteins not localized to peroxisome or mitochondrion were classified as cytosolic and were included in the metabolic reconstruction for the completion of central carbon metabolism. Metabolic connections among the peroxisomal, mitochondrial, and cytosolic compartments were examined by tracing the carbon flow in the metabolic network using the findprimarypairs function in the PSAMM software package version 1.0 (Steffensen et al., 2016).
Binding sites relevant to the function of Complex II were analyzed based on multiple sequence alignments (MSA), generated using the PROfile Multiple Alignment with predicted Local Structures and 3D constraints (PROMALS3D) (Pei et al., 2008). Each subunit of Complex II was aligned to reference sequences from the protein data bank, marine microbial eukaryotic transcriptome sequencing project (MMETSP) (Keeling et al., 2014), Swiss-Prot (Boeckmann et al., 2003), NCBI RefSeq non-redundant protein database (O’Leary et al., 2016), and from eukaryotes known to live in anoxic conditions (Gawryluk and Stairs, 2021). Three-dimensional protein structures of the Complex II from Ascaris suum (PDB ID 3VR8), Sus scrofa (PDB ID 3FSD), and Gallus gallus (PDB ID 2H89) were used in PROMALS3D (Pei et al., 2008) to guide the MSA construction for each subunit. Alignments of the four core subunits (CII-SDHA, CII-SDHB, CII-SDHC/QFRC, CII-SDHD) were concatenated and used as input for the phylogenetic reconstruction using RAxML version 8.2 (Stamatakis, 2014). A concatenated alignment of all subunits resulted in an alignment with 39.28% gaps and 1677 distinct alignment positions. No trimming was performed on the MSA. An optimal substitution model was selected by RAxML using the automatic model selection under Akaike Information Criterion (AIC) (Burnham and Anderson, 2007). The best model, LG substitution with the GAMMA model of rate heterogeneity and empirical base frequencies, was applied for the phylogenetic reconstruction of Complex II using 100 bootstraps. Specific amino acid residues relevant to the functions of QFR and SDH were identified following previous studies on the quinone-binding pocket of these proteins (Del Borrello et al., 2019).
Differentially expressed genes were identified using representative transcripts from the assembly after clustering at 99% minimum amino acid sequence identity (described above). Quality controlled sequencing reads from each sample were mapped to these representative transcripts using BBMap from the BBTools package (Bushnell, 2014). Briefly, read mapping files from each sample were sorted, indexed with the SAMtools package version 1.7 (Danecek et al., 2021), and used for the calculation of transcript abundance using the Pileup function in BBTools version 37.36 (Bushnell, 2014). The species-specific mapping of transcripts was achieved by counting the abundance of a transcript across all treatments of a species. A denoising step was applied to remove sporadic mappings of reads by exclusively assigning each transcript to a species (i.e. N. stella or B. argentea) if the read mapping was dominated (e.g. with a 10 times higher abundance) by samples from that species. If a transcript received a similar number of mapped reads from both species, the transcript was assigned to both species if greater than 60 reads were mapped across all samples of each species. Transcripts mapped to less than 60 reads in both species were removed from further consideration, as having less than 60 reads would on average represent only around one read mapped per sample in each species. Count data were summed for all transcripts mapped to the same species and annotated as the same gene, and this combined count data were used as inputs for the differential gene expression analysis using DESeq2 version 1.26.0 in R (Love et al., 2014). Each combination of chemical amendments and oxygen conditions was used as an experimental treatment for the modeling of differential gene expression. Additionally, a variable was introduced to the statistical model with two factor levels to control for the potential variation introduced from the use of two different library preparation techniques: (1) polyA selection that targets the polyadenylated mRNA from eukaryotes, and (2) total RNA preparation that targets all mRNA molecules from eukaryotes and prokaryotes (Gomaa et al., 2021). Statistical significance was based on the Wald test by estimating variation in log-fold change (LFC) using differences between experimental treatments. Results from the Wald test were further considered if the two sets of samples represented the same oxygenation treatment but different chemical amendment, or if they represented the same chemical amendment but different oxygenation treatments. For example, the non-amended hypoxic treatment and the amended hypoxic treatment reflected a comparison of chemical-driven responses. In contrast, the amended hypoxic treatment and the amended anoxic treatment reflected a comparison of oxygen-driven responses. A significance threshold less than 0.05 was used on the adjusted p-values based on a Benjamin Hochberg correction for false discovery rates. The expression of genes in a treatment was calculated by taking the mean of the median ratio normalization (MRN) values across all biological replicates of a given treatment followed by log2 transformations. Heatmaps were created based on the log2 transformed MRN values using ggplot2, version 3.3.2 (Ginestet, 2011). Differential expression of genes was similarly plotted using ggplot2.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA714124. Assemblies, annotations, and abundance mapping of the (meta)transcriptomes can be accessed following doi: 10.6084/m9.figshare.16632259.
JB, YZ, CH, and VE conceived the study. JB, DB, CH, SW, and HF contributed to sample collection. FG, JB, and DB performed the experiment. CP, YZ, FG, EB, DU, SW, CH, and JB analyzed and interpreted the data. CP, YZ, FG, and JB composed the manuscript. All authors gave feedback on the manuscript.
This project was funded by the U.S. National Science Foundation IOS 1557430 and 1557566. CP acknowledges partial support from the U.S. National Science Foundation DBI 1553211. EB acknowledges support from the URI MARC U*STAR grant from the National Institute of General Medical Sciences of the National Institutes of Health under grant number T34GM131948 (Niall G. Howlett, PI). HF acknowledges support from the Swedish Research Council VR (grant number 2017-04190). JB acknowledges partial support from the Investment in Science Program at WHOI.
The authors acknowledge that this work has appeared previously online as a preprint at biorxiv (DOI: https://doi.org/10.1101/2022.07.20.500910). We thank the captain, crew, and science parties of the R/V Robert Gordon Sproul cruises SP1703, SP1718, and SP1811. Figures 2, 5, S3, S4, and S5 were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.1010319/full#supplementary-material
Andrews S. (2010) FastQC: a quality control tool for high throughput sequence data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Antonenkov V. D., Hiltunen J. K. (2012). Transfer of metabolites across the peroxisomal membrane. Biochim. Biophys. Acta 1822, 1374–1386. doi: 10.1016/j.bbadis.2011.12.011
Armenteros J. J. A., Salvatore M., Emanuelsson O., Winther O., Von Heijne G., Elofsson A., et al. (2019). Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2:e201900429. doi: 10.26508/lsa.201900429
Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bernert A. C., Jacobs E. J., Reinl S. R., Choi C. C. Y., Roberts Buceta P. M., Culver J. C., et al. (2019). Recombinant RquA catalyzes the in vivo conversion of ubiquinone to rhodoquinone in Escherichia coli and Saccharomyces cerevisiae. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 1226–1234. doi: 10.1016/j.bbalip.2019.05.007
Bernhard J. M. (2003). Potential symbionts in bathyal foraminifera. Science 299, 861. doi: 10.1126/science.1077314
Bernhard J. M., Bowser S. S. (1999). Benthic foraminifera of dysoxic sediments: chloroplast sequestration and functional morphology. Earth-Sci. Rev. 46, 149–165. doi: 10.1016/S0012-8252(99)00017-3
Bernhard J. M., Bowser S. S. (2008). Peroxisome proliferation in foraminifera inhabiting the chemocline: an adaptation to reactive oxygen species exposure? J. Eukaryot. Microbiol. 55, 135–144. doi: 10.1111/j.1550-7408.2008.00318.x
Bernhard J. M., Buck K. R., Farmer M. A., Bowser S. S. (2000). The Santa Barbara basin is a symbiosis oasis. Nature 403, 77–80. doi: 10.1038/47476
Bernhard J. M., Casciotti K. L., McIlvin M. R., Beaudoin D. J., Visscher P. T., Edgcomb V. P. (2012a). Potential importance of physiologically diverse benthic foraminifera in sedimentary nitrate storage and respiration. J. Geophys. Res. 117:G03002. doi: 10.1029/2012jg001949
Bernhard J. M., Edgcomb V. P., Casciotti K. L., McIlvin M. R., Beaudoin D. J. (2012b). Denitrification likely catalyzed by endobionts in an allogromiid foraminifer. ISME J. 6, 951–960. doi: 10.1038/ismej.2011.171
Bernhard J. M., Habura A., Bowser S. S. (2006). An endobiont-bearing allogromiid from the Santa Barbara basin: Implications for the early diversification of foraminifera. J. Geophys. Res. 111:G03002. doi: 10.1029/2005jg000158
Bernhard J. M., Martin J. B., Rathburn A. E. (2010). Combined carbonate carbon isotopic and cellular ultrastructural studies of individual benthic foraminifera: 2. toward an understanding of apparent disequilibrium in hydrocarbon seeps. Paleoceanography 25:PA4206. doi: 10.1029/2010pa001930
Bernhard J. M., Sen Gupta B. K., Borne P. F. (1997). Benthic foraminiferal proxy to estimate dysoxic bottom-water oxygen concentrations; Santa Barbara basin, US pacific continental margin. J. Foraminiferal Res. 27, 301–310. doi: 10.2113/gsjfr.27.4.301
Bernhard J. M., Visscher P. T., Bowser S. S. (2003). Submillimeter life positions of bacteria, protists, and metazoans in laminated sediments of the Santa Barbara basin. Limnol. Oceanogr. 48, 813–828. doi: 10.4319/lo.2003.48.2.0813
Boeckmann B., Bairoch A., Apweiler R., Blatter M.-C., Estreicher A., Gasteiger E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Brown L.-A., Baker A. (2003). Peroxisome biogenesis and the role of protein import. J. Cell. Mol. Med. 7, 388–400. doi: 10.1111/j.1582-4934.2003.tb00241.x
Buchfink B., Reuter K., Drost H.-G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Burki F., Corradi N., Sierra R., Pawlowski J., Meyer G. R., Abbott C. L., et al. (2013). Phylogenomics of the intracellular parasite Mikrocytos mackini reveals evidence for a mitosome in rhizaria. Curr. Biol. 23, 1541–1547. doi: 10.1016/j.cub.2013.06.033
Burnham K. P., Anderson D. R. (2007). Model selection and multimodel inference: A practical information-theoretic approach (New York, NY, USA: Springer Science & Business Media).
Bushmanova E., Antipov D., Lapidus A., Prjibelski A. D. (2019). rnaSPAdes: A de novo transcriptome assembler and its application to RNA-seq data. Gigascience 8:giz100. doi: 10.1093/gigascience/giz100
Bushnell B. (2014) BBTools software package. Available at: http://sourceforge.net/projects/bbmap.
Calvo S. E., Clauser K. R., Mootha V. K. (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257. doi: 10.1093/nar/gkv1003
Chaudhuri M., Ott R. D., Hill G. C. (2006). Trypanosome alternative oxidase: from molecule to function. Trends Parasitol. 22, 484–491. doi: 10.1016/j.pt.2006.08.007
Choquel C., Geslin E., Metzger E., Filipsson H. L., Risgaard-Petersen N., Launeau P., et al. (2021). Denitrification by benthic foraminifera and their contribution to n-loss from a fjord environment. Biogeosciences 18, 327–341. doi: 10.5194/bg-18-327-2021
Danecek P., Bonfield J. K., Liddle J., Marshall J., Ohan V., Pollard M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10:giab008. doi: 10.1093/gigascience/giab008
DeLaca T. E., Karl D. M., Lipps J. H. (1981). Direct use of dissolved organic carbon by agglutinated benthic foraminifera. Nature 289, 287–289. doi: 10.1038/289287a0
Del Borrello S., Lautens M., Dolan K., Tan J. H., Davie T., Schertzberg M. R., et al. (2019). Rhodoquinone biosynthesis in C. elegans requires precursors generated by the kynurenine pathway. Elife 8:e48165. doi: 10.7554/eLife.48165
Eastmond P. J., Graham I. A. (2001). Re-examining the role of the glyoxylate cycle in oilseeds. Trends Plant Sci. 6, 72–78. doi: 10.1016/S1360-1385(00)01835-5
Eddy S. R. (2011). Accelerated profile HMM searches. PloS Comput. Biol. 7, e1002195. doi: 10.1371/journal.pcbi.1002195
Eme L., Gentekaki E., Curtis B., Archibald J. M., Roger A. J. (2017). Lateral gene transfer in the adaptation of the anaerobic parasite Blastocystis to the gut. Curr. Biol. 27, 807–820. doi: 10.1016/j.cub.2017.02.003
Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fukasawa Y., Tsuji J., Fu S.-C., Tomii K., Horton P., Imai K. (2015). MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol. Cell. Proteomics 14, 1113–1126. doi: 10.1074/mcp.M114.043083
Fu L., Niu B., Zhu Z., Wu S., Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gabaldón T., Ginger M. L., Michels P. A. M. (2016). Peroxisomes in parasitic protists. Mol. Biochem. Parasitol. 209, 35–45. doi: 10.1016/j.molbiopara.2016.02.005
Gawryluk R. M. R., Chisholm K. A., Pinto D. M., Gray M. W. (2014). Compositional complexity of the mitochondrial proteome of a unicellular eukaryote (Acanthamoeba castellanii, supergroup amoebozoa) rivals that of animals, fungi, and plants. Proteomics J. 109, 400–416. doi: 10.1016/j.jprot.2014.07.005
Gawryluk R. M. R., Kamikawa R., Stairs C. W., Silberman J. D., Brown M. W., Roger A. J. (2016). The earliest stages of mitochondrial adaptation to low oxygen revealed in a novel rhizarian. Curr. Biol. 26, 2729–2738. doi: 10.1016/j.cub.2016.08.025
Gawryluk R. M. R., Stairs C. W. (2021). Diversity of electron transport chains in anaerobic protists. Biochim. Biophys. Acta Bioenerg. 1862, 148334. doi: 10.1016/j.bbabio.2020.148334
Gill S. S., Anjum N. A., Hasanuzzaman M., Gill R., Trivedi D. K., Ahmad I., et al. (2013). Glutathione and glutathione reductase: a boon in disguise for plant abiotic stress defense operations. Plant Physiol. Biochem. 70, 204–212. doi: 10.1016/j.plaphy.2013.05.032
Ginestet C. (2011). ggplot2: elegant graphics for data analysis. J. R Stat. Soc. Ser. A Stat. Soc. 174, 245–245. doi: 10.1111/j.1467-985X.2010.00676_9.x
Gomaa F., Utter D. R., Powers C., Beaudoin D. J., Edgcomb V. P., Filipsson H. L., et al. (2021). Multiple integrated metabolic strategies allow foraminiferan protists to thrive in anoxic marine sediments. Sci. Adv. 7:eabf1586. doi: 10.1126/sciadv.abf1586
Gooday A. J., Bernhard J. M., Levin L. A., Suhr S. B. (2000). Foraminifera in the Arabian Sea oxygen minimum zone and other oxygen-deficient settings: taxonomic composition, diversity, and relation to metazoan faunas. Deep-Sea Res. II: Top. Stud. Oceanogr. 47, 25–54. doi: 10.1016/s0967-0645(99)00099-5
Grant C. E., Bailey T. L., Noble W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064
Gray M. W., Burger G., Derelle R., Klimeš V., Leger M. M., Sarrasin M., et al. (2020). The draft nuclear genome sequence and predicted mitochondrial proteome of Andalucia godoyi, a protist with the most gene-rich and bacteria-like mitochondrial genome. BMC Biol. 18. doi: 10.1186/s12915-020-0741-6
Grzymski J., Schofield O. M., Falkowski P. G., Bernhard J. M. (2002). The function of plastids in the deep-sea benthic foraminifer, Nonionella stella. Limnol. Oceanogr. 47, 1569–1580. doi: 10.4319/lo.2002.47.6.1569
Gualdrón-López M., Brennand A., Hannaert V., Quiñones W., Cáceres A. J., Bringaud F., et al. (2012). When, how and why glycolysis became compartmentalised in the kinetoplastea. a new look at an ancient organelle. Int. J. Parasitol. 42, 1–20. doi: 10.1016/j.ijpara.2011.10.007
Haanstra J. R., van Tuijl A., Kessler P., Reijnders W., Michels P. A. M., Westerhoff H. V., et al. (2008). Compartmentation prevents a lethal turbo-explosion of glycolysis in trypanosomes. PNAS 105, 17718–17723. doi: 10.1073/pnas.0806664105
Hansel C. M., Diaz J. M. (2021). Production of extracellular reactive oxygen species by marine biota. Ann. Rev. Mar. Sci. 13, 177–200. doi: 10.1146/annurev-marine-041320-102550
Huerta-Cepas J., Szklarczyk D., Heller D., Hernández-Plaza A., Forslund S. K., Cook H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 doi: doi: 10.1093/nar/gky1085.
Jedelský P. L., Doležal P., Rada P., Pyrih J., Šmíd O., Hrdý I., et al. (2011). The minimal proteome in the reduced mitochondrion of the parasitic protist Giardia intestinalis. PloS One 6, e17285. doi: 10.1371/journal.pone.0017285
Katoh K., Misawa K., Kuma K.-I., Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Keeling P. J., Burki F., Wilcox H. M., Allam B., Allen E. E., Amaral-Zettler L. A., et al. (2014). The marine microbial eukaryote transcriptome sequencing project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PloS Biol. 12, e1001889. doi: 10.1371/journal.pbio.1001889
Koch J., Pranjic K., Huber A., Ellinger A., Hartig A., Kragler F., et al. (2010). PEX11 family members are membrane elongation factors that coordinate peroxisome proliferation and maintenance. J. Cell Sci. 123, 3389–3400. doi: 10.1242/jcs.064907
Krueger F. (2015). Trim galore. a wrapper tool around cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files 516, 517. Available at: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
Kuwabara J. S., van Geen A., McCorkle D. C., Bernhard J. M. (1999). Dissolved sulfide distributions in the water column and sediment pore waters of the Santa Barbara basin. Geochim. Cosmochim. Acta 63, 2199–2209. doi: 10.1016/s0016-7037(99)00084-8
Lee M. S., Mullen R. T., Trelease R. N. (1997). Oilseed isocitrate lyases lacking their essential type 1 peroxisomal targeting signal are piggybacked to glyoxysomes. Plant Cell 9, 185–197 doi: doi: 10.1105/tpc.9.2.185.
Leger M. M., Kolisko M., Kamikawa R., Stairs C. W., Kume K., Čepička I., et al. (2017). Organelles that illuminate the origins of Trichomonas hydrogenosomes and Giardia mitosomes. Nat. Ecol. Evol. 1:0092. doi: 10.1038/s41559-017-0092
LeKieffre C., Bernhard J. M., Mabilleau G., Filipsson H. L., Meibom A., Geslin E. (2018a). An overview of cellular ultrastructure in benthic foraminifera: New observations of rotalid species in the context of existing literature. Mar. Micropaleontol. 138, 12–32. doi: 10.1016/j.marmicro.2017.10.005
LeKieffre C., Jauffrais T., Geslin E., Jesus B., Bernhard J. M., Giovani M.-E., et al. (2018b). Inorganic carbon and nitrogen assimilation in cellular compartments of a benthic kleptoplastic foraminifer. Sci. Rep. 8:10140. doi: 10.1038/s41598-018-28455-1
Levin L. A., Ekau W., Gooday A. J., Jorissen F., Middelburg J. J., Naqvi S. W. A., et al. (2009). Effects of natural and human-induced hypoxia on coastal benthos. Biogeosciences 6, 2063–2098. doi: 10.5194/bg-6-2063-2009
Le T., Žárský V., Nývltová E., Rada P., Harant K., Vancová M., et al. (2020). Anaerobic peroxisomes in Mastigamoeba balamuthi. PNAS 117, 2065–2075. doi: 10.1073/pnas.1909755117
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Ma Z., Marsolais F., Bernards M. A., Sumarah M. W., Bykova N. V., Igamberdiev A. U. (2016). Glyoxylate cycle and metabolism of organic acids in the scutellum of barley seeds during germination. Plant Sci. 248, 37–44. doi: 10.1016/j.plantsci.2016.04.007
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. doi: 10.14806/ej.17.1.200
Mattiazzi Ušaj M., Brložnik M., Kaferle P., Žitnik M., Wolinski H., Leitner F., et al. (2015). Genome-wide localization study of yeast Pex11 identifies peroxisome–mitochondria interactions through the ERMES complex. J. Mol. Biol. 427, 2072–2087. doi: 10.1016/j.jmb.2015.03.004
Michels P. A. M., Moyersoen J., Krazy H., Galland N., Herman M., Hannaert V. (2005). Peroxisomes, glyoxysomes and glycosomes (review). Mol. Membr. Biol. 22, 133–145. doi: 10.1080/09687860400024186
Millenaar F. F., Benschop J. J., Wagner A. M., Lambers H. (1998). The role of the alternative oxidase in stabilizing the in vivo reduction state of the ubiquinone pool and the activation state of the alternative oxidase. Plant Physiol. 118, 599–607. doi: 10.1104/pp.118.2.599
Mindthoff S., Grunau S., Steinfort L. L., Girzalsky W., Hiltunen J. K., Erdmann R., et al. (2016). Peroxisomal Pex11 is a pore-forming protein homologous to TRPM channels. Biochim. Biophys. Acta 1863, 271–283. doi: 10.1016/j.bbamcr.2015.11.013
Morales J., Hashimoto M., Williams T. A., Hirawake-Mogi H., Makiuchi T., Tsubouchi A., et al. (2016). Differential remodelling of peroxisome function underpins the environmental and metabolic adaptability of diplonemids and kinetoplastids. Proc. Biol. Sci. 283:20160520. doi: 10.1098/rspb.2016.0520
Müller M., Hogg J. F., De Duve C. (1968). Distribution of tricarboxylic acid cycle enzymes and glyoxylate cycle enzymes between mitochondria and peroxisomes in Tetrahymena pyriformis. J. Biol. Chem. 243, 5385–5395. doi: 10.1016/S0021-9258(18)91961-7
Müller M., Mentel M., van Hellemond J. J., Henze K., Woehle C., Gould S. B., et al. (2012). Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 76, 444–495. doi: 10.1128/MMBR.05024-11
Nomaki H., Bernhard J. M., Ishida A., Tsuchiya M., Uematsu K., Tame A., et al. (2016). Intracellular isotope localization in ammonia sp. (Foraminifera) of oxygen-depleted environments: Results of nitrate and sulfate labeling experiments. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00163
O’Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Orsi W. D., Morard R., Vuillemin A., Eitel M., Wörheide G., Milucka J., et al. (2020). Anaerobic metabolism of foraminifera thriving below the seafloor. ISME J. 14, 2580–2594. doi: 10.1038/s41396-020-0708-1
Oshima Y., Kamigaki A., Nakamori C., Mano S., Hayashi M., Nishimura M., et al. (2008). Plant catalase is imported into peroxisomes by Pex5p but is distinct from typical PTS1 import. Plant Cell Physiol. 49, 671–677. doi: 10.1093/pcp/pcn038
Pei J., Kim B.-H., Grishin N. V. (2008). PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300. doi: 10.1093/nar/gkn072
Petriv O. I., Tang L., Titorenko V. I., Rachubinski R. A. (2004). A new definition for the consensus sequence of the peroxisome targeting signal type 2. J. Mol. Biol. 341, 119–134. doi: 10.1016/j.jmb.2004.05.064
Petsalaki E. I., Bagos P. G., Litou Z. I., Hamodrakas S. J. (2006). PredSL: a tool for the n-terminal sequence-based prediction of protein subcellular localization. Genomics Proteomics Bioinf. 4, 48–55. doi: 10.1016/S1672-0229(06)60016-8
Piña-Ochoa E., Høgslund S., Geslin E., Cedhagen T., Revsbech N. P., Nielsen L. P., et al. (2010). Widespread occurrence of nitrate storage and denitrification among foraminifera and gromiida. PNAS 107, 1148–1153. doi: 10.1073/pnas.0908440107
Reimers C. E., Ruttenberg K. C., Canfield D. E., Christiansen M. B., Martin J. B. (1996). Porewater pH and authigenic phases formed in the uppermost sediments of the Santa Barbara basin. Geochim. Cosmochim. Acta 60, 4037–4057. doi: 10.1016/S0016-7037(96)00231-1
Risgaard-Petersen N., Langezaal A. M., Ingvardsen S., Schmid M. C., Jetten M. S. M., Op den Camp H. J. M., et al. (2006). Evidence for complete denitrification in a benthic foraminifer. Nature 443, 93–96. doi: 10.1038/nature05070
Rottensteiner H., Kramer A., Lorenzen S., Stein K., Landgraf C., Volkmer-Engert R., et al. (2004). Peroxisomal membrane proteins contain common Pex19p-binding sites that are an integral part of their targeting signals. Mol. Biol. Cell 15, 3406–3417. doi: 10.1091/mbc.e04-03-0188
Rucktäschel R., Girzalsky W., Erdmann R. (2011). Protein import machineries of peroxisomes. Biochim. Biophys. Acta 1808, 892–900. doi: 10.1016/j.bbamem.2010.07.020
Saier M. H., Reddy V. S., Moreno-Hagelsieb G., Hendargo K. J., Zhang Y., Iddamsetty V., et al. (2021). The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 49, D461–D467. doi: 10.1093/nar/gkaa1004
Salinas G., Langelaan D. N., Shepherd J. N. (2020). Rhodoquinone in bacteria and animals: Two distinct pathways for biosynthesis of this key electron transporter used in anaerobic bioenergetics. Biochim. Biophys. Acta Bioenerg. 1861, 148278. doi: 10.1016/j.bbabio.2020.148278
Schlüter A., Real-Chicharro A., Gabaldón T., Sánchez-Jiménez F., Pujol A. (2010). PeroxisomeDB 2.0: an integrative view of the global peroxisomal metabolome. Nucleic Acids Res. 38, D800–D805 doi: doi: 10.1093/nar/gkp935.
Shiflett A. M., Johnson P. J. (2010). Mitochondrion-related organelles in eukaryotic protists. Annu. Rev. Microbiol. 64, 409–429. doi: 10.1146/annurev.micro.62.081307.162826
Škodová-Sveráková I., Záhonová K., Juricová V., Danchenko M., Moos M., Baráth P., et al. (2021). Highly flexible metabolism of the marine euglenozoan protist Diplonema papillatum. BMC Biol. 19, 251. doi: 10.1186/s12915-021-01186-y
Stairs C. W., Eme L., Muñoz-Gómez S. A., Cohen A., Dellaire G., Shepherd J. N., et al. (2018). Microbial eukaryotes have adapted to hypoxia by horizontal acquisitions of a gene involved in rhodoquinone biosynthesis. Elife 7:e34292. doi: 10.7554/eLife.34292
Stairs C. W., Leger M. M., Roger A. J. (2015). Diversity and origins of anaerobic metabolism in mitochondria and related organelles. Philos. Trans. R. Soc Lond. B Biol. Sci. 370, 20140326. doi: 10.1098/rstb.2014.0326
Stairs C. W., Táborský P., Salomaki E. D., Kolisko M., Pánek T., Eme L., et al. (2021). Anaeramoebae are a divergent lineage of eukaryotes that shed light on the transition from anaerobic mitochondria to hydrogenosomes. Curr. Biol. 31, 5605–5612.e5. doi: 10.1016/j.cub.2021.10.010
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steffensen J. L., Dufault-Thompson K., Zhang Y. (2016). PSAMM: A portable system for the analysis of metabolic models. PloS Comput. Biol. 12, e1004732. doi: 10.1371/journal.pcbi.1004732
Steinegger M., Söding J. (2018). Clustering huge protein sequence sets in linear time. Nat. Commun. 9, 2542. doi: 10.1038/s41467-018-04964-5
Sun Q., Zybailov B., Majeran W., Friso G., Olinares P. D. B., van Wijk K. J. (2009). PPDB, the plant proteomics database at Cornell. Nucleic Acids Res. 37, D969–D974. doi: 10.1093/nar/gkn654
Tang S., Lomsadze A., Borodovsky M. (2015). Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43, e78. doi: 10.1093/nar/gkv227
Verner Z., Žárský V., Le T., Narayanasamy R. K., Rada P., Rozbeský D., et al. (2021). Anaerobic peroxisomes in Entamoeba histolytica metabolize myo-inositol. PloS Pathog. 17, e1010041. doi: 10.1371/journal.ppat.1010041
Vishwakarma A., Kumari A., Mur L. A. J., Gupta K. J. (2018). A discrete role for alternative oxidase under hypoxia to increase nitric oxide and drive energy production. Free Radic. Biol. Med. 122, 40–51. doi: 10.1016/j.freeradbiomed.2018.03.045
Wang M., Jia Y., Xu Z., Xia Z. (2016). Impairment of sulfite reductase decreases oxidative stress tolerance in Arabidopsis thaliana. Front. Plant Sci. 7, 1843. doi: 10.3389/fpls.2016.01843
Williams C., Opalinski L., Landgraf C., Costello J., Schrader M., Krikken A. M., et al. (2015). The membrane remodeling protein Pex11p activates the GTPase Dnm1p during peroxisomal fission. Proc. Natl. Acad. Sci. U. S. A. 112, 6377–6382. doi: 10.1073/pnas.1418736112
Wiwatwattana N., Landau C. M., Cope G. J., Harp G. A., Kumar A. (2007). Organelle DB: an updated resource of eukaryotic protein localization and function. Nucleic Acids Res. 35, D810–D814. doi: 10.1093/nar/gkl1000
Woehle C., Roy A.-S., Glock N., Wein T., Weissenbach J., Rosenstiel P., et al. (2018). A novel eukaryotic denitrification pathway in foraminifera. Curr. Biol. 28, 2536–2543.e5. doi: 10.1016/j.cub.2018.06.027
Záhonová K., Treitli S. C., Le T., Škodová-Sveráková I., Hanousková P., Čepička I., et al. (2022). Anaerobic derivates of mitochondria and peroxisomes in the free-living amoeba Pelomyxa schiedti revealed by single-cell genomics. BMC Biol. 20. doi: 10.1186/s12915-022-01247-w
Zhang Z., Schwartz S., Wagner L., Miller W. (2000). A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214. doi: 10.1089/10665270050081478
Zhao R.-Z., Jiang S., Zhang L., Yu Z.-B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 44, 3–15. doi: 10.3892/ijmm.2019.4188
Keywords: protists, mitochondria, peroxisomes, chemocline, anoxia, benthic foraminifera, Santa Barbara Basin
Citation: Powers C, Gomaa F, Billings EB, Utter DR, Beaudoin DJ, Edgcomb VP, Hansel CM, Wankel SD, Filipsson HL, Zhang Y and Bernhard JM (2022) Two canonically aerobic foraminifera express distinct peroxisomal and mitochondrial metabolisms. Front. Mar. Sci. 9:1010319. doi: 10.3389/fmars.2022.1010319
Received: 02 August 2022; Accepted: 09 November 2022;
Published: 02 December 2022.
Edited by:
Carolina Madeira, NOVA University Lisbon, PortugalReviewed by:
Michael Ginger, University of Huddersfield, United KingdomCopyright © 2022 Powers, Gomaa, Billings, Utter, Beaudoin, Edgcomb, Hansel, Wankel, Filipsson, Zhang and Bernhard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhang, eWluZ3poYW5nQHVyaS5lZHU=; Joan M. Bernhard, amJlcm5oYXJkQHdob2kuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.