 Na Yao
Na Yao Yaqun Zhang
Yaqun Zhang Yangzhen Li
Yangzhen Li Yuanri Hu
Yuanri Hu Hengde Li
Hengde Li- 1National Demonstration Center for Experimental Fisheries Science Education, Shanghai Ocean University, Shanghai, China
- 2Key Laboratory of Aquatic Genomics, Ministry of Agriculture and Rural Affairs, Chinese Academy of Fishery Sciences, Beijing, China
- 3Yellow Sea Fishery Research Institute, Chinese Academy of Fishery Sciences, Qingdao, China
Chinese tongue sole (Cynoglossus semilaevis) is an economically important marine fish in China. Generally, the eyeless side of the Chinese tongue sole is white and the side with eyes is brown after metamorphosis, hypermelanosis may still occur in the eyeless side in certain individuals after metamorphosis, which greatly decreases consumer acceptance and market price. In order to study the possibility of genetic improvement, we determined genomic markers in Chinese tongue sole using the genotyping-by-sequencing method and analyzed their association with hypermelanosis area. Genetic analysis showed that hypermelanosis was a complicated quantitative trait, and the estimated heritability for hypermelanosis incidence and area ratio were 0.16 and 0.21, respectively. Genomic selection analysis showed that selection based on hypermelanosis incidence and area ratio had similar reliabilities and prediction accuracies, indicating the feasibility of genetic improvement. Nine loci were significantly associated with hypermelanosis, few of which included genes or flanked genes potentially associated with skin disease, indicating the potential complicated genetic mechanisms underlying hypermelanosis in the Chinese tongue sole.
1 Introduction
The Chinese tongue sole (Cynoglossus semilaevis) is an economically important marine flatfish found in the coastal waters of the Yellow Sea and Bohai Sea regions of China (Dou, 1995; Gibson, 1997). Chinese tongue sole is largely popular owing to its taste and is widely cultivated owing to its fast growth rate. Usually, the eyeless side of the Chinese tongue sole is white and the side with eyes is brown after metamorphosis. Larval pigmentation is symmetrically distributed in the larval stage, during which the eye side presents a uniform distribution in the metamorphosis stage, leading to the appearance of a black brown-colored eye side. Normally, larval pigmentation on the eyeless side completely disappears, resulting in the eyeless side appearing pure white. However, pigmentation still occurs in the eyeless side in certain individuals after metamorphosis (Estevez et al., 2001; Zhu et al., 2005; Amiya et al., 2008; Yamada et al., 2011; Isojima et al., 2013a; Matsuda et al., 2018). In Chinese tongue soles, a relatively high proportion (10–90%) of adult melanin is locally transformed into adult melanin, which results in irregular black patches of skin on the eyeless side (Li et al., 2021). Usually, hypermelanosis in the eyeless side starts in the area under the gills of the tongue sole. Hypermelanosis in the eyeless side also occurs in other flatfish; however, the location of pigmentation varies. For example, pigmentation usually starts in the caudal peduncle area in Japanese flounder (Paralichthys olivaceus) (Isojima et al., 2013b; Kang et al., 2014).
Hypermelanosis greatly affects consumer acceptance of flatfish in the market, and the price of Chinese tongue sole showing hypermelanosis is at least 20% lower than that of fish with normal skin. To meet market demand, farmers intend to cultivate fish without hypermelanosis in the eyeless side. Studies have explored the causes of hypermelanosis in flatfish, including Chinese tongue sole and Japanese flounder, and have attempted to reduce the development of hypermelanosis by changing the background color of the breeding pond, adjusting the light period and intensity, or adjusting the feeding time (Stickney and White, 1975; Isojima et al., 2014; Nakata et al., 2017; Yamanome et al., 2005). However, these approaches diminish or alleviate the development of hypermelanosis in the eyeless side in flatfish to a certain extent and do not completely inhibit development. Apart from the environment, physiological and genetic factors are also thought to be related to hypermelanosis (Kang and Kim, 2015; Takahashi et al., 2004; Zhang et al., 2021; Li et al., 2022). Pedigree analysis revealed that the variation in hypermelanosis is heritable in the Chinese tongue sole, indicating the potential to alleviate or eliminate the occurrence of hypermelanosis via genetic improvement (Li et al., 2021).
Genetic analysis using genomic markers has gradually become the default method in plant and animal breeding owing to its high prediction accuracy (Morris et al., 2011; Zhang et al., 2013; Feng et al., 2016; Martin et al., 2016; Owens et al., 2019) and ability to identify genetic markers underlying the traits of interest. However, so far, no report is found to study the genetic mechanism of hypermelanosis of Chinese tongue sole using genomic markers. In this study, genomic markers of Chinese tongue sole were determined using the genotyping-by-sequencing (GBS) method and their association with hypermelanosis incidence and area were analyzed. The feasibility of genomic selection for avoiding hypermelanosis was investigated which will benefit future breeding in tongue sole aquaculture.
2 Materials and methods
2.1 Materials and phenotyping
A total of 320 one-year-old Chinese tongue soles were sampled from Caofeidian, Hebei Province, China, in August 2021, of which half of the sample population had hypermelanosis on the eyeless side and the other half had normal skin. Tongue soles with hypermelanosis were photographed on the eyeless side, and their images were processed using Adobe Photoshop 2021 to obtain the pixel values of the pigmented parts and whole eyeless side. The ratios of pigmented area to the entire eyeless area, which was a continuous trait, were recorded. Whether a fish had hypermelanosis was a binary trait, the fish with hypermelanosis were recoded as 1 and the others with normal skin were recoded as 0. Caudal fins of all individuals were sampled and preserved in anhydrous ethanol for DNA extraction.
2.2 Genotyping
DNA was extracted using a TIANamp Marine Animals DNA Kit (Tiangen Biotech Co., Ltd, Beijing), digested using enzymes Pst1 (NEB, Beijing) and Msp1 (NEB, Beijing) simultaneously at 37°C for 2 h, and then ligated with barcodes and adaptors at 22°C for 2 h. The ligated products were used as templates and amplified using polymerase chain reaction (Qi et al., 2018). The pooled GBS libraries (100 ng) were sequenced on an Illumina Nova platform with paired-end 150 bp reads. Raw reads were first separated by barcodes using the module ‘process_radtags’ via Stacks v2.1 (options: -r –rensz-1, -adapter_mm 1) (Catchen et al., 2013), and the quality of reads was checked and filtered using fastp (Chen et al., 2018). The clean reads were aligned to the genome reference database using bwa (Li, 2013) (v0.7.17) with default parameters. Using sequencing, a total of 2,629,256,786 reads were produced, averaging 8,216,427 reads per sample; a total of 2,517,131,004 clean reads were obtained, accounting for 95.74% of the total reads. GATK (v4.1.3) (McKenna et al., 2010) was used to call out all the single nucleotide polymorphisms (SNPs). To obtain robust results in subsequent analyses, the following criteria were applied for variant filtering using vcftools (v0.1.16) (Danecek et al., 2011): loci with sequencing depth< 4, SNPs with a minor allele frequency (MAF)< 0.01, and SNPs that were missing in more than 20% of the samples were removed. A total of 464,732 SNPs were obtained for all individuals. After quality control based on an MAF of ≥0.05 and a call rate of ≥0.8, 89,670 SNP loci remained (Table 1). Based on our previous study, the missing genotypes were imputed according to linkage disequilibrium between markers (Jiang and Li, 2016).
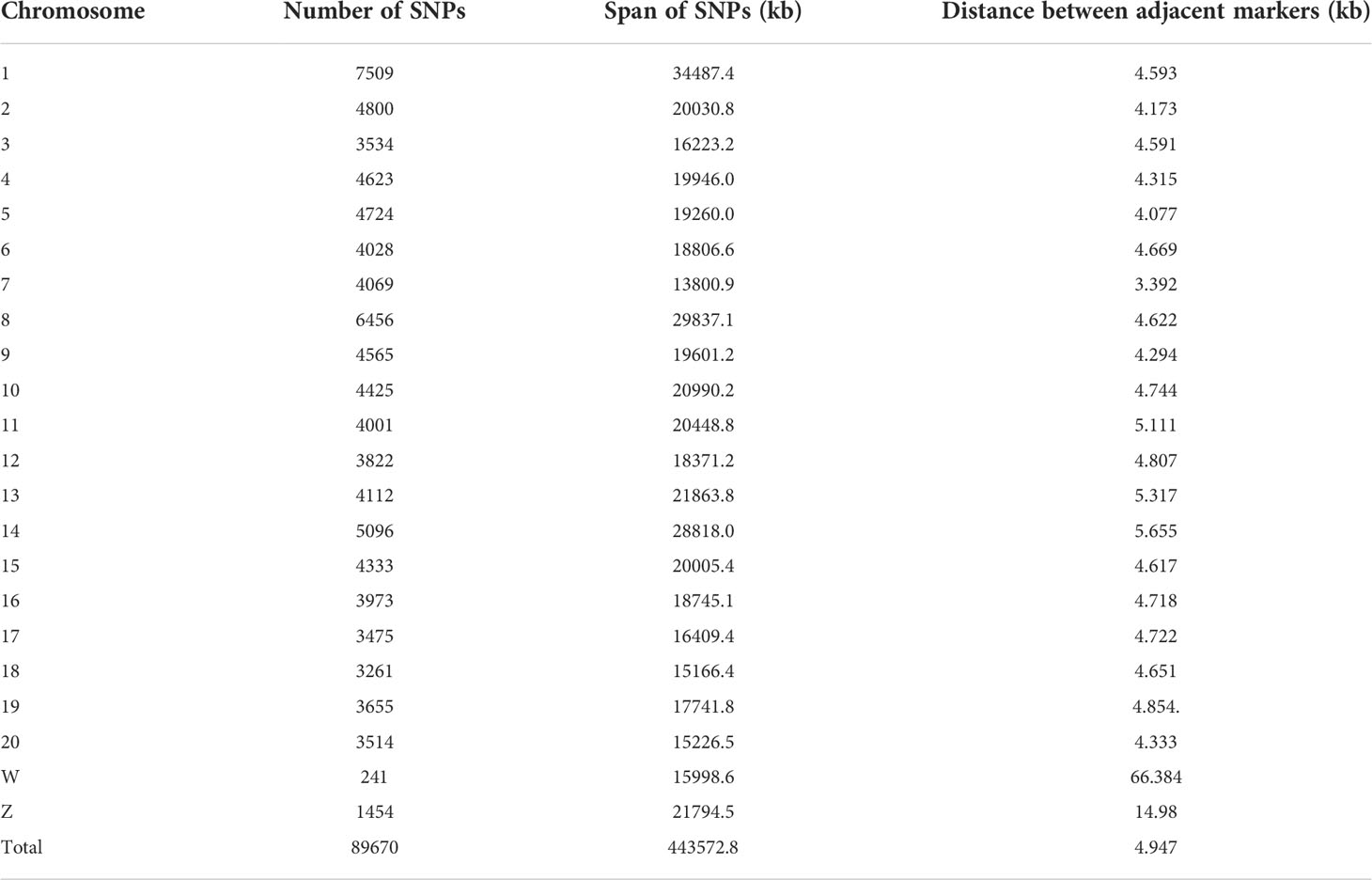
Table 1 Single nucleotide polymorphism (SNP) information.
2.3 Statistical analysis
2.3.1 Genomic selection
For continuous traits (portion of the hypermelanosis area), the genomic best linear unbiased prediction (GBLUP) model (VanRaden, 2008) was used for genomic selection analysis. The model is expressed as follows:
where y is the vector of continuous traits; b is the vector of fixed effects, including population mean, sex, and pond effects; g is the vector of additive genetic effects explained by polygenes, , where G is the realized genetic relationship matrix calculated from genomic markers (VanRaden, 2008) and is the genetic variance explained by the polygenes; X and Z are the corresponding design matrices for b and g, respectively; e is the vector of residuals; and , is the variance of random errors. The heritability was calculated as follows:
For binary traits (incidence of hypermelanosis), the generalized linear mixed model (GLMM) was used as follows:
where y* indicates binary traits, logit(y*) =log(p/(1-p)), and p is the frequency of incidence of hypermelanosis.
The mixed model equations of models (1) and (2) are as follows:
and
Variance components were estimated using the DMU software (Madsen and Jensen, 2013). For binary traits, the residual variance was restricted as a unit. Ten-fold cross validation was performed to study the reliability of the genomic predictions (Li et al., 2017; Zhang et al., 2020).
2.3.2 Genome-wise association study
The model of the genome-wise association study for hypermelanosis area (3) and hypermelanosis incidence (4) is as follows:
where m is the vector of genotypes of each marker, which were coded as 0 for homozygotes of the first allele, 1 for heterozygotes, and 2 for homozygotes of the alternative allele, and β is the effect of the corresponding marker.
To improve the computation speed, a method similar to that of the efficient multilocus mixed model was used (Segura et al., 2012). First, the corresponding phenotypic covariance matrix for the model was calculated as: . Subsequently, the linear mixed model was transformed to a simple linear regression via Cholesky decomposition of the phenotypic covariance structure V using the following equations:
For continuous traits:
For binary traits:
β is equivalent to that in the mixed model and can be quickly tested using a simple linear model.
After a genome scan, critical threshold values were determined (Piepho, 2001).Ten-fold cross validations were performed for each model, the 10% individuals were set as missing, and predicted using the other 90% individuals, the correlation between real values and predicted values were used for the model evaluation. The cross validation was replicated 50 times.
3 Results
3.1 Descriptive statistics
For individuals with an incidence of hypermelanosis in the eyeless side, the portion of pigmented area accounted for the whole eyeless side ranged from 0.004–0.714. More than 70% of the individuals showed pigmentation in less than 10% of the area on the eyeless side (Figure 1).
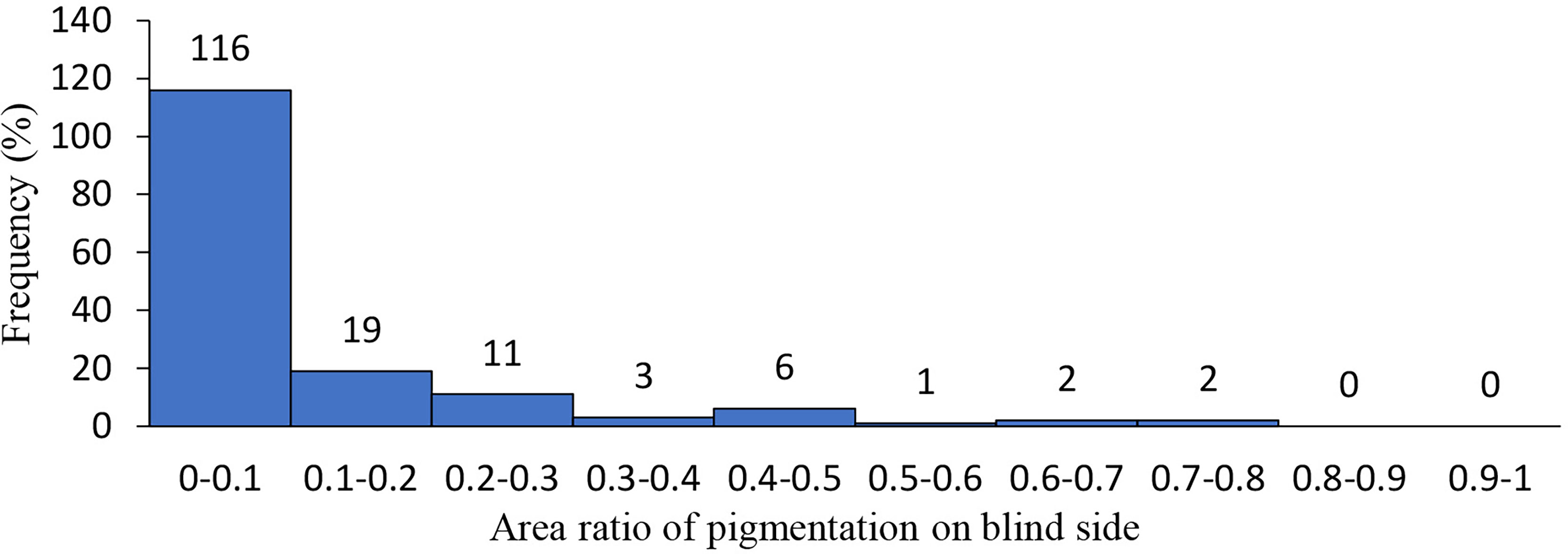
Figure 1 Distribution of hypermlanosis area in Chinese tongue sole.
3.2 Genomic selection
The results obtained from hypermelanosis analysis using the GLMM was slightly different from those using the GBLUP models. The heritability calculated using the GLMM model was 0.16 (for binary traits) and that calculated using the GBLUP model was 0.21 (for continuous traits). Cross-validation showed that the reliabilities of genomic prediction for hypermelanosis area (continuous trait) and incidence (binary trait) were 0.29 ± 0.14 (mean and standard deviation) and 0.27 ± 0.11, respectively (Table 2).
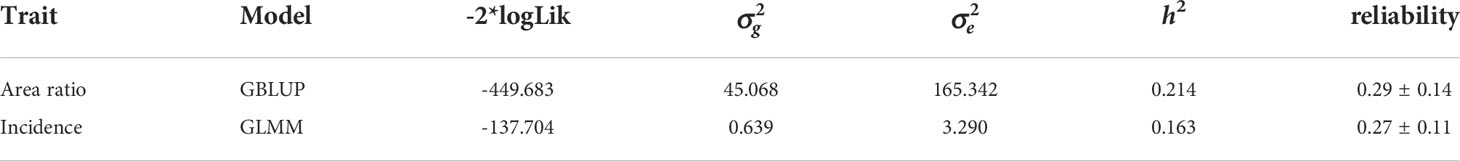
Table 2 variance component estimation and cross validation of hypermelanosis.
3.3 Genome-wide association study
With model (3), eight SNPs were significantly associated with the hypermelanosis area and were distributed on chromosomes 4, 5, 6, 7, 9, 11, and 20 (Figure 2A). Although the two SNPs on chromosome 4 were very close, no data suggested that they were present on the same locus because the correlation between the genotypes of these two SNPs was only 0.3. With model (4), only one locus was significantly associated with hypermelanosis incidence and was located on chromosome 9 (Figure 2B) at a different position from the locus associated with hypermelanosis area present on the same chromosome. Nearly all loci explained approximately 10% of the total phenotypic variance (Table 3), indicating that they were potential quantitative train loci.
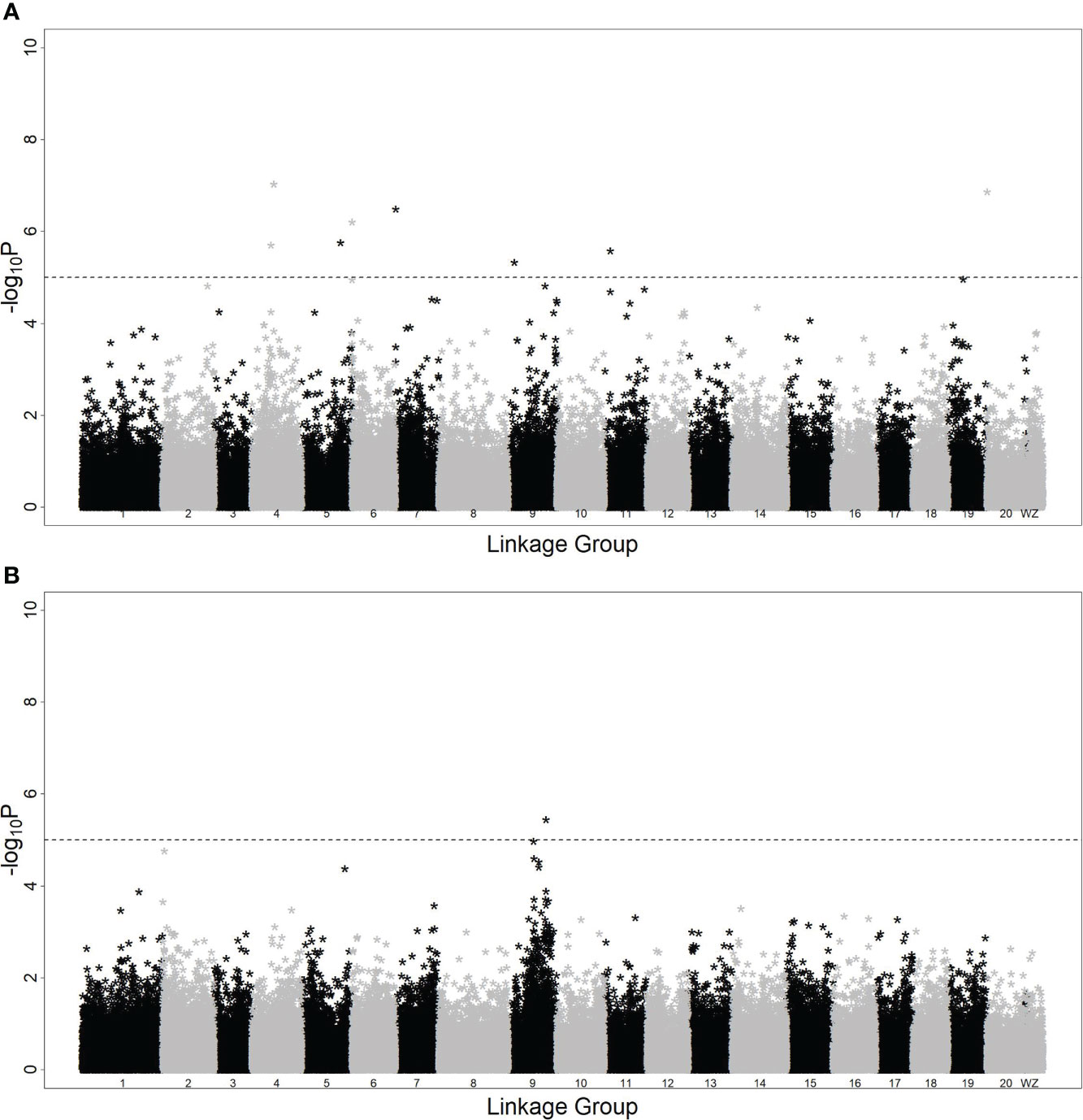
Figure 2 Genome-wide association study of hypermelanosis of Chinese tongue sole. (A): hypermelanosis area; (B): hypermelanosis incidence *:the negative logarithm of P-values at the corresponding position.
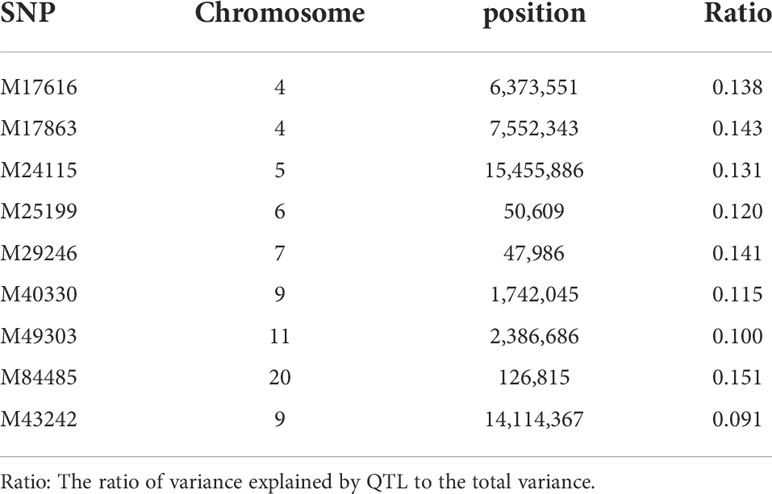
Table 3 SNPs associated with hypermelanosis in Chinese tongue sole.
4 Discussion
In contrast to features in ornamental fish, abnormal body color is an undesirable characteristic in flatfish aquaculture (Padowicz and Harpaz, 2007; Lewbart 2016; Atsumi and Koizumi, 2018). Hypermelanosis is influenced by genetic and epigenetic factors, as well as environmental conditions, such as nutrition (Haga et al., 2004), tank color (Matsumoto and Seikai, 2008), and density (Takahashi, 1994). Genetic selection is a fundamental and effective approach for breeding Chinese tongue soles with preferred body color. Based on the data of a large number of families and generations, the heritability of hypermelanosis in the eyeless side is reported to be moderate (Liu et al., 2016; Li et al., 2021) in the Chinese tongue sole. In our study, the heritabilities were both low for the hypermelanosis area and incidence, which were significantly lower than those reported previously. It is usually considered that heritability estimated using genomic markers is lower than that estimated from pedigree, that is, the so-called missing heritability (Eichler et al., 2010). It is possible that certain markers in linkage disequilibrium with hypermelanosis had not been genotyped using our method. Considering the heritable variation, hypermelanosis may be a complex quantitative trait with low or moderate heritability. Genetic evaluation using genomic markers is a feasible and effective approach to decrease the incidence and extent of hypermelanosis.
Usually, a quantitative trait has a higher reliability than that of a binomial trait (Bangera et al., 2017). However, in our study, either the GBLUP or the GLMM model was used for genomic selection and they had similar reliabilities. Since the relationship between the genomic prediction accuracy (r) and the reliability (R) is r=R/h , the genomic prediction accuracies for both hypermelanosis area ratio and incidence were approximately 65%, indicating the feasibility of genomic selection and a possible long duration for improvement.
The use of markers associated with traits of interest is an effective approach to increasing genetic merits, and marker-assisted selection has been used successfully in few species (Yue, 2014). In the present study, several potential loci were found to be associated with hypermelanosis in the Chinese tongue sole. Among these loci, SNPs M17616 and M24115 were located in an intergenic region, whereas the others were located in intronic regions. The flanking genes of SNP M17616 are AP-1 complex subunit sigma-3 (ap1s3) and potassium voltage-gated channel subfamily E member 4 (kcne4), where ap1s3 encodes a member of the adaptor-related protein complex 1 and ap1s3 knockout disrupts keratinocyte autophagy and causes pustular skin disease (Mahil et al., 2016; Mössner et al., 2018); kcne4 regulates potassium voltage-gated channel stability (Solé et al., 2009). The flanking genes of SNP M24115 have not been characterized. SNP M25199 was located in an intronic region of gene alpha-kinase 3a, which was predicted to enable ATP-binding activity and possibly be involved in cardiac muscle cell development. SNP M49303 was located in an intronic region of gene bicaudal D homolog 2, which encodes an adaptor protein that functions as an intracellular cargo transport cofactor that regulates the microtubule-based loading of cargo onto the dynein motor complex. SNP M29246 is located in the intronic region of solute carrier family 16 member 10 gene, which mediates the Na(+)-independent transport of aromatic amino acids across the plasma membrane; it is associated with the occurrence of melanoma in skin (He et al., 2020). Notably, the adjacent gene ribosome production factor 2 homolog is associated with vitiligo, which occurs when the skin lacks normal melanocytes and leads to color patches, which is very similar to the appearance of hypermelanosis (Derakhshani et al., 2020). SNP M43242 was located in an intronic region of gene SH3 and PX domains 2Aa, which was predicted to enable superoxide-generating NADPH oxidase activator activity and it plays a role in neural crest cell migration and posterior lateral line neuromast primordium migration (Gallardo et al., 2015). SNPs M17863, M40330, and M84485 were located in the intronic regions of LOC103377888, LOC103383278, and LOC103395801, respectively; however, these loci are uncharacterized and no evidence showed that they and their nearby genes were associated with skin disease.
Hundreds of genes have been found to be differentially expressed between tongue soles with normal skin and hypermelanosis (Li et al., 2022); however, no shared gene was found in our study, indicating the complicated genetic mechanism of hypermelanosis in the tongue sole. The genes activating transcription factor 4, inositol 1,4,5-trisphosphate receptor type 2, and adenylate cyclase type 6 have been found to be associated with hypermelanosis in Japanese flounder (Peng et al., 2019; Zhang et al., 2021); however, they were not detected in tongue sole, indicating that the genetic mechanism of hypermelanosis in Chinese tongue sole differed from that in Japanese flounder because the color of hypermelanosis in Japanese flounder is dark, whereas it is brown in Chinese tongue sole.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://figshare.com/, https://doi.org/10.6084/m9.figshare.20365533.
Ethics statement
The animal study was reviewed and approved by Chinese Academy of Fishery Sciences.
Author contributions
NY and YZ performed the experiment and analyzed the data. YL and YH collected the samples. HL designed the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Special Scientific Research Funds for Central Non-profit Institutes [grant number CAFS: 2020TD24]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amiya N., Amano M., Yamanome T., Yamamori K., Takahashi A. (2008). Effects of background color on GnRH and MCH levels in the barfin flounder brain. Gen. Comp. Endocrinol. 155, 88–93. doi: 10.1016/j.ygcen.2007.03.007
Atsumi K., Koizumi I. (2018). Correction to: Web image search revealed large-scale variations in breeding season and nuptial coloration in a mutually ornamented fish, tribolodon hakonensis. Ecol. Res. 32, 567–578. doi: 10.1007/s11284-018-1598-9
Bangera R., Correa K., Lhorente J. P., Figueroa R., Yáñez J. M. (2017). Genomic predictions can accelerate selection for resistance against piscirickettsia salmonis in Atlantic salmon (Salmo salar). BMC Genom. 18, 121. doi: 10.1186/s12864-017-3487-y
Catchen J., Hohenlohe P. A., Bassham S., Amores A., Cresko W. A. (2013). Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. doi: 10.1111/mec.12354
Chen S. F., Zhou Y. Q., Chen Y. R., Gu J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics. 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Derakhshani A., Mollaei H., Parsamanesh N., Fereidouni M., Miri-Moghaddam E., Nasseri S., et al. (2020). Gene Co-expression network analysis for identifying modules and functionally enriched pathways in vitiligo disease: A systems biology study. Iran. J. Allergy Asthma Immunol. 19, 517–528. doi: 10.18502/ijaai.v19i5.4467
Dou S. Z. (1995). Life history cycles of flatfish species in the bohai Sea, China. Neth. J. Sea Res. 34, 195–210. doi: 10.1016/0077-7579(95)90027-6
Eichler E. E., Flint J., Gibson G., Kong A., Leal S. M., Moore J. H., et al. (2010). Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450. doi: 10.1038/nrg2809
Estevez A., Kaneko T., Seikai T., Dores R. M., Tagawa M., Tanaka M. (2001). Ontogeny of ACTH and MSH cells in Japanese flounder (Paralichthys olivaceus) in relation to albinism. Aquaculture. 202, 131–143. doi: 10.1016/S0044-8486(01)00571-3
Feng J. Y., Wen Y. J., Zhang J., Zhang Y. M. (2016). Advances on methodologies for genome-wide association studies in plants. Acta Agron. Sin. 42, 945–956. doi: 10.3724/sp.j.1006.2016.00945
Gallardo V. E., Varshney G. K., Lee M., Bupp S., Xu L. S., Shinn P., et al. (2015). Phenotype-driven chemical screening in zebrafish for compounds that inhibit collective cell migration identifies multiple pathways potentially involved in metastatic invasion. Dis. Models Mech. 8, 565–576. doi: 10.1242/dmm.018689
Gibson R. N. (1997). Behaviour and the distribution of flatfishes. J. Sea Res. 37, 241–256. doi: 10.1016/s1385-1101(97)00019-1
Haga Y., Takeuchi T., Murayama Y., Ohta K., Fukunaga T. (2004). Vitamin D3 compounds induce hypermelanosis on the blind side and vertebral deformity in juvenile Japanese flounder Paralichthys olivaceus. Fish. Sci. 70, 59–67. doi: 10.1111/j.1444-2906.2003.00771.x
He C. C., Zhang Y. J., Jiang H. H., Niu X. L., Qi R. Q., Gao X. H. (2020). Identification of differentially expressed methylated genes in melanoma versus nevi using bioinformatics methods. PeerJ. 8, e9273. doi: 10.7717/peerj.9273
Isojima T., Makino N., Miyama Y., Tagawa M. (2014). Effects of time and duration of rearing with bottom sand on the occurrence and expansion of staining-type hypermelanosis in the Japanese flounder paralichthys olivaceus. Fish. Sci. 80, 785–794. doi: 10.1007/s12562-014-0755-5
Isojima T., Makino N., Takakusagi M., Tagawa M. (2013b). Progression of staining-type hypermelanosis on the blind side in normally metamorphosed juveniles and pigmentation progression in pseudoalbino juveniles of the Japanese flounder Paralichthys olivaceus using individual identification. Fish. Sci. 79, 787–797. doi: 10.1007/s12562-013-0655-0
Isojima T., Tsuji H., Masuda R., Tagawa M. (2013a). Formation process of staining-type hypermelanosis in Japanese flounder juveniles revealed by examination of chromatophores and scales. Fish. Sci. 79, 231–242. doi: 10.1007/s12562-013-0600-2
Jiang L., Li H. D. (2016). ). single locus maintains Large variation of sex reversal in half-smooth tongue sole (Cynoglossus semilaevis). G3. 7, 583–589. doi: 10.1534/g3.116.036822
Kang D. Y., Kim H. C. (2015). Functional relevance of three proopiomelanocortin (POMC) genes in darkening camouflage, blind-side hypermelanosis, and appetite of Paralichthys olivaceus. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 179, 44–56. doi: 10.1016/j.cbpb.2014.09.002
Kang D. Y., Kim H. C., Kang H. S. (2014). The functional relevance of prepro-melanin concentrating hormone (pMCH) to skin color change, blind-side malpigmentation and feeding of Oliver flounder paralichthys olivaceus. Fish. Aquat. Sci. 17, 325–337. doi: 10.5657/fas.2014.0325
Lewbart G.A. (2016). Ornamental fishes and aquatic invertebrates: Self-assessment color review. Boca Raton, USA. doi: 10.1201/9781315381497
Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, pp. arXiv preprint arXiv. 1303, 3997. doi: 10.48550/arXiv.1303.3997
Li Y. Z., Hu Y. R., Cheng P., Chen S. L. (2022). Identification of potential blind-side hypermelanosis-related lncRNA-miRNA-mRNA regulatory network in a flatfish species, Chinese tongue sole (Cynoglossus semilaevis). Front. Genet. 12. doi: 10.3389/fgene.2021.817117
Li Y. Z., Hu Y. R., Zheng W. W., Cui Z. K., Cheng J. Y., Cheng P., et al. (2021). Insights into the heritable variation of hypermelanosis in Chinese tongue sole (Cynoglossus semilaevis): Potential for future selective breeding. Aquaculture. 539, 736617. doi: 10.1016/j.aquaculture.2021.736617
Li H. D., Su G. S., Jiang L., Bao Z. M. (2017). An efficient unified model for genome-wide association studies and genomic selection. Genet. Sel. Evol. 49, 64–71. doi: 10.1186/s12711-017-0338-x
Liu F., Yang Y. M., Li Y. Z., Guo H., Dai H., Gao J., et al. (2016). Phenotypic and genetic parameter estimation of juvenile growth and bottom color traits in half-smooth tongue sole, Cynoglossus semilaevis. Acta Oceanol. Sin. 35, 83–87. doi: 10.1007/s13131-016-0888-8
Madsen P., Jensen J. (2013) A user's guide to DMU. a package for analyzing multivariate mixed models. Available at: http://dmu.agrsci.dk/DMU/Doc/ (Accessed 7 July, 2017).
Mahil S. K., Twelves S., Farkas K., Setta-Kaffetzi N., Burden A. D., Gach J. E., et al. (2016). AP1S3 mutations cause skin autoinflammation by disrupting keratinocyte autophagy and up-regulating IL-36 production. J. Invest. Dermatol. 136, 2251–2259. doi: 10.1016/j.jid.2016.06.618
Martin P. M., Palhière I., Ricard A., Tosser-Klopp G., Rupp R. (2016). Correction: Genome wide association study identifies new loci associated with undesired coat color phenotypes in saanen goats. PloS One 11, e0152426. doi: 10.1371/journal.pone.0152426
Matsuda N., Kasagi S., Nakamaru T., Masuda R., Takahashi A., Tagawa M. (2018). Left-right pigmentation pattern of Japanese flounder corresponds to expression levels of melanocortin receptors (MC1R and MC5R), but not to agouti signaling protein 1 (ASIP1) expression. Gen. Comp. Endocrinol. 262, 90–98. doi: 10.1016/j.ygcen.2018.03.019
Matsumoto J., Seikai T. (2008). Asymmetric pigmentation and pigment disorders in pleuronectiformes (Flounders). Pigm. Cell Res. 3, 275–282. doi: 10.1111/j.1600-0749.1990.tb00385.x
McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Morris G. P., Borevitz J. O., Brachi B. (2011). Genome-wide association studies in plants: the missing heritability is in the field. Genome Biol. 12, 232. doi: 10.1186/gb-2011-12-10-232
Mössner R., Wilsmann-Theis D., Oji V., Gkogkolou P., Löhr S., Schulz P., et al. (2018). The genetic basis for most patients with pustular skin disease remains elusive. Br. J. Dermatol. 178, 740–748. doi: 10.1111/bjd.15867
Nakata K., Yamamoto I., Miyama Y., Nakamaru T., Masuda R., Tagawa M. (2017). Undulated flooring in the rearing tank decreases hypermelanosis in Japanese flounder Paralichthys olivaceus. Fish. Sci. 83, 1027–1035. doi: 10.1007/s12562-017-1135-8
Owens B. F., Mathew D., Diepenbrock C. H., Tiede T., Wu D., Mateos-Hernandez M., et al. (2019). Genome-wide association study and pathway-level analysis of kernel color in maize. G3. 9, 1945–1955. doi: 10.1534/g3.119.400040
Padowicz D., Harpaz S. (2007). Color enhancement in the ornamental dwarf cichlid microgeophagus ramirezi by addition of plant carotenoids to the fish diet. Isr. J. Aquac. 59, 195–200. doi: 10.46989/001c.20536
Peng K. K., Zhang B., Xu J., Zhao N., Jia L., Che J. Y., et al. (2019). Identification of SNPs related to hypermelanosis of the blind side by transcriptome profiling in the Japanese flounder (Paralichthys olivaceus). Aquaculture. 519, 734906. doi: 10.1016/j.aquaculture.2019.734906
Piepho H. P. (2001). A quick method for computing approximate thresholds for quantitative trait loci detection. Genetics. 157, 425–432. doi: 10.1093/genetics/157.1.425
Qi P., Gimode D., Saha D., Schröder S., Chakraborty D., Wang X. W., et al. (2018). UGbS-flex, a novel bioinformatics pipeline for imputation-free SNP discovery in polyploids without a reference genome: finger millet as a case study. BMC Plant Biol. 18, 117. doi: 10.1186/s12870-018-1316-3
Segura V., Vilhjálmsson B. J., Platt A., Korte A., Seren Ü., Long Q., et al. (2012). An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830. doi: 10.1038/ng.2314
Solé L., Roura-Ferrer M., Pérez-Verdaguer M., Oliveras A., Calvo M., Fernández-Fernández J. M., et al. (2009). KCNE4 suppresses Kv1.3 currents by modulating trafficking, surface expression and channel gating. J. Cell Sci. 122, 3738–3748. doi: 10.1242/jcs.056689
Stickney R. R., White D. B. (1975). Ambicoloration in tank cultured flounder, Paralichthys dentatus. Trans. Am. Fish. Soc 104, 158–160. doi: 10.1577/1548-8659(1975)104<158:aitcfp>2.0.co;2
Takahashi Y. (1994). Influence of stocking density and food at late phase of larval period on hypermelanosis on the blind body side in juvenile Japanese flounder. Nippon Suisan Gakkaishi. 60, 593–598. doi: 10.2331/suisan.60.593
Takahashi A., Tsuchiya K., Yamanome T., Amano M., Yasuda A., Yamamori K., et al. (2004). Possible involvement of melanin-concentrating hormone in food intake in a teleost fish, barfin flounder. Peptides. 25, 1613–1622. doi: 10.1016/j.peptides.2004.02.022
VanRaden P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423. doi: 10.3168/jds.2007-0980
Yamada T., Donai H., Okauchi M., Tagawa M., Araki K. (2011). Induction of ambicoloration by exogenous cortisol during metamorphosis of spotted halibut Verasper variegatus. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 160, 174–180. doi: 10.1016/j.cbpb.2011.08.004
Yamanome T., Amano M., Takahashi A. (2005). White background reduces the occurrence of staining, activates melanin-concentrating hormone and promotes somatic growth in barfin flounder. Aquaculture. 244, 323–329. doi: 10.1016/j.aquaculture.2004.11.020
Yue G. H. (2014). Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish (Oxf). 15, 376–396. doi: 10.1111/faf.12020
Zhang Y. Q., Liu Z. J., Li H. D. (2020). Genomic prediction of columnaris disease resistance in catfish. Mar. Biotechnol. 22, 145–151. doi: 10.1007/s10126-019-09941-7
Zhang B., Peng K. K., Che J. Y., Zhao N., Jia L., Zhao D. K., et al. (2021). Single-nucleotide polymorphisms responsible for pseudo-albinism and hypermelanosis in Japanese flounder (Paralichthys olivaceus) and reveal two genes related to malpigmentation. Fish Physiol. Biochem. 47, 339–350. doi: 10.1007/s10695-020-00916-3
Zhang H., Wang Z. P., Wang S. Z., Li H. (2013). Retraction: progress of genome wide association study in domestic animals. J. Anim. Sci. Biotechnol. 4, 3. doi: 10.1186/2049-1891-4-3
Keywords: Chinese tongue sole (Cynoglossus semilaevis), hypermelanosis, genomic selection, genome-wide association study, continuous traits, binary traits
Citation: Yao N, Zhang Y, Li Y, Hu Y and Li H (2022) Genetic analysis of hypermelanosis in Chinese tongue sole (Cynoglossus semilaevis). Front. Mar. Sci. 9:1002292. doi: 10.3389/fmars.2022.1002292
Received: 25 July 2022; Accepted: 11 October 2022;
Published: 24 October 2022.
Edited by:
Xiaotong Wang, Ludong University, ChinaReviewed by:
Jinqiang Huang, Gansu Agricultural University, ChinaGuanpin Yang, Ocean University of China, China
Xiangshan Ji, Shandong Agricultural University, China
Copyright © 2022 Yao, Zhang, Li, Hu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hengde Li, aGVuZ2RlLmxpQGNhZnMuYWMuY24=