 Jessica Pearce
Jessica Pearce Matthew W. Fraser
Matthew W. Fraser Ana M. M. Sequeira
Ana M. M. Sequeira Parwinder Kaur
Parwinder Kaur
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Mar. Sci. , 12 October 2021
Sec. Marine Biotechnology and Bioproducts
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.744986
Chondrichthyan species (sharks, rays, skates, and chimeras) are a class of high ecological, economic, and cultural significance, and yet they are the most threatened taxa in the marine environment. The creation of reference chromosome-length genome assemblies allows for conservation genomics methods, such as population and ecological genomics, to be utilized. Despite being greatly threatened and of great importance in maintaining ecosystem function, chondrichthyan species have been repeatedly absent from conservation-based genome sequencing projects. Less than 1% of these species have a genome sequence, despite their almost 50% either threatened or Data Deficient conservation status. Most notably, there are seven orders within this class without any genome representation. In this review, we identify gaps in chondrichthyan genomic resources and demonstrate how the lack of genomic resources for this major taxonomic class is limiting the conservation of these already difficult to conserve species. We highlight other applications for chondrichthyans genomics, such as evolutionary and developmental biology. Likely, the mismatching sampling protocols and limited computational skills and communication between fields have been preventing the integration of marine and molecular sciences. Here, we propose that this field is in dire need to move forward quickly to increase protection for marine species and ecosystems through improved collaboration between marine, molecular, and computer sciences.
The marine environment is vital in providing a multitude of ecosystem services and is responsible for the function of our global climate (Barbier, 2017; Gattuso et al., 2018). Marine ecosystems display extensive signs of human interference that are reflected at multiple levels, ranging from primary producers that provide habitat (e.g., seagrasses and corals) to apex predators like sharks (Hoegh-Guldberg and Bruno, 2010; Smale et al., 2019). Cumulative pressures and impacts of multiple stressors (Sequeira et al., 2019) are likely contributing to species extinction rates which have been rapidly increasing far above historical averages, and exceeding threshold boundaries, resulting in the sixth mass extinction in the earth’s history (Rockstrom, 2009; Ceballos et al., 2015). The conservation of species is of critical importance to maintain Earth’s biodiversity and its ecological, economic, and social benefits (Hoffmann et al., 2015; Shafer et al., 2015). Novel techniques will be required to assist environmental stewardship in the face of increasing threats.
Emerging genomic technologies are powerful tools to assist conservation research (Corlett, 2017). For instance, ecological and environmental genomics uses genomic data to investigate relationships between organisms and their environment to predict species’ responses and adaptions to environmental change (Matz, 2018; Ruegg et al., 2018). However, these methods require high-quality reference genomes to compare study-specific genetic and genomic data against to have accurate results (Shafer et al., 2015). Genomics technology has recently become widely accessible to a range of fields outside the traditional molecular sciences through increased power and affordability (Goodwin et al., 2016; Worley et al., 2017). Historically, genome sequencing involved generating many short reads (contigs) and piecing them together based on sequence overlap (contigging), where longer sequence reads allow more overlap (Schatz et al., 2010). These larger contiguous sequences can then be linked to create scaffolds and ordered according to the predicted genome structure (Dudchenko et al., 2017). However, due to regions of high repeat content in genomes or low sequencing coverage, consequently resulting in gaps between contigs, within and between scaffolds, most genome assemblies are highly fragmented (Kadota et al., 2020). Recent developments in long-read sequencing technology, such as Pacific Biosciences (PacBio) or the Oxford Nanopore, have made advancements in overcoming this issue by generating very long contigs initially (Lang et al., 2020). This creates higher sequence overlap and therefore less gaps with higher genome completeness.
Novel high-throughput chromosome conformation capture (Hi-C) technology, combined with contigging, can generate high-quality full chromosome-length genome assemblies in a relatively inexpensive and quick manner (Dudchenko et al., 2017). Hi-C data are produced based on the principle that the closer two DNA locations are to each other, the more frequently they will come into contact and so a proximity matrix of DNA locations can be created. Then, through a series of bioinformatics pipelines, chromosome-length scaffolds can be produced (Lieberman-Aiden et al., 2009; Rao et al., 2014; Dudchenko et al., 2018). The storage and analysis of biological data using computer sciences, termed “bioinformatics,” is a fast-developing field in molecular sciences with its ability to deal with big and complex data sets such as genetic/genomic data (Baxevanis et al., 2020). Bioinformatics pipelines can transfer data through multiple functions within and between software and programs. The combination of Hi-C data and bioinformatics pipelines has allowed chromosome-length genome assembly to become largely automated.
The addition of Hi-C data to traditional genome sequencing methods has allowed the production of high-quality genomes in a timely and cost-effective manner (Dudchenko et al., 2017). This development has led to multiple global projects aiming to sequence the genomes of Earth’s biodiversity for a multitude of purposes. Ambitious consortiums like the Earth BioGenome Project (EBP) (Lewin et al., 2018), which aims to sequence the genomes of all eukaryotic species, work on a worldwide scale of networks and partnering organizations, such as the DNA Zoo (Hoencamp et al., 2021) and the Genome 10k/Vertebrate Genomes Project (VGP) (Koepfli et al., 2015). Despite almost 40 affiliated projects globally, there has been a heavy focus on terrestrial species, leading to a lack of representation of marine species. In particular, there is a severe lack of cartilaginous fish, such as sharks and rays (Class: Chondrichthyes), with less than 1% of these species sequenced, in spite of their high ecological, social, and economic importance (Figure 1).
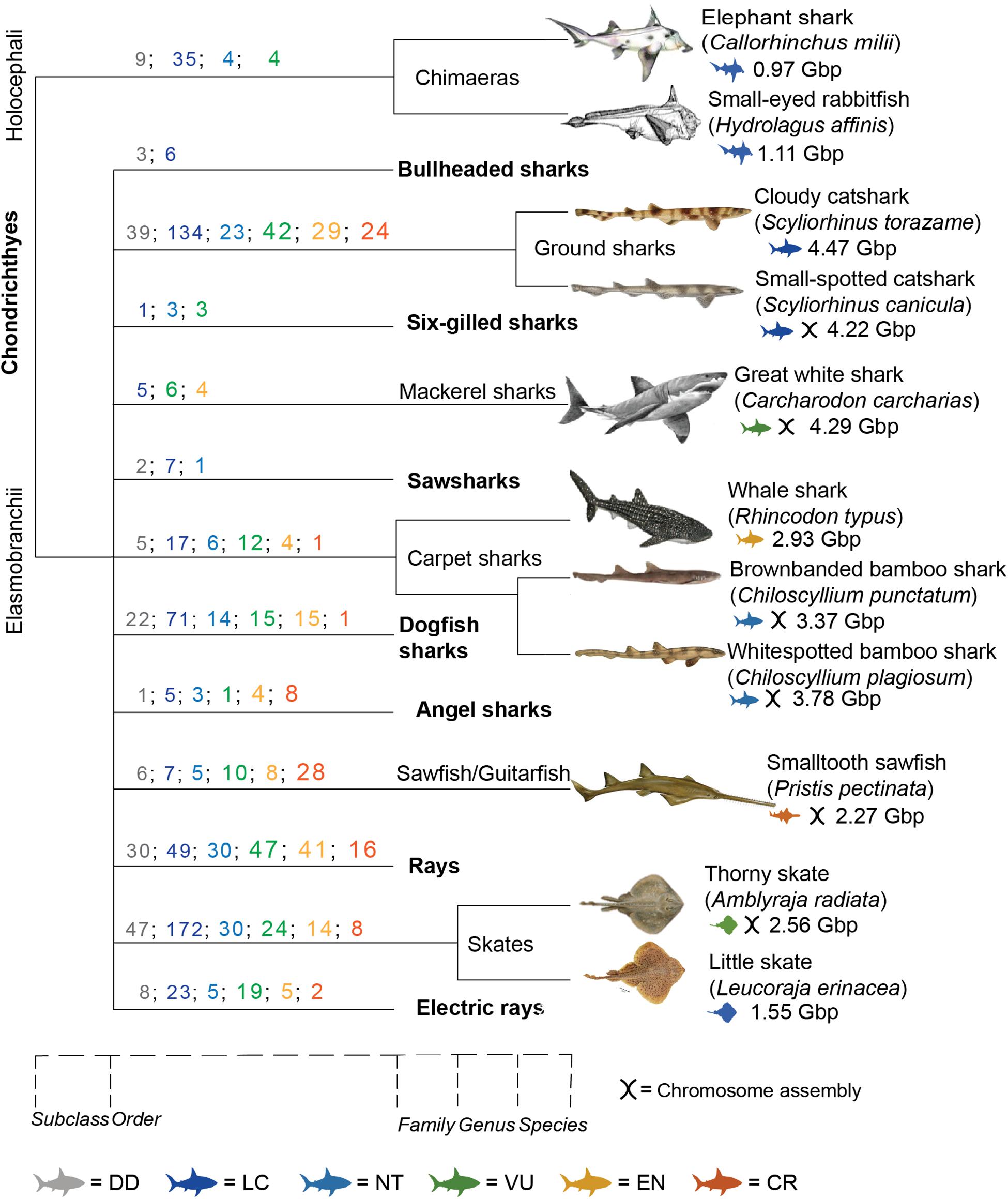
Figure 1. Taxonomic distribution of chondrichthyan genomic resources. It displays species with available genome assemblies; Callorhinus milii (Venkatesh et al., 2014; NCBI, 2021a), Hydrolagus affinis (Fonseca et al., 2020), Scyliorhinus torazame (Hara et al., 2018), Scyliorhinus canicular (NCBI, 2021c), Carcharodon carcharias (Marra et al., 2019; NCBI, 2021b), Rhincodon typus (Read et al., 2017; Weber et al., 2020), Chiloscyllium punctatum (Hara et al., 2018; Hoencamp et al., 2021), Chiloscyllium plagiosum (Zhang et al., 2020), Pristis pectinata (NCBI, 2019b), Amblyraja radiata (Rhie et al., 2020), and Leucoraja erinacea (Wyffels et al., 2014). Scale at bottom displays where different classifications diverged. Colored numbers represent the number of species in that order for each IUCN status. Colored icons represent the IUCN status for that species. DD, data deficient; LC, least concern; NT, near threatened; VU, vulnerable; EN, endangered; CR, critically endangered (IUCN Red List, 2021).
Sharks and rays have vital roles in maintaining ecosystem health and function through top-down regulation (Yagnesh et al., 2020; Flowers et al., 2021). However, these chondrichthyan species have an extinction risk well above most other vertebrate classes on average with an estimated over one-third of species threatened by extinction (Dulvy et al., 2014, 2021). Despite their high-risk status and significance to ecosystem function, which should make these species high priority for conservation, an extensive gap in conservation-based genetic and genomic resources exists (Johri et al., 2019). Additionally, chondrichthyan species present opportunities for investigation into other potential applications, including biomedical research (Marra et al., 2019). The integration of genomics in the conservation and study of chondrichthyan species will provide more effective and efficient methods of protection through accurate population assessments (Attard et al., 2018; Hohenlohe et al., 2021). In this review, we discuss the state of chondrichthyan genomics research to date along with clear applications for conservation as well as other research fields. We indicate the obstacles currently hindering swift integration of molecular marine sciences in marine conservation and provide recommendations for a way forward through improved protocols and communication between fields.
The class of Chondrichthyes is split into two subclasses, one containing sharks, rays, and skates (Elasmobranchii) and the other chimeras (Holocephali). To date, only 11 chondrichthyan genomes have been sequenced out of ∼1,200 species, i.e., less than 1% (Figure 1). The first genome published for this class was of the Elephant “Shark” (Callorhinchus milii), which is in fact a chimera, member of the Holocephali subclass (Venkatesh et al., 2014). This was the first chondrichthyan genome chosen to be sequenced due to its small size [0.975 gigabase pairs (Gbp)] and potential to become a model organism for evolution and development studies (Venkatesh et al., 2005). Indeed, that study, which was published almost two decades ago, has prompted recent research into chondrichthyan genomes and their utilization in evolutionary and developmental biology as well as biomedical insights (Hara et al., 2018; Marra et al., 2019; Figure 1). Recently, a higher quality genome assembly for the elephant shark was made publicly available, however it is still only at scaffold level (NCBI, 2021a). The small-eyed rabbitfish (Hydrolagus affinis) is the only other species of chimera published (Fonseca et al., 2020).
Other published chondrichthyan genomes are from the Elasmobranchii subclass and are currently limited to two skates: little skate (Leucoraja erinacea) (Wyffels et al., 2014) and thorny skate (Amblyraja radiata) (Rhie and Project, 2021), and five sharks: whale shark (Rhincodon typus) (Read et al., 2017; Weber et al., 2020), brownbanded bamboo shark (Chiloscyllium punctatum) (Hara et al., 2018; Hoencamp et al., 2021), cloudy catshark (Scyliorhinus torazame) (Hara et al., 2018), great white shark (Carcharodon carcharias) (Marra et al., 2019), and whitespotted bamboo shark (Chiloscyllium plagiosum) (Zhang et al., 2020; Figure 1). Draft genomes for other elasmobranch species are publicly available online but have not yet been formally published, including the smalltooth sawfish (Pristis pectinata) (NCBI, 2019b), small-spotted catshark (Scyliorhinus canicula) (NCBI, 2021c), and chromosome-length white shark assembly (NCBI, 2021b; Figure 1).
Multiple genome assemblies have been published for a few chondrichthyan species, such as the whale shark and white shark, which demonstrate differences in sequencing technology and assembly techniques. A second de novo (from scratch) genome assembly for the whale shark was recently published (Weber et al., 2020) with a lower percentage of missing BUSCOs (Benchmarking Universal Single-Copy Orthologs), meaning more complete genes, despite the higher fragmentation compared to the older scaffold assembly for this species. The difference in these two assemblies is due to differences in the sequencing technologies used: Roche 454, Illumina, and Pacific Biosciences platforms were used originally (Read et al., 2017), compared with Illumina HiSeq 2500m platform (Weber et al., 2020) used in the recent assembly (Table 1). The different quality of these genome assemblies highlights how both sequencing technology and assembly techniques can impact the final genome. A genome assembly with higher completeness can be utilized much more effectively and investigated deeper than a limited incomplete assembly (Hoffmann et al., 2015; Rhie and Project, 2021). For example, investigation into the white shark’s ability to regulate cell growth to prevent tumors was hindered by the original incomplete genome, as a large section of the specific gene of interest was missing and therefore could not be investigated further (Marra et al., 2019). Therefore, the desired output quality should be considered during the sequencing process and technology choices made accordingly.
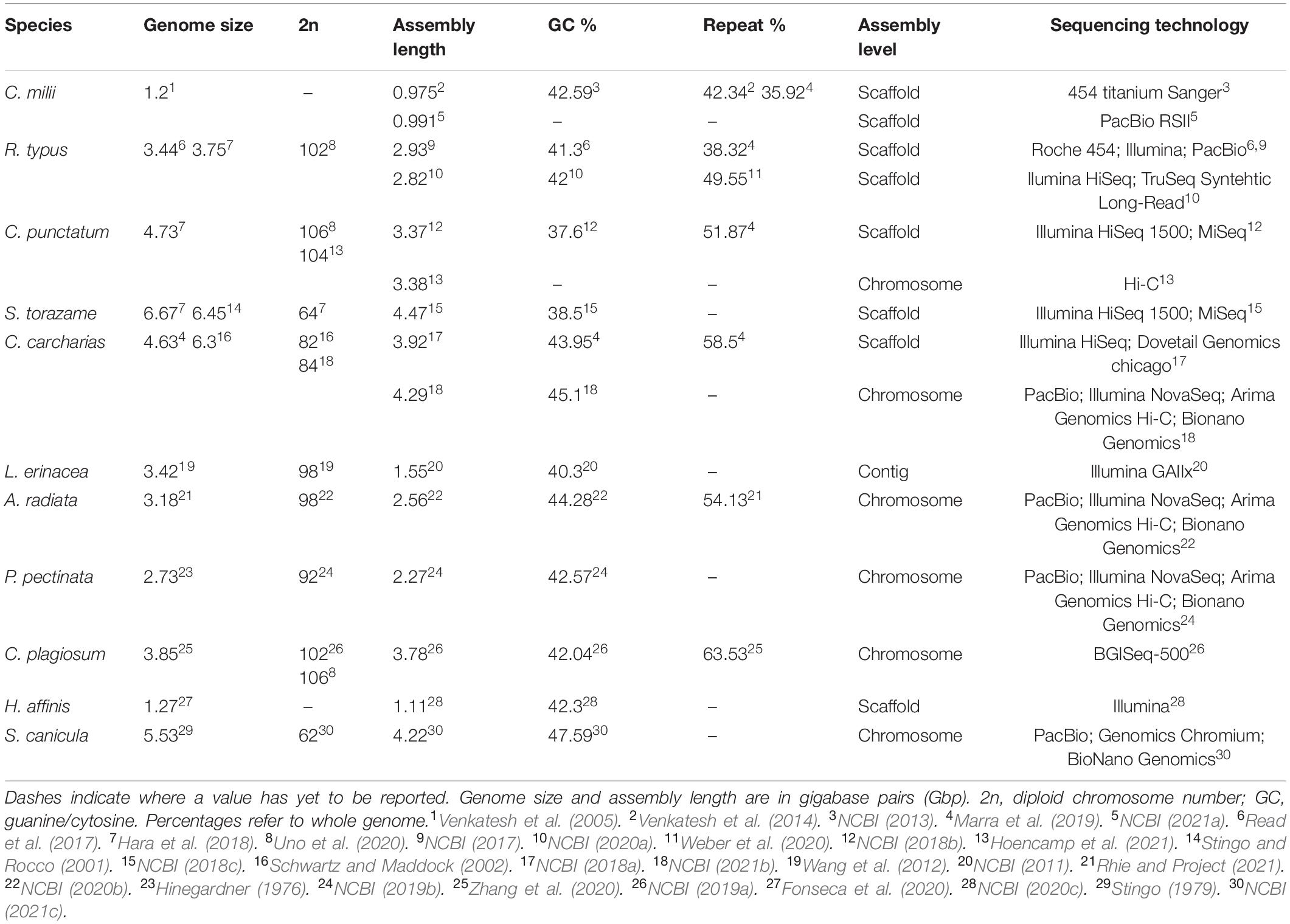
Table 1. Current knowledge of chondrichthyan genomes to date.
Consistent comparisons between chondrichthyan species are made difficult due to discrepancies between sequenced genomes (Table 1). Many of these discrepancies, such as assembly size, may be due to the use of different methods; sequencing technologies, like Illumina or PacBio, and bioinformatics software, such as varying pipelines used when assembling genomes. Some of these methods may now be outdated and less accurate than current technology, or simply use different algorithms, and so produce varying results (Table 1). Notably, one of the major differences between these genomes is the level of assembly. While most recently released assemblies are at chromosome-length, earlier genomes, limited by the technology of the time, are only to a scaffold level and therefore more likely to be incomplete and fragmented. For example, the original 2019 scaffold genome assembly of the white shark is smaller, compared to the recent 2021 chromosome-length assembly, as it is less complete (Table 1). This comparison between two assemblies of the same species demonstrates the difference quality can have for a reference genome. This highlights the issues associated with comparing genomes of different species of varying qualities, when lower quality assemblies are likely to limit downstream analysis due to missing fragments of genomic data and could end up impacting results.
Chondrichthyan species are an exceptionally difficult class for applying conservation strategies (Dulvy et al., 2017). This is likely due to their large geographical range, fishing pressure, and slow life-history traits such as reproduction rate. Conservation genomics has extensive applications to assist investigating and protecting challenging species like chondrichthyans, through studies such as population genomics which can accurately assess effective population sizes and structures (Johri et al., 2019; Oleksiak and Rajora, 2020). These methods, however, optimally require high-quality chromosome-length genome assemblies to function as important references from which to compare and build investigations (Matz, 2018; Cordier et al., 2020). For example, low coverage whole-genome resequencing (WGR) requires a species-specific reference assembly to align to individual’s genomes, in order to assess relatedness and genetic structure within and between populations (Fuentes-Pardo and Ruzzante, 2017; Lou et al., 2020). Without a reference genome, other methods such as polymorphism analyses have lower resolution and less accurate results (Galla et al., 2019; Johri et al., 2019). Reference genomes are particularly useful for marine non-model species, such as chondrichthyans which are lacking prior genetic or genomic data, as it creates a baseline resource (Oleksiak and Rajora, 2020). Additionally, in the event of species extinction, a reference genome can assist with preserving genetic diversity and information for posterior studies. Hence, sequencing reference genomes for target conservation species provides the optimal and most comprehensive approach to conservation genomics (Oleksiak and Rajora, 2020).
Significant advances in genomics have been made over the last decade, yet uptake in the conservation and management of species continues to be limited. Two branches of genomics are particularly relevant for conservation: ecological genomics and environmental genomics. Ecological aims to investigate the genome structure and function to understand the relationship between organisms and their environment. Environmental genomics aims to predict organism responses and adaptations to environmental changes (Martin, 2008; Ungerer et al., 2008; Van Straalen and Roelofs, 2011). Environmental and ecological genomics has the potential to detect species presence, local and potential future genetic adaptations, and responses to anthropogenic influences, such as climate change and ocean acidification (Ungerer et al., 2008; Savolainen et al., 2013; Ruegg et al., 2018; Cordier et al., 2020). Additionally, environmental genomics can investigate, monitor, and manage non-indigenous species in the marine environment through studying genetic traits that allow species to become highly adaptable and invasive (Bourne et al., 2018). Both offer high value to conservation efforts; environmental genomics has value for assessing species composition through presence detection, whereas ecological genomics is more valuable for investigating species responses and adaption through genome function. These applications for a single genome alone have vast potential for investigating adaptations or negative responses in chondrichthyan species, and other non-model organisms, to climate change and other threats (Matz, 2018). The inclusion of environmental and ecological genomics in applied conservation management, however, requires high-quality genomic resources (e.g., chromosome-length assemblies), again highlighting the need for their development.
With a chromosome-length genome assembly and the accompanying fine-scale genetic data it is also possible to track an individual’s movements, population sizes (Ne), and kin relationships on a global scale (Oleksiak and Rajora, 2020; Hohenlohe et al., 2021). As many shark species are highly migratory and cosmopolitan, investigation at the global scale is essential to assess the effective protection of these high-risk species (Dulvy et al., 2017; Attard et al., 2018). Methods such as documenting genetic polymorphisms on a genome-scale, close-kin mark-recapture, and kinship analysis, have already been successful with marine species (Cipriano and Palumbi, 1999; Attard et al., 2018; Hillary et al., 2018). While genome-wide polymorphism analyses do not necessarily require a reference genome, only with a high-quality genome can fine-scale resolution of genetic data be investigated, in particular adaptive genetic variation (Pazmiño et al., 2017; Galla et al., 2019; Oleksiak and Rajora, 2020). The ability to detect adaptive genetic variation, as opposed to only neutral variation, is one of the main advances of population genomics utilizing reference genomes over traditional genetic techniques (Funk et al., 2012; Attard et al., 2018; Hohenlohe et al., 2021). Additionally, the availability of a reference genome allows calculation of population Ne based on linkage disequilibrium, which is more accurate and effective than other methods, and the reconstruction of demographic history (Hendricks et al., 2018; Hohenlohe et al., 2021). These techniques can be implemented in genomics-based ecosystem monitoring and informing management practices (Cordier et al., 2020; Hohenlohe et al., 2021) to assist the conservation of ecologically significant species and associated habitats.
Moreover, genomics’ ability to monitor and assess populations leads to extensive applications in fisheries management and aquaculture (Bernatchez et al., 2017; Houston et al., 2020). Approximately one-third of chondrichthyan species are threatened due to overfishing as many are commercially exploited or caught accidently as by-catch (Dulvy et al., 2017, 2021). For instance, the elephant shark (chimera) (Finucci et al., 2021), brownbanded bamboo shark (Fahmi et al., 2020), whitespotted bamboo shark (Chen et al., 2007), catshark, smalltooth sawfish (Yan et al., 2021), little skate (Curtis and Sosebee, 2015), and thorny skate (Pennino et al., 2019) (all species with available genomes), are commercially fished species which require management to avoid overexploitation. Molecular methods are fast becoming a mainstream tool in fisheries management and have already shown underrepresented or unregulated catch of chondrichthyan species through DNA barcoding and analyzing genetic population structure (Vargas-Caro et al., 2017; Kuguru et al., 2018). Population genomics methods, such as utilizing reference genomes to estimate effective population size or genetic structure, could become critical in ensuring sustainable fisheries management. Therefore, the slow development and utilization of chondrichthyan genomics is currently limiting potential application in sustainable resource management.
There are 12 extant orders within the Elasmobranchii subclass (and one within Holocephali), yet only six have genome representation (Table 2). If the purpose of mass genome sequencing projects is to comprehensively act as a seed bank of genomes for conservation purposes, then it is imperative that this gap be addressed. Chondrichthyans are a class of highly unique evolution, including many keystone species that exert top-down control on marine ecosystems, provide economic and cultural values, and are greatly threatened by a multitude of stressors on global, regional, and local scales (Dulvy et al., 2014, 2017; Stein et al., 2018). These factors alone should make them a target for biodiversity conservation regardless. Out of the ∼1,200 chondrichthyan species, 180 (14.7) are classified as Vulnerable, 124 (10.1%) are Endangered, and 89 (7.3%) are Critically Endangered on the International Union for Conservation of Nature (IUCN) Red List (IUCN Red List, 2021). Strikingly, 172 species (14%) within this major class are listed as Data Deficient (Table 2; IUCN Red List, 2021). This means that almost 50% of all chondrichthyan species are either threatened by extinction or are of unknown conservation status. In these cases, a reference genome would provide a baseline from which a multitude of analyses could be conducted and greatly assist in conservation status assessment utilizing methods previously discussed. Hence, conservation genomics has the potential to provide researchers and management with vast applications in the field of chondrichthyan conservation where there is limited baseline knowledge (Johri et al., 2019).
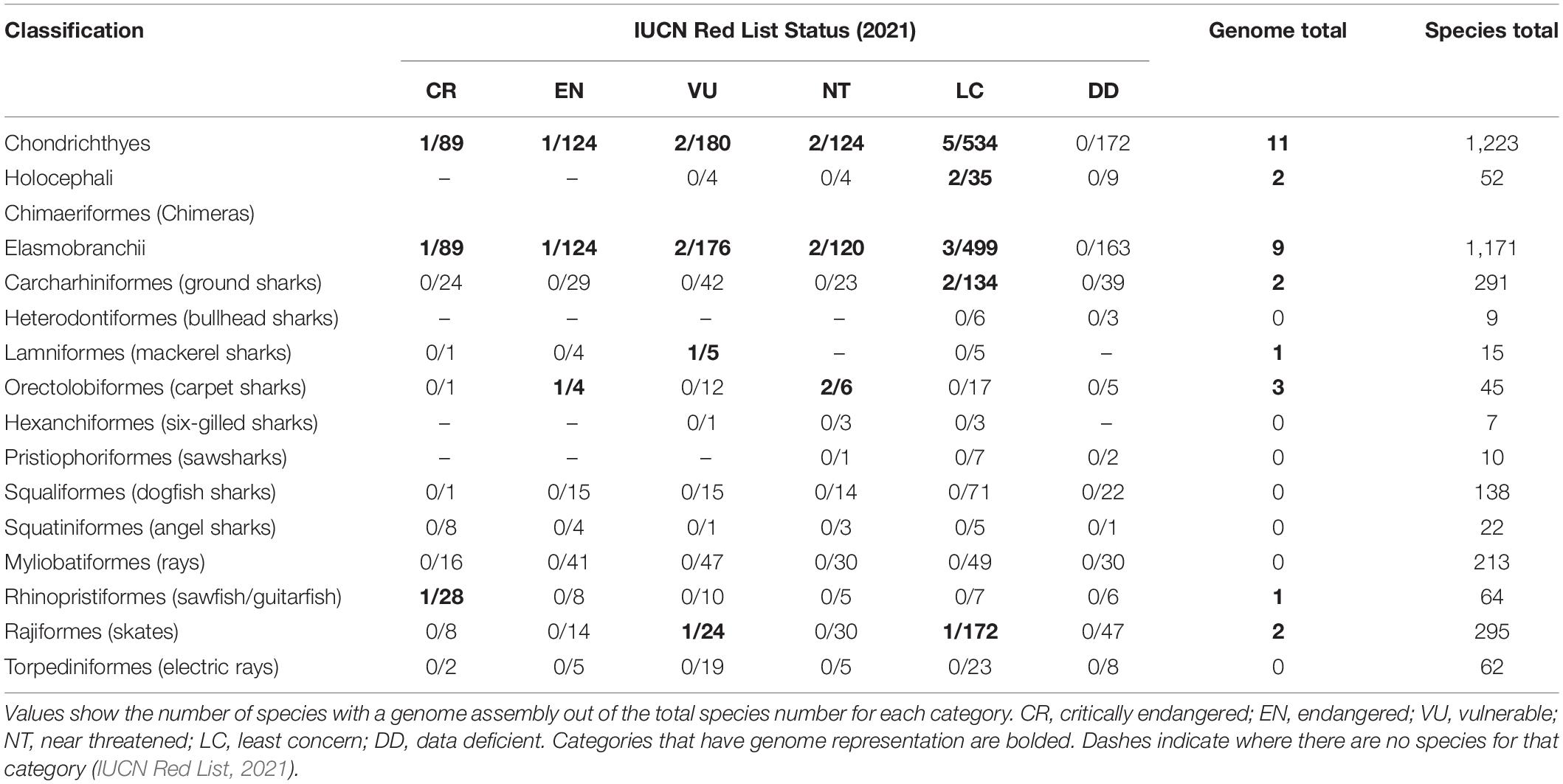
Table 2. IUCN Red List conservation status of species within the Chondrichthyes class, divided into subclasses and orders.
Despite the high percentage of threatened or Data Deficient species, there are many orders of elasmobranch species without genome representation (Table 2). For instance, the order Squatiniformes (angel sharks – subclass Elasmobranchii) has a total of 22 species, of which 8 (36.4%) are Critically Endangered, 13 (59.1%) threatened overall, and 1 (4.5%) is Data Deficient (Table 2), while none have genomic representation. Another order of Elasmobranchii, the Rhinopristiformes (sawfish and guitarfish), has only a single genome represented in the total of 64 species although 43.8% of these species are Critically Endangered, 71.9% threatened and 9.4% are Data Deficient (Table 2). The contrast between the percentage of Endangered species and the proportion of species with genomes within this class is alarming, given the significant role genomic resources can have in conservation. For instance, in the event of species extinction, a reference genome can assist with preserving genetic diversity and information for posterior studies. Conservation genomics methods could assist in preventing these high-risk species from becoming extinct, through previously mentioned methods to assess effective population sizes, species distributions, adaptive capacity, and responses to stress, and incorporating these into management solutions such as genetic rescues (Attard et al., 2018; Chattopadhyay et al., 2019; Hohenlohe et al., 2021). For example, through genome-wide population analysis, the Critically Endangered Siamese crocodile was found to have extremely low genetic diversity and effective population size and allowed conservation efforts to pick individuals with the correct genetics for captive breeding programs and others that were reintroduced into the wild (Chattopadhyay et al., 2019).
Chondrichthyan species are distinctly unique in their evolution. They represent an ancient lineage with a high degree of evolutionary divergence between the two subclasses and different orders (refer to Figure 1 for taxonomic relationships). It is estimated the class diverged roughly 473 million years ago (MYA) from other Gnathostomes (jawed vertebrates), with the two subclasses, Holocephali (chimeras) and Elasmobranchii (sharks, rays, and skates), diverging roughly 399 MYA (Hedges and Kumar, 2009; Inoue et al., 2010; Chen et al., 2012; Licht et al., 2012; Betancur-R et al., 2015). Chondrichthyans are a very distinct class as they are the only vertebrate clade to possess a cartilaginous skeleton, rather than bone (Boisvert et al., 2019). They are also noted for their ability to lose and replaced teeth continuously throughout their lifespan, particularly in sharks and rays (Boisvert et al., 2019). The availability of multiple species’ genome allows for the reconstruction of clade phylogenetic relationships and demographic history (Hohenlohe et al., 2021). Phylogenomics has risen as the optimal method for resolving the phylogeny of species, as simply using traditional gene analysis has led to inconsistency and ambiguity in the evolution of species (Johri et al., 2019). This has highlighted the need for genome-wide investigations to increase accuracy in these studies (Johri et al., 2019).
The first chondrichthyan genome (elephant shark) was sequenced for evolutionary and developmental investigation (Venkatesh et al., 2005), but led to the surprising revelation that the elephant shark had a higher degree of sequence similarity to humans than teleost fish genomes. This is despite humans being more closely related to teleost (bony) fish (Venkatesh et al., 2007). The elephant shark genome also displayed the lowest evolutionary rate of any vertebrate species yet investigated, when compared to teleost and tetrapod (bony) species, which likely stems from their slow neutral nucleotide mutation rate and very little change to the organization of their genes (Venkatesh et al., 2007, 2014). The sequencing of the genome for this species alone provided insights into the applications for developing chondrichthyan genomic resources, as it has subsequently been used in a range of studies, including embryogenesis, evolutionary and developmental biology, and functional genetics (Venkatesh et al., 2014; Hara et al., 2018; Marra et al., 2019).
In these following recent investigations, chromosome-length chondrichthyan genome assemblies have allowed the reconstruction of ancestral vertebrate chromosomes, chromosomal rearrangements, and breakages as well as the evolution of immune responses that show potential for human disease treatments (Zhang et al., 2020). Additionally, investigation of the small-eyed rabbitfish genome showed the evolution of the endocrine systems in early jawed vertebrates through a comparative analysis of nuclear receptors (NRs) in these chondrichthyan genomes (Fonseca et al., 2020). These investigations provide an example of how chondrichthyan genomes can be utilized to solve evolutionary mysteries into the evolution and development of vertebrates, jawed vertebrates, and chondrichthyan species (Zhang et al., 2020). These studies show a glimpse of fundamental advances in the field of evolutionary biology that can be made from chondrichthyan genomes.
Chondrichthyan genomics also has applications in biomedical science and physiology, as demonstrated by the great white shark and whale shark genomes (Marra et al., 2019; Weber et al., 2020). Compared to other vertebrate species, both genomes displayed positive selection and enrichment for molecular adaptations relating to genome stability and wound healing (Marra et al., 2019). Additionally, they had markers for cancer resistance through enhanced DNA damage response and repair mechanisms, genome, and telomere maintenance (Marra et al., 2019; Weber et al., 2020). Whale sharks possess expanded neurodegeneration genes, among other neural connectivity genes, again illustrating chondrichthyan evolutionary resistance (Weber et al., 2020). Without the addition of chondrichthyan species to a wide range of other classes, it would not be possible to assess physiological trends across all animals. For example, comparative genomics revealed that body weight, lifespan, and metabolic rate are all positively correlated across all vertebrates (Weber et al., 2020). This clearly demonstrates the broader context of chondrichthyan species as an important component to address overarching questions across all vertebrate classes.
As demonstrated above, a high-quality chromosome-length scaffold assembly can have a multitude of applications spanning various disciplines (Lewin et al., 2018; Kadota et al., 2020). A single species genome can serve as a baseline for research into various aspects of shark biology, physiology, and therefore conservation, which can continue to be built upon and improved. Genomics studies of chondrichthyan species have contributed new knowledge to many fields of research and have provided a foundation for further in-depth study. An abundance of different research areas could utilize these reference genomes, including but not limited to conservation genetics, transcriptomics, proteomics, comparative genetics/genomics, functional genetics/genomics, and evolution and development studies (Worley et al., 2017; Lewin et al., 2018).
To develop genomic resources efficiently and effectively for chondrichthyan species, there are necessary obstacles to overcome to achieve the best research and conservation outcomes. Genome assemblies at the chromosome length are typically of higher quality and completeness (Dudchenko et al., 2017; Kadota et al., 2020; Rhie and Project, 2021). Therefore, they are likely to be more informative in downstream analyses, such as conservation and population genomics. Chondrichthyan genomes, particularly larger ones such as that of the catsharks (>4 Gbp), require long-read sequencing (e.g., Pacific Biosciences HiFi reads) to produce an accurate chromosome-length assembly due to the high level of heterozygosity and repeat content (Kelley et al., 2016; Rhie and Project, 2021). High percentages of repetitive elements throughout a genome lead to a higher number of gaps, making chromosome-length scaffolds extremely difficult to assemble (Rhie and Project, 2021). Although utilizing long-read sequencing technology assists in overcoming this issue, it is not a perfect solution due to the higher degree of sequence inaccuracy. Scaffold gaps remain unavoidable, meaning that accurate genome assembly for these species requires manual curation, in addition to automated pipelines (Rhie and Project, 2021). This presents a challenge to developing genomic resources for this at-risk class of species as both long-read sequencing and manual curation are expensive and time intensive. Long-range sequencing remains the ideal approach for chondrichthyan species, as this sequencing technology will continue to increase in accuracy and decrease in cost. Developing this as a standard practice should be a priority to maximize effectiveness of genomic resources.
Another major challenge for developing molecular resources of any kind for this class is obtaining suitable species samples. Many chondrichthyan species are notoriously difficult to collect samples from, mostly due to their elusiveness and habitat choice making them hard to find (Kelley et al., 2016). For example, chimeras inhabit the deep sea, while many pelagic species of sharks spend their time in the open ocean. For cosmopolitan shark species often found near-shore, such as the great white shark, another point of difficulty is safety while collecting biopsy samples. Conversely, Hammerhead shark species exhibit high capture stress and post-release mortality, making sampling a delicate task (Drymon and Wells, 2017). These factors make collecting fresh tissue samples, necessary for genome sequencing, a complicated, resource-intensive, and time-consuming activity. We recommend that all future opportunities where sharks are already being handled, for research or government tagging purposes, collect samples suitable for genetic study, preferably in a standardized approach according to molecular practices.
Obtaining suitable samples for genomic DNA extraction, Hi-C preparation, and sequencing from previously collected samples can also be problematic. Extracting RNA, which is necessary for proper genome annotation, essentially requires completely undegraded and uncontaminated DNA samples. Historically in biological sciences, biopsy samples were preserved in ethanol. However, ethanol prevents Hi-C techniques from detecting DNA contacts and so these samples are often unusable for sequencing genomes. Ideally, for a good quality chromosome-length genome assembly, tissue samples must be flash frozen at −80°C or liquid nitrogen. Although freezing samples has become more popular in recent years, storage in ethanol or at −20°C is common, resulting in many samples with DNA that is still too degraded to utilize for genomics as high molecular weight DNA is essential (Matz, 2018). Biological sciences in general, particularly in marine sciences, must move toward biobanking and storing species samples in accordance with standardized molecular techniques to improve sample quality critical for developing genomic resources (Wong et al., 2012; Rhie and Project, 2021).
An emerging challenge lies not only in generating genomic data but also in processing it (Dupont et al., 2007; Schweizer et al., 2021). The advances in sequencing technology and lack of computational software for bioinformatics particularly has led to a “bottleneck,” where data are being produced much faster than can be analyzed (Corlett, 2017; Hohenlohe et al., 2021; Schweizer et al., 2021). Additionally, genomics realistically requires high-performance computing to process and analyze full genomes, often gigabytes of data. Many bioinformatics pipelines and programs require a reasonably high level of computer programming knowledge, therefore outsourcing bioinformatic and computational assistance is a common way to overcome this issue (Corlett, 2017). Whilst there are increasing resources, such as workshops, seminars, and online courses, now available for researchers to upskill on emerging techniques, these are currently underutilized (Hendricks et al., 2018; Schweizer et al., 2021). Increasing the user-friendliness of bioinformatic tools would accelerate the integration of bioinformatics and computer sciences in marine sciences, which will be essential for the efficient development of genomic resources for species conservation (Hohenlohe et al., 2021).
This review highlights the limited genomic resources for chondrichthyan species, with less than 1% having a sequenced genome, as well as the prominent gaps, most notably the seven orders in this class lacking genome representation. Many of the few available genomes are only at a fragmented scaffold level that is insufficient for providing full potential for conservation of these highly threatened taxa. Chromosome-length genome assemblies have higher gene completeness and are therefore more informative in analysis compared to scaffolds, proving to be a more valuable resource. The development of genomic resources, in particular the creation of chromosome-length assemblies, can aid in the conservation and management of chondrichthyan species, such as through population and ecological genomics methods. So far, however, the development of chondrichthyan genomes has largely been for other purposes, such as the study of laboratory species (e.g., bamboo and catsharks), resulting in the majority of the available genomes for this class being species with conservation status of Least Concern or not Endangered. This is important due to the vast applications for chondrichthyan genomes beyond conservation, including evolutionary and developmental biology and biomedical sciences.
Despite the progress of chondrichthyan genomics over the past decade and the potential to quickly generate new knowledge that could directly aid in the conservation of these threatened species, genomic resources for this class remain very limited. There is still a long way to go to match the resources available for other marine clades, and even further to match terrestrial taxa. In molecular science, chondrichthyan species have been underutilized, and likewise, genomics technologies remain limited in marine sciences. Previous investigations and reviews have highlighted the great applications for non-model organism genomic data (Ellegren, 2014; Matz, 2018; Rhie and Project, 2021). Others have clearly demonstrated the need for more effective conservation strategies for sharks and rays, which remain difficult to manage (Dulvy et al., 2017; Stein et al., 2018). However, there has been very limited discussion of utilizing genomic data in the context of chondrichthyan species conservation (Johri et al., 2019).
This review identifies the urgent needs and the prioritization of orders most lacking in genomic resources that also have high percentages of Endangered species, such as the sawfish and guitarfish (Rhinopristiformes) and angel sharks (Squatiniformes). At minimum, baseline data should first be established to include a representative chromosome-length genome for every order within the Chondrichthyes class, considering how different they are from other vertebrate classes, and then for Critically Endangered species. This is a critical first step; however, the full applications for these genomes could have much further potential, such as comprehensive genomic population assessments of chondrichthyan species. This will assist with targets for conservation and increase management effectiveness and efficiency, with resulting conservation assessments able to inform policy decisions.
Going forward, it will be imperative to work across disciplinary boundaries to address emerging global issues like climate change and biodiversity loss. In particular, developing suitable sample collection and storage for genetic study and long-read sequencing as standard practice, as well as taking advantage of all opportunities for sampling will be vital in integrating these biological and molecular fields. The increase in bioinformatic resources becoming available is a promising sign of progress toward multidisciplinary utilization. If these resources become more user-friendly for other disciplines, then these molecular techniques that require high levels of bioinformatic skillsets and computational power for analyses will have higher utility in marine science. Increasing accessibility of molecular and bioinformatic skills will assist in decreasing the resistance and hesitation to fully integrating these fields across different biological sciences. If marine, molecular and computer sciences come together to develop genomic resources for chondrichthyan species, there is hope to conserve their high ecological significance and unique evolutionary history before extinction takes place.
JP: writing. JP, MF, AS, and PK: review and editing. MF, AS, and PK: supervision. All authors have read and agreed to the published version of the manuscript.
AS was supported by a 2020 Pew Fellowship in Marine Conservation and an ARC DP210103091. MF was supported by the Robson and Robertson Research Fellowship. MF and AS were also partly supported by the Gatthaagudu Animal Tracking project with funding from the Jock Clough Marine Foundation and by the Integrated Coastal Analyses and Sensor Technology (ICoAST) project with funding from the Indian Ocean Marine Research Centre, a joint partnership between the University of Western Australia (UWA), the Australian Institute of Marine Science (AIMS), the Commonwealth Scientific and Industrial Research Organisation (CSIRO), and the Department of Primary Industries and Regional Development (DPIRD), Western Australia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We gratefully acknowledge the resources provided by the University of Western Australia and additional computational resources and support from the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia. We are thankful to the Jock Cough Marine Foundation, Mr. and Mrs. James and Marion Taylor, and another donor to the University of Western Australia.
Attard, C. R. M., Beheregaray, L. B., Sandoval-Castillo, J., Jenner, K. C. S., Gill, P. C., Jenner, M. N. M., et al. (2018). From conservation genetics to conservation genomics: a genome-wide assessment of blue whales (Balaenoptera musculus) in australian feeding aggregations. R. Soc. Open Sci. 5:170925. doi: 10.1098/rsos.170925
Baxevanis, A. D., Bader, G. D., and Wishart, D. S. (2020). Bioinformatics. Hoboken, NJ: John Wiley & Sons.
Bernatchez, L., Wellenreuther, M., Araneda, C., Ashton, D. T., Barth, J. M. I., Beacham, T. D., et al. (2017). Harnessing the power of genomics to secure the future of seafood. Trends Ecol. Evol. 32, 665–680. doi: 10.1016/j.tree.2017.06.010
Betancur-R, R., Ortí, G., and Pyron, R. A. (2015). Fossil-based comparative analyses reveal ancient marine ancestry erased by extinction in ray-finned fishes. Ecol. Lett. 18, 441–450. doi: 10.1111/ele.12423
Boisvert, C. A., Johnston, P., Trinajstic, K., and Johanson, Z. (2019). “Chondrichthyan evolution, diversity, and senses,” in Heads, Jaws, and Muscles, eds J. Ziermann, R. Diaz, and Jr. R. Diogo (Cham: Springer), 65–91. doi: 10.1007/978-3-319-93560-7_4
Bourne, S. D., Hudson, J., Holman, L. E., and Rius, M. (2018). “Marine invasion genomics: revealing ecological and evolutionary consequences of biological invasions,” in Population Genomics: Marine, ed. O. P. Rajora (Berlin: Springer), 363–398. doi: 10.1007/13836_2018_21
Ceballos, G., Ehrlich, P. R., Barnosky, A. D., García, A., Pringle, R. M., and Palmer, T. M. (2015). Accelerated modern human-induced species losses: entering the sixth mass extinction. Sci. Adv. 1:e1400253. doi: 10.1126/sciadv.1400253
Chattopadhyay, B., Garg, K. M., Soo, Y. J., Low, G. W., Frechette, J. L., and Rheindt, F. E. (2019). Conservation genomics in the fight to help the recovery of the critically endangered Siamese crocodile Crocodylus siamensis. Mol. Ecol. 28, 936–950. doi: 10.1111/mec.15023
Chen, M., Zou, M., Yang, L., and He, S. (2012). Basal jawed vertebrate phylogenomics using transcriptomic data from Solexa sequencing. PLoS One 7:e36256. doi: 10.1371/journal.pone.0036256
Chen, W. K., Chen, P. C., Liu, K. M., and Wang, S. B. (2007). Age and growth estimates of the whitespotted bamboo shark, Chiloscyllium plagiosum, in the northern waters of Taiwan. Zool. Stud. 46, 92–102.
Cipriano, F., and Palumbi, S. R. (1999). Genetic tracking of a protected whale. Nature 397, 307–308. doi: 10.1038/16823
Cordier, T., Alonso-Sáez, L., Apothéloz-Perret-Gentil, L., Aylagas, E., Bohan, D. A., Bouchez, A., et al. (2020). Ecosystems monitoring powered by environmental genomics: a review of current strategies with an implementation roadmap. Mol. Ecol. 30, 2937–2958. doi: 10.1111/mec.15472
Corlett, R. T. (2017). A bigger toolbox: biotechnology in biodiversity conservation. Trends Biotechnol. 35, 55–65. doi: 10.1016/j.tibtech.2016.06.009
Curtis, T. H., and Sosebee, K. A. (2015). Landings composition of the Northeast U.S. Skate, Rajidae, wing fishery and the effectiveness of prohibited species regulations. Mar. Fish. Rev. 77, 1–8. doi: 10.7755/mfr.77.4.1
Drymon, J. M., and Wells, R. J. D. (2017). Double tagging clarifies post-release fate of great hammerheads (Sphyrna mokarran). Anim. Biotelemetry 5:28.
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Dudchenko, O., Shamim, M. S., Batra, S. S., Durand, N. C., Musial, N. T., Mostofa, R., et al. (2018). The juicebox assembly tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. Biorxiv [Preprint] doi: 10.1101/254797
Dulvy, N. K., Fowler, S. L., Musick, J. A., Cavanagh, R. D., Kyne, P. M., Harrison, L. R., et al. (2014). Extinction risk and conservation of the world’s sharks and rays. Elife 3:e00590.
Dulvy, N. K., Pacoureau, N., Rigby, C. L., Pollom, R. A., Jabado, R. W., Ebert, D. A., et al. (2021). Overfishing drives over one-third of all sharks and rays toward a global extinction crisis. Curr. Biol. 31, 1–15.
Dulvy, N. K., Simpfendorfer, C. A., Davidson, L. N. K., Fordham, S. V., Bräutigam, A., Sant, G., et al. (2017). Challenges and priorities in shark and ray conservation. Curr. Biol. 27, R565–R572.
Dupont, S., Wilson, K., Obst, M., Sköld, H., Nakano, H., and Thorndyke, M. C. (2007). Marine ecological genomics: when genomics meets marine ecology. Mar. Ecol. Prog. Ser. 332, 257–273. doi: 10.3354/meps332257
Ellegren, H. (2014). Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 29, 51–63. doi: 10.1016/j.tree.2013.09.008
Fahmi, A., Oktaviyani, S., Bennett, M. B., Dudgeon, C. L., and Tibbetts, I. R. (2020). Reproductive biology of a bamboo shark as a framework for better fisheries management. Mar. Freshw. Res. 72, 964–977.
Finucci, B., Cheok, J., Ebert, D. A., Herman, K., Kyne, P. M., and Dulvy, N. K. (2021). Ghosts of the deep – biodiversity, fisheries, and extinction risk of ghost sharks. Fish Fish. 22, 391–412. doi: 10.1111/faf.12526
Flowers, K. I., Heithaus, M. R., and Papastamatiou, Y. P. (2021). Buried in the sand: uncovering the ecological roles and importance of rays. Fish Fish. 22, 105–127. doi: 10.1111/faf.12508
Fonseca, E., Machado, A. M., Vilas-Arrondo, N., Gomes-Dos-Santos, A., Veríssimo, A., Esteves, P., et al. (2020). Cartilaginous fishes offer unique insights into the evolution of the nuclear receptor gene repertoire in gnathostomes. Gen. Comp. Endocrinol. 295, 113527–113527. doi: 10.1016/j.ygcen.2020.113527
Fuentes-Pardo, A. P., and Ruzzante, D. E. (2017). Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol. Ecol. 26, 5369–5406. doi: 10.1111/mec.14264
Funk, W. C., Mckay, J. K., Hohenlohe, P. A., and Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 27, 489–496. doi: 10.1016/j.tree.2012.05.012
Galla, S. J., Forsdick, N. J., Brown, L., Hoeppner, M. P., Knapp, M., Maloney, R. F., et al. (2019). Reference genomes from distantly related species can be used for discovery of single nucleotide polymorphisms to inform conservation management. Genes 10:9. doi: 10.3390/genes10010009
Gattuso, J.-P., Magnan, A. K., Bopp, L., Cheung, W. W., Duarte, C. M., Hinkel, J., et al. (2018). Ocean solutions to address climate change and its effects on marine ecosystems. Front. Mar. Sci. 5:337. doi: 10.3389/fmars.2018.00337
Goodwin, S., Mcpherson, J. D., and Mccombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Hara, Y., Yamaguchi, K., Onimaru, K., Kadota, M., Koyanagi, M., Keeley, S. D., et al. (2018). Shark genomes provide insights into elasmobranch evolution and the origin of vertebrates. Nat. Ecol. Evol. 2, 1761–1771. doi: 10.1038/s41559-018-0673-5
Hendricks, S., Anderson, E. C., Antao, T., Bernatchez, L., Forester, B. R., Garner, B., et al. (2018). Recent advances in conservation and population genomics data analysis. Evol. Appl. 11, 1197–1211. doi: 10.1111/eva.12659
Hillary, R. M., Bravington, M. V., Patterson, T. A., Grewe, P., Bradford, R., Feutry, P., et al. (2018). Genetic relatedness reveals total population size of white sharks in eastern Australia and New Zealand. Sci. Rep. 8:2661.
Hinegardner, R. (1976). The cellular DNA content of sharks, rays and some other fishes. Comp. Biochem. Physiol. B 55, 367–370. doi: 10.1016/0305-0491(76)90305-9
Hoegh-Guldberg, O., and Bruno, J. F. (2010). The impact of climate change on the world’s marine ecosystems. Science 328, 1523–1528. doi: 10.1126/science.1189930
Hoencamp, C., Dudchenko, O., Elbatsh, A. M. O., Brahmachari, S., Raaijmakers, J. A., Schaik, T. V., et al. (2021). 3D genomics across the tree of life reveals condensin II as a determinant of architecture type. Science 372, 984–989.
Hoffmann, A., Griffin, P., Dillon, S., Catullo, R., Rane, R., Byrne, M., et al. (2015). A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim. Change Resp. 2:1.
Hohenlohe, P. A., Funk, W. C., and Rajora, O. P. (2021). Population genomics for wildlife conservation and management. Mol. Ecol. 30, 62–82.
Houston, R. D., Bean, T. P., Macqueen, D. J., Gundappa, M. K., Jin, Y. H., Jenkins, T. L., et al. (2020). Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 21, 389–409. doi: 10.1038/s41576-020-0227-y
Inoue, J. G., Miya, M., Lam, K., Tay, B.-H., Danks, J. A., Bell, J., et al. (2010). Evolutionary origin and phylogeny of the modern holocephalans (Chondrichthyes: Chimaeriformes): a mitogenomic perspective. Mol. Biol. Evol. 27, 2576–2586. doi: 10.1093/molbev/msq147
IUCN Red List (2021). The IUCN Red List of Threatened Species. Available online at: https://www.iucnredlist.org (accessed October 20, 2021).
Johri, S., Doane, M. P., Allen, L., and Dinsdale, E. A. (2019). Taking advantage of the genomics revolution for monitoring and conservation of chondrichthyan populations. Diversity 11:49. doi: 10.3390/d11040049
Kadota, M., Nishimura, O., Miura, H., Tanaka, K., Hiratani, I., and Kuraku, S. (2020). Multifaceted Hi-C benchmarking: what makes a difference in chromosome-scale genome scaffolding? Gigascience 9:giz158.
Kelley, J. L., Brown, A. P., Therkildsen, N. O., and Foote, A. D. (2016). The life aquatic: advances in marine vertebrate genomics. Nat. Rev. Genet. 17, 523–534. doi: 10.1038/nrg.2016.66
Koepfli, K.-P., Paten, B., and O’brien, S. J. (2015). The genome 10K project: a way forward. Annu. Rev. Anim. Biosci. 3, 57–111. doi: 10.1146/annurev-animal-090414-014900
Kuguru, G., Maduna, S., Da Silva, C., Gennari, E., Rhode, C., and Bester-Van Der Merwe, A. (2018). DNA barcoding of chondrichthyans in South African fisheries. Fish. Res. 206, 292–295. doi: 10.1016/j.fishres.2018.05.023
Lang, D., Zhang, S., Ren, P., Liang, F., Sun, Z., Meng, G., et al. (2020). Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. GigaScience 9:giaa123.
Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., Crandall, K. A., et al. (2018). Earth biogenome project: sequencing life for the future of life. Proc. Natl. Acad. Sci. U.S.A. 115, 4325–4333. doi: 10.1073/pnas.1720115115
Licht, M., Schmuecker, K., Huelsken, T., Hanel, R., Bartsch, P., and Paeckert, M. (2012). Contribution to the molecular phylogenetic analysis of extant holocephalan fishes (Holocephali, Chimaeriformes). Organ. Divers. Evol. 12, 421–432. doi: 10.1007/s13127-011-0071-1
Lieberman-Aiden, E., Berkum, N. L. V., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 33292, 289–294. doi: 10.1126/science.1181369
Lou, R. N., Jacobs, A., Wilder, A. P., and Therkildsen, N. O. (2020). A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol. doi: 10.1111/mec.16077 [Epub ahead of print].
Marra, N. J., Stanhope, M. J., Jue, N. K., Wang, M., Sun, Q., Bitar, P. P., et al. (2019). White shark genome reveals ancient elasmobranch adaptations associated with wound healing and the maintenance of genome stability. Proc. Natl. Acad. Sci. U.S. A. 116, 4446–4455. doi: 10.1073/pnas.1819778116
Matz, M. V. (2018). Fantastic beasts and how to sequence them: ecological genomics for obscure model organisms. Trends Genet. 34, 121–132. doi: 10.1016/j.tig.2017.11.002
NCBI (2011). LER_WGS_1. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_000238235.1/ (accessed June 28, 2021)
NCBI (2013). Callorhinchus_milii-6.1.3. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCF_000165045.1/ (accessed June 28, 2021)
NCBI (2017). ASM164234v2. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCF_001642345.1/ (accessed June 28, 2021)
NCBI (2018a). ASM360424v1. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_003604245.1/ (accessed June 28, 2021)
NCBI (2018b). Cpunctatum_v1.0. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_003427335.1/ (accessed June 28, 2021)
NCBI (2018c). Storazame_v1.0. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_003427355.1/ (accessed June 28, 2021)
NCBI (2019b). sPriPec2.1.pri. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_009764475.2/ (accessed June 28, 2021)
NCBI (2019a). ASM401019v1. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_004010195.1/ (accessed June 28, 2021)
NCBI (2020a). RhiTyp_1.0. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_013626285.1/ (accessed June 28, 2021)
NCBI (2020b). sAmbRad1.1.pri. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCF_010909765.2/ (accessed June 28, 2021)
NCBI. (2020c). UP_Haf. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_012026655.1/ (accessed June 28, 2021)
NCBI. (2021a). IMCB_Cmil_1.0. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_018977255.1/ (accessed June 20, 2021)
NCBI (2021b). sCarCar2.pri. National Center of Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCF_017639515.1/ (accessed June 20, 2021)
NCBI (2021c). sScyCan1.2. National Center for Biotechnology Information (NCBI). Available online at: https://www.ncbi.nlm.nih.gov/assembly/GCA_902713615.2/ (accessed June 28, 2021)
Oleksiak, M. F., and Rajora, O. P. (2020). Population Genomics: Marine Organisms. Berlin: Springer International Publishing.
Pazmiño, D. A., Maes, G. E., Simpfendorfer, C. A., Salinas-De-León, P., and Van Herwerden, L. (2017). Genome-wide SNPs reveal low effective population size within confined management units of the highly vagile Galapagos shark (Carcharhinus galapagensis). Conserv. Genet. 18, 1151–1163. doi: 10.1007/s10592-017-0967-1
Pennino, M. G., Guijarro-García, E., Vilela, R., Del Río, J. L., and Bellido, J. M. (2019). Modeling the distribution of thorny skate (Amblyraja radiata) in the southern grand banks (Newfoundland, Canada). Can. J. Fish. Aquat. Sci. 76, 2121–2130. doi: 10.1139/cjfas-2018-0302
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Read, T. D., Petit, R. A., Joseph, S. J., Alam, M. T., Weil, M. R., Ahmad, M., et al. (2017). Draft sequencing and assembly of the genome of the world’s largest fish, the whale shark: Rhincodon typus Smith 1828. BMC Genomics 18:532. doi: 10.1186/s12864-017-3926-9
Rhie, A., Mccarthy, S. A., Fedrigo, O., Damas, J., Formenti, G., London, S. E., et al. (2020). Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746.
Rhie, A., and Project, V. G. (2021). Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746.
Ruegg, K., Bay, R. A., Anderson, E. C., Saracco, J. F., Harrigan, R. J., Whitfield, M., et al. (2018). Ecological genomics predicts climate vulnerability in an endangered southwestern songbird. Ecol. Lett. 21, 1085–1096. doi: 10.1111/ele.12977
Savolainen, O., Lascoux, M., and Merilä, J. (2013). Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820. doi: 10.1038/nrg3522
Schatz, M. C., Delcher, A. L., and Salzberg, S. L. (2010). Assembly of large genomes using second-generation sequencing. Genome Res. 20, 1165–1173. doi: 10.1101/gr.101360.109
Schwartz, F. J., and Maddock, M. B. (2002). Cytogenetics of the elasmobranchs: genome evolution and phylogenetic implications. Mar. Freshw. Res. 53, 491–502. doi: 10.1071/mf01139
Schweizer, R. M., Saarman, N., Ramstad, K. M., Forester, B. R., Kelley, J. L., Hand, B. K., et al. (2021). Big data in conservation genomics: boosting skills, hedging bets, and staying current in the field. J. Hered. 112, 313–327. doi: 10.1093/jhered/esab019
Sequeira, A. M. M., Hays, G. C., Sims, D. W., Eguíluz, V. M., Rodríguez, J. P., Heupel, M. R., et al. (2019). Overhauling ocean spatial planning to improve marine megafauna conservation. Front. Mar. Sci. 6:639. doi: 10.3389/fmars.2019.00639
Shafer, A. B. A., Wolf, J. B. W., Alves, P. C., Bergström, L., Bruford, M. W., Brännström, I., et al. (2015). Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 30, 78–87.
Smale, D. A., Wernberg, T., Oliver, E. C. J., Thomsen, M., Harvey, B. P., Straub, S. C., et al. (2019). Marine heatwaves threaten global biodiversity and the provision of ecosystem services. Nat. Clim. Chang. 9, 306–312. doi: 10.1038/s41558-019-0412-1
Stein, R. W., Mull, C. G., Kuhn, T. S., Aschliman, N. C., Davidson, L. N. K., Joy, J. B., et al. (2018). Global priorities for conserving the evolutionary history of sharks, rays and chimaeras. Nat. Ecol. Evol. 2, 288–298. doi: 10.1038/s41559-017-0448-4
Stingo, V. (1979). The chromosomes of the cartilaginous fishes. Genetica 50, 227–239. doi: 10.1007/bf00122048
Ungerer, M. C., Johnson, L. C., and Herman, M. A. (2008). Ecological genomics: understanding gene and genome function in the natural environment. Heredity 100, 178–183. doi: 10.1038/sj.hdy.6800992
Uno, Y., Nozu, R., Kiyatake, I., Higashiguchi, N., Sodeyama, S., Murakumo, K., et al. (2020). Cell culture-based karyotyping of orectolobiform sharks for chromosome-scale genome analysis. Commun. Biol. 3, 652–652.
Van Straalen, N. M., and Roelofs, D. (2011). An Introduction to Ecological Genomics. Oxford: Oxford University Press.
Vargas-Caro, C., Bustamante, C., Bennett, M. B., and Ovenden, J. R. (2017). Towards sustainable fishery management for skates in South America: the genetic population structure of Zearaja chilensis and Dipturus trachyderma (Chondrichthyes, Rajiformes) in the south-east Pacific Ocean. PLoS One 12:e0172255. doi: 10.1371/journal.pone.0172255
Venkatesh, B., Kirkness, E. F., Loh, Y. H., Halpern, A. L., Lee, A. P., Johnson, J., et al. (2007). Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 5:e101. doi: 10.1371/journal.pbio.0050101
Venkatesh, B., Lee, A. P., Ravi, V., Maurya, A. K., Lian, M. M., Swann, J. B., et al. (2014). Elephant shark genome provides unique insights into gnathostome evolution. Nature 505, 174–179. doi: 10.1038/nature12826
Venkatesh, B., Tay, A., Dandona, N., Patil, J. G., and Brenner, S. (2005). A compact cartilaginous fish model genome. Curr. Biol. 15, 82–83.
Wang, Q., Arighi, C. N., King, B. L., Polson, S. W., Vincent, J., Chen, C., et al. (2012). Community annotation and bioinformatics workforce development in concert-little skate genome annotation workshops and jamborees. Database 2012:bar064. doi: 10.1093/database/bar064
Weber, J. A., Park, S. G., Luria, V., Jeon, S., Kim, H. M., Jeon, Y., et al. (2020). The whale shark genome reveals how genomic and physiological properties scale with body size. Proc. Natl. Academy Sci. U.S.A. 117, 20662–20671. doi: 10.1073/pnas.1922576117
Wong, P. B. Y., Wiley, E. O., Johnson, W. E., Ryder, O. A., O’brien, S. J., Haussler, D., et al. (2012). Tissue sampling methods and standards for vertebrate genomics. Gigascience 1:8.
Worley, K. C., Richards, S., and Rogers, J. (2017). The value of new genome references. Exp. Cell Res. 358, 433–438. doi: 10.1016/j.yexcr.2016.12.014
Wyffels, J., King, B. L., Vincent, J., Chen, C., Wu, C. H., and Polson, S. W. (2014). SkateBase, an elasmobranch genome project and collection of molecular resources for chondrichthyan fishes. F1000Res. 3:191. doi: 10.12688/f1000research.4996.1
Yagnesh, M., Durga, F., Raj, D., Rehanavaz, M., and Poojaben, T. (2020). Importance of sharks in ocean ecosystem. J. Entomol. Zool. Stud. 8, 611–613.
Yan, H. F., Kyne, P. M., Jabado, R. W., Leeney, R. H., Davidson, L. N. K., Derrick, D. H., et al. (2021). Overfishing and habitat loss drives range contraction of iconic marine fishes to near extinction. Sci. Adv. 7, 1–11.
Keywords: Chondrichthyes, sharks, rays, conservation, genomics, molecular science, marine management
Citation: Pearce J, Fraser MW, Sequeira AMM and Kaur P (2021) State of Shark and Ray Genomics in an Era of Extinction. Front. Mar. Sci. 8:744986. doi: 10.3389/fmars.2021.744986
Received: 21 July 2021; Accepted: 22 September 2021;
Published: 12 October 2021.
Edited by:
Sílvia C. Gonçalves, Polytechnic Institute of Leiria, PortugalReviewed by:
Nathaniel Jue, California State University, Monterey Bay, United StatesCopyright © 2021 Pearce, Fraser, Sequeira and Kaur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parwinder Kaur, cGFyd2luZGVyLmthdXJAdXdhLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.