 Wirulda Pootakham1*
Wirulda Pootakham1* Chutima Sonthirod1Chaiwat Naktang1Narinratana Kongjandtre2†Lalita Putchim3†Duangjai Sangsrakru1Thippawan Yoocha1
Chutima Sonthirod1Chaiwat Naktang1Narinratana Kongjandtre2†Lalita Putchim3†Duangjai Sangsrakru1Thippawan Yoocha1 Sithichoke Tangphatsornruang1
Sithichoke Tangphatsornruang1- 1National Omics Center, National Science and Technology Development Agency, Khlong Luang, Thailand
- 2Department of Aquatic Science, Faculty of Science, Burapha University, Chonburi, Thailand
- 3Marine and Coastal Resources Research Center (Upper Eastern Gulf of Thailand), Bangpakong, Thailand
Background
Coral reefs are one of the most productive ecosystems on the planet, supporting the productivity of ~25% of marine fisheries (Moberg and Folke, 1999). Scleractinian corals constitute the primary framework of coral ecosystems. Unfortunately, they are highly sensitive to changes in the water temperature (Hoegh-Guldberg and Bruno, 2010; Mumby Peter and van Woesik, 2014). Ocean warming has caused coral bleaching and posed a global threat to coral health and survival. Reef corals have suffered major declines as the frequency and severity of periodic ocean warming have increased (Loya et al., 2001). Coral reefs in Southeast Asia especially in the Andaman Sea and Gulf of Thailand were severely affected during the 2010 mass bleaching event (Tun et al., 2010). The scleractinian coral Platygyra sinensis is one of the dominant reef-builders commonly found in the Gulf of Thailand and Andaman Sea. Previous studies have shown that Platygyra corals in Southeast Asia were extensively bleached during the major thermal anomaly in May 2010 (McClanahan, 2004; Guest et al., 2012). Little is known about the responses of the brain coral genus Platygyra to heat stress at the molecular level due to the lack of genomic/transcriptomic resources. The availability of genomic and transcriptomic data from a number of coral species allows us to probe the molecular stress response of the organisms to biotic and abiotic stress conditions (Shinzato et al., 2011; Traylor-Knowles et al., 2011; Kenkel et al., 2013; Kitchen et al., 2015; Davies et al., 2016; Buitrago-López et al., 2020). Here, we report the first reference genome assembly for the brain coral P. sinensis using long-read Pacific Biosciences sequencing technology. We hope that our genomic resource will help the coral research community gain insights into genetic factors driving responses to thermal stress and other biotic/abiotic stresses and help promote the conservation of this species.
Data Description
Sample Collection
Gamete bundles from Platygyra sinensis colonies were collected in situ from the inshore reef in Sattahip district (Samaesarn subdistrict) located in the Gulf of Thailand (12°35′556″N, 100°57′508″E), following the guideline in (Edwards et al., 2010). Prior to the collection date, we conducted several night dives to assess proximity to spawning. Once we were able to predict the onset of spawning, we placed collecting devices over three mature P. sinensis colonies that were not in the vicinity of other coral species. The collecting devices consisted of a funnel made of 100-μM plankton mesh with a 500-mL transparent plastic container attached at the mouth of the net. The collecting devices were attached to the surrounding substrata using stainless steel nails, and drawstrings were used to tighten the base of the net around the colony (so that the net enclosed the entire colony). When spawning was finished, we collected and closed the container underwater and immediately transported the gamete bundles to the lab station (The Marine Science Camp and Conservation, Samaesarn, Chonburi).
Upon returning to the lab, eggs and sperm bundles were separated using Pasteur pipettes and plankton mesh sieves, and the eggs were transferred to 2-mL screw-capped tubes, immediately frozen and stored in liquid nitrogen until use. Even though we collected the samples from three individual colonies, we chose to sequence one colony from which we could retrieve the largest amount of eggs from the collecting device (to ensure we could isolate sufficient amount of genomic DNA required for sequencing). Small coral fragments from that same colony were also collected (for RNA sequencing) and placed in sterile disposable 15-mL tubes submerged in seawater. Seawater was removed upon returning to the lab station, and the samples were frozen and stored in liquid nitrogen until use. Access to the field site was authorized and the permit for coral sample collection was issued by the Department of Marine and Coastal Resources, Ministry of National Resources and Environment (Thailand), following the Nagoya protocol (permit number 20210304).
DNA/RNA Extraction and Sequencing
High molecular weight genomic DNA was isolated from the eggs using the MagAttract HMW DNA kit (Qiagen, Hilden, Germany) following the manufacturer's instruction. The DNA sample was quantified using Qubit fluorometer (Thermo Fisher Scientific, Waltham, USA), and its integrity was assessed using the Pippin Pulse Electrophoresis System (Sage Science, Beverly, USA). SMRTbell libraries with an insert size of 10,000 nt were constructed for the Pacific Biosciences (PacBio) Sequel sequencing system. Sequencing was performed with the Sequel Binding Kit 2.0 using a 20-h movie collection time according to manufacturer's protocols (Pacific Biosciences, Menlo Park, USA).
To isolate RNA for library preparation and sequencing, frozen samples were homogenized in liquid nitrogen with sterile mortars and pestles, and the CTAB buffer (2% CTAB, 1.4 M NaCl, 2% PVP, 20 mM EDTA pH 8.0, 100 mM Tris-HCl pH 8.0, 0.4% SDS) was added. RNA was extracted from the aqueous phase twice using 24:1 chloroform:isoamylalcohol and precipitated in 1/3 volume of 8 M LiCl overnight. RNA pellets were washed with 70% ethanol, air-dried and resuspended in RNase-free water. Poly(A) mRNAs were enriched from total RNA samples using the Dynabeads mRNA purification kit (Thermo Fisher Scientific, Waltham, USA). RNA integrity was assessed with a Fragment Analyzer System (Agilent, Santa Clara, CA, USA). The RNA library was prepared using 200 ng of poly(A) mRNA according to the MGIEasy RNA Library Prep Kit V3.0 protocol (MGI, Shenzen, China). The library was sequenced on the MGISEQ-2000RS using the MGISEQ-2000RS Sequencing Flow Cell V3.0 (MGI, Shenzen, China).
Genome Assembly
A total of 11,092,922 PacBio raw reads totaling 93.32 Gb were subjected to read correction and trimming by MECAT2 version 2020.02.28 (https://github.com/xiaochuanle/MECAT2). Prior to assembling the corrected reads, we identified the taxonomic origin of those reads by aligning each of them to the Symbiodiniaceae database (http://speedus.sampgr.org.cn/) and prokaryotic RefSeq genome database from NCBI (downloaded on March 4, 2021) using BLASTN as the specimens used in this study might contain DNA from P. sinensis-associated Symbiodiniaceae and prokaryotes. Any corrected read that matched the sequences in the database with an e-value of <10−5 and bit-scores higher than 50 was discarded. After filtering out Symbiodiniaceae and bacterial associated reads, we carried out an assembly of P. sinensis using 7,960,302 corrected PacBio reads (totaling 61.98 Gb; N50 of the corrected reads = 10,727 bases), representing ~61X coverage calculated from the estimated genome size of 1,001 Mb based on the analysis of k-mer depth distribution using the BBmap software version 38.90 (kmercountexact.sh; Supplementary Figure S1) (Bushnell, 2014). De novo assembly was performed using the Flye software version 2.8.3 (Kolmogorov et al., 2019) with the following parameter setting: genomeSize = 1 g –iterations 3, and -m 3000. The polishing was carried out using the GenomicConsensus package in the SMRT Analysis software suite version 2.3 with –algorithm =arrow (https://github.com/PacificBiosciences/GenomicConsensus). The draft genome assembly was 1.1 Gb with an N50 length of 32.4 kb and contained 56,352 contigs larger than 1 kb (Table 1). Given the high coverage of the long-read PacBio sequences used, the contiguity of the assembly was lower than expected, and this is likely due to the repetitive nature of the genome.
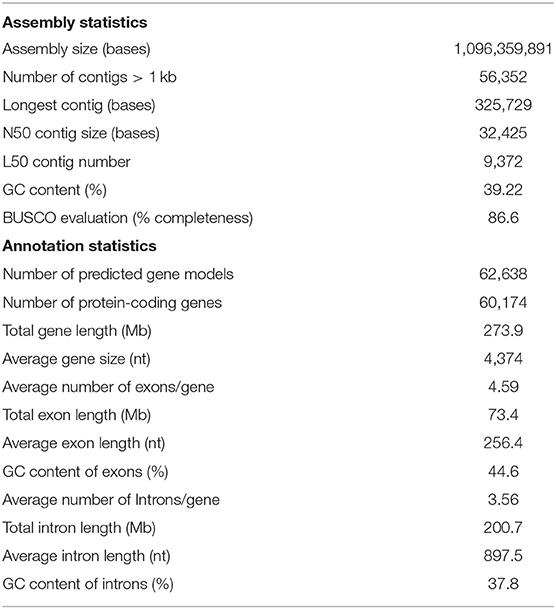
Table 1. Assembly and annotation statistics for P. sinensis genome.
Identification of Repetitive Sequences
To analyze the repetitive sequences in P. sinensis, we employed RepeatModeler version 2.0.1 to predict transposable elements and generate a de novo repeat library and RECON version 1.08 and RepeatScout version 1.0.5 to identify the boundaries of repetitive elements and to build consensus models of interspersed repeats. We discovered that 50.03% of the genome assembly was occupied by repetitive elements, most of which were unclassified repeats (29.28%) and DNA elements (10.79%). The proportion of repetitive sequences in the brain coral was higher than the figures reported for A. millepora (34.55%) (Ying et al., 2019) and P. verrucosa (41.22%) (Buitrago-López et al., 2020) but comparable to the number reported for Pachyseris speciosa (52.5%) (Bongaerts et al., 2021).
Genome Quality Assessment
To evaluate the quality of the assembly, we aligned short-read RNA-seq sequences from this study to the genome using BLASTN at an e-value cutoff of 10−10. Prior to performing the alignment, any short RNA sequence read that was mapped to the Symbiodiniaceae database with an e-value of <10−5 and bit-scores higher than 50 was discarded. We found that 87.92% of the P. sinensis RNA-seq reads could be mapped back to the genome assembly. To further assess the completeness of the final assembly, we employed the Benchmarking Universal Single-Copy Orthologs (BUSCO) software version 3 using the Metazoa OrthoDB release 10 (Kriventseva et al., 2015; Simão et al., 2015). Gene predictions based on the assembly revealed the presence of 954 highly conserved orthologs in the metazoan gene set, with 66.8% identified as complete and single-copy, 12.1% as complete and duplicated, 7.7% as fragmented, and 13.4% as missing. We also ran the HaploMerger2 (Huang et al., 2017) software to resolve allelic relations in the assembly. The size of the resulting assembly (1.04 Gb) was very similar to the original assembly with an N50 contig length of 33,443 nt. When we evaluated the completeness of the gene space of this merged assembly, 65.8, 11.6, 8.1, and 14.5% of the highly conserved orthologs in the metazoan gene set were identified as complete and single-copy, complete and duplicated, fragmented and missing, respectively. We decided to present the original unmerged assembly in this study since that version appeared to be slightly more complete with regard to the gene space assessed by BUSCO.
Genome Annotation
Evidence from transcriptome-based prediction, ab initio gene prediction and homology-based prediction were combined to predict protein-coding sequences in the unmasked P. sinensis genome using EvidenceModeler (EVM) version 1.1.1 r2015-07-03 (Haas et al., 2008). Short-read RNA-seq data associated with Symbiodiniaceae were filtered out as mentioned earlier. The remaining P. sinensis RNA-seq data were mapped to the assembly during the initial step of annotation using the PASA2 pipeline version 2.0.1 (Haas et al., 2008). Protein sequences from Acropora digitifera, Acropora millepora, Montipora capitata, Orbicella faveolata, Pocillopora damicornis, Pocillopora verrucosa, and Stylophora pistillata obtained from public databases were aligned to the unmasked genome using AAT version 1.52 (Huang et al., 1997). An ab initio gene predictor was run on the unmasked assembly. Protein-coding gene predictions were obtained with Augustus version 3.2.1 (Stanke et al., 2004) trained with A. digitifera, A. millepora, M. capitata, O. faveolata, P. damicornis, P. verrucosa, and S. pistillata, and PASA transcriptome alignment using P. sinensis RNA-seq (Chen et al., 2015) alignment files as inputs. All gene predictions were integrated by EVM to generate consensus gene models using the following weight for each evidence type: PASA2−1, AAT−0.3, and Augustus−0.3. The positions of annotate genes were cross-checked with those of known repeats, and any gene that had more than 20% overlapping sequence with repetitive elements were excluded from the list of annotated genes. In addition, RNA-seq data were mapped to the predicted gene set using HISAT2 version 2.2.0 (Kim et al., 2019), and any predicted genes with no mapped reads were excluded from the annotation list. The genome annotation contained 62,638 predicted gene models, of which 60,174 were protein-coding genes (Table 1; Supplementary Table S1). There were on average 4.59 exons per gene, and the mean transcript length was 4,374 bp. The GC contents of the coding sequences and the introns were 44.6 and 37.8%, respectively. Predicted protein-coding genes were assigned functions by aligning them to the best matches in the NCBI Genbank nr protein database, and the BLASTP results were used to map and retrieve gene ontology (GO) annotation (Figure 1). Among genes annotated to cellular component, the largest category was integral component of membrane, followed by membrane and nucleus. The most prevalent GO terms associated with molecular function were ATP binding, metal ion binding and nucleic acid binding. DNA integration, proteolysis and protein phosphorylation were among the largest categories associated with biological function. We compared the top 10 most prevalent GO terms identified in P. sinensis, S. pistillata and A. digitifera and found that phosphorylation was among the largest category associated with biological process in all three species (Supplementary Table S2). Nucleus, cytoskeleton and endoplasmic reticulum membrane were the common top 10 cellular component annotations in P. sinensis, S. pistillata, and A. digitifera while DNA and nucleotide binding appeared to be among the most prevalent GO term in the molecular function category.
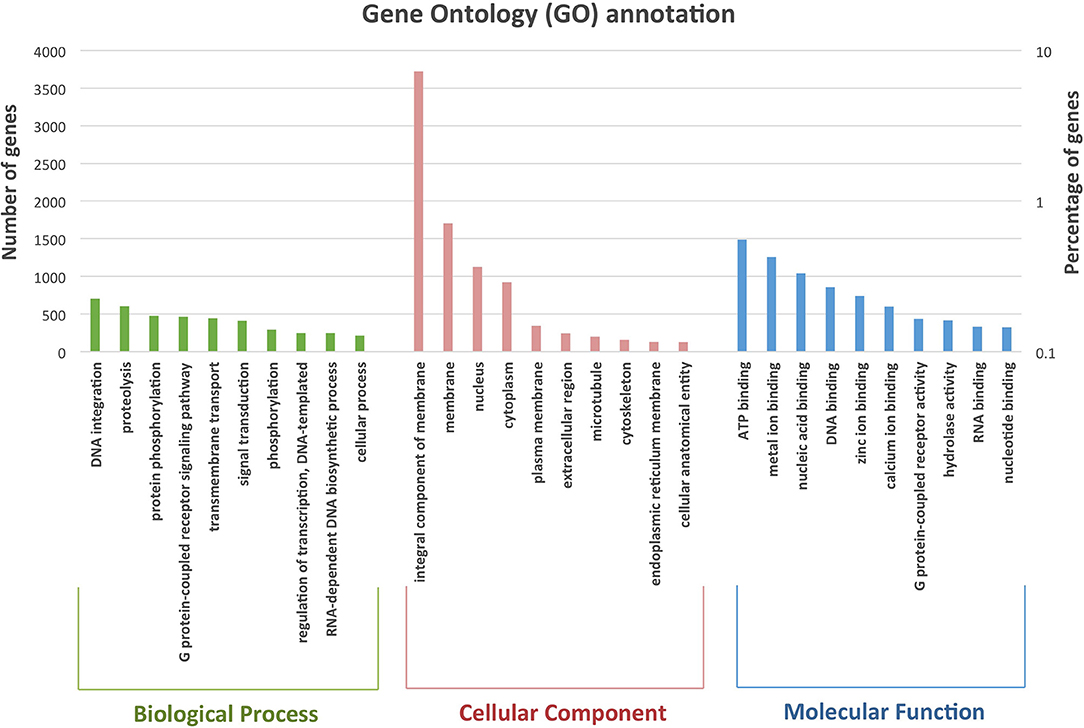
Figure 1. Gene Ontology (GO) annotation of P. sinensis genes in the genome assembly. Results are summarized in three main categories: cellular components, molecular functions, and biological processes. A total of 23,240 genes have been assigned GO terms.
Re-use Potential
We report the first draft assembly of the P. sinensis genome using the PacBio long-read single molecule real-time sequencing technology. The availability of this genome assembly and annotation enable the coral research community to study thermal stress responses and gain a better understanding of how P. sinensis copes with elevated ocean temperature. This knowledge may be useful for future reef conservation and restoration programs.
Data Availability Statement
P. sinensis genome assembly and RNA-seq data have been submitted to the NCBI Genbank databases under the BioProject number PRJNA736579 (NCBI accession number JAHPZR000000000 for P. sinensis genome assembly). The annotation files (gff, cds, and protein) are available at https://www.nstda.or.th/noc/research-development/80-about-us/102-our-genome-assemblies.html.
Author Contributions
WP and ST conceived and designed the experiment. NK and LP collected coral samples. TY and DS performed DNA and RNA extraction and sequencing. CS and CN carried out bioinformatics analyses. WP wrote and revised the manuscript. All authors have read and approved the final manuscript.
Funding
This study was funded by the National Science and Technology Development Agency (NSTDA), Thailand (grant number: 1000221) and the L'Oréal-UNESCO for Women in Science Fellowship (awarded to WP).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the Marine Science Camp and Conservation (Samaesarn, Chonburi) for sharing their lab facilities. We would also like to thank Bawornnan Jitphong, Piyasak Sangpaiboon, Phumiphat Jaronvannaying, and Chiraphan Raengdi for their assistance with sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.732650/full#supplementary-material
References
Bongaerts, P., Cooke, I. R., Ying, H., Wels, D., den Haan, S., Hernandez-Agreda, A., et al. (2021). Morphological stasis masks ecologically divergent coral species on tropical reefs. Curr. Biol. 31, 2286–2298.e8. doi: 10.1016/j.cub.2021.03.028
Buitrago-López, C., Mariappan, K. G., Cárdenas, A., Gegner, H. M., and Voolstra, C. R. (2020). The genome of the cauliflower coral Pocillopora verrucosa. Genome Biol. Evol. 12, 1911–1917. doi: 10.1093/gbe/evaa184
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. Berkeley, CA: Lawrence Berkeley National Lab (LBNL).
Chen, X., Tan, T., Xu, C., Huang, S., Tan, J., Zhang, M., et al. (2015). Genome-wide transcriptome profiling reveals novel insights into Luffa cylindrica browning. Biochem. Biophys. Res. Commun. 463, 1243–1249. doi: 10.1016/j.bbrc.2015.06.093
Davies, S. W., Marchetti, A., Ries, J. B., and Castillo, K. D. (2016). Thermal and pCO2 stress elicit divergent transcriptomic responses in a resilient coral. Front. Mar. Sci.. 3:112. doi: 10.3389/fmars.2016.00112
Edwards, A., Guest, J., Shafir, S., Fisk, D., Gomez, E., Rinkevich, B., et al. (2010). Reef Rehabilitation Manual. ed A. Edwards (Brisbane QLD: The Coral Reef Targeted Research & Capacity Building for Management Program), 1–166.
Guest, J. R., Baird, A. H., Maynard, J. A., Muttaqin, E., Edwards, A. J., Campbell, S. J., et al. (2012). Contrasting patterns of coral bleaching susceptibility in 2010 suggest an adaptive response to thermal stress. PLoS One 7:e33353. doi: 10.1371/journal.pone.0033353
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9:R7. doi: 10.1186/gb-2008-9-1-r7
Hoegh-Guldberg, O., and Bruno, J. F. (2010). The impact of climate change on the world's marine ecosystems. Science 328, 1523–1528. doi: 10.1126/science.1189930
Huang, S., Kang, M., and Xu, A. (2017). HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33, 2577–2579. doi: 10.1093/bioinformatics/btx220
Huang, X., Adams, M. D., Zhou, H., and Kerlavage, A. R. (1997). A tool for analyzing and annotating genomic sequences. Genomics 46, 37–45. doi: 10.1006/geno.1997.4984
Kenkel, C. D., Meyer, E., and Matz, M. V. (2013). Gene expression under chronic heat stress in populations of the mustard hill coral (Porites astreoides) from different thermal environments. Mol. Ecol. 22, 4322–4334. doi: 10.1111/mec.12390
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Kitchen, S. A., Crowder, C. M., Poole, A. Z., Weis, V. M., and Meyer, E. (2015). De novo assembly and characterization of four anthozoan (Phylum cnidaria) transcriptomes. G3 5, 2441–2452. doi: 10.1534/g3.115.020164
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kriventseva, E. V., Tegenfeldt, F., Petty, T. J., Waterhouse, R. M., Simao, F. A., Pozdnyakov, I. A., et al. (2015). OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 43(Database issue), D250–D256. doi: 10.1093/nar/gku1220
Loya, Y., Sakai, K., Yamazato, K., Nakano, Y., Sambali, H., and van Woesik, R. (2001). Coral bleaching: the winners and the losers. Ecol. Lett. 4, 122–131. doi: 10.1046/j.1461-0248.2001.00203.x
McClanahan, T. R. (2004). The relationship between bleaching and mortality of common corals. Mar. Biol. 144, 1239–1245. doi: 10.1007/s00227-003-1271-9
Moberg, F., and Folke, C. (1999). Ecological goods and services of coral reef ecosystems. Ecol. Econ. 29, 215–233. doi: 10.1016/S0921-8009(99)00009-9
Mumby Peter, J., and van Woesik, R. (2014). Consequences of ecological, evolutionary and biogeochemical uncertainty for coral reef responses to climatic stress. Curr. Biol. 24, R413–R423. doi: 10.1016/j.cub.2014.04.029
Shinzato, C., Shoguchi, E., Kawashima, T., Hamada, M., Hisata, K., Tanaka, M., et al. (2011). Using the Acropora digitifera genome to understand coral responses to environmental change. Nature 476, 320–323. doi: 10.1038/nature10249
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., Steinkamp, R., Waack, S., and Morgenstern, B. (2004). AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32(Suppl. 2), W309–W312. doi: 10.1093/nar/gkh379
Traylor-Knowles, N., Granger, B. R., Lubinski, T. J., Parikh, J. R., Garamszegi, S., Xia, Y., et al. (2011). Production of a reference transcriptome and transcriptomic database (PocilloporaBase) for the cauliflower coral, Pocillopora damicornis. BMC Genomics 12:585. doi: 10.1186/1471-2164-12-585
Tun, K., Chou, L. M., Low, J., Yeemin, T., Phongsuwan, N., Setiasih, N., . (eds.). (2010). “The 2010 coral bleaching event in Southeast Asia - A regional overview,” 22nd Pacific Science Congress (Kuala Lumpur).
Keywords: genome assembly, PacBio, Platygyra sinensis, transcriptome, annotation, brain coral
Citation: Pootakham W, Sonthirod C, Naktang C, Kongjandtre N, Putchim L, Sangsrakru D, Yoocha T and Tangphatsornruang S (2021) De novo Assembly of the Brain Coral Platygyra sinensis Genome. Front. Mar. Sci. 8:732650. doi: 10.3389/fmars.2021.732650
Received: 29 June 2021; Accepted: 24 September 2021;
Published: 18 October 2021.
Edited by:
Michael Sweet, University of Derby, United KingdomReviewed by:
Ira Cooke, James Cook University, AustraliaYi Jin Liew, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Copyright © 2021 Pootakham, Sonthirod, Naktang, Kongjandtre, Putchim, Sangsrakru, Yoocha and Tangphatsornruang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wirulda Pootakham, d2lydWxkYUBhbHVtbmkuc3RhbmZvcmQuZWR1
†These authors have contributed equally to this work