
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 02 December 2021
Sec. Marine Biotechnology and Bioproducts
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.763622
This article is part of the Research Topic High Throughput Omics Technologies for Understanding Molecular Stress Response and Devising Mitigation Actions View all 8 articles
 Lize San1,2
Lize San1,2 Baosuo Liu1,3,4
Baosuo Liu1,3,4 Bo Liu1,3
Bo Liu1,3 Huayang Guo1,3,5
Huayang Guo1,3,5 Liang Guo1,3
Liang Guo1,3 Nan Zhang1,3
Nan Zhang1,3 Kecheng Zhu1,3,4,5
Kecheng Zhu1,3,4,5 Shigui Jiang1,3
Shigui Jiang1,3 Dianchang Zhang1,3,4,5*
Dianchang Zhang1,3,4,5*
Golden pompano (Trachinotus ovatus) is one of the most economically critical marine fish in South China. Low oxygen stress has resulted in substantial economic losses to the aquaculture of T. ovatus. However, the molecular responses of fish gills to hypoxia challenge remain unclear. To understand the mechanism underlying adaption to hypoxia, we analyzed the transcriptome of T. ovatus gills in response to hypoxic stress in the normal oxygen group, hypoxic group, and hypoxia treatment after oxygen recovery group. This study found that hypoxia for 8 h was the critical time of hypoxic stress and corresponded to the largest number of differentially expressed genes. After hypoxic stress, genes for chemokines, chemokine receptors, interleukins, complement factors, and other cytokines were significantly downregulated, which may be why fish are vulnerable to pathogen infection in a hypoxic environment. According to a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, many downregulated genes were significantly enriched in the steroid biosynthesis, focal adhesion, and the extracellular matrix (ECM)-receptor interaction signal pathways, which affected cell signal transduction, adhesion, and apoptosis. Compared with the hypoxic group, the amounts of upregulated genes related to phagocytosis and protein degradation were upregulated in the dissolved oxygen recovery group. These results indicated that after the recovery of dissolved oxygen, the fish body repaired the stress-induced damage by rapidly removing misfolded proteins. These findings provide a better understanding of the hypoxia response mechanism of fish and represent a useful resource for the genetic breeding of T. ovatus.
The life activities of fish are easily affected by various environmental factors, among which the dissolved oxygen content in water is critical. Under natural conditions, the flow of water and physical agitation by wind and waves cause a large amount of oxygen to dissolve in water. Oxygen produced by photosynthesis by aquatic organisms is also the primary source of dissolved oxygen. When seawater exchange is not timely or pollution induces red tides, dissolved oxygen is reduced in the water, resulting in the lack of oxygen for fish. After a red tide burst, large amounts of dead algae are decomposed in a process that consumes large amounts of oxygen (Paerl and Otten, 2013). In addition, parasitic infections can also cause oxygen deprivation in fish. Some parasites, such as Cryptocaryon irritans and Amyloodinium ocellatum, destroy gill tissue, which prevents fish from exchanging gas and leads to the eventual death of the fish (Yin et al., 2015; Moreira et al., 2017).
A decrease in dissolved oxygen in the water affects fish, including their growth, metabolism, cell apoptosis and other biological processes, and causes changes in swimming behavior (Onukwufor and Wood, 2018). For example, in studies on Nile tilapia, low oxygen has been shown to reduce its appetite and affect its growth (Obirikorang et al., 2020). A study of hypoxia in rainbow trout found that it swims slower under low oxygen conditions (Poulsen et al., 2011). In addition to affecting the behavior and feeding of fish, a hypoxic environment also affects the normal development of embryos. Prolonged hypoxia mitigates the growth and development of Atlantic salmon eggs and even leads to embryo deformities (Wood et al., 2020). Moreover, the morphological structure of the gill tissue is changed under hypoxic stress. In crucian carp (Carassius carassius), goldfish (C. auratus), and Megalobrama amblycephala, the functional surface area of the gills is increased in response to hypoxic stress based on the reconstruction of gill tissue to expose the gill lamina (Sollid et al., 2003; Mitrovic et al., 2009). Hypoxia also causes a different degree of apoptosis in fish tissues. For example, in a study of hypoxia in largemouth bass, hypoxic environments promoted apoptosis in gill and liver tissues (Wu et al., 2017).
To cope with the hypoxic environment, fish have adapted to the hypoxic environment in terms of their morphological structure and physiological and biochemical characteristics. Previous studies have shown that most fish react behaviorally in response to acute hypoxia (Nilsson and Renshaw, 2004; Mitrovic and Perry, 2009). In the hypoxic environment, fish will present an increased respiratory rate, reduced movement, and surface breathing to solve hypoxic stress (Petersen and Gamperl, 2010; Capossela et al., 2012; Killen et al., 2012). The biochemical responses of fish to low oxygen levels are more elaborate than their behavioral responses. Fish have a complete stress response regulation system. Previous studies have found that fish meet their energy needs by changing their energy supply sources in hypoxic environments (Pan et al., 2020; Liu et al., 2021; Pang et al., 2021). Under prolonged hypoxic conditions, fish develop auxiliary respiratory organs. Lepidocephalichthys and Gobionellus oceanicus rely on the skin, intestines, oral mucosa, and other organs to assist in gas exchange (Ghosh et al., 2011; Aguilar et al., 2018).
Transcriptome sequencing refers to high-throughput sequencing after the reverse transcription of all mRNA in a cell or tissue at a specific time and space. RNA sequencing (RNA-Seq) methods have the advantages of high efficiency, low cost, and high throughput, and the sequencing results are generally considered a reliable sequencing technology for the study of non-model organisms. Progress has been made in the study of hypoxic stress in fish by transcriptome analyses. In a study of silver sillago (Sillago sihama) under hypoxic stress, transcriptome sequencing technology was used to explore the different expression patterns. Many genes related to steroid and amino acid biosynthesis have been reported (Saetan et al., 2020). A comparative transcriptome analysis showed that the Gymnocypris ecklon reduces energy consumption in response to hypoxic stress by inhibiting cell growth and proliferation (Qi et al., 2018), and mRNA sequencing of large yellow croaker liver tissue showed that carbohydrate metabolism plays a crucial role in energy supply and amino acid metabolism is an important supplement to cope with energy supply (Ding et al., 2020).
Trachinotus ovatus, also known as golden pompano, is widely distributed in tropical and subtropical waters of Southeast Asia and the Mediterranean Sea. It is an essential economic fish in coastal breeding in southern China. However, hypoxia caused by long-distance transportation, parasites, and other reasons easily leads to death. Therefore, this study aims to detect the transcriptome response of T. ovatus to hypoxic stress by RNA-Seq. The transcriptome characteristics of gill tissues between the restored dissolved oxygen group, hypoxic group, and normoxic group were compared and analyzed through an enrichment analysis of differential genes. This study provides a new perspective for understanding the molecular mechanism of antihypoxic stress in T. ovatus.
Trachinotus ovatus was obtained from the Tropical Fisheries Research and Development Centre, Lingshui, Hainan Province, China. Before the experiment, the experimental fish were acclimated to the water tank (DO 8.8 ± 0.2 mg/L, pH 8.1–8.3) for 1 month. During the acclimation period, the fish were fed twice daily using a compound feed, the feeding weight was 5% of the body weight of the fish, and the feeding was stopped the day before the start of the experiment. Before the experiment, 50 fish were selected for the pre-experiment to measure the asphyxiation point. In this study, we defined the asphyxiation point of T. ovatus as the dissolved oxygen level at the time when the fish first rolled to the bottom of the barrel and stopped swimming within 10 s. Fifty T. ovatus (weight: 78.4 ± 4.1 g) were placed in a tank of 250 L seawater sealed off from the air with plastic. A dissolved oxygen sensor was used to measure the dissolved oxygen in the water whenever a fish rolled to the bottom. The pre-experiment found that the asphyxiation points of different pompano individuals were different, indicating that different individuals had different tolerances to hypoxic stress. The asphyxiation point of T. ovatus was 1.0–1.2 mg/L.
The asphyxiation point of T. ovatus was measured at approximately 1.0 mg/L in the pre-experiment; therefore, the dissolved oxygen level was maintained at 1.5 ± 0.3 mg/L in the experiment. Four hundred and fifty T. ovatus (weight: 78.4 ± 4.1 g) were randomly selected from among the temporary breeding fish and placed into three tanks filled with 500 L seawater, with 150 in each group. Before the hypoxic stress experiment, three fish were selected from each tank as a normal oxygen group. Then, nitrogen was charged into the water, and the dissolved oxygen reached 1.5 ± 0.3 mg/L after 1 h. The DO of the water was measured using a WTW dissolved oxygen meter (Multi 3620, Germany), and the dissolved oxygen level was maintained for 24 h. After hypoxic stress, the water tank was filled with oxygen and the dissolved oxygen recovered to 8.8 ± 0.2 mg/L after 30 min. Three fish were taken from each group at 4, 8, 12, and 24 h after hypoxia treatment and 12 and 24 h after recovery of dissolved oxygen. The T. ovatus in the normoxic, hypoxic stress, and hypoxia recovery groups were placed on ice, and the gill tissue was rapidly separated, transferred to a tube with RNAlater, frozen with liquid nitrogen, and stored at −80°C for subsequent RNA extraction.
The total RNA of the T. ovatus gill tissue was extracted according to the instructions of the RNA extraction kit. The extracted RNA was subjected to 1% agarose gel electrophoresis to detect the integrity of the total RNA. The concentration of RNA and the OD260/OD280 ratio were detected by a NanoDrop-2000 ultraviolet imaging system. The extracted RNA was stored at −80°C. The NebNext® UltraTM RNA Library Prep Kit for Illumina® Kit was used to build the library. mRNA with oligo (dT) was enriched by the magnetic bead method, and then the mRNA was randomly broken by divalent cations in fragmentation buffer. First-strand cDNA was synthesized with a random primer from the broken mRNA, and RNaSeH was used to degrade the RNA strand. Then, the second cDNA strand was synthesized by the DNA polymerase I system using dNTPs as raw material. A cDNA fragment of approximately 370–420 bp was screened for PCR amplification, and then library construction was completed. Transcriptome sequencing was performed on an Illumina HiSeq 6000, and approximately 150 bp paired-end (PE) reads were obtained.
To ensure the reliability of the data, the original data must be filtered with FASTQ software (Chen et al., 2018). Clean reads were obtained by removing reads containing adapters and poly-N and low-quality reads from the raw data. Sequenced fragments are randomly interrupted mRNAs. To determine the genes that transcribed these fragments, the filtered clean reads were compared to the reference genome. Clean reads and the reference genome were compared quickly and accurately with HisAT2 software (Mortazavi et al., 2008) to obtain the positioning information of reads on the reference genome. Stringtie (Pertea et al., 2015) was used to predict new genes. Based on the location information of the gene alignment on the reference genome, featureCounts (Liao et al., 2014) was used to calculate the readings mapped to each gene. Thus, the number of reads covered from initiation to termination of each gene was counted. Because of the influence of sequencing depth and gene length, the gene expression of RNA-Seq is generally expressed in fragments per kilobase per million mapped reads (FPMM) instead of read counts. The FPKM value was corrected for sequencing depth and gene length. DESeq2 software (Love et al., 2014) was used to identify differentially expressed genes (DEGs), and an adjusted P-value < 0.05 and | log2 (FoldChange)| > 1 were used as the threshold for significant differential expression. Through the software Clusterprofiler (Yu et al., 2012), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were carried out for the differentially obtained gene sets obtained from the difference significance analysis. The threshold value of significant enrichment for GO and KEGG enrichment was set to an adjusted P-value < 0.05.
Five DEGs involved in steroid biosynthesis, lysine degradation, focal adhesion, glycine, serine and threonine metabolism, and extracellular matrix (ECM)-receptor interaction signaling pathways were selected for quantitative real-time PCR (qRT-PCR) to determine the accuracy of the RNA-Seq data. The primers were designed using Primer Premier 5 software (Premier Biosoft International, United States) (Table 1). qRT-PCR was performed using a Roche LightCycler® 480II (Roche) with gill cDNA samples from different hypoxic stress times as templates. The qRT-PCR kit was a SYBR Green Premix Pro Taq HS qPCR Kit (Regen, Guangzhou, China). The reaction program for the 12.5 μl PCR system was as follows: 95°C for 30 s; 40 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 20 s. Three biological replicates were performed for each group, and all samples were examined in triplicate for the same DEGs. The elongation factor 1-alpha 1 (EF1A) gene was selected as the internal reference gene, and the 2–ΔΔCt method was applied to determine the relative expression levels of target genes.

Table 1. Primers used for actin, non-muscle (ACTIN), farnesyl-diphosphate farnesyltransferase 1 (FDFT1), L-threonine 3-dehydrogenase (TDH), collagen type IV alpha 2 (COL4A2), and fibronectin 1b (FN1B) gene expression analysis.
In this study, a total of 948,411,128 raw reads were obtained. There were 930,888,066 clean reads after quality control filtering. The error rate was 1.8%. As shown in Table 2, the Q30 of all libraries was greater than 90%, indicating that the sequencing data were reliable. Approximately 88–92% of clean reads were compared to the genome, and 75–78% of clean reads were compared to the exons in each library.
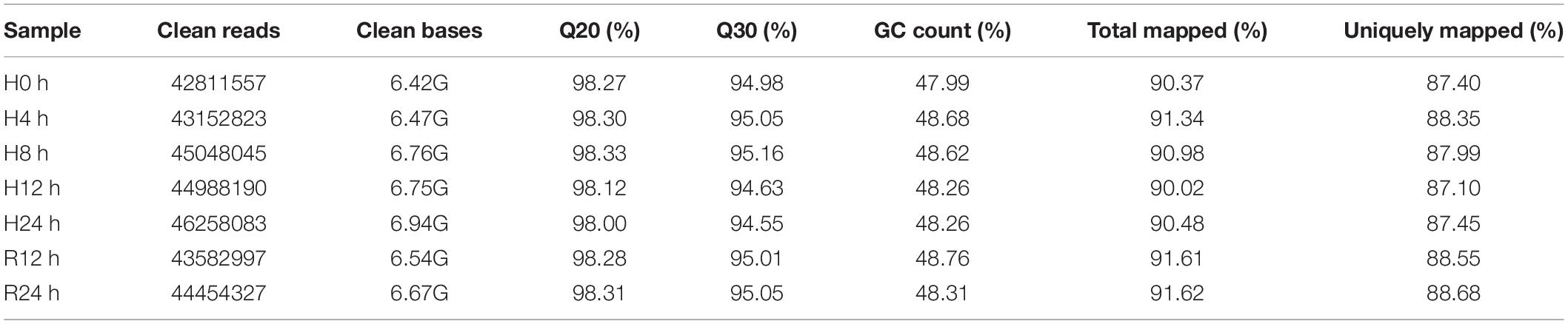
Table 2. Illumina sequencing and mapping statistics of the gill transcriptomes of T. ovatus.
After the hypoxic treatment, the results of gill transcriptome sequencing showed that compared with the normoxic group, a total of 5294 genes were identified as DEGs in the hypoxia 4 h, hypoxia 8 h, hypoxia 12 h, hypoxia 24 h, reoxygenation 12 h, and reoxygenation 24 h groups. Compared to the normoxic group, 1594 genes were obtained after 8 h of hypoxic treatment, of which 572 were upregulated and 1022 were downregulated (Figure 1). The number of DEGs among the six groups initially increased and then decreased, reaching a peak at hypoxia for 8 h, and the number of downregulated genes (DRGs) in each group was greater than that of the upregulated genes (URGs). The UpSet plot showed that 90 identical DEGs were expressed between the four groups treated with hypoxia (Figure 2). There were 43 identical URGs and 47 identical DRGs. The number of DEGs in the reoxygenation group was greater than that in the hypoxic group, with 533 DEGs, 433 DRGs, and 100 URGs.
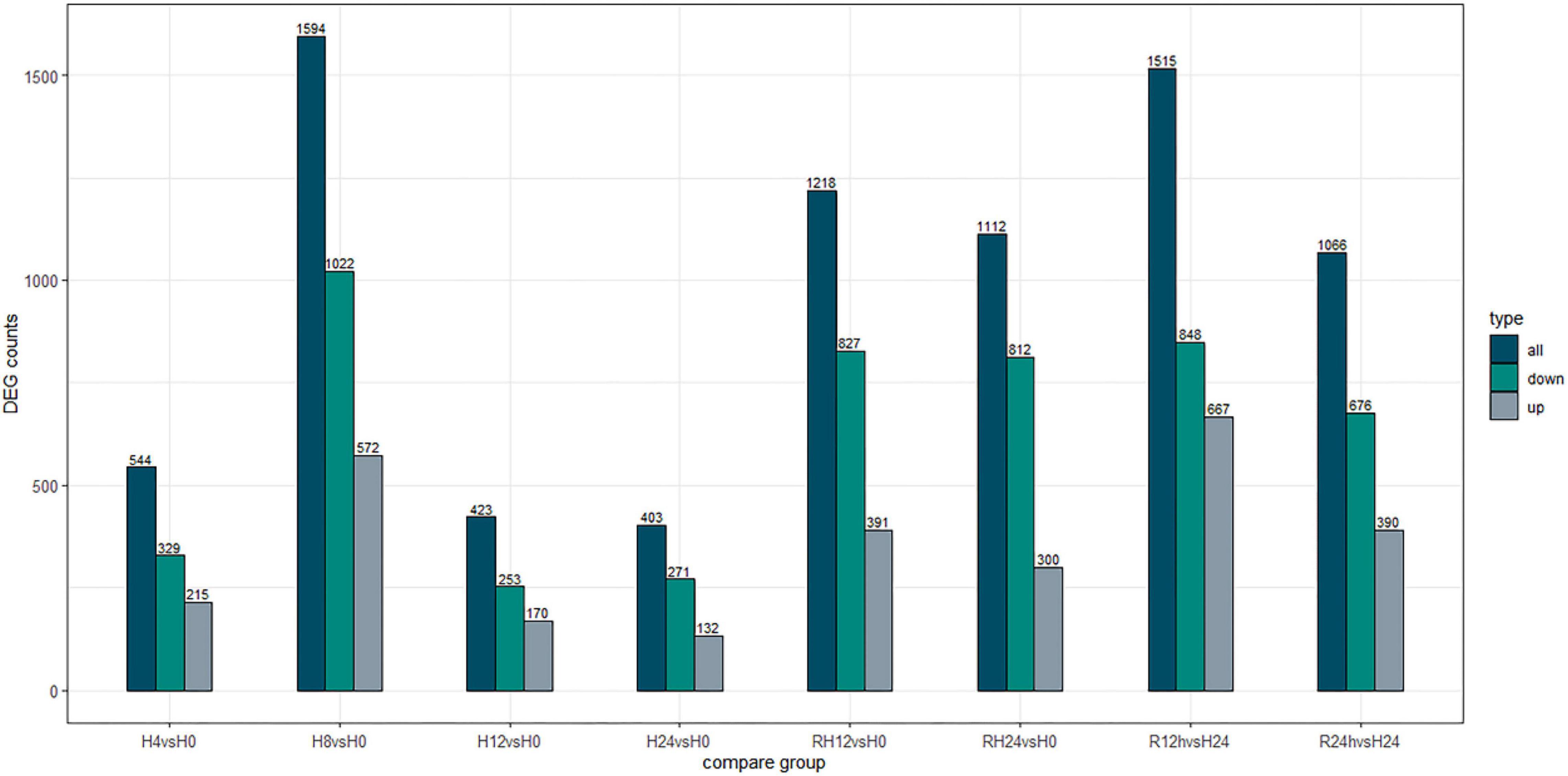
Figure 1. Number of differentially expressed genes under different hypoxic stress time.
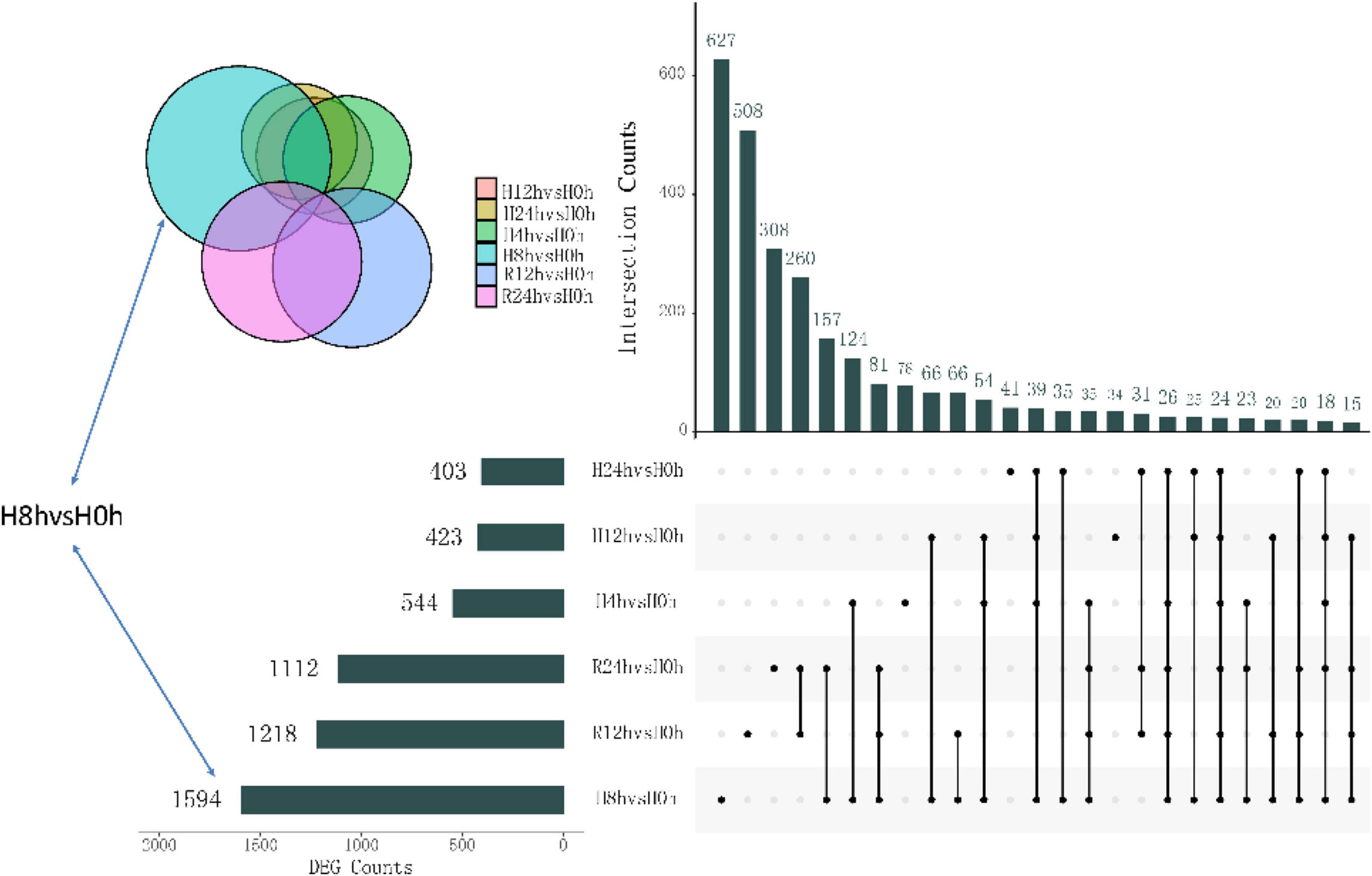
Figure 2. Upset diagram showing the unique and shared DEGs of different hypoxic stress time.
A total of 544 DEGs were obtained by comparative transcriptome analysis between the hypoxic 4 h group and the normoxic group, of which 215 were URGs and 329 were DRGs. The URGs were not significantly enriched in any GO terms but were significantly enriched in the steroid biosynthesis signaling pathway (Figure 3A). The DRGs were significantly enriched in 20 GO terms, including 4 BPs, 15 MFs, and 1 CC. Four GO terms were related to the small molecule metabolic process, one term was related to the extracellular region, and the remaining 15 GO terms were associated with enzyme regulator activity (Figure 4A). The KEGG pathway enrichment analysis indicated that the genes were enriched in the focal adhesion, glycine, serine, and threonine metabolism pathways and the ECM-receptor interaction signal pathway. The KEGG and GO enrichment analyses identified enrichment in amino acid metabolism, which may be inhibited under hypoxic stress.
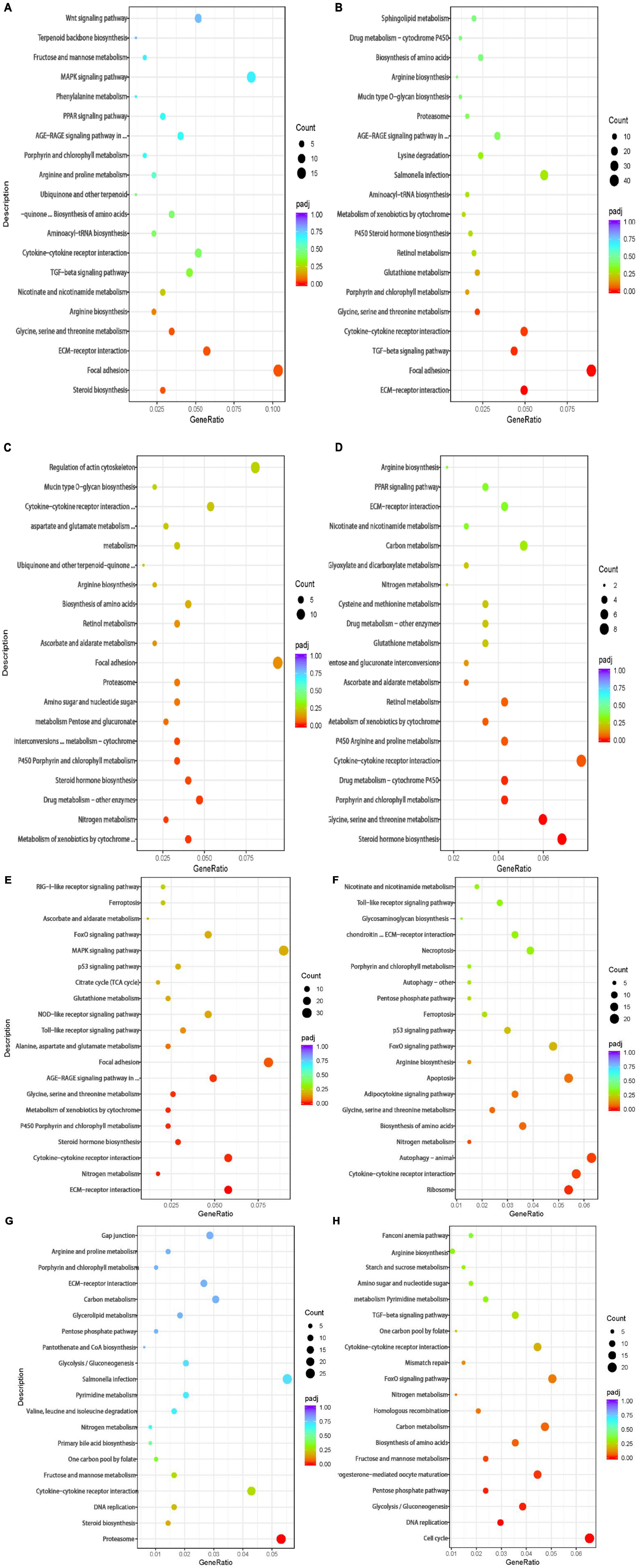
Figure 3. Bubble chart of KEGG pathway enrichment of the DEGs in the (A) 4 vs. 0 h, (B) 8 vs. 0 h, (C) 12 vs. 0 h, (D) 24 vs. 0 h,(E) R12 vs. 0 h, (F) R24 vs. 0 h, (G) R12 vs. H24 h, and (H) R24 vs. H24 h. The vertical axis represents the pathway categories, and the horizontal axis shows the gene ratio. The point size shows the number of DEGs enriched to the KEGG pathway. The point color shows different Q values as indicated on the right.
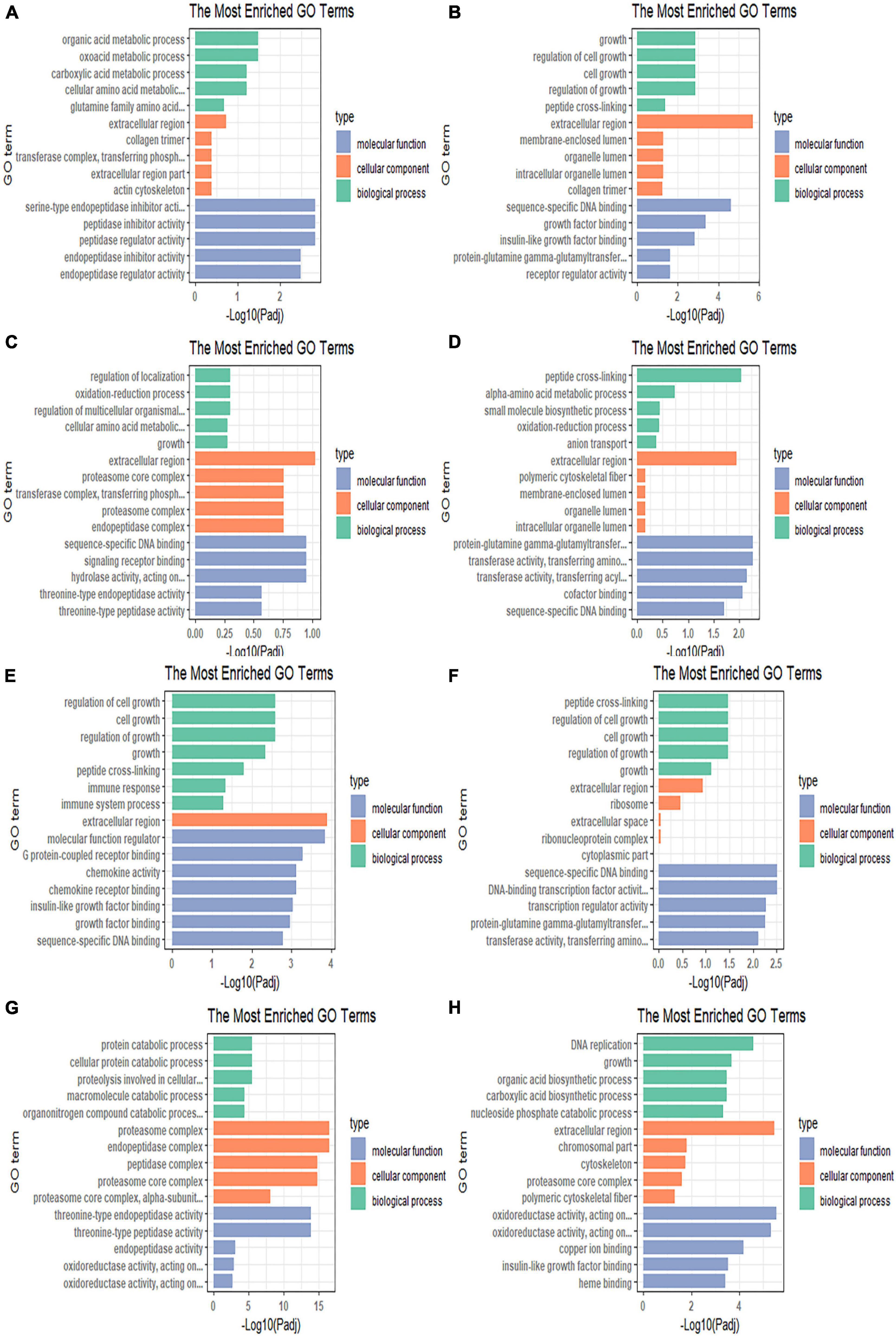
Figure 4. Gene Ontology enrichment bar chart illustrating the differentially expressed genes enriched in GO terms and the counts of genes for each GO terms in the (A) 4 vs. 0 h, (B) 8 vs. 0 h, (C) 12 vs. 0 h, (D) 24 vs. 0 h, (E) R12 vs. 0 h, (F) R24 vs. 0 h, (G) R12 vs. H24 h, and (H) R24 vs. H24 h.
Compared to the normoxic group, 1594 DEGs were detected in the hypoxia 8 h group, including 572 URGs and 1022 DRGs. URGs were significantly enriched in seven GO terms related to the membrane-enclosed lumen. In addition, the lysine degradation and melanogenesis pathways were significantly enriched among the URGs, which proves that external stimulation can cause the fish to turn black. These DRGs were significantly enriched in total of 44 GO terms, including 13 BPs, 2 CCs, and 29 MFs. The most significantly enriched terms were mainly related to growth and growth factor binding (Figure 4B). The DRGs were significantly enriched in three KEGG pathways, including focal adhesion, glycine, serine, and threonine metabolism and the ECM-receptor interaction signal pathway (Figure 3B). These signaling pathways were consistent with DRG enrichment under hypoxia for 4 h.
The comparison of the transcriptomes of the hypoxia 12 h and normoxic groups generated 423 DEGs, including 170 URGs and 253 DRGs. No significantly enriched GO terms or KEGG pathways were found for the URGs. Regulation of localization, cellular amino acid metabolic processes, and catalytic complex-related GO terms were downregulated under hypoxia for 12 h (Figure 4C). The DRGs were significantly enriched in thirteen KEGG pathways. Most DRGs were enriched in the metabolism of xenobiotics by cytochrome P450 and nitrogen metabolism signaling pathways (Figure 3C).
Compared to the normoxic group, 403 DEGs were detected in the hypoxia 24 h group, including 132 URGs and 271 DRGs. The URGs were significantly enriched six GO terms. The most significantly enriched GO term was monooxygenase activity. No KEGG pathways were found for the URGs. A total of six GO terms, including three BPs and three MFs, were significantly enriched for the DRGs. The GO terms associated with small-molecule metabolic process and transferase activity were significantly enriched (Figure 4D). The DRGs were significantly enriched in six KEGG pathways. The glycine, serine, and threonine metabolism signaling pathways were significantly downregulated in the hypoxia 24 h group, which was similar to that observed in the hypoxia 4 h and hypoxia 8 h groups (Figure 3D).
A total of 2330 genes were differentially expressed in the reoxygenation 12 h and reoxygenation 24 h groups compared with the normoxic group. Among these DEGs, 1218 showed differential expression between the reoxygenation 12 h and normoxic groups, including 391 URGs and 827 DRGs in the reoxygenation 12 h group; and 1112 DEGs were identified between the reoxygenation 24 h and normoxic groups, including 300 URGs and 812 DRGs. The enrichment analysis showed that the regulation of Rho protein signal transduction, Rho GTPase binding and guanyl-nucleotide exchange factor activity were severely impacted in the reoxygenation 12 h group compared with the normoxic group (Figures 4E,F). The URGs after reoxygenation for 12 h were not significantly enriched in any GO terms. In the reoxygenation 12 h ECM-receptor interaction, the NOD-like receptor signaling pathway was significantly enriched by the URGs. Compared with reoxygenation at 12 h, the URGs at 24 h of reoxygenation were significantly enriched in the lysine degradation signaling pathway (Figures 3E,F). The DRGs at both reoxygenation for 12 h and reoxygenation for 24 h were significantly enriched in the regulation of cell growth and sequence-specific DNA binding. The DRGs at reoxygenation for 12 h were significantly enriched in the steroid hormone biosynthesis and glycine, serine, and threonine metabolism signaling pathways. At 12 h of reoxygenation, genes related to the ribosome and biosynthesis of amino acid signaling pathways were significantly downregulated.
Compared to the hypoxia 24 h group, 1515 DEGs were detected in the reoxygenation 12 h group, including 667 URGs and 848 DRGs, and 1066 DEGs were detected in the reoxygenation 24 h group, including 390 URGs and 676 DRGs. The GO terms related to proteolysis involved in cellular protein catabolic process, proteasome core complex, alpha-subunit complex, and threonine-type endopeptidase activity were upregulated in the reoxygenation 12 h group (Figure 4G). Genes related to the biological functions of copper ion binding, monocarboxylic acid, and nucleotide biosynthetic processes were downregulated. Proteasome and DNA replication were significantly enriched for the URGs after reoxygenation for 12 h (Figure 3G). Compared with the hypoxia 24 h group, the URGs were significantly enriched in the biological process of DNA replication and the DRGs were significantly enriched in the glycolytic process at 24 h of reoxygenation (Figure 4H). At 24 h of reoxygenation, genes associated with the glycolysis pathway were downregulated, suggesting that the fish’s energy supply occurred via an anaerobic pathway (Figure 3H).
To verify the accuracy of RNA-Seq, five genes were randomly selected and analyzed by qRT-PCR to obtain the expression patterns of these genes in different groups. The qRT-PCR results showed that the expression patterns of these genes in different groups were consistent with the results of RNA-Seq analysis (Figure 5), indicating the specificity and accuracy of the transcriptome expression analysis.

Figure 5. Comparison of gene expression data between RNA-seq and quantitative real-time PCR (qRT-PCR). The expression level of actin, non-muscle (A), farnesyl-diphosphate farnesyltransferase 1 (B), L-threonine 3-dehydrogenase (C), collagen type IV alpha 2 (D), and fibronectin 1b (E) in different groups.
The gill is a crucial gas exchange organ in fish that plays an essential role in resisting hypoxic stress. Therefore, we compared the transcriptomes of three treatment groups (normoxic, hypoxic stress, and reoxygenation groups). Compared to the normoxic group, most DEGs were detected in the hypoxia 8 h group, of which a total of 1594 DEGs were identified, including 572 upregulated and 1022 DRGs. In all treatment groups, the number of DRGs was higher than that of URGs. A comparative transcriptome analysis of the response of sillago gill tissue to hypoxia indicated that the number of URGs was greater than that of DRGs in different hypoxic treatment groups (Saetan et al., 2020). The number of URGs increased with increasing hypoxic stress duration, contrary to the results of this study. However, in the transcriptome study of the gill tissue of large yellow croak after hypoxia treatment, the results were consistent with those observed here: within 24 h of stress, the number of DRGs was greater than that of URGs, the number of DEGs was the highest in the hypoxia 12 h group, and the number of DFGs decreased in the subsequent hypoxia 24 h group (Ding et al., 2020). These results indicate that different fish have different hypoxia response mechanisms. Hypoxic stress caused the downregulation of many genes in T. ovatus, which may be the reason for its low hypoxic tolerance.
Gill-associated lymphoid tissues (GIALTs) are active immune tissues composed of immune factors and active immune cells (Salinas et al., 2011). The mucus secreted by GIALT is rich in various immune-related factors, including interleukins, chemokines, and chemokine receptors. In this study, the gene encoding the chemokine CLC25 and its receptor (ACKR4) were significantly downregulated in gill tissue. CCL25 is currently involved in T-cell development, but it also combines with ACKR4 to reduce availability through internalization. In addition, ACKR4 plays an essential role in controlling the migration of immune cells expressing chemokine receptors CCR7 and CCR9 (Ibanez et al., 2014). Three genes encoding cytokine receptors were also significantly downregulated, i.e., IL1R, IL10R, and IL18R. The combination of IL8R and IL8 was conducive to the production of cytokines. In this study, many genes related to immune factors were significantly downregulated after hypoxia treatment and reached a maximum at 8 h after treatment. Moreover, after 8 h of hypoxia treatment, the gene encoding the Toll-like receptor family (TLR22 and TLR14) was also significantly downregulated. TLR22 is a member of the TLR11 subfamily located on the cell membrane, and it is responsible for identifying RNAs, thus playing an essential role in the immune response after a virus or bacterial infection (Palti, 2011). The analysis found that genes associated with immune factors were suppressed until the end of the experiment, and restoring dissolved oxygen did not change this. Moreover, the expression of genes encoding complement factors (C6 and C7) was significantly downregulated and remained significantly downregulated after oxygen was restored for 12 h. A similar expression pattern was found in Atlantic salmon and large yellow croaker (Eslamloo et al., 2020; Mu et al., 2020). These results suggest that the low expression of these genes suppresses the innate immune system of fish. Fish under low oxygen conditions may be more susceptible to pathogen infection due to innate immune suppression. Previous studies have also found that when fish are exposed to multiple pathogens, their immunity declines with a decrease in dissolved oxygen. Of course, hypoxic stress does not entirely break down the fish’s innate immune system. TNF receptor (TNFR) is mainly a transmembrane protein involved in inflammation, apoptosis, autoimmunity, and other physiological processes (Wajant and Scheurich, 2001). Tnfrsf10 and tnfrsf19 were highly expressed in gill tissues after hypoxic treatment and induced apoptosis. This immune-suppressing apoptotic response pattern is replicated in the gill tissue of largemouth bass (Micropterus salmoides) (Sun et al., 2020). The differential gene analysis between the restored dissolved oxygen group and the hypoxic group after 24 h showed that the significantly downregulated expression of immune factor genes was increased compared with the normal oxygen group, which indicated that the immunity of the fish was also restored after the restoration of dissolved oxygen, although it could not be restored to the normal level within a short time.
Through the KEGG enrichment analysis, after 4 and 8 h of hypoxia treatment, we found that the DRGs were significantly enriched in the ECM receptor interaction, focal adhesion, glycine, serine, and threonine metabolism signaling pathways. The ECM includes proteins such as collagen and laminin, which interact with transmembrane molecules to directly or indirectly control cell activities (Wang and Luo, 2010). After hypoxia treatment for 4 h, the expression of collagen type IV alpha 5 chain (COL4A5), collagen type VI alpha 6 chain (COL6A6), and collagen type IV alpha 2 chain (COL4A2) was downregulated. These genes encode different types of collagen subunits. With prolonged hypoxia time, the expression of the laminin alpha 2 (LAMA2) and laminin subunit beta 3 (LAMB3) genes encoding laminin was also downregulated after 8 h of hypoxia treatment. Previous studies found that defects or deletion of the LAMA2 protein leads to human muscle weakness (Koutsoulidou and Phylactou, 2020; Fan et al., 2021). Therefore, the decrease in LAMA2 gene expression may be one of the reasons for the decrease in the swimming speed of T. ovatus after hypoxic stress. We found that DRGs enriched in the focal adhesion signaling pathway included genes encoding integrin subunits (Figure 6). Integrin interacts with the ECM and transmits signals downstream through focal adhesion kinase (FAK), thus affecting cell adhesion and signal transduction (Calderwood et al., 2013). Recent studies have found that the apoptosis of human cardiomyocytes is related to the loss of integrin (Su et al., 2018). According to the downregulation results of genes encoding ECM and integrin, it is speculated that hypoxic stress leads to the loss of adhesion between gill tissue cells and ECM, resulting in cell apoptosis. In the glycine, serine, and threonine metabolism pathways, glycine is mainly synthesized by the serine pathway, and serine is mainly synthesized by 3-phosphoglyceric acid. Serine can enhance antioxidants (Parker and Metallo, 2016). After hypoxic stress, we found that the expression of phosphoserine phosphotase (PSPH) and phosphoserine aminotransferase 1 (PSAT1) was downregulated. The proteins encoded by these two genes are the rate-limiting enzymes of the serine synthesis pathway. The downregulation of the expression levels of these two genes resulted in the obstruction of the synthesis of serine, thus resulting in the impairment of the antioxidant capacity of T. ovatus.
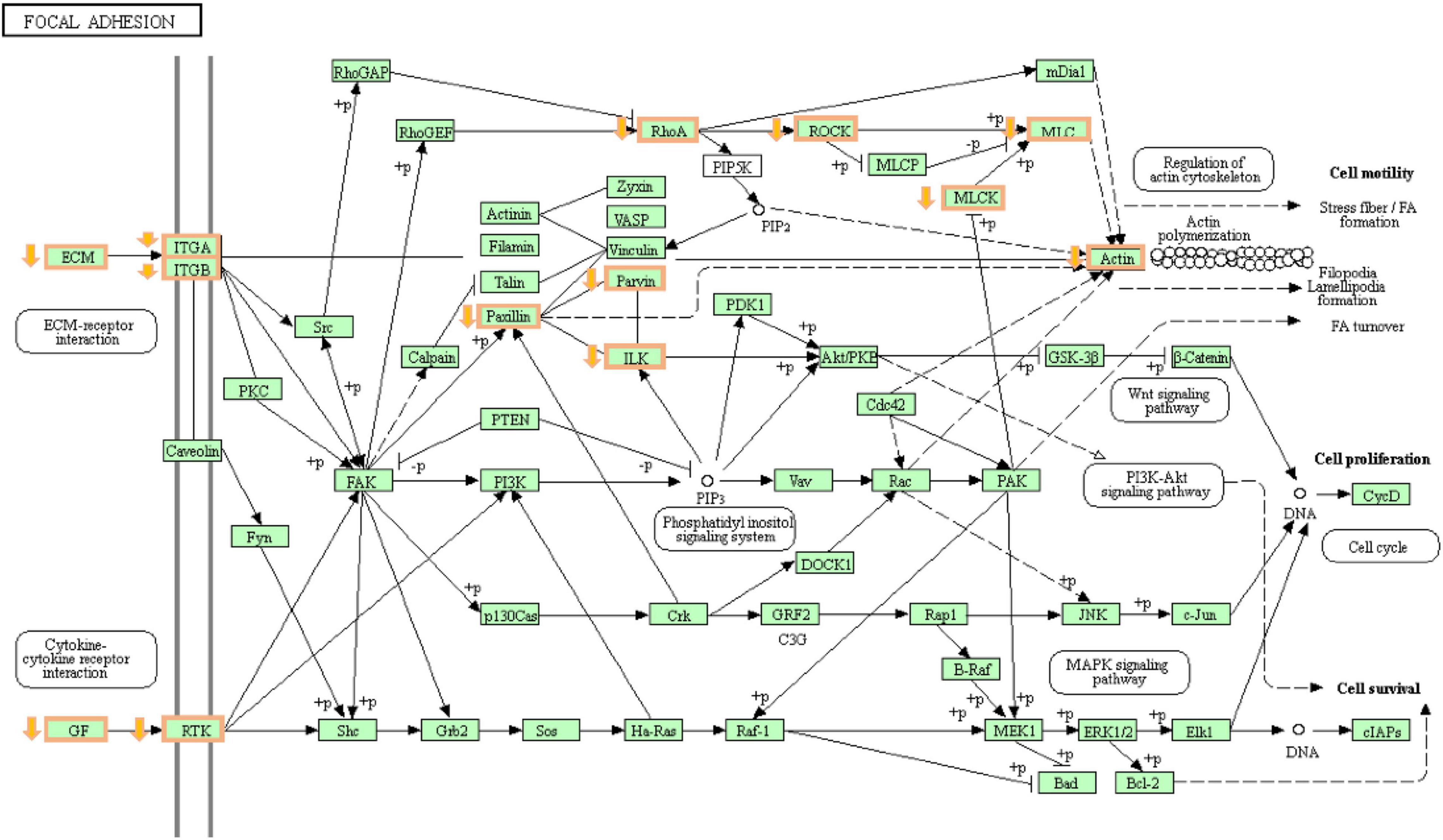
Figure 6. The changes of focal adhesion pathways during hypoxic stress 8 h. Orange boxes are genes with decreased expression.
To better understand the transcriptomic response of T. ovatus after the recovery of dissolved oxygen, the recovered dissolved oxygen group was compared not only with the normal oxygen group but also with the hypoxic 24 h group. Compared with 24 h of hypoxia, many URGs were enriched in the proteasome (Figure 7), phagosome, ubiquitin-mediated proteolysis, and apoptosis pathways. The ubiquitin–proteasome system (UPS) and autophagy–lysosome system are two major protein degradation pathways in eukaryotic cells. The proteasome removes excess damaged proteins during proteolytic activity (Nath and Shadan, 2009). Autophagy degrades and retrieves cellular components by converting defective organelles and misfolded proteins into two-membrane autophagosomes and transferring them to lysosomes (Lin et al., 2013). These results suggest that proteasome and autophagy reactions play an essential role in the recovery process of T. ovatus after hypoxic stress.
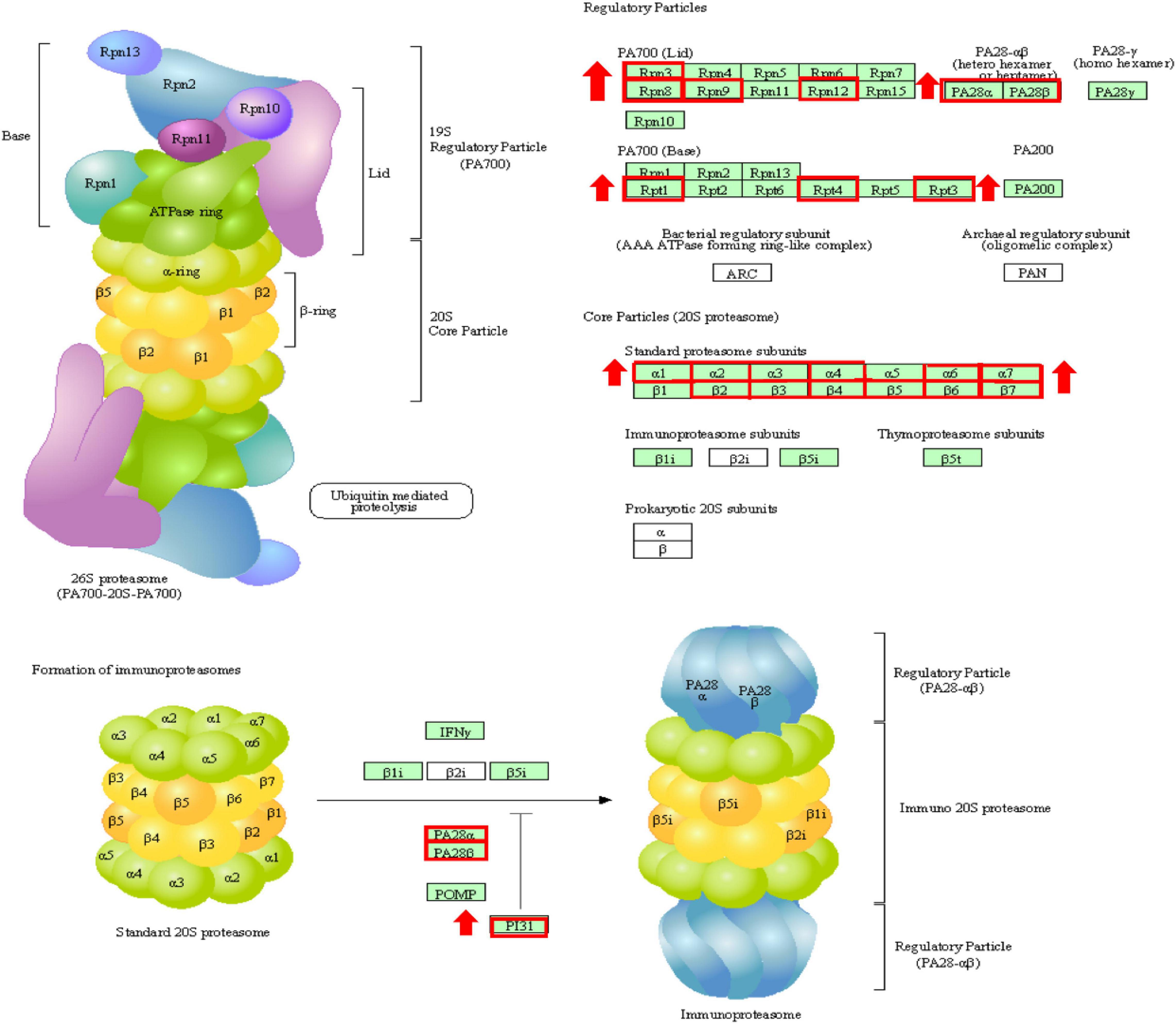
Figure 7. The changes of proteasome during reoxygenation 12 h. Red boxes are genes with increased expression.
In the present study, the transcriptome analysis showed that hypoxic stress for 8 h was the critical time at which gene expression changed the most. The differential gene enrichment analysis found that genes related to immunity, signal transduction, adhesion, and apoptosis were downregulated. It was speculated that hypoxic stress affected the immune function and signal-transduction efficiency of T. ovatus. By comparing the transcriptome of the hypoxic group and dissolved oxygen recovery group, it was found that T. ovatus may repair body damage after dissolved oxygen recovery by removing misfolded proteins. Future investigations will focus on screening genes related to hypoxia tolerance among DEGs to improve survival in a hypoxic environment. In conclusion, these findings provide new insights into the molecular mechanisms of T. ovatus in response to hypoxic stress.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: SRR14949116–SRR14949135).
The animal study was reviewed and approved by the Animal Experiment Committee of South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences. Written informed consent was obtained from the owners for the participation of their animals in this study.
DZ and SJ conceived and designed the experiments. BoL, HG, and NZ performed the experiments. NZ and LS contributed to sample collection. BaL and LG analyzed the data and wrote the manuscript. LS and KZ contributed to real-time analysis. LS and BaL assisted with writing and proofreading. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (U20A2064), Central Public-Interest Scientific Institution Basal Research Fund, CAFS (2020TD29), supported by China Agriculture Research System of MOF and MARA (CARS-47), Central Public-Interest Scientific Institution Basal Research Fund of South China Sea Fisheries Research Institute, CAFS (2021SD12), Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams (2019KJ143), Financial Fund of the Ministry of Agriculture and Rural Affairs, China (NHYYSWZZZYKZX2020), National Key R&D Program of China (2018YFD0900301), National Key R&D Program of China (2018YFD0901204), Science and Technology Infrastructure Construction Project of Guangdong Province (2019B030316030), and National Marine Genetic Resource Center, China-ASEAN Maritime Cooperation Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aguilar, L., Leite, R. N., Ferreira, C. A., and da Cruz, A. L. (2018). The integument of the nonamphibious goby Gobionellus oceanicus: its functional morphology and respiratory capacity. J. Morphol. 279, 1548–1558. doi: 10.1002/jmor.20848
Calderwood, D. A., Campbell, I. D., and Critchley, D. R. (2013). Talins and kindlins: partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 14, 503–517. doi: 10.1038/nrm3624
Capossela, K. M., Brill, R. W., Fabrizio, M. C., and Bushnell, P. G. (2012). Metabolic and cardiorespiratory responses of summer flounder Paralichthys dentatus to hypoxia at two temperatures. J. Fish Biol. 81, 1043–1058. doi: 10.1111/j.1095-8649.2012.03380.x
Chen, S. F., Zhou, Y. Q., Chen, Y. R., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890. doi: 10.1093/bioinformatics/bty560
Ding, J., Liu, C., Luo, S., Zhang, Y., Gao, X., Wu, X., et al. (2020). Transcriptome and physiology analysis identify key metabolic changes in the liver of the large yellow croaker (Larimichthys crocea) in response to acute hypoxia. Ecotoxicol. Environ. Saf. 189:109957. doi: 10.1016/j.ecoenv.2019.109957
Eslamloo, K., Caballero-Solares, A., Inkpen, S. M., Emam, M., Kumar, S., Bouniot, C., et al. (2020). Transcriptomic profiling of the adaptive and innate immune responses of Atlantic Salmon to Renibacterium salmoninarum infection. Front. Immunol. 11:567838. doi: 10.3389/fimmu.2020.567838
Fan, Y. B., Tan, D. D., Song, D. Y., Zhang, X., Chang, X. Z., Wang, Z. X., et al. (2021). Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J. Med. Genet. 58, 326–333. doi: 10.1136/jmedgenet-2019-106671
Ghosh, S. K., Ghosh, B., and Chakrabarti, P. (2011). Fine anatomical structures of the intestine in relation to respiratory function of an air-breathing loach, Lepidocephalichthys guntea (Actinopterygii: Cypriniformes: Cobitidae). Acta Ichthyol. Et Piscator. 41, 1–5. doi: 10.3750/aip2011.41.1.01
Ibanez, L. G., Cook, S. L., Zhang, Y., Yam-Puc, J. C., Brown, G., Antal, R., et al. (2014). Atypical chemokine receptor 4 (ACKR4) in B cell activation. Immunology 143, 183–183. doi: 10.1002/JLB.2MA1119-300R
Killen, S. S., Marras, S., Ryan, M. R., Domenici, P., and McKenzie, D. J. (2012). A relationship between metabolic rate and risk-taking behaviour is revealed during hypoxia in juvenile European sea bass. Funct. Ecol. 26, 134–143. doi: 10.1111/j.1365-2435.2011.01920.x
Koutsoulidou, A., and Phylactou, L. A. (2020). Circulating biomarkers in muscular dystrophies: disease and therapy monitoring. Mol. Ther. Methods Clin. Dev. 18, 230–239. doi: 10.1016/j.omtm.2020.05.017
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Lin, N. Y., Beyer, C., Giessl, A., Kireva, T., Scholtysek, C., Uderhardt, S., et al. (2013). Autophagy regulates TNF alpha-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 72, 761–768. doi: 10.1136/annrheumdis-2012-201671
Liu, Z. F., Ma, A. J., Yuan, C. H., Zhao, T. T., Chang, H. W., and Zhang, J. S. (2021). Transcriptome analysis of liver lipid metabolism disorders of the turbot Scophthalmus maximus in response to low salinity stress. Aquaculture 534:7. doi: 10.1016/j.aquaculture.2020.736273
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15:38. doi: 10.1186/s13059-014-0550-8
Mitrovic, D., Dymowska, A., Nilsson, G. E., and Perry, S. F. (2009). Physiological consequences of gill remodeling in goldfish (Carassius auratus) during exposure to long-term hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R224–R234. doi: 10.1152/ajpregu.00189.2009
Mitrovic, D., and Perry, S. F. (2009). The effects of thermally induced gill remodeling on ionocyte distribution and branchial chloride fluxes in goldfish (Carassius auratus). J. Exp. Biol. 212, 843–852. doi: 10.1242/jeb.025999
Moreira, M., Schrama, D., Soares, F., Wulff, T., Pousao-Ferreira, P., and Rodrigues, P. (2017). Physiological responses of reared sea bream (Sparus aurata Linnaeus, 1758) to an Amyloodinium ocellatum outbreak. J. Fish Dis. 40, 1545–1560. doi: 10.1111/jfd.12623
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Mu, Y. N., Li, W. R., Wei, Z. Y., He, L. H., Zhang, W. N., and Chen, X. H. (2020). Transcriptome analysis reveals molecular strategies in gills and heart of large yellow croaker (Larimichthys crocea) under hypoxic stress. Fish Shellfish Immunol. 104, 304–313. doi: 10.1016/j.fsi.2020.06.028
Nilsson, G. E., and Renshaw, G. M. C. (2004). Hypoxic survival strategies in two fishes: extreme anoxia tolerance in the North European crucian carp and natural hypoxic preconditioning in a coral-reef shark. J. Exp. Biol. 207, 3131–3139. doi: 10.1242/jeb.00979
Obirikorang, K. A., Acheampong, J. N., Duodu, C. P., and Skov, P. V. (2020). Growth, metabolism and respiration in Nile tilapia (Oreochromis niloticus) exposed to chronic or periodic hypoxia. Comp. Biochem. Physiol. Mol. Integr. Physiol. 248:9. doi: 10.1016/j.cbpa.2020.110768
Onukwufor, J. O., and Wood, C. M. (2018). The osmorespiratory compromise in rainbow trout (Oncorhynchus mykiss): the effects of fish size, hypoxia, temperature and strenuous exercise on gill diffusive water fluxes and sodium net loss rates. Comp. Biochem. Physiol. Mol. Integr. Physiol. 219, 10–18. doi: 10.1016/j.cbpa.2018.02.002
Paerl, H. W., and Otten, T. G. (2013). Harmful cyanobacterial blooms: causes, consequences, and controls. Microb. Ecol. 65, 995–1010. doi: 10.1007/s00248-012-0159-y
Palti, Y. (2011). Toll-like receptors in bony fish: from genomics to function. Dev. Comp. Immunol. 35, 1263–1272. doi: 10.1016/j.dci.2011.03.006
Pan, Y., Ai, C. X., Zeng, L., Liu, C., and Li, W. C. (2020). Modulation of copper-induced antioxidant defense, Cu transport, and mitophagy by hypoxia in the large yellow croaker (Larimichthys crocea). Fish Physiol. Biochem. 46, 997–1010. doi: 10.1007/s10695-020-00765-0
Pang, M., Wang, Y., Tang, Y., Dai, J., Tong, J., and Jin, G. (2021). Transcriptome sequencing and metabolite analysis reveal the toxic effects of nanoplastics on tilapia after exposure to polystyrene. Environ. Pollut. 277:116860. doi: 10.1016/j.envpol.2021.116860
Parker, S. J., and Metallo, C. M. (2016). Chasing one-carbon units to understand the role of serine in epigenetics. Mol. Cell 61, 185–186. doi: 10.1016/j.molcel.2016.01.006
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Petersen, L. H., and Gamperl, A. K. (2010). Effect of acute and chronic hypoxia on the swimming performance, metabolic capacity and cardiac function of Atlantic cod (Gadus morhua). J. Exp. Biol. 213, 808–819. doi: 10.1242/jeb.033746
Poulsen, S. B., Jensen, L. F., Nielsen, K. S., Malte, H., Aarestrup, K., and Svendsen, J. C. (2011). Behaviour of rainbow trout Oncorhynchus mykiss presented with a choice of normoxic and stepwise progressive hypoxia. J. Fish Biol. 79, 969–979. doi: 10.1111/j.1095-8649.2011.03069.x
Qi, D., Chao, Y., Wu, R., Xia, M., Chen, Q., and Zheng, Z. (2018). Transcriptome analysis provides insights into the adaptive responses to hypoxia of a Schizothoracine fish (Gymnocypris eckloni). Front. Physiol. 9:1326. doi: 10.3389/fphys.2018.01326
Saetan, W., Tian, C., Yu, J., Lin, X., He, F., Huang, Y., et al. (2020). Comparative transcriptome analysis of gill tissue in response to hypoxia in Silver Sillago (Sillago sihama). Animals 10:628. doi: 10.3390/ani10040628
Salinas, I., Zhang, Y. A., and Sunyer, J. O. (2011). Mucosal immunoglobulins and B cells of teleost fish. Dev. Comp. Immunol. 35, 1346–1365. doi: 10.1016/j.dci.2011.11.009
Sollid, J., De Angelis, P., Gundersen, K., and Nilsson, G. E. (2003). Hypoxia induces adaptive and reversible gross morphological changes in crucian carp gills. J. Exp. Biol. 206, 3667–3673. doi: 10.1242/jeb.00594
Su, Y. F., Tian, H., Wei, L. J., Fu, G. H., and Sun, T. (2018). Integrin beta 3 inhibits hypoxia-induced apoptosis in cardiomyocytes. Acta Biochim. Et Biophys. Sin. 50, 658–665. doi: 10.1093/abbs/gmy056
Sun, J. L., Zhao, L. L., He, K., Liu, Q., Luo, J., Zhang, D. M., et al. (2020). MiRNA-mRNA integration analysis reveals the regulatory roles of miRNAs in the metabolism of largemouth bass (Micropterus salmoides) livers during acute hypoxic stress. Aquaculture 526:735362. doi: 10.1016/j.aquaculture.2020.735362
Wajant, H., and Scheurich, P. (2001). Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int. J. Biochem. Cell Biol. 33, 19–32. doi: 10.1016/s1357-2725(00)00064-9
Wang, W., and Luo, B. H. (2010). Structural basis of integrin transmembrane activation. J. Cell. Biochem. 109, 447–452. doi: 10.1002/jcb.22427
Wood, A. T., Clark, T. D., Elliott, N. G., Frappell, P. B., and Andrewartha, S. J. (2020). The effects of constant and cyclical hypoxia on the survival, growth and metabolic physiology of incubating Atlantic salmon (Salmo salar). Aquaculture 527:735449. doi: 10.1016/j.aquaculture.2020.735449
Wu, C.-B., Liu, Z.-Y., Li, F.-G., Chen, J., Jiang, X.-Y., and Zou, S.-M. (2017). Gill remodeling in response to hypoxia and temperature occurs in the hypoxia sensitive blunt snout bream (Megalobrama amblycephala). Aquaculture 479, 479–486. doi: 10.1016/j.aquaculture.2017.06.020
Yin, F., Gong, H., Ke, Q. Z., and Li, A. X. (2015). Stress, antioxidant defence and mucosal immune responses of the large yellow croaker Pseudosciaena crocea challenged with Cryptocaryon irritans. Fish Shellfish Immunol. 47, 344–351. doi: 10.1016/j.fsi.2015.09.013
Keywords: Trachinotus ovatus, hypoxia stress, transcriptome, gene expression, gills
Citation: San L, Liu B, Liu B, Guo H, Guo L, Zhang N, Zhu K, Jiang S and Zhang D (2021) Transcriptome Analysis of Gills Provides Insights Into Translation Changes Under Hypoxic Stress and Reoxygenation in Golden Pompano, Trachinotus ovatus (Linnaeus 1758). Front. Mar. Sci. 8:763622. doi: 10.3389/fmars.2021.763622
Received: 24 August 2021; Accepted: 04 November 2021;
Published: 02 December 2021.
Edited by:
Akbar John, International Islamic University Malaysia, MalaysiaReviewed by:
Gianfranco Santovito, University of Padua, ItalyCopyright © 2021 San, Liu, Liu, Guo, Guo, Zhang, Zhu, Jiang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dianchang Zhang, emhhbmdkY2hAc2NzZnJpLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.