 Vinaya Kumar Katneni1*†
Vinaya Kumar Katneni1*† Mudagandur Shashi Shekhar1†
Mudagandur Shashi Shekhar1† Ashok Kumar Jangam1
Ashok Kumar Jangam1 Karthic Krishnan1
Karthic Krishnan1 Sudheesh K. Prabhudas1
Sudheesh K. Prabhudas1 Nimisha Kaikkolante1
Nimisha Kaikkolante1 Dushyant Singh Baghel2
Dushyant Singh Baghel2 Vijayan K. Koyadan1
Vijayan K. Koyadan1 Joykrushna Jena3Trilochan Mohapatra3
Joykrushna Jena3Trilochan Mohapatra3
- 1Nutrition Genetics and Biotechnology Division, Indian Council of Agricultural Research-Central Institute of Brackishwater Aquaculture, Chennai, India
- 2Nucleome Informatics Pvt. Ltd., Hyderabad, India
- 3Indian Council of Agricultural Research, New Delhi, India
Penaeid shrimp fishery and culture is a commercial enterprise contributing to employment, nutritional security and foreign exchange of developing countries. The genetic improvement programs being operated in shrimp benefit hugely from genomic resources. We report here a high-quality genome assembly for a penaeid shrimp, Penaeus indicus, which is the only Crustacean assembly to meet the reference standards of 1 and 10 Mb N50 lengths for contigs and scaffolds, respectively, among genomes of >1.5 Gb assembly length. The assembly is 1.93 Gb length (34.4 Mb scaffold N50) with 28,720 protein-coding genes and 49.31% repeat elements. The P. indicus assembly has 31.99% of simple sequence repeats, the highest among sequenced animal genomes. In comparison to other shrimp genomes having short contig lengths, the P. indicus assembly has 346 un-gapped contigs of over 1 Mb length and betters other shrimp genomes on sequence contiguity. This contiguous genome revealed 15,563 coding single nucleotide polymorphisms (SNPs) of which 2,572 are non-synonymous. The assembly and the SNP data resources have applications to genetic improvement programs, evolutionary studies and stock management.
Introduction
Farmed shrimp are important contributors of seafood, provide nutritional security, support employment opportunities and are an export commodity earning foreign exchange for many developing countries. About 83% of the 6.55 million tonnes of global farmed shrimp production in 2019 (FAO, 2020) is contributed by a single species, Penaeus vannamei. Though P. vannamei is not a native species, several shrimp producing countries are importing the broodstock of this species to breed locally and produce post-larvae required for commercial cultures. Availability of genetically improved and specific pathogen free stocks is the main reason in choosing P. vannamei for shrimp production. Such global dependence on a single species is not an ideal scenario for sustainability of shrimp farming industry. There is a need to develop and promote other shrimp species that have natural distribution in shrimp producing countries. For example, Penaeus indicus has wide natural distribution in the Indo-West Pacific: East and South East Africa to South China, New Guinea and North Australia (Holthuis et al., 1980). Development of local shrimp species brings diversity required for sustainability and prevents inter-country disease spread through shrimp movement. The future genetic improvement programs with focus on species like P. indicus would benefit global aquaculture with increased productivity and sustainability. The shrimp genetic improvement programs benefit hugely from genomic resources. With the genomics revolution, there is great interest to decipher the whole genome sequence with an aim to integrate genomic information into breeding programs being operated to improve desired economic traits. In this line, we have already developed full-length transcript data for P. indicus, a valuable resource for functional studies (Katneni et al., 2020).
Few challenges like large genome size ranging from 2.14 to 2.91 Gb (Swathi et al., 2018), large number of chromosomes (Chow et al., 1990), high percentage of repetitive sequences (∼80%) and high genome heterozygosity (Yu et al., 2015) might be the reasons for delay in deciphering a shrimp genome till 2019. Also, there was difficulty in preparation of high quality genomic DNA and large-insert bacterial artificial chromosome (BAC) libraries due to presence of mucopolysaccharides, alkaline phosphatase, and other secondary metabolites in shrimp (Zhang et al., 2010). It was only very recently that the genome assemblies of three shrimp, P. vannamei (Zhang X. et al., 2019; Yuan et al., 2021), Penaeus monodon (Uengwetwanit et al., 2021), and Penaeus chinensis (Yuan et al., 2021) were successfully reported. The assembly of P. vannamei genome illustrated the high repetitive content in shrimp genome and utility of assembly tools like WTDBG (Ruan and Li, 2020) for such cases. The assembly presented for genome of P. monodon has merit in covering >90% of full length. All these three reported genomes of P. vannamei, P. monodon, and P. chinensis contains shorter contigs with contig N50 values of 58, 79, and 59 Kb, respectively and some of the assemblies for example P. vannamei was later refined and updated for better parameters (Yuan et al., 2021). Only the genome assembly of P. monodon contained un-gapped contigs (n = 3) of over 1 Mb length. As reflected from contig N50 lengths, the available shrimp genomes are low in sequence contiguity, which is essential for accurate prediction of repetitive elements and protein-coding gene models in the genome. Benefits of contiguous assembly in repeat/gene annotation and resolution of complex regions/polymorphisms have been demonstrated in animal and plant genomes (Kalbfleisch et al., 2018; Michael et al., 2018; Low et al., 2019; Perumal et al., 2020). Therefore, taking advantage of the developments in long-read sequencing technologies and assembly algorithms, the present study was conducted with an aim to generate a contiguous genome assembly for P. indicus shrimp.
We report here a very high quality genome assembly of P. indicus covering 1.93 Gb with contig N50 of 1.4 Mb having very high number of 346 un-gapped contigs of over 1 Mb length and scaffold N50 of 34.4 Mb. Considering only the large genomes of >1.5 Gb length, the assembly presented for P. indicus is the only Crustacean genome and one among the only nine Invertebrate genomes sequenced so far, to meet the reference standard of 1 Mb contig N50 and 10 Mb scaffold N50 lengths (Reference Standard For Genome Biology, 2018). The assembly was generated with Pacbio subreads, polished for indels with Illumina paired-end reads and scaffolded with HiC chromatin interaction data. We also report 2,572 high quality, non-synonymous coding single nucleotide polymorphisms (SNPs) identified for the first time in a finished genome assembly of shrimp. The contiguous assembly and the non-synonymous substitution data resources presented here have applications to genetic improvement programs, stock management and ecology and evolutionary studies in species of commercial significance.
Materials and Methods
Pacbio Library Preparation and Sequencing
The high molecular weight genomic DNA was isolated from muscle tissue of a single shrimp using QIAGEN genomic-tip 100/G kit (Qiagen, Germany). The size selection was done according to the protocol described under “Procedure and Checklist–20 Kb template preparation using BluePippin size selection system.” The sequencing libraries were prepared using SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, United States) and assessed for quality and quantity using the Pippin pulse field inversion gel electrophoresis system (Sage Science, United States). The sequencing was performed on Pacific Biosciences Sequel system using magnetic bead loading and 600-min movies. The Pacbio subreads of 5 Kb and longer were used to generate primary contig-level assembly.
Illumina Sequencing
For short-reads sequence data, 15 paired-end sequencing libraries (5 each with 350, 550, and 650 bp insert size) were prepared using Illumina Truseq Nano DNA Library Prep Kit (Illumina, United States). The PCR enriched libraries were sequenced on Illumina NextSeq500 using 2 × 150 bp chemistry in paired-end mode. The raw reads were trimmed for adapters and poor-quality bases/reads using a sliding window algorithm in paired-end mode as implemented in Trimmomatic V0.36 (Bolger et al., 2014). The trimmed reads equal to or longer than 75 bases only were further used for polishing (correction of base errors and indels) the contigs in primary genome assembly.
Arima HiC Data Generation
For HiC data, the library was prepared using Arima Hi-C kit (Arima Genomics, United States). Tissue crosslinking and proximity ligation was performed following Arima Hi-C animal tissue protocol. The Illumina sequencing library was prepared from proximally ligated DNA using Swift Accel-NGS 2S plus kit (Swift Biosciences, United States) following manufacture’s guidelines. The library was sequenced on Illumina NovaSeq6000 platform with 150 bp paired-end mode. The HiC reads were used for scaffolding the contigs in primary assembly.
RNA Sequencing
Total RNA was extracted from the gill, hepatopancreas, muscle, pleopod and heart tissues using TRIzol method and utilized for cDNA synthesis. The cDNA sequencing libraries were prepared following the protocol of “Sure select strand-specific RNA library prep for Illumina multiplexed sequencing” (Agilent Technologies, United States). Sequencing was performed on Illumina NextSeq500 with 2 × 150 bp paired-end chemistry. Similar procedures were followed to generate pooled-RNAseq datasets for gill, hepatopancreas and muscle tissues, wherein, each sample was derived by pooling cDNA from nine different shrimp (three each from Chennai, Kanyakumari, and Puri Coast of India). The Pacific Biosciences Iso-Sequencing data for gills, hepatopancreas, muscle and pooled larvae of P. indicus as described previously (Katneni et al., 2020) along with the RNAseq data were used for genome annotation and the pooled-RNAseq data were used for identification of SNPs in candidate transcripts.
Genome Assembly
The Pacbio subreads with a minimum length of 5 Kb at 73× coverage were used in WTDBG2.5 (Ruan and Li, 2020) to generate a de novo contig-level assembly. To improve the quality of the genome, the contigs were polished in two steps, using Pacbio subreads in first step and Illumina short-reads in the second. In first step, polishing of contigs was performed with the Arrow algorithm of variantCaller tool1 using Pacbio subreads. In the second step, POLCA tool (Zimin and Salzberg, 2020) was used with Illumina short-reads to correct error bases and indels in contigs. The polished contigs were then scaffolded with 3D-DNA pipeline (Dudchenko et al., 2017) using HiC reads to generate the final draft assembly.
Assessing Quality of Assembled Genome
The quality of the final assembly was evaluated by aligning the Pacbio subreads and Illumina short-reads on to the genome scaffolds using bwa-mem v0.7.15-r1140 (Li and Durbin, 2009) and bowtie2 v2.3.4.3 (Langmead and Salzberg, 2012), respectively. Assembly evaluation based on alignment statistics was also performed with Illumina RNAseq reads and Pacbio IsoSeq transcripts using Hisat2 v2.2.0 (Kim et al., 2019) and GMAP v2020-06-30 (Wu and Watanabe, 2005), respectively. In addition, the genome was assessed for completeness by benchmarking against the arthropoda_odb10 (September 10, 2020) dataset of BUSCO single-copy orthologs (Seppey et al., 2019). Further, the assembly of P. indicus was compared with other shrimp genomes for assessment of sequence contiguity based on un-gapped contig length distribution and number of gaps in the finished genomes.
Repeat Annotation and Masking
Homology-based annotation of repeat elements was performed with Penaeidae subset (Taxonomy ID:6685) of RepBase library using the RMBlast search of RepeatMasker (Jurka et al., 2005)2 module implemented in OmicsBox v1.3.11 (Bioinformatics, 2019). For utility in gene prediction, the genome was soft masked for repeat regions with the same procedure excluding the low complexity and simple repeats.
Protein-Coding Gene Prediction and Annotation
Structural annotation of protein-coding regions in P. indicus genome was carried out by combining the gene models obtained from short-read RNAseq data, long-read IsoSeq data and proteins from related species (Supplementary Table 1), with ab initio gene predictions. The ab initio gene models were predicted on masked genome using Augustus v3.3.3 (Stanke et al., 2006) and GeneMark-ES v4.59 (Lomsadze et al., 2005) self training module. While predicting gene models in Augustus, hints generated by aligning IsoSeq data on to the genome using GMAP v2020-06-30 (Wu and Watanabe, 2005) were also given as input. The PASA v2.4.1 (Haas et al., 2003) was used to generate valid gene structures from IsoSeq data which utilizes near perfect alignments made by GMAP v2020-06-30 and BLAT v36 (Kent, 2002; Wu and Watanabe, 2005) to the genome. Similarly RNAseq data was aligned in a splice aware manner to genome using Hisat2 v2.2.0 (Kim et al., 2019) and gene models were generated using StringTie v2.1.4 (Pertea et al., 2015), from which likely coding sequences were identified using TransDecoder v5.5.0.3 Proteins from the related species were aligned to the genome using GenomeThreader v1.7.3 (Gremme, 2012) to derive valid gene models. All these ab initio and evidence based predicted gene models were combined as a weighted matrix in Evidence Modeler (Haas et al., 2008) to obtain the final set of non-redundant consensus gene models. Homology based annotation was performed using blastx against non-redundant protein database of Genbank and UniProt database to obtain functional description of the genes. The Interproscan and EggNOG Mapper module of OmicsBox v1.3.11 were used to obtain annotations of protein domains and orthology based annotations respectively (Bioinformatics, 2019). The annotations were merged and the final gene ontology annotations were obtained using OmicsBox v1.3.11. Pathway maps for the annotated genes were generated by mapping against the KEGG database (Kanehisa and Goto, 2000).
Gene Family and Phylogenetic Analysis
Protein-coding gene sets of 17 species in the phyllum, Arthropoda (Supplementary Table 2) including the P. indicus were subjected to gene family analysis using OrthoMCL v2.0.9 (Fischer et al., 2011). For this analysis, other Metazoan species belonging to the phylla, Mollusca, Chordata, and Echinodermata were also included. The datasets downloaded from NCBI4 were filtered based on length (minimum 50 amino acids) and selection of the longest isoform. Good protein list from 21 species was subjected to an all-versus-all search using blastp (Altschul et al., 1990) and then single copy orthologous gene (SCOG) sequences were extracted from orthomcl groups. MUSCLE v3.8.1551 (Edgar, 2004) was used to generate multiple sequence alignments which were then trimmed using “trimAl” v1.4 (Capella-Gutiérrez et al., 2009) tool. The FASconCAT v1.04 (Kück and Meusemann, 2010) was used to concatenate the alignments, ProtTest v3.0 (Darriba et al., 2011) was used to find the best evolutionary model and RAxML v8.2.12 (Stamatakis, 2014) was used to build Maximum Likelihood tree. Visualization of the tree was done using FigTree v1.4.4 (Rambaut, 2009).
Variant Calling in Coding Sequences
Pooled-individual RNAseq datasets were trimmed with Trimmomatic v0.39 (Bolger et al., 2014) to remove poor quality reads and bases. Good quality reads were aligned using TopHat v2.1.1 (Trapnell et al., 2012) on to the gene sequences indexed with bowtie2 v2.3.4.3 (Langmead and Salzberg, 2012). The generated bam file was sorted using SAMtools v1.2 (Li et al., 2009) and then processed in bcftools-1.3.1 (Li, 2011) to generate variant call format (VCF) file. Those SNPs with a raw read depth of ≥20 at SNP site, at least 10 reads each supporting the reference and alternative alleles and phred quality scores of ≥100 were extracted from VCF file as good quality SNPs. The non-synonymous coding SNPs were analyzed for the possible functional impact on the proteins harboring them, using a standalone version of the PANTHER Coding SNP Analysis tool, PANTHER PSEP v1.01 (Tang and Thomas, 2016). The tool analyses the SNP by first identifying the Panther family of the protein using blastp (Altschul et al., 1990), followed by retrieving the ancestor sequence of the family and tracing the query protein though evolution, then reporting how long the SNP position was conserved in Millions of years. Finally based on the predicted values the results are classified as probably benign, possibly damaging or probably damaging if the values are <180, ≥180, or ≥380, respectively.
Results
Genome Assembly
Flow cytometry analysis using propidium iodide stained hemocytes indicated a genome size of 2.47 Gb for P. indicus (Swathi et al., 2018). Early attempts made by us to assemble P. indicus genome with 145× coverage of Illumina short reads sequence data were unsuccessful (Supplementary Note). Various assemblers like SOAPdenovo2 (Luo et al., 2015), CLC Genomics Workbench v10.0.1 (CLCbio, Denmark), and Platanus v1.2.4 (Kajitani et al., 2014) produced primary assemblies with large number of contigs. A final assembly reduced with Redundans v0.14a (Pryszcz and Gabaldón, 2016) contained 358,878 contigs covering about a quarter of genome length (607.78 Mb) with N50 of 1698 bases. Though short-reads could not generate a quality genome, k-mer analysis on them indicated high repetitive nature of P. indicus genome thereby suggesting the necessity of long sequence reads to assemble repeat-rich P. indicus genome.
About 73× coverage of PacBio Sequel long reads (Supplementary Table 3) generated using DNA of a P. indicus female shrimp were processed in WTDBG2.5 (Ruan and Li, 2020) to generate primary contigs. These contigs were corrected for error bases/indels in POLCA tool (Zimin and Salzberg, 2020) using Illumina reads (Supplementary Table 4). The assembled genome was of 1.98 Gb length and consisted of 12,051 contigs with N50 of 1.4 Mb (Table 1). Among large crustacean genomes of more than 1 Gb size, only the genome of Eriocheir sinensis (Tang et al., 2020) has a better contig N50 length (3.16 Mb) than obtained for P. indicus. Compared to the other shrimp genomes generated for P. vannamei (Zhang X. et al., 2019; Yuan et al., 2021), P. monodon (Uengwetwanit et al., 2021), and P. chinensis (Yuan et al., 2021), the assembly obtained for P. indicus has 24–, 24–, and 18-fold improvement, respectively for contig N50 length.
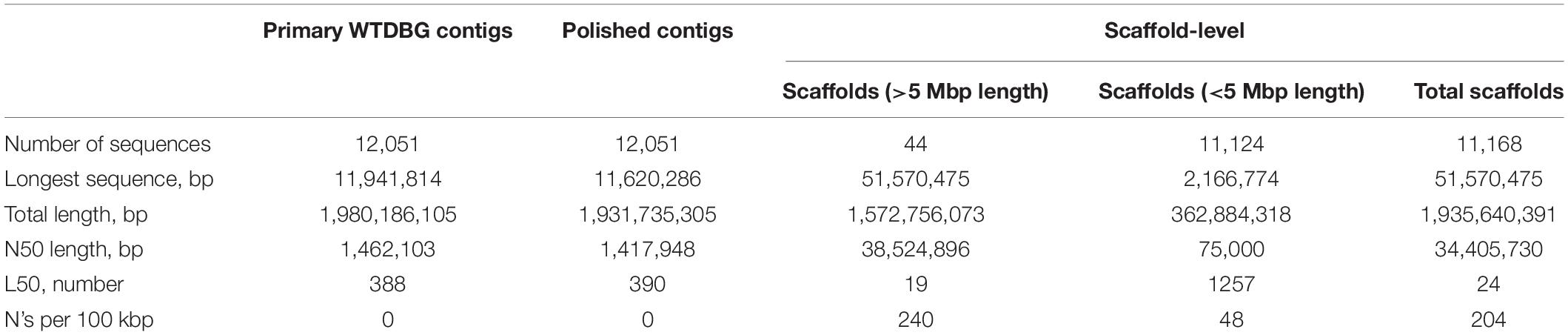
Table 1. Summary statistics for assembled genome of P. indicus.
The scaffolding of polished contigs with 104× coverage (1.7 billion reads/258 Gb) of HiC reads in 3D-DNA (Dudchenko et al., 2017) resulted in an assembly with 11,168 scaffolds. The final assembly was of 1.935 Gb length (∼78% of genome) with N50 and longest scaffold length of 34.4 and 51.57 Mb, respectively (Table 1). Final assembly obtained for P. indicus had 44 scaffolds that were longer than 5 Mb, which equaled the reported haploid chromosome number (Figure 1). These 44 longest scaffolds assumed as pseudochromosomes span about 1.57 Gb length and cover about 81.3% of the assembly (Supplementary Table 5). The genome length in P. indicus that is accounted by the pseudochromosomes alone is almost same as the total genome length presented for P. vannamei and P. chinensis (Yuan et al., 2021). The assembly obtained for P. indicus genome in the present study is the only shrimp genome that meets the reference standard of 1 Mb contig N50 for primary contigs and 10 Mb scaffold N50 length for final scaffolds (Figure 2A).

Figure 1. Assembly of P. indicus genome. (A) Circos plot depicting the properties of 44 pseudochromosomes. Track 1 (outer-most): the 44 pseudochromosomes. Track 2: line diagram of contigs with peaks indicating the size of contig. Track 3: protein-coding genes plotted as heatmap with progressive colors (yellow–orange–red) indicating length of genes. Track 4: protein-coding genes on positive strand. Track 5: protein-coding genes on negative strand. Track 6 (innermost): GC content of the genome as a histogram in gray color, here GC values <30 are shown in red and >40 are shown in blue. (B) The longest scaffolds representing the pseudochromosomes as obtained with 3D-DNA tool.
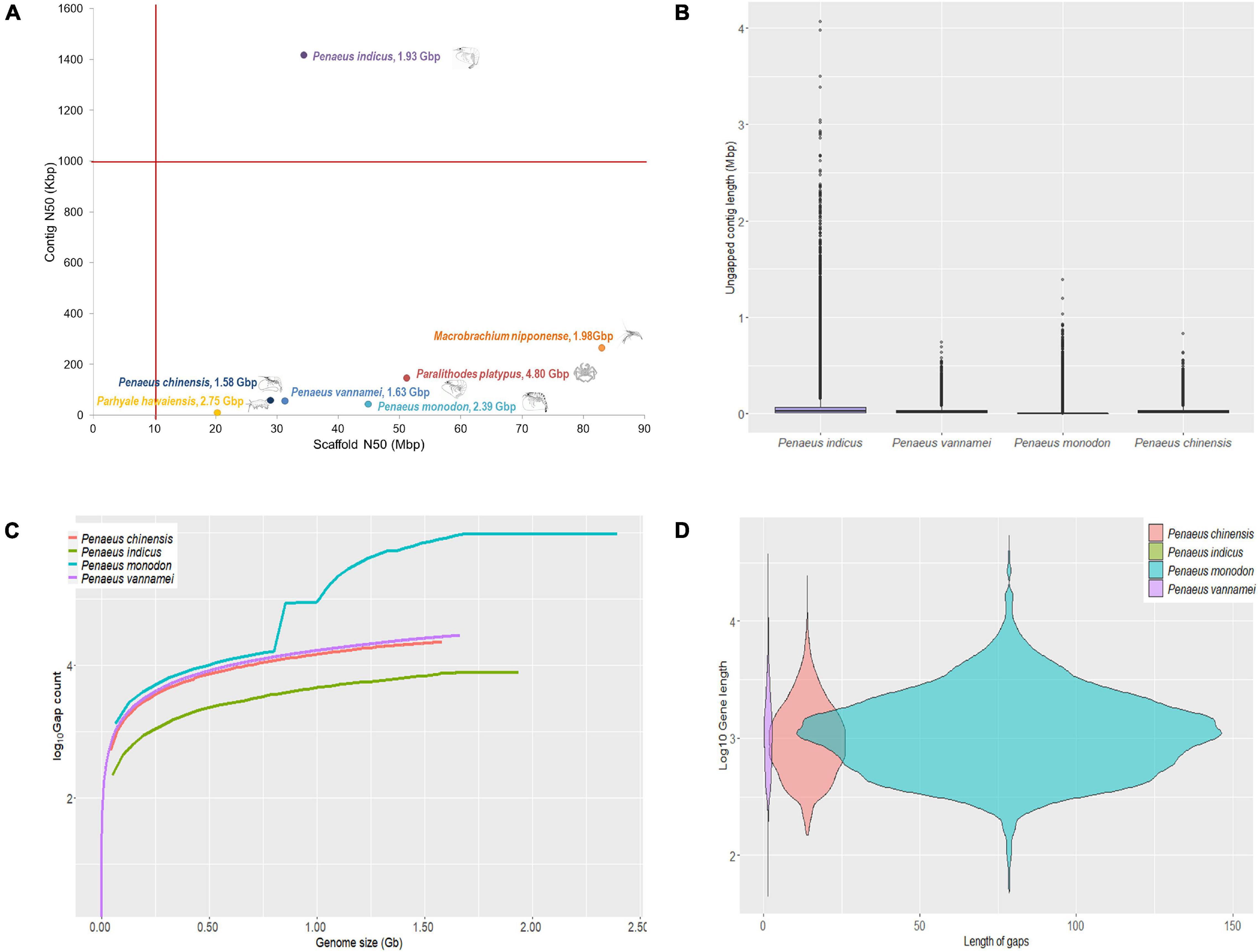
Figure 2. Quality assessment of assembly generated for P. indicus in comparison to the other shrimp assemblies. (A) Plot of contig N50 and scaffold N50 lengths of finished large (>1.5 Gb) crustacean genomes. The P. indicus is the only large crustacean genome assembly that has >1 Mb contig N50 and >10 Mb scaffold N50 lengths. (B) Un-gapped contig length distribution for finished shrimp genomes. Only the genome assembly of P. indicus has contigs longer than 1 Mb length. (C) Plot of the number of gaps (log10 scale) in the finished genomes of shrimp species. The X-axis is the cumulative genome size by counting the individual scaffold lengths in decreasing order. The Y-axis is the total number of gaps within the scaffolds contributing to the corresponding genome size. Notice that the assembly of P. indicus has less number of N’s than others. (D) Violin plot of number of N’s in gene sequences from finished genomes of shrimp plotted against the corresponding gene lengths (log10 scale). Notice that the assembly of P. indicus has no N’s whereas the assembly of P. monodon has highest number of N’s in gene sequences.
Genome Assembly Validation
Benchmarking of the P. indicus genome with arthropoda_odb10 (September 10, 2020) BUSCO gene groups (n = 1013) indicated the presence of 77% of complete orthologs in the genome. About 3.5% of BUSCO orthologs were fragmented and the remaining 19.5% were missing. Against the same dataset, the P. vannamei genome (Zhang X. et al., 2019) had 76% complete, 4.1% fragmented, and 19.6% missing BUSCO orthologs. In comparison, the proportion of missing genes reduced to 14.4% if odb9 dataset (n = 1066) was used for benchmarking. On the other hand, the P. indicus genome when assessed with gVolante 1.2.1 (Nishimura et al., 2017) against CEG ortholog set in CEGMA pipeline (Parra et al., 2007), has a completeness score of 98.39%. We observed that the completeness scores changed with the orthologous gene set and the data release associated with it. We suggest exercising caution to draw conclusions while directly comparing quality of genomes based on different gene sets. Moreover, with the shrinkage of orthologous gene datasets due to increased number of sequenced genomes, we opine that their usage soon becomes debatable.
Read mapping statistics indicated high quality of the assembly with high percentage of reads aligning on to the genome (Supplementary Table 6). For DNA data, about 98.89% of Pacbio reads and 95.4% of Illumina reads could be aligned back to the genome scaffolds. For RNA-based data, about 94.29% of RNAseq reads and 99.25% of Pacbio IsoSeq transcripts could be mapped on to the genome. High quality of the P. indicus genome assembly presented in this study has been demonstrated with high mapping statistics and BUSCO completeness scores as comparable to other shrimp genomes.
The sequence contiguity assessed based on un-gapped contig lengths and the number of gaps in assembly is another quality metric that we used in this study to assess the quality of P. indicus in relation to other shrimp genomes. A distribution of un-gapped contig lengths (Figure 2B) indicated that P. monodon is the only shrimp genome other than the P. indicus that contained un-gapped contigs (n = 3) of over 1 Mb length. The P. indicus assembly has 346 un-gapped contigs of 1 Mb or higher length. Again, a plot of the number of gaps (Figure 2C) in the finished assembly also tables P. indicus genome over other shrimp genomes on assembly quality. Interestingly, the chromosome-scale assembly presented for P. monodon was found to have high gap number in the finished assembly. As observed in the Figure 2C, the intermittent elevations in P. monodon line plot indicate the presence of high number of gaps in some scaffolds. The coding sequences also were observed to contain N’s in P. monodon assembly while none existed in the P. indicus assembly (Figure 2D).
Repeat Content
The repeat elements as derived on the basis of the number of bases masked, constituted 49.31% (954 Mb) of the assembled genome (Table 2). One of the prominent features was a high proportion of simple sequence repeats (SSR) which spanned 31.99% of the genome. The proportion of SSRs reported here for P. indicus was found to be the highest amongst all sequenced genomes in the animal kingdom. The role of SSRs in adaptive evolution was recently demonstrated for shrimp (Yuan et al., 2021). Other major repeat classes include LINEs (5.8%) and low complexity regions (4.57%). The satellites (1.36%), LTR elements (0.31%), DNA transposons (0.2%), and small RNA (0.07%) were the other minor repeat families observed in the P. indicus genome assembly.
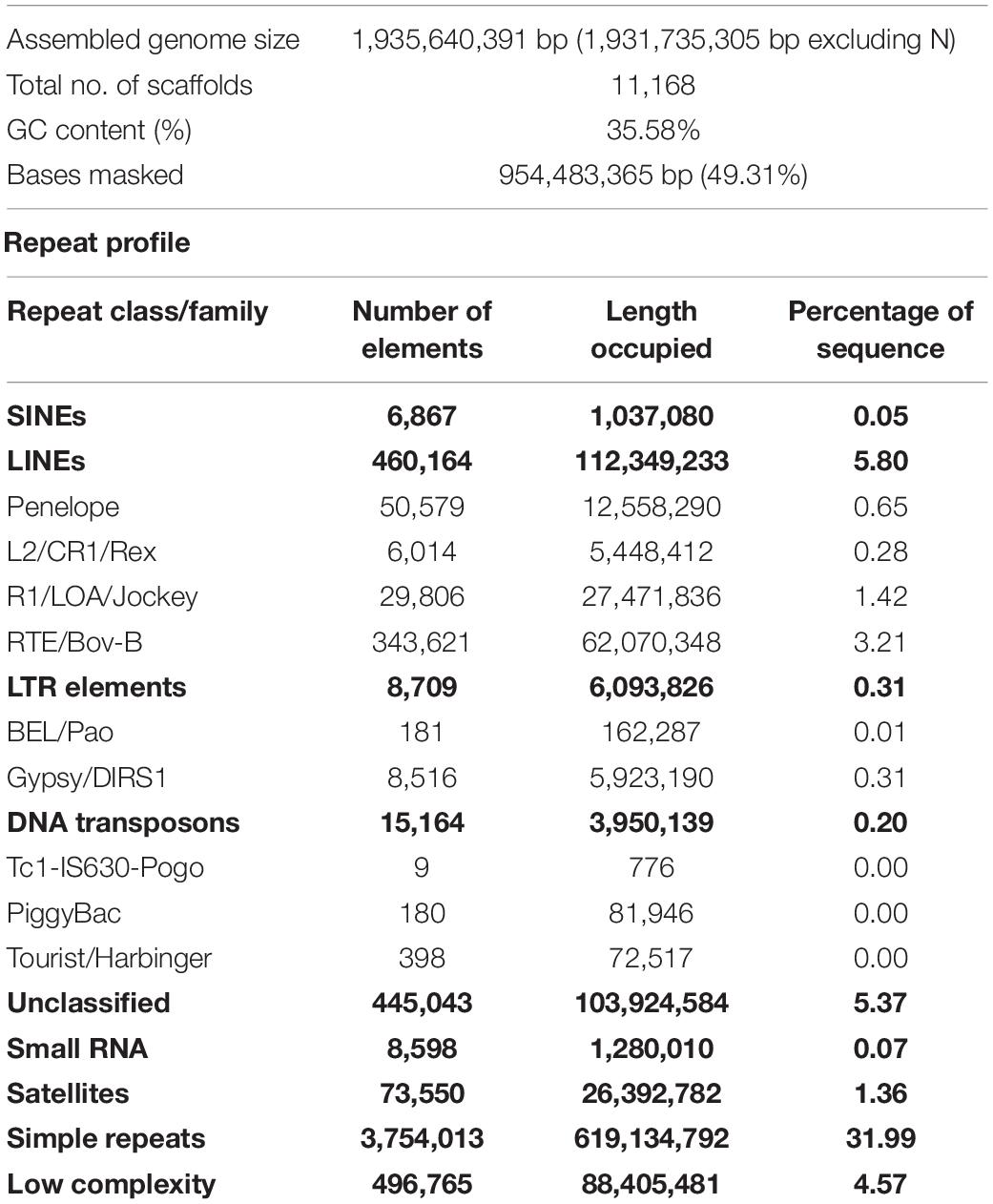
Table 2. Repeat profile in assembled P. indicus genome.
Gene Prediction and Annotation
Combined evidence from ab initio gene prediction, Illumina RNAseq data, Pacbio Iso-Sequencing data and protein sequences from related species, identified 28,720 protein-coding genes in the P. indicus genome (Figure 3). The predicted protein-coding gene number was higher than P. chinensis (26,343) and P. vannamei (25,596) genomes but lower when compared to P. monodon (30,038) genome. The mean exon and intron lengths were 259 and 2315 bp, respectively. The longest gene, exon and intron lengths were 98,168, 14,941, and 76,392 bp, respectively (Table 3). Overall, 81.79% of the predicted genes had evidence from RNAseq data or IsoSeq data or proteins from related species. Functional annotations yielded results for 98.36% of the predicted genes using Interproscan, non-redundant protein database of Genbank and the UniProt database. For majority of the genes, the P. vannamei was the top hit species showing homology (Supplementary Figure 1).
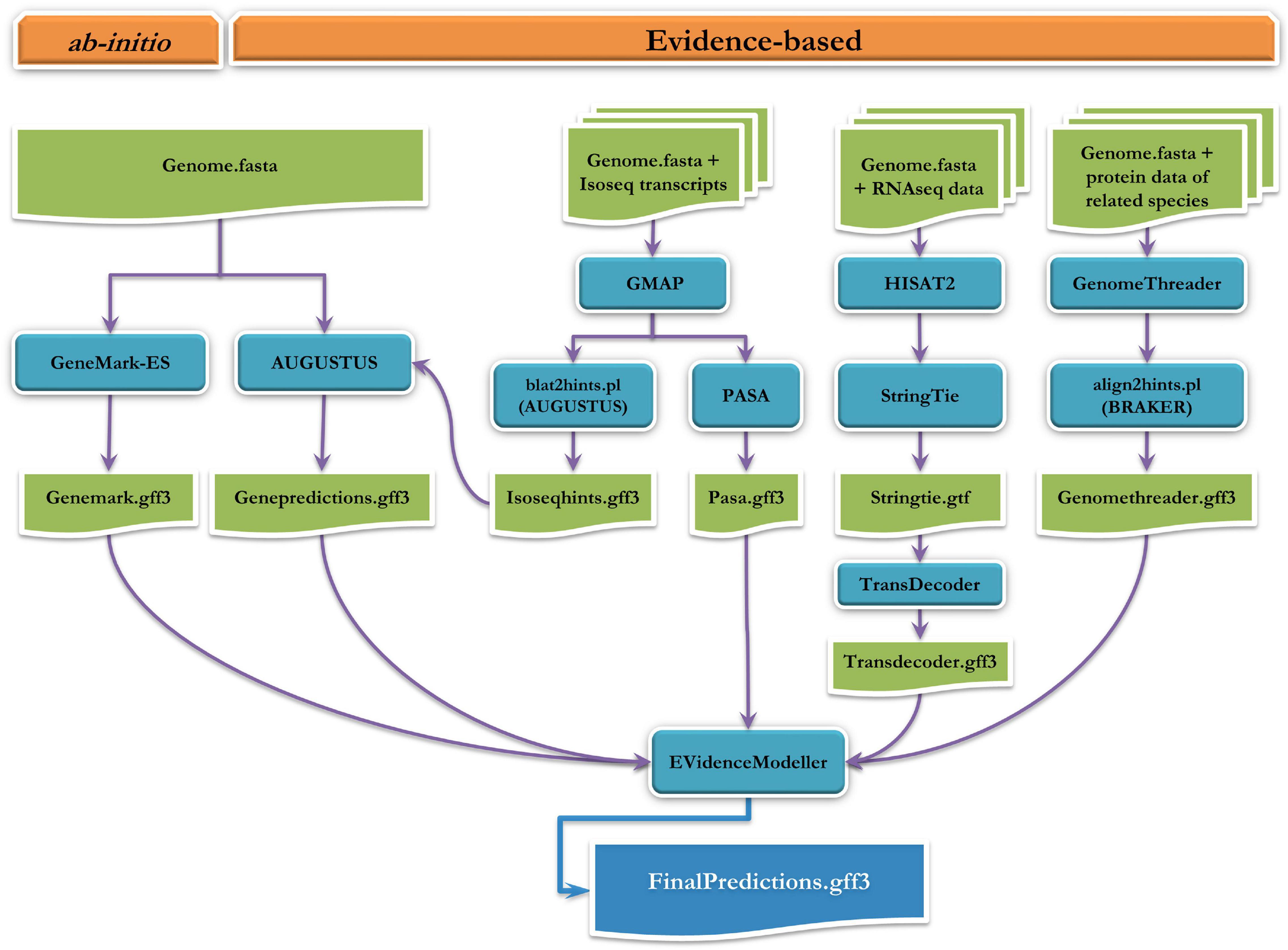
Figure 3. Workflow followed for predicting protein-coding genes in assembly of P. indicus genome.

Table 3. Properties of predicted genes in P. indicus genome.
Gene Family Analyses and Phylogenetic Relations
The gene family analyses with protein sequences of 21 species including P. indicus identified 148 single-copy orthologous genes amongst them. Out of 399,313 genes subjected to the analyses, 81.75% (326,455) were clustered into 35,611 orthogroups and 18.25% (72,858) were singletons (Supplementary Figure 2). About 1,504 orthogroups were shared by all the 21 species. In P. indicus, of the 9595 orthologous gene families, maximum number were found sharing with P. monodon (8387) followed by P. vannamei (8255) and P. chinensis (7928). We found that 6,722 orthologous gene families were shared among the four shrimp species and 1,987 gene families only among them. The phylogenetic tree generated with sequences of single-copy orthologous genes depicted three distinct clades representative of Chelicerates, Crustaceans, and Hexapods (Figure 4).
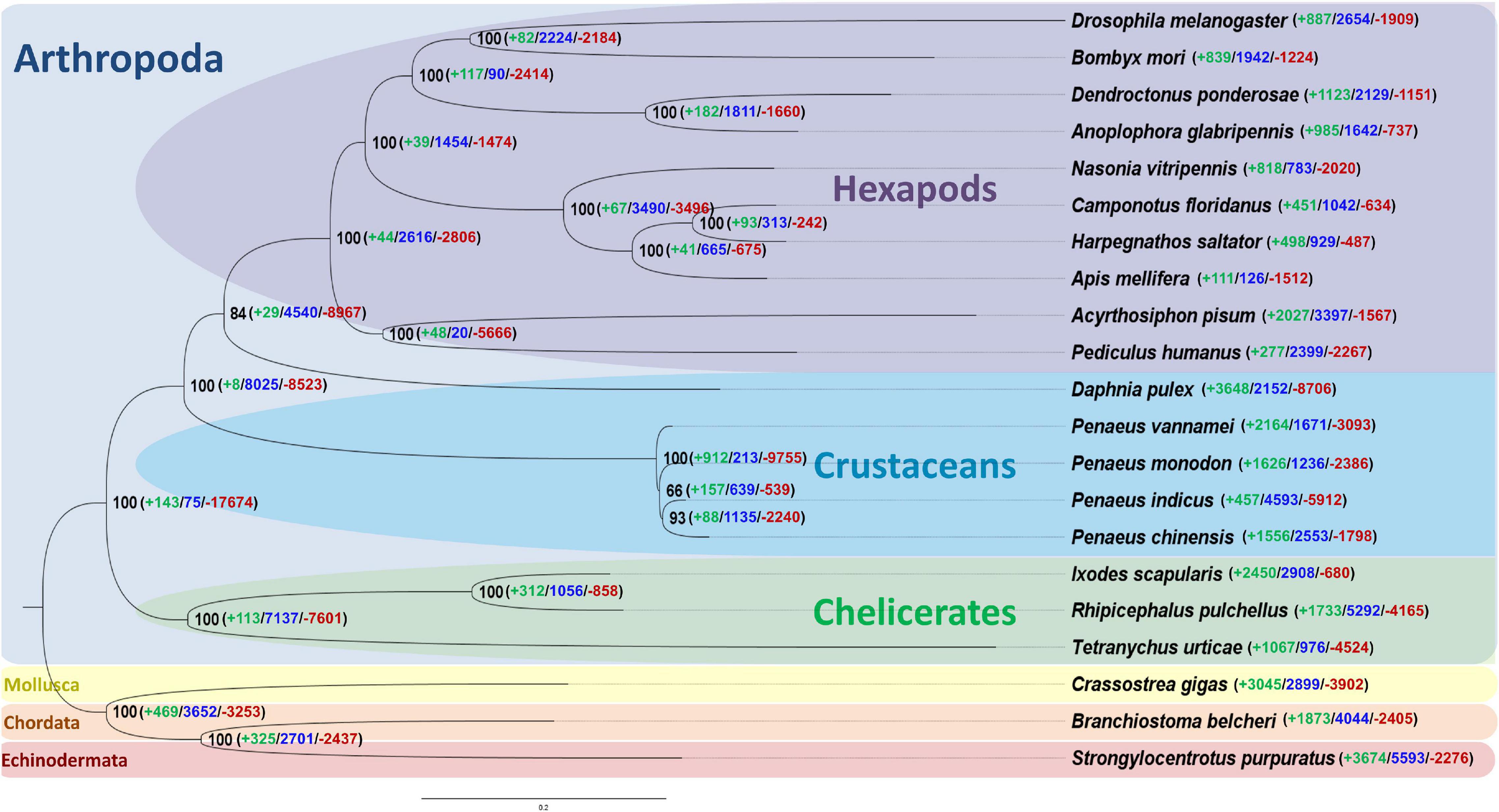
Figure 4. Phylogenetic placement of Penaeus indicus among Arthropods. Numbers at the nodes are bootstrap values and those given in the parenthesis indicate number of gene families that were expanding (green), rapidly evolving (blue), and contracting (red).
Coding Single Nucleotide Polymorphisms
Exploiting the pooled-sample RNA sequencing approach, the study reports 15,554 coding SNPs present in 3,965 different protein-coding genes in P. indicus genome (Supplementary Table 7). Minimum raw-read depth of 20, minimum evidence of 10 reads for each of the alleles and minimum phred quality of 100 were the criteria followed for short-listing the good quality SNPs. About one-third of the genes (n = 1185) had only one SNP and 283 genes had ≥10 SNP positions (Supplementary Figure 3). Majority of the SNPs were transversions (n = 13,655) than transitions (n = 1,899). Of 15,554 only 2,571 SNPs situated in 1,262 unique gene sequences were non-synonymous in nature contributing to amino acid polymorphic sites in the resulting proteins. Among the non-synonymous SNPs, majority were observed to be transversions (n = 2038) rather transitions (533). Of the total SNPs, 28% of transition and 15% of transversion substitutions were observed to be non-synonymous in nature (Table 4). As observed, a transition substitution has more chances of becoming non-synonymous than transversion. The PANTHER PSEP v1.01 (Tang and Thomas, 2016) tool classified 76 of these non-synonymous SNPs into probably benign (19), possibly damaging (11), and probably damaging (46) as shown in Supplementary Table 8 and Supplementary Figure 4. The tool was unable to determine the score for the remaining non-synonymous SNPs either due to a mismatch of the amino acid at the mentioned position between the query sequence and the panther family sequence or the absence of the panther family to the given query sequence.

Table 4. Single nucleotide polymorphisms (SNP’s) present in protein coding genes of P. indicus.
Discussion
One of the foremost requirements suggested for benchmarking genome assemblies (Reference Standard For Genome Biology, 2018) is to have a N50 size of at least 1 Mb for contigs and 10 Mb for scaffolds, in addition to other quality metrics concerning base error rates, structural variants and chromosome level phased assembly. It is interesting to observe a very few of the available Crustacean genomes satisfy this benchmark based on N50 statistics (Supplementary Table 9). These include the genomes assembled for P. indicus (1.93 Gb, this study), E. sinensis (1.27 Gb), Lepeophtheirus salmonis (0.67 Gb), and Eulimnadia texana (0.12 Gb). The assembly presented for P. indicus in this study is the largest Crustacean genome as on date to meet these quality standards. There are other genomes of crustaceans that are superior to P. indicus assembly in terms of scaffold N50 metric but no other large Crustacean genome (of >1.5 Gbp) has a contig N50 of >1 Mb which is the minimum requirement as per the suggested standards. A combination of primary assembly with Pacbio Sequel subreads, error correction with high quality Illumina reads and scaffolding with Arima HiC reads resulted in a highly contiguous assembly reported so far for a shrimp species. Considering only the large genomes with assembly length of >1.5 Gb, the P. indicus assembly is one among the only nine Invertebrate genomes sequenced so far to meet the reference standard of 1 Mb contig N50 and 10 Mb scaffold N50 lengths (Supplementary Tables 10, 11).
Previous attempts to assemble genome with short reads in other shrimp species such as P. monodon (Yuan et al., 2018; Van Quyen et al., 2020) and Marsupenaeus japonicus (Yuan et al., 2018) also produced a highly fragmented assembly like that of P. indicus. For example, the genome of P. monodon consisting of over a million scaffolds with N50 of 1756 bp and covering just above 60% of genome length was reported to have a BUSCO score of 96.8% (Van Quyen et al., 2020). Recently, the chromosome scale genome assembly (92% coverage and N50 of 44.86 Mb) presented for P. monodon has 94.7% BUSCO score using Eukaryota odb9 dataset which is lower than the score obtained for a highly fragmented assembly. Similarly for P. vannamei assembly (Zhang X. et al., 2019), the missing BUSCO orthologs were 19.6% when benchmarked against arthropoda_odb10 whereas the missing orthologs were only 5.2% if arthropoda_odb9 dataset was used (Supplementary Figure 5). Therefore, we suggest not emphasizing BUSCO completeness scores for fragmented assemblies. Similar opinion was expressed while comparing the latest chromosome scale assembly of water buffalo genome against the previous highly fragmented assembly (Low et al., 2019).
The repeat content in the four shrimp genomes (including of P. indicus in this study) assembled so far ranged from 48.58 to 62.50% (Supplementary Table 12). It can be firmly established that shrimp genomes are characterized by high proportion of simple repeats whose origin and role remains intriguing. The genomes of P. chinensis and P. vannamei have higher proportion of DNA transposons and low complexity repeats than other shrimp. Whereas P. monodon contains more genome length spanning SINEs, LINEs, LTR elements and unclassified repeats compared to other shrimp genomes. As the assembly length varies (1.58–2.39 Gb) among shrimp genomes, a comparison in terms of number of bases would be more appropriate rather on proportions. Among shrimp, though the repeat content varied between 768 and 1498 Mb, the non-repeat portion of the genome remained uniform between 813 and 981 Mb. The genome of P. monodon with the largest assembly length also has higher repeat length. The added evidence about presence of higher orthologous genes content in 1.66 and 1.58 Gb length of P. vannamei and P. chinensis genomes, respectively might indicate a higher proportion of repeat elements in the unassembled portion of these genomes. Nevertheless, assessing on proportion of assembled length or on actual base count, the P. indicus genome has the highest length of simple repeats among shrimp genomes. The high SSRs in the genome of P. indicus may be attributed to the sequence contiguity as shorter repeats get resolved in longer contigs. It is fascinating to observe high SSR content within the coding genes of P. indicus in comparison to other shrimp genomes (Supplementary Table 13 and Supplementary Figure 6). The SSR spans about 7.56% of coding sequences in P. indicus as against 1.12–2.29% in other shrimp. The demonstrated role of SSRs in genomic plasticity of P. vannamei and P. chinensis shrimp (Yuan et al., 2021) would suggest the influence of high SSR on certain species-specific adaptive functions of P. indicus, which needs to be explored.
In shrimp, the role of SNPs was demonstrated for use in construction of linkage maps (Baranski et al., 2014; Yu et al., 2015; Jones et al., 2017) trait-specific association studies (Robinson et al., 2014; Santos et al., 2018; Zhang Q. et al., 2019; Janpoom et al., 2020), genetic characterization (Perez-Enriquez et al., 2018; Vu et al., 2020) and parentage testing (Henshall et al., 2014; Sellars et al., 2014). In this study, we report 2,572 non-synonymous SNPs, of which 46 might have potential to impact the functions of coding proteins. Earlier in P. monodon, majority of coding SNPs identified through pooled-sequencing approach proved to be real polymorphic sites and useful for QTL finding (Robinson et al., 2014). Therefore, we believe that the SNPs identified in this study with further stringent criteria would be real SNP sites with potential applications in genome-wide association studies.
Conclusion
We report the assembly of P. indicus genome which is the largest Crustacean genome assembly reported so far to meet the 1 Mb contig N50 and 10 Mb scaffold N50 quality metrics. The protein-coding gene prediction strategy followed in the current study which combines evidence from RNAseq, IsoSeq, ab initio methods and proteins from related species, has general application to other genomes. The contiguous assembly presented here would serve as reference for future genome-guided assemblies. Continuous improvements in sequencing technologies and bioinformatics approaches shall lead to a better understanding of abundant repetitive sequences especially of SSRs in shrimp genomes. The identified non-synonymous SNPs would be a valuable resource to construct custom genotyping panels useful in genome-wide association studies.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA495105; https://www.ncbi.nlm.nih.gov/, PRJNA494937; https://www.ebi.ac.uk/ena, PRJEB38936; https://www.ncbi.nlm.nih.gov/genbank/, JAGYIC000000000; https://figshare.com/, https://doi.org/10.6084/m9.figshare.14806308.v2.
Author Contributions
TM, JJ, VKo, and MS conceived and designed the study. DB generated the sequence data. VKa, AJ, KK, SP, and NK performed the genome assembly, repeat masking, genome annotation, and cSNP identification. AJ and NK performed the gene family analyses and phylogenetic analyses. MS and VKa wrote the manuscript with inputs from all other authors. All authors have reviewed the manuscript and accepted the final version.
Funding
The work is funded by the ICAR-CRP on Genomics, Indian Council of Agricultural Research, New Delhi, India.
Conflict of Interest
DB is employed by Nucleome Informatics Pvt. Ltd., Hyderabad, India.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.808354/full#supplementary-material
Footnotes
- ^ https://github.com/PacificBiosciences/GenomicConsensus
- ^ http://repeatmasker.org
- ^ https://github.com/TransDecoder
- ^ www.ncbi.nlm.nih.gov
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Baranski, M., Gopikrishna, G., Robinson, N. A., Katneni, V. K., Shekhar, M. S., Shanmugakarthik, J., et al. (2014). The development of a high density linkage map for black tiger shrimp (Penaeus monodon) based on cSNPs. PLoS One 9:85413. doi: 10.1371/journal.pone.0085413
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chow, S., Dougherty, W. J., and Sandifer, P. A. (1990). Meiotic chromosome complements and nuclear DNA contents of four species of shrimps of the genus Penaeus. J. Crustac. Biol. 10, 29–36. doi: 10.1163/193724090X00221
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
FAO (2020). Food Agric. Organ. United Nations. http://www.fao.org/
Fischer, S., Brunk, B. P., Chen, F., Gao, X., Harb, O. S., Iodice, J. B., et al. (2011). Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinforma. 2011, 1–19. doi: 10.1002/0471250953.bi0612s35
Gremme, G. (2012). Computational gene structure prediction. dissertation Hamburg: University of Hamburg.
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22. doi: 10.1186/gb-2008-9-1-r7
Henshall, J. M., Dierens, L., and Sellars, M. J. (2014). Quantitative analysis of low-density SNP data for parentage assignment and estimation of family contributions to pooled samples. Genet. Sel. Evol. 46, 1–17. doi: 10.1186/s12711-014-0051-y
Janpoom, S., Kaewduang, M., Prasertlux, S., Rongmung, P., Ratdee, O., Lirdwitayaprasit, T., et al. (2020). A SNP of the hemocyanin gene (LvHc) is a marker for high growth and ammonia-tolerance in Pacific white shrimp Litopenaeus vannamei. Fish Shellf. Immunol. 106, 491–501. doi: 10.1016/j.fsi.2020.07.058
Jones, D. B., Jerry, D. R., Khatkar, M. S., Raadsma, H. W., Steen, H., Van Der Prochaska, J., et al. (2017). A comparative integrated gene-based linkage and locus ordering by linkage disequilibrium map for the Pacific white shrimp. Litopen. Vannamei. Sci. Rep. 7, 1–16. doi: 10.1038/s41598-017-10515-7
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kalbfleisch, T. S., Rice, E. S., DePriest, M. S., Walenz, B. P., Hestand, M. S., Vermeesch, J. R., et al. (2018). Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 1, 1–8. doi: 10.1038/s42003-018-0199-z
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Katneni, V. K., Shekhar, M. S., Jangam, A. K., Prabhudas, S. K., Krishnan, K., Kaikkolante, N., et al. (2020). Novel isoform sequencing based full-length transcriptome resource for indian white shrimp. Penaeus Indicus. Front. Mar. Sci. 7, 1–4. doi: 10.3389/fmars.2020.605098
Kent, W. J. (2002). BLAT—The BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Kück, P., and Meusemann, K. (2010). FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 56, 1115–1118. doi: 10.1016/j.ympev.2010.04.024
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357. doi: 10.1038/nmeth.1923
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079.
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506. doi: 10.1093/nar/gki937
Low, W. Y., Tearle, R., Bickhart, D. M., Rosen, B. D., Kingan, S. B., Swale, T., et al. (2019). Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-018-08260-0
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2015). Erratum to SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 4:30. doi: 10.1186/s13742-015-0069-2
Michael, T. P., Jupe, F., Bemm, F., Motley, S. T., Sandoval, J. P., Lanz, C., et al. (2018). High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 9, 1–8. doi: 10.1038/s41467-018-03016-2
Nishimura, O., Hara, Y., and Kuraku, S. (2017). GVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics 33, 3635–3637. doi: 10.1093/bioinformatics/btx445
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Perez-Enriquez, R., Robledo, D., Houston, R. D., and Llera-Herrera, R. (2018). SNP markers for the genetic characterization of Mexican shrimp broodstocks. Genomics 110, 423–429.
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Perumal, S., Koh, C. S., Jin, L., Buchwaldt, M., Higgins, E. E., Zheng, C., et al. (2020). A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 6, 929–941. doi: 10.1038/s41477-020-0735-y
Pryszcz, L. P., and Gabaldón, T. (2016). Redundans: An assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113. doi: 10.1093/nar/gkw294
Rambaut, A. (2009). FigTree. Tree Figure Drawing Tool. Available online at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed November 25, 2018).
Reference Standard For Genome Biology (2018). A reference standard for genome biology. Nat. Biotechnol. 36:1121.
Robinson, N. A., Gopikrishna, G., Baranski, M., Katneni, V. K., Shekhar, M. S., Shanmugakarthik, J., et al. (2014). QTL for white spot syndrome virus resistance and the sex-determining locus in the Indian black tiger shrimp (Penaeus monodon). BMC Genomics 15:731.
Ruan, J., and Li, H. (2020). Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158.
Santos, C. A., Andrade, S. C. S., and Freitas, P. D. (2018). Identification of SNPs potentially related to immune responses and growth performance in Litopenaeus vannamei by RNA-seq analyses. PeerJ 2018, 1–19. doi: 10.7717/peerj.5154
Sellars, M. J., Dierens, L., Mcwilliam, S., Little, B., Murphy, B., Coman, G. J., et al. (2014). Comparison of microsatellite and SNP DNA markers for pedigree assignment in Black Tiger shrimp, Penaeus monodon. Aquac. Res. 45, 417–426. doi: 10.1111/j.1365-2109.2012.03243.x
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). BUSCO: assessing genome assembly and annotation completeness in Gene prediction. New York, NY: Springer, 227–245.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313.
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: A b initio prediction of alternative transcripts. Nucleic Acids Res. 34, 435–439. doi: 10.1093/nar/gkl200
Swathi, A., Shekhar, M. S., Katneni, V. K., and Vijayan, K. K. (2018). Genome size estimation of brackishwater fishes and penaeid shrimps by flow cytometry. Mol. Biol. Rep. 45, 951–960.
Tang, B., Wang, Z., Liu, Q., Zhang, H., Jiang, S., Li, X., et al. (2020). High-quality genome assembly of eriocheir japonica sinensis reveals its unique genome evolution. Front. Genet. 10:1–9. doi: 10.3389/fgene.2019.01340
Tang, H., and Thomas, P. D. (2016). PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 32, 2230–2232. doi: 10.1093/bioinformatics/btw222
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Uengwetwanit, T., Pootakham, W., Nookaew, I., Sonthirod, C., Angthong, P., Sittikankaew, K., et al. (2021). A chromosome-level assembly of the black tiger shrimp (Penaeus monodon) genome facilitates the identification of growth-associated genes. Mol. Ecol. Resour. 2021, 1–21. doi: 10.1111/1755-0998.13357
Van Quyen, D., Gan, H. M., Lee, Y. P., Nguyen, D. D., Nguyen, T. H., Tran, X. T., et al. (2020). Improved genomic resources for the black tiger prawn (Penaeus monodon). Mar. Genomics 52:100751. doi: 10.1016/j.margen.2020.100751
Vu, N. T. T., Zenger, K. R., Guppy, J. L., Sellars, M. J., Silva, C. N. S., Kjeldsen, S. R., et al. (2020). Fine-scale population structure and evidence for local adaptation in Australian giant black tiger shrimp (Penaeus monodon) using SNP analysis. BMC Genomics 21:1–18. doi: 10.1186/s12864-020-07084-x
Wu, T. D., and Watanabe, C. K. (2005). GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875. doi: 10.1093/bioinformatics/bti310
Yu, Y., Zhang, X., Yuan, J., Li, F., Chen, X., Zhao, Y., et al. (2015). Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp Litopenaeus vannamei. Sci. Rep. 5, 1–14. doi: 10.1038/srep15612
Yuan, J., Zhang, X., Liu, C., Yu, Y., Wei, J., Li, F., et al. (2018). Genomic resources and comparative analyses of two economical penaeid shrimp species, Marsupenaeus japonicus and Penaeus monodon. Mar. Genomics 39, 22–25. doi: 10.1016/j.margen.2017.12.006
Yuan, J., Zhang, X., Wang, M., Sun, Y., Liu, C., Li, S., et al. (2021). Simple sequence repeats drive genome plasticity and promote adaptive evolution in penaeid shrimp. Commun. Biol. 4, 1–14. doi: 10.1038/s42003-021-01716-y
Zhang, Q., Yu, Y., Wang, Q., Liu, F., Luo, Z., Zhang, C., et al. (2019). Identification of single nucleotide polymorphisms related to the resistance against acute hepatopancreatic necrosis disease in the pacific white shrimp litopenaeus vannamei by target sequencing approach. Front. Genet. 10:1–11. doi: 10.3389/fgene.2019.00700
Zhang, X., Yuan, J., Sun, Y., Li, S., Gao, Y., Yu, Y., et al. (2019). Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat. Commun. 10, 1–14.
Zhang, X., Zhang, Y., Scheuring, C., Zhang, H.-B., Huan, P., Wang, B., et al. (2010). Construction and characterization of a bacterial artificial chromosome (BAC) library of Pacific white shrimp, Litopenaeus vannamei. Mar. Biotechnol. 12, 141–149.
Keywords: Indian white shrimp, complete genome, sequence contiguity, Penaeus indicus, genome annotation, coding SNPs
Citation: Katneni VK, Shekhar MS, Jangam AK, Krishnan K, Prabhudas SK, Kaikkolante N, Baghel DS, Koyadan VK, Jena J and Mohapatra T (2022) A Superior Contiguous Whole Genome Assembly for Shrimp (Penaeus indicus). Front. Mar. Sci. 8:808354. doi: 10.3389/fmars.2021.808354
Received: 03 November 2021; Accepted: 14 December 2021;
Published: 21 January 2022.
Edited by:
Taewoo Ryu, Okinawa Institute of Science and Technology Graduate University, JapanReviewed by:
Jianbo Yuan, Institute of Oceanology, Chinese Academy of Sciences (CAS), ChinaMengqiang Wang, Ocean University of China, China
Camilla Alves Santos, University of São Paulo, Brazil
Copyright © 2022 Katneni, Shekhar, Jangam, Krishnan, Prabhudas, Kaikkolante, Baghel, Koyadan, Jena and Mohapatra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinaya Kumar Katneni, VmluYXlhLkthdG5lbmlAaWNhci5nb3YuaW4=
†These authors have contributed equally to this work