 Amaro Saco
Amaro Saco Antonella Panebianco
Antonella Panebianco Sofía Blanco
Sofía Blanco Beatriz Novoa
Beatriz Novoa Angel P. Diz
Angel P. Diz Antonio Figueras
Antonio Figueras
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 05 October 2021
Sec. Marine Molecular Biology and Ecology
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.735309
In recent years, the immune response of mussels (Mytilus galloprovincialis) has been studied at the transcriptomic level against several bacterial infections. As a result, different immune mechanisms have been revealed, including both conserved essential innate pathways and particularities of the mussel immune response according to its nature and environment. However, there is often a lack of functional verification because mussels are a non-model species and because transcriptomic and proteomic information is not always well correlated. In the current study, a high-throughput quantitative proteomics study coupled to LC-MS/MS analysis using isobaric tandem mass tags (TMTs) for protein labeling was employed to study the mussel gill immune response to a Vibrio splendidus bath (waterborne) infection at a functional protein level. A total of 4,242 proteins were identified and quantified, of which 226 were differentially expressed (DEPs) after infection, giving to the study a depth that was lacking in previous proteomic studies of the bivalve immune response. Modulated proteins evidenced an important cytoskeletal disruption caused by bacterial infection. A conserved network of associated proteins was modulated, regulating oxidative stress and NF-kB inflammatory responses and leading to innate immunity effectors. Proteomic results were submitted to an integrated analysis with those obtained in a previous transcriptomic approach with the same infection. Half of all the quantified proteins had a concordant transcriptomic expression trend, but this concordance increased when focusing on the DEPs. The correlation was higher within the immune-related DEPs, and the activation of the conserved NF-kB pro-inflammatory pathway was the main response in both approaches. The results of both techniques could be integrated to obtain a more complete vision of the response.
Mussels (Mytilus galloprovincialis) have great importance in scientific research for different reasons. These bivalves are among the most produced species in aquaculture, having a non-negligible impact on the European economy (Avdelas et al., 2020). Mussels are filter feeders in contact with large amounts and diverse potential pathogens, and despite this, they do not commonly suffer severe mortality episodes. Mussels can use their efficient innate immune system to deal with different pathogenic agents in a specific way (Costa et al., 2009; Balbi et al., 2013). This attribute is supported by the vast genetic variability that characterizes this species, with several expanded immune gene families as immune recognition C1q factors (Gerdol et al., 2011) or pro-inflammatory cytokines as IL-17 (Saco et al., 2021). The recent discovery of mussel genomic organization as a pan-genome revealed presence/absence phenomena in the genetic repertoires of these animals, where a core set of genes was accompanied by great amounts of dispensable and variable genes (Gerdol et al., 2020). Gene families with immune importance are enriched in the dispensable and variable genomic fraction, such as the antimicrobial peptide myticins (Rey-Campos et al., 2020).
To reveal how mussels use their immune system to deal with infections, transcriptomic approaches have been quite successful (Figueras et al., 2019). Classical approaches have studied the transcriptome of the main immune cells, hemocytes, against in vitro and in vivo stimulation with different pathogenic bacteria or pathogen-associated molecular patterns (PAMPs) (Rey-Campos et al., 2019a, b; Moreira et al., 2020). In addition to hemocytes, mucosal immunity should have great importance in filtering bivalves such as mussels. Transcriptomic studies indeed revealed interesting immune expression patterns in more tissues beyond hemocytes (Moreira et al., 2015). A transcriptomic study performed in mussel gills against a bacterial bath (waterborne) infection tried to mimic how mussels recognize an infection from the surrounding water as in natural conditions. The modulation of different pathogen recognition and immune triggering genes revealed that gills may play a natural first step in the activation of the immune response in these animals (Saco et al., 2020). These transcriptomic studies described both essential key immune mechanisms that would therefore be conserved during evolution up to higher vertebrates, as well as particularities adapted to the mussel environment, such as the high variability of certain genes (Rey-Campos et al., 2020), to survive in the ever-changing marine environment.
In addition to transcriptomic studies, more functional approaches have been used to improve our understanding of the mussel immune system. Traditional proteomic analyses consist of two-dimensional electrophoresis (2-DE)-based methods and have proven to be useful in several analyses on mussels and other non-model species. However, high-throughput and more effective techniques emerged, allowing the identification of many more proteins in each analysis and a more accurate relative quantification, such as iTRAQ (isobaric tag for relative and absolute quantitation) and TMT (tandem mass tag) approaches, both coupled to liquid chromatography-tandem mass spectrometry (LC-MS/MS). They allow much larger identification rates, higher precision and accuracy in relative quantification than traditional 2-DE gel assays (Aslam et al., 2017).
Different proteomic studies have been conducted with mussels and other bivalves subjected to different stimuli. Mussel and other bivalve gills have been used in proteomic studies on environmental stress or in studying the bioaccumulation and toxic effects of different water pollutants. Traditional 2-DE analyses identified a limited number of differentially expressed proteins (DEPs) after the analysis of all the protein spots (Manduzio et al., 2005; Ji et al., 2013b; Song et al., 2016; Chen H. et al., 2018). A big leap in the magnitude of the analyses and the number of identified and quantified proteins has been reported with the most recent LC-MS/MS technological advances (Zhang et al., 2015; Sánchez-Marín et al., 2021).
Proteomics has also been employed for the study of the immune response in bivalves, as in the scallop hepatopancreas infected by Vibrio (Huan et al., 2011), for characterization studies of the immune proteins expressed in hemocytes from different bivalves (Campos et al., 2015; Leprêtre et al., 2020), for the study of the immune response in mussel gills to the intramuscular injection of bacteria (Ji et al., 2013a), among other studies (Castellanos-Martínez et al., 2014; Dinguirard et al., 2018; Jiang et al., 2018; Smits et al., 2020). Most likely, the most studied infection model in bivalve proteomics is oyster infection by herpesvirus and virus-like stimuli (Corporeau et al., 2014; Masood et al., 2016; Leprêtre et al., 2021).
Few immune proteomic responses to bacterial infections have been reported in mussels or in other bivalves, and the evolution of proteomic techniques since the 2DE analyses caused some past studies to have severe limitations. The objective of the current work was to carry out a high-throughput proteomic study in gills of the marine mussel Mytilus galloprovincialis subjected to a waterborne infection with Vibrio splendidus, simulating natural infection conditions by a bloom of certain infective bacteria. The infection model followed the same design as a previous transcriptomic experiment, which had revealed key mechanisms in gills in terms of pathogen recognition and activation of the immune response (Saco et al., 2020). Multiplexing of TMT-labeled protein samples coupled to liquid chromatography-tandem mass spectrometry (LC-MS/MS) was applied to this proteomic study, and the results were also complemented with those obtained by the previous transcriptomic approach with the objective of gaining a more complete understanding of the marine mussel response to this infection model.
Adult mussels (Mytilus galloprovincialis) were obtained in May from a mussel farm (Vigo, Galicia, NW Spain) and maintained in open-circuit filtered seawater (FSW) tanks at 15°C with aeration. The mussels were fed daily with Phaeodactylum tricornutum and Isocrhysis galbana. The mussels were used for the experiments after at least 1 week of acclimatization.
Vibrio splendidus (reference strain LGP32) was cultured in tryptic soy agar (TSA) plates at 22°C for 24 h before use. Bacterial suspensions were prepared in FSW before infection. Serial dilutions were made to calculate the number of colony forming units (CFU)/mL 24 h after inoculation.
Waterborne infections were performed by treating mussels with a concentration of 108 CFU/mL V. splendidus in tank water. Control mussels were maintained in another tank with only filtered seawater (FSW). Gill samples from 5 control and 5 infected mussels were taken 24 h after the infection. To perform the sampling, gills were cautiously separated from gonads and mantle and frozen directly at −80°C.
Gill samples were homogenized with several ultrasonication cycles (5-s pulses, with 30-s ice stops and 20% amplitude) in lysis buffer consisting of 7 M urea, 2 M thiourea and 4% CHAPS. Once the tissue was correctly disaggregated and proteins released and solubilized in the buffer, samples were centrifuged (14,000 g, 60 min, 4°C), and the supernatants were collected in 50-μL aliquots and frozen at −80°C.
Protein quantification was performed following the Bradford method in microplates. Both BSA standards and samples were analyzed in triplicate, and the absorbance was read at 595 nm in a BIO-RAD iMarkTM Microplate Reader. After quantification, samples were diluted to an equal concentration of 0.5 μg/μL.
To perform the precipitation step, 600 μL of prechilled acetone (−20°C) was added to 100 μL of each sample. Samples were incubated for 2 h at −20°C to ensure precipitation. After the incubation step, the samples were centrifuged (8,000 g, 10 min, 4°C). The supernatant consisting of acetone was discarded, and the pellet was dried for a maximum of 10 min before adding 100 μL of 100 mM TEAB (triethylammonium bicarbonate). A brief bath sonication was performed to resuspend the pellet. Then, 5 μL of TCEP (Bond-Breaker TCEP Solution) was added, and samples were incubated under soft shaking conditions (300 rpm) for 1 h at 55°C. To alkylate the samples, 5 μL of 375 mM iodoacetamide (diluted in 100 mM TEAB) was added, and the samples were incubated for 30 min at room temperature and under dark conditions. Finally, 10 μL of trypsin (0.1 μg μL–1 in 100 mM TEAB) was added, and the samples were incubated at 37°C overnight with soft shaking at 300 rpm. Because we began protein precipitation with 100 μL of homogenized protein sample at 0.5 μg/μL, we expected to precipitate a maximum of 50 μg of proteins per sample.
Labeling of peptides was performed using the TMT 10plexTM Isobaric Mass Tagging Kit (Thermo Fisher Scientific, Waltham, MA, United States). Each of the 10 TMT tags was used for each of the 10 samples: 5 control (C1–C5) and 5 infected (I1–I5) samples (126 label: C1; 127N label: C2; 127C label: C3; 128N label: C4; 128C label: C5; 129N label: I1; 129C label: I2; 130N label: I3; 130C label: I4; 131 label: I5). Tags (0.8 mg) were dissolved in 90 μL of anhydrous acetonitrile, and 41 μL of each equilibrated tag was added to their corresponding sample after incubation with trypsin. After incubation for 1 h at room temperature, 8 μL of 5% hydroxylamine was added. Samples contained 50 μg of peptides in a volume of 169 μL.
The 10 already labeled samples were combined in the same tube at equal concentrations, for which 10.3 μL was added from each sample, and then the final volume was raised to 300 μL with TFA (trifluoroacetic acid) 0, 1%. The mix consisting of 300 μL with an estimated peptide amount of 30 μg was submitted to peptide fractionation using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific) following the kit protocol. After conditioning and washing the column, a 300-μL sample mix was applied to the column. Elution solutions consisted of a series of 8 solutions with increasing acetonitrile concentrations from 12.5 to 50% supplemented with 0.1% triethylamine. Therefore, 8 fractions or elutions were obtained, and each one of them was divided in 2 aliquots of 150 μL with approximately 2 μg of peptides.
Each 150 μL aliquot was dried using a SpeedVack (Thermo Fisher Scientific), and 2 μg of peptides from each aliquot was suspended in 13 μL of 0.5% formic acid. A brief bath sonication for 10 min was followed by centrifugation (14,000 rpm, 1 min, 10°C). From each aliquot, 10 μL corresponding to 1.5 μg of peptides was analyzed with LC–MS/MS using a Proxeon EASY-nLC II liquid chromatography system (Thermo Fisher Scientific) coupled to an LTQ-Orbitrap Elite mass spectrometer (Thermo Fisher Scientific).
To identify the proteins using the MS/MS spectra, a customized protein database was generated from the six-frame translation of the 162,167 contig sequences from the mussel gill transcriptome performed under the same bacterial bath infection stimulus (Saco et al., 2020). MS/MS spectra were searched against this database using PEAKS Studio v8.0 software (Bioinformatics Solutions Inc., Waterloo, Canada). A contaminant database (CRAPome) was included, as well as a decoy sequence database to calculate the false discovery rate (FDR) in the peptide identification, which was set to 1% FDR for PSMs (Peptide-Spectrum Matches). Trypsin was indicated to be the enzyme for protein cleavage, and only one missed cleavage was allowed. For peptide identification, the precursor and fragment ion mass tolerances were set to 10 ppm and 0.02 Da, respectively. Carbamidomethylation of cysteine and TMT 10 plex were set as fixed modifications, and oxidation on methionine was set as a variable modification. For protein identification, the number of matched peptide sequences was set to ≥ 2, the number of unique peptides to ≥ 1 and the protein identification PEAKS score to ≥ 20.
In addition to the mentioned self-built protein database, for comparative purposes, the same procedure (both the identification step and the downstream analyses) was repeated using the NCBI non-redundant protein database restrained to Mollusca (23/05/2019; 749,599 sequences).
For protein quantification, only unique peptides were considered. The spectrum filter for the reporter ions was set to FDR 1% and quality ≥ 10. Intra-experiment normalization was performed by calculating an equalizer factor to normalize the reporter ion intensities for each TMT 10plex label using the total ion counts per channel. After intra-normalization, a logarithmic transformation was applied to the normalized protein abundance data to meet the normality and homoscedasticity assumptions necessary to carry out parametric statistics, which is a standard procedure for proteomic datasets. Differential expression analysis was performed with a t-test followed by a multi-test correction. These corrections were calculated using SGoF + software v.3.8 (Carvajal-Rodriguez and de Uña-Alvarez, 2011) following the strategy previously described in Diz et al. (2011). Adjusted p-values for false discovery rate (q-values, Storey, 2003) were obtained and set to ≤ 0.05 as the criterion to define DEPs.
Annotation of the identified and quantified proteins was derived from the annotation of the transcriptome used to construct the identification database. This annotation was performed using a Blastx approach on OmicsBox (BioBam Bioinformatics, Valencia, Spain) (Götz et al., 2008) against UniProt/Swiss-Prot with an e-value of 10–3. The annotation of DEPs was manually verified with Blastp against the NCBI non-redundant protein database.
Two enrichment analyses, both of which were performed with generic GO slim terms and with specific GO terms, were performed with Fisher’s exact test and FDR ≤ 0.05. The first analysis considered all the identified and quantified proteins as the test set against the reference set (identification database) to analyze which types of proteins were identified in the proteome (a sort of functional clustering). The other enrichment analysis was performed with the modulated proteins against the reference (all the identified proteins) to associate enriched terms with the response to the infection. Enrichment analyses were represented using a ratio between the weight of each term in the test set and the weight in the reference set. Thus, processes with better test/reference ratio will be of interest as they are more enriched in the studied test set than in the reference set.
A protein interaction network was built using the STRING algorithm (Szklarczyk et al., 2015) and the modulated proteins. Default parameters were employed, including the use of human homologs to the mussel proteins as a reference, a common procedure with non-model species that are not included in the server. Only high confidence interactions were shown (0.7 minimum required interaction score) and disconnected nodes were removed. Enriched biological processes in the network were also analyzed with the STRING algorithm. A second STRING network was constructed, following the same criteria (0.7 minimum score and no disconnected nodes), using homolog proteins from a phylogenetically closer species, the marine invertebrate Strongylocentrotus purpuratus. Both analyses were then compared to check which interactions are recovered using the homologs from each species and which ones are shared by both analyses.
Heatmaps and PCA were constructed using ClustVis (Metsalu and Vilo, 2015). For the PCA, singular value decomposition was used, and for the heatmaps, distances were calculated with the unit variance scaling method. Clustering was performed based on average correlation. Schemes were created with BioRender.com.
Transcriptome analysis performed on gills from the same marine mussel species (M. galloprovincialis) after waterborne V. splendidus infection (Saco et al., 2020) was employed both as a database for proteomic analysis and to retrieve transcriptomic expression of the detected proteins. The experimental design was the same for both approaches, with the same concentration of bacteria in the infection (108 CFU/mL V. splendidus) and the same sampling time (24 h). The transcriptomic analysis was performed with 3 biological replicates for each control and infected group instead of the 5 biological replicates employed in the proteome analysis. Reads accessible from Bioproject PRJNA638821 under the accessions SRR11996464, SRR11996686, and SRR11996723 for control samples and SRR11996734, SRR11996735, and SRR11996743 for infected samples were assembled with the parameters indicated in Saco et al. (2020). Expression of transcriptomic contigs equivalent to the modulated proteins in the proteome was retrieved in transcripts per million (TPMs). Log2 transformation of the fold change values was used for the correlation analyses between proteomic quantification and transcriptomic expression. Linear regression algorithm analyses were made in R.
The use of the specific mussel gill transcriptome (Saco et al., 2020) allowed us to achieve better protein identification rates in the analysis than the NCBI database (Table 1). After the LC-MS/MS analysis, 245,047 MS/MS spectra were obtained, and using the mentioned mussel gill transcriptome as a database, 35,217 peptide spectrum matches (PSMs) were retrieved. A total of 4,388 proteins were identified from 28,141 peptide sequences based on the parameters used. Considering only unique peptides that contained signals from reporter (TMT) ions, 4,242 proteins could be quantified. Protein relative abundance data could be retrieved for those proteins.

Table 1. Protein identification and determined DEPs using the transcriptome database and the mollusc NCBI nr protein database.
Enrichment analyses were performed with all the identified and quantified proteins to reveal a general characterization of what type of proteins dominate the mussel gill proteome. For this purpose, GO Slim terms were employed. This broad overview of the proteome is shown in Figure 1A for the molecular functions, Figure 1B for the cellular components and Figure 1C for the biological processes. More than half of the proteins were related to binding functions, both inside the cell and in the plasma membrane. These proteins are implicated in functions such as response to stress, signal transduction and immune processes, among others. Biological processes of the general proteome were also studied with specific GO terms, without GO slim generalization, specifying the importance of gill proteins in the recognition of exogenous antigens and in the transduction of C-type lectin, Fc-epsilon receptor, interleukin or Wnt signaling pathways, among others (Figure 1D).
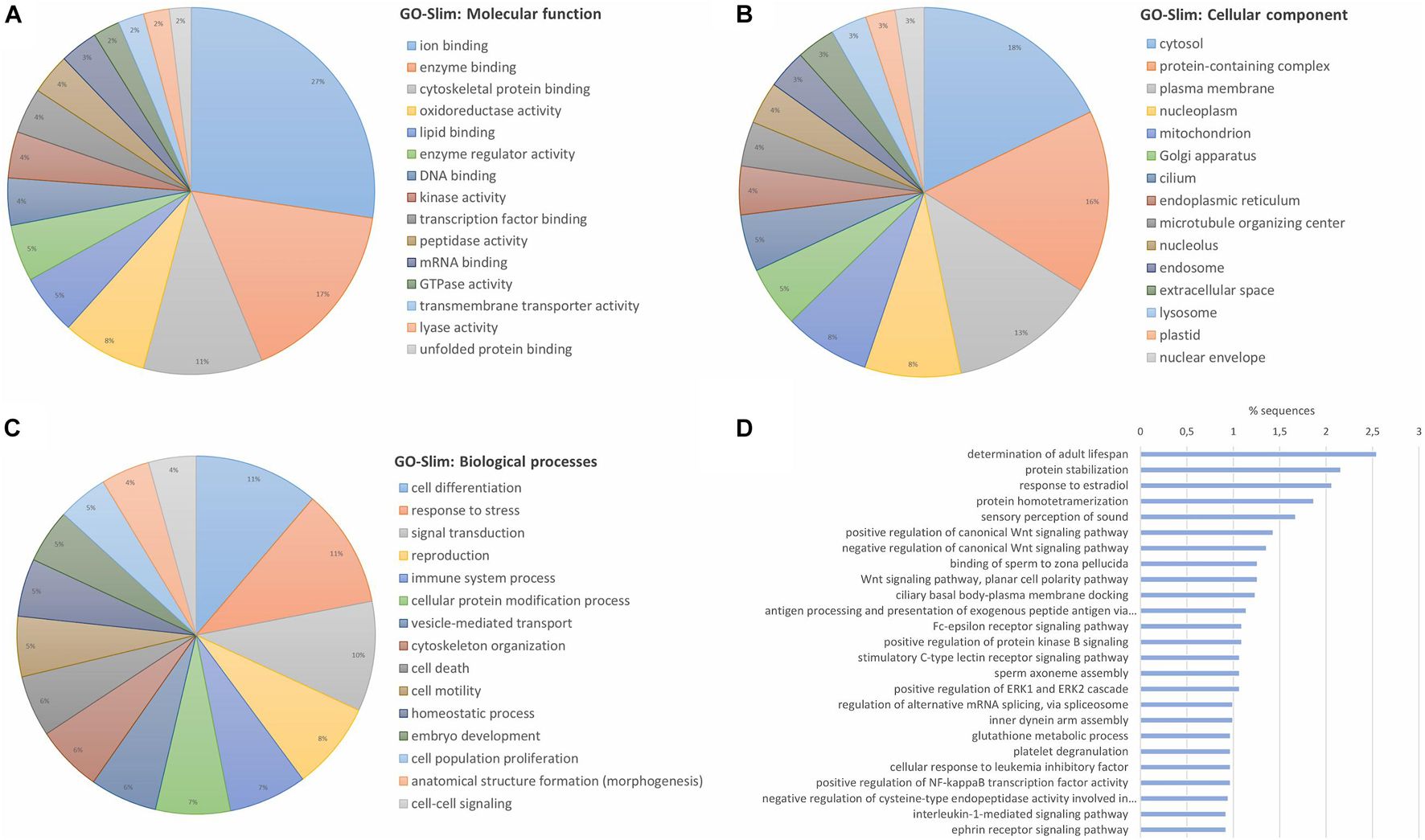
Figure 1. General characterization of gill proteome from the marine mussel (M. galloprovincialis) based on the confidently identified proteins. Identified proteins in the proteomic analysis were submitted to an enrichment analysis with GO-Slim terms to obtain a generic view of the type of proteins that were identified in the gill proteome. The top 15 groups of GO-Slim terms with the highest number of representative proteins are shown for (A) “molecular function”; (B) “cellular component”; and (C) “biological processes.” (D) The most present specific biological processes are also represented without GO-Slim generalization.
Based on the quantification data, after applying the statistical methodology explained, a final number of 226 DEPs were obtained (q-value ≤ 0.05). The DEPs and the rest of the quantified proteins are displayed in Supplementary Table 1. Modulated proteins displayed a clear difference between the two conditions (Figure 2A), and no great variability was observed between biological replicates within the control and infected groups of samples (Figure 2B).
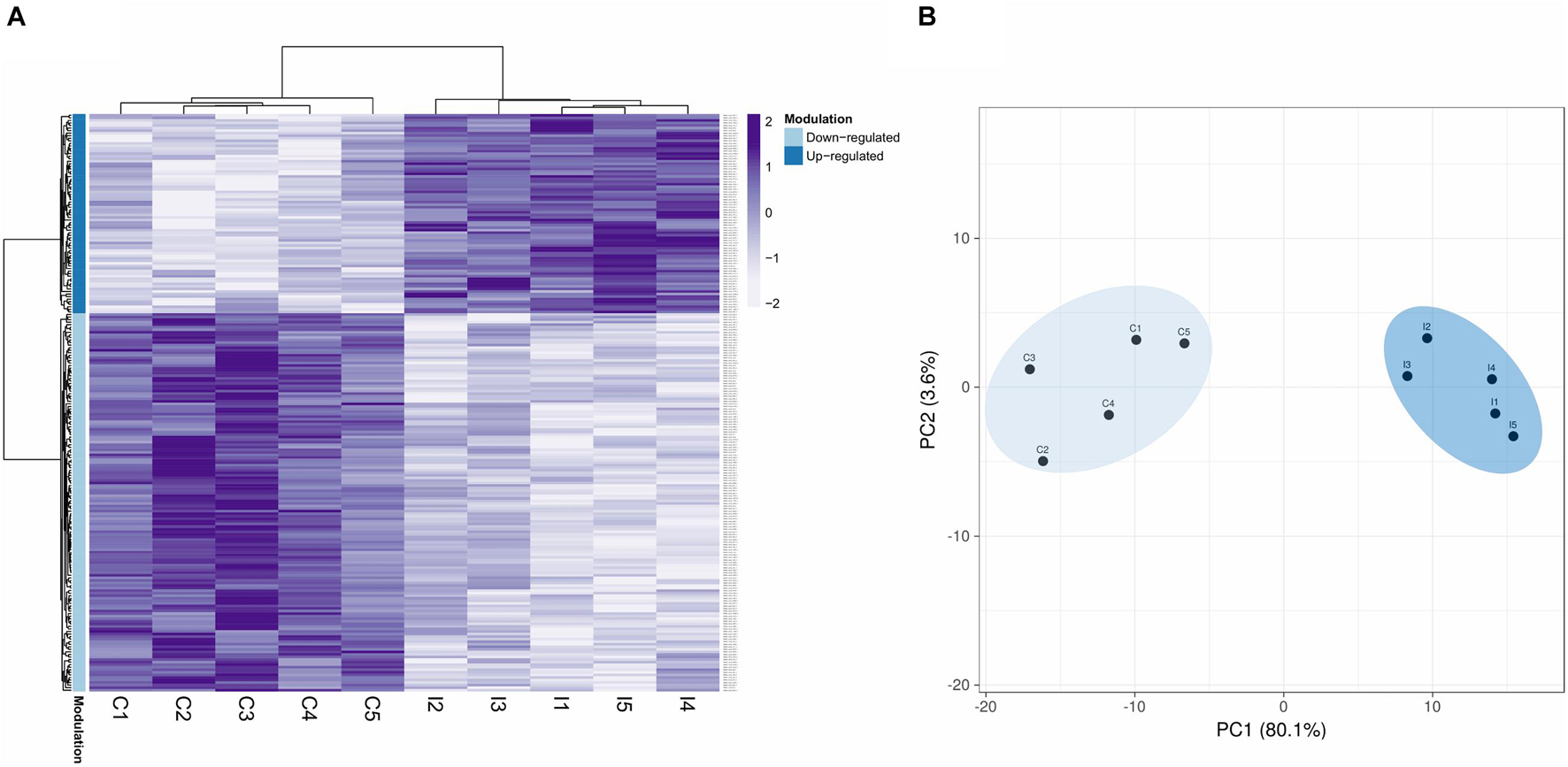
Figure 2. Proteomic modulated response to a bacterial bath infection in mussel gills. (A) Heatmap showing the 226 differentially expressed protein DEPs (q-value < 0.05). Upmodulated and downregulated proteins are indicated with different colors in the legend. (B) Principal component analysis (PCA) shows clear differences between the control (C1–5) and infected (I1–5) groups for these 226 DEPs.
The biological processes of the modulated proteins were first revealed with enrichment analyses of the upregulated and downregulated DEPs. Functions such as activation of innate immune cells/response and response to oxidative stress are among the most common upregulated proteins against infection (Figure 3A). Other apparently less-related immune terms, more focused on regulatory functions, response to ions, and regulation of gene expression, are also directly related to the response to the infection. Cilia movement, as expected in the gills, involved in cell motility was the dominant process in the downregulated proteins (Figure 3B). Immune processes downregulated with the infection were response to cytokine stimulus, response to external stimulus and oxidation-reduction processes.
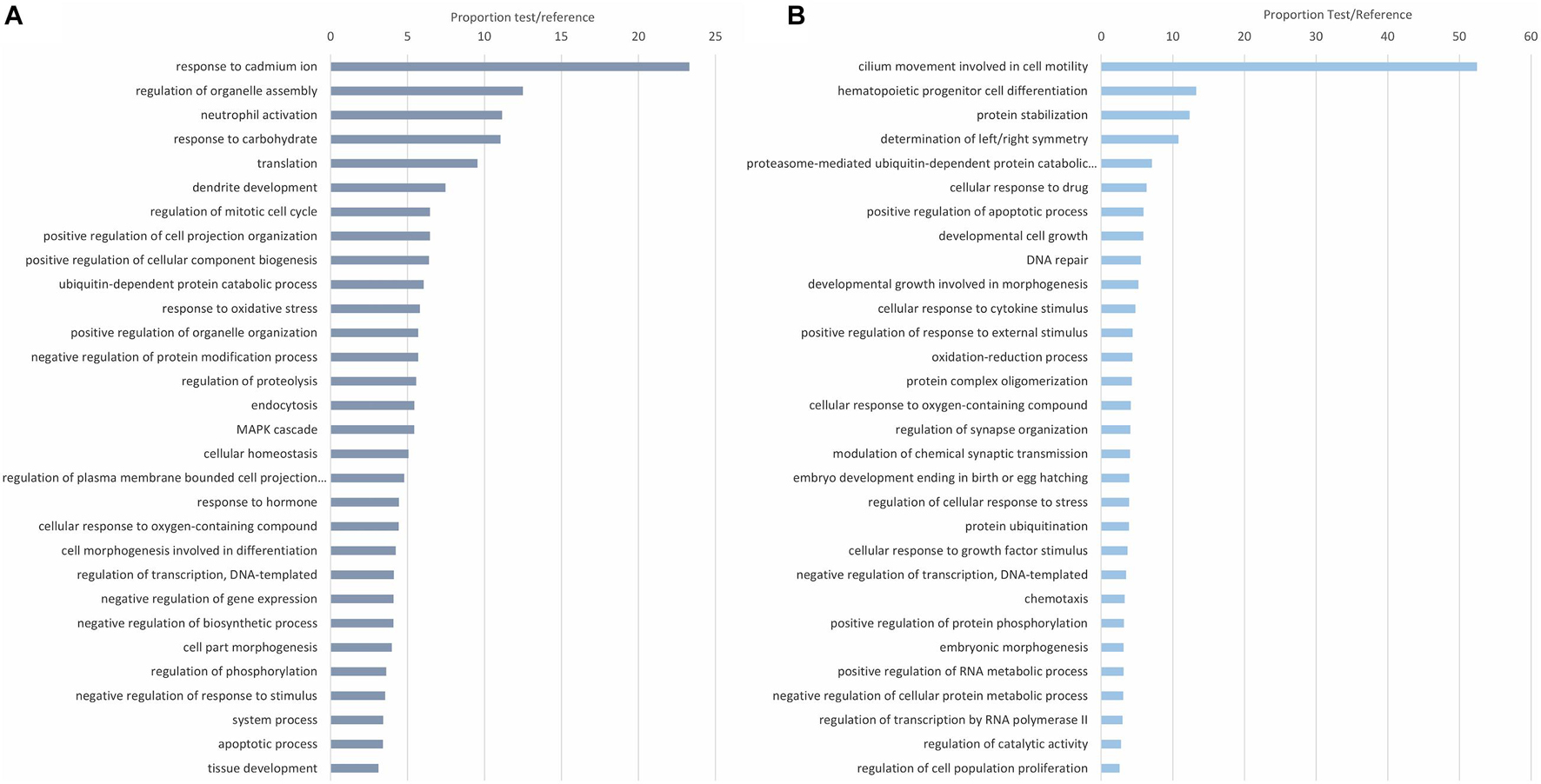
Figure 3. Enrichment analysis showing the top 30 enriched biological processes in the upregulated (A) and downregulated (B) differentially expressed proteins (DEPs). The 30 most overrepresented biological processes (Fisher’s exact test, FDR < 0.05) were represented for both the upregulated (A) and downregulated (B) DEPs. Biological processes are represented in accordance with their test/reference proportion (test = presence of a particular process in the studied DEPs; reference = presence of a particular process in the whole proteome).
Focusing on specific functions of the modulated proteins, many of them were related to the cytoskeleton or to immune pathway regulation, as revealed by the GO Slim biological processes represented in Figure 4A. Table 2 also displays the identity of the DEPs specifically related to the mentioned cytoskeleton-related and immune-related functions that dominated the response. All the cytoskeletal proteins that were modulated (main components such as actin and tubulin, transport proteins such as dynein and many regulators) evidenced general cytoskeletal disruption and reorganization with the infection, which will be discussed in the following sections. DEPs with immune implications were involved mainly in the regulation of the NF-kB pro-inflammatory response and the response to oxidative stress. An interaction net was constructed with human homolog proteins to the mussel DEPs (Figure 4B). The net was significant (PPI enrichment p-value < 1.0e-16), meaning that proteins modulated in the analysis showed a significantly higher degree of interactions within each other than random proteins. The core of the interaction network was enriched in cytoskeletal and immune DEPs (Figure 4B and Supplementary Table 2). The co-modulation of this net of associated proteins in the gill mussel proteome could suggest a conservation of these protein interactions in the mussel innate immune response with respect to the higher vertebrates in which protein interactions are usually described. Using homolog proteins from a phylogenetically closer species, the sea urchin (Strongylocentrotus purpuratus), another significant interaction net was constructed (Supplementary Figure 1A). Interactions detected using sea urchin were mostly shared with the human homologs analysis, since sea urchin interactions information mainly relied on data from homologs in the more studied model species (Supplementary Figure 1B and Supplementary Table 3).
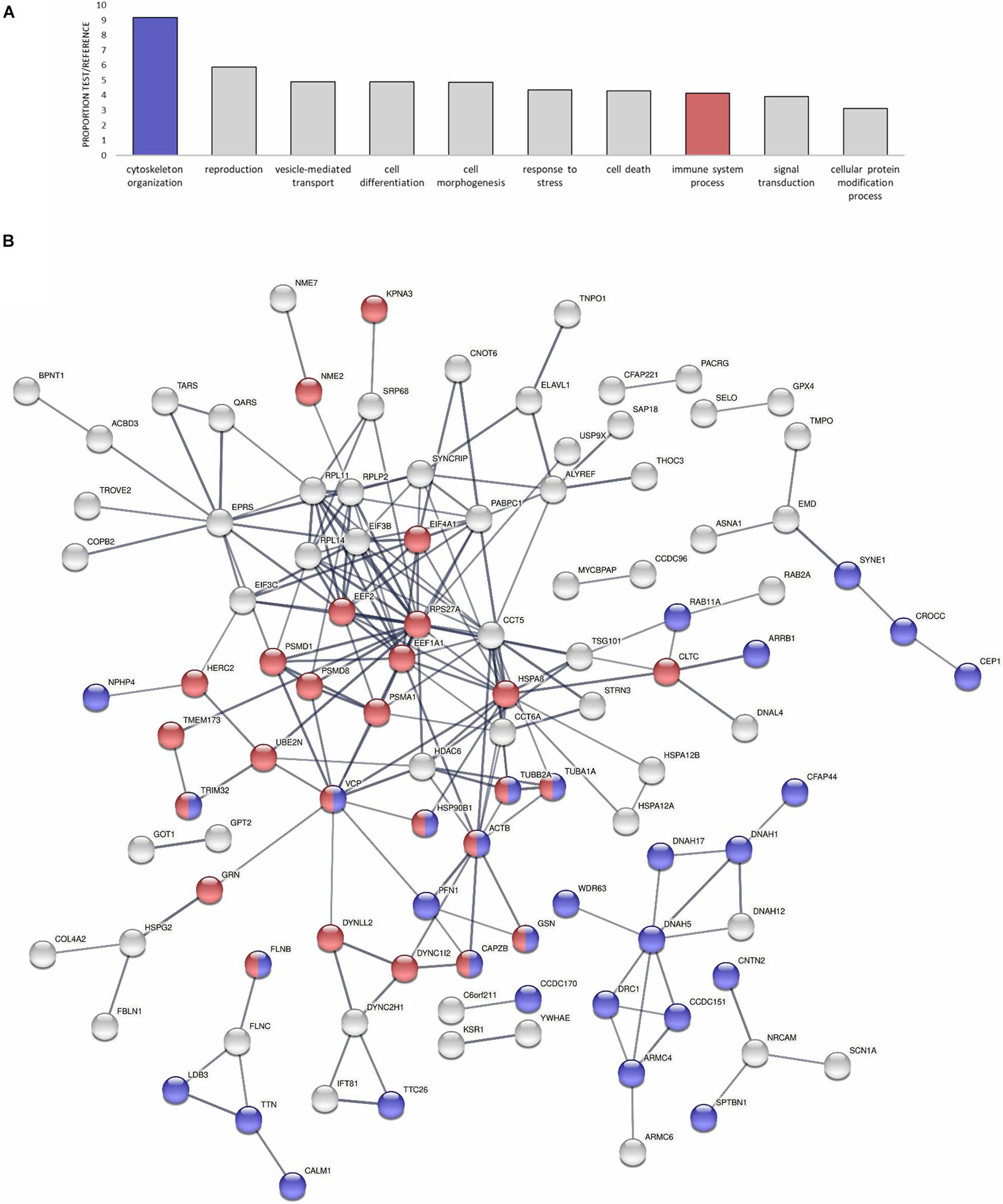
Figure 4. Modulated proteins (DEPs) enriched in cytoskeletal and immune functions could conform to a conserved protein interaction network. (A) GO-Slim Enrichment Analysis of the most present biological processes in the modulated proteins. Biological processes are represented in accordance with their test/reference proportion (test = presence of a particular process in the studied DEPs; reference = presence of a particular process in the whole proteome). Cytoskeletal and immune functions are highlighted because of their importance in the proteomic response. (B) Significant protein interaction net conformed by human homologs to the modulated proteins in the analysis (DEPs). The interaction network is enriched in cytoskeletal and immune-related proteins, which are highlighted in the same color legend as in (A). The thickness of the lines represents the confidence of each interaction. The legend that indicates to which proteins the symbols of the net correspond is in Supplementary Table 2.

Table 2. Differentially expressed proteins (DEPs, q-value < 0.05) and non-modulated proteins of interest.
An effort was made to summarize how the modulated proteins regulate the NF-kB immune response according to their function (Figure 5). The NF-kB inflammatory response is initiated after the recognition of pathogen molecular patterns with pro-inflammatory cytokine signaling. Then, an intracellular pathway liberates NF-kB from its inhibitory factor and allows it to express pro-inflammatory and innate immune effectors. The trend was clear: proteins that can positively regulate the pathway were mainly upregulated, while inhibitory proteins were all downregulated. Figure 5 also displays the response related to oxidative stress, comprising ROS- and NOS-related proteins, as well as the calcium-regulatory response that affects them. This redox stress response caused by the infection would also affect the NF-kB regulatory net, as both responses are associated, mainly through the Pirin protein.
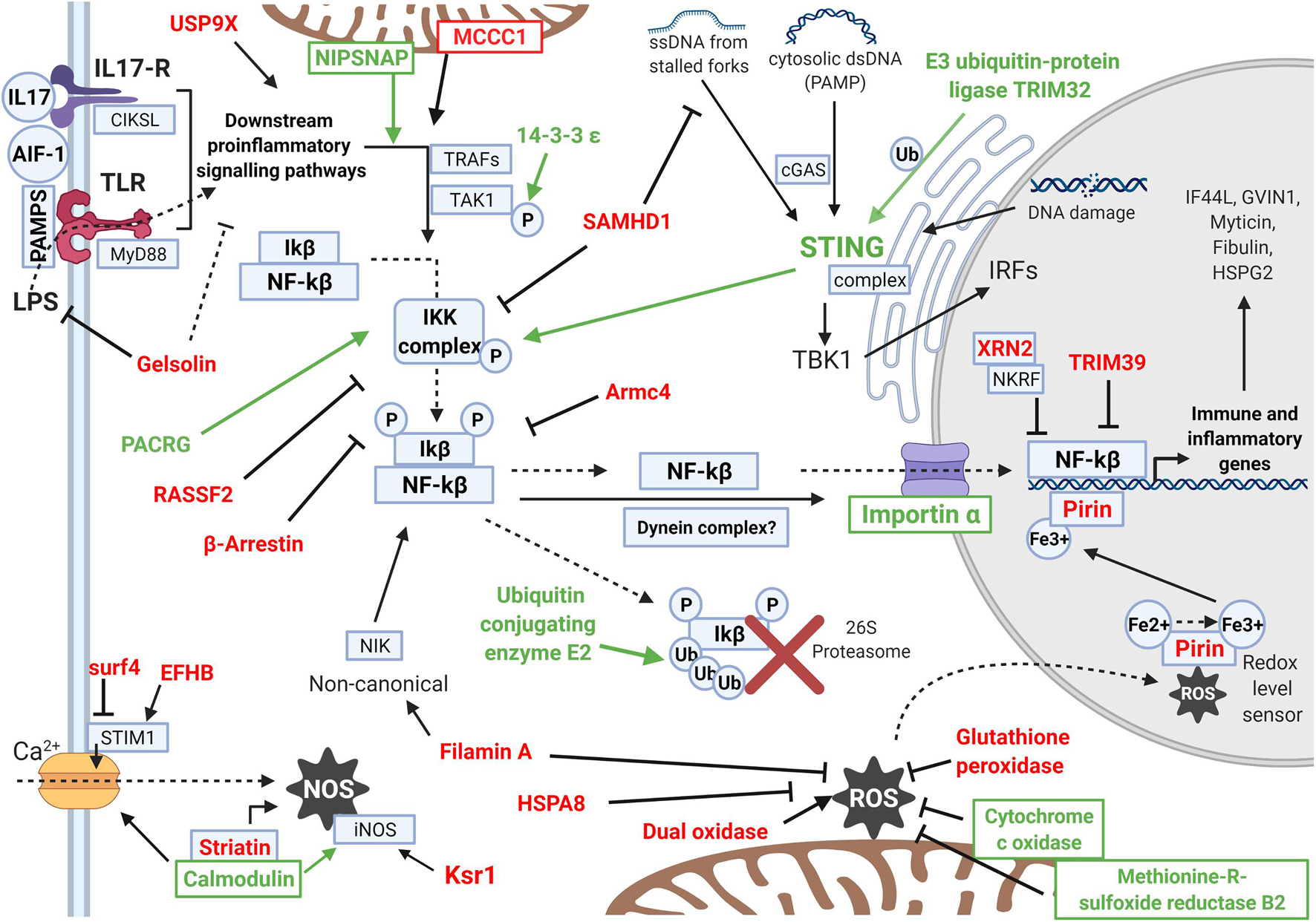
Figure 5. Scheme showing the differentially expressed proteins (DEPs) that regulate the intracellular inflammatory NF-kB pathway and the oxidative and calcium regulation response. The names of the upregulated DEPs are indicated in green, while downregulated DEPs are indicated in red. The activity that each protein exerts is represented with arrows if it is an activation or with an inhibition sign if it is an inhibitory protein. The pathway represented in the scheme follows the same order that is explained in the Discussion. Proteins with functions enhancing the NF-kB pathway were generally upregulated (green), while inhibitory proteins were downregulated in the experiment (red). The scheme shows how the NF-kB response is activated and the oxidative stress is maintained under control.
Other immune-related proteins were also modulated, affecting different functions than previously seen NF-kB and ROS. These proteins are also listed in Table 2 and display more effector roles against infection: Tubby protein homolog stimulates phagocytosis, alanine and aspartate aminotransferases, which are related to apoptosis under bacterial infection, Kinesin-like proteins related to antibacterial lytic granule secretion, Histone deacetylase 6, and granulin, among others.
Gene expression was retrieved from the equivalent transcriptomic analysis (Saco et al., 2020) for all the proteins detected in the proteome. The expression of the equivalent contigs is represented for immune-related DEPs in Figure 6A. There was a general correlation between these modulated immune proteins and their transcriptomic expression trends. Furthermore, the transcriptomic modulated response agreed with the proteomic study in key processes, as will be discussed later. Transcriptomic and proteomic comparison can shed some light on the putative reasons why some expected terms do not appear in the proteomic study, i.e., the great transcriptomic response related to pathogen recognition and cytokine signaling (Saco et al., 2020) was not modulated in the proteomic experiment. Figure 6B shows how contigs corresponding to the detected proteins have higher average expression than the overall transcriptome. The depth of both approaches is a factor to consider as well since the proteome retrieved 4,388 proteins and the transcriptome retrieved 162,167 contigs.
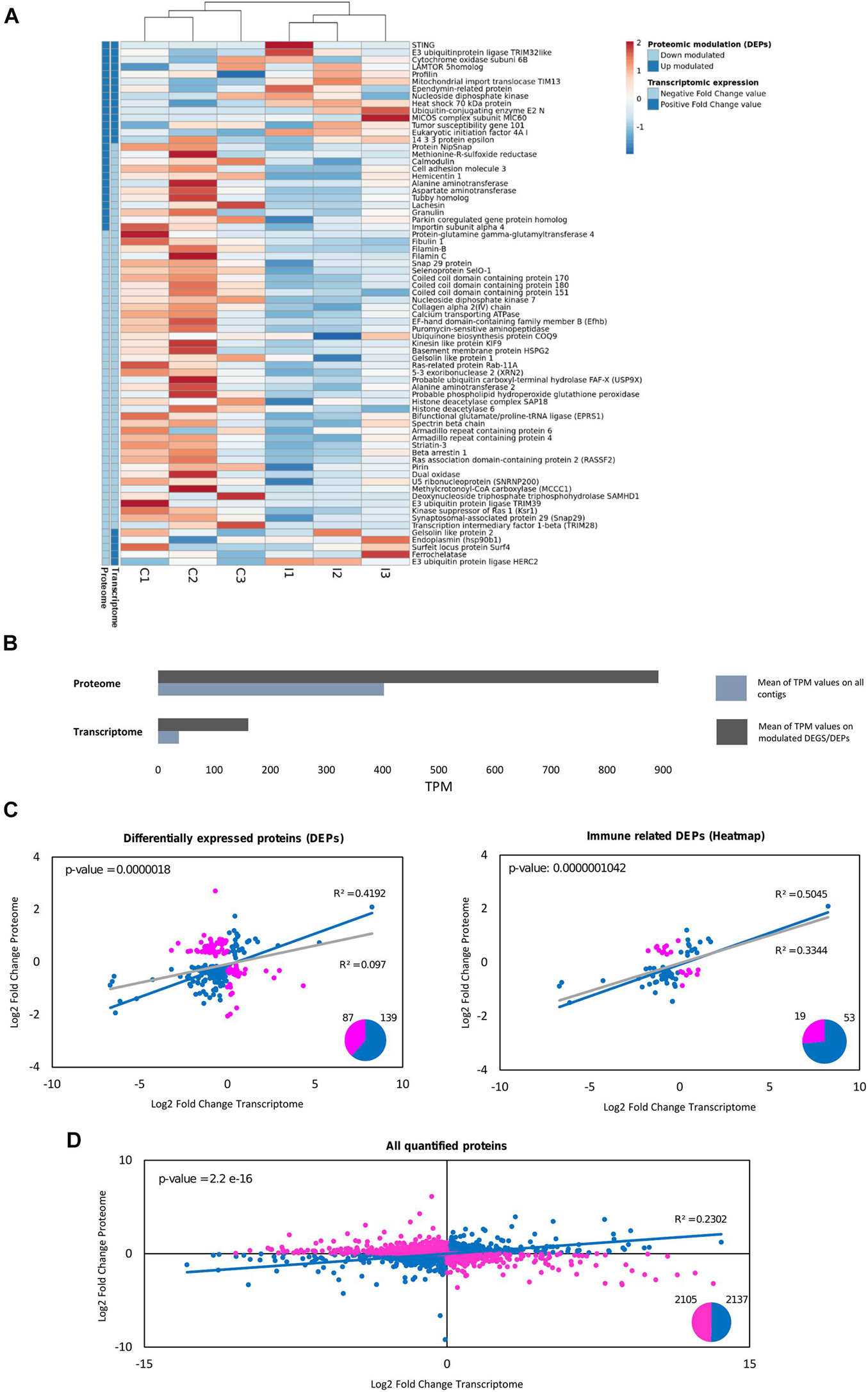
Figure 6. Comparative analysis between proteomic quantification and transcriptomic expression for the differentially expressed proteins (DEPs). (A) For the immune-related DEPs, the mRNA expression of their equivalent contigs was retrieved from a previous transcriptomic study in gills against the same bath infection, which was also used as a database for proteomic identification (Saco et al., 2020). The heatmap shows the expression in TPMs (transcripts per million) for these immune DEPs in each individual sample used for the transcriptomic analysis (three biological replicates for each group: control and infected). The legend in the heatmap indicates for every protein if their fold change values (both in the proteome and in the transcriptome) are positive (dark blue) or negative (light blue). (B) Average TPM values are represented for the whole transcriptome and for the differentially expressed contigs, as well as for the equivalent contigs to the proteins quantified in the proteome and the DEPs. How the proteins detected in the proteome correspond to contigs with higher average expression than the overall transcriptome (it is more possible to identify in proteomics a term with high transcriptomic expression than one with low expression) can be seen. (C) Correlation analysis between the log2-fold change values of the proteomic quantification and the transcriptomic expression of the DEPs. From the total number of 226 DEPs, 87 were discordant gene-protein pairs, indicated in pink (different trend between both approaches), and 139 (61%) were concordant gene-protein pairs, indicated in blue (same expression trend between both approaches). The R-squared value when considering all the DEPs (gray line) and when considering the concordant gene-protein pairs (blue line) are represented, as well as the p-value of the fold change value correlation for the concordant gene-protein pairs. The same correlation analysis was performed considering only the immune-related DEPs, the same ones that were used in (A). How the correlation increases with certain types of modulated genes (immune functions) can be seen, while it is highly decreased in other functions (cytoskeletal-related DEPs, with R2 = 0.0641; data not shown). (D) Correlation analysis between the log2-fold change values of the proteomic quantification and the transcriptomic expression for all the quantified proteins in the proteome. The regression analysis considering the whole proteome was not significant. From the total number of 4,242 quantified proteins, 2,105 were discordant gene-protein pairs (pink), while 2,137 were concordant gene-protein pairs (blue). All the concordant gene-protein pairs (blue dots) were used for the represented regression (blue line), and the R2 and p-value are indicated for this analysis with concordant terms.
Correlation analyses between the quantitative proteomic and RNA-Seq transcriptomic experiments revealed that 139 out of 226 DEPs (60%) were concordant gene-protein interactions (gene expression and protein quantification fold change values with the same trend). Fold change values for these concordant DEPs had a significant correlation of R2 = 0.4192 (Figure 6C). The correlation was more noticeable within the modulated immune proteins (Figure 6C), which is especially important since DEPs related to other functions, such as the cytoskeleton, showed null correlation (R2 = 0.0641). When focusing on the entire proteome characterization instead of the modulated response, the agreement decreased, with half the terms concordant (2,137 out of 4,242 quantified proteins) and with less correlation in the magnitudes of their fold changes (Figure 6D). The complementarity between proteomic and transcriptomic experiments for the immune implicated DEPs supposes a cross-validation for the implication of certain biological responses to the infection, as NF-kB and the oxidative responses. Other groups of proteins/transcripts did not show complementarity, as the cytoskeletal DEPs in the proteome or the modulated immune recognition response in the transcriptome. Immune recognition proteins of interest were identified and quantified in the proteome but they were not modulated in the study (listed in Table 2). Relative quantification of some of these important but non-modulated proteins is shown in Supplementary Figure 2, along with their corresponding transcriptomic expression: AIF-1, Myd88 and a toll-interacting protein.
After the LC-MS/MS analysis, a mussel gill proteome with high protein identification and quantification rates was obtained. Identification rates in proteomic analyses, especially in non-model species, are improved by using customized databases from appropriate and specific transcriptomes, as we did, rather than using general databases such as NCBI (Romero et al., 2019). The number of identified proteins is similar to the number of proteins reported in other studies in bivalves following high-throughput proteomic approaches based on ITRAQ or TMT sample labeling (Zhang et al., 2015; Sánchez-Marín et al., 2021)., far above the numbers obtained in traditional 2-DE assays (Manduzio et al., 2005; Ji et al., 2013a; Song et al., 2016; Chen H. et al., 2018). General characterization of the mussel gill proteome revealed functional information on terms that are typically found in gill transcriptomes, in particular the biological processes related to recognition and signaling pathways (Moreira et al., 2015). The proteins identified corroborated and expanded the results of a previous 2-DE proteomic characterization of the M. edulis mussel gill (Rocher et al., 2015).
In the current work, the study for the first time of the proteomic modulated response to a bacterial waterborne infection with Vibrio splendidus in mussel gills demonstrated that modulation against the infection occurs not only at the transcriptomic level but also at the protein scale. The study of the same infection model by the combination of proteomic and transcriptomic approaches reported different agents taking part in the same response: the main modulated genes in the transcriptome were focused on pathogen recognition terms and cytokines activating the NF-kB inflammatory response (Saco et al., 2020), while the proteome was dominated by proteins that positively regulate the activation of the NF-kB pathway at an intracellular level.
Agreement between transcriptomic and proteomic analyses is not always obtained. High-throughput proteomic analysis of oyster response to viral-like simulation with dsRNA revealed disparities between transcriptomic and proteomic analyses (Masood et al., 2016). Discrepancies had been observed before in gills of mussels with bacteria injected in the muscle but in a lower number of proteins/genes in the comparison because of the 2D analyses (Ji et al., 2013b). Other studies have reported a generally good correlation between transcriptomic and proteomic results, such as in the genes/proteins expressed in the sperm of different mussel species, even though not for all cases (Romero et al., 2019). In our study, because of using the indicated transcriptome as a database for protein identification, it was possible to recover the transcriptomic expression of the contigs that corresponded exactly to the proteins identified and quantified. There was a greater concordance in terms of expression trends between both techniques when focusing on DEPs (60% concordant terms with R2 = 0.4) than with the complete proteome (50% concordant terms with R2 = 0.2). When classifying these DEPs according to their function, the correlation was observed to be much more important for certain functions, such as immune proteins (70% concordant terms with R2 = 0.5), while it was null for the cytoskeleton-related DEPs (60% concordant terms with R2 = 0.0641). The fact that the transcriptomic-proteomic complementarity within a certain experiment is better for some specific functions is consistent with what has been observed in other studies (Xiong et al., 2015; Romero et al., 2019) and points to a functional cross-validation of certain biological responses to the stimulus.
The modulation of cytoskeletal proteins was a clear result in the proteomic analysis and was not among the modulated responses in the transcriptomic study of the same infection model. The presence of these proteins in the analysis, according to what the lack of agreement with the transcriptomic results points out, would not be due to a modulation in the gene transcription and subsequent protein production. These proteins would be modulated because of the physical effects of infective bacteria disrupting the cells and defensive cells (hemocytes) migrating to the infection site (Wu et al., 2013; de Souza Santos and Orth, 2015). Cytoskeleton disruption is a direct consequence of infective bacteria entering host cells (Barbieri et al., 2002; Yoshida and Sasakawa, 2003). The Vibrio splendidus strain employed has been described to use OmpU porins to attach and invade mollusc cells, which would subvert the host cell actin cytoskeleton, causing this general cytoskeleton disruption (Duperthuy et al., 2011). Revising mussel and other bivalve proteomes studying Vibrio and other different infection stimuli, the detection of cytoskeletal proteins among the modulated proteins is recurrent (Huan et al., 2011; Wu et al., 2013; Corporeau et al., 2014; Zhang et al., 2014; Wang et al., 2017). Within the cytoskeletal-related DEPs in our work, associated proteins that could interact with each other were found, as the homolog proteins interaction network revealed. Tubulin is modulated and is known to be the major binding site of bacterial peptidoglycans, which can regulate its polymerization (Dziarski et al., 2000). Tubulin-related proteins such as microtubule organizing center proteins and tubulin polymerization-promoting proteins were also modulated. Modulated dynein proteins have also been described to interact with bacteria, particularly with the Pilin protein of gram-negative bacteria, such as Vibrio splendidus (Kausar et al., 2013). However, most of the cytoskeletal DEPs were related to actin modulation, i.e., thymosin (a regulator of actin polymerization), nephrocystin-4 (actin network regulation in multi-ciliated epithelial cells, such as mussel gills), Cdc42-interacting protein 4 (which reorganizes the actin cytoskeleton during endocytosis), F actin capping protein and even cytoplasmic actin protein. Profilin is another DEP regulator of actin polymerization that has been directly related to the actin-based intracellular motility of different bacteria (Geese et al., 2000; Mimuro et al., 2000; Liverman et al., 2007), so it has been typically modulated with bacterial challenges in a skin mucus cod proteome (Rajan et al., 2013) and in bivalve transcriptomes (Somboonwiwat et al., 2006; Araya et al., 2010).
Cytoskeletal disruption caused by infection affects adhesion, phagocytosis and oxidative burst responses in host cells (Tanguy et al., 2013). Modulated proteins related to all these functions were found. In a proteomic study of oysters infected with OsHV-1 herpesvirus, general cytoskeletal disruption (actin, dynein, F actin capping protein) was associated with oxidative stress (modulation of catalase, glutathione-S-transferase, superoxide dismutase). Actin could be related to that oxidative response, since the carbonylation and glutathionylation of this protein have been related in Mytilus edulis with a recognition system of oxidative stress (McDonagh et al., 2005). It has been reported that “decreased actin dynamics cause depolarization of the mitochondrial membrane and an increase in reactive oxygen species (ROS) production” (Gourlay et al., 2004). In accordance with these previous reports, our proteomic study on infected mussel gills also reported an important response related to oxidative stress along with cytoskeletal modulation. Upmodulated cytochrome c oxidase subunit 12 and methionine-R-sulfoxide reductase B2 proteins could also be implicated in the regulation of ROS. Oxidative proteins such as dual oxidase were downregulated, as were antioxidant proteins such as hydroperoxide glutathione peroxidase. Additionally, related to oxidative stress situations is the alteration of calcium homeostasis (Görlach et al., 2015). The modulation of oxidative/antioxidant and calcium-related proteins is common in previous bivalve proteomic studies, not only under infection conditions but also against stress stimuli of different kinds (Manduzio et al., 2005; Ji et al., 2013b; Zhang et al., 2015; Chen H. et al., 2018; Franco-Martínez et al., 2018). Calmodulin is an upregulated DEP that controls the function of numerous enzymes, ion channels and other proteins through Ca2+ regulation. The Ca2+ flux regulated by calmodulin is necessary for NOS production in the immune response (Tagliazucchi and Conte, 2005; Ma et al., 2008). However, Striatin-3, an associated protein that directly binds calmodulin and is responsible for the activation of endothelial NO synthase (Lu et al., 2004), was downregulated. The kinase suppressor of Ras Ksr1 was also downregulated in infected samples. Ksr1 is responsible for the assembly of inducible nitric oxide (NO) synthase (iNOS) upon bacterial infections (Zhang et al., 2011). NO production would therefore be inhibited to avoid increasing oxidative stress conditions. Proteins related to the regulation of STIM1-dependent calcium entry (Surfeit locus protein 4 and EF-hand domain-containing family member B) were downregulated as well.
As mentioned before, NF-kB regulation was the key response detected in the analysis to the infective situation caused by the bacteria. The results showed how many of those modulated proteins could interact with each other in what would be an NF-kB regulatory network, also highlighting associations with other DEPs implicated in different functions. The regulation of the NF-kB inflammatory response is essential for the immune response; therefore, it is conserved from ancestral molluscs as mussels to the innate component of the immune response in superior vertebrates. Several pathogen recognition proteins that could recognize the bacteria and initiate this response were modulated in the transcriptome (Saco et al., 2020) and were identified but not modulated in the proteome: fibrinogen-like proteins or FREPs, different types of lectins, C1-q containing proteins, and allograft inflammatory factor-1 (AIF-1). AIF-1 is a pro-inflammatory cytokine implicated in immune activation in molluscs (De Zoysa et al., 2010; Zhang et al., 2013) and can activate the NF-kB response (Egaña-Gorroño et al., 2019; Zhou et al., 2019). Immune recognition and signaling terms dominated the modulated response in the transcriptomic study of the same infection model, but they were not modulated in the current proteomic approach. The lack of concordance between both approaches concerning this aspect of the response can be due to the different depth of each technique, to the different technique that is used to detect each type of molecule (proteins and transcripts) or to biological reasons.
Despite this lack of concordance, the modulated DEPs completely covered the entire pathway of the NF-kB response. Downmodulated Gelsolin-like protein 1 and Gelsolin-like protein 2 are calcium-regulated and actin-associated proteins. Gelsolin proteins can bind LPS from the gram-negative Vibrio membrane, resulting in neutralization of LPS effects, including cytoskeletal actin remodeling and NF-kB response (Bucki et al., 2005, 2008). The clear downmodulation of gelsolin would support the responses observed in the proteome. In an unstimulated context, NF-kB is captured in the cytoplasm by IκB proteins (inhibitors of NF-kB). The NF-kB activation pathway is activated after pathogens are recognized (TLR recognition) or inflammatory signals arrive at the cells (inflammatory cytokines such as AIF-1 and IL-17) (Liu et al., 2017). Then, the NF-kB pathway is activated by different transductor proteins also identified in the proteome, such as MyD88 and TRAF proteins. Scaffold mitochondrial proteins implicated in pathway transduction were modulated, such as NIP-SNAP and MCC1 (Cao et al., 2016; Yamamoto et al., 2017). The phosphorylation of TAK-1 is another signaling step, and it is enhanced by the upregulation of the 14-3-3 protein, which is directly associated with the upregulation of calmodulin (Luk et al., 1999; Somboonwiwat et al., 2010; Zuo et al., 2010).
The transduction pathway results in the phosphorylation and subsequent activation of IκB kinase (IKK). IKK phosphorylates two serine residues of the IκB protein, leading to ubiquitination and degradation by the proteasome and releasing NF-kB. This crucial step is activated in the proteomic response, since proteins that would enhance the process are upregulated (PACRG and the associated proteins STING and TRIM32) (Albor et al., 2006; Dunphy et al., 2018; Meschede et al., 2020) and inhibitory proteins of the phosphorylation and degradation of IκB are all downregulated: SAMHD1, RASSF2, β-Arrestin (Witherow et al., 2004; Song et al., 2012; Chen S. et al., 2018). The upregulated ubiquitin-conjugating enzyme E2 is implicated in the elimination of phosphorylated IκB in the proteasome, and its implication in the NF-kB response has already been reported in bivalves (Hatada et al., 2000; Liu et al., 2019). Released NF-kB is transported to the nucleus, and this process in certain cell types is reported to be carried by a dynein complex (Mikenberg et al., 2007). Translocation of released NF-kB into the nucleus is regulated by importin α, which is also upregulated in the proteome, in a nuclear localization signal (NLS)-dependent process (Fagerlund et al., 2005, 2008). Importin α is an adapter protein of KPNB1, the major nuclear receptor for NF-kB import (Liang et al., 2013).
Once NF-kB has been translocated into the nucleus, we found modulation of the Pirin protein, a transcriptional co-regulator of NF-kB. Pirin facilitates NF-kB binding to target genomic regions in a redox-state-dependent manner: it is more active when oxidized in its ferric form (Liu et al., 2013) under oxidative conditions as the infection would unleash. Modulation of Pirin quantity has also been associated with NF-kB activity inside the nucleus (Brzóska and Kruszewski, 2010; Licciulli et al., 2010). Two proteins implicated in the inhibition of NF-kB transcriptional activity are downregulated in the proteome: the E3 ubiquitin-protein ligase TRIM39 and 5′-3′ exoribonuclease 2 (XRN2) (Rother et al., 2016; Suzuki et al., 2016).
Proteins corresponding to typical downstream genes whose expression can be regulated by NF-kB were identified as well: HSPG2, Fibulin-1, IF44L, and GVIN1. These proteins are implicated in effector functions against infection. Myticins were also identified in the proteome. Myticins are antimicrobial peptides showing cytokine-like functions and are the most highly expressed genes in mussel defensive cells and hemocytes (Mitta et al., 1999; Balseiro et al., 2011). Antimicrobial peptide expression in bivalves has been demonstrated to be regulated by NF-kB (Oyanedel et al., 2016). More effector proteins were found to be modulated in the experiment. Adhesion-related proteins, commonly implicated in bivalve immune responses and related to cell migration at the site of infection, were modulated: fibulin-1, hemicentin, cell adhesion molecule 3, lachesin (Gueguen et al., 2003; Xu and Faisal, 2008; Xu et al., 2018) and filamin A, which are also implicated in NF-kB and ROS responses (Muscolini et al., 2011; Uotila et al., 2017). Phagocytosis-related Tubby protein homolog and granulin are also implicated in effector actions against infection (Caberoy et al., 2010; Wu et al., 2018; Hambrook et al., 2019).
The proteomic approach contributed with a complementary vision to the transcriptomic results, adding a functional validation of the processes indicated by the modulation of gene expression. Proteomic results described effects caused by bacterial infection in the host cytoskeleton that were previously undetected in the transcriptomic analysis. Furthermore, a net of proteins acting together was described, linking cytoskeletal disruption with the main modulated responses: the regulation of oxidative stress and the inflammatory NF-kB axis. The induction of the NF-kB response after the pathogen recognition steps is in agreement with previous results at the transcriptomic level in a similar experiment. In fact, the correlation between proteomic quantification and transcriptomic expression was higher within these immune DEPs, validating this response at two different levels of gene expression. Discrepancies were also observed, since the modulated response with each approach was based on different terms that would take part in the same response pathway: the transcriptome reported receptors and cytokines that would activate the NF-kB response, while in the proteome, a network of proteins that would activate the intracellular pathway was obtained. Because of this, an enriched and more complete overall vision of the mussel response to this natural infection model was obtained with the combination of both approaches.
The datasets presented in this study can be found in online repositories. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Deutsch et al., 2020) via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD028043 and 10.6019/PXD028043.
The Mediterranean mussel, M. galloprovincialis, is not considered an endangered or protected species in any international species catalog, including the CITES list (www.cites.org), and it is not included in the list of species regulated by the EC Directive 2010/63/EU. Therefore, no specific authorization is required to work on mussel samples.
BN and AF conceived and designed the project. APD, AS, SB, and AP performed the proteomic analyses. APD, AS, and AF performed the bioinformatic analyses. AS wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the Spanish AEI/EU-FEDER (RTI2018-095997-B-I00); the European Regional Development Fund (ERDF) Interreg V Spain—Portugal (0474_BLUEBIOLAB); the Consellería de Economía, Emprego e Industria—GAIN, Xunta de Galicia, project IN607B 2019/01 and EU H2020; project VIVALDI (678589); Xunta de Galicia (ED431C 2020/05) and FONDOS FEDER. AS was supported by a Spanish AEI/EU-FSE predoctoral contract PRE2019-090760.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.735309/full#supplementary-material
Albor, A., El-Hizawi, S., Horn, E. J., Laederich, M., Frosk, P., Wrogemann, K., et al. (2006). The interaction of piasy with Trim32, an E3-Ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H. promotes piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFκB. J. Biol. Chem. 281, 25850–25866. doi: 10.1074/jbc.M601655200
Araya, M. T., Markham, F., Mateo, D. R., McKenna, P., Johnson, G. R., Berthe, F. C. J., et al. (2010). Identification and expression of immune-related genes in hemocytes of soft-shell clams, mya arenaria, challenged with vibrio splendidus. Fish Shellfish Immunol. 29, 557–564. doi: 10.1016/j.fsi.2010.05.017
Aslam, B., Basit, M., Nisar, M. A., Khurshid, M., and Rasool, M. H. (2017). Proteomics: technologies and their applications. J. Chromatogr. Sci. 55, 182–196. doi: 10.1093/chromsci/bmw167
Avdelas, L., Avdic-Mravlje, E., Borges Marques, A. C., Cano, S., Capelle, J. J., Carvalho, N., et al. (2020). The decline of mussel aquaculture in the European Union: causes, economic impacts and opportunities. Rev. Aquac. 13, 91–118. doi: 10.1111/raq.12465
Balbi, T., Fabbri, R., Cortese, K., Smerilli, A., Ciacci, C., Grande, C., et al. (2013). Interactions between Mytilus galloprovincialis hemocytes and the bivalve pathogens Vibrio aestuarianus 01/032 and Vibrio splendidus LGP32. Fish Shellfish Immunol. 35, 1906–1915. doi: 10.1016/j.fsi.2013.09.027
Balseiro, P., Falcó, A., Romero, A., Dios, S., Martínez-López, A., Figueras, A., et al. (2011). Mytilus galloprovincialis Myticin C: a chemotactic molecule with antiviral activity and immunoregulatory properties. PLoS One 6:e23140. doi: 10.1371/journal.pone.0023140
Barbieri, J. T., Riese, M. J., and Aktories, K. (2002). Bacterial toxins that modify the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 18, 315–344. doi: 10.1146/annurev.cellbio.18.012502.134748
Brzóska, K., and Kruszewski, M. (2010). Impact of pirin protein expression level on NF−κB signaling pathway activation. AIP Conf. Proc. 1204, 260–261. doi: 10.1063/1.3295671
Bucki, R., Byfield, F. J., Kulakowska, A., McCormick, M. E., Drozdowski, W., Namiot, Z., et al. (2008). Extracellular gelsolin binds lipoteichoic acid and modulates cellular response to proinflammatory bacterial wall components. J. Immunol. 181, 4936L–4944. doi: 10.4049/jimmunol.181.7.4936
Bucki, R., Georges, P. C., Espinassous, Q., Funaki, M., Pastore, J. J., Chaby, R., et al. (2005). Inactivation of endotoxin by human plasma gelsolin. Biochemistry 44, 9590–9597. doi: 10.1021/bi0503504
Caberoy, N. B., Maiguel, D., Kim, Y., and Li, W. (2010). Identification of tubby and tubby-like protein 1 as eat-me signals by phage display. Exp. Cell Res. 316, 245–257. doi: 10.1016/j.yexcr.2009.10.008
Campos, A., Apraiz, I., da Fonseca, R. R., and Cristobal, S. (2015). Shotgun analysis of the marine mussel Mytilus edulis hemolymph proteome and mapping the innate immunity elements. Proteomics 15, 4021–4029. doi: 10.1002/pmic.201500118
Cao, Z., Xia, Z., Zhou, Y., Yang, X., Hao, H., Peng, N., et al. (2016). Methylcrotonoyl-CoA carboxylase 1 potentiates RLR-induced NF-κB signaling by targeting MAVS complex. Sci. Rep. 6:33557. doi: 10.1038/srep33557
Carvajal-Rodriguez, A., and de Uña-Alvarez, J. (2011). Assessing significance in high-throughput experiments by sequential goodness of fit and q-value estimation. PLoS One 6:e24700. doi: 10.1371/journal.pone.0024700
Castellanos-Martínez, S., Diz, A. P., Álvarez-Chaver, P., and Gestal, C. (2014). Proteomic characterization of the hemolymph of Octopus vulgaris infected by the protozoan parasite Aggregata octopiana. J. Proteomics 105, 151–163. doi: 10.1016/j.jprot.2013.12.008
Chen, H., Diao, X., Wang, H., and Zhou, H. (2018). An integrated metabolomic and proteomic study of toxic effects of Benzo[a]pyrene on gills of the pearl oyster Pinctada martensii. Ecotoxicol. Environ. Saf. 156, 330–336. doi: 10.1016/j.ecoenv.2018.03.040
Chen, S., Bonifati, S., Qin, Z., St. Gelais, C., Kodigepalli, K. M., Barrett, B. S., et al. (2018). SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. U.S.A. 115, E3798L–E3807. doi: 10.1073/pnas.1801213115
Corporeau, C., Tamayo, D., Pernet, F., Quéré, C., and Madec, S. (2014). Proteomic signatures of the oyster metabolic response to herpesvirus OsHV-1 μVar infection. J. Proteomics 109, 176–187. doi: 10.1016/j.jprot.2014.06.030
Costa, M. M., Prado-Alvarez, M., Gestal, C., Li, H., Roch, P., Novoa, B., et al. (2009). Functional and molecular immune response of Mediterranean mussel (Mytilus galloprovincialis) haemocytes against pathogen-associated molecular patterns and bacteria. Fish Shellfish Immunol. 26, 515–523. doi: 10.1016/j.fsi.2009.02.001
de Souza Santos, M., and Orth, K. (2015). Subversion of the cytoskeleton by intracellular bacteria: lessons from Listeria, Salmonella and Vibrio. Cell. Microbiol. 17, 164–173. doi: 10.1111/cmi.12399
De Zoysa, M., Nikapitiya, C., Kim, Y., Oh, C., Kang, D.-H., Whang, I., et al. (2010). Allograft inflammatory factor-1 in disk abalone (Haliotis discus discus): molecular cloning, transcriptional regulation against immune challenge and tissue injury. Fish Shellfish Immunol. 29, 319–326. doi: 10.1016/j.fsi.2010.04.006
Deutsch, E. W., Bandeira, N., Sharma, V., Perez-Riverol, Y., Carver, J. J., Kundu, D. J., et al. (2020). The ProteomeXchange consortium in 2020: enabling “big data” approaches in proteomics. Nucleic Acids Res. 48, D1145–D1152. doi: 10.1093/nar/gkz984
Dinguirard, N., Cavalcanti, M. G. S., Wu, X.-J., Bickham-Wright, U., Sabat, G., and Yoshino, T. P. (2018). Proteomic analysis of biomphalaria glabrata hemocytes during in vitro encapsulation of schistosoma mansoni sporocysts. Front. Immunol 9:2773. doi: 10.3389/fimmu.2018.02773
Diz, A. P., Carvajal-Rodríguez, A., and Skibinski, D. O. F. (2011). Multiple hypothesis testing in proteomics: a strategy for experimental work. Mol. Cell. Proteomics 10:M110.004374. doi: 10.1074/mcp.M110.004374
Dunphy, G., Flannery, S. M., Almine, J. F., Connolly, D. J., Paulus, C., Jønsson, K. L., et al. (2018). Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB signaling after nuclear DNA damage. Mol. Cell 71, 745.e–760.e. doi: 10.1016/j.molcel.2018.07.034
Duperthuy, M., Schmitt, P., Garzón, E., Caro, A., Rosa, R. D., Le Roux, F., et al. (2011). Use of OmpU porins for attachment and invasion of Crassostrea gigas immune cells by the oyster pathogen Vibrio splendidus. Proc. Natl. Acad. Sci. U.S.A. 108, 2993L–2998. doi: 10.1073/pnas.1015326108
Dziarski, R., Rasenick, M. M., and Gupta, D. (2000). Bacterial peptidoglycan binds to tubulin. Biochim. Biophys. Acta 1524, 17–26. doi: 10.1016/S0304-4165(00)00137-9
Egaña-Gorroño, L., Chinnasamy, P., Casimiro, I., Almonte, V. M., Parikh, D., Oliveira-Paula, G. H., et al. (2019). Allograft inflammatory factor-1 supports macrophage survival and efferocytosis and limits necrosis in atherosclerotic plaques. Atherosclerosis 289, 184–194. doi: 10.1016/j.atherosclerosis.2019.07.022
Fagerlund, R., Kinnunen, L., Köhler, M., Julkunen, I., and Melén, K. (2005). NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J. Biol. Chem. 280, 15942–15951. doi: 10.1074/jbc.M500814200
Fagerlund, R., Melén, K., Cao, X., and Julkunen, I. (2008). NF-kappaB p52. RelB and c-Rel are transported into the nucleus via a subset of importin alpha molecules. Cell. Signal. 20, 1442–1451. doi: 10.1016/j.cellsig.2008.03.012
Figueras, A., Moreira, R., Sendra, M., and Novoa, B. (2019). Genomics and immunity of the mediterranean mussel Mytilus galloprovincialis in a changing environment. Fish Shellfish Immunol. 90, 440–445. doi: 10.1016/j.fsi.2019.04.064
Franco-Martínez, L., Martínez-Subiela, S., Escribano, D., Schlosser, S., Nöbauer, K., Razzazi-Fazeli, E., et al. (2018). Alterations in haemolymph proteome of Mytilus galloprovincialis mussel after an induced injury. Fish Shellfish Immunol. 75, 41–47. doi: 10.1016/j.fsi.2018.01.038
Geese, M., Schlüter, K., Rothkegel, M., Jockusch, B. M., Wehland, J., and Sechi, A. S. (2000). Accumulation of profilin II at the surface of Listeria is concomitant with the onset of motility and correlates with bacterial speed. J. Cell Sci. 113, 1415–1426.
Gerdol, M., Manfrin, C., De Moro, G., Figueras, A., Novoa, B., Venier, P., et al. (2011). The C1q domain containing proteins of the Mediterranean mussel Mytilus galloprovincialis: a widespread and diverse family of immune-related molecules. Dev. Comp. Immunol. 35, 635–643. doi: 10.1016/j.dci.2011.01.018
Gerdol, M., Moreira, R., Cruz, F., Gómez-Garrido, J., Vlasova, A., Rosani, U., et al. (2020). Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 21:275. doi: 10.1186/s13059-020-02180-3
Görlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Gourlay, C. W., Carpp, L. N., Timpson, P., Winder, S. J., and Ayscough, K. R. (2004). A role for the actin cytoskeleton in cell death and aging in yeast. J. Cell Biol. 164, 803–809. doi: 10.1083/jcb.200310148
Gueguen, Y., Cadoret, J. P., Flament, D., Barreau-Roumiguière, C., Girardot, A. L., Garnier, J., et al. (2003). Immune gene discovery by expressed sequence tags generated from hemocytes of the bacteria-challenged oyster, Crassostrea gigas. Gene 303, 139–145. doi: 10.1016/s0378-1119(02)01149-6
Hambrook, J. R., Gharamah, A. A., Pila, E. A., Hussein, S., and Hanington, P. C. (2019). Biomphalaria glabrata granulin increases resistance to schistosoma mansoni infection in several biomphalaria species and induces the production of reactive oxygen species by haemocytes. Genes (Basel). 11:38. doi: 10.3390/genes11010038
Hatada, E. N., Krappmann, D., and Scheidereit, C. (2000). NF-kappaB and the innate immune response. Curr. Opin. Immunol. 12, 52–58. doi: 10.1016/s0952-7915(99)00050-3
Huan, P., Wang, H., and Liu, B. (2011). Comparative proteomic analysis of challenged Zhikong scallop (Chlamys farreri): a new insight into the anti-Vibrio immune response of marine bivalves. Fish Shellfish Immunol. 31, 1186–1192. doi: 10.1016/j.fsi.2011.10.014
Ji, C., Wu, H., Wei, L., Zhao, J., Wang, Q., and Lu, H. (2013a). Responses of Mytilus galloprovincialis to bacterial challenges by metabolomics and proteomics. Fish Shellfish Immunol. 35, 489–498. doi: 10.1016/j.fsi.2013.05.009
Ji, C., Wu, H., Wei, L., Zhao, J., and Yu, J. (2013b). Proteomic and metabolomic analysis reveal gender-specific responses of mussel Mytilus galloprovincialis to 2,2’,4,4’-tetrabromodiphenyl ether (BDE 47). Aquat. Toxicol. 14, 449–457. doi: 10.1016/j.aquatox.2013.07.009
Jiang, S., Qiu, L., Wang, L., Jia, Z., Lv, Z., Wang, M., et al. (2018). Transcriptomic and quantitative proteomic analyses provide insights into the phagocytic killing of hemocytes in the oyster Crassostrea gigas. Front. Immunol. 9:1280. doi: 10.3389/fimmu.2018.01280
Kausar, S., Asif, M., Bibi, N., and Rashid, S. (2013). Comparative molecular docking analysis of cytoplasmic dynein light chain DYNLL1 with pilin to explore the molecular mechanism of pathogenesis caused by Pseudomonas aeruginosa PAO. PLoS One 8:e76730. doi: 10.1371/journal.pone.0076730
Leprêtre, M., Almunia, C., Armengaud, J., Le Guernic, A., Salvador, A., Geffard, A., et al. (2020). Identification of immune-related proteins of Dreissena polymorpha hemocytes and plasma involved in host-microbe interactions by differential proteomics. Sci. Rep. 10:6226. doi: 10.1038/s41598-020-63321-z
Leprêtre, M., Faury, N., Segarra, A., Claverol, S., Degremont, L., Palos-Ladeiro, M., et al. (2021). Comparative Proteomics of Ostreid Herpesvirus 1 and pacific oyster interactions with two families exhibiting contrasted susceptibility to viral infection. Front. Immunol. 11:621994. doi: 10.3389/fimmu.2020.621994
Liang, P., Zhang, H., Wang, G., Li, S., Cong, S., Luo, Y., et al. (2013). KPNB1. XPO7 and IPO8 mediate the translocation ofNF-κB/p65 into the nucleus. Traffic 14, 1132–1143. doi: 10.1111/tra.12097
Licciulli, S., Cambiaghi, V., Scafetta, G., Gruszka, A. M., and Alcalay, M. (2010). Pirin downregulation is a feature of AML and leads to impairment of terminal myeloid differentiation. Leukemia 24, 429–437. doi: 10.1038/leu.2009.247
Liu, F., Rehmani, I., Esaki, S., Fu, R., Chen, L., de Serrano, V., et al. (2013). Pirin is an iron-dependent redox regulator of NF-κB. Proc. Natl. Acad. Sci. U.S.A. 110, 9722L–9727. doi: 10.1073/pnas.1221743110
Liu, R., Cheng, Q., Song, X., Wang, H., Wang, X., Wang, L., et al. (2019). A vital ubiquitin-conjugating enzyme CgUbe2g1 participated in regulation of immune response of Pacific oyster Crassostrea gigas. Dev. Comp. Immunol. 91, 132–142. doi: 10.1016/j.dci.2018.10.014
Liu, T., Zhang, L., Joo, D., and Sun, S.-C. (2017). NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2:17023. doi: 10.1038/sigtrans.2017.23
Liverman, A. D. B., Cheng, H.-C., Trosky, J. E., Leung, D. W., Yarbrough, M. L., Burdette, D. L., et al. (2007). Arp2/3-independent assembly of actin by Vibrio type III effector VopL. Proc. Natl. Acad. Sci. U.S.A. 104, 17117L–17122. doi: 10.1073/pnas.0703196104
Lu, Q., Pallas, D. C., Surks, H. K., Baur, W. E., Mendelsohn, M. E., and Karas, R. H. (2004). Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor α. Proc. Natl. Acad. Sci. U. S. A 101, 17126L–17131. doi: 10.1073/pnas.0407492101
Luk, S. C., Ngai, S. M., Tsui, S. K., Fung, K. P., Lee, C. Y., and Waye, M. M. (1999). In vivo and in vitro association of 14-3-3 epsilon isoform with calmodulin: implication for signal transduction and cell proliferation. J. Cell. Biochem. 73, 31–35. doi: 10.1002/(sici)1097-4644(19990401)73:1<31::aid-jcb4<3.0.co;2-x
Ma, W., Smigel, A., Tsai, Y.-C., Braam, J., and Berkowitz, G. A. (2008). Innate immunity signaling: cytosolic ca elevation is linked to downstream nitric oxide generation through the action of calmodulin or a calmodulin-like protein. Plant Physiol. 148, 818L–828. doi: 10.1104/pp.108.125104
Manduzio, H., Cosette, P., Gricourt, L., Jouenne, T., Lenz, C., Andersen, O.-K., et al. (2005). Proteome modifications of blue mussel (Mytilus edulis L.) gills as an effect of water pollution. Proteomics 5, 4958–4963. doi: 10.1002/pmic.200401328
Masood, M., Raftos, D. A., and Nair, S. V. (2016). Two oyster species that show differential susceptibility to virus infection also show differential proteomic responses to generic dsRNA. J. Proteome Res. 15, 1735–1746. doi: 10.1021/acs.jproteome.5b00615
McDonagh, B., Tyther, R., and Sheehan, D. (2005). Carbonylation and glutathionylation of proteins in the blue mussel Mytilus edulis detected by proteomic analysis and Western blotting: actin as a target for oxidative stress. Aquat. Toxicol. 73, 315–326. doi: 10.1016/j.aquatox.2005.03.020
Meschede, J., Šadiæ, M., Furthmann, N., Miedema, T., Sehr, D. A., Müller-Rischart, A. K., et al. (2020). The parkin-coregulated gene product PACRG promotes TNF signaling by stabilizing LUBAC. Sci. Signal. 13:eaav1256. doi: 10.1126/scisignal.aav1256
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Mikenberg, I., Widera, D., Kaus, A., Kaltschmidt, B., and Kaltschmidt, C. (2007). Transcription Factor NF-κB Is transported to the nucleus via cytoplasmic dynein/dynactin motor complex in hippocampal neurons. PLoS One 2:e589. doi: 10.1371/journal.pone.0000589
Mimuro, H., Suzuki, T., Suetsugu, S., Miki, H., Takenawa, T., and Sasakawa, C. (2000). Profilin is required for sustaining efficient intra- and intercellular spreading of Shigella flexneri. J. Biol. Chem. 275, 28893–28901. doi: 10.1074/jbc.M003882200
Mitta, G., Hubert, F., Noël, T., and Roch, P. (1999). Myticin, a novel cysteine-rich antimicrobial peptide isolated from haemocytes and plasma of the mussel Mytilus galloprovincialis. Eur. J. Biochem. 265, 71–78. doi: 10.1046/j.1432-1327.1999.00654.x
Moreira, R., Pereiro, P., Canchaya, C., Posada, D., Figueras, A., and Novoa, B. (2015). RNA-Seq in Mytilus galloprovincialis: comparative transcriptomics and expression profiles among different tissues. BMC Genomics 16:728. doi: 10.1186/s12864-015-1817-5
Moreira, R., Romero, A., Rey-Campos, M., Pereiro, P., Rosani, U., Novoa, B., et al. (2020). Stimulation of Mytilus galloprovincialis hemocytes with different immune challenges induces differential transcriptomic, mirnomic, and functional responses. Front. Immunol. 11:606102. doi: 10.3389/fimmu.2020.606102
Muscolini, M., Sajeva, A., Caristi, S., and Tuosto, L. (2011). A novel association between filamin A and NF-κB inducing kinase couples CD28 to inhibitor of NF-κB kinase α and NF-κB activation. Immunol. Lett. 136, 203–212. doi: 10.1016/j.imlet.2011.01.011
Oyanedel, D., Gonzalez, R., Flores-Herrera, P., Brokordt, K., Rosa, R. D., Mercado, L., et al. (2016). Molecular characterization of an inhibitor of NF-κB in the scallop Argopecten purpuratus: first insights into its role on antimicrobial peptide regulation in a mollusk. Fish Shellfish Immunol. 52, 85–93. doi: 10.1016/j.fsi.2016.03.021
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Rajan, B., Lokesh, J., Kiron, V., and Brinchmann, M. F. (2013). Differentially expressed proteins in the skin mucus of Atlantic cod (Gadus morhua) upon natural infection with Vibrio anguillarum. BMC Vet. Res. 9:103. doi: 10.1186/1746-6148-9-103
Rey-Campos, M., Moreira, R., Gerdol, M., Pallavicini, A., Novoa, B., and Figueras, A. (2019a). Immune tolerance in Mytilus galloprovincialis hemocytes after repeated contact with Vibrio splendidus. Front. Immunol 10:1894. doi: 10.3389/fimmu.2019.01894
Rey-Campos, M., Moreira, R., Valenzuela-Muñoz, V., Gallardo-Escárate, C., Novoa, B., and Figueras, A. (2019b). High individual variability in the transcriptomic response of Mediterranean mussels to Vibrio reveals the involvement of myticins in tissue injury. Sci. Rep. 9:3569. doi: 10.1038/s41598-019-39870-3
Rey-Campos, M., Novoa, B., Pallavicini, A., Gerdol, M., and Figueras, A. (2020). Comparative genomics reveals a significant sequence variability of myticin genes in Mytilus galloprovincialis. Biomolecules 10:943. doi: 10.3390/biom10060943
Rocher, B., Bultelle, F., Chan, P., Foll, F. L., Letendre, J., Monsinjon, T., et al. (2015). 2-DE Mapping of the Blue Mussel Gill Proteome: the Usual Suspects Revisited. Proteomes 3, 3–41. doi: 10.3390/proteomes3010003
Romero, M. R., Pérez-Figueroa, A., Carrera, M., Swanson, W. J., Skibinski, D. O. F., and Diz, A. P. (2019). RNA-seq coupled to proteomic analysis reveals high sperm proteome variation between two closely related marine mussel species. J. Proteomics 192, 169–187. doi: 10.1016/j.jprot.2018.08.020
Rother, S., Bartels, M., Schweda, A. T., Resch, K., Pallua, N., and Nourbakhsh, M. (2016). NF-κB-repressing factor phosphorylation regulates transcription elongation via its interactions with 5′→3′ exoribonuclease 2 and negative elongation factor. FASEB J. 30, 174–185. doi: 10.1096/fj.15-270256
Saco, A., Rey-Campos, M., Novoa, B., and Figueras, A. (2020). Transcriptomic response of mussel gills after a vibrio splendidus infection demonstrates their role in the immune response. Front. Immunol. 11:3273. doi: 10.3389/fimmu.2020.615580
Saco, A., Rey-Campos, M., Rosani, U., Novoa, B., and Figueras, A. (2021). The evolution and diversity of interleukin-17 highlight an expansion in marine invertebrates and its conserved role in mucosal immunity. Front. Immunol. 12:692997. doi: 10.3389/fimmu.2021.692997
Sánchez-Marín, P., Vidal-Liñán, L., Fernández-González, L. E., Montes, R., Rodil, R., Quintana, J. B., et al. (2021). Proteomic analysis and biochemical alterations in marine mussel gills after exposure to the organophosphate flame retardant TDCPP. Aquat. Toxicol. 230:105688. doi: 10.1016/j.aquatox.2020.105688
Smits, M., Artigaud, S., Bernay, B., Pichereau, V., Bargelloni, L., and Paillard, C. (2020). A proteomic study of resistance to brown ring disease in the manila clam, Ruditapes philippinarum. Fish Shellfish Immunol. 99, 641–653. doi: 10.1016/j.fsi.2020.02.002
Somboonwiwat, K., Chaikeeratisak, V., Wang, H.-C., Fang Lo, C., and Tassanakajon, A. (2010). Proteomic analysis of differentially expressed proteins in Penaeus monodon hemocytes after Vibrio harveyi infection. Proteome Sci. 8:39. doi: 10.1186/1477-5956-8-39
Somboonwiwat, K., Supungul, P., Rimphanitchayakit, V., Aoki, T., Hirono, I., and Tassanakajon, A. (2006). Differentially expressed genes in hemocytes of vibrio harveyi-challenged shrimp penaeus monodon. J. Biochem. Mol. Biol. 39, 26–36. doi: 10.5483/bmbrep.2006.39.1.026
Song, H., Kim, H., Lee, K., Lee, D.-H., Kim, T.-S., Song, J. Y., et al. (2012). Ablation of Rassf2 induces bone defects and subsequent haematopoietic anomalies in mice. EMBO J. 31, 1147–1159. doi: 10.1038/emboj.2011.480
Song, Q., Chen, H., Li, Y., Zhou, H., Han, Q., and Diao, X. (2016). Toxicological effects of benzo(a)pyrene. DDT and their mixture on the green mussel Perna viridis revealed by proteomic and metabolomic approaches. Chemosphere 144, 214–224. doi: 10.1016/j.chemosphere.2015.08.029
Storey, J. D. (2003). The positive false discovery rate: a Bayesian interpretation and the q-value. Ann. Stat. 31, 2013–2035. doi: 10.1214/aos/1074290335
Suzuki, M., Watanabe, M., Nakamaru, Y., Takagi, D., Takahashi, H., Fukuda, S., et al. (2016). TRIM39 negatively regulates the NFκB-mediated signaling pathway through stabilization of Cactin. Cell. Mol. Life Sci. 73, 1085–1101. doi: 10.1007/s00018-015-2040-x
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Tagliazucchi, D., and Conte, A. (2005). Calcium/calmodulin dependence of nitric oxide synthase from Viviparus ater immunocytes. Invertebr. Surviv. J. 2, 54–59.
Tanguy, M., McKenna, P., Gauthier-Clerc, S., Pellerin, J., Danger, J.-M., and Siah, A. (2013). Functional and molecular responses in Mytilus edulis hemocytes exposed to bacteria, Vibrio splendidus. Dev. Comp. Immunol. 39, 419–429. doi: 10.1016/j.dci.2012.10.015
Uotila, L. M., Guenther, C., Savinko, T., Lehti, T. A., and Fagerholm, S. C. (2017). Filamin a regulates neutrophil adhesion, production of reactive oxygen species, and neutrophil extracellular trap release. J. Immunol. 199, 3644–3653. doi: 10.4049/jimmunol.1700087
Wang, T., Zhao, M., Liang, D., Bose, U., Kaur, S., McManus, D. P., et al. (2017). Changes in the neuropeptide content of Biomphalaria ganglia nervous system following Schistosoma infection. Parasit. Vectors 10:275. doi: 10.1186/s13071-017-2218-1
Witherow, D. S., Garrison, T. R., Miller, W. E., and Lefkowitz, R. J. (2004). beta-Arrestin inhibits NF-kappaB Activity by Means of Its Interaction with the kappaB Inhibitor IkappaBalpha. Proc. Natl. Acad. Sci. U.S.A 101, 8603–8607.
Wu, H., Ji, C., Wei, L., Zhao, J., and Lu, H. (2013). Proteomic and metabolomic responses in hepatopancreas of Mytilus galloprovincialis challenged by Micrococcus luteus and Vibrio anguillarum. J. Proteomics 94, 54–67. doi: 10.1016/j.jprot.2013.09.001
Wu, S.-H., Lin, H.-J., Lin, W.-F., Wu, J.-L., and Gong, H.-Y. (2018). A potent tilapia secreted granulin peptide enhances the survival of transgenic zebrafish infected by Vibrio vulnificus via modulation of innate immunity. Fish Shellfish Immunol. 75, 74–90. doi: 10.1016/j.fsi.2018.01.044
Xiong, Q., Feng, J., Li, S., Zhang, G., Qiao, Z., Chen, Z., et al. (2015). Integrated transcriptomic and proteomic analysis of the global response of Synechococcus to high light stress. Mol. Cell. Proteomics 14, 1038–1053. doi: 10.1074/mcp.M114.046003
Xu, W., and Faisal, M. (2008). Putative identification of expressed genes associated with attachment of the zebra mussel (Dreissena polymorpha). Biofouling 24, 157–161. doi: 10.1080/08927010801975345
Xu, Y., Shi, J., Hao, W., Xiang, T., Zhou, H., Wang, W., et al. (2018). iTRAQ-based quantitative proteomic analysis of Procambarus clakii hemocytes during Spiroplasma eriocheiris infection. Fish Shellfish Immunol. 77, 438–444. doi: 10.1016/j.fsi.2018.04.005
Yamamoto, S., Ogasawara, N., Yamamoto, K., Uemura, C., Takaya, Y., Shiraishi, T., et al. (2017). Mitochondrial proteins NIP-SNAP-1 and -2 are a target for the immunomodulatory activity of clarithromycin, which involves NF-κB-mediated cytokine production. Biochem. Biophys. Res. Commun. 483, 911–916. doi: 10.1016/j.bbrc.2016.12.100
Yoshida, S., and Sasakawa, C. (2003). Exploiting host microtubule dynamics: a new aspect of bacterial invasion. Trends Microbiol. 11, 139–143. doi: 10.1016/S0966-842X(03)00023-4
Zhang, P., Li, C., Zhang, P., Jin, C., Pan, D., and Bao, Y. (2014). iTRAQ-Based proteomics reveals novel members involved in pathogen challenge in sea cucumber Apostichopus japonicus. PLoS One 9:e100492. doi: 10.1371/journal.pone.0100492
Zhang, Y., Li, J., Yu, F., He, X., and Yu, Z. (2013). Allograft inflammatory factor-1 stimulates hemocyte immune activation by enhancing phagocytosis and expression of inflammatory cytokines in Crassostrea gigas. Fish Shellfish Immunol. 34, 1071–1077. doi: 10.1016/j.fsi.2013.01.014
Zhang, Y., Li, X., Carpinteiro, A., Goettel, J. A., Soddemann, M., and Gulbins, E. (2011). Kinase suppressor of Ras-1 protects against pulmonary Pseudomonas aeruginosa infections. Nat. Med. 17, 341–346. doi: 10.1038/nm.2296
Zhang, Y., Sun, J., Mu, H., Li, J., Zhang, Y., Xu, F., et al. (2015). Proteomic basis of stress responses in the gills of the Pacific Oyster Crassostrea gigas. J. Proteome Res. 14, 304–317. doi: 10.1021/pr500940s
Zhou, Y., Li, X., Yuan, X., and Hao, L. (2019). Role of allograft inflammatory factor-1 in the regulation of inflammation and oxidative stress in primary peritoneal mesothelial cells. Cell Biol. Int. 43, 495–505. doi: 10.1002/cbin.11115
Zuo, S., Xue, Y., Tang, S., Yao, J., Du, R., Yang, P., et al. (2010). 14-3-3 epsilon dynamically interacts with key components of mitogen-activated protein kinase signal module for selective modulation of the TNF-alpha-induced time course-dependent NF-kappaB activity. J. Proteome Res. 9, 3465–3478. doi: 10.1021/pr901137
Keywords: proteomic, transcriptomic, immune, mussel, bacterial infection, mollusc
Citation: Saco A, Panebianco A, Blanco S, Novoa B, Diz AP and Figueras A (2021) Integration of Transcriptomics and Proteomics Improves the Characterization of the Role of Mussel Gills in a Bacterial Waterborne Infection. Front. Mar. Sci. 8:735309. doi: 10.3389/fmars.2021.735309
Received: 05 July 2021; Accepted: 09 September 2021;
Published: 05 October 2021.
Edited by:
Ciro Rico, Institute of Marine Sciences of Andalusia (ICMAN), SpainReviewed by:
Alberto Pallavicini, University of Trieste, ItalyCopyright © 2021 Saco, Panebianco, Blanco, Novoa, Diz and Figueras. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angel Pérez Diz, YW5nZWwucC5kaXpAdXZpZ28uZXM=; Antonio Figueras, YW50b25pb2ZpZ3VlcmFzQGlpbS5jc2ljLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.