 Ashok Kumar Jangam1,2*
Ashok Kumar Jangam1,2* Vinaya Kumar Katneni1
Vinaya Kumar Katneni1 Akshaya Panigrahi1
Akshaya Panigrahi1 Karyath Palliyath Gangaraj1
Karyath Palliyath Gangaraj1 Suganya Nathamuni1
Suganya Nathamuni1 Jesudhas Raymond Jani Angel1
Jesudhas Raymond Jani Angel1 Mudagandur Shashi Shekhar1
Mudagandur Shashi Shekhar1- 1ICAR-Central Institute of Brackish Water Aquaculture, Chennai, India
- 2Aquaculture and Genetics, University of Madras, Chennai, India
Introduction
Indian white shrimp, Penaeus indicus, is an important candidate species and can be considered as an alternative species for sustainable aquaculture growth along with tiger shrimp Penaeus monodon and Pacific white shrimp Penaeus vannamei. Many shrimp-producing countries import specific pathogen-free broodstock of P. vannamei for seed production to be used for aquaculture. For many of these countries, P. vannamei is not a native species. Such dependence on single exotic species is not an ideal scenario for many developing coastal countries. The culture of native species like P. indicus, has many advantages in restricting the entry of exotic pathogens via import of exotic species and thereby minimizing the economic loss to the aquaculture industry (Vijayan, 2019). The growth performance of this species was reported to be comparable with P. vannamei which showed its potential as an alternate or complementary to the Pacific white shrimp for culture production (Panigrahi et al., 2020).
Advances in next-generation sequencing technologies have contributed to the significant rise in genomic resources of several organisms. However, there has been a slow pace in building such information generally for crustaceans and specifically for shrimps due to the complexities involved in the genetic makeup of the organism. Among shrimps, whole genomes of Pacific white shrimp, black tiger shrimp, and Chinese shrimp have been published recently (Zhang et al., 2019; Uengwetwanit et al., 2021; Wang et al., 2021) and a total of 25,527, 32,900, and 26,343 encoding genes were reported for these species, respectively. Each of these species showed considerable variations with respect to the reported genomic content which could be the main reason for their physiological differences. With respect to P. indicus, limited information is available at present on the genetic composition and there is an immediate need to build such resources to have an in-depth understanding of this commercially important shrimp. In addition, building resources on intestinal microbiota is also important as it is found to play a major role in the health and development of shrimps (Tello, 2020). The study aimed to understand primarily the gene content in the hepatopancreas of juvenile Indian white shrimps along with the intestinal microbial communities, which would act as a useful resource for future studies.
Methods
Sample Collection and RNA Extraction
In this study, six juvenile P. indicus shrimp were collected from the demonstration ponds located at Muttukadu experimental station (12.80°N, 80.24°E) of ICAR-Central Institute of Brackish water Aquaculture, Chennai. At the time of sampling, the stage of the crop was at 60 days of culture. Animals having body weights ranging from 3.4 to 5.1 grams with a mean weight of 4.12 g were sampled. Hepatopancreas tissue was selected for RNA sequencing as it is the metabolically active site of the shrimp. The tissue sample from each shrimp was dissected, quickly frozen in liquid nitrogen, and preserved at −80°C until RNA extraction. Total RNA from each juvenile shrimp sample was isolated using the conventional TRIzol method. The quality and quantity of each isolated RNA sample were checked on NanoDrop (Thermo Scientific, Brea, CA, United States) followed by denatured agarose gel.
Library Preparation and Sequencing
The RNA-seq paired-end sequencing libraries were prepared with good quality RNA using Illumina TruSeq Stranded mRNA sample preparation kit (Illumina, San Diego, CA, United States). Initially, mRNA was enriched from the total RNA using poly-T attached magnetic beads, followed by enzymatic fragmentation. Afterward, 1st strand cDNA was converted using superscript II and Act-D mix containing Random Hexamer to facilitate RNA dependent synthesis. The 1st strand cDNA was then synthesized to the second strand using a second strand mix. The dscDNA was then purified using AMPure XP beads (Beckman Coulter, Brea, CA, United States) followed by A-tailing adapter ligation and then enriched by a limited number of PCR cycles. The PCR enriched libraries were analyzed on a 4200 TapeStation system Agilent Technologies (Santa Clara, CA, United States) using high sensitivity D1000 screen tape as per the manufacturer’s protocol. After obtaining the Qubit concentration for the libraries and the mean peak sizes from the Agilent TapeStation profile, the paired-end (PE) high-quality libraries were loaded onto the Illumina NovaSeq6000 platform (San Diego, CA, United States) for cluster generation and sequencing. On average 35 million paired reads of length 150 bp were generated for each of the six samples.
De novo Transcriptome Assembly
The quality checking of raw RNA-seq reads was carried out using FastQC version 0.11.8.1 The contaminant adapters, poor quality reads, and bases were trimmed using Trimmomatic version 0.39 (Bolger et al., 2014). The good quality reads were then assembled using the Trinity version 2.4 (Haas et al., 2013) and the assembly statistics are presented in Table 1. Furthermore, the unigenes were obtained from assembled transcripts following the procedure suggested by Chabikwa et al. (2020). Briefly, the Trinity transcripts were clustered using cd-hit-est version 4.8.1 to filter the redundant transcripts. Then, the longest open reading frames (ORFs) predicted using TransDecoder version 5.5 from the clustered transcripts were considered as unigenes. Overall, the assembly contained 28,115 unigenes accounting for a total length of 14,029,914 bp with an average length of 499 bp.
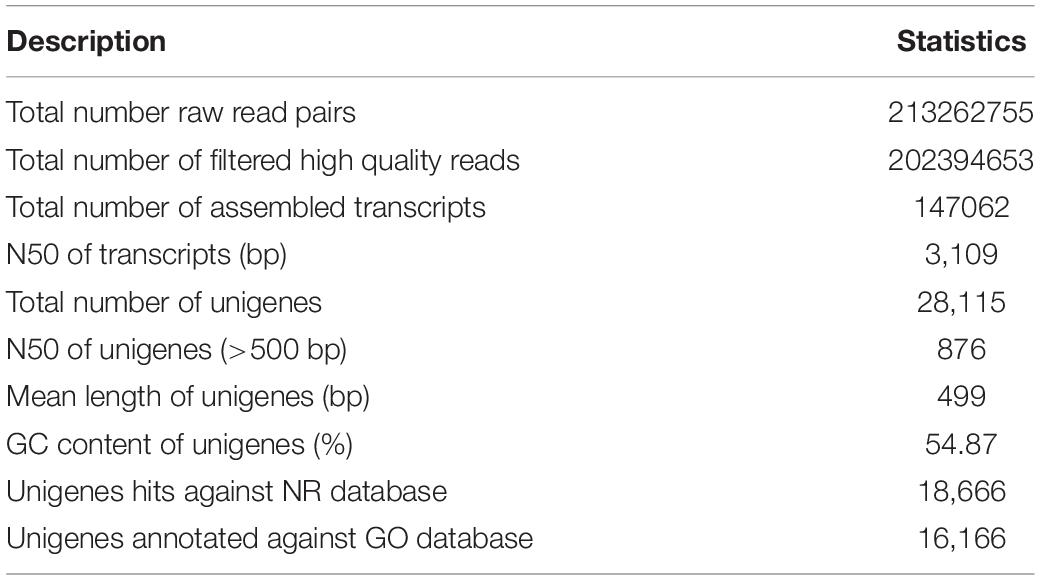
Table 1. Transcriptome assembly and annotation statistics.
Functional Annotation of Unigenes
Annotation of unigenes was carried out using OmicsBox version 2.0.24 software (OmicsBox, 2019).2 Briefly, sequence homology was performed using blastx search against non-redundant (nr) protein database (Pruitt et al., 2005) and mapping was performed with OmicsBox. Gene ontology (GO) terms were then obtained for unigenes through a search against InterPro and EggNOG databases (Huerta-Cepas et al., 2019; Blum et al., 2021). Afterward, GO terms were merged and final annotations along with enzyme codes and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were identified for unigenes. Overall, functional annotations were obtained for 16,166 (57.5%) of unigenes (Figure 1A). For about 31% of unigenes (8,796), blastx search (Altschul et al., 1990) did not yield a positive hit to any entry of the nr protein database at an e-value threshold of 10e−5. The P. vannamei is the top-hit species for a majority of unigenes during blastx search (Supplementary Figure 1). The InterProScan (IPS) search resulted in the assignment of family/domain information for 99.92% of IPS annotations. The predominant IPS domain, family, repeats and sites were ABC transporter-like ATP-binding domain, P-loop containing nucleoside triphosphate hydrolase, WD40 repeat and ABC transporter-like conserved site, respectively (Supplementary Figure 2). GO analysis revealed that the majority of the ontologies were assigned to Molecular Function (87%, 14,225) followed by Biological Process (76%, 12,337) and Cellular Component (59%, 9,607) (Supplementary Table 1). Organic cyclic compound binding, Organic substance metabolic process and intracellular anatomical structure were among the top assignments for Molecular Function, Biological Process, and Cellular Component categories, respectively (Figure 1B). The GO annotations for about 31% of unigenes were obtained with the EggNOG database (Supplementary File 1). The hydrolases and transferases were the dominant enzyme code classes followed by the oxidoreductases and translocases (Supplementary Figure 3). About 10,119 of the 16,166 annotated unigenes were linked to 331 KEGG pathways. Major KEGG pathway representations include carbohydrate metabolism (24.82%), amino acid metabolism (18.44%), energy metabolism (10.76%), and metabolism of cofactor/vitamins (9.67%) (Figure 1C).
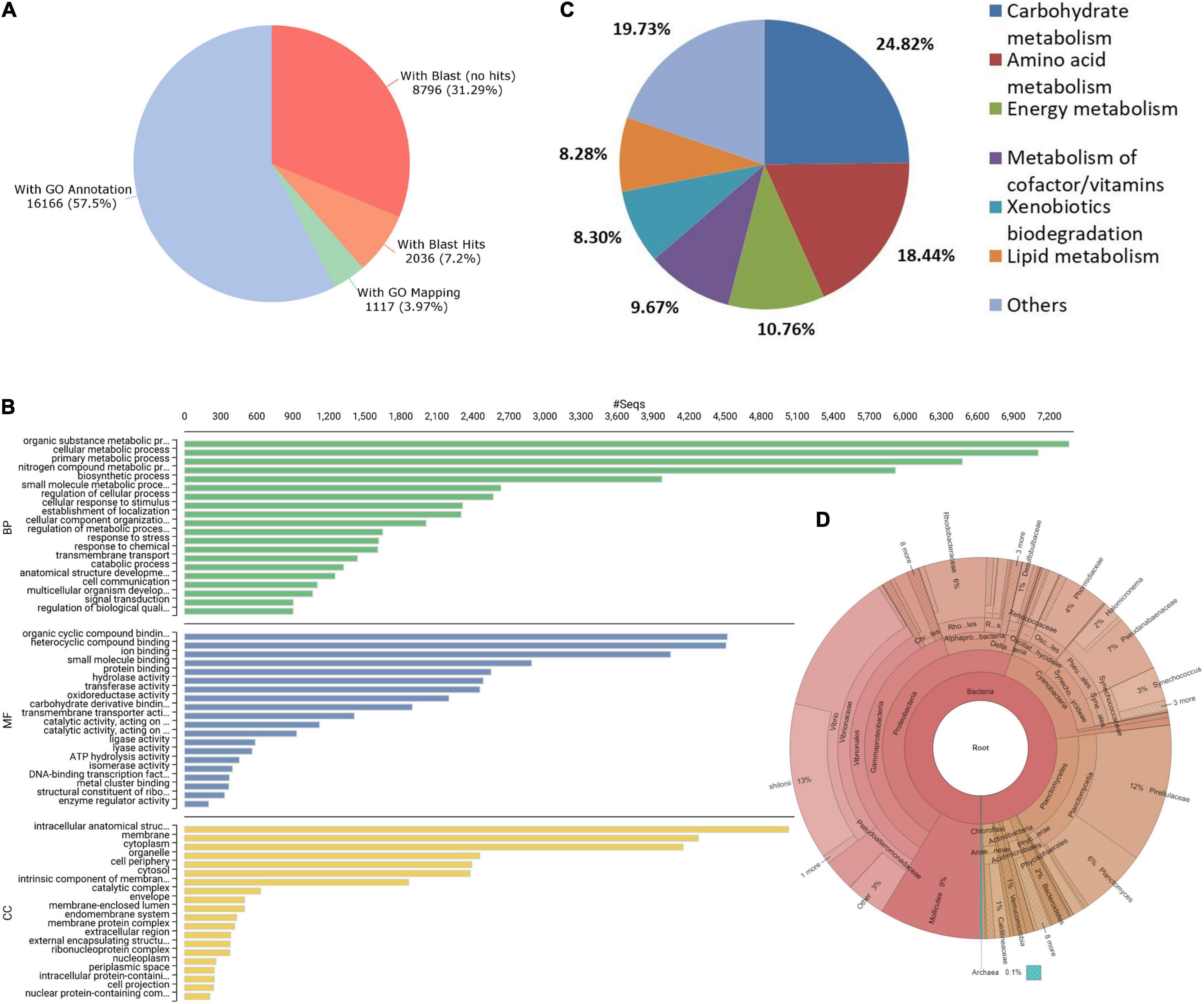
Figure 1. (A) Annotation summary of unigenes. (B) Distribution of most abundant Gene ontology (GO) terms. (C) Represented Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of unigenes. (D) Representation of intestinal microbial composition through Krona plot.
Simple Sequence Read Identification
Simple sequence reads (SSRs) in the unigene set were identified using MicroSAtellite (MiSA) tool (Beier et al., 2017). The stand-alone version of MiSA was run with the default parameters of di-nucleotide repeats >6 tri-nucleotide to hexanucleotide repeats >5 and the maximum length of sequence between SSRs at 100. In the unigenes, a total of 2,344 SSRs were detected. There were 1,284 SSR-containing sequences in which 575 sequences contained more than one SSRs. Tri-nucleotide repeats were found to be the most abundant class (1,001) followed by mono (608) and di-nucleotide (571). Among the di-nucleotide motifs, AG/CT followed by AC/GT was found to be most abundant and for tri-nucleotides, it is AAG/CTT. These were also reported to be the most abundant repeats in two other shrimp species F. chinensis and L. vannamei in a genome-wide comparison study of SSRs (Yuan et al., 2021).
Intestinal Microbiome of Juvenile Shrimp
To understand the intestinal microbiota of P. indicus juveniles, metagenomic reads of V3-V4 16s rRNA regions were generated from pooled gut contents of six juveniles. Briefly, the pooled gut contents were subject to c-TAB Phenol: chloroform method followed by RNase A treatment to isolate the metagenomic DNA, and the quality of DNA was checked using NanoDrop 2000 (Thermo Scientific, Brea, CA, United States). Amplicon libraries were prepared using the NextEra XT Index kit Illumina Inc. (San Diego, CA, Uinted States). The libraries were subject to quality check on the Agilent 4200 Tapestation (Santa Clara, CA, United States). A total of 1,85,653 paired-end reads were generated using the 2 × 300 MiSeq library on the Illumina sequencing platform (San Diego, CA, United States). The microbial communities were identified through the analytical pipeline QIIME (Caporaso et al., 2010). Proteobacteria, Planctomycetes, Cyanobacteria, and Tenericutes were found to be major phylum level associations, while Vibrio, Planctomyces, and Synechococcus were among the identified highly abundant genera (Figure 1D). Microbial associations identified for P. indicus juveniles are similar to the intestinal microbiota of other shrimps such as P. monodon, Penaeus japonicus, and P. vannamei (Fan et al., 2019; Angthong et al., 2020; Zhang et al., 2021). Complete microbial associations of P. indicus gut at phylum, class, order, family, genus, and species levels are available in Supplementary Table 2.
Conclusion
The transcriptomic and metagenomics resources of juvenile P. indicus are generated using Illumina NovaSeq6000 and MiSeq platforms, respectively. The data generated in this study will be a useful resource for ongoing and future research projects related to the discovery and expression profiling of genes and the gut microbiome of P. indicus.
Data Availability Statement
The RNA-seq datasets and shrimp gut metagenome datasets generated from the current research were deposited in the NCBI database under BioProject IDs PRJNA750258 and PRJNA773565, respectively. All the files generated with the analytical procedures followed in this study are available on FigShare at https://doi.org/10.6084/m9.figshare.16863124.
Author Contributions
AJ and VK conceived the idea and collected the animal samples. KG and SN performed the bioinformatics analysis. AJ wrote the manuscript by taking inputs from VK, AP, JA, and MS. All authors read and approved the final version of the manuscript.
Funding
This research was supported under the Network Project on Agricultural Bioinformatics and Computational Biology, New Delhi.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are thankful to the funding agency for financial support. The authors are also thankful to the Director, ICAR-CIBA, for providing the necessary approval for conducting this research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.809720/full#supplementary-material
Footnotes
References
Angthong, P., Uengwetwanit, T., Arayamethakorn, S., Chaitongsakul, P., Karoonuthaisiri, N., and Rungrassamee, W. (2020). Bacterial analysis in the early developmental stages of the black tiger shrimp (Penaeus monodon). Sci. Rep. 10:4896. doi: 10.1038/s41598-020-61559-1
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Blum, M., Chang, H.-Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchell, A., et al. (2021). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354. doi: 10.1093/nar/gkaa977
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chabikwa, T. G., Barbier, F. F., Tanurdzic, M., and Beveridge, C. A. (2020). De novo transcriptome assembly and annotation for gene discovery in avocado, macadamia and mango. Sci. Data 7:9. doi: 10.1038/s41597-019-0350-9
Fan, L., Wang, Z., Chen, M., Qu, Y., Li, J., Zhou, A., et al. (2019). Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 657, 1194–1204. doi: 10.1016/j.scitotenv.2018.12.069
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314. doi: 10.1093/nar/gky1085
ltschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
OmicsBox (2019). Bioinformatics Made Easy, BioBam Bioinformatics, Available online at: https://www.biobam.com/omicsbox [acessed on Mar 3, 2019].
Panigrahi, A., Sivakumar, M. R., Sundaram, M., Saravanan, A., Das, R. R., Katneni, V. K., et al. (2020). Comparative study on phenoloxidase activity of biofloc-reared pacific white shrimp Penaeus vannamei and Indian white shrimp Penaeus indicus on graded protein diet. Aquaculture 518:734654. doi: 10.1016/j.aquaculture.2019.734654
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2005). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504. doi: 10.1093/nar/gki025
Tello, M. (2020). “Application of Metagenomics to Chilean Aquaculture,” in Metagenomics - Basics, Methods and Applicationsed, Chap. 8, ed. N. Valdes (Rijeka: IntechOpen), doi: 10.5772/intechopen.86302
Uengwetwanit, T., Pootakham, W., Nookaew, I., Sonthirod, C., Angthong, P., Sittikankaew, K., et al. (2021). A chromosome-level assembly of the black tiger shrimp (Penaeus monodon) genome facilitates the identification of growth-associated genes. Mol. Ecol. Resour. 21, 1620–1640. doi: 10.1111/1755-0998.13357
Vijayan, K. K. (2019). Domestication and Genetic Improvement of Indian White Shrimp. J. Coast. Res. 86, 270–276. doi: 10.2112/SI86-040.1
Wang, Q., Ren, X., Liu, P., Li, J., Lv, J., Wang, J., et al. (2021). Improved genome assembly of Chinese shrimp (Fenneropenaeus chinensis) suggests adaptation to the environment during evolution and domestication. Mol. Ecol. Resour. 22, 334–344. doi: 10.1111/1755-0998.13463
Yuan, J., Zhang, X., Wang, M., Sun, Y., Liu, C., Li, S., et al. (2021). Simple sequence repeats drive genome plasticity and promote adaptive evolution in penaeid shrimp. Commun. Biol. 4:186. doi: 10.1038/s42003-021-01716-y
Zhang, X., Li, X., Lu, J., Qiu, Q., Chen, J., and Xiong, J. (2021). Quantifying the importance of external and internal sources to the gut microbiota in juvenile and adult shrimp. Aquaculture 531:735910. doi: 10.1016/j.aquaculture.2020.735910
Keywords: Indian white shrimp (Penaeus indicus), Brackish water, transcriptome de novo assembly, metagenome, unigenes
Citation: Jangam AK, Katneni VK, Panigrahi A, Gangaraj KP, Nathamuni S, Angel JRJ and Shekhar MS (2022) Hepatopancreas Transcriptome and Gut Microbiome Resources for Penaeus indicus Juveniles. Front. Mar. Sci. 08:809720. doi: 10.3389/fmars.2021.809720
Received: 05 November 2021; Accepted: 20 December 2021;
Published: 03 February 2022.
Edited by:
Parin Chaivisuthangkura, Srinakharinwirot University, ThailandReviewed by:
Saengchan Senapin, National Center for Genetic Engineering and Biotechnology (BIOTEC), ThailandJitendra Kumar Sundaray, Central Institute of Freshwater Aquaculture, Indian Council of Agricultural Research, India
Copyright © 2022 Jangam, Katneni, Panigrahi, Gangaraj, Nathamuni, Angel and Shekhar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashok Kumar Jangam, QXNob2suSmFuZ2FtQGljYXIuZ292Lmlu