 Shijie Bai
Shijie Bai Hengchao Xu
Hengchao Xu- Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences, Sanya, China
The microbial communities of the hydrothermal Scaly-foot Snails (SFSs) from independent hydrothermal vent fields have not been investigated in depth. In this study, we collected SFSs from two different hydrothermal environments located on the Central Indian Ridge (CIR) and the Southwest Indian Ridge (SWIR), the Kairei and Longqi vent fields, respectively. Additionally, one SFS collected from the Kairei vent field was reared for 16 days with in situ deep-sea seawater. The epibiotic and internal samples of SFSs, including ctenidium, esophageal gland, visceral mass, shells, and scales, were examined for microbial community compositions based on the 16S rRNA gene. Our results revealed significant differences in microbial community composition between SFSs samples collected from Kairei and Longqi vent fields. Moreover, the microbial communities of epibiotic and internal SFS samples also exhibited significant differences. Epibiotic SFS samples were dominated by the bacterial lineages of Sulfurovaceae, Desulfobulbaceae, Flavobacteriaceae, and Campylobacteraceae. While in the internal SFS samples, the genus Candidatus Thiobios, affiliated with the Chromatiaceae, was the most dominant bacterial lineage. Furthermore, the core microbial communities of all samples, which accounted for 78 ∼ 92% of sequences, were dominated by Chromatiaceae (27 ∼ 49%), Sulfurovaceae (10 ∼ 35%), Desulfobulbaceae (2 ∼ 7%), and Flavobacteriaceae (3 ∼ 7%) at the family level. Based on the results of random forest analysis, we also found the genera Desulfobulbus and Sulfurovum were the primary bacterial lineages responsible for the dissimilarity of microbial communities between the SFS samples collected from the Kairei and Longqi vent fields. Our results indicated that the microbial lineages involved in the sulfur cycle were the key microorganisms, playing a crucial role in the hydrothermal vent ecosystems. Our findings expand current knowledge on microbial diversity and composition in the epibiotic and internal microbial communities of SFS collected from different hydrothermal vent fields.
Introduction
Deep sea hydrothermal vents are oases of life in the deep seafloor (Brazelton, 2017). Typically, hydrothermal vents are located at seafloor spreading centers, such as the Mid Ocean Ridges. However, some hydrothermal vents were also been proven to exist in some other geological settings (Dick, 2019). The cold bottom seawater percolates through the fractured and porous basement rock, and then the seawater encountered the geothermal heat of the melting zone underneath the spreading center. Afterward, the subsurface heated seawater ultimately changed its chemical composition through water-rock interaction, leaching of sulfur and metals from the subsurface rocks. Under extremely high pressure and temperature, the hot, buoyant hydrothermal fluid rises constantly along the channels, emerging from hydrothermal vent orifices, and rapidly mixing with cold surrounding ambient seawater, resulting in different redox interfaces at which chemical sources of energy support hydrothermal vent ecosystems, such as the dissolved metal sulfides, H2S, and CO2 (Teske, 2009; Dick, 2019).
Deep sea hydrothermal vent ecosystems are mainly composed of chemosynthetic bacteria and archaea, these microorganisms are profiting from the chemical disequilibrium, caused by the reducing hydrothermal fluids and ambient oxidizing seawater, and these lithoautotrophs are reaping chemical energy to fix inorganic carbon into their biomass. Vent microbial communities, including vent animal symbionts, are fueled by chemosynthesis, and these organic carbons fixed through the chemosynthesis process support dense animal communities mainly through symbiotic relationships. Even there was a study that proposed the Pompeii worms, Alvinella pompejana inhabit an up to 60°C high-temperature environment. However, the rapid mixing of hot vent fluids and cold ambient seawater can result in a dynamic thermal regime that is difficult to ascertain the thermotolerance of vent animals via in situ measurements. Therefore, the current agreement is that the vent animals can not be tolerated sustained temperatures above 55°C (Cary et al., 1998; Girguis and Lee, 2006). The shrimp, crabs, bivalves, tubeworms, and snails constitute the animal communities of deep sea hydrothermal vent ecosystems. These animals have to poise themselves on the balance of harvesting reducing chemicals from hydrothermal fluids for symbiotic bacteria, and tolerating physiological challenges, such as the high-temperature stress, toxic sulfide, heavy metals, and low oxygen levels. Vent animals and their symbiotic bacteria have to adapt to different hydrothermal vent environments, displaying strong biogeography (Van Dover et al., 2001; Childress and Girguis, 2011).
The scaly-foot snail (SFS), Chrysomallon squamiferum, is different from all known mollusks in that its foot is covered by scales of mineralized iron sulfide on conchiolin, was first discovered at Kairei hydrothermal vent field on the Central Indian Ridge (CIR; Van Dover et al., 2001; Warén et al., 2003; Chen et al., 2015c). Since that initial discovery, a total of three morphotypes of SFSs have been reported in the Indian Ocean, each with its distinguishing characteristics (Nakamura et al., 2012; Chen et al., 2015a). The SFSs first discovered at the Kairei were dark morphotypes, with dark shells and scales (Van Dover et al., 2001). A white morphotype of SFSs was observed at the Solitaire hydrothermal field, 750 km to the north of Kairei on the CIR (Nakamura et al., 2012). A third SFSs morphotype, with brown shells and dark scales, was observed at Longqi, a hydrothermal vent field located on the ultraslow-spreading Southwest Indian Ridge (SWIR; Zhou et al., 2018). Despite the color discrepancy, genetic and morphological results reveal that all three morphotypes are genetically the same species (Chen et al., 2015b).
The SFSs have metal-rich sclerites and unique enlarged esophageal gland, which results in the morphologically and presumably physiologically different from other gastropod hosts of chemoautotrophic symbionts. Molecular study of the symbionts utilizing the 16S rRNA gene indicated that two distinct assemblages of bacteria associate with SFS: a diverse community of epibionts, which involved in the formation of the pedal sclerites with iron sulfide minerals, and a single endosymbiont that contribute to the nutritional demands of the snail (Goffredi et al., 2004). Even so, owing to difficulties with the sampling of SFSs around black smoker chimneys located in CIR and SWIR, we limited our focus to the microbiome of SFSs in Kairei and Longqi vent fields to answer the following questions: Whether the microbial communities of epibiotic and internal of SFS are different? Considering the SFS from Kairei and Longqi vent fields are the same species, both with dark shells and scales (Chen et al., 2015c), what are the similarities and differences of microbial community structure and composition? Moreover, after reared with deep-sea in situ seawater (prokaryotic cells and particles were excluded) for 16 days, did the microbial communities changed? For instance, the distribution of two sulfur-oxidizing bacteria, Sulfurovum and Sulfurimonas, which are affiliated with Epsilonproteobacteria, is mostly determined by the concentrations of oxygen and sulfur (Meier et al., 2017). Obviously, the rearing experiment on board will increase the concentration of oxygen.
The goal of this study was to compare microbial communities from multiple locations both within (ctenidium, esophageal gland, and visceral mass) (Nakagawa et al., 2014; Chen et al., 2015a), and on the surface (shell and scales) of SFS collected from two different hydrothermal vent locations, the Kairei and Longqi vent fields. We also analyzed the microbial communities of a single SFS from the Kairei black smoker chimney, which was reared with deep-sea in situ seawater for 16 days until it died.
Materials and Methods
Sample Collection
The identified hydrothermal fields of the SWIR are mainly centered on two ridge sections. One ridge section is located between the Indomed and Gallieni transform faults of the SWIR, from 49°E to 53°E, where six hydrothermal fields have been discovered, including Yuhuang (37°56′S, 49°16′E), Longqi (37°47′S, 49°39′E), Duanqiao (37°39′S, 50°24′E), Changbaishan carbonate field (37°37′S, 50°56′E), and two other unnamed fields (37°27′S, 51°19′E and 36°60′S, 53°15′E) (Tao et al., 2014). The other section of the hydrothermal field lies on the SWIR between the Melville transform fault and Rodriguez triple junction (RTJ) from 63°E to 64°E, where Tiancheng (27°51′S, 63°55′E) and Tianzuo (27°57′S, 63°33′E) hydrothermal fields were discovered (Tao et al., 2014). The Kairei vent field (25°19′S, 70°02′E) was discovered in 2000 on the Central Indian Ridge (CIR), approximately 22 km north of the Rodriguez Triple junction (Hashimoto et al., 2001).
Scaly-foot Snails were collected from the Longqi (water depth = 2773 m, assigned as Lq) and Kairei (water depth = 2495 m, assigned as Kr) vent fields at the SWIR and CIR, respectively (Supplementary Figure 1), during the TS10 expedition in the Indian Ocean from November 2018 to March 2019. The scaly-foot snails were grabbed by the mechanical arms of the manned submersible Shenhaiyongshi and then recovered in the sample basket. The Snail samples were kept in a lockable PVC bio-box on the sample basket to avoid contamination. The bio-box was closed during the process of diving and surfacing. Only when the samples were collected, the cover lib could be open, and after sampling, the bio-box will be closed again. Once onboard the supporting vessel R/V Tansuoyihao, these samples were stored at −80°C. A single scaly-foot snail retrieved from the Kairei vent field was reared for 16 days in a covered tank containing 20 L deep sea in situ seawater filtered with 0.22 μm polycarbonate membrane to remove all protozoa and the vast majority of prokaryotic cells (assigned as Kr16), filter samples were stored at −80°C ultra-low freezer. After the expedition, the samples were transferred on dry ice to the laboratory of the Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences (IDSSE, CAS). The dissection of the SFSs was conducted in a Vertical Flow Clean Bench with sterilized scissors and tweezers. Samples of the ctenidium (Ct), esophageal gland (Gl), visceral mass (Vm), shells (Sh), and scales (Sc; Nakagawa et al., 2014; Chen et al., 2015a), were collected for DNA extraction. The esophageal gland was divided into three parts, the fore-end part (assigned as gl_1), which connected with the ctenidium, the middle part of the esophageal gland (assigned as gl_2), and the tail end (assigned as gl_3), which connected with the visceral mass.
DNA Extraction and Sequencing
DNA was extracted from the different body site samples of SFSs from Longqi and Kairei vent fields (three extraction blank control samples were used) with MoBio PowerSoil extraction kits (Mo Bio Laboratories, Carlsbad, CA, United States), according to the manufacturer’s instructions. The extracted DNA was quantified with a Qubit fluorometer (Invitrogen Inc., Manufacturer: Life Technologies Holdings Pte., Ltd., Singapore) and used for amplification of the V4 region of the 16S rRNA gene with the primer pair 515f Modified and 806r Modified (Walters et al., 2015). The PCR cycling conditions were as follows: denaturation at 95°C for 3 min, followed by 27 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 45 s and a final extension at 72°C for 10 min. Triplicate PCR amplicons were combined after purification using a TaKaRa purification kit (TaKaRa, Japan). The PCR products were prepared for library construction with the TruSeq DNA sample preparation kit (Illumina, San Diego, CA, United States), according to the manufacturer’s instructions. The libraries were sequenced at MajorBio Co., Ltd. (Shanghai, China) using the HiSeq platform (Illumina) with a paired-end 250 bp sequence read run.
Microbial Community Analysis
After sequencing, the raw reads were categorized according to their barcodes and forward and reverse primers (one mismatch each was allowed). Paired-end reads of sufficient length were combined with at least a 30 bp overlap into full-length sequences by using FLASH program version 1.2.8 (Magoč and Salzberg, 2011). The average fragment length was 253 bp. Btrim program version 0.2.0 was used to filter out low-quality sequences. The quality score was set to >20 with a 5-base window size as the standard; any sequences containing Ns or <200 bp were discarded. The sequences with lengths of 245–260 bp were retained (Kong, 2011). UPARSE (Edgar, 2013) was used to remove chimeras and cluster the sequences into 97% identical operational taxonomic units (OTUs). Singletons were excluded from further analysis. We also used UNOISE3 to correct sequencing errors to determine real biological sequences at single-nucleotide resolution by generating amplicon sequence variants (ASVs) with default settings (Edgar, 2016). A representative sequence from each OTU/ASV was selected for taxonomic annotation by comparison with the SILVA 132 database (Quast et al., 2013), which includes bacterial, archaeal, and eukaryotic sequences. The OTUs/ASVs were randomly subsampled to normalize the reads of each sample. The raw sequencing reads of all samples were deposited to the NCBI database1 under BioProject accession number: PRJNA679429.
Statistical Analysis
The diversity of the microbial communities from the different body site samples of SFSs from Longqi and Kairei vent fields were determined by statistical analysis of the α-diversity indices. The Shannon and Inverse Simpson indices were calculated using the vegan package in R language version 3.4.3 (R Core Team, 2018). The Chao1 values (Chao, 1984) were generated using the Mothur program (Schloss et al., 2009). The random forest analysis was conducted using the randomForest package in R, and β-diversity-based statistical tools, non-metric multidimensional scaling (NMDS), were used to test the differences within the microbial community structure. DNA extraction and data analysis were followed by the method previously described (Bai and Hou, 2020). Data comparison between different groups was performed by the Wilcoxon rank-sum test using IBM SPSS Statistics 19.
Results
Sequencing Statistics and Microbial Diversity
A total of 932,551 sequences were obtained from 18 samples after quality assessment, including seven SFS samples from the Kairei vent field, seven SFS samples from the Longqi vent field, but only four samples from the reared Kairei vent field SFS were successfully sequenced for their 16S rRNA gene. Since the Kr16_gl_1, Kr16_gl_2, and Kr16_ct samples were not generated enough PCR products after PCR amplification, as well as the three blank control samples, no sequencing actions were taken for these samples. An average of 51,808 ± 11,163 sequences were obtained from each sample. We randomly resampled to 26,217 sequences per sample for the next analyses of microbial diversity, composition, and structure. The α-diversities of microbial communities from different samples were calculated (Supplementary Table 1). As shown in Figure 1, if all samples from a single SFS were treated as an individual, the observed richness and Chao1 indices all indicated that the α-diversity of the microbiome from Kairei field was higher than that of Longqi field (Figures 1C,D). Moreover, if the samples were divided into two groups, an internal group (including the samples of Ct, Gl, and Vm) and an external epibiotic group (shells and scales), it was observed that the α-diversity of the epibiotic group was significantly higher than that of the internal group, especially the Shannon and Inverse Simpson indices (Wilcoxon rank-sum test, p < 0.01) (Figures 1E,F). Moreover, the alpha diversity results based on the ASV table was also showed the same trends (Supplementary Figure 2).
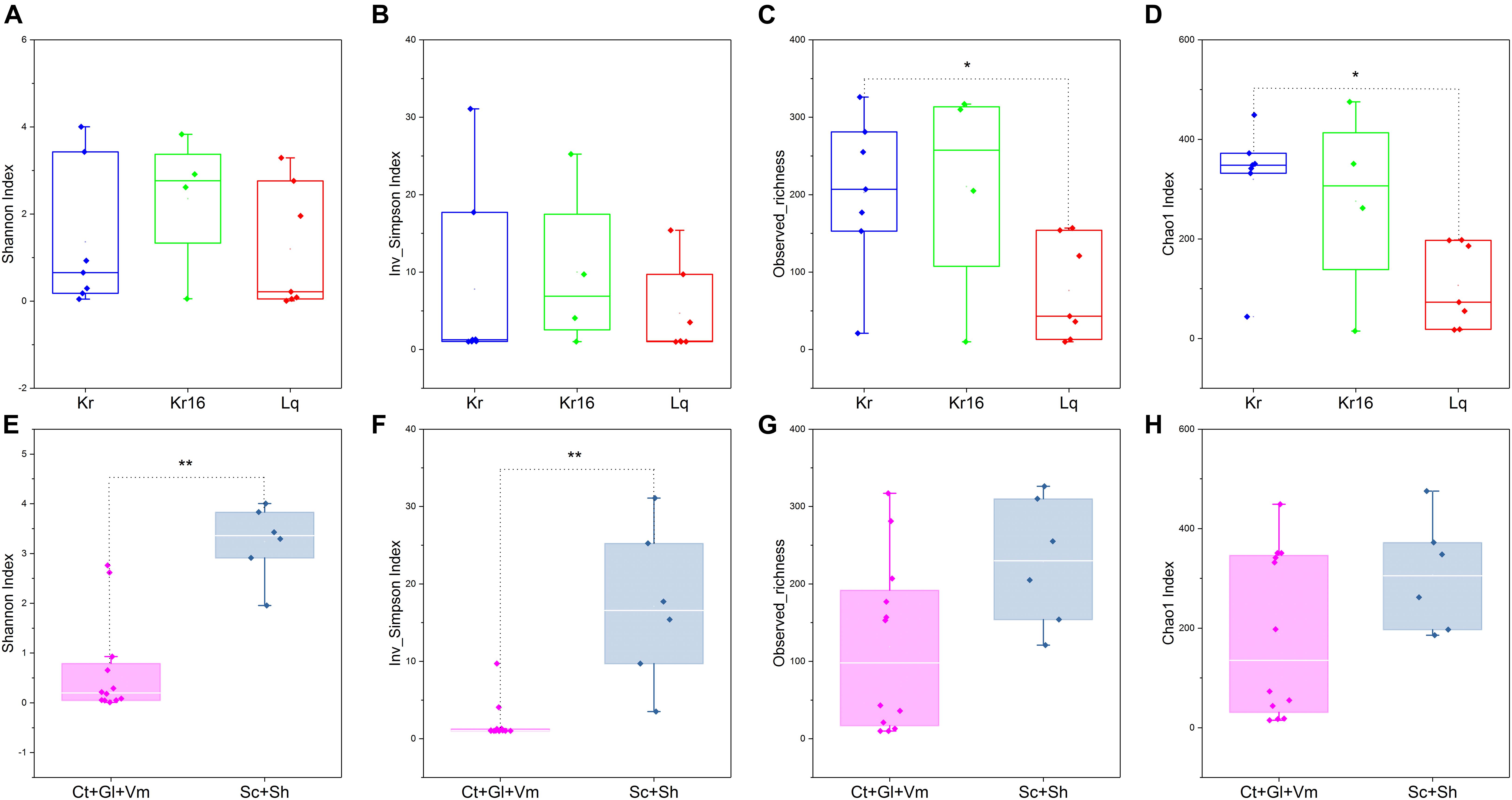
Figure 1. Comparisons of four α-diversity indices, Shannon index (A,E), Inverse Simpson index (B,F), observed richness (C,G), and Chao1 index (D,H), of the SFS samples. Kr refers to SFS samples collected from the Kairei vent field; Kr16 refers to samples collected from the Kairei vent field SFS reared for 16 days; Lq refers to SFS samples collected from the Longqi vent field; The group of Ct, Gl, and Vm refers to the internal samples of SFSs; The group of Sc and Sh refers to the epibiotic samples of SFSs. *Difference is significant at 0.05 level; **Difference is significant at 0.01 level, based on Wilcoxon rank-sum test. The results based on the OTUs datasets.
Non-metric multidimensional scaling analysis of microbial communities clearly separated theses samples into two principal groups, one consisting of the SFS samples obtained from the Longqi vent field, and another composed completely of all SFS samples collected from the Kairei vent field, including the SFS samples of reared one (Figures 2A,B,D). To compare the microbial communities of the SFS directly collected from the Kairei vent field and the one reared for 16 days with in situ deep-sea seawater on aboard, these SFS samples were picked out for further NMDS analysis. The results revealed that although the rearing treatment have changed the microbial communities, however, a significant difference was not observed (Figure 2C). The NMDS analysis based on the ASVs datasets was also support these results (Supplementary Figure 3).
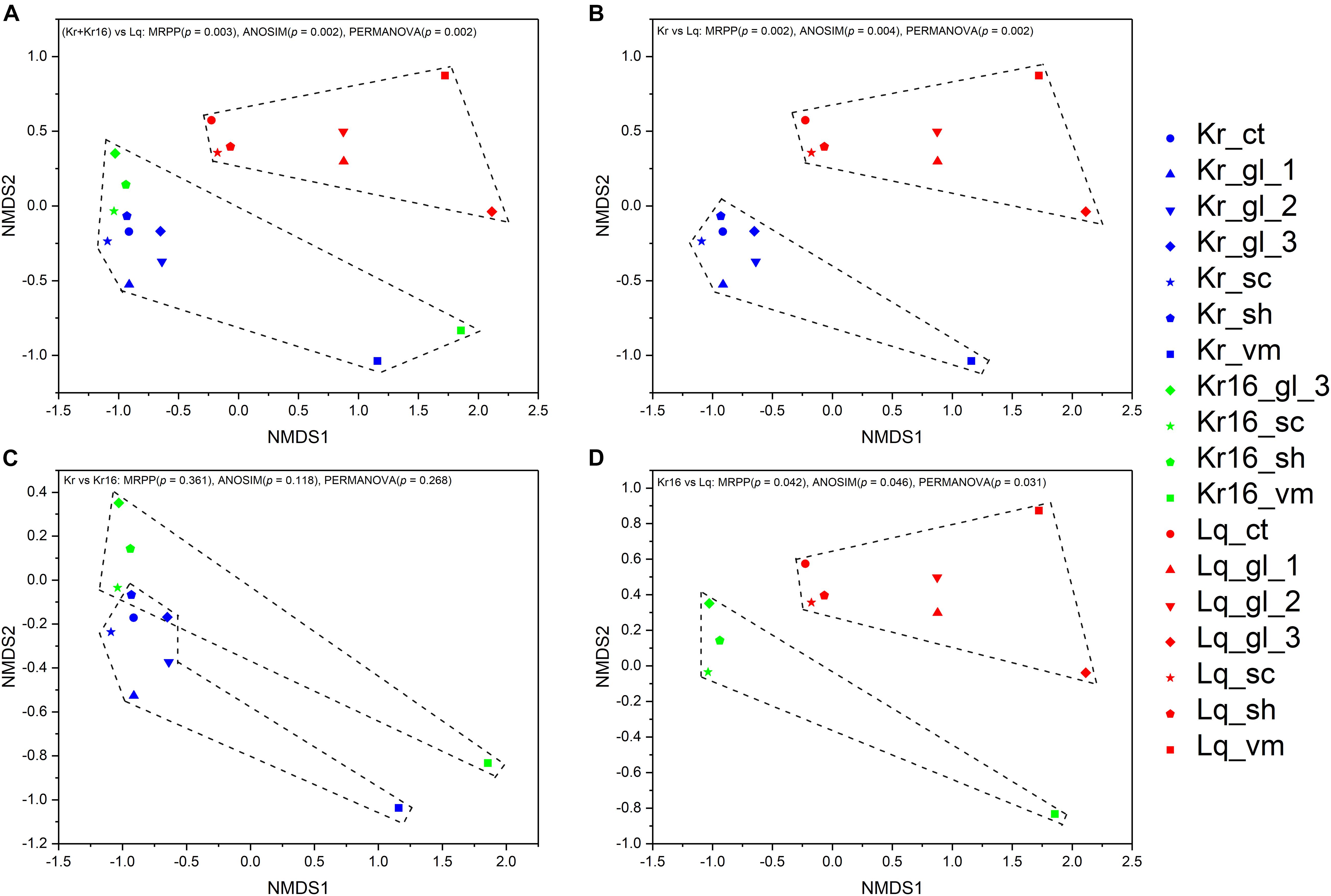
Figure 2. NMDS analysis of the microbial communities separated the samples into two principal groups, one composed of the samples of SFSs collected from the Kairei vent field, and another group composed of SFS samples collected from the Longqi vent field (A). Although the samples of Kr and Kr16 grouped together, they were in a loose pattern with no significant difference (C). However, the microbial community structures of Kr and Kr16 were clearly separated from the microbial community of Lq (B,D). The results based on the OTUs datasets.
The Composition and Statistical Analysis of Microbial Communities
Gammaproteobacteria were the dominant bacterial lineage, being found in the internal (Ct, Gl, and Vm) samples of SFS collected in the Kairei vent field, ranging from 88 ∼ 98% (Figure 3A). Gammaproteobacteria also dominated the internal samples of Longqi SFS, accounted for 99 ∼ 100%, but with one exception, the sample of Lq_ct, which harbored Epsilonbacteraeota as the most dominant bacterial lineage (57%), followed by Bacteroidetes (16%) and Gammaproteobacteria (12%). By contrast, after 16 days of rearing, the dominant bacterial lineages of Kr16_gl_3 had switched to Epsilonbacteraeota, accounting for 65% of sequences, with the exception of sample Kr16_vm, in which Gammaproteobacteria continued to account for almost 100% of sequences. Among the epibiotic samples (Sh and Sc), Epsilonbacteraeota, Bacteroidetes, and Deltaproteobacteria were the dominant bacterial lineages in the Kairei vent field samples, including the reared SFS. However, the Longqi epibiotic samples were dominated by Gammaproteobacteria, Bacteroidetes, and Epsilonbacteraeota. Deltaproteobacteria were not the dominant bacterial lineage in the samples of Lq_sc and Lq_sh, accounting for only 0.2 ∼ 9% (Figure 3A). At the order and family taxonomic levels, the Chromatiaceae, which are affiliated with the Chromatiales, displayed a universal distribution, with high relative abundance, in the majority of CT, Gl, and Vm SFS samples, except for the samples of Kr16_gl_3 and Lq_ct, which were dominated by Sulfurovaceae and Campylobacterales, respectively (Figures 3B,C). In the epibiotic samples (Sc and Sh), the dominant bacterial members from the Kairei vent field consisted of Sulfurovaceae, Desulfobulbaceae, Flavobacteriaceae, and Campylobacteraceae, which belonged to Campylobacterales, Desulfobacterales, Flavobacteriales, and Campylobacterales, respectively. While Lq_sc and Lq_sh showed some heterogeneity, in sample Lq_sc, Flavobacteriaceae and Sulfurovaceae were the dominant bacterial lineages, but Flavobacteriaceae was not dominant in the Lq_sh sample, being replaced by Chromatiaceae. However, regardless of which vent field the SFS were collected from, Sulfurovaceae was the dominant bacterial lineage within all Sc and Sh samples (Figures 3B,C). Furthermore, the genus level was dominated by Candidatus Thiobios (classified to endosymbiont of unidentified scaly snail isolate Monju at species level) in the internal SFS samples, accounting for 88 ∼ 100% of sequences, with the exception of sample Kr16_gl_3, which dominated by Sulfurovum (61%). Sulfurovum was the dominant bacterial genus of all epibiont samples collected from the Kairei and Longqi vent fields (Figure 3D). The Campylobacter and Cocleimonas were among the dominant genera present at higher abundance in the sh, but not sc, SFS samples of the Kairei vent field. Moreover, Desulfobulbus was one of the dominant bacterial lineages in sc and sh SFS samples collected from the Kairei vent field, but not the samples of sc and sh collected from the Longqi vent field samples. The taxonomic annotation of ASVs was also conducted to compare the microbial community composition generated by OTUs and ASVs (Supplementary Figure 4). Our results showed that the biological conclusions based on microbial relative abundance were not affected by the choice of denoising or not. Thereby, we only use the datasets generated by OTUs for the rest of the analysis.
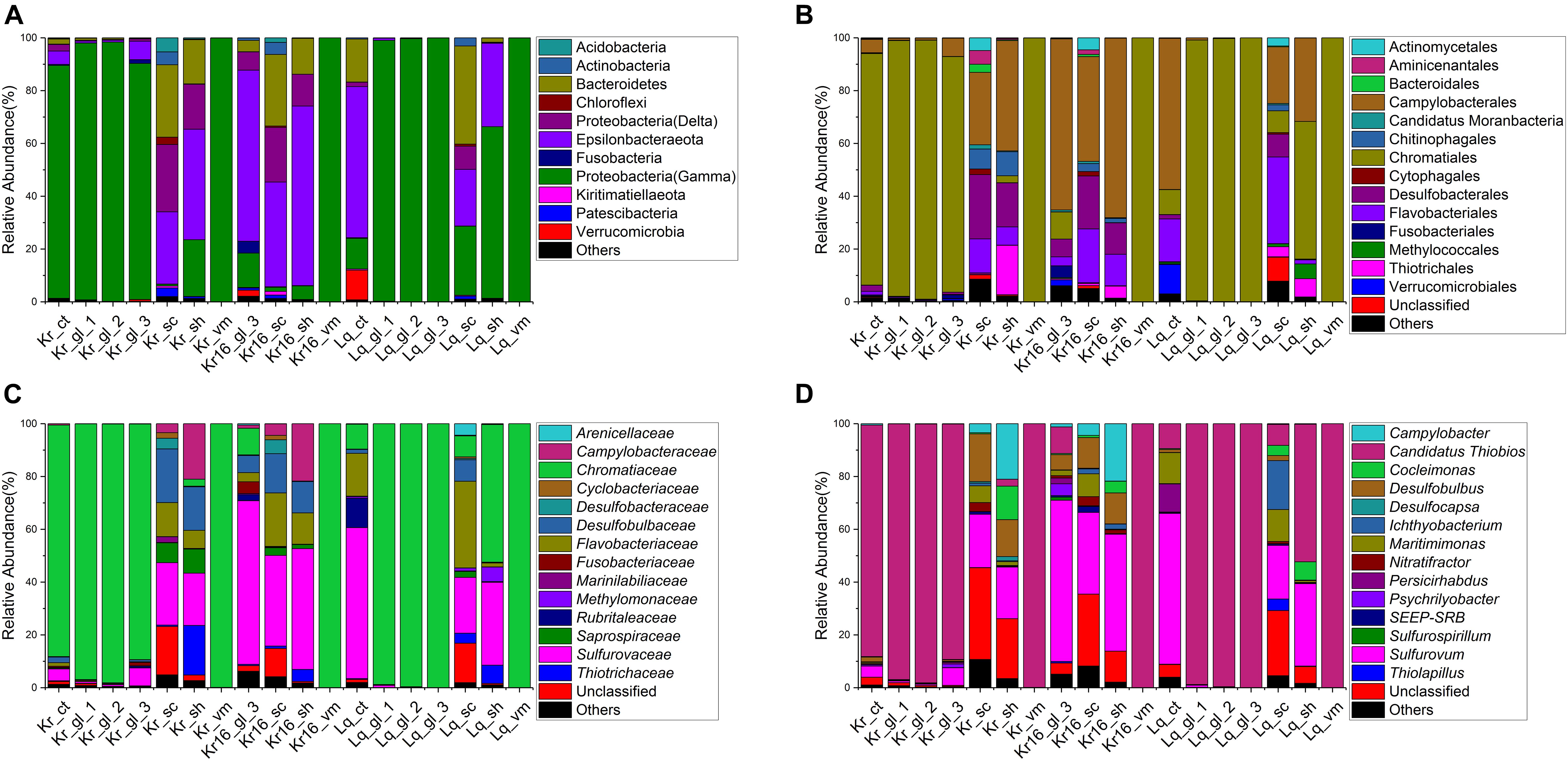
Figure 3. Stacked bar chart showing the relative abundance of microbial communities from all samples at phylum and class levels (A), order level (B), family level (C), and genus level (D). The results based on the OTUs datasets.
The Shared and Key Microbial Communities From Different Scaly-Foot Snail Samples
To better understand the composition of the core microbial community, we explored the detailed differences at the OTU level between different SFS samples. Quality control and random resampling of the 18 samples were conducted, and the sequence reads were clustered into 770 OTUs at 97% similarity level. As shown in Figure 4, 187, 158, and 41 unique OTUs were detected in the samples of Kr, Kr16, and Lq, respectively. 331 OTUs were shared between Kr and Kr16, 182 OTUs were shared between Kr and Lq, and 158 OTUs were shared between Kr16 and Lq (Figure 4A). The 146 OTUs detected in all groups of Kr, Kr16, and Lq, formed the core microbial communities of SFS samples collected from the Kairei and Longqi vent fields. This core microbial community, which accounted for 78 ∼ 92% of sequences, was dominated by Chromatiaceae (27 ∼ 49%), Sulfurovaceae (10 ∼ 35%), Desulfobulbaceae (2 ∼ 7%), and Flavobacteriaceae (3 ∼ 7%) at the family level (Figure 4B). Moreover, Candidatus Thiobios (27 ∼ 49%), Sulfurovum (9 ∼ 33%), Desulfobulbus (1 ∼ 6%), and Maritimimonas (2 ∼ 5%) were the dominant bacterial lineages of this core community. Interestingly, the relative abundance of Sulfurovum dramatically increased after the SFS (Kr16) that was reared for 16 days. By contrast, compared to the samples of Kr, the proportion of Candidatus Thiobios decreased in the samples of Kr16.
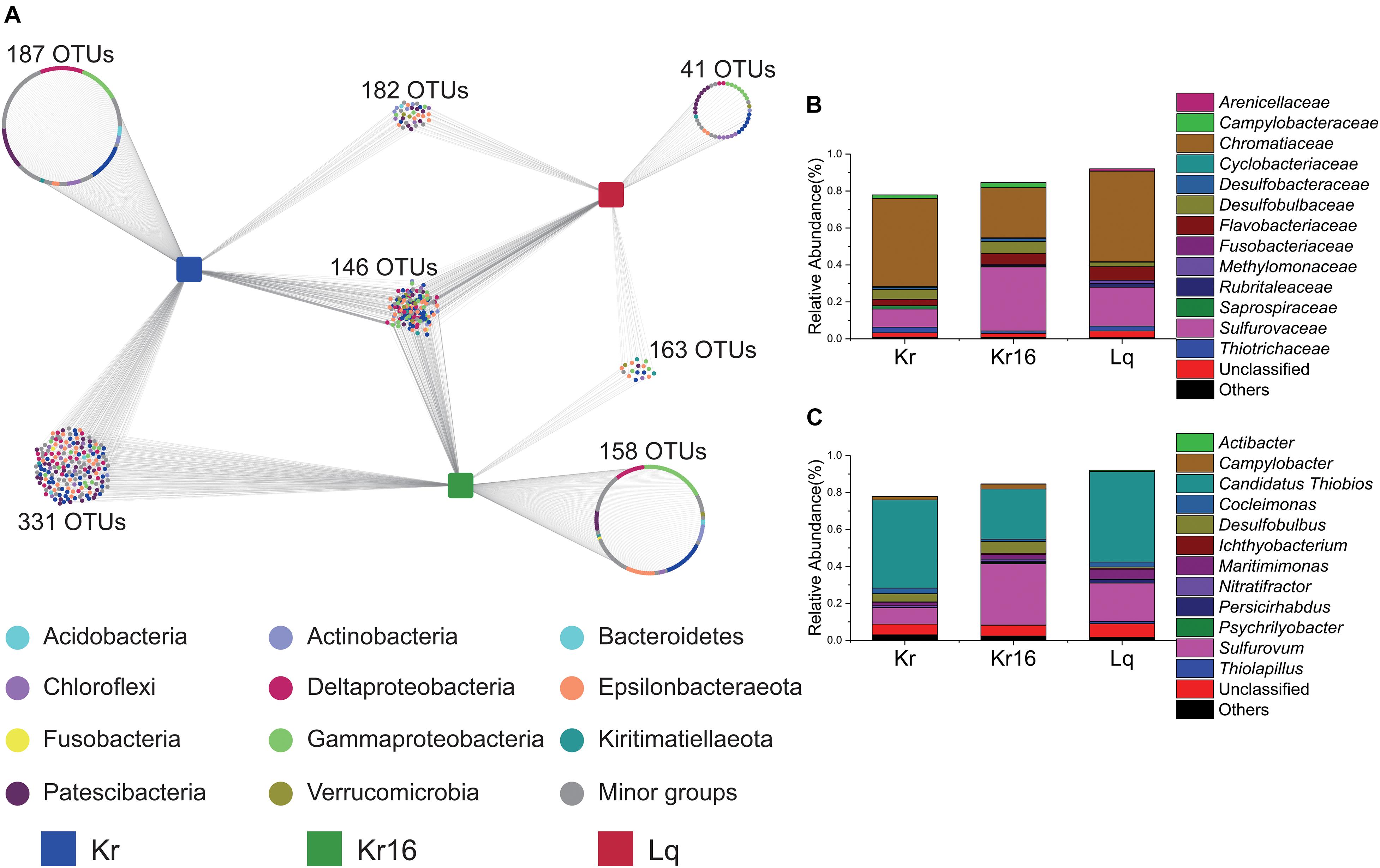
Figure 4. Distribution of OTUs in the samples of Kr, Kr16, and Lq (A), and the stacked bar chart showing the relative abundance of the microbial communities consisting of the shared OTUs from all samples at the level of family (B), and genus (C). Kr refers to SFS samples collected from the Kairei vent field; Kr16 refers to SFS samples collected from the Kairei vent field, and reared for 16 days; Lq refers to SFS samples collected from the Longqi vent field.
The microbial community members unique to the Kr, Kr16, and Lq sample sets were revealed by random forest analysis. According to indices such as the Mean Decrease Accuracy and Mean Decrease Gini, the 20 most important OTUs were delineated (Figure 5A). The five most important OTUs were OTU_6, OTU_40, OTU_181, OTU_46, and OTU_1090, which were assigned to Desulfobulbus (Desulfobulbaceae), unclassified (unclassified, Candidatus Moranbacteria), Sulfurovum (Sulfurovaceae), unclassified (unclassified, Candidatus Moranbacteria), and Sulfurovum (Sulfurovaceae), respectively. The heatmap consisting of the 20 most important OTUs indicated the SFS samples of Lq separated from the SFS samples collected from the Kairei vent field, except for the samples of Kr_vm and Kr16_vm, which were almost entirely dominated by a single bacterial lineage, Candidatus Thiobios, affiliated with Chromatiaceae, Chromatiales, just like sample Lq_vm (Figure 5B). Moreover, OTU_6, OTU_1090, OTU_40, OTU_26, and OTU_54, the top five OTUs (Supplementary Figure 5A), that played an important role in shaping the differences in community structure between Kr and Lq. The heatmap results of bacterial communities, which constituted by the top 20 most important OTUs, also displayed a clear separation into two principal groups based upon hydrothermal vent site (Supplementary Figure 5B). The five OTUs most crucial to the differences between the microbial communities of Kr16 and Lq were OTU_46, OTU_40, OTU_181, OTU_1090, and OTU_4 (Supplementary Figure 6A). Furthermore, these 20 most important OTUs can also distinguish the microbial communities of Kr16 and Lq (Supplementary Figure 6B). Based on these results, OTU_6, OTU_40, OTU_181, OTU_46, OTU_1090, OTU_26, OTU_54, and OTU_1090 were the most important OTUs, distinguishing the microbial community between the SFS samples collected from Kairei and Lq vent fields, most of which were affiliated with bacterial lineages involved in deep sea sulfur cycling. Even there were no significant differences between the microbial communities of Kr and Kr16, we still attempted to ascertain the most important OTUs between these two groups. According to the results of random forest analysis, OTUs affiliated with the Arcobacter (Arcobacteraceae) and unclassified Vibrionaceae, Sulfurovum (Sulfurovaceae), Sulfurospirillum (Sulfurospirillaceae) were the key bacterial lineages, which that potentially resulted in differences between the microbial communities of Kr and Kr16 (Supplementary Figure 7).
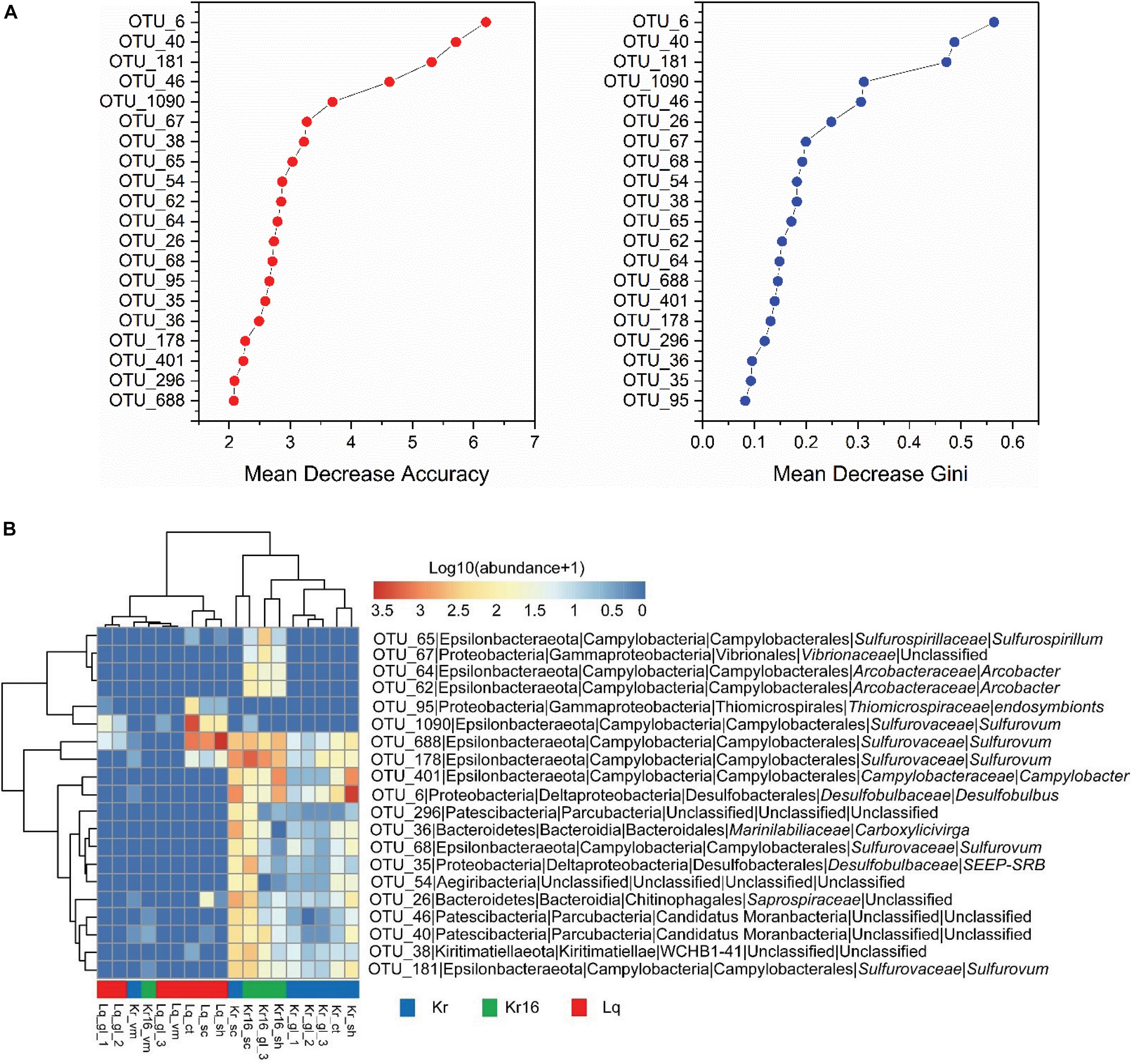
Figure 5. The importance (A) and the heatmap (B) of the 20 most important OTUs, which distinguish the samples of Kr, Kr16, and Lq. The microbial abundance was scaled with log transformation in the heatmap. Kr refers to SFS samples collected from the Kairei vent field; Kr16 refers to SFS samples collected from the Kairei vent field, and reared for 16 days; Lq refers to SFS samples collected from the Longqi vent field.
Discussion
Characterize the Microbial Communities of Scaly-Foot Snails From Kairei and Longqi Vent Fields
In our study, samples were collected from different parts of scaly-foot snails, which were captured from the Kairei, and Longqi vent fields. The Kairei vent field is located in the CIR and the Longqi vent field is located in the SWIR, indicating these SFSs were from different hydrothermal environments. Furthermore, on the basis of the end-member Vent fluid compositions, there were some geochemical differences between the two vent fields, for instance, the end-member Fe and H2 concentrations of the vent fluids from the Kairei field were higher than those of the Longqi vent field while the concentration of CH4 from Longqi vent fluid was far higher than that of the Kairei vent fluid (Gallant and Von Damm, 2006; Ji et al., 2017). Moreover, one of the SFSs collected from the Kairei vent field was reared in a tank with in situ deep seawater for 16 days at 4°C, the in situ deep seawater were filtered to remove all prokaryotic cells, it should be noted that the filtrate would have still contained in situ deep-sea viruses, which could not be filtered by 0.22 μm filter membranes. The hydrothermal fluid discharging out into seawater causes intense turbulence, which can lead to microorganisms from the ambient near bottom seawater being mixed in Jiang and Breier (2014) and Sheik et al. (2015). Since the SFSs were perched on the root of the hydrothermal chimney, and the shells and scales were exposed to the surrounding seawater, thereby, the microbial communities of seawater samples taken in close vicinity to the hydrothermal chimneys should show some similarities to the shell and scale samples of SFSs. Our results confirmed that the main bacterial lineages of SFS shell and scale samples, including Thiotrichaceae, Sulfurovaceae, Desulfobulbaceae, and Flavobacteriaceae, within the phyla (class) Gammaproteobacteria, Epsilonbacteraeota, Deltaproteobacteria, and Bacteroidetes, respectively, were in line with those of a previous study, which found that the members of these four phyla were the dominant bacterial lineages in SFS shells and scales (Goffredi et al., 2004). Moreover, these four lineages were also present in the near bottom seawater samples (Li et al., 2020). Even hydrothermal systems inflict a strong influence on the habitats of SFS, the microbial communities of SFS shells and scales still showed some peculiarities. For instance, while the (hyper)thermophilic bacterial lineages, Aquificaceae, Hydrogenothermaceae, and Desulfurobacteriaceae within the phylum Aquificae, dominate the samples of the initial root of hydrothermal plumes, but they were not observed in the shell and scale samples of SFSs. Moreover, another thermophilic lineage, the genus Caminibacter, affiliated with Nautiliales, also was not detected in SFS shells or scales, but was found in the ambient near bottom seawater samples of the vent (Li et al., 2020). On the contrary, Cocleimonas, Campylobacter, Desulfobulbus, and Maritimimonas, affiliated with Thiotrichaceae, Sulfurovaceae, Desulfobulbaceae, and Flavobacteriaceae, respectively, dominated the samples of SFS shells and scales. Cocleimonas was previously been detected in the bottom water sample of the Longqi hydrothermal vent, but at extremely low relative abundance (Li et al., 2020). However, it was one of the predominant bacterial lineages in the shells and scales. Very little is known about the genus Cocleimonas, with only one strain (capable of sulfur oxidation) isolated from a sand snail that lives on the sediments of the Japan sea (Tanaka et al., 2011). Subsequently, Cocleimonas related organisms have been found in samples of endemic caridean shrimp living in the deep-sea hydrothermal environment on the Mid Atlantic Ridge (Apremont et al., 2018), and a reared hydrothermal vent squat lobster from the Iheya North hydrothermal field in the Okinawa Trough, Japan (Watsuji et al., 2018). All these lineages of Cocleimonas were known to be the chemoautotrophic sulfur-oxidizers. The genus Campylobacter are very common bacteria in both animal digestive tracts and at hydrothermal vents, whose powerful flagellar motility is very important in nutrient acquisition, whereby bacterial cells attach to surfaces and rotate their flagella to increase nutrient flux for metabolism (Beeby, 2015). Moreover, single-cell-based activity measurements and 16S rRNA-gene analysis have previously shown that autotrophic Campylobacter dominate carbon fixation in oxygen/nitrate limited deep sea hydrothermal environments (McNichol et al., 2018). The family Flavobacteriaceae is comprised of more than 45 genera that are distributed across diverse environments, including freshwater, marine, soil, and epibenthic fauna. However, the genus Maritimimonas, which dominated the shells and scales of SFS collected from Kairei and Longqi vent fields, is not widely reported in the microbial communities of hydrothermal environments. Only one strain of Maritimimonas was isolated from a veined rapa whelk, Rapana venosa, collected from the south sea off the coast of the South Korea (Park et al., 2009). Desulfobulbus, as one of the sulfate-reducing bacteria (SRB), are widely distributed in different environments (Suzuki et al., 2007; Sorokin et al., 2012; Kharrat et al., 2017). These bacterial lineages have an important role in both sulfur and carbon cycling, utilizing sulfate as a terminal electron acceptor in their respiratory metabolism, facilitating the degradation of organic matter, and additionally, bind heavy metal ions from surrounding water into insoluble sulfides (Jørgensen, 1982; El Houari et al., 2017). According to these data, we can infer that the microorganisms present on the shells and scales of SFS are mainly involved in the cycling of sulfur and carbon, with organic matter mineralization. By contrast, different microbial communities were observed in the internal SFS samples. The Candidatus Thiobios of Chromatiaceae was the most dominant bacterial lineage, accounting for nearly 100% sequences of the internal SFS samples. These findings broadly support the work of previous studies (Goffredi et al., 2004; Nakagawa et al., 2014). Candidatus Thiobios is a thiotrophic symbiont that utilizes hydrogen sulfide or other reduced sulfur compounds, which are typically produced either biologically by anaerobic sulfate-reducing bacteria or geothermally at hydrothermal vents, to gain energy for carbon fixation (Dubilier et al., 2008). The complete genome analysis of the scaly snail endosymbiont also confirmed that these bacteria are mainly involved in the functions of sulfur/hydrogen oxidation and carbon fixation (Nakagawa et al., 2014). Previously, the chemoautotrophic Candidatus Thiobios was thought to be unable to survive without its symbionts (Bauer-Nebelsick et al., 1996). However, an autotrophic Chromatiaceae bacterium was found to exist in the water column at 1500 m water depth in the southern Pacific Ocean, based on the phylogenetic analysis of 16S rRNA genes, this bacterium was closely related to the endosymbiont of scaly snails, and showed similar functionality (Sass et al., 2020). Furthermore, cultivation experiments of the thiotrophic symbiosis between Zoothamnium niveum and Candidatus Thiobios zoothamnicoli showed that the symbiosis was not able to survive without sulfide. Intriguingly, a high concentration of sulfide harmed the symbiotic association (Rinke et al., 2007). These findings may help us to understand why Scaly-foot Snails are only present at hydrothermal vent fields, and always maintain a specific distance from the vent orifice, perhaps acquiring the appropriate sulfide concentration for metabolic activities.
Key Microbial Lineages to Distinguish the Microbial Community Structure of Scaly-Foot Snail From Kairei and Longqi Vent Field
Different microbial community structures were observed in the SFS samples collected from Kairei and Longqi vent fields. Based on the NMDS statistical analysis, the microbial communities of all samples were separated into two principal groups by location (p = 0.003, MRPP; p = 0.002, ANOSIM; p = 0.002, PERMANOVA). According to the description of operator who collected these SFSs, the intensity of discharged hydrothermal fluid from the Longqi vent was much stronger than that of Kairei, perhaps this is one of the reasons why the microbial community structure was different between the SFS samples of the two vent fields. In addition, the α-diversity of the microbiome from Longqi field was lower than that of the microbiome from Kairei field. As previously known, once the hot, anoxic hydrothermal fluid discharged from the orifices, the hydrothermal fluids are progressively diluted by cold oxygenated ambient seawater, which exert an important influence on the changes in chemical and microbial features (Elderfield and Schultz, 1996; Marbler et al., 2010; Anantharaman et al., 2016). Thereby, the degree of intense turbulence caused by hydrothermal fluid mixed with surrounding seawater may result in greatly varying chemical conditions, and these different, steep gradients can generate a wide range of geochemical niches and different energy sources for microorganisms, and thus alter the microbial communities. Moreover, based on the random forest analysis, there are several important differences between the SFS samples of Kairei and Longqi at the OTU level, most of the key OTUs were affiliated with bacterial lineages involved in sulfur cycling, such as Desulfobulbus (Desulfobulbaceae) and Sulfurovum (Sulfurovaceae). The genus Sulfurovum is a chemolithoautotroph bacterial lineage, commonly found in many hydrothermal vent fields (Inagaki et al., 2004; Mino et al., 2014; Giovannelli et al., 2016; Mori et al., 2018). Deep-sea hydrothermal vent ecosystems are primarily supported by microbial chemosynthesis (Jannasch, 1985), and these chemolithoautotrophs can use a range of reductive substrates as energy sources (Campbell et al., 2006; Fisher et al., 2007; Sievert and Vetriani, 2012), for instance, chemolithoautotrophs can gain energy from the redox reactions occurred at the interface of cold, oxidized seawater, which carries O2, NO3–, Fe3+, SO42–, and CO2, and hot, anoxic hydrothermal fluid, which contains H2, H2S, CH4, Fe2+, and formate (Fisher et al., 2007; Sievert and Vetriani, 2012). Based on the characteristics of Desulfobulbus and Sulfurovum isolated as pure cultures from deep-sea hydrothermal vents, the genus Sulfurovum mainly involved in sulfur-oxidation, thiosulfate-reduction, nitrate-reduction, and hydrogen oxidation (Inagaki et al., 2004; Mino et al., 2014; Giovannelli et al., 2016; Mori et al., 2018). The genus Desulfobulbus is primarily considered to be composed of sulfate-reducers (Suzuki et al., 2007; Sorokin et al., 2012), with some potentially capable of propionate-oxidation and mercury methylation (Moreau et al., 2015; El Houari et al., 2017). Taken together, these findings suggest that the dissimilarity of microbial communities from the SFS samples of Kairei and Longqi vent fields were likely to due to the steep chemical gradients caused by the mixture of varying hydrothermal fluid and ambient seawater. Moreover, after being reared for 16 days, the microbial communities of Kr16 were altered compared with the microbial communities of Kr. However, no significant difference was observed (p = 0.361, MRPP; p = 0.118, ANOSIM; p = 0.268, PERMANOVA). The results of random forest analysis indicated that Arcobacter (Arcobacteraceae), Sulfurovum (Sulfurovaceae), and Sulfurospirillum (Sulfurospirillaceae) were the main bacterial lineages responsible for the difference between microbial communities. The genera Arcobacter and Sulfurospirillum were previously observed to be common autotrophic bacterial lineages in the hydrothermal vent environment, that acquire energy by sulfur-oxidation (Djurhuus et al., 2017; Zhang et al., 2017; Li et al., 2018, 2020). It should be noted that samples Kr_ct, Kr16_gl_1, and Kr16_gl_2 could not be sequenced due to the inability to generate enough DNA after 16S rRNA gene amplification, indicating the extremely low microbial biomass after the host was reared for 16 days. From this perspective, the SFS may die from a lack of enough symbiotic bacteria to support their metabolic requirements.
Conclusion
In this study, we collected SFS samples from the Kairei and Longqi vent fields. Different epibiotic and internal samples were obtained, including samples of the ctenidium, esophageal gland, visceral mass, shells, and scales. The results show that there is a significant difference in microbial community compositions among the SFS samples of Kairei and Longqi vent fields, and this may attributed to the different chemical gradients in the habitats caused by the varying intensity of hydrothermal fluid mixing with the ambient bottom seawater. The lower alpha diversity of microbial communities of the Longqi vent field SFS samples, indicated strong turbulent mixing of hydrothermal fluids and seawater in this region with a harsher environment compared with the Kairei vent field. Candidatus Thiobios, Sulfurovum, Desulfobulbus, and Maritimimonas, affiliated with the Chromatiaceae, Sulfurovaceae, Desulfobulbaceae, and Flavobacteriaceae, respectively, were the dominant bacterial lineages, constituting the core microbial communities of all SFS samples. Moreover, the genus Desulfobulbus and Sulfurovum were the primary bacterial lineages responsible for the dissimilarity of microbial communities between the SFS samples collected from the two vent fields. Additionally, since the deep sea in situ seawater for the rearing were filtered with 0.22 μm membrane, many of the in situ viruses were still present in the filtrate, and perhaps some specific lytic viruses led to the symbiotic bacteria cell lysis and indirectly caused the death of reared SFS (We have detected extreme more phages from the esophageal gland of the Kr16 than other two SFSs, data not shown). While many published papers detail the functions of the main microbial lineages present within hydrothermal vent environments, many bacteria and archaea still lack such functional information. From this perspective, to better understand the role of microorganisms in the epibiotic and internal samples of SFS, 16S rRNA gene sequencing and metagenomics should be conducted on different SFS samples collected from hydrothermal vent fields located around the globe, to reveal more detailed microbial community composition and functional information. Moreover, viral communities should also be addressed.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA679429.
Author Contributions
XP and HX designed the experiments and took the samples. HX was responsible for the experiment of dissection. SB did the DNA extraction, treated and analyzed the sequence data, and wrote the manuscript with contributions from all other authors. PCR amplification and sequencing were conducted by MajorBio Co., Ltd. (Shanghai, China). All authors have read and approved the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Grant Nos. 41906059 and 42006061) and Youth Innovation Promotion Association CAS (to HX).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are appreciative of the editor and the reviewers for their constructive comments and suggestions that improved the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.764000/full#supplementary-material
Supplementary Figure 1 | Map of deep-sea hydrothermal vent fields where snail samples were collected. SWIR, South West Indian Ridge; CIR, Central Indian Ridge; SEIR, South East Indian Ridge.
Supplementary Figure 2 | Comparisons of four α-diversity indices, Shannon index (A,E), Inverse Simpson index (B,F), observed richness (C,G), and Chao1 index (D,H), of the SFS samples. Kr refers to SFS samples collected from the Kairei vent field; Kr16 refers to samples collected from the Kairei vent field SFS reared for 16 days; Lq refers to SFS samples collected from the Longqi vent field; The group of Ct, Gl, and Vm refers to the internal samples of SFSs; The group of Sc and Sh refers to the epibiotic samples of SFSs. ∗Difference is significant at 0.05 level; ∗∗Difference is significant at 0.01 level, based on Wilcoxon rank-sum test. The results based on the ASVs datasets.
Supplementary Figure 3 | NMDS analysis of the microbial communities separated the samples into two principal groups, one composed of the samples of SFSs collected from the Kairei vent field, and another group composed of SFS samples collected from the Longqi vent field (A). Although the samples of Kr and Kr16 grouped together, they were in a loose pattern with no significant difference (C). However, the microbial community structures of Kr and Kr16 were clearly separated from the microbial community of Lq (B,D). The results based on the ASVs datasets.
Supplementary Figure 4 | Stacked bar chart showing the relative abundance of microbial communities from all samples at phylum and class levels (A), order level (B), family level (C), and genus level (D). The results based on the ASVs datasets.
Supplementary Figure 5 | The importance (A) and the heatmap (B) of the 20 most important OTUs, which distinguished the samples of Kr and Lq. The microbial abundance was scaled with log transformation in the heatmap. Kr refers to SFS samples collected from the Kairei vent field; Lq refers to SFS samples collected from the Longqi vent field.
Supplementary Figure 6 | The importance (A) and the heatmap (B) of the 20 most important OTUs, which distinguished the samples of Kr16 and Lq. The microbial abundance was scaled with log transformation in the heatmap. Kr16 refers to SFS samples collected from the Kairei vent field, and reared for 16 days; Lq refers to SFS samples collected from the Longqi vent field.
Supplementary Figure 7 | The importance (A) and the heatmap (B) of the 20 most important OTUs, which distinguished the samples of Kr and Kr16. The microbial abundance was scaled with log transformation in the heatmap. Kr refers to SFS samples collected from the Kairei vent field; Kr16 refers to SFS samples collected from the Kairei vent field, and reared for 16 days.
Footnotes
References
Anantharaman, K., Breier, J. A., and Dick, G. J. (2016). Metagenomic resolution of microbial function in deep-sea hydrothermal plumes across the Eastern Lau spreading center. ISME J. 10, 225–239. doi: 10.1038/ismej.2015.81
Apremont, V., Cambon-Bonavita, M. A., Cueff-Gauchard, V., François, D., Pradillon, F., Corbari, L., et al. (2018). Gill chamber and gut microbial communities of the hydrothermal shrimp Rimicaris chacei Williams and Rona 1986: a possible symbiosis. PLoS One 13:e0206084. doi: 10.1371/journal.pone.0206084
Bai, S., and Hou, G. (2020). Microbial communities on fish eggs from Acanthopagrus schlegelii and Halichoeres nigrescens at the XuWen coral reef in the Gulf of Tonkin. PeerJ 8:e8517. doi: 10.7717/peerj.8517
Bauer-Nebelsick, M., Bardele, C. F., and Ott, J. A. (1996). Redescription of Zoothamnium niveum (Hemprich & Ehrenberg, 1831) Ehrenberg, 1838 (Oligohymenophora, Peritrichida) a ciliate with ectosymbiotic, chemoautotrophic bacteria. Eur. J. Prostistol. 32, 18–30. doi: 10.1016/S0932-4739(96)80036-8
Beeby, M. (2015). Motility in the epsilon-proteobacteria. Curr. Opin. Microbiol. 28, 115–121. doi: 10.1016/j.mib.2015.09.005
Campbell, B. J., Engel, A. S., Porter, M. L., and Takai, K. (2006). The versatile epsilon-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4, 458–468. doi: 10.1038/nrmicro1414
Cary, S., Shank, T., and Stein, J. (1998). Worms bask in extreme temperatures. Nature 391, 545–546. doi: 10.1038/35286
Chao, A. (1984). Nonparametric-estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270.
Chen, C., Linse, K., Copley, J. T., and Rogers, A. D. (2015c). The ‘scaly-foot gastropod’: a new genus and species of hydrothermal vent-endemic gastropod (Neomphalina: Peltospiridae) from the Indian Ocean. J. Molluscan Stud. 81, 322–334. doi: 10.1093/mollus/eyv013
Chen, C., Copley, J. T., Linse, K., and Rogers, A. D. (2015a). Low connectivity between ‘scaly-foot gastropod’ (Mollusca: Peltospiridae) populations at hydrothermal vents on the Southwest Indian Ridge and the Central Indian Ridge. Org. Divers. Evol. 15, 663–670. doi: 10.1007/s13127-015-0224-8
Chen, C., Copley, J. T., Linse, K., Rogers, A. D., and Sigwart, J. D. (2015b). The heart of a dragon: 3D anatomical reconstruction of the ‘scaly-foot gastropod’ (Mollusca: Gastropoda: Neomphalina) reveals its extraordinary circulatory system. Front. Zool. 12:13. doi: 10.1186/s12983-015-0105-1
Childress, J. J., and Girguis, P. R. (2011). The metabolic demands of endosymbiotic chemoautotrophic metabolism on host physiological capacities. J. Exp. Biol. 214, 312–325. doi: 10.1242/jeb.049023
Dick, G. J. (2019). The microbiomes of deep-sea hydrothermal vents: distributed globally, shaped locally. Nat. Rev. Microbiol. 17, 271–283. doi: 10.1038/s41579-019-0160-2
Djurhuus, A., Mikalsen, S. O., Giebel, H. A., and Rogers, A. D. (2017). Cutting through the smoke: the diversity of microorganisms in deep-sea hydrothermal plumes. R. Soc. Open Sci. 4:160829. doi: 10.1098/rsos.160829
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C. (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv [Preprint] doi: 10.1101/081257
El Houari, A., Ranchou-Peyruse, M., Ranchou-Peyruse, A., Dakdaki, A., Guignard, M., Idouhammou, L., et al. (2017). Desulfobulbus oligotrophicus sp. nov., a sulfate-reducing and propionate-oxidizing bacterium isolated from a municipal anaerobic sewage sludge digester. Int. J. Syst. Evol. Microbiol. 67, 275–281. doi: 10.1099/ijsem.0.001615
Elderfield, H., and Schultz, A. (1996). Mid-ocean ridge hydrothermal fluxes and the chemical composition of the ocean. Annu. Rev. Earth Planet. Sci. 24, 191–224. doi: 10.1146/annurev.earth.24.1.191
Fisher, C. R., Takai, K., and Le Bris, N. (2007). Hydrothermal vent ecosystems. Oceanography 20, 14–23. doi: 10.5670/oceanog.2007.75
Gallant, R. M., and Von Damm, K. L. (2006). Geochemical controls on hydrothermal fluids from the Kairei and Edmond Vent Fields, 23°–25°S, Central Indian Ridge. Geochem. Geophys. Geosyst. 7:Q06018. doi: 10.1029/2005GC001067
Giovannelli, D., Chung, M., Staley, J., Starovoytov, V., Le Bris, N., and Vetriani, C. (2016). Sulfurovum riftiae sp. nov., a mesophilic, thiosulfate-oxidizing, nitrate-reducing chemolithoautotrophic epsilonproteobacterium isolated from the tube of the deep-sea hydrothermal vent polychaete Riftia pachyptila. Int. J. Syst. Evol. Microbiol. 66, 2697–2701. doi: 10.1099/ijsem.0.001106
Girguis, P. R., and Lee, R. W. (2006). Thermal preference and tolerance of alvinellids. Science 312:231. doi: 10.1126/science.1125286
Goffredi, S. K., Warén, A., Orphan, V. J., Van Dover, C. L., and Vrijenhoek, R. C. (2004). Novel forms of structural integration between microbes and a hydrothermal vent gastropod from the Indian Ocean. Appl. Environ. Microbiol. 70, 3082–3090. doi: 10.1128/AEM.70.5.3082-3090.2004
Hashimoto, J., Ohta, S., Gamo, T., Chiba, H., Yamaguchi, T., Tsuchida, S., et al. (2001). First hydrothermal vent communities from the Indian Ocean discovered. Zool. Sci. 18, 717–721. doi: 10.2108/zsj.18.717
Inagaki, F., Takai, K., Nealson, K. H., and Horikoshi, K. (2004). Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the epsilon-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 54, 1477–1482. doi: 10.1099/ijs.0.03042-0
Jannasch, H. W. (1985). Review lecture: the chemosynthetic support of life and the microbial diversity at deep-sea hydrothermal vents. Proc. R. Soc. Lond. B. Biol. Sci. 225, 277–297. doi: 10.1098/rspb.1985.0062
Ji, F., Zhou, H., Yang, Q., Gao, H., Wang, H., and Lilley, M. D. (2017). Geochemistry of hydrothermal vent fluids and its implications for subsurface processes at the active longqi hydrothermal field, southwest indian ridge. Deep Sea Res. I 122, 41–47. doi: 10.1016/j.dsr.2017.02.001
Jiang, H., and Breier, J. A. (2014). Physical controls on mixing and transport within rising submarine hydrothermal plumes: a numerical simulation study. Deep Sea Res. I 92, 41–45. doi: 10.1016/j.dsr.2014.06.006
Jørgensen, B. (1982). Mineralization of organic matter in the sea bed-the role of sulphate reduction. Nature 296, 643–645. doi: 10.1038/296643a0
Kharrat, H., Karray, F., Bartoli, M., Ben Hnia, W., Mhiri, N., Fardeau, M. L., et al. (2017). Desulfobulbus aggregans sp. nov., a Novel sulfate reducing bacterium isolated from marine sediment from the Gulf of Gabes. Curr. Microbiol. 74, 449–454. doi: 10.1007/s00284-017-1211-4
Kong, Y. (2011). Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153. doi: 10.1016/j.ygeno.2011.05.009
Li, J., Yang, J., Sun, M., Su, L., Wang, H., Gao, J., et al. (2020). Distribution and succession of microbial communities along the dispersal pathway of hydrothermal plumes on the Southwest Indian Ridge. Front. Mar. Sci. 7:581381. doi: 10.3389/fmars.2020.581381
Li, Y., Tang, K., Zhang, L., Zhao, Z., Xie, X., Chen, C. A., et al. (2018). Coupled Carbon, Sulfur, and Nitrogen Cycles mediated by microorganisms in the water column of a shallow-water hydrothermal ecosystem. Front. Microbiol. 9:2718. doi: 10.3389/fmicb.2018.02718
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Marbler, H., Koschinsky, A., Pape, T., Seifert, R., Weber, S., Baker, E. T., et al. (2010). Geochemical and physical structure of the hydrothermal plume at the ultramafic-hosted Logatchev hydrothermal field at 14°45′N on the Mid-Atlantic Ridge. Mar. Geol. 271, 187–197. doi: 10.1016/j.margeo.2010.01.012
McNichol, J., Stryhanyuk, H., Sylva, S. P., Thomas, F., Musat, N., Seewald, J. S., et al. (2018). Primary productivity below the seafloor at deep-sea hot springs. Proc. Natl. Acad. Sci. U.S.A. 115, 6756–6761. doi: 10.1073/pnas.1804351115
Meier, D. V., Pjevac, P., Bach, W., Hourdez, S., Girguis, P. R., Vidoudez, C., et al. (2017). Niche partitioning of diverse sulfur-oxidizing bacteria at hydrothermal vents. ISME J. 11, 1545–1558. doi: 10.1038/ismej.2017.37
Mino, S., Kudo, H., Arai, T., Sawabe, T., Takai, K., and Nakagawa, S. (2014). Sulfurovum aggregans sp. nov., a hydrogen-oxidizing, thiosulfate-reducing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent chimney, and an emended description of the genus Sulfurovum. Int. J. Syst. Evol. Microbiol. 64, 3195–3201. doi: 10.1099/ijs.0.065094-0
Moreau, J. W., Gionfriddo, C. M., Krabbenhoft, D. P., Ogorek, J. M., DeWild, J. F., Aiken, G. R., et al. (2015). The effect of natural organic matter on mercury methylation by Desulfobulbus propionicus 1pr3. Front. Microbiol. 6:1389. doi: 10.3389/fmicb.2015.01389
Mori, K., Yamaguchi, K., and Hanada, S. (2018). Sulfurovum denitrificans sp. nov., an obligately chemolithoautotrophic sulfur-oxidizing epsilonproteobacterium isolated from a hydrothermal field. Int. J. Syst. Evol. Microbiol. 68, 2183–2187. doi: 10.1099/ijsem.0.002803
Nakagawa, S., Shimamura, S., Takaki, Y., Suzuki, Y., Murakami, S., Watanabe, T., et al. (2014). Allying with armored snails: the complete genome of gammaproteobacterial endosymbiont. ISME J. 8, 40–51. doi: 10.1038/ismej.2013.131
Nakamura, K., Watanabe, H., Miyazaki, J., Takai, K., Kawagucci, S., Noguchi, T., et al. (2012). Discovery of new hydrothermal activity and chemosynthetic fauna on the Central Indian Ridge at 18°–20°S. PLoS One 7:e32965. doi: 10.1371/journal.pone.0032965
Park, S. C., Baik, K. S., Kim, D., and Seong, C. N. (2009). Maritimimonas rapanae gen. nov., sp. nov., isolated from gut microflora of the veined rapa whelk, Rapana venosa. Int. J. Syst. Evol. Microbiol. 59, 2824–2829. doi: 10.1099/ijs.0.010504-0
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
R Core Team (2018). R: a Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rinke, C., Lee, R., Katz, S., and Bright, M. (2007). The effects of sulphide on growth and behaviour of the thiotrophic Zoothamnium niveum symbiosis. Proc. Biol. Sci. 274, 2259–2269. doi: 10.1098/rspb.2007.0631
Sass, K., Güllert, S., Streit, W. R., and Perner, M. (2020). A hydrogen-oxidizing bacterium enriched from the open ocean resembling a symbiont. Environ. Microbiol. Rep. 12, 396–405. doi: 10.1111/1758-2229.12847
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sheik, C. S., Anantharaman, K., Breier, J. A., Sylvan, J. B., Edwards, K. J., and Dick, G. J. (2015). Spatially resolved sampling reveals dynamic microbial communities in rising hydrothermal plumes across a back-arc basin. ISME J. 9, 1434–1445. doi: 10.1038/ismej.2014.228
Sievert, S. M., and Vetriani, C. (2012). Chemoautotrophy at deep-sea vents: past, present, and future. Oceanography 25, 218–233. doi: 10.5670/oceanog.2012.21
Sorokin, D. Y., Tourova, T. P., Panteleeva, A. N., and Muyzer, G. (2012). Desulfonatronobacter acidivorans gen. nov., sp. nov. and Desulfobulbus alkaliphilus sp. nov., haloalkaliphilic heterotrophic sulfate-reducing bacteria from soda lakes. Int. J. Syst. Evol. Microbiol. 62, 2107–2113. doi: 10.1099/ijs.0.029777-0
Suzuki, D., Ueki, A., Amaishi, A., and Ueki, K. (2007). Desulfobulbus japonicus sp. nov., a novel Gram-negative propionate-oxidizing, sulfate-reducing bacterium isolated from an estuarine sediment in Japan. Int. J. Syst. Evol. Microbiol. 57, 849–855. doi: 10.1099/ijs.0.64855-0
Tanaka, N., Romanenko, L. A., Iino, T., Frolova, G. M., and Mikhailov, V. V. (2011). Cocleimonas flava gen. nov., sp. nov., a gammaproteobacterium isolated from sand snail (Umbonium costatum). Int. J. Syst. Evol. Microbiol. 61, 412–416. doi: 10.1099/ijs.0.020263-0
Tao, C., Li, H., Deng, X., Lei, J., Wang, Y., Zhang, K., et al. (2014). “Hydrothermal Activity on ultraslow spreading ridge: new hydrothermal fields found on the Southwest Indian ridge,” in Proceedings of the 2014 AGU Fall Meeting, (Washington, DC: American Geophysical Union).
Teske, A. (2009). “Deep-sea hydrothermal vents,” in Encyclopedia of Microbiology, 3rd Edn, ed. M. Schaechter (Cambridge: Academic Press). doi: 10.1016/B978-012373944-5.00276-5
Van Dover, C. L., Humphris, S. E., Fornari, D., Cavanaugh, C. M., Collier, R., Goffredi, S. K., et al. (2001). Biogeography and ecological setting of Indian Ocean hydrothermal vents. Science 294, 818–823. doi: 10.1126/science.1064574
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., et al. (2015). Improved Bacterial 16S rRNA Gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. 1:e00009-15. doi: 10.1128/mSystems.00009-15
Warén, A., Bengtson, S., Goffredi, S. K., and Van Dover, C. L. (2003). A hot-vent gastropod with iron sulfide dermal sclerites. Science 302, 1007–1007. doi: 10.1126/science.1087696
Watsuji, T. O., Motoki, K., Hada, E., Nagai, Y., Takaki, Y., Yamamoto, A., et al. (2018). Compositional and functional shifts in the epibiotic bacterial community of Shinkaia crosnieri Baba & Williams (a Squat Lobster from Hydrothermal Vents) during methane-fed rearing. Microbes Environ. 33, 348–356. doi: 10.1264/jsme2.ME18072
Zhang, N., Song, C., Wang, M., Liu, Y., Hui, M., and Cui, Z. (2017). Diversity and characterization of bacteria associated with the deep-sea hydrothermal vent crab Austinograea sp. comparing with those of two shallow-water crabs by 16S ribosomal DNA analysis. PLoS One 12:e0187842. doi: 10.1371/journal.pone.0187842
Zhou, Y., Zhang, D., Zhang, R., Liu, Z., Tao, C., Lu, B., et al. (2018). Characterization of vent fauna at three hydrothermal vent fields on the Southwest Indian Ridge: implications for biogeography and interannual dynamics on ultraslow-spreading ridges. Deep Sea Res. I 137, 1–12. doi: 10.1016/j.dsr.2018.05.001
Keywords: scaly-foot snails, epibiotic samples, internal samples, microbial communities, hydrothermal vent fields, Southwest Indian Ridge
Citation: Bai S, Xu H and Peng X (2021) Microbial Communities of the Hydrothermal Scaly-Foot Snails From Kairei and Longqi Vent Fields. Front. Mar. Sci. 8:764000. doi: 10.3389/fmars.2021.764000
Received: 24 August 2021; Accepted: 27 September 2021;
Published: 13 October 2021.
Edited by:
Qingyun Yan, Sun Yat-sen University, ChinaReviewed by:
Zhifei Li, Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences, ChinaChongqing Wen, Guangdong Ocean University, China
Copyright © 2021 Bai, Xu and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaotong Peng, eHRwZW5nQGlkc3NlLmFjLmNu