 Elisa Banchi
Elisa Banchi Paola Del Negro
Paola Del Negro Mauro Celussi
Mauro Celussi Francesca Malfatti
Francesca Malfatti- 1National Institute of Oceanography and Applied Geophysics – OGS, Trieste, Italy
- 2Department of Life Sciences, University of Trieste, Trieste, Italy
Sediment microbial communities play essential roles in marine ecosystem functioning. Their study is crucial to understand how environmental conditions affect microbial diversity and biogeochemical cycles. The Venice Lagoon, one of the largest Adriatic transitional systems, is subjected to different natural and anthropogenic stressors. In this study, surface sediments microbial communities were finely characterized using 16S rRNA gene amplicon sequencing and metagenomics. Samples were collected seasonally in 2019 and 2020 at different sites within Lagoon sub-basins. Our results indicated a stable spatial distribution of the sediment microbiome: salinity, grain size, and total organic carbon were found to be important drivers in shaping prokaryotic communities, while temperature had a minor role. We detected high microbial diversity at all stations, mainly due to low abundant taxa: bacteria represented the vast majority of the reads (∼96.1%), with Woeseia the most abundant genus (∼4.4%). The metagenomic analysis highlighted significant differences among sites in terms of biogeochemical processes (e.g., C, N, Fe, and S metabolism), and cell-cell interaction strategies (e.g., mobilome, regulations and cell signaling). Chioggia, a sandy site subjected to marine influence, presented the highest abundance of ammonia-oxidizing archaeon Candidatus Nitrosopumilus, in accordance with the highest amount of ammonia monooxygenase subunit genes. At the same site, sulfate-reducing bacteria (Desulfobacteria and Desulfobacterales) and sulfur-related genes were found in lower abundance. Marghera and Tresse, the most polluted sites, showed higher abundance of sewage-related bacteria and antibiotic and toxic compound resistance genes. Furthermore, these sites showed higher amount of genes related to cell-cell interaction, such as pathogenicity islands, transposable-elements, and biofilm formation. Our findings highlighted that sediment features and human-related activities have profound and long-term impacts on the surface sediment microbial communities of the Venice Lagoon.
Introduction
Globally, marine microbes living in the top 10–50 cm account for 1.7–5.0 × 1028 cells (Whitman et al., 1998; Danovaro et al., 2015; Flemming and Wuertz, 2019). Within transitional shallow-water benthic ecosystems, the surface sediment is a highly spatially and temporally dynamic habitat. It is subjected to frequent resuspension and deposition events, mixing, transport, and it is shaped by significant biotic activities of macro- to microorganisms (Daumas, 1990; Mayer, 1993). The surface sediment is highly heterogeneous and characterized by high water content, steep physical and chemical gradients that structure the ecology, the interactions, and the metabolism of microorganisms (e.g., light, oxygen, and organic matter) (Zinger et al., 2011). The microbes within this layer play an essential role in the decomposition of organic matter, nutrient cycling, benthic food webs, thus governing the ecosystem functioning (Schallenberg and Kalff, 1993; Azam and Malfatti, 2007). In respect to the microbial diversity and functions of the pelagic realm, fewer information is available on benthic microbial communities (Probandt et al., 2017). Surface sediments usually present higher microbial abundance and diversity in terms of taxa, potential functions, and metabolisms (Zinger et al., 2011) in comparison to the deeper ones. This microbial environment is influenced by the water column biogeochemistry, and on the other side, from the deeper sediment biogeochemistry, via porewater fluxes (e.g., anoxic conditions, reduced state of organic matter, and contaminants). The study of the microbial distribution patterns within “sediment skin” is fundamental to understand and predict the responses of the marine ecosystem to environmental changes (Zinger et al., 2011). In contrast to the water environment (with the exception of marine snow particles), microbial abundances reach 109 cells for cm3 of sediment and within these universes there is harsh microscale competition for resources (e.g., energy and nutrients) and space (Petro et al., 2017).
Microbes exert their ecosystem role at the microscale (Azam and Malfatti, 2007; Stocker et al., 2008) displaying a great variety of adaptive strategies from dormancy to exchanging electrons and specialized behaviors. Specifically, motility, two-component systems, antiviral systems, the propensity to exchange genetic material, secretion system, and production of redox active antibiotics may promote overall their persistence and success in such crowded environment.
The Venice Lagoon (northern Adriatic Sea), with a surface of 550 km2, is one of the largest transitional systems in the Mediterranean Sea (Madricardo et al., 2019). This lagoon includes different environments, such as salt marshes, channels, fish farms, natural and artificial islands. It is subjected to different and extensive natural and anthropogenic stressors (Solidoro et al., 2010) such as sea-level rising and subsidence, elevated touristic pressure on Venice city (Seraphin et al., 2018), and the presence of industrial-chemical areas (Bellucci et al., 2002). The essence to be a lagoon relies on the tight coupling between the water and sediments; the sediment environment provides information on the dynamic history of these transitional areas. In the Venice Lagoon, several contaminants have been detected, coming mainly from industrial processes, incinerators, drainages, and urban wastes (Solidoro et al., 2010). These include heavy metals, toxins, polychlorinated biphenyls (PCBs), and others (Han et al., 2007; Borin et al., 2009; Solidoro et al., 2010; Gieskes et al., 2015). These pollutants were discharged in the water column and then they accumulated in the sediments, by the interaction with the downward fluxes of particulate matter (DePinto et al., 2010). The quality of the Venice Lagoon sediments has been evaluated via different risk assessment screenings: several contaminants exceed sediment quality guidelines thresholds, with the highest screening risk level in the north and central part of the lagoon, near the two hotspots of contamination Porto Marghera and Venice city canals (Apitz et al., 2007).
Even if the Venice Lagoon is one of the most studied coastal ecosystems (Solidoro et al., 2004), more effort needs to be done by employing molecular approaches focusing on the microbial role within this transitional area. So far, a handful of high-profile studies have been carried out, but none of them have employed the metagenomic approach. In 2007, Danovaro and Pusceddu used automated ribosomal intergenic spacer analysis (ARISA) to study microbial diversity in some Mediterranean coastal lagoons, including Venice. They found no relationships between the environmental features of the lagoon and the bacterial diversity, but, on the other hand, the latter was significantly and positively correlated with the ecosystem functioning and efficiency (Danovaro and Pusceddu, 2007). Borin et al. (2009) used 16S rRNA gene clone libraries to assess microbial community structure and diversity in the different sub-basins of the Lagoon. They highlighted that the sediments, mainly anoxic, were colonized by microbial communities that presented a species richness correlated with total elemental sulfur and acid-volatile sulfide (Borin et al., 2009). More recently, Quero et al. (2017) applied DNA metabarcoding to Venice Lagoon planktonic and benthic bacterial assemblages to explore diversity patterns and the role of environmental gradients. They found evidence of different temporal dynamics in the pelagic and benthic domains (Quero et al., 2017).
This study aims to provide new insights into the spatial and temporal distribution and dynamics of microbes living in the surface sediment of the Venice Lagoon in terms of diversity, functions, and metabolisms. The metagenomic approach was employed for the first time in the Lagoon, enhancing our knowledge of the behavior of the surface sediment microbial communities. Our data contribute to the understanding of the importance of the surface sediment within the microbial biogeochemical cycles of carbon, nutrients, metals, pollutants, antibiotics, and xenobiotics from the large to the micro-scale. Moreover, we have used the sediment microenvironment to test hypotheses on the microscale functional adaptations of microbes, shedding light on the potential behavior in such a complex and heterogeneous matrix.
Materials and Methods
Sampling
The sampling was carried out seasonally in 2019 (Supplementary Table 1) at five sites of the Venice lagoon, Italy (Figure 1): Chioggia (C), Marghera (M), Palude della Rosa, (P), Sacca Sessola (S), and Tresse (T). Furthermore, C, M, and P were sampled with the same time frequency in 2020. The sites were chosen to cover the four sub-basins, defined according to risk analyses following the international sediment quality guideline of the Lagoon (Apitz et al., 2007; Supplementary Table 2). They are located in the southern (C), in the central (S), in the central-north (M and T), and in the northern (P) sub-basins. The Chioggia site is near the city of Chioggia, very close to the southern lagoon inlet and more subjected to marine influence. The Sacca Sessola site, situated in the center of the Lagoon, south of Venice city, is potentially affected by urban contamination (Picone et al., 2016). The Marghera site is inside the active industrial area of Porto Marghera and is characterized by high anthropogenic impact. The Tresse site is next to an artificial island created as a dumping site for urban and industrial waste. Finally, Palude della Rosa site is less impacted by human activities (Picone et al., 2016).
Figure 1. Sampling sites in the Venice Lagoon. The maps were created with the software Ocean Data View (Schlitzer, 2018).
In Chioggia, Marghera, and Palude della Rosa samples were collected with a Van Veen grab (0.045 m2), while in Sacca Sessola and Tresse with manually operated corers (10 cm diameter). The first centimeter of sediment was collected with a sterile spatula. The sampling was performed in three independent replicates. Water temperature and salinity were recorded with the multiparametric probes HI98128 (HANNA Instruments, Italy) and AP-2000 (Aquasearch, Italy) (Supplementary Table 3).
Sediment Characterization and Bacterial Abundance Measurement
The water content was estimated by weighing fresh and dried (105°C for 24 h) sediment aliquots (2 cm3). The grain size composition was determined with a laser Particle Size Analyzer (BECKMAN COULTER LS 13 320) after treating 1 g of wet sediment with H2O2. The sediment was described using Shepard’s classification (Shepard, 1954) and the data were expressed as percentages of sand, silt, and clay. The limit between silt and clay was fixed to 4 μm, following the Udden-Wentworth classification (Wentworth, 1922).
For Total Organic Carbon (TOC) estimation, freeze-dried sediment was grounded in a ceramic mortar and sieved on a 250 μm iron steel sieve (Endecotts LTD., United Kingdom). Triplicate subsamples of about 8–12 mg were weighed on a micro ultrabalance with an accuracy of 0.1 μg. Before TOC determination, subsamples were treated directly into capsules with increasing concentrations of HCl (0.1 N and 1 N) to remove the carbonate fraction (Nieuwenhuize et al., 1994). Carbon content was determined using a CHNO-S elemental analyzer ECS 4010, Costech, Italy) according to Pella and Colombo (1973). Standard acetanilide (Costech, purity ≥ 99.5%) was used to calibrate the instrument and empty capsules were also analyzed to correct for blank. Measurement quality control was performed using internal standards and it was also verified for carbon against the certified marine sediment reference material PACS-2 (National Research Council Canada). Significant differences in TOC were calculated in the R environment (v. 4.0.3, R Core Team, 2019) by Kruskal-Wallis and Wilcoxon non-paired tests. False discovery rate (FDR) was used for p-value correction. Results with q-value < 0.05 were considered significant.
The prokaryotic abundance was estimated by flow cytometry following Deng et al. (2019) with modifications, using a FACSCanto II (Becton Dickinson) instrument equipped with an air-cooled laser at 488 nm and standard filter setup. A sediment slurry was subjected to mechanical shaking, ultrasonication, and hydrofluoridric acid (1%, final concentration) treatment. Slurry mixed with the Stop solution was filtered through a 10 μm polycarbonate filter, diluted, and stained with SYBRGreen I (2×, final concentration). Negative controls were prepared by autoclaving sediments and running unstained sediments. The acquisition threshold was set to green fluorescence and stained prokaryotic cells were identified in the side scatter versus green fluorescence plot. The flow rate was calibrated daily, by running distilled water and weighing it before and after the run (at least five replicates). Data were acquired and processed with the FACSDiva software (Becton Dickinson). Prokaryotic cells per gram of dry sediment were calculated using the acquired cell counts, the flow rates and the respective sample water content, and statistical analyses were run as described for TOC data.
A Principal component analysis (PCA) was performed to explore and visualize similarities among the samples based on environmental variables (temperature, salinity, grain size, TOC and prokaryotic abundance) with the R package FactoMineR (Le et al., 2008), factoextra (Kassambara, 2015), and ggplot2 (Wickham et al., 2016).
DNA Extraction and Sequencing
DNA was extracted with DNeasy PowerSoil Pro Kit (Qiagen) following the manufacturer’s instructions. Quality and quantity of the extracted DNA was assessed with Nanodrop spectrometer (Thermo Fisher Scientific) and with Qubit Fluorimeter (Thermo Fisher Scientific).
For the amplicon sequencing analysis, sediments from both 2019 (at each site, one sample per season for a total of 20, in three replicates) and 2020 (at C, M, and P sites, one sample per season for a total of 12, in three replicates) were considered. The V4-V5 region of 16S rRNA gene was amplified using 515-Y (5′-GTGYCAGCMGCCGCGGTAA-3′) and 926R (5′-CGYCAATTYMTTTRAGTTT-3′) primers (Parada et al., 2016). Libraries were prepared following the 16S Metagenomic Sequencing Library Preparation protocol and run on an Illumina MiSeq System for a read length of 2 × 250 bp at the genetic and epigenetic ARGO Open Lab Platform, Area Science Park, Trieste, Italy.
For the metagenomic analysis, sediments from 2019 (at each site, one sample per season for a total of 20, in three replicates) were considered. Libraries were prepared following the Illumina Nextera DNA Flex Library Prep protocol and run on an Illumina NovaSeq 6000 System for a read length of 2 × 300 bp at the genetic and epigenetic ARGO Open Lab Platform, Area Science Park, Trieste, Italy.
16S Bioinformatic Pipeline
Bioinformatic analyses were performed with QIIME2 2020.6 (Bolyen et al., 2019). Raw sequences were quality filtered and denoised with DADA2 (Callahan et al., 2016). Alpha-diversity metrics were estimated after samples were rarefied. To assess the reliability and robustness of the replicates, a correlation analysis (Spearman’s ρ) on the amplicon sequence variants (ASVs) table was performed with the R package ggplot2 (Wickham et al., 2016). Taxonomy was assigned to ASVs using the sklearn naïve Bayes taxonomy classifier (Bokulich et al., 2018) against the Silva 138 99% reference database with 7-level taxonomy (Quast et al., 2013). Reads belonging to Eukarya, mitochondria, chloroplast, and with frequency < 2 (singletons) were removed.
Statistical analyses were conducted in the R environment (v. 4.0.3, R Core Team, 2019). Rarefaction curves on ASVs table were plotted with the R package vegan (Oksanen et al., 2019). Significant differences in alpha diversity metrics were calculated by Kruskal-Wallis H test. FDR was used for p-value correction. Results with q-value < 0.05 were considered significant.
To visualize similarity patterns of prokaryotic communities, a Principal Coordinates Analysis (PcoA) using Bray-Curtis dissimilarity matrices was constructed using normalized read abundances in the R package vegan (Oksanen et al., 2019). A permutational multivariate analysis of variance (PERMANOVA) with 4999 permutations was computed on normalized ASVs table to investigate the effect of site and season using the function adonis in the R package vegan (Oksanen et al., 2019).
To further explore the environmental drivers of the community, we used a distance-based redundancy analysis (dbRDA) using the capscale function in the R package vegan (Oksanen et al., 2019). The environmental variables (temperature, salinity, grain size, and TOC) were normalized using a z-score transformation and variance partitioning analysis was used to quantify the contribution made by each variable using the varpart function in the R package vegan (Oksanen et al., 2019). The significance of the variables was tested with ANOVA and 4999 permutations after building a reduced model. As the grain size components (sand, silt, and clay) present multicollinearity, only one component (sand) was included in the analysis.
An indicator species analysis was performed at site level with the R package indicspecies (De Cáceres and Legendre, 2009). The results were visualized as networks by mean of the R package igraph (Csard and Nepusz, 2006). Moreover, an analysis of composition of microbiomes (ANCOM; Mandal et al., 2015) was used to test for differences at the genus level. Microbial diversity visualization at different taxonomic level was done using the average normalized abundance of the three replicates of each sample, with R package phyloseq (McMurdie and Holmes, 2012) and ggplot2 (Wickham et al., 2016). Microbial signatures of fecal- and sewage-associated bacteria (McLellan et al., 2010; Newton et al., 2013; Paliaga et al., 2017) were also evaluated. Statistical Analyses of Metagenomic Profiles 2.1.3 (STAMP; Parks and Beiko, 2010) was used to assess differences in the relative proportion of sequences belonging to the different taxa considering the whole dataset. Significant differences were calculated by ANOVA and Tukey-Kramer post hoc test. FDR was used for p-value correction. Results with q-value < 0.001 were considered significant.
Metagenomic Analysis
The number and quality of reads were checked with Fast QC (Andrews, 2010). Reads were assembled into contigs using MEGAHIT-1.2.9 (Li et al., 2015) combining the three replicates of each sample after testing the significance of their correlations. Gene prediction was performed with Prodigal 2.6.3 (Hyatt et al., 2010) on contigs longer than 1,000 bp (Van der Walt et al., 2017).
The contigs > 1,000 bp were submitted to Metagenome Analysis Rapid Annotation using Subsystem Technology (MG-RAST; Keegan et al., 2016) for a similarity search using the SEED Subsystem (Overbeek et al., 2005) and the Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto, 2000) databases, using a cut-off E-value of 1e–5, minimum identity of 60% and a minimum alignment length of 15 bp. SEED and KEGG annotations were normalized by the number of reads assigned to the prokaryotic single copy gene recA (Acinas et al., 2021) prior to calculating the relative abundances.
Key oceanic marker genes (Sunagawa et al., 2015) and key sediment metabolic genes (Dombrowski et al., 2018) were evaluated to provide an overview of the main biochemical cycling metabolisms present. We have created a novel ad hoc specific microscale marker function list to explore the microbial potential at the small-scale in the sediment environment.
Annotation results were visualized using Statistical Analyses of Metagenomic Profiles 2.1.3 (STAMP; Parks and Beiko, 2010) to assess differences in their relative proportion considering the whole dataset. Significant differences among samples were calculated by ANOVA and Tukey-Kramer post hoc test. FDR was used for p-value correction. Results with q-value < 0.001 were considered significant.
Results
Sediment Characterization
The five stations were characterized by different percentages of the grain size classes of sand, silt, and clay (Supplementary Table 4), in agreement with previous studies (Borin et al., 2009; Picone et al., 2016; Zonta et al., 2018). Based on Shepard’s classification, Chioggia was categorized as sand, Marghera as clayey silt, Palude della Rosa as silty-loam, Sacca Sessola as loam, and Tresse as silty sand.
Total Organic Carbon ranged from 0.87 to 32.8 mg gd–1 (Supplementary Table 5). Marghera (16.4 ± 7.5) and Palude della Rosa (14.8 ± 7.5) showed significantly higher concentrations than Sacca Sessola (6.6 ± 0.9), Tresse (5.5 ± 1.0), and Chioggia (3.3 ± 3.2). No significant differences were detected among seasons.
The average sediment prokaryotic abundance was 25.7 ± 21.5 × 109 Cells gd–1 (Supplementary Figure 1). Palude della Rosa showed significantly higher abundances (45.4 ± 32.5 × 109 Cells gd–1) than Marghera (27.7 ± 24.6 × 109 Cells gd–1; p < 0.05), Chioggia (8.3 ± 7.5 × 109 Cells gd–1; p < 0.001), Sacca Sessola (7.8 ± 4.3 × 109 Cells gd–1; p < 0.001), and Tresse (4.5 ± 4.1 × 109 Cells gd–1; p < 0.01). Overall, Autumn data (30.2 ± 26.9 × 109 Cells gd–1) were significantly higher than Spring (10.1 ± 8.5 × 109 Cells gd–1; p < 0.05) and Summer ones (16.0 ± 12.5 × 109 Cells gd–1; p < 0.05). The PCA (Figure 2) showed a separation of sampling sites on PC1 (54.35% variance), mainly due to grain size features and TOC concentration. The effect of seasonality was also observed, even though the projection of temperature on PC2 explained a lower percentage of the total variance (18.16%).
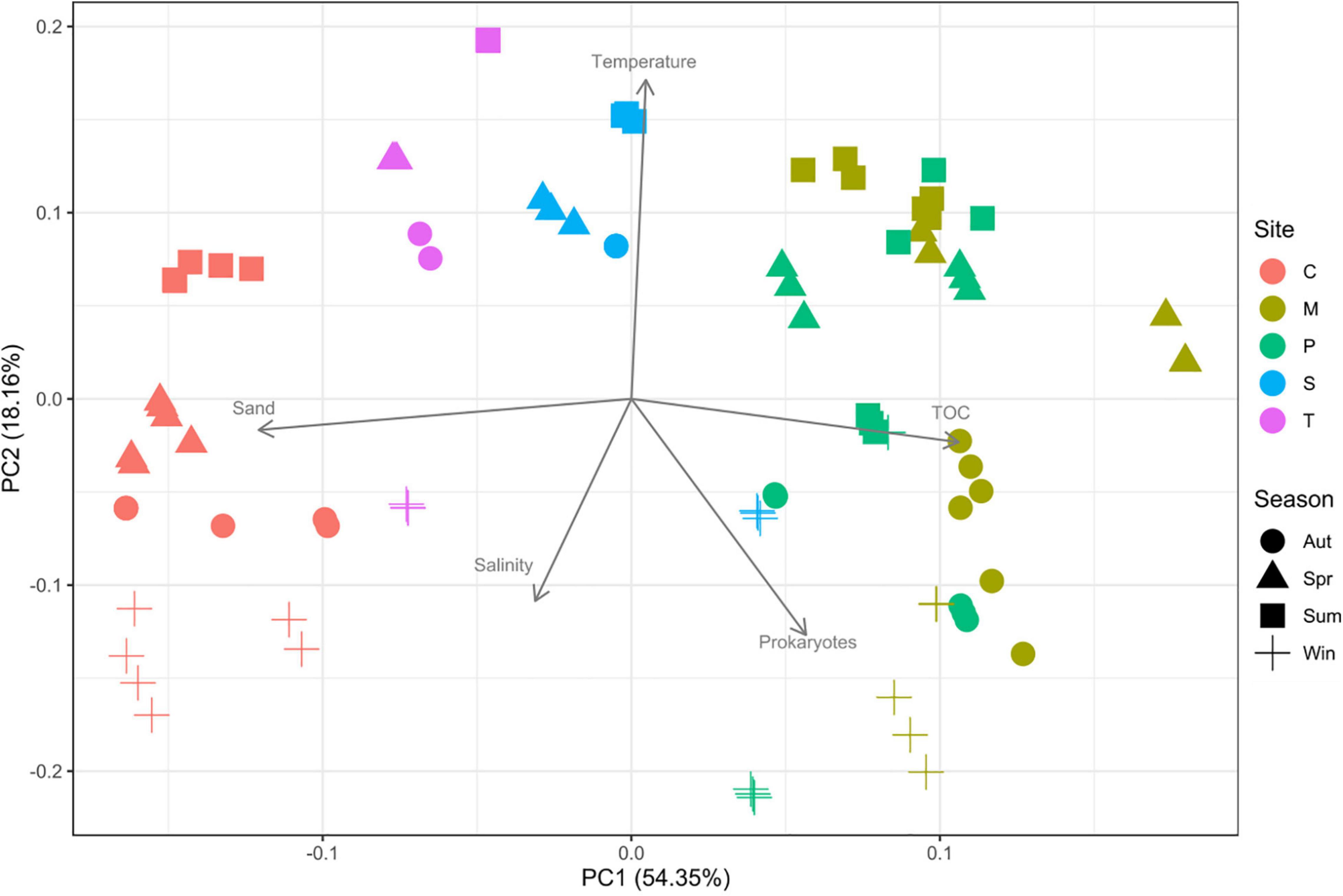
Figure 2. Principal component analysis (PCA) of the environmental parameters measured. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse. TOC = total organic carbon.
Prokaryotic Diversity and Community Composition
A total of 31,189,504 raw sequences were generated. After the denoising procedure, 20,191,333 were retained with an average of 219,471 ± 92,086 sequences per sample. The average number of ASVs was 5,230 ± 1,536. The rarefaction curves indicated that the sequencing effort was enough to assess the sample biodiversity (Supplementary Figure 2). The replicates significantly correlated (p < 0.05) with an average ρ of 0.86 ± 0.09.
Shannon’s diversity index showed an average of 10.7 ± 0.4 and Pielou’s evenness showed an average of 0.89 ± 0.02 (Supplementary Figure 3). Overall, Autumn had a significantly higher index than the other seasons. No significant differences in biodiversity indices were detected among sites and site-season combinations.
The PERMANOVA highlighted that site, season, and their interaction had a significant effect (p < 0.001) on the prokaryotic community structure (Supplementary Table 6). The highest amount of variance was explained by the site (37%), followed by season-site interaction (11%) and season (6%).
The PCoA (Supplementary Figure 4), in agreement with the PERMANOVA, grouped the samples by site.
The variance partitioning analysis showed that environmental parameters explained ∼20% of the variance (Supplementary Table 7). The dbRDA (Figure 3) revealed that grain size, TOC, and salinity had a significant role (p < 0.001) in shaping the communities, while temperature was not a significant variable (p > 0.05).
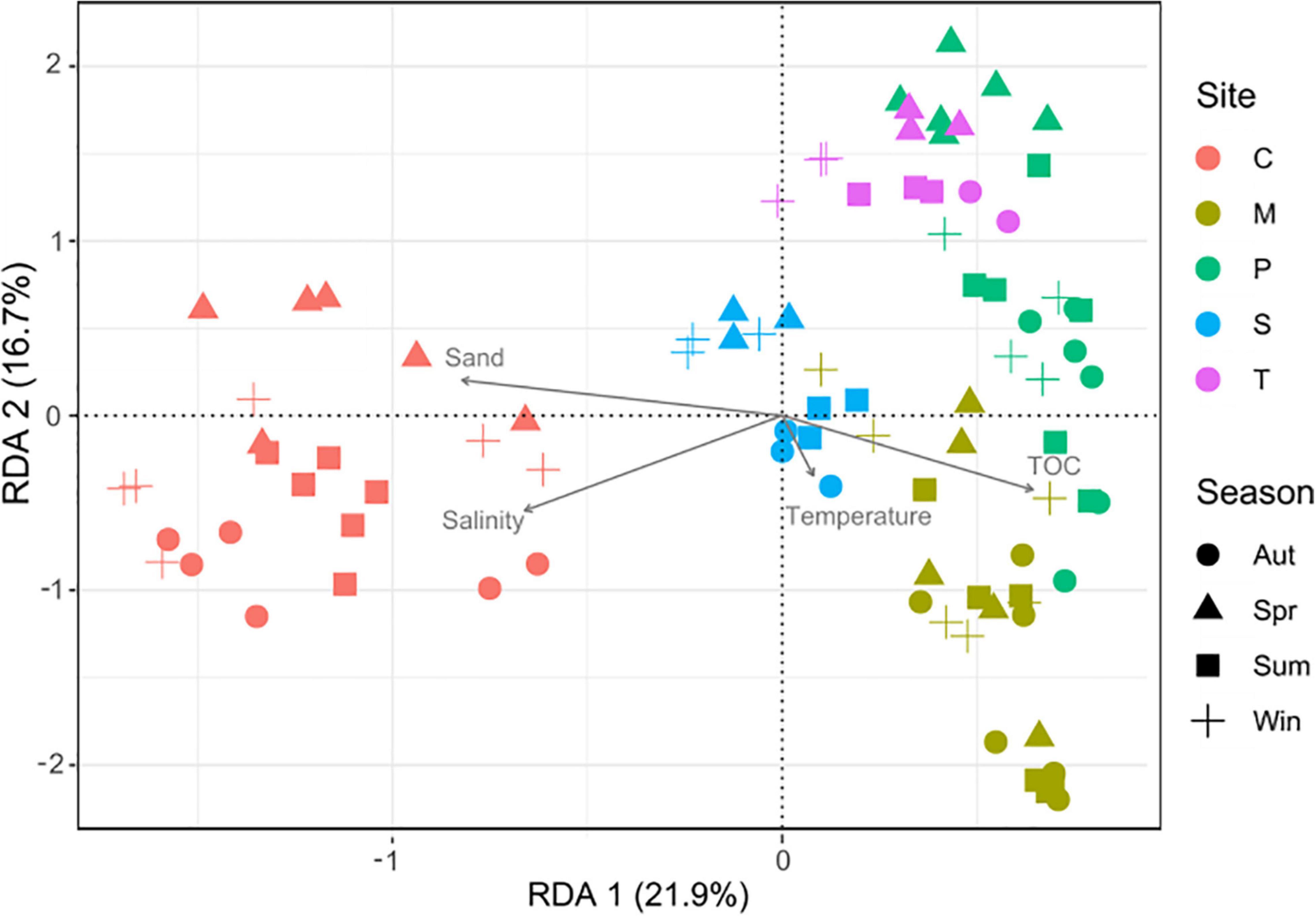
Figure 3. Distance-based redundancy analysis (dbRDA) based on Bray-Curtis dissimilarity in community composition. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse. TOC = total organic carbon.
Regarding the taxonomic composition, at the domain level most reads were assigned to Bacteria (average on the overall dataset 96.1 ± 2.6%), and a smaller percentage (3.8 ± 2.6%) to Archaea.
For Bacteria (Figure 4), the most represented phyla were Proteobacteria (29.3 ± 4.4%), followed by Bacteroidota (15.5 ± 4.4%) (Figure 4). Desulfobacterota were significantly less abundant in Chioggia than the other sites (9.2 ± 2.4% vs. 12.8 ± 0.8%), while Actinobacteriota presented the opposite distribution pattern (4.8 ± 1.2% vs. 2.2 ± 0.5%) (Figure 4). The most represented classes were Gammaproteobacteria (23.9 ± 3.7%), followed by Bacteroida (14.7 ± 4.3%) (Supplementary Figure 5). The most represented orders were the bacterial Flavobacteriales (6.8 ± 3.8%), Desulfobacterales (6.1 ± 2.2%), and Pirellulales (5.2 ± 2.3%) (Supplementary Figure 5). The most represented families were Flavobacteriaceae (6.2 ± 3.6%), Desulfosarcinaceae (5.2 ± 1.8%), and Pirellulaceae (4.5 ± 0.9%). The most abundant genus was Woeseia (4.4 ± 0.9%) (Figure 4).
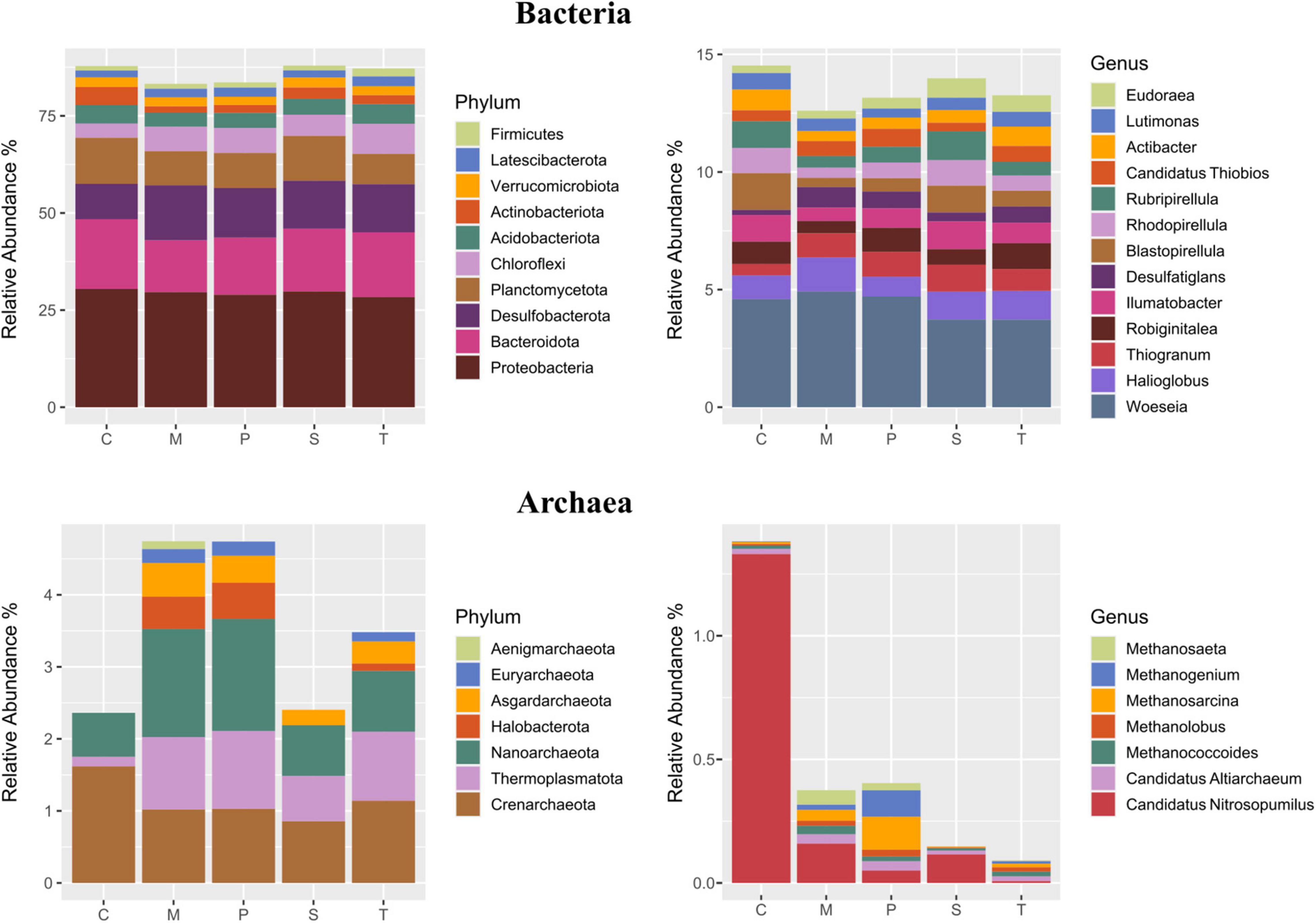
Figure 4. Taxonomic composition (average relative abundance) of Bacteria (upper panel) and Archaea (lower panel) at the phylum (left panel) and the genus (right panel) levels in the five sampling sites. For Bacteria, taxa representing > 1 and 0.5% are shown, respectively. For Archaea, taxa representing > 0.1% are shown. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse.
For Archaea (Figure 4), the most represented phyla were Crenarchaeota (1.3 ± 0.6%), followed by Thermoplasmatota that were lower in Chioggia in respect to the other sites (0.14 ± 0.08% vs. 0.9 ± 0.6%). The ammonia-oxidizing archaeon Candidatus Nitrosopumilus was significantly higher in Chioggia (1.2 ± 0.7% vs. 0.1 ± 0.1%) (Figure 4).
Among the genera related to microscale processes, we detected the cable bacteria Candidatus Electrothrix at all sites (0.1 ± 0.2%); these bacteria have long, multicellular filaments that can conduct electric currents over centimeter-scale distances (Trojan et al., 2016). Likewise, lignin-degrader genera (Bacillus, Novosphingobium, Pseudomonas, Rhodococcus, and Streptomyces; Lee et al., 2019) were found at all sites (0.006 ± 0.008%). The Indicator Species analysis showed that each site hosted a set of indicator bacterial genera (Figure 5, Supplementary Table 8), with Chioggia showing the highest number (n = 21). On the contrary, excluding Chioggia, the highest number of genera (n = 27) were shared among Marghera, Palude della Rosa, Sacca Sessola and Tresse. Chioggia was characterized by the presence of aerobic (or strictly aerobic), halophilic bacteria such as Aquimarina, Aureisphaera, and Pseudoalteromonas and by the absence of genera of sulfate-reducing Desulfobacteraceae that were shared among the other sites, like Desulfoconvexum, Desulfonema, and Desulfobacter. ANCOM analysis (Supplementary Figure 6) confirmed the significant higher abundance ammonia-oxidizing archaeon Candidatus Nitrosopomilus in Chioggia and a lower presence of the Desulfobacteraceae member Desulfobacter. Moreover, it highlighted a higher presence of the nitrite-oxidizing Nitrospira and of the aerobic Ahrensia and Granulosicoccus. Palude della Rosa showed significantly higher presence of the halophile Halochromatium and the glycogen-accumulating Candidatus Competibacter.
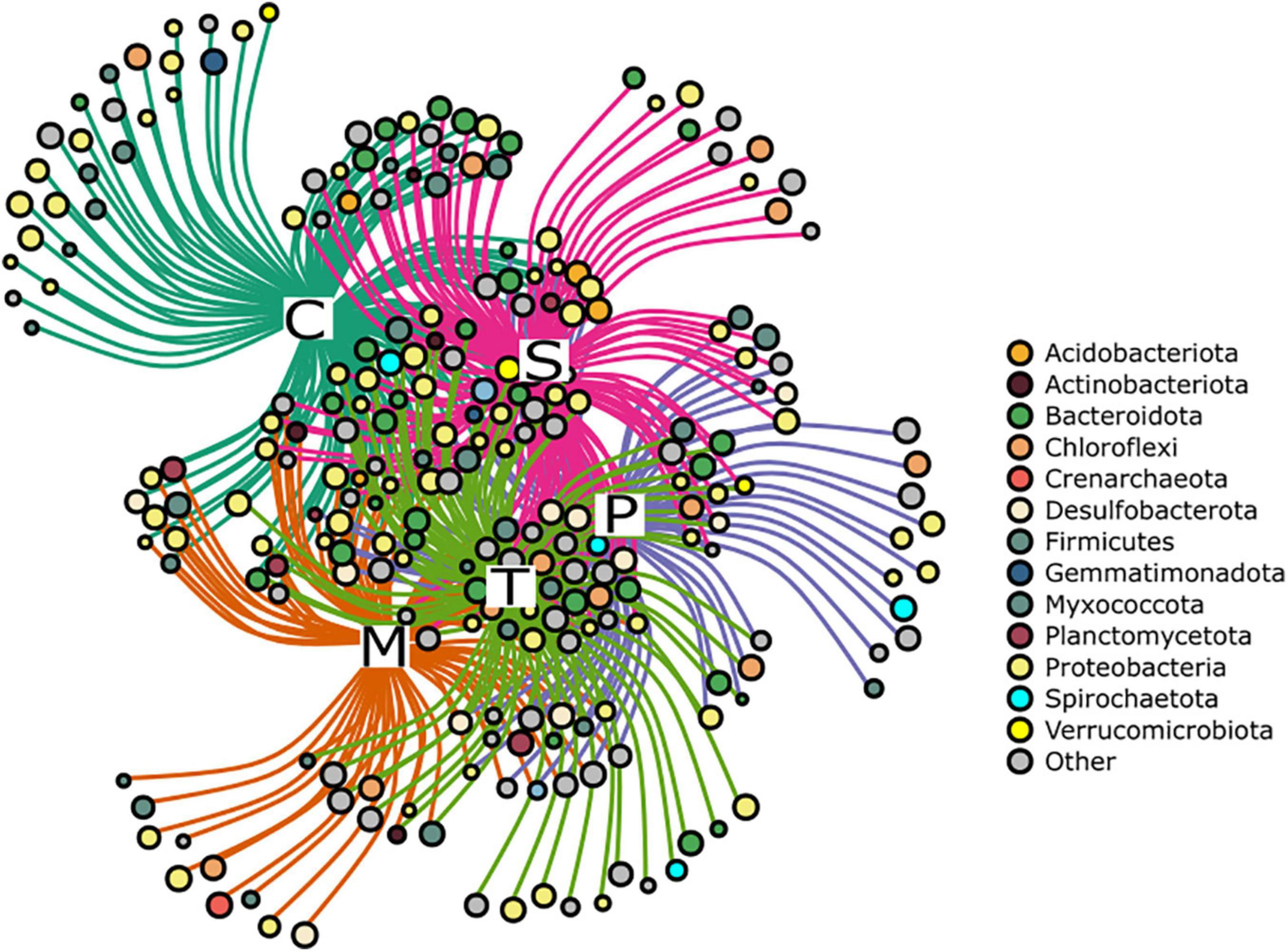
Figure 5. Network representation of the Indicator Species analysis at the genus level. Each node represents a genus, and the node size is scaled based on the fidelity to the sampling site. Colours refer to the phylum to which each genus belongs. Grey nodes represent low abundant phyla (< 1%). C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse.
Microbial signatures of selected anthropogenic-related taxa (Figure 6) presented overall 0.1–0.3% relative abundance for fecal-associated bacteria and 0.01–0.04% for sewage-associated bacteria. In Sacca Sessola and Chioggia, fecal-associated microbes showed the highest presence, mainly due to Clostridiaceae, while Marghera showed the highest proportion of Bacteroidaceae and Ruminococcaceae. For the sewage-associated bacteria, Marghera showed on average the highest proportion of reads due to Acinetobacter and Trichococcus while Palude della Rosa showed the lowest proportion.
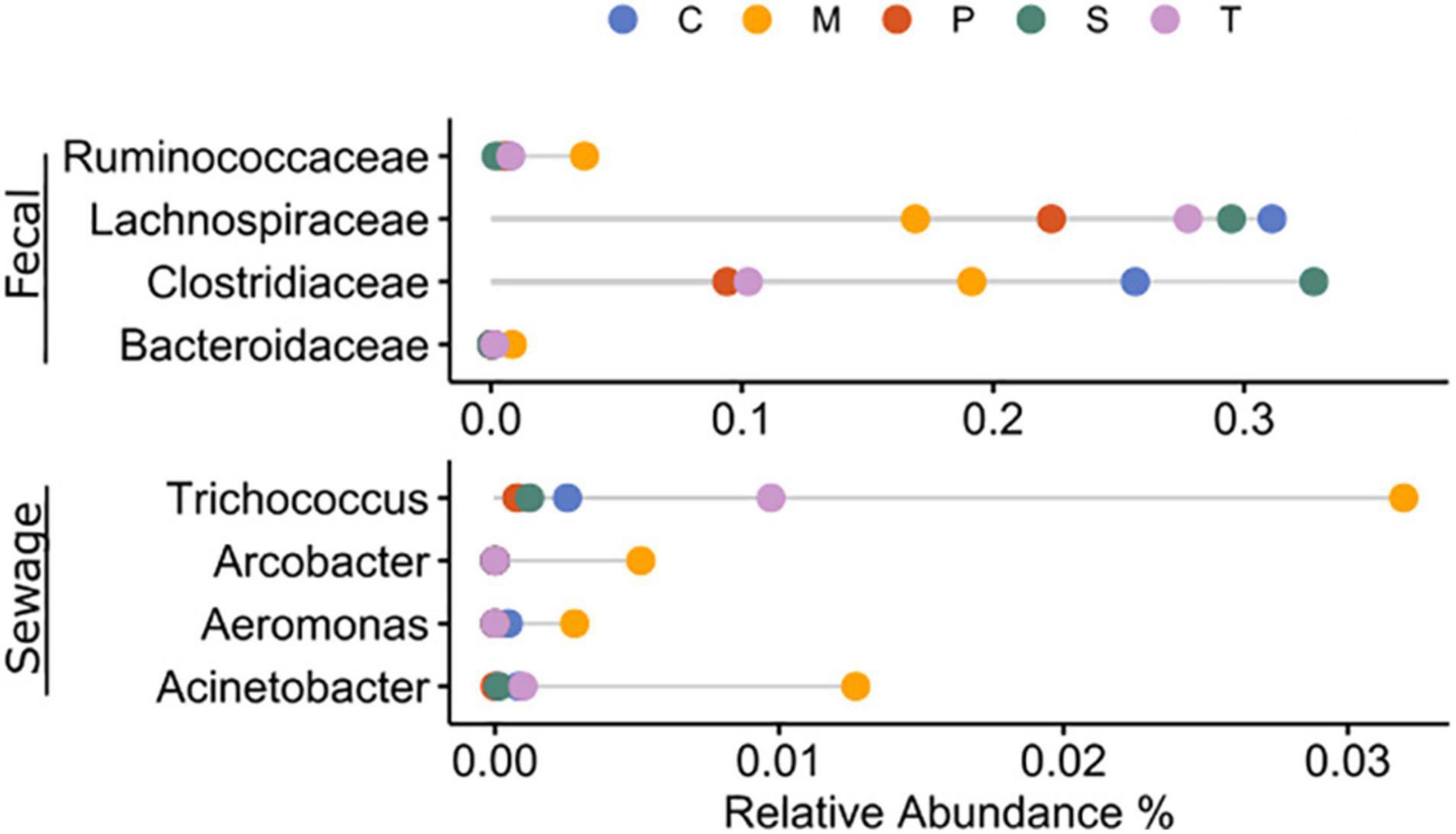
Figure 6. Average relative abundance of fecal- and sewage-associated taxa in the sampling sites. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse.
Metagenomic Analyses and Functional Characterization
A total ∼120 million of raw reads (30 Gbp) were generated, specifically 14.8 ± 3.7 millions of paired-end reads with quality score of 35 ± 2 for each metagenome. On average, contigs derived from the three co-assembled replicates for each sample were 5,208,341 ± 376,210 contigs > 1,000 bp, and 652,755 ± 174,565 coding sequences.
In the KEGG functional characterization (Supplementary Figure 7), the most represented level 1 categories were Metabolism (63.2 ± 0.4% on the overall dataset), Genetic Information Processing (17.0 ± 0.2%), and Environmental Information Processing (14.2 ± 0.5%); at level 2, Amino acid metabolism (21.5 ± 0.6%), Carbohydrate metabolism (12.9 ± 0.3%), and Translation (7.9 ± 0.1%); at level 3, ATP-binding cassette (ABC) transporters (6.3 ± 0.4%), Two-component system (4.7 ± 0.2%), and Aminoacyl-tRNA biosynthesis (4.2 ± 0.1%).
Key oceanic marker genes (Sunagawa et al., 2015) and key sediment metabolic genes (Dombrowski et al., 2018) have been selected and clustered as follows: Carbon fixation, Energy and metabolism, and Carbon-nutrient coupling.
The category of Carbon fixation included: oxygenic photosynthesis, anoxygenic photosynthesis, and other autotrophic pathways (Wood-Ljungdahl, Hydroxypropionate bicycle, hydroxypropionate/hydroxybutyrate, C1-metabolism). The category of Energy and metabolism included: aerobic respiration, fermentation, manganese-related metabolism, nitrogen metabolism, iron-related metabolism, sulfur metabolism, methanogenesis, butane metabolism, and hydrogenase. The category of Carbon-nutrient coupling included: phosphorus metabolism, peptide degradation, motility and chemotaxis, transport, and fatty acids degradation. We also assessed Anthropogenic-related metabolisms regarding hydrocarbons and xenobiotics (Figure 7, Supplementary Table 9).
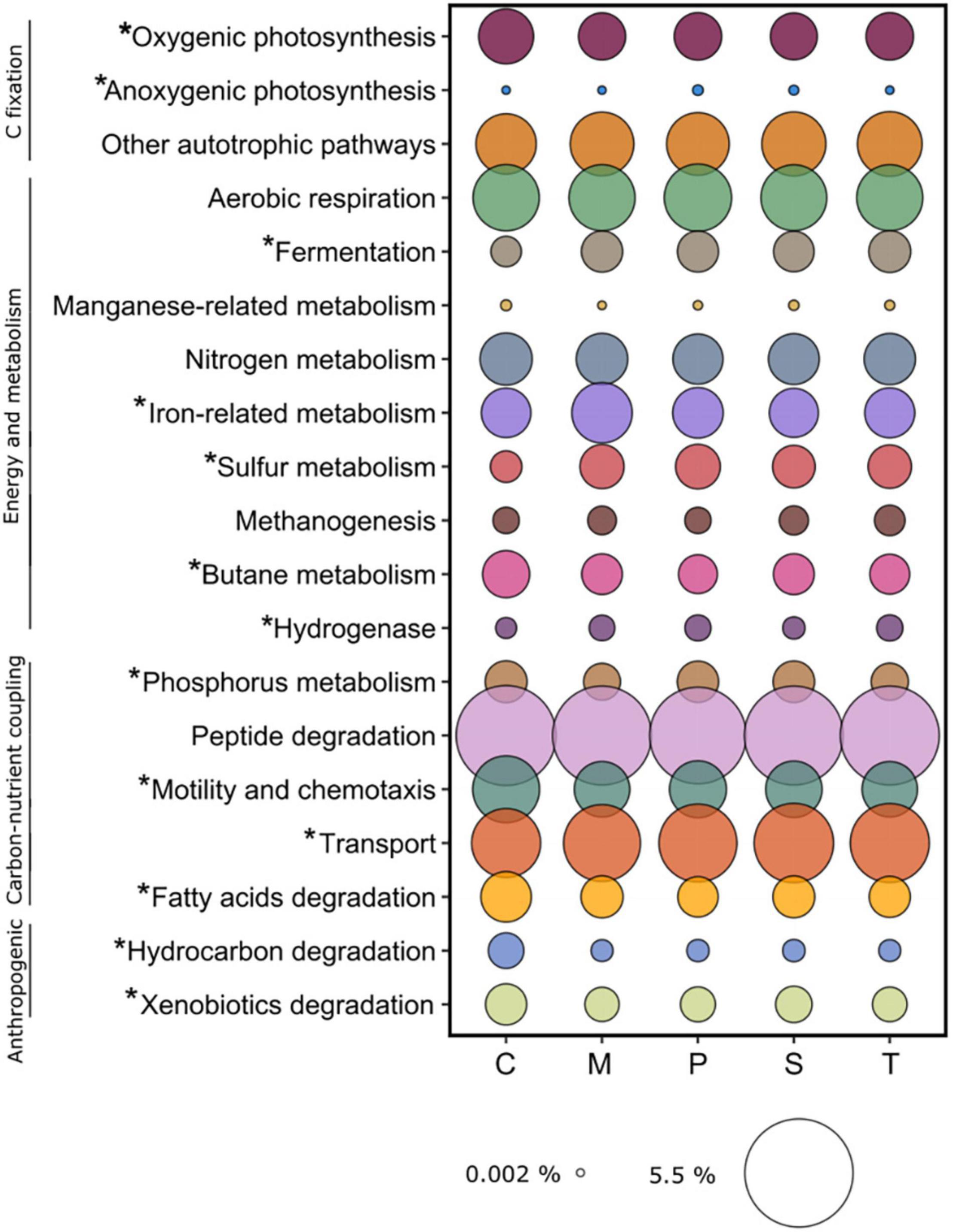
Figure 7. Average relative abundance of functions related to ecosystem-scale processes identified in the sampling sites. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse. Asterisks indicate significant differences among sites.
Overall, in the sediment microbial environment, Peptide degradation was the most represented category (4.5 ± 0.1%), followed by Transport (2.7 ± 0.1%), Aerobic respiration (1.8 ± 0.1), Other autotrophic pathways (1.7 ± 0.01%), and Motility and chemotaxis (1.3 ± 0.1%). Here we present the significant differences detected in the above-mentioned categories across the sites (Figure 7). Within Carbon fixation, Oxygenic photosynthesis was higher in Chioggia (0.93 ± 0.02% vs. 0.86 ± 0.01%), while Anoxygenic photosynthesis was higher in Palude della Rosa (0.008 ± 0.001% vs. 0.003 ± 0.002%).
Within Energy and metabolism, Chioggia presented a lower number of genes coding for functions related to Fermentation (0.50 ± 0.03% vs. 0.72 ± 0.03%), Sulfur metabolism (0.62 ± 0.01% vs. 0.72 ± 0.03%), and Butane metabolism (0.66 ± 0.01% vs. 0.58 ± 0.03%) in respect to the other sites. Marghera was higher in Iron-related metabolism (1.13 ± 0.03% vs. 0.97 ± 0.03%) whereas Palude della Rosa, Tresse and Marghera harbored the highest number of Hydrogenase genes (0.190 ± 0.005% vs. 0.147 ± 0.01%). Within the Carbon-nutrient coupling category, Phosphorus metabolism functions were less present in Marghera and Tresse (0.582 ± 0.007% and 0.692 ± 0.008%, respectively). Regarding Nitrogen metabolism, we detected significant amounts of genes coding for ammonia monooxygenase subunits (amoA, amoB, and amoC) in Chioggia in respect to the other sites (0.0007 ± 0.0003% and 0.00008 ± 0.00002%, respectively), in accordance with the presence of Candidatus Nitrosopumilus (Walker et al., 2010; Bayer et al., 2019). Chioggia presented the highest number of genes related to Fatty acid degradation (0.713 ± 0.001% vs. 0.662 ± 0.002%). Furthermore, in the Anthropogenic-related metabolisms category: Hydrocarbon degradation was significantly higher in Chioggia (0.183 ± 0.009% vs. 0.118 ± 0.003%), mostly due to hydrolases involved in the degradation of chlorocyclohexane, chlorobenzene fluorobenzoate, including toluene. The Xenobiotic metabolism and degradation in the same site was also significantly higher (0.524 ± 0.051% vs. 0.410 ± 0.002%), mostly due to haloalkane dehalogenase (dhaA).
This sandy site was significantly different from the others for Motility and chemotaxis (1.5 ± 0.2% vs. 1.3 ± 0.3%, flagellum-related genes), and Transport (2.5 ± 0.1% vs. 2.7 ± 0.1%, lipid and vitamin).
In the SEED functional characterization (Supplementary Figure 8), the most represented level 1 categories were Clustering-based subsystems (14.7 ± 0.1%; functional coupling evidence with unknown function), Carbohydrates (11.0 ± 0.3%), Amino Acids and Derivatives (8.8 ± 0.1%), and Protein metabolism (8.6 ± 0.1%), whereas at the level 2, Plant-Prokaryote DOE project (6.8 ± 0.1%), Protein biosynthesis (5.3 ± 0.1%), and Central carbohydrate metabolism (3.7 ± 0.1%).
Regarding anthropogenic-related functions, the “Virulence, Disease and Defense” category was significantly higher in Marghera and Tresse in respect to the other sites (3.36 ± 0.02% vs. 3.04 ± 0.03%), mainly due to a significantly higher presence of genes in the “Resistance to antibiotics and toxic compounds” group (3.08 ± 0.04% vs. 2.72 ± 0.03%).
Antibiotic resistance annotation (Figure 8) showed that Multidrug and Beta-lactamase resistance were the most present at all sites (0.93 ± 0.01% and 0.58 ± 0.03%, respectively), with Vancomycin resistance being significantly higher in Chioggia (0.016 ± 0.002% vs. 0.007 ± 0.002%).
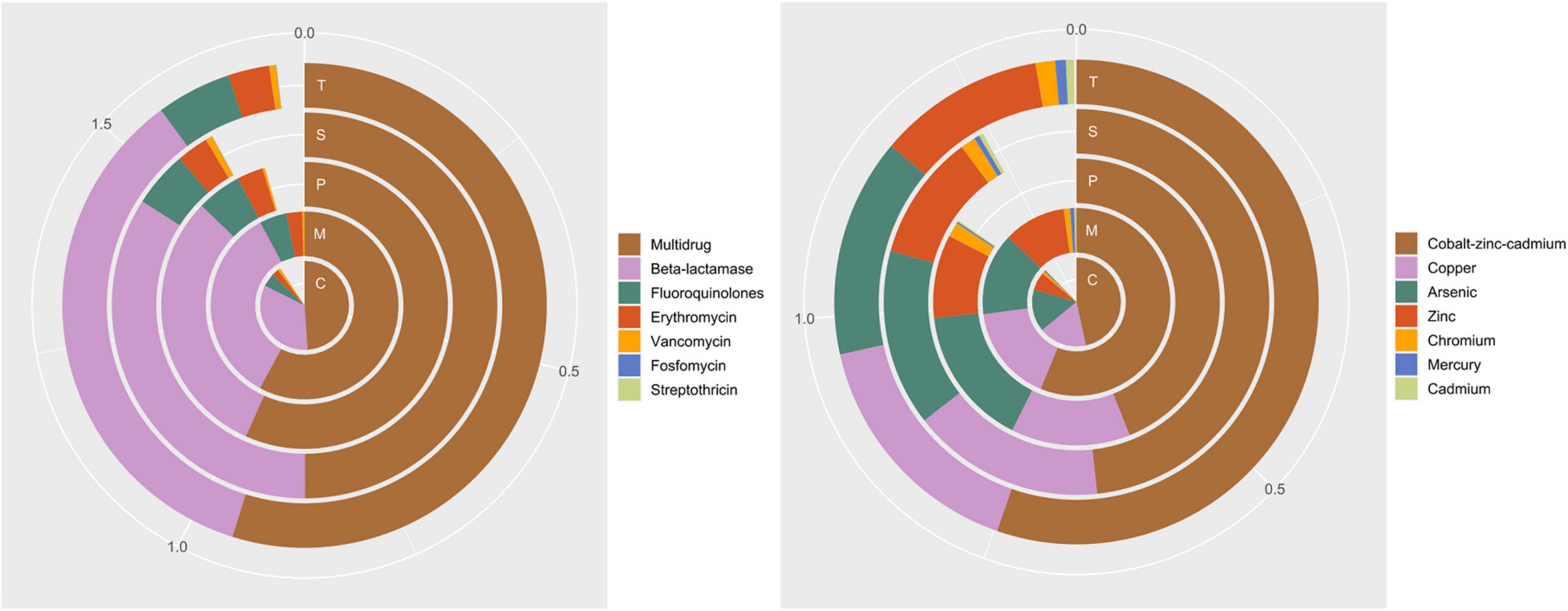
Figure 8. Average relative abundance of genes coding for resistance to antibiotics (left panel) and toxic compounds (right panel) in the sampling sites. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse.
Among Toxic compound resistance annotation, Cobalt-zinc-cadmium was the most present at all sites (0.676 ± 0.002%), with Mercury resistance being significantly higher in Marghera and Tresse (0.009 ± 0.001% vs. 0.004 ± 0.002), and Arsenic resistance being significantly higher in Chioggia and Palude della Rosa (0.221 ± 0.002% vs. 0.196 ± 0.004).
The key specific microscale marker functions, identified with the SEED database, have been clustered as follows: DNA metabolism (CRISPs and DNA uptake and competence), Mobilome (Gene Transfer Agents, integrons, pathogenicity islands, phages and prophages, plasmids, and transposable elements), Regulation and Cell Signaling (biofilm formation, phenazine biosynthesis, osmotic stress, sporulation, siderophores, toxic-antitoxic systems, and two-component systems) and Nanomachines (protein secretion system Type IV and Type VI) (Figure 9).
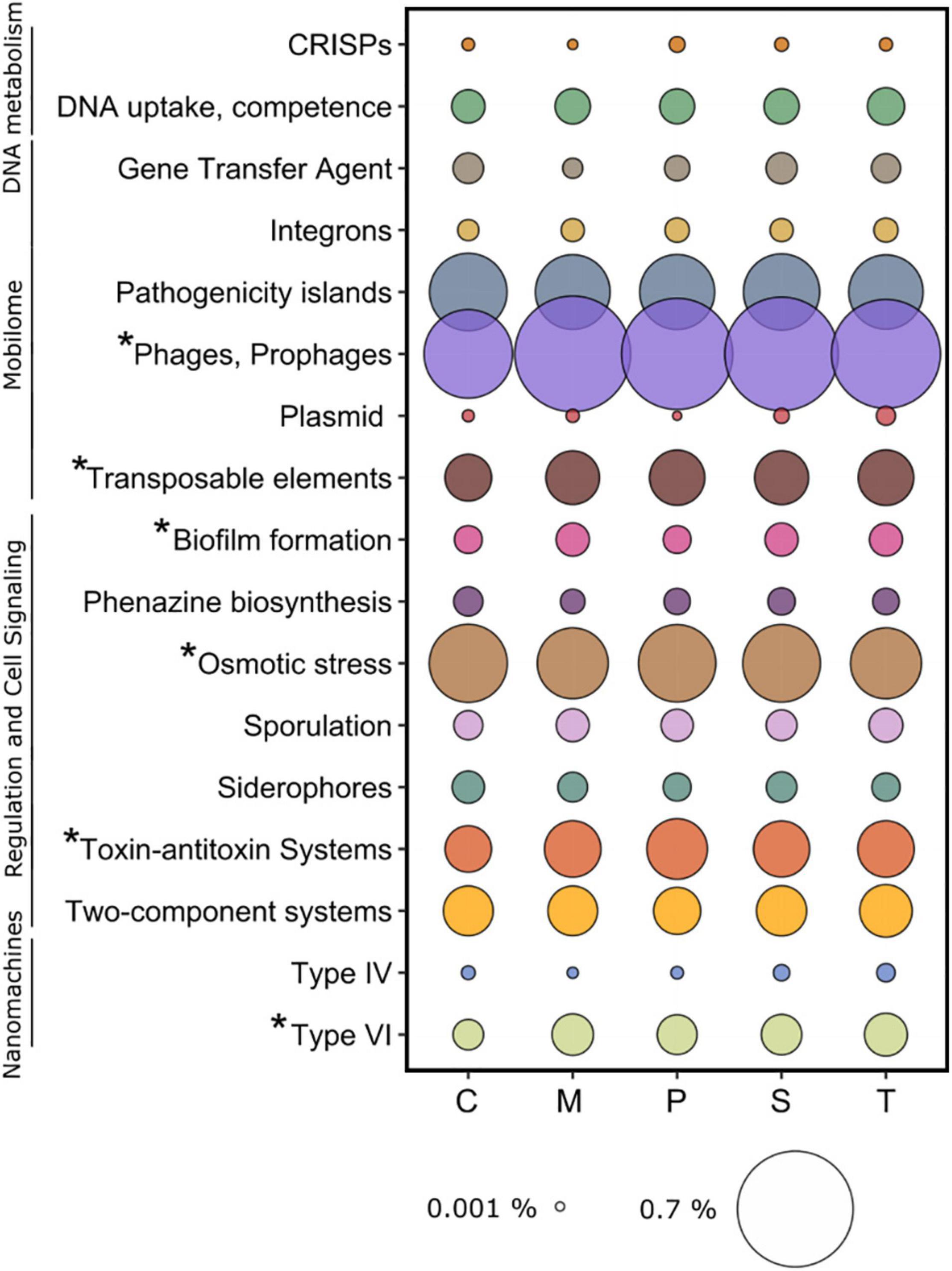
Figure 9. Average relative abundance of functions related to microscale processes identified in the sampling sites. C: Chioggia; M: Marghera; P: Palude della Rosa; S: Sacca Sessola; T: Tresse. Asterisks indicate significant differences among sites.
Overall, in the sediment microbial environment, Phages and prophages was the most represented category (0.58 ± 0.07%), followed by Osmotic stress (0.27 ± 0.01%), Pathogenicity islands (0.26 ± 0.01), Toxic-antitoxic systems (0.13 ± 0.02%), Transposable elements (0.12 ± 0.09%), and Two-component systems (0.01 ± 0.09%) (Figure 9).
Here we present the significant differences detected in the above-mentioned categories across the sites. Within Mobilome, Phages and prophages and Transposable elements were lower in Chioggia (0.47 ± 0.02% vs. 0.62 ± 0.04% and 0.1 ± 0.01% vs. 0.12 ± 0.01%, respectively). Within Regulation and Cell Signaling, Biofilm formation was higher in Marghera, Sacca Sessola, and Tresse (0.04 ± 0.01% vs. 0.02 ± 0.01%), Osmotic stress was higher in Chioggia, Palude della Rosa, and Sacca Sessola (0.28 ± 0.01% vs. 0.25 ± 0.01%), and Toxic-antitoxic systems was lower in Chioggia (0.08 ± 0.01% vs. 0.14 ± 0.01%). Within Nanomachines, Protein secretion system type VI was lower in Chioggia (0.03 ± 0.01% vs. 0.06 ± 0.01%).
Discussion
Microbial Community Diversity and Structure Are Shaped by Site Features
Microbial communities in marine sediment are extremely diverse due to the combination of different factors such as habitat temporal stability, high niche diversity, and resource partitioning due to the complex physical-chemical gradients (Zinger et al., 2011; Acosta-Gonzalez and Marques, 2016). As such, they represent a major reservoir of genetic variability in the marine environment (Polymenakou et al., 2005).
Our analysis detected a pronounced diversity in the communities, in agreement with 16S studies in the Venice Lagoon (Borin et al., 2009; Quero et al., 2017) and in other transitional and coastal benthic systems (Bianchelli et al., 2020; Miksch et al., 2021). The sampling site specific features (grain size, salinity and TOC content, Figure 2) were important drivers in shaping the prokaryotic assemblages, while seasonality had a significant but minor role (Figure 3). Depending on the area, seasonality in the sediment microbial communities is not as regularly and clearly found as in the water column (Tšertova et al., 2011; Gobet et al., 2012; Miksch et al., 2021), even in the Venice Lagoon (Quero et al., 2017). Quero et al. (2017), in a study including pelagic and benthic communities in the central part of the Venice Lagoon, found seasonality in both compartments but less pronounced in surface sediments than in the overlaying water, where this feature is well documented (Celussi et al., 2009).
In marine sediments, it is now known that physical-chemical characteristics including salinity, chlorophyll a, TOC content, grain size, and benthic fauna can influence the microbial communities (Sapp et al., 2010; Pala et al., 2018; Hoshino et al., 2020). Generally, the composition of the sediment bacterial communities can present high dissimilarities between sites, reflecting the heterogeneity of these environments, mostly due to limited physical mixing and the geographical distance (Acosta-Gonzalez and Marques, 2016).
We found that salinity, grain size, and TOC significantly contributed to the structure of the microbial communities in the different sites of the Venice Lagoon (Figure 3).
Salinity is recognized as a major environmental determinant of microbial composition in benthic communities (Lozupone and Knight, 2007; Bolhuis et al., 2013) in coastal (Bolhuis and Stal, 2011; Severin et al., 2012; Bolhuis et al., 2013) and lagoon systems (Tsuboi et al., 2013; Pavloudi et al., 2016; Behera et al., 2017). In our study, the more marine-influenced site (Chioggia, located at the southern inlet of the Venice Lagoon) was confirmed as the most diverse one, forming a separated group in the multivariate analyses (see Figure 3).
It is known that grain size can affect microbial abundance and activity by providing different surface properties (e.g., fine sediments provide greater areas that can be colonized) and influencing permeability, water chemistry, hydraulic conductivity, and pore space between sediment particles (Dale, 1974; Zhang et al., 1998). A positive correlation between bacterial abundance, sediment grain size, and organic carbon content is well established (Dale, 1974) and reported in different studies (Santmire and Leff, 2007; Legg et al., 2012; Tsuboi et al., 2013; Fazi et al., 2020). In another Italian Adriatic lagoon, it was found that lowest salinity, higher percentage of fine grain sediment and organic matter corresponded to the highest microbial abundances (Cibic et al., 2019). This was confirmed also in our study, in which the sites enriched in silt, Marghera and Palude della Rosa, presented the highest prokaryotic abundance and TOC concentration.
Sediment grain type influences indirectly the microbial assemblage at the microscale, due to the variable water fluxes (Ahmerkamp et al., 2020) that changes the redox conditions thus driving more diverse and able-to-adapt communities in sandy sites compared to the clay and silty ones.
Being and important source of energy for heterotrophs the organic carbon (TOC) pool is an important structuring agent for benthic microbial communities (Oni et al., 2015). As such, in our study the content of TOC was important in structuring the differences among the communities (e.g., Chioggia vs. other sites), even though we did not analyze its quality (biopolymers’ concentration, labile vs. recalcitrant matrices, etc.), which is also known to affect the composition of bacterial communities (e.g., Fazi et al., 2020). Likewise, ecological factors that were not investigated in this study can contribute to the differentiation of the sites: interspecific bacteria competition, grazing pressure, viral lysis, vegetation, contaminants, and also stochastic events (Böer et al., 2009; Luna et al., 2013; Acosta-Gonzalez and Marques, 2016; Bianchelli et al., 2020).
The Rare Biosphere Supports Microbial Diversity
The microbial communities were characterized by few dominant taxonomic groups, the core microbiome, and a high number of low abundant taxa, the so-called rare biosphere (Sogin et al., 2006), that accounted for most of the phylogenetic diversity.
For instance, among the 84 phyla detected in the whole dataset, 49 were considered rare, as represented by an average relative abundance < 0.01% (Galand et al., 2009; Gobet et al., 2012), as well as for 1181 out of the 1587 detected genera. The presence of such rare biosphere is of ecological importance as these taxa, often overlooked, can have an over-proportional role in biogeochemical cycles and can represent a hidden driver of ecosystem functions (Jousset et al., 2017).
Even if we detected significant differences among sites in high abundant taxa, from the phylum to the genus level, the numerous low-abundant taxonomic units were those that more contributed to their site-specific fingerprint (see Figures 3, 5). These taxa were mostly uncharacterized (e.g., belong to uncultured bacteria typically detected only with molecular techniques and are still not defined in terms of physiology and metabolism), and more studies are needed to define their ecological roles.
In accordance with Jousset et al. (2017) we suggest that the rare biosphere in the Venice Lagoon can be considered as an ensemble of keystone species that provide stability and seeds for ecosystem functions, specifically in terms of water purification, nutrients, and global climate regulations (da Mosto et al., 2020).
Regarding the most abundant taxa, the globally distributed (Gamma)Proteobacteria were confirmed to be prevalent at all sites (see Figure 4), as generally detected in sediment in both the costal and the deep seafloor (Fry et al., 2008; Zinger et al., 2011; Acosta-Gonzalez and Marques, 2016), including the Venice Lagoon one (Borin et al., 2009; Quero et al., 2017). Among these, the ubiquitous genus Woeseia was the most abundant (see Figure 4). Recent molecular characterization suggested that it is likely to grow on proteinaceous matter, potentially derived from detrital cell membranes, cell walls, and other organic remnants (Mußmann et al., 2017; Hoffmann et al., 2020).
Several significant differences in taxonomic composition were highlighted by ANOVA/Tukey-Kramer and ANCOM analyses in sulfur-related bacteria (Desulfobacteria, Desulfobacterales) involved in sulfate reduction. In our study, these taxa were less represented in Chioggia where, differently form the inner area of the Lagoon where the sediment is mostly anoxic, hypoxic conditions are commonly found (Borin et al., 2009). Chioggia sandy sediment is in fact more exposed to the effect of winds, tidal currents, and ship movements, that favor partial oxygenation (Borin et al., 2009). Permeable sandy sediment pore space is constantly flushed by the overlying water and could trap detritus and living cells from the water column (Boudreau et al., 2001; Gobet et al., 2012). In the other sites, the higher amount of organic matter contributes to the oxygen depletion and to the activity of sulfate-reducing bacteria in sulfide production (Zaggia et al., 2007). This is further supported by the metagenomics analysis, which also highlighted a significantly lower number of genes related to sulfur metabolism in Chioggia (see section “Wide Range of Carbon Fixation and Carbon Flow Strategies”).
Wide Range of Carbon Fixation and Carbon Flow Strategies
Sediment bacteria play a key ecological and biogeochemical role in marine ecosystems. They present high abundance and genetic variability, critical role in the transformation and speciation of major bioactive elements (e.g., carbon, nitrogen, phosphorus, oxygen, and sulfur), and in the degradation of organic pollutants (Polymenakou et al., 2005; Wu et al., 2008). The metagenomics approach adopted in this study allowed the identification of the genes involved in biogeochemical cycles providing insights into the potential metabolic functioning of the microbial communities (Doney et al., 2004; Dombrowski et al., 2018; Acinas et al., 2021). Following the “carbon currency” in the microbial sediment compartment we were able to map the carbon fixation processes, the energy and metabolism processes, the carbon-nutrient coupling, the carbon reworking processes, which includes the degradative processes, and the behavior and anthropogenic-related metabolisms (see Figure 7).
In our sites, we found that oxygenic photosynthesis (by Prokaryotes) was the main carbon fixation pathway. Anoxygenic photosynthesis was also detected together with five additional carbon fixation pathways which differ in reducing compounds, energy source, and oxygen sensitivity of enzymes alternative pathways (Wood-Ljungdahl, Hydroxypropionate bicycle, hydroxypropionate/hydroxybutyrate, C1-metabolism). These findings highlighted the metabolic flexibility of the community in fixing CO2 from different sources thus suggesting high diversity in C fixation modes in the sediment. Once the carbon is turned into biomass and organic carbon, it can be metabolized via aerobic and anaerobic respiration or fermentation. The surface sediment is affected by the light diurnal cycle and by the presence of oxygen (as a by-product of photosynthesis and from the oxygenated seawater column) (Petersen et al., 1994; Nealson, 1997). Aerobic respiration was the main pathway to respire the organic matter and secondly, anaerobic respiration (N, Mg, Fe, S; i.e., changing the terminal electron acceptor according to their availably, Nealson, 1997) was detected. Furthermore, often understudied metabolisms, such as methanogenesis, butane, proton degradation (hydrogenase, with a great diversity of enzymes), and fermentation were also important in enriching the metabolic repertoire of the microbial communities across sites (Falkowski et al., 2008). Within the concept of shared metabolisms, cable bacteria (family Desulfobulbaceae, Kjeldsen et al., 2019) were present in our samples. These microbes are formed by centimeter long filaments that span from the oxic water-sediment interface to the sulphidic deeper sediment layer. Within their filament, they couple sulfide oxidation with oxygen or nitrate reduction via long-distance electron transport.
Sulfur-related genes were found in high abundance in our sites with the exception of the sandy site of Chioggia. This is possibly due to the sediment texture that is more permeable to water and oxygen thus causing the community metabolism to switch more often from anoxic to oxic functionalities. Sulfur is essential for building amino acids (e.g., methionine and cysteine), cellular components (sulfolipids) and metabolites (DMSP) (Wasmund et al., 2017). It has a paramount role as electron acceptor (e.g., sulfate and thiosulphate) and electron donor (sulfite and elemental sulfur) (Jørgensen et al., 2019). We found genes related to sulfate-reduction and sulfur-oxidation processes that are coupled in the surface sediment. Overall, sulfur cycling is interconnected with the other elements, the availability of organic matter and redox species present in the lagoon system. In a high-temperature and low-oxygen lagoon scenario, sulfur-related metabolisms might become dominant in the ecosystem functioning.
Phosphorus-cycle-related genes were also highly abundant in our samples, being especially linked to the involvement of nucleic acids in biofilm structures (Karygianni et al., 2020) and organic P degradation. Furthermore, approximately 4.5% of genes (KEGG annotation) in the metagenomes were addressed to peptide degradation. Proteolysis is in fact the fastest hydrolytic activity within surface brackish and coastal sediments, aiming to provide prokaryotes with both organic C and N via amino acids, as highlighted by direct measurements (e.g., Cibic et al., 2012; Franzo et al., 2019). Together with carbohydrates, proteins and lipids are indeed the major constituents of the labile organic matter in the sediments (Pusceddu et al., 1999) and microbes are adapted to utilize these compounds heterotrophically (Oni et al., 2015), thus supporting the concept of sediments as hot spots for organic matter degradation (Lipka et al., 2018).
Cell-Cell Microscale Interactions
Within this framework of reference, we have mined our data set to test hypotheses on the putative functional microscale behavior of the microbes in the lagoon surface sediments (see Figure 9). Starting from DNA, we have asked questions about whether the community was able to display adaptive strategies such as competence and gene exchange to improve their persistence in the environment. Among the mobile genetic elements, what were the most abundant, and was it connected with the site features?
DNA is an integral part of the sediment, carrying genetic information and providing 3D structure by binding to other molecules (Dell’Anno and Corinaldesi, 2004; Corinaldesi et al., 2008; Torti et al., 2015). It is known that viruses are abundant in the sediment and are major structuring agents, keeping the microbial community under check (Danovaro et al., 2001; Breitbart et al., 2004; Breitbart and Rohwer, 2005). Among the microbial defense systems (Makarova et al., 2013), we detected the CRISPR-Cas (Doudna and Charpentier, 2014) associated genes at all the stations, despite the sediment grain size. This suggests that there is a persistent strategy of Bacteria and Archaea to fight off the viral invasions using this adaptive immunity against foreign genetic elements while creating a viral data bank for further viral encounters. Recent studies showed that in Global Ocean Sampling Expedition (GOS) and Tara Ocean data set, CRISPR-Cas were highly diverse and specific to the viral architecture (Cai et al., 2013; Nasko et al., 2019). In addition to CRISPR-Cas, Competence and Sporulation related functions were found throughout the Lagoon. DNA can be taken up during the competence window (Lennon, 2007; Dubnau and Blokesch, 2019), while a population of microbes is undergoing major environmental stress such as oxygen depletion/enrichment or nutrient scarcity. Among that population, a few cells can promote the lysis of the other individuals exploiting the released DNA for ensuring success in the future. Temporally subsequent to competence is the sporulation, an important unique microbial mechanism in marine sediment for surviving burial (Wörmer et al., 2019; Morono et al., 2020).
An ability that microbes use to move information relies on the highly diverse architecture of the mobile genetic elements, the Mobilome (Gillings, 2013; Carr et al., 2021). Prophages and Transposons were lower in Chioggia, possibly due to the site features such as higher water flow and lower TOC that might select against these mobilome strategies. Overall, mapping the sediment mobilome could give insights on rapid adaptation processes to the presence of pollutants in marine habitat due to anthropogenic activities.
Within the Regulation and cell signaling and Nanomachines categories, we have mined and detected genes related to physiology, adaptations and interactions of the microbial communities and consortia in the sediment. Biofilm in the marine environment is a pervasive structuring feature (de Carvalho, 2018). Biofilm formation is essential in keeping the microenvironment stable for the establishing of the microbial communities thus providing protection from grazers and phages, toxic substances (e.g., antibiotics), and accumulating nutrients (Flemming et al., 2016). Toxin-antitoxin (TA) systems regulate dormancy and persistence in microbes (Spoering and Lewis, 2001; Keren et al., 2004; Shah et al., 2006). TA genes are abundant on plasmids, phages, and chromosomes. They are formed by a toxin, that arrests growth by interfering with a vital cellular process, and a cognate antitoxin, that neutralizes the toxin activity to resume normal growth (Page and Peti, 2016). TA plays a major role in supporting microbial resistance to antibiotics by developing a subpopulation of persistent cells. It is interesting to note that in Chioggia, biofilm and TA were lower than the other sites, suggesting that these strategies were not favored in a sandy sediment habitat and or were not so successful as in the other sites. Despite it is known that in sandy sediments, microbes colonize grains via biofilms (Probandt et al., 2017), the biofilm might be more widespread in muddy sediment to create an organic bridge among clay and silt particles.
In terms of competition for space, nutrient, energy sources and ability to resist osmotic stress, phenazine (Price-Whelan et al., 2007; Schiessl et al., 2019), siderophores (Sandy and Butler, 2009; D’Onofrio et al., 2010), two-component systems (Held et al., 2019), osmotic stress genes, and nanomachines for cell-cell warfare were identified in the data set. Microbes constantly under siege could express phenazine, a redox-active pigment produced by Pseudomonas aeruginosa that affect gene expression, metabolic flux, and redox balancing to promote their success. Pseudomonas despite being a multi-drug resistant, biofilm former, quorum sensing human pathogenic microbe is highly versatile and has been found in the marine environment (Kimata et al., 2004). Siderophores are structures produced by microbes that chelate ions, such as Fe to facilitate the acquisition of the insoluble species thus harshening the competition for this element. We identified, within the two-component signal transduction systems (Yamamoto et al., 2005) single transduction for anaerobic respiration, nitrate and nitrite utilization, nitrogen assimilation metals efflux system, fumarate respiration, outer membrane proteins, phosphate assimilation, magnesium transport, and osmotic and envelop stress response, chemotaxis modulation, and flagellar motor switch. This great diversity found in the sediment microbial communities highlighted the adaptive strategies that microbes need to exploit while sensing the external environment at the microscale in order to activate genes that promote their success in the microniche. In the water column, a high abundance of these regulative network systems was found to be linked to copiotrophy and diazotrophy (Held et al., 2019).
The last microscale features we have identified in our dataset were the Type IV (Craig et al., 2019) and Type VI (Cianfanelli et al., 2016) secretion nanomachines, that are involved in cell-cell interactions by allowing nucleic information being shuttle from one individual to the other and attaching with effectors the other organisms. Type IV was found in microbes from acetate-stimulated aquifer sediments (Kantor et al., 2013), thus suggesting conjugation being potentially important to exchange genes and associated metabolic potentials. In the ocean, Type VI secretion systems were found to be globally distributed and associated with particle-attached microbes (Kempnich and Sison-Mangus, 2020).
Finally, motility and chemotaxis patterns, highly abundant in Chioggia, suggested a specific strategy in sandy sediments, that are more permeable to water in comparison to clayey and silty one and where a more diverse microenvironment can develop within the grains.
In sum, microscale-related genes highlight a very elaborated fine structure of interactions and behavior in the surface sediment microbial communities that then results in the large-scale biogeochemical processes on the lagoon system.
Anthropogenic-Related Pressures Influence Benthic Microbial Communities
DNA metabarcoding and metagenomic analyses allowed the exploration of microbial features related to different anthropogenic pressures, of particular importance in the monitoring of fecal and sewage bacteria, antibiotic and metal resistant, hydrocarbon degraders, and xenobiotic degraders microbes.
The use of DNA-based approach is increasingly applied to microbial profiling in wastewater plants, coastal areas, and in tracking and identifying microbial signatures in the environment; such studies contributed to the identification of bacterial taxa associated with sewage- and fecal- contamination (Newton et al., 2013; Tan et al., 2015). Marine sediments are a reservoir of fecal bacteria (Luna et al., 2010) and have the potential to favor the persistence of fecal microbes and to contaminate the overlying water through resuspension, which likely poses important public health and environmental threats (Luna et al., 2016). In our study, microbial signatures showed the highest proportion of sewage-associated taxa in Marghera and the lowest proportion in Palude della Rosa, considered less impacted by human activities. Sacca Sessola and Chioggia had a higher percentage of fecal-associated taxa, being mostly affected by urban contamination (Picone et al., 2016). Overall, we detected several indicators of fecal and sewage contamination (see Figure 6), and their relative abundance and taxonomic assignment are in line with a previous study in central part of the Venice Lagoon (Luna et al., 2016), confirming a diffuse contamination and accumulation of these bacteria in the Lagoon sediment.
Antibiotic resistance spread is one of the main threats to global human, fish, shellfish, and in overall ecosystem health (Yang et al., 2012; Ventola, 2015; Mackenzie and Jeggo, 2019; Helsens et al., 2020). For this reason, the detection, tracking, and assessment of the increasing antibiotic resistant microbes and genes, is necessary in terms of risk management (Heß et al., 2018). The marine environment is considered a global reservoir of antibiotic resistance genes, but we still have little understanding of their presence and diversity in both pelagic and benthic environments (Chen et al., 2013; Hatosy and Martiny, 2015). Several antibiotic resistance genes were detected in our dataset (see Figure 8), with a significantly higher presence in the two more impacted sites of Marghera and Tresse. The Multi-drugs and Beta-lactamase resistance genes were found as the most abundant at all sites, as previously detected in seawater (Hatosy and Martiny, 2015) and sediment (Huang et al., 2019).
Surface sediment metal contamination is another consequence of anthropogenic pressure in transitional environments, representing a potential threat to both public health and marine ecosystems (Han et al., 2011). Metals not only accumulate in the sediment but can be subjected to complex cycling that can make toxic compounds available to benthic organisms; sediment mobilization and resuspension under particular pH and redox conditions may release them in the water column becoming a secondary source of pollution (Turner and Millward, 2002; Eggleton and Thomas, 2004; Zonta et al., 2018). Furthermore, microbes that are antibiotic resistant can carry information for metal resistance and vice versa, thus exacerbating the problem of resistance spreading (Chen et al., 2019). Several toxic compound resistance genes were detected in our dataset, with a significantly higher presence in the two more impacted sites of Marghera and Tresse. These sites presented the highest number of genes related to Cobalt-zinc-cadmium resistance, and to Mercury resistance, due to the release of such metals from industrial plants. In particular, the mercury contamination mainly originates from the chloralkali discharge in Porto Marghera, which occurred until the 1980s (Bloom et al., 2004). Mercury is one of the most severe pollutants, and its flux to Venice Lagoon water is mostly due to the resuspension of contaminated sediment (Bloom et al., 2004), underlying the importance of its regulated management. Chioggia and Palude della Rosa presented the highest number of genes coding for Arsenic resistance. Even if the presence of this contaminant in the lagoon is mainly due to the geochemical characteristics of the sediments, human activities can contribute to its accumulation (Zonta et al., 2018).
Chioggia presented the highest number of genes related to hydrocarbon degradation. Interestingly, in this site, both surface and sub-surface sediment were recently found to harbor the highest concentrations of PAH and PBC in the whole Venice Lagoon (Cassin et al., 2018), due to the important urban waste discharge, road/boat traffic, and being located close to the harbor of the second Italian largest fishing fleet.
It was worth noting that among the identified hydrocarbon degraders some taxa were also able to utilize lignin. Lignin is a very complex and hard-to-degrade polymer (Hedges and Mann, 1979) and Bacteria, Archaea and Eukaryotes have evolved adaptive strategies to gain energy from it (Cragg et al., 2015). In the Venice Lagoon, wood has been historically and extensively used for maritime needs, like mooring piles and navigation channel delimitation (named “briccole,” approximately 7,000, formed by two or three poles) (Ghirardini et al., 2010). Briccole are subjected to physical and biotic degradation, the latter mainly due to marine borers (Ghirardini et al., 2010). Since our data highlighted that the sediment represents a reservoir of potential lignin-degrader bacteria, we hypothesize a role of these taxa (alone or in consortia with other microorganisms) in the turnover/recycling of human-introduced wood structures in the Lagoon.
Conclusion and Future Perspectives
Our analyses showed a clear spatial distribution of the highly biodiverse surface sediment microbial communities in the different sub-basins of the Venice Lagoon, mainly due to low abundant taxa. Salinity, grain size, and TOC concentration were significant drivers in shaping community structure and diversity. Further analysis will be necessary to depict the environmental drivers of microbial dynamics at the small scale.
Metagenomic approach allowed for the first time to assess the potential functions and metabolism of these communities at different levels. At both the ecosystem- and the micro-scale, subtle but significant differences were detected in the diverse sites, including the main biogeochemical cycles. The focus on anthropogenic-related functions highlighted that chronically polluted areas are hotspots of resistance genes, with implications on sediment management in the Lagoon. Furthermore, our data indicate that an enhanced understanding of microbial microscale features is important to better describe how interactions play out in structuring the community and thus dictating the ecosystem functioning.
In the near future, the metagenome-assembled genomes (MAGs) analysis will give more detail about the link between taxonomy and functions of interest, especially important for the still uncharacterized taxa. This study represents the initial, essential step toward the application of other “omics” approaches in the Venice Lagoon and other transitional sites. Among them, RNA-based approaches (e.g., metatranscriptomics) will benefit from these data to identify the functions and metabolisms that are actually active in the sediment microbial communities. This will allow inferring the microbial response to environmental changes including the increase of temperature, acidification, and extreme flood events.
Data Availability Statement
The 16S amplicon sequences generated for this study can be found in Sequence Reads Archive (SRA) at NCBI with the accession number PRJNA750327. The metagenomes generated for this study can be found in MG-RAST server with the sample accessions mgs826787, mgs826814, mgs826826, mgs826790, mgs826829, mgs826811, mgs826817, mgs826820, mgs826838, mgs826835, mgs826823, mgs826841, mgs826796, mgs826793, nmgs826799, mgs826808, mgs826844, mgs826802, mgs826805, mgs826832, and at EMBL-EBI European Nucleotide Archive (ENA) with the sample accessions ERS5426915–ERS5426934 and analysis accession MGYA00581082–MGYA00581101. All the datasets are available on request to the corresponding author.
Author Contributions
FM conceptualized the study. PDN acquired the funding. FM and MC performed the management of the project activities. EB performed the laboratory and bioinformatic analyses, wrote the manuscript with the help, and inputs of all co-authors. All authors contributed to the article and approved the submitted version.
Funding
Scientific activity performed with the contribution of the Provveditorato for the Public Works of Veneto, Trentino Alto Adige, and Friuli Venezia Giulia, provided through the concessionary of State Consorzio Venezia Nuova and coordinated by CORILA within Venezia2021 framework. This work benefited from access to SBR-Station Biologique de Roscoff, an EMBRC-FR and EMBRC-ERIC. Financial support was provided by EMBRC-ERIC and the European Marine Research Network (EuroMarine) through their joint 2020 call targeting early career researchers (EB). This work benefited from CINECA-ISCRA C resources (HP10CS2I1S; PI: EB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Alessandro Vezzi (UNIPD, Italy), and Dr. Daniele Cassin, Dr. Simone Leoni, Dr. Giorgia Manfè, Dr. Roberto Zonta (CNR-ISMAR, Italy) for the support in sediment sampling, Prof. Alberto Pallavicini (UNITS, Italy) for the availability of laboratory facilities, Dr. Erwan Corre (Station Biologique de Roscoff, France) for the advice in metagenome analyses, and Dr. Luca Zoccarato (IGB Berlin, Germany) for help with network visualization. We are grateful to the OGS colleagues Dr. Cecilia Balestra for the support in flow cytometry, Dr. Annalisa Franzo and Dr. Federica Cerino for help in sample processing, Dr. Viviana Fonti and Dr. Vincenzo Manna for the stimulating discussions, Dr. Matteo Bazzaro for the grain size analysis and Dr. Federica Relitti for TOC analysis. We acknowledge the CINECA award under the ISCRA initiative (Italy) and Roscoff Bioinformatics platform ABiMS (France) for the availability of High Performance Computing (HCP) resources and support. The constructive comments of the reviewers helped to improve the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.762292/full#supplementary-material
References
Acinas, S. G., Sánchez, P., Salazar, G., Cornejo-Castillo, F. M., Sebastián, M., Logares, R., et al. (2021). Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Commun. Biol. 4, 1–15. doi: 10.1038/s42003-021-02112-2
Acosta-Gonzalez, A., and Marques, S. (2016). Bacterial diversity in oil-polluted marine coastal sediments. Curr. Opin. Biotechnol. 38, 24–32. doi: 10.1016/j.copbio.2015.12.010
Ahmerkamp, S., Marchant, H. K., Peng, C., Probandt, D., Littmann, S., Kuypers, M. M. M., et al. (2020). The effect of sediment grain properties and porewater flow on microbial abundance and respiration in permeable sediments. Sci. Rep. 10:3573. doi: 10.1038/s41598-020-60557-7
Andrews, S. (2010). FastQC: A Quality Control Tool For High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed October 5, 2020).
Apitz, S. E. A., Barbanti, A., Bocci, M., Carlin, A., Montobbio, L., and Bernestein, A. G. (2007). The sediments of the Venice lagoon (Italy) evaluated in a screening risk assessment approach: part I e application of International Sediment Quality Guidelines. Integr. Environ. Assess. Manag. 3:393e414. doi: 10.1002/ieam.5630030310
Azam, F., and Malfatti, F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 10, 782–791. doi: 10.1038/nrmicro1747
Bayer, B., Vojvoda, J., Reinthaler, T., Reyes, C., Pinto, M., and Herndl, G. J. (2019). Nitrosopumilus adriaticus sp. nov. and Nitrosopumilus piranensis sp. nov., two ammonia-oxidizing archaea from the Adriatic Sea and members of the class Nitrososphaeria. Int. J. Syst. Evol. Microbiol. 69, 1892–1902. doi: 10.1099/ijsem.0.003360
Behera, P., Mahapatra, S., Mohapatra, M., Kim, J. Y., Adhya, T. K., Raina, V., et al. (2017). Salinity and macrophyte drive the biogeography of the sedimentary bacterial communities in a brackish water tropical coastal lagoon. Sci. Total Environ. 595, 472–485. doi: 10.1016/j.scitotenv.2017.03.271
Bellucci, L. G., Frignani, M., Paolucci, D., and Ravanelli, M. (2002). Distribution of heavy metals in sediments of the Venice Lagoon: the role of the industrial area. Sci. Total. Environ. 295, 5–49. doi: 10.1016/s0048-9697(02)00040-2
Bianchelli, S., Nizzoli, D., Bartoli, M., Viaroli, P., Rastelli, E., and Pusceddu, A. (2020). Sedimentary organic matter, prokaryotes, and meiofauna across a river-lagoon-sea gradient. Diversity 12:189. doi: 10.3390/d12050189
Bloom, N. S., Moretto, L. M., Scopece, P., and Ugo, P. (2004). Seasonal cycling of mercury and monomethyl mercury in the Venice Lagoon (Italy). Mar. Chem. 91, 85–99. doi: 10.1016/j.marchem.2004.06.002
Böer, S., Hedtkamp, S., van Beusekom, J., Fuhrman, J. A., Boetius, A., and Remette, A. (2009). Time- and sediment depth-related variations in bacterial diversity and community structure in subtidal sands. ISME J. 3, 780–791. doi: 10.1038/ismej.2009.29
Bokulich, N. A., Dillon, M. R., Zhang, Y., Rideout, J. R., Bolyen, E., Li, H., et al. (2018). q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. mSystems 3, e219–e318. doi: 10.1128/mSystems.00219-18
Bolhuis, H., and Stal, L. J. (2011). Analysis of bacterial and archaeal diversity in coastal microbial mats using massive parallel 16S rRNA gene tag sequencing. ISME J. 5, 1701–1712. doi: 10.1038/ismej.2011.52
Bolhuis, H., Fillinger, L., and Stal, L. J. (2013). Coastal microbial mat diversity along a natural salinity gradient. PLoS One 8:e63166. doi: 10.1371/journal.pone.0063166
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Borin, S., Brusetti, L., Daffonchio, D., Delaney, E., and Baldi, F. (2009). Biodiversity of prokaryotic communities in sediments of different sub-basins of the Venice lagoon. Res. Microbiol. 160, 307–314. doi: 10.1016/j.resmic.2009.04.005
Boudreau, B. P., Huettel, M., Forster, S., Jahnke, R. A., McLachlan, A., Middelburg, J. J., et al. (2001). Permeable marine sediments: overturning an old paradigm. Eos. Trans. AGU 82, 133–136. doi: 10.1029/EO082i011p00133-01
Breitbart, M., and Rohwer, F. (2005). Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13, 278–284. doi: 10.1016/j.tim.2005.04.003
Breitbart, M., Felts, B., Kelley, S., Mahaffy, J. M., Nulton, J., Salamon, P., et al. (2004). Diversity and population structure of a near-shore marine-sediment viral community. Proc. Biol. Sci. 271, 565–574. doi: 10.1098/rspb.2003.2628
Cai, F., Axen, S. D., and Kerfeld, C. A. (2013). Evidence for the widespread distribution of CRISPR-Cas system in the Phylum Cyanobacteria. RNA Biol. 10, 687–693. doi: 10.4161/rna.24571
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carr, V. R., Shkoporov, A., Hill, C., Mullany, P., and Moyes, D. L. (2021). Probing the mobilome: discoveries in the dynamic microbiome. Trends Microbiol. 29, 158–170. doi: 10.1016/j.tim.2020.05.003
Cassin, D., Dominik, J., Botter, M., and Zonta, R. (2018). PAH and PCB contamination in the sediments of the Venice Lagoon (Italy) before the installation of the MOSE flood defence works. Environ. Sci. Pollut. Res. Int. 25, 24951–24964. doi: 10.1007/s11356-018-2524-y
Celussi, M., Pugnetti, A., and Del Negro, P. (2009). Structural dynamics of bacterioplankton assemblages in the Lagoon of Venice. Estuar. Coast. Shelf Sci. 84, 154–160. doi: 10.1016/j.ecss.2009.05.028
Chen, B., Yang, Y., Liang, X., Yu, K., Zhang, T., and Li, X. (2013). Metagenomic profiles of antibiotic resistance genes (ARGs) between human impacted estuary and deep ocean sediments. Environ. Sci. Technol. 47, 12753–12760. doi: 10.1021/es403818e
Chen, J., Li, J., Zhang, H., Shi, W., and Liu, Y. (2019). Bacterial heavy-metal and antibiotic resistance genes in a copper tailing dam area in northern China. Front. Microbiol. 10:1916. doi: 10.3389/fmicb.2019.01916
Cibic, T., Franzo, A., Celussi, M., and Fabbro, C.Del Negro, P. (2012). Benthic ecosystem functioning in hydrocarbon and heavy-metal contaminated sediments of an Adriatic lagoon. Mar. Ecol. Prog. Ser. 458, 69–87. doi: 10.3354/meps09741
Cianfanelli, F. R., Monlezun, L., and Coulthurst, S. J. (2016). Aim, load, fire: the type VI secretion system, a bacterial nanoweapon. Trends Microbiol. 24, 51–62. doi: 10.1016/j.tim.2015.10.005
Cibic, T., Fazi, S., Nasi, F., Pin, L., Alvisi, F., Berto, D., et al. (2019). Natural and anthropogenic disturbances shape benthic phototrophic and heterotrophic microbial communities in the Po river delta system. Estuar. Coast. Shelf Sci. 222, 168–182. doi: 10.1016/j.ecss.2019.04.009
Corinaldesi, C., Beolchini, F., and Dell’Anno, A. (2008). Damage and degradation rates of extracellular DNA in marine sediments: implications for the preservation of gene sequences. Mol. Ecol. 17, 3939–3951. doi: 10.1111/j.1365-294X.2008.03880.x
Cragg, S. M., Beckham, G. T., Bruce, N. C., Bugg, T. D., Distel, D. L., Dupree, P., et al. (2015). Lignocellulose degradation mechanisms across the Tree of Life. Curr. Opin. Chem. Biol. 29, 108–119. doi: 10.1016/j.cbpa.2015.10.018
Craig, L., Forest, K. T., and Maier, B. (2019). Type IV pili: dynamics, biophysics and functional consequences. Nat. Rev. Microbiol. 17, 429–440. doi: 10.1038/s41579-019-0195-4
Csard, G., and Nepusz, T. (2006). The igraph software package for complex network research. Int. J. Complex Syst. 1695, 1–9. doi: 10.5281/zenodo.3630268
D’Onofrio, A., Crawford, J. M., Stewart, E. J., Witt, K., Gavrish, E., Epstein, S., et al. (2010). Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem. Biol. 17, 254–264. doi: 10.1016/j.chembiol.2010.02.010
da Mosto, J., Bertolini, C., Markandya, A., Spencer, T., Palaima, A., and Onofri, L. (2020). “Rethinking Venice from an ecosystem services perspective,” in Proceedings of the FEEM Working Paper 23.2020, Milano, Italy. doi: 10.2139/ssrn.3749939
Dale, N. G. (1974). Bacteria in intertidal sediments: factors related to their distribution. Limnol. Oceanogr. 19, 509–518. doi: 10.4319/lo.1974.19.3.0509
Danovaro, R., and Pusceddu, A. (2007). Biodiversity and ecosystem functioning in coastal lagoons: does microbial diversity play any role? Estuar. Coast. Shelf Sci. 75, 4–12. doi: 10.1016/j.ecss.2007.02.030
Danovaro, R., Corinaldesi, C., Rastelli, E., and Dell Anno, A. (2015). Towards a better quantitative assessment of the relevance of deep-sea viruses, Bacteria and Archaea in the functioning of the ocean seafloor. Aquat. Microb. Ecol. 75, 81–90. doi: 10.3354/ame01747
Danovaro, R., Dell’Anno, A., Trucco, A., Serresi, M., and Vanucci, S. (2001). Determination of virus abundance in marine sediments. Appl. Environ. Microbiol. 67, 1384–1387. doi: 10.1128/AEM.67.3.1384-1387.2001
Daumas, R. (1990). Contribution of the water-sediment interface to the transformation of biogenic substances: application to nitrogen compounds. Hydrobiologia 207, 15–29. doi: 10.1007/BF00041436
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
de Carvalho, C. C. (2018). Marine biofilms: a successful microbial strategy with economic implications. Front. Mar. Sci. 5:126. doi: 10.3389/fmars.2018.00126
Dell’Anno, A., and Corinaldesi, C. (2004). Degradation and turnover of extracellular DNA in marine sediments: ecological and methodological considerations. Appl. Environ. Microbiol. 70, 4384–4386. doi: 10.1128/AEM.70.7.4384-4386.2004
Deng, L., Fiskal, A., Han, X., Dubois, N., Bernasconi, S. M., and Lever, M. A. (2019). Improving the accuracy of flow cytometric quantification of microbial populations in sediments: importance of cell staining procedures. Front. Microbiol. 10:720. doi: 10.3389/fmicb.2019.00720
DePinto, J., McCulloch, R., Redder, T., Wolfe, J., and Dekker, T. (2010). “Deposition and resuspension of particles and the associated chemical transport across the sediment–water interface,” in The Handbook Of Chemical Mass Transport In The Environment, ed. D. Mackay (Boca Raton, FL: CRC Press), 253–299.
Dombrowski, N., Teske, A. P., and Baker, B. J. (2018). Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat. Commun. 27:4999. doi: 10.1038/s41467-018-07418-0
Doney, S., Abbott, M., Cullen, J., Karl, D., and Rothstein, L. (2004). From genes to ecosystems: the ocean’s new frontier. Front. Ecol. Environ. 2, 457–466. doi: 10.2307/3868334
Doudna, J. A., and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096
Dubnau, D., and Blokesch, M. (2019). Mechanisms of DNA uptake by naturally competent bacteria. Annu. Rev. Genet. 53, 217–237. doi: 10.1146/annurev-genet-112618-043641
Eggleton, J., and Thomas, K. V. (2004). A review of factors affecting the release and bioavailability of contaminants during sediment disturbance events. Environ. Int. 30, 973–980. doi: 10.1016/j.envint.2004.03.001
Falkowski, P. G., Fenchel, T., and Delong, E. F. (2008). The microbial engines that drive Earth’s biogeochemical cycles. Science 320, 1034–1039. doi: 10.1126/science.1153213
Fazi, S., Baldassarre, L., Cassin, D., Quero, G. M., Pizzetti, I., Cibic, T., et al. (2020). Prokaryotic community composition and distribution in coastal sediments following a Po River flood event (northern Adriatic Sea. Italy). Estuar. Coast Shelf Sci. 233:106547. doi: 10.1016/j.ecss.2019.106547
Flemming, H. C., and Wuertz, S. (2019). Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 17, 247–260. doi: 10.1038/s41579-019-0158-9
Flemming, H. C., Wingender, J., Szewzyk, U., Steinberg, P., Rice, S. A., and Kjelleberg, S. (2016). Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 14, 563–575. doi: 10.1038/nrmicro.2016.94
Franzo, A., Celussi, M., Bazzaro, M., Relitti, F., and Del Negro, P. (2019). Microbial processing of sedimentary organic matter at a shallow LTER site in the northern Adriatic Sea: an 8-year case study. Nat. Conserv. 34:397. doi: 10.3897/natureconservation.34.30099
Fry, J. C., Parkes, R. J., Cragg, B. A., Weightman, A. J., and Webster, G. (2008). Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 66, 181–196. doi: 10.1111/j.1574-6941.2008.00566.x
Galand, P. E., Casamayor, E. O., Kirchman, D. L., and Lovejoy, C. (2009). Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. U.S.A. 109, 22427–22432. doi: 10.1073/pnas.0908284106
Ghirardini, A. V., Losso, C., Libralato, G., Zanella, M., Keppel, E., Sigovini, M., et al. (2010). “Sea water piling: traditional or alternative materials? An integrated biological and ecotoxicological evaluation in Venice Lagoon (Italy),” in Proceedings of the 39th Mediterranean Science Commission (CIESM) Congress (Venice: CIESM).
Gieskes, J. M., Han, S., Rathburn, A., Rothwell, G., Pérez, M. E., Porrachia, M., et al. (2015). Anthropogenic contaminants in Venice Lagoon sediments and their pore fluids: results from the SIOSED Projet. Mar. Chem. 174, 73–84. doi: 10.1016/j.marchem.2015.05.008
Gillings, M. R. (2013). Evolutionary consequences of antibiotic use for the resistome, mobilome and microbial pangenome. Front. Microbiol. 4:4. doi: 10.3389/fmicb.2013.00004
Gobet, A., Böer, S., Huse, S. M., Van Beusekom, J. E., Quince, C., Sogin, M. L., et al. (2012). Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 6, 542–553. doi: 10.1038/ismej.2011.132
Han, S., Gieskes, J., Obraztsova, A., Deheyn, D. D., and Tebo, B. M. (2011). Relocation effects of dredged marine sediments on mercury geochemistry: Venice Lagoon, Italy. Estuar. Coast. Shelf Sci. 93, 7–13. doi: 10.1016/j.ecss.2011.03.004
Han, S., Obraztsova, A., Pretto, P., Choe, K. Y., Gieskes, J., Deheyn, D. D., et al. (2007). Biogeochemical factors affecting mercury methylation in sediments of the Venice Lagoon, Italy. Environ. Toxicol. Chem. 26, 655–663. doi: 10.1897/06-392r.1
Hatosy, S. M., and Martiny, A. C. (2015). The ocean as a global reservoir of antibiotic resistance genes. Appl. Environ. Microbiol. 81, 7593–7599. doi: 10.1128/AEM.00736-15
Hedges, J. I., and Mann, D. C. (1979). The characterization of plant tissues by their lignin oxidation products. Geochim. Cosmochim. Acta 43, 1803–1807. doi: 10.1016/0016-7037(79)90028-0
Held, N. A., McIlvin, M. R., Moran, D. M., Laub, M. T., and Saito, M. A. (2019). Unique patterns and biogeochemical relevance of two-component sensing in marine bacteria. mSystems 4, e317–e318. doi: 10.1128/mSystems.00317-18
Helsens, N., Calvez, S., Prevost, H., Bouju-Albert, A., Maillet, A., Rossero, A., et al. (2020). Antibiotic resistance genes and bacterial communities of farmed rainbow trout fillets (Oncorhynchus mykiss). Front. Microbiol. 11:3070. doi: 10.3389/fmicb.2020.590902
Heß, S., Berendonk, T. U., and Kneis, D. (2018). Antibiotic resistant bacteria and resistance genes in the bottom sediment of a small stream and the potential impact of remobilization. FEMS Microbiol. Ecol. 94:fiy128. doi: 10.1093/femsec/fiy128
Hoffmann, K., Bienhold, C., Buttigieg, P. L., Knittel, K., Laso-Pérez, R., Rapp, J. Z., et al. (2020). Diversity and metabolism of Woeseiales bacteria, global members of marine sediment communities. ISME J. 14, 1042–1056. doi: 10.1038/s41396-020-0588-4
Hoshino, T., Doi, H., Uramoto, G. I., Wörmer, L., Adhikari, R. R., Xiao, N., et al. (2020). Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. U.S.A. 117, 27587–27597. doi: 10.1073/pnas.1919139117
Huang, Z., Zhao, W., Xu, T., Zheng, B., and Yin, D. (2019). Occurrence and distribution of antibiotic resistance genes in the water and sediments of Qingcaosha Reservoir, Shanghai, China. Environ. Sci. Eur. 31:81. doi: 10.1186/s12302-019-0265-2
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 8, 111–119. doi: 10.1186/1471-2105-11-119
Jørgensen, B. B., Findlay, A. J., and Pellerin, A. (2019). The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 10:849. doi: 10.3389/fmicb.2019.00849
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., Kurm, V., et al. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.174
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kantor, R. S., Wrighton, K. C., Handley, K. M., Sharon, I., Hug, L. A., Castelle, C. J., et al. (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 4, e708–e713. doi: 10.1128/mBio.00708-13
Karygianni, L., Ren, Z., Koo, H., and Thurnheer, T. (2020). Biofilm matrixome: extracellular components in structured microbial communities. Trends Microbiol. 28, 668–681. doi: 10.1016/j.tim.2020.03.016
Kassambara, A. (2015). factoextra: Visualization of the Outputs of a Multivariate Analysis. R Package Version.
Keegan, K. P., Glass, E. M., and Meyer, F. (2016). MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol. Biol. 1399, 207–233. doi: 10.1007/978-1-4939-3369-3_13
Kempnich, M. W., and Sison-Mangus, M. P. (2020). Presence and abundance of bacteria with the Type VI secretion system in a coastal environment and in the global oceans. PLoS One 15:e0244217. doi: 10.1371/journal.pone.0244217
Keren, I., Shah, D., Spoering, A., Kaldalu, N., and Lewis, K. (2004). Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004
Kimata, N., Nishino, T., Suzuki, S., and Kogure, K. (2004). Pseudomonas aeruginosa isolated from marine environments in Tokyo Bay. Microb. Ecol. 47, 41–47. doi: 10.1007/s00248-003-1032-9
Kjeldsen, K. U., Schreiber, L., Thorup, C. A., Boesen, T., Bjerg, J. T., and Yang, T. (2019). On the evolution and physiology of cable bacteria. Proc. Natl. Acad. Sci. U.S.A. 116, 19116–19125. doi: 10.1073/pnas.1903514116
Le, S., Josse, J., and Husson, F. (2008). FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25, 1–18. doi: 10.18637/jss.v025.i01
Lee, S., Kang, M., Bae, J. H., Sohn, J. H., and Sung, B. H. (2019). Bacterial valorization of lignin: strains, enzymes, conversion pathways, biosensors, and perspectives. Front. Bioeng. Biotechnol. 3:209. doi: 10.3389/fbioe.2019.00209
Legg, T. M., Zheng, Y., Simone, B., Radloff, K. A., Mladenov, N., González, A., et al. (2012). Carbon, metals, and grain size correlate with bacterial community structure in sediments of a high arsenic aquifer. Front. Microbiol. 3:82. doi: 10.3389/fmicb.2012.00082
Lennon, J. T. (2007). Diversity and metabolism of marine bacteria cultivated on dissolved DNA. Appl. Environ. Microbiol. 73, 2799–2805. doi: 10.1128/AEM.02674-06.
Li, D., Liu, C.M., Luo, R., Sadakane, K., and Lam, T.W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 15, 1674–1676. doi: 10.1093/bioinformatics/btv033
Lipka, M., Woelfel, J., Gogina, M., Kallmeyer, J., Liu, B., Morys, C., et al. (2018). Solute reservoirs reflect variability of early diagenetic processes in temperate brackish surface sediments. Front. Mar. Sci. 5:413. doi: 10.3389/fmars.2018.00413
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U.S.A. 104, 11436–11440. doi: 10.1073/pnas.0611525104
Luna, G. M., Corinaldesi, C., Rastelli, E., and Danovaro, R. (2013). Patterns and drivers of bacterial alpha- and beta-diversity across vertical profiles from surface to subsurface sediments. Environ. Microbiol. Rep. 5, 731–739. doi: 10.1111/1758-2229.12075
Luna, G. M., Quero, G. M., and Perini, L. (2016). Next generation sequencing reveals distinct fecal pollution signatures in aquatic sediments across gradients of anthropogenic influence. Adv. Oceanogr. Limnol. 7, 115–124. doi: 10.4081/aiol.2016.5948
Luna, G. M., Vignaroli, C., Rinaldi, C., Pusceddu, A., Nicoletti, L., Gabellini, M., et al. (2010). Extraintestinal Escherichia coli carrying virulence genes in coastal marine sediments. Appl. Environ. Microbiol. 76, 5659–5668. doi: 10.1128/AEM.03138-09
Mackenzie, J. S., and Jeggo, M. (2019). The one health approach-why is it so important? Trop. Med. Infect. Dis. 4:88. doi: 10.3390/tropicalmed4020088
Madricardo, F., Foglini, F., Campiani, E., Grande, V., Catenacci, E., Petrizzo, A., et al. (2019). Assessing the human footprint on the sea-floor of coastal systems: the case of the Venice Lagoon. Italy. Sci. Rep. 9:6615. doi: 10.1038/s41598-019-43027-7
Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2013). Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 41, 4360–4377. doi: 10.1093/nar/gkt157
Mandal, S., Van Treuren, W., White, R. A., Eggesbø, M., Knight, R., and Peddada, S. D. (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26:27663. doi: 10.3402/mehd.v26.27663
Mayer, L. M. (1993). “Organic matter at the sediment-water interface,” in Organic Geochemistry, eds M. H. Engel and S. A. Macko (Boston, MA: Springer), 171–184.
McLellan, S. L., Huse, S. M., Mueller-Spitz, S. R., Andreishcheva, E. N., and Sogin, M. L. (2010). Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 12, 378–392. doi: 10.1111/j.1462-2920.2009.02075.x
McMurdie, P. J., and Holmes, S. (2012). Phyloseq: a bioconductor package for handling and analysis of high-throughput phylogenetic sequence data. Pac. Symp. Biocomput. 2012, 235–246.
Miksch, S., Meiners, M., Meyerdierks, A., Probandt, D., Wegener, G., Titschack, J., et al. (2021). Bacterial communities in temperate and polar coastal sands are seasonally stable. ISME Commun. 1:29. doi: 10.1038/s43705-021-00028-w
Morono, Y., Ito, M., Hoshino, T., Terada, T., Hori, T., Ikehara, M., et al. (2020). Aerobic microbial life persists in oxic marine sediment as old as 101.5 million years. Nat. Commun. 11:3626. doi: 10.1038/s41467-020-17330-1
Mußmann, M., Pjevac, P., Krüger, K., and Dyksma, S. (2017). Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 11, 1276–1281. doi: 10.1038/ismej.2016.185
Nasko, D. J., Ferrell, B. D., Moore, R. M., Bhavsar, J. D., Polson, S. W., and Wommack, K. E. (2019). CRISPR spacers indicate preferential matching of specific virioplankton genes. MBio 10, e2651–e2718. doi: 10.1128/mBio.02651-18
Nealson, K. H. (1997). Sediment bacteria: who’s there, what are they doing, and what’s new? Annu. Rev. Earth Planet Sci. 25, 403–434. doi: 10.1146/annurev.earth.25.1.403
Newton, R. J., Bootsma, M. J., Morrison, H. G., Sogin, M. L., and McLellan, S. L. (2013). A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of Lake Michigan. Microb. Ecol. 65, 1011–1023. doi: 10.1007/s00248-013-0200-9
Nieuwenhuize, J., Maas, E. M., and Middelburg, J. J. (1994). Rapid analysis of organic carbon and nitrogen in particulate materials. Mar. Chem. 45, 217–224. doi: 10.1016/0304-4203(94)90005-1
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). Vegan: Community Ecology Package. R Package Version 2.5-6. Available online at: https://CRAN.R-project.org/package=vegan (accessed February 25, 2020).
Oni, O. E., Schmidt, F., Miyatake, T., Kasten, S., Witt, M., Hinrichs, K. U., et al. (2015). Microbial communities and organic matter composition in surface and subsurface sediments of the Helgoland mud area, North Sea. Front. Microbiol. 6:1290. doi: 10.3389/fmicb.2015.01290
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Page, R., and Peti, W. (2016). Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214. doi: 10.1038/nchembio.2044
Pala, C., Molari, M., Nizzoli, D., Bartoli, M., Viaroli, P., and Manini, E. (2018). Environmental drivers controlling bacterial and archaeal abundance in the sediments of a Mediterranean lagoon ecosystem. Curr. Microbiol. 75, 1147–1155. doi: 10.1007/s00284-018-1503-3
Paliaga, P., Korlević, M., Ivančić, I., and Najdek, M. (2017). Limited influence of primary treated sewage waters on bacterial abundance, production and community composition in coastal seawaters. Mar. Environ. Res. 131, 215–226. doi: 10.1016/j.marenvres.2017.09.012
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Parks, D. H., and Beiko, R. G. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721. doi: 10.1093/bioinformatics/btq041
Pavloudi, C., Oulas, A., Vasileiadou, K., Sarropoulou, E., Kotoulas, G., and Arvanitidis, C. (2016). Salinity is the major factor influencing the sediment bacterial communities in a Mediterranean lagoonal complex (Amvrakikos Gulf. Ionian Sea). Mar. Genomics 28, 71–81. doi: 10.1016/j.margen.2016.01.005
Pella, E., and Colombo, B. (1973). Study of carbon, hydrogen and nitrogen determination by combustion-gas chromatography. Mikrochim. Acta 61, 697–719. doi: 10.1007/BF01218130
Petersen, N. R., Rysgaard, S., Nielsen, L. P., and Revsbech, N. P. (1994). Diurnal variation of denitrification and nitrification in sediments colonized by benthic microphytes. Limnol. Oceanogr. 39, 573–579. doi: 10.1007/s00248-004-0274-5
Petro, C., Starnawski, P., Schramm, A., and Kjeldsen, K. U. (2017). Microbial community assembly in marine sediments. Aquat. Microb. Ecol. 79, 177–195. doi: 10.3354/ame01826
Picone, M., Bergamin, M., Losso, C., Delaney, E., Novelli, A. A., and Ghirardini, A. V. (2016). Assessment of sediment toxicity in the Lagoon of Venice (Italy) using a multi-species set of bioassays. Ecotoxicol. Environ Saf. 123, 32–44. doi: 10.1016/j.ecoenv.2015.09.002
Polymenakou, P. N., Bertilsson, S., Tselepides, A., and Stephanou, E. G. (2005). Links between geographic location, environmental factors, and microbial community composition in sediments of the Eastern Mediterranean Sea. Microb. Ecol. 49, 367–378.
Price-Whelan, A., Dietrich, L. E., and Newman, D. K. (2007). Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J. Bacteriol. 189, 6372–6381. doi: 10.1128/JB.00505-07
Probandt, D., Knittel, K., Tegetmeyer, H. E., Ahmerkamp, S., Holtappels, M., and Amann, R. (2017). Permeability shapes bacterial communities in sublittoral surface sediments. Environ. Microbiol. 19, 1584–1599. doi: 10.1111/1462-2920.13676
Pusceddu, A., Sarà, G., Armeni, M., Fabiano, M., and Mazzola, A. (1999). Seasonal and spatial changes in the sediment organic matter of a semi-enclosed marine system (W-Mediterranean Sea). Hydrobiologia 397, 59–70. doi: 10.1023/A:1003690313842
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Quero, G. M., Perini, L., Pesole, G., Manzari, C., Lionetti, C., Bastianini, M., et al. (2017). Seasonal rather than spatial variability drives planktonic and benthic bacterial diversity in a microtidal lagoon and the adjacent open sea. Mol. Ecol. 26, 5961–5973. doi: 10.1111/mec.14363
R Core Team (2019). R: A Language And Environment For Statistical Computing [Computer software]. Vienna: R Foundation for Statistical Computing.
Sandy, M., and Butler, A. (2009). Microbial iron acquisition: marine and terrestrial siderophores. Chem. Rev. 109, 4580–4595. doi: 10.1021/cr9002787
Santmire, J. A., and Leff, L. G. (2007). The effect of sediment grain size on bacterial communities in streams. J. North Am. Benthol. Soc. 26, 601–610. doi: 10.1899/06-130.1
Sapp, M., Parker, E. R., Teal, L. R., and Schratzberger, M. (2010). Advancing the understanding of biogeography–diversity relationships of benthic microorganisms in the North Sea. FEMS Microbiol. Ecol. 74, 410–429. doi: 10.1111/j.1574-6941.2010.00957.x
Schallenberg, M., and Kalff, J. (1993). The ecology of sediment bacteria in lakes and comparisons with other aquatic ecosystems. Ecology 74, 919–934. doi: 10.2307/1940816
Schiessl, K. T., Hu, F., Jo, J., Nazia, S. Z., Wang, B., Price-Whelan, A., et al. (2019). Phenazine production promotes antibiotic tolerance and metabolic heterogeneity in Pseudomonas aeruginosa biofilms. Nat. commun. 10:762. doi: 10.1038/s41467-019-08733-w
Schlitzer, R. (2018). Ocean Data View. Available online at: https://odv.awi.de, 2018 (accessed May 2, 2019).
Seraphin, H., Sheeran, P., and Pilato, M. (2018). Over-tourism and the fall of Venice as a destination. J. Destin. Mark. Manag. 9, 374–376. doi: 10.1016/j.jdmm.2018.01.011
Severin, I., Confurius-Guns, V., and Stal, L. J. (2012). Effect of salinity on nitrogenase activity and composition of the active diazotrophic community in intertidal microbial mats. Arch. Microbiol. 194, 483–491. doi: 10.1007/s00203-011-0787-5
Shah, D., Zhang, Z., Khodursky, A., Kaldalu, N., Kurg, K., and Lewis, K. (2006). Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6:53. doi: 10.1186/1471-2180-6-53
Shepard, F. P. (1954). Nomenclature based on sand-silt-clay ratios. J. Sediment Petrol. 24, 151–158. doi: 10.1306/D4269774-2B26-11D7-8648000102C1865D
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Solidoro, C., Bandelj, V., Bernardi, F. A., Camatti, E., Ciavatta, S., Cossarini, G., et al. (2010). “Response of Venice lagoon ecosystem to natural and anthropogenic pressures over the last 50 years,” in Coastal Lagoons: Critical Habitats And Environmental Change, eds M. Kennish and H. Paerl (Boca Raton, FL: CRC Press), 483–511.
Solidoro, C., Pastres, R., Cossarini, G., and Ciavatta, S. (2004). Seasonal and spatial variability of water quality parameters in the lagoon of Venice. J. Mar. Syst. 51, 7–18. doi: 10.1016/J.JMARSYS.2004.05.024
Spoering, A. L., and Lewis, K. (2001). Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183, 6746–6751. doi: 10.1128/JB.183.23.6746-6751.2001
Stocker, R., Seymour, J. R., Samadani, A., Hunt, D. E., and Polz, M. F. (2008). Rapid chemotactic response enables marine bacteria to exploit ephemeral microscale nutrient patches. Proc. Natl. Acad. Sci. U.S.A. 105, 4209–4214. doi: 10.1073/pnas.0709765105
Sunagawa, S., Coelho, L. P., Chaffron, S., Kultima, J. R., Labadie, K., Salazar, G., et al. (2015). Ocean plankton. Structure and function of the global ocean microbiome. Science 22:1261359. doi: 10.1126/science.1261359
Tan, B., Ng, C., Nshimyimana, J. P., Loh, L. L., Gin, K. Y., and Thompson, J. R. (2015). Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front. Microbiol. 6:1027. doi: 10.3389/fmicb.2015.01027
Torti, A., Lever, M. A., and Jørgensen, B. B. (2015). Origin, dynamics, and implications of extracellular DNA pools in marine sediments. Mar. Genomics 24, 185–196. doi: 10.1016/j.margen.2015.08.007
Trojan, D., Schreiber, L., Bjerg, J. T., Bøggild, A., Yang, T., Kjeldsen, K. U., et al. (2016). A taxonomic framework for cable bacteria and proposal of the candidate genera Electrothrix and Electronema. Syst. Appl. Microbiol. 39, 297–306. doi: 10.1016/j.syapm.2016.05.006
Tšertova, N., Kisand, A., Tammert, H., and Kisand, V. (2011). Low seasonal variability in community composition of sediment bacteria in large and shallow lake. Environ. Microbiol. Rep. 3, 270–277. doi: 10.1111/j.1758-2229.2010.00221.x
Tsuboi, S., Amemiya, T., Seto, K., Itoh, K., and Rajendran, N. (2013). The ecological roles of bacterial populations in the surface sediments of coastal lagoon environments in Japan as revealed by quantification and qualification of 16S rDNA. World J. Microbiol. Biotechnol. 29, 759–774. doi: 10.1007/s11274-012-1231-y
Turner, A., and Millward, G. E. (2002). Suspended particles: their role in estuarine biogeochemical cycles. Estuar. Coast. Shelf Sci. 55, 857–883. doi: 10.1006/ecss.2002.1033
Van der Walt, A. J., van Goethem, M. W., Ramond, J. B., Makhalanyane, T. P., Reva, O., and Cowan, D. A. (2017). Assembling metagenomes, one community at a time. BMC Genomics 18:521. doi: 10.1186/s12864-017-3918-9
Walker, C. B., de la Torre, J. R., Klotz, M. G., Urakawa, H., Pinel, N., Arp, D. J., et al. (2010). Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. U.S.A. 107, 8818–8823. doi: 10.1073/pnas.0913533107
Wasmund, K., Mußmann, M., and Loy, A. (2017). The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ. Microbiol. Rep. 9, 323–344. doi: 10.1111/1758-2229.12538
Wentworth, K. (1922). A scale of grade and class terms for clastic sediments. J. Geol. 30, 377–392. doi: 10.1086/622910
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583. doi: 10.1073/pnas.95.12.6578
Wickham, H., Chang, W., and Wickham, M. H. (2016). Package “ggplot2”. Create Elegant Data Visualisations Using the Grammar of Graphics. Available online at: https://cran.r-project.org/web/packages/reshape2/index.html (accessed February 25, 2020).
Wörmer, L., Hoshino, T., Bowles, M. W., Viehweger, B., Adhikari, R. R., Xiao, N., et al. (2019). Microbial dormancy in the marine subsurface: global endospore abundance and response to burial. Sci. Adv. 5:eaav1024. doi: 10.1126/sciadv.aav1024
Wu, L., Kellogg, L., Devol, A. H., Tiedje, J. M., and Zhou, J. (2008). Microarray-based characterization of microbial community functional structure and heterogeneity in marine sediments from the Gulf of Mexico. Appl. Environ. Microbiol. 74, 4516–4529. doi: 10.1128/AEM.02751-07
Yamamoto, K., Hirao, K., Oshima, T., Aiba, H., Utsumi, R., and Ishihama, A. (2005). Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 280, 1448–1456. doi: 10.1074/jbc.M410104200
Yang, J., Wang, C., Shu, C., Liu, L., Geng, J., Hu, S., et al. (2012). Marine sediment bacteria harbor antibiotic resistance genes highly similar to those found in human pathogens. Microb. Ecol. 65, 975–981. doi: 10.1007/s00248-013-0187-2
Zaggia, L., Rosso, J., and Zonta, R. (2007). Sulphate reduction in the sediment of the Venice canals (Italy). Mar. Pollut. Bull. 55, 415–424. doi: 10.1016/j.marpolbul.2007.09.004
Zhang, C., Palumbo, A. V., Phelps, T. J., Beauchamp, J. J., Brockman, F. J., Murray, C. J., et al. (1998). Grain size and depth constraints on microbial variability in coastal plain subsurface sediments. Geomicrobiol. J. 15, 171–185. doi: 10.1080/01490459809378074
Zinger, L., Amaral-Zettler, L. A., Fuhrman, J. A., Horner-Devine, M. C., Huse, S. M., Welch, D. B., et al. (2011). Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS One 6:e24570. doi: 10.1371/journal.pone.0024570
Keywords: water-sediment interface, metagenome, metabarcoding, 16S rRNA gene, antibiotic resistance, microscale, mobilome, mercury
Citation: Banchi E, Del Negro P, Celussi M and Malfatti F (2021) Sediment Features and Human Activities Structure the Surface Microbial Communities of the Venice Lagoon. Front. Mar. Sci. 8:762292. doi: 10.3389/fmars.2021.762292
Received: 21 August 2021; Accepted: 25 November 2021;
Published: 14 December 2021.
Edited by:
Yong Wang, Institute of Deep-Sea Science and Engineering, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Gurdeep Rastogi, Chilika Development Authority, IndiaXupeng Cao, Dalian Institute of Chemical Physics, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Banchi, Del Negro, Celussi and Malfatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Celussi, bWNlbHVzc2lAaW5vZ3MuaXQ=; Francesca Malfatti, Zm1hbGZhdHRpQGlub2dzLml0