 Songqian Huang1*
Songqian Huang1* Yuki Ichikawa1Kazutoshi Yoshitake1Shigeharu Kinoshita1
Yuki Ichikawa1Kazutoshi Yoshitake1Shigeharu Kinoshita1 Md Asaduzzaman2Fumito Omori3Kaoru Maeyama3Kiyohito Nagai4
Md Asaduzzaman2Fumito Omori3Kaoru Maeyama3Kiyohito Nagai4 Shugo Watabe5Shuichi Asakawa1*
Shugo Watabe5Shuichi Asakawa1*- 1Graduate School of Agricultural and Life Sciences, The University of Tokyo, Bunkyo-ku, Japan
- 2Department of Marine Bioresources Science, Faculty of Fisheries, Chittagong Veterinary and Animal Sciences University, Chittagong, Bangladesh
- 3Mikimoto Pharmaceutical Co., Ltd., Mie, Japan
- 4Pearl Research Laboratory, K. MIKIMOTO & Co., Ltd., Mie, Japan
- 5School of Marine Biosciences, Kitasato University, Sagamihara, Japan
PIWI proteins and PIWI-interacting RNAs (piRNAs) suppress transposon activity in animals, thus safeguarding the genome from detrimental insertion mutagenesis. Recent studies revealed additional targets and functions of piRNAs in various animals. piRNAs are ubiquitously expressed in somatic tissues of the pearl oyster Pinctada fucata, however, the role of somatic piRNAs has not well characterized. This study reports the PIWI/piRNA pathway, including piRNA biogenesis and piRNA-mediated transposon silencing, and gene regulation in P. fucata. The biogenesis factors of PIWI, Zucchini, and HEN1, which are ubiquitous in somatic and gonadal tissues, were first identified in P. fucata using transcriptome analysis. Bioinformatics analyses suggested that different populations of piRNAs participate in the ping-pong amplification loop in a tissue-specific manner. In addition, a total of 69 piRNA clusters were identified in the genome of P. fucata based on the expression of piRNAs, which contained 26% transposons and enhanced for DNA/Crypton, LINE/CR1, SINE/Deu, and DNA/Academ. The expression patterns of the piRNAs and piRNA clusters in somatic tissues were not substantially different, but varied significantly between the somatic and gonadal tissues. Furthermore, locked-nucleic-acid modified oligonucleotide (LNA-antagonist) was used to silence single piRNA (piRNA0001) expression in P. fucata. Hundreds of endogenous genes were differentially expressed after piRNA silencing in P. fucata. Target prediction showed that some endogenous genes were targeted by piRNA0001, including twelve upregulated and nine downregulated genes after piRNA0001 silencing. The results indicated that piRNAs from somatic tissues may be related to gene regulation, whereas piRNAs from gonadal tissues are more closely associated to transposon silencing. This study will enhance our understanding of the role of piRNAs in mollusks, transposon silencing, and the regulatory function of the PIWI/piRNA pathway on protein-coding genes outside of germ line cells in P. fucata.
Introduction
Animal species express three types of endogenous silencing-instigating small RNAs: microRNAs (miRNAs), endogenous siRNAs (endo-siRNAs), and PIWI-interacting RNAs (piRNAs), based on their biogenesis mechanism and type of Argonaute-binding partners (Kim et al., 2009). miRNAs and endo-siRNAs, which are usually 20–23 nucleotides (nt) in length, are generated from double-stranded precursors by Dicer (Peters and Meister, 2007; Kim et al., 2009), whereas piRNAs are generated from single-stranded precursors independent of RNase III enzymes (Houwing et al., 2007), which are necessary for miRNA and endo-siRNA biogenesis. piRNAs are associated with PIWI subfamily members of the Argonaute family of proteins, whereas miRNAs and endo-siRNAs are associated with AGO subfamily members. The large Argonaute superfamily is characterized by a conserved PAZ domain, which is a single-stranded nucleic acid binding motif (Cerutti et al., 2000), and the PIWI domain, which implements RNase H slicing activity (Liu et al., 2004; Song et al., 2004; Tolia and Joshua-Tor, 2007).
The mechanisms underlying piRNA biogenesis and function remain largely unknown, mainly because the process has few similarities with the miRNA and endo-siRNA pathways. However, significant progress has recently been made, particularly in the area of piRNA biogenesis (Izumi et al., 2020). A comprehensive computational analysis of piRNA populations generated two models for piRNA biogenesis in various animals: the primary biogenesis pathway and the amplification loop or ping-pong cycle (Ishizu et al., 2012; Ross et al., 2014). In the primary biogenesis pathway, long piRNA precursors are transcribed from specific genomic loci called piRNA clusters, cleaved, and modified by intricate factors in the cytoplasm before being transported into the nucleus by specific transcription factor complexes (Izumi and Tomari, 2014; Ross et al., 2014). Primary piRNAs undergo an amplification process to induce high piRNA expression, known as the amplification loop or ping-pong cycle (Ishizu et al., 2012). The Zucchini (Zuc) endonuclease potentially forms the piRNA 5′ end, whereas the 3′ end is 2′-O-methylated by HEN1/Pimet, associated with PIWI proteins (Saito et al., 2007; Ipsaro et al., 2012). In addition, an uncharacterized 3′-5′ exonuclease such as Trimmer was observed to trim the 3′ end of piRNAs in silkworms (Kawaoka et al., 2011; Izumi et al., 2016). Hsp83/Shu may play a role in the PIWI loading step, and Hsp90/FKBP6 plays a role in secondary piRNA production (Ishizu et al., 2012). Despite the recent characterization of several factors in the piRNA biogenesis pathway, it remains poorly understood.
PIWI proteins and their associated piRNAs suppress transposon activity in various animals, thereby safeguarding the genome from detrimental insertion mutagenesis (Aravin et al., 2001, 2007; Carmell et al., 2007; Yang and Xi, 2017). However, many animals produce piRNAs that do not match transposon sequences determined in Caenorhabditis elegans (Zhang et al., 2018) and mouse genome (Aravin et al., 2007), suggesting additional piRNA targets and functions. Nucleic small RNA-mediated silencing has gained significant attention in recent years, and numerous advances have been made in this field. Recent findings describing the role of transposon transcriptional regulation downstream of piRNAs have provided a paradigm for studying transcriptional regulation by small RNAs in animals. The use of C. elegans, Drosophila melanogaster, and mice as simple model systems, can offer important new insights into this process (Weick and Miska, 2014; Zhang et al., 2018). For example, Fas3 was postulated to be regulated by piRNAs generated from the 3′ untranslated regions (3′ UTR) of the traffic jam transcript (Saito et al., 2009), whereas traffic jam protein levels were elevated in PIWI mutants, indicating a cis-regulatory mechanism for piRNA action in D. melanogaster (Robine et al., 2009).
Mollusks are one of the largest groups of marine animals, of which Pinctada fucata is well studied due to its economic potential for pearl production and a model organism to investigate the fascinating biology of mollusks. Recent studies have revealed that PIWI proteins are ubiquitously expressed in mollusks (Rajasethupathy et al., 2012; Ma et al., 2017; Jehn et al., 2018). Moreover, abundant piRNA-sized small RNAs were observed in the oysters Crassostrea gigas (Xu et al., 2014; Zhou et al., 2014), Pinctada martensii (Jiao et al., 2014), and the scallop Chlamys farreri (Chen et al., 2014). Our previous study characterized the ubiquitous expression of piRNAs in somatic and gonadal tissues (Huang et al., 2019a). However, piRNA biogenesis and their functions in P. fucata remain largely unknown. This study aimed to demonstrate the biogenesis and function of PIWI/piRNA, and investigate its transposon silencing and endogenous gene regulation in P. fucata. Several key piRNA biogenesis factors (PIWI, Zucchini, and HEN1) have been identified in P. fucata, and the primary and secondary biogenesis pathways were determined by comprehensive computational analysis. To understand piRNA-regulated endogenous gene expression, we examined the effect of silencing of one of the most abundant piRNAs (piRNA0001) on potential target genes, such as miRNAs (predicted by a bioinformatics algorithm), using an LNA-antagonist.
Materials and Methods
Transcriptomic Analysis of piRNA Biogenesis Factors in P. fucata
Six (female: male = 1: 1) 1-year-old and six (female: male = 1: 13-year-old pearl oysters were collected from the Mikimoto Pearl Research Institution Base, Mie Prefecture, Japan in May 2018. Adductor muscle (Ad), gill (Gi), mantle (Ma), and gonad (Go) tissues were collected and individually transferred to 2-mL tubes containing RNA later (QIAGEN, Maryland, United States) separately. The samples were stored overnight at 4°C and preserved at −80°C until further analysis. Total RNA was isolated from each sample using the RNeasy Mini Kit (QIAGEN), according to the manufacturer’s instructions. After assessing RNA quality and quantity using the Agilent 2200 TapeStation (Agilent Technologies, Waldbronn, Germany), three isolated total RNA samples were mixed at equivalent concentrations to construct an RNA sequencing library for each tissue sample at different ages. A total of 2 μg of mixed total RNA was used for library construction according to the manufacturer’s protocol and subjected to paired-end sequencing on the Illumina HiSeq 4,000 platform. High-quality reads were required for de novo assembly analysis. Before assembly, raw reads were trimmed by removing adapter sequences and low-quality reads. Clean reads were assembled into unigenes using the Trinity software (Grabherr et al., 2011), and transcriptome annotation was performed using the Trinotate pipeline1 (Bryant et al., 2017). The edgeR package in R software was used to identify and draw the significant differentially expressed genes (DEGs) between two different libraries with the following threshold: P-value < 0.05, and folds > 2 (Robinson et al., 2010).
The consensus sequences of piRNA biogenesis factors were assembled using CLC Genomics Workbench version 8.0.1 (QIAGEN) and confirmed by published homolog sequences using BLASTp. Open reading frames (ORFs) were identified using the ORF finder available at NCBI.2 The structural features of the deduced protein sequences were analyzed via SMART webtool3 (Letunic and Bork, 2017). The predicted molecular weights and isoelectric points were determined using ExPASy (Gasteiger et al., 2005). All nucleotide and deduced amino acid sequences were electronically edited using BioEdit (Hall, 1999). Sequence similarity of the deduced proteins was performed using BLAST.4 Homologous sequences of piRNA biogenesis factors were downloaded from NCBI Protein databases and the alignment of amino acid sequences was performed using MEGA5.0 (Tamura et al., 2014). The neighbor-joining method from MEGA 5.0 was used for building phylogenic trees with 1,000 bootstrap replications.
piRNA Processing in P. fucata
Eight small RNA libraries from the adductor muscle, gill, ovary, and mantle tissues of P. fucata were sequenced using the Ion Proton system in our laboratory (the sequencing raw data were stored in the DNA Data Bank of Japan (DDBJ) under accession number DRA006953). Reads that did not produce a match to known non-coding RNAs were considered putative piRNAs and were merged for piRNA cluster prediction using proTRAC (v2.4.2) with default settings (Jehn et al., 2018). piRNA-sized molecules, approximately 30 nt in length, were widely expressed in all tissues and were used for ping-pong amplification analysis in P. fucata. Small RNA reads were pooled from the same tissues to perform piRNA annotation by the unitas (v1.5.3), which was run with the option -pp (Gebert et al., 2017). To compare ping-pong signatures and the number of sequence reads within different tissue types, PPmeter (v0.4) was used to quantify and compare the extent of ongoing ping-pong amplification (Jehn et al., 2018). Pseudo-replicates were generated by repeated bootstrapping (default = 100) of a fixed number of sequence reads (default = 1,000,000) from a set of original small RNA sequence datasets. Thereafter, the ping-pong signature of each pseudo-replicate was then calculated, and the number of sequence reads that participated in the ping-pong amplification loop was counted. The resulting parameter (ping-pong reads per million bootstrapped reads, ppr-mbr) were subsequently used to quantify and compare ping-pong activity in different tissue datasets. Both tools were operated using default parameter settings.
Transposon Element Annotation
We performed a de novo prediction of repetitive elements in the genome of P. fucata using RepeatMasker (v4.0.7) (Chen, 2004) based on a de novo repeat library, constructed using RepeatModeler (v1.0.11) (Flynn et al., 2020). An analysis was also conducted with the entire repeat data set and repeats localized in predicted piRNA clusters in P. fucata. The enrichment of transposon categories was compared between the genome and piRNA clusters. The relative expression patterns of piRNA clusters were calculated as mapped reads normalized for piRNA cluster length according to transcripts per million reads (TPM) (Li and Dewey, 2011). ggplot2 and pheatmap packages in R were used to perform principal component analysis (PCA) and heat maps of piRNA and piRNA cluster expression performance in the somatic and gonadal tissues. A t-test was used to compare the transposon percentages among different piRNA clusters between the somatic and gonadal tissues.
piRNA0001 Silencing in P. fucata and Quantification of piRNA Expression
A single piRNA with the sequence 5′-UACUUUAACAUG GCACAGAUAUAAUGACCU-3′ (piRNA0001) showed the highest expression in P. fucata somatic tissues, which was not annotated in any piRNA clusters. To explore its function, an LNA-antagonist was used to silence its expression in P. fucata. Eight P. fucata oysters (approximately 3 years old) were randomly divided into two groups: the LNA-antagonist group (LNA) and the control group (Con). Oysters from the LNA group were injected with 100 μL of 5 mg mL–1 LNA-antagonist probe solution into their body cavity. In contrast, those in the Con group were injected with isotonic PBS solution. The sequence of the high-affinity LNA-antagonist was 5′-AggTcaTtaTatCtgTgcCa-3′ (LNA in capitals) (Elmén et al., 2008). 2 weeks after injection, all individuals were subjected to tissue collection. Somatic tissues, including the adductor muscle, gill, and mantle, and gonadal tissues were collected and stored separately in 2-mL tubes containing RNA later (QIAGEN) overnight at 4°C, and preserved at –80°C until further use. Stem-loop RT-PCR was used for piRNA0001 relative expression analysis using U6 sRNA as a reference gene. This process was performed as described in our previous study (Huang et al., 2019b).
Transcriptomic Analysis of Gene Expression Profiles After piRNA0001 Silencing in P. fucata
Twenty-four libraries of somatic tissues, including adductor muscle, gill, and mantle, with four replicates for each tissue type from the LNA and Con groups, were constructed for RNA sequencing in 2017. Library construction, sequencing, de novo assembly, and functional annotation were performed as described above. Mapped fragments were normalized for RNA length according to the TPM method (Li and Dewey, 2011), facilitating the comparison of transcript levels between libraries. The analysis of DEGs between the Con and LNA groups was the same as in the previous method using edgeR.
Prediction of piRNA0001 Targeting Sites in P. fucata
piRNAs possess second to eighth nucleotide seed sequence that requires near-perfect complementarity between the piRNA and target genes, such as miRNAs (Gou et al., 2014). Additional base-pairing outside of the seed is also important for piRNA targeting. piRNAs can only tolerate a few mismatches outside the seed region (Shen et al., 2018; Zhang et al., 2018). In this study, 3′ UTR sequences of DEGs were used for piRNA target site predictions. No piRNA target rules have been explored in mollusks, therefore, we used miRNA-like pairing rules to predict potential target sites (Wang et al., 2018). These were predicted between piRNA and mRNA 3′ UTR using miRanda tools5 (Enright et al., 2003) with an alignment score > 150 and energy < –10 kcal mo–1.
Gene Expression Analysis by RT-PCR
To validate PIWI gene expression in P. fucata, the early development stage was achieved by artificial fertilization, and the somatic and gonadal tissues were dissected from 1- to 3-years-old oysters. The corresponding primers were designed using Primer5.0, based on the complete sequence with the following parameters: primer length between 18 and 22 bp, melting temperatures (Tm) between 55 and 65°C, product size between 80 and 150 bp, and avoidance of primer dimers or hairpin structure (Supplementary Table 1). β-actin was used as reference gene for PIWI relative expression calculations. First-stand cDNA was synthesized using the PrimerScript RT Master Mix reagent Kit (TaKaRa, Shiga, Japan), according to the manufacturer’s instructions. To determine the expression patterns of PIWI genes in various tissues at different developmental stages, all samples were analyzed using the Applied Biosystem 7,300 Fast Real-Time PCR System (Life Technologies, California, United States). RT-PCR analysis was performed in a 96-well plate, in which each well contained 20 μL of reaction mixture consisting of 10 μL SYBR Premix Ex Taq II (TaKaRa), 0.4 μL ROX Reference Dye (50×) (TaKaRa), 0.8 μL of each primer (10 μM), 2 μL of complementary DNA (cDNA) template and 6 μL of sterilized double-distilled water. RT-PCR conditions were as follows: pre-denaturation at 95°C for 30 s, followed by 40 cycles of amplification at 95°C for 5 s and 60°C for 31 s, a dissociation stage with one cycle at 95°C for 15 s, 60°C for 60 s, and 95°C for 15 s after amplification. Each sample was tested in triplicate. The average value per gene was calculated from three replicates.
Nine DEGs, predicted to be targeted by piRNA0001, were also selected for relative expression analysis by RT-PCR as described above, with total RNA consistent with RNA_Seq2 libraries. The resulting PCR products contained the predicted interaction sites on the mRNA 3′ UTR. The relative expression of each mRNA quantified by RT-PCR was presented as n = 4, from three replicates. Ct values were represented by the mean values of three independent replicates. The relative expression level of each gene was calculated using the 2(-△△Ct method (Pfaffl, 2011). A t-test was used to compare the differences in the relative expression of LNA/Con between RNA-seq and RT-PCR.
Results
piRNA Biogenesis Pathway in P. fucata
Following clean-up and quality checks, sixteen transcriptomic libraries with 191.11 million reads, with a total length of 28.67 Gb, were used for de novo assembly (Supplementary Table 2). A total of 324,779 transcripts were assembled, with a mean length of 564 bp (Table 1, RNA_Seq1). Complete sequences of PIWI (Piwil1 and Piwil2), Zucchini (Zuc), and HEN1 were assembled based on the annotation results of transcripts, and their respective lengths are listed in Table 2. PIWI possessed the conserved PAZ and PIWI domains as homologs in other organisms, whereas the Zuc protein contained the PLDc domain (Figure 1). The conserved identities of the PAZ and PIWI domains were 51.37 and 60.54%, respectively. Piwil1 and Piwil2 clustered into two categories of the PIWI subfamily (Supplementary Figure 1).
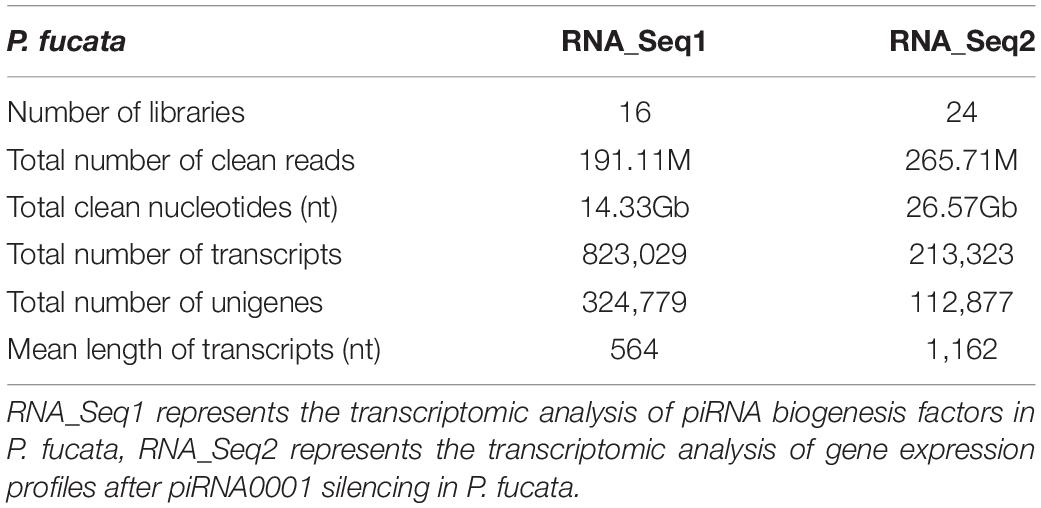
Table 1. Summary of the mixed-assembling results of RNA sequencing in P. fucata.
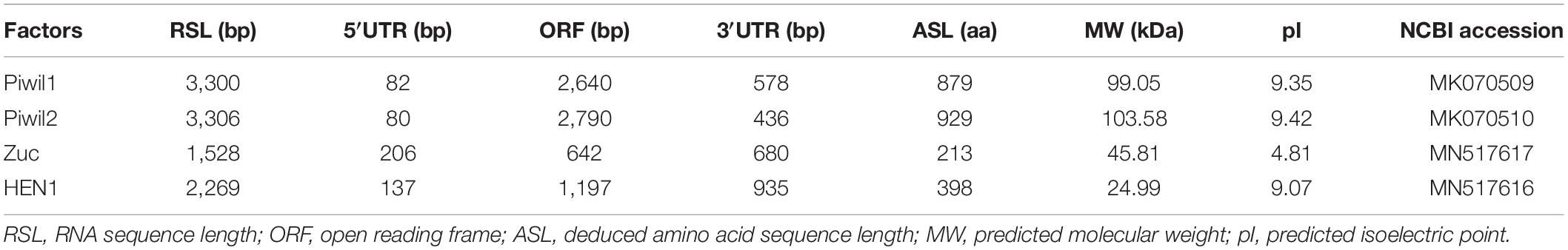
Table 2. piRNA biogenesis factors assembly in P. fucata.
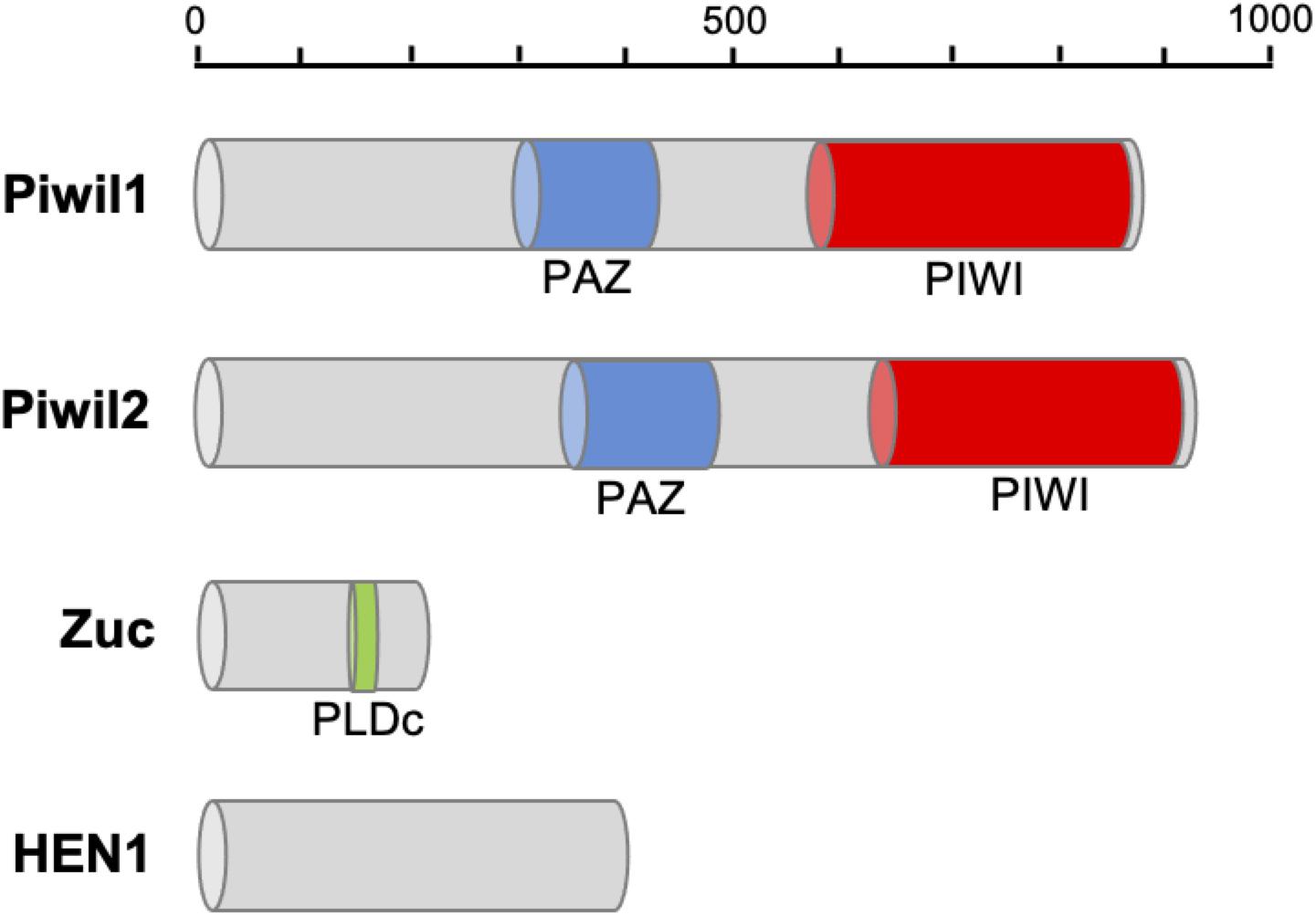
Figure 1. Protein domain structure of piRNA biogenesis factors in P. fucata. The PAZ domain contributes to specific and productive incorporation of miRNAs, endo-siRNAs, and piRNAs into the RNAi pathway; the PIWI domain can be inferred to cleave single-stranded RNA, such as mRNA should be guided by single-stranded small RNAs; PIWI domains place the guide uniquely in the proper position in Argonaute-RNA complexes. PLDc produces phosphatidic acid from phosphatidylcholine, which may be essential for the formation of certain types of transport vesicles or involved in constitutive vesicular transport via signal transduction pathways.
The somatic and gonadal tissues of 1- and 3-years-old individuals were used for transcriptome analysis to study the expression profiles of the piRNA biogenesis factors. Gene expression was calculated as transcripts per million (TPM) using transcriptomic analysis. Piwil1 and Piwil2 were widely expressed in the P. fucata somatic and gonadal tissues, with significantly higher expression in the gonadal tissues at 1- and3-years-old stages (Figures 2A,B). Piwil1 expression levels were significantly higher than those in Piwil2. Zuc was highly expressed in the gonadal tissues, particularly in3-years-old females (Figure 2C), and HEN1 was expressed in all examined tissues (Figure 2D). Furthermore, transcriptome and early development stage samples were used for gene expression verification. RT-PCR analysis validated Piwil1 and Piwil2 expression levels in larvae, juveniles, and adult P. fucata, which were high in fertilized eggs (0 hpf), significantly decreased in embryos (0.5 hpf), and weak from the blastula (4 hpf) to spat (30 dpf) stages (Supplementary Figures 2A,B). Piwil1 and Piwil2 were ubiquitously expressed in the somatic and gonadal tissues and were highly expressed in female P. fucata, both at the 1- and 3-years-old stages. These piRNA biogenesis factors are highly expressed in the gonadal tissues of other animals; however, they are weakly but ubiquitously expressed in somatic tissues, indicating their contribution to piRNA biogenesis in P. fucata somatic tissues.
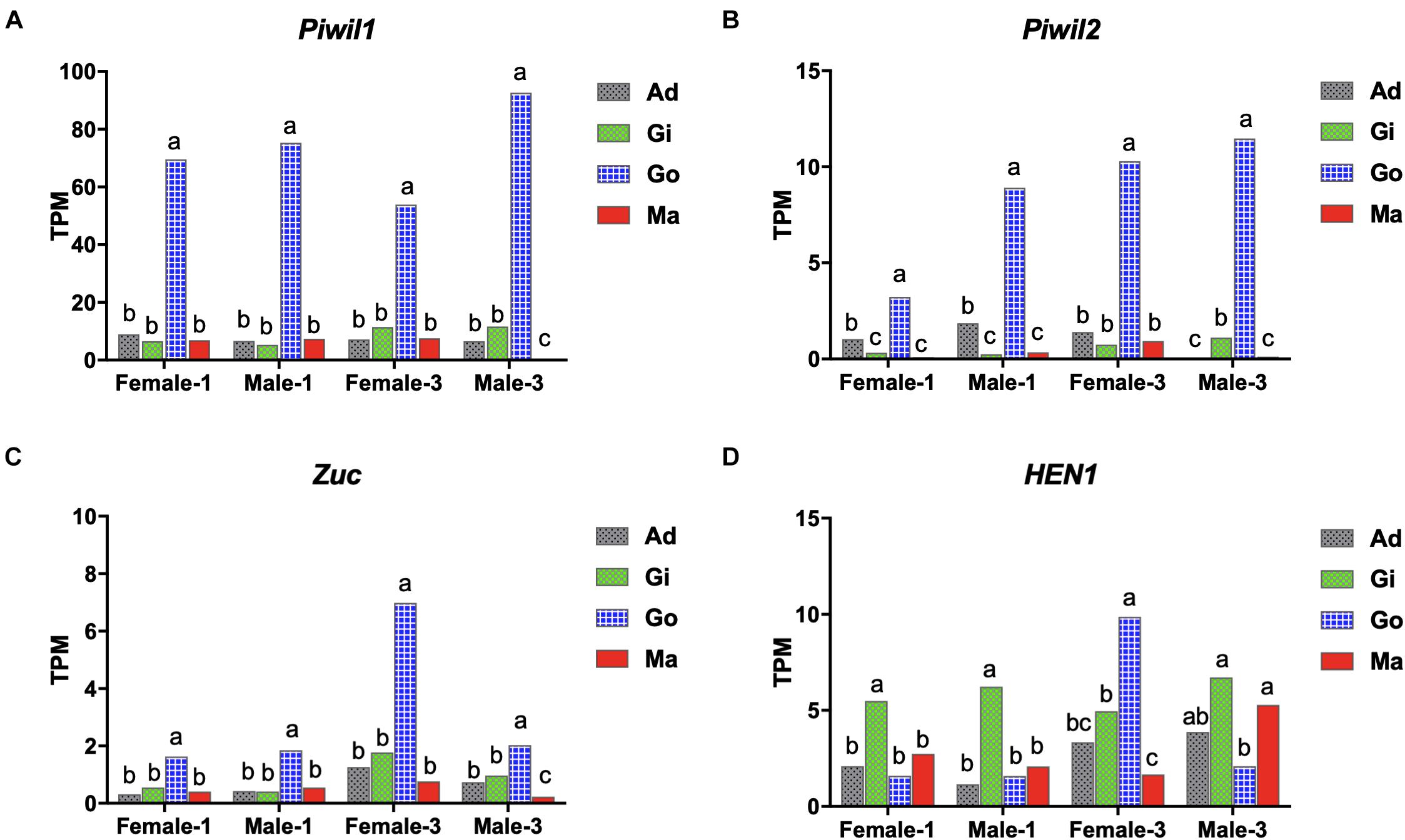
Figure 2. Expression performance of piRNA biogenesis factors in P. fucata at different developmental stages analyzed by RNA-seq. (A) PIWI-like 1 gene (Piwil1). (B) PIWI-like 2 gene (Piwil2). (C) Zucchini gene (Zuc). (D) HEN1 gene. Different lowercase letters represent differentially expressed genes (P < 0.05). TPM, transcripts per million; Ad, adductor muscle; Gi, gill; Go, gonad; Ma, mantle.
P. fucata encodes two ubiquitously expressed PIWI proteins, therefore, we further analyzed the participation of distinct piRNA populations in the ping-pong amplification loop. We employed a bioinformatics approach, under the premise that Piwil1 and Piwil2 bind to piRNAs with different length profiles, similar to their corresponding mouse homologs Miwi and Mili, which particularly bind to 29/30 nt and 26/27 nt long piRNAs, respectively (Vourekas et al., 2012). We analyzed pairs of P. fucata sequenced reads with a 10-bp 5′ overlap (ping-pong pairs), which is the typical sequence length of each ping-pong partner (Figure 3A). A significant 10 nt overlap between sense and antisense strands was detected in the small RNAs derived from P. fucata, which is consistent with piRNAs generated by the ping-pong amplification mechanism (Supplementary Figure 3). In somatic tissues, ping-pong pairs combine 29/30 nt long piRNAs, suggesting Piwil1-Piwil1-dependent homotypic ping-pong amplification. In the gonadal tissues, most ping-pong pairs combine piRNAs predominantly 29/30 nt and 25/26 nt long, suggesting both Piwil1-Piwil2-dependent heterotypic and Piwil1-Piwil1-dependent homotypic ping-pong amplification, as shown in Figure 3B. Furthermore, 29/30 nt long piRNAs that presumably bound to Piwil1 were heavily biased for a 5′ uridine (U), whereas 25/26 nt long piRNAs that presumably bound to Piwil2 showed a stronger bias for an adenine (A) at position 10.
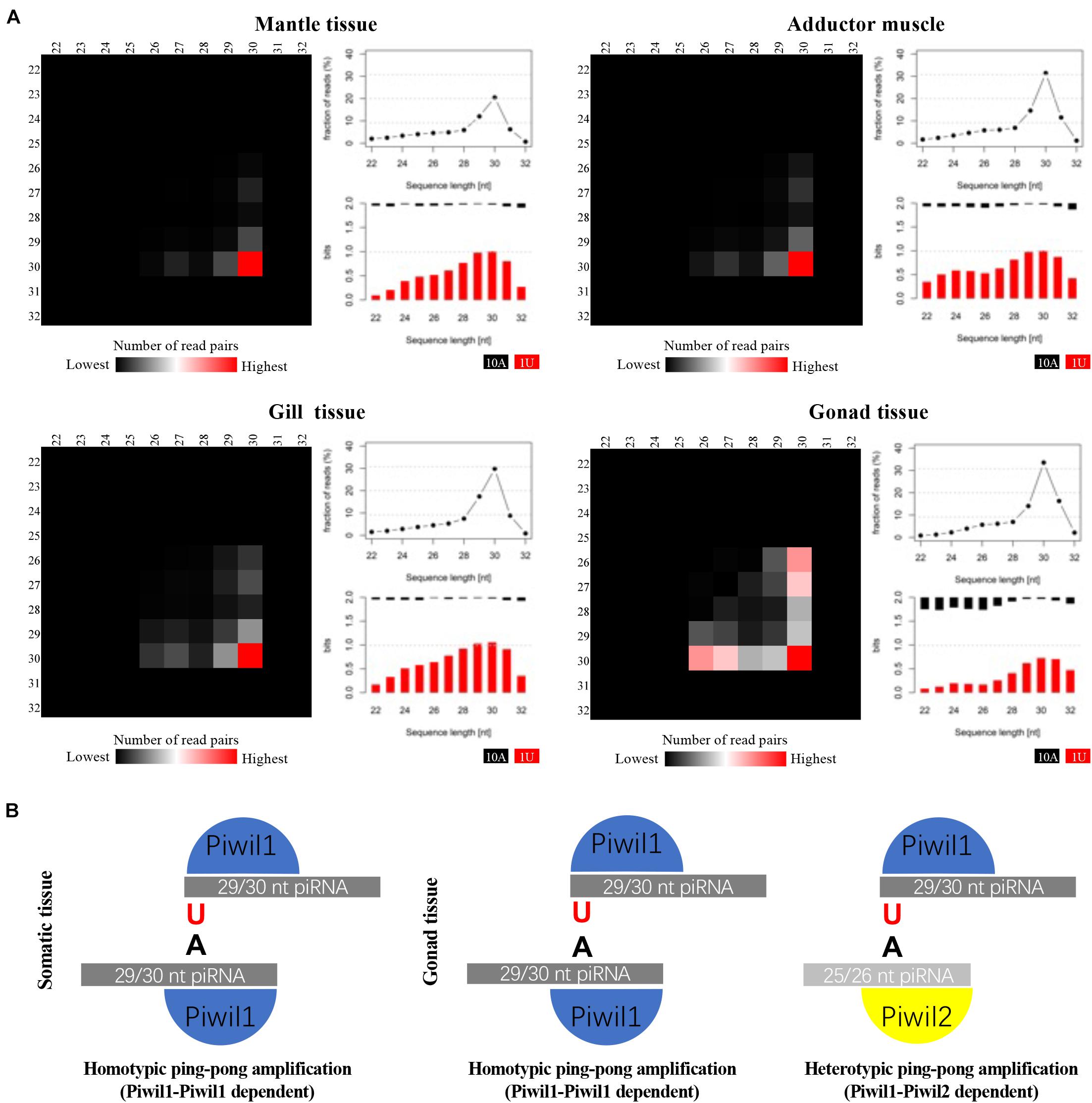
Figure 3. Analysis of putative piRNAs that participate in the ping-pong amplification loop in P. fucata. (A) Ping-pong matrices illustrate frequent length-combinations of ping-pong pairs (sequences with a 10-bp 5′ overlap). Sequence read length distribution and 1U/10A bias [bits] for ping-pong sequences are shown. (B) Proposed ping-pong amplification model in somatic (left) and gonadal (right) tissues of P. fucata.
piRNA-Mediated Transposon Silencing in P. fucata
Small RNA sequencing was performed to identify putative piRNAs in P. fucata (Huang et al., 2019a). Reads that did not produce a match to known non-coding RNAs were considered as putative piRNAs and were merged for piRNA processing. A total of 69 piRNA clusters (total size 0.58 Mb, 0.056% of the genome) were identified in the genome of P. fucata (Figure 4A and Supplementary Table 3). The P. fucata genome contains 539.3 Mb repetitive sequences accounting for 52% of the genome; however, 23.3% of repetitive sequences are classified as unknown (Figure 4B). In line with the TE suppressive role of piRNAs, the identified piRNA clusters also showed a lower enrichment for transposon sequences compared to the whole genome situation, which contained 15% known and 11.06% unknown transposons (Figure 4C). The composition of transposons in piRNA clusters does not at all reflect to the transposon landscape of the whole genome. Therefore, piRNA clusters were enriched for DNA/Crypton, LINE/CR1, SINE/Deu, and DNA/Academ showing up to 28-fold enrichment in piRNA clusters (Figure 4D). In addition, a total of 43,266 unigenes (13.32% of the total) from RNA-Seq1 were annotated with transposon consensus (Supplementary Figure 4). However, thousands of distinct piRNAs do not map to the transposons in P. fucata.
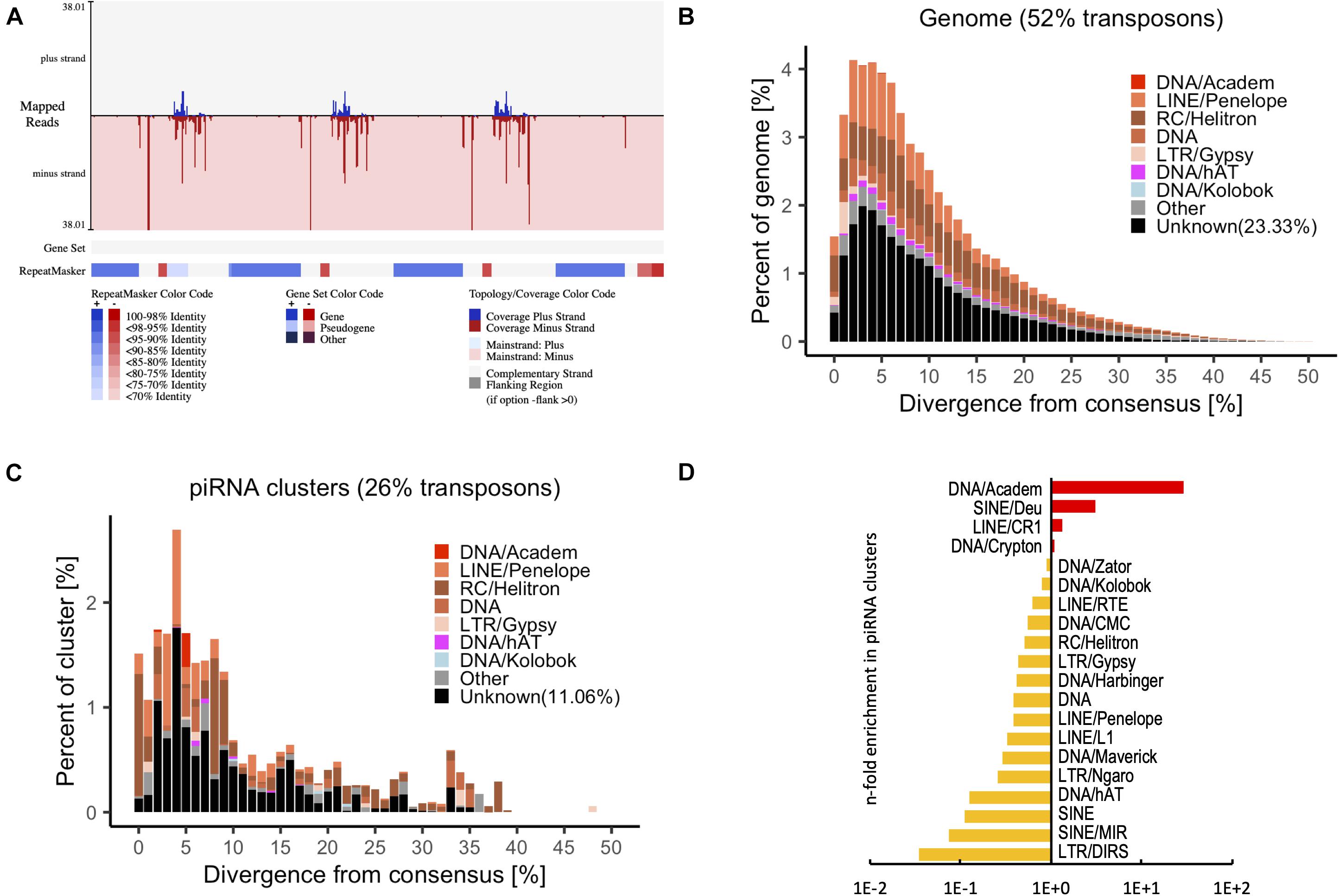
Figure 4. Transposon annotation in the genome and piRNA clusters from putative piRNAs in P. fucata samples. (A) Example of a piRNA cluster predicted by proTRAC with annotation of transposon consensus sequences and gene set. Transposon sequences were detected among most of the piRNA clusters. (B) Representation of transposons in the genome of P. fucata plotted by divergence [%] from transposon consensus. (C) Representation of transposons within piRNA clusters of P. fucata plotted by divergence [%] from transposon consensus. (D) Prominent transposons that are enriched or depleted in P. fucata piRNA clusters.
It is well known that piRNAs suppress transposons in germ line (Ishizu et al., 2012; Yang and Xi, 2017). The expression performance of putative piRNAs was compared between the somatic and gonadal tissues. The PCA of putative piRNA expression patterns from all examined libraries clustered the somatic tissues together and differentiated the somatic and gonadal tissues (Figure 5A). All putative piRNA reads were re-mapped with piRNA clusters to calculate the relative expression performance using the TPM method. The expression performance of these piRNA clusters was also differentiated the somatic and gonadal tissues; however, there was small difference within different somatic tissues (Figure 5B). In addition, two different categories of piRNA clusters were clustered in the somatic and gonadal tissues (Figure 5B). The piRNA clusters of Class 1 were highly expressed in the gonadal tissues, whereas a piRNA clusters of Class 2 were highly expressed in the somatic tissues. The transposon element percentage of each piRNA cluster was compared between Class 1 and Class 2. The piRNA clusters of Class 1 had an average transposon percentage of 32.16%, which was significantly higher than that of the piRNA clusters of Class 2 (1.85×, P < 0.01) (Figure 5C).
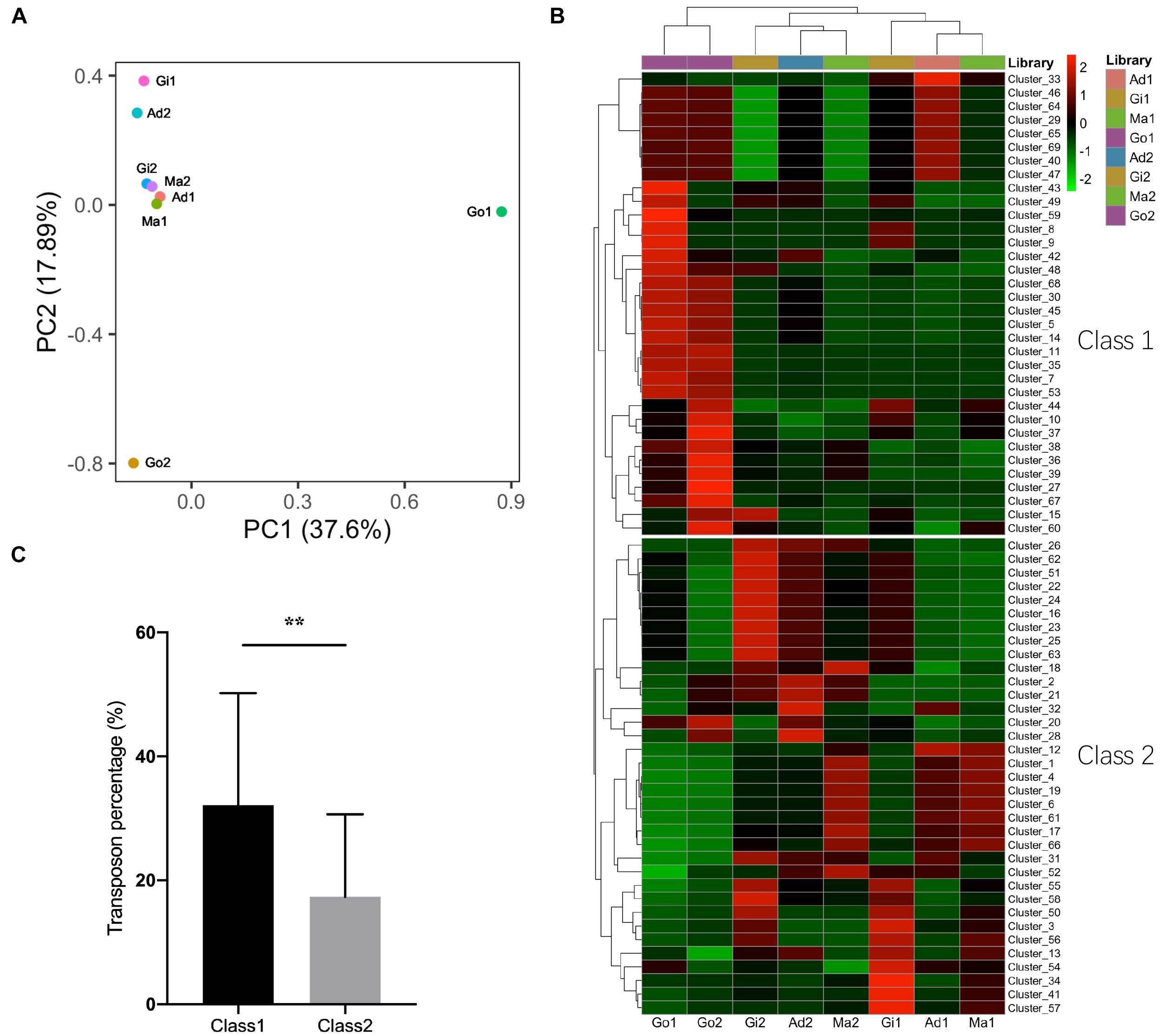
Figure 5. Expression profiles of piRNAs and piRNA clusters in P. fucata. (A) PCA analysis of the expression patterns of putative piRNAs in P. fucata somatic and gonadal tissues. (B) Heat map of expression levels of piRNA clusters in P. fucata somatic and gonadal tissues. (C) Difference in transposon percentage in different piRNA cluster categories. Class1 and Class2 represent different piRNA clusters in (B). Double-asterisk (**P < 0.01) indicates significant differences in transposon percentage between Class1 and Class2 piRNA clusters. Ma, Mantle tissue; Ad, Adductor muscle; Gi, Gill tissue; Go, Gonad.
piRNA-Mediated Gene Regulation in P. fucata
LNA nucleotides can be hybridized with DNA or RNA residues in the oligonucleotide to form stable heteroduplexes between the LNA-antagonist and small RNA to silence its expression (Elmén et al., 2008). A modified LNA-antagonist was used to silence the expression of piRNA0001 in P. fucata. All samples were dissected for tissue collection 2 weeks after the injection. Following RNA extraction, stem-loop RT-PCR was used for the relative quantitative analysis of piRNAs. piRNA0001 expression levels in LNA-introduced pearl oysters were decreased in the adductor muscle (0.04×, p < 0.01), gill (0.19×, p < 0.01), gonad (0.28×, p < 0.01), and mantle tissues (0.62×, p < 0.05) compared to those of the control group (Figure 6A). Moreover, the piRNA0001 precursor detected by transcriptomic analysis of gene expression profiles showed no significant differences in the expression between the LNA and Con groups (Supplementary Figure 5).
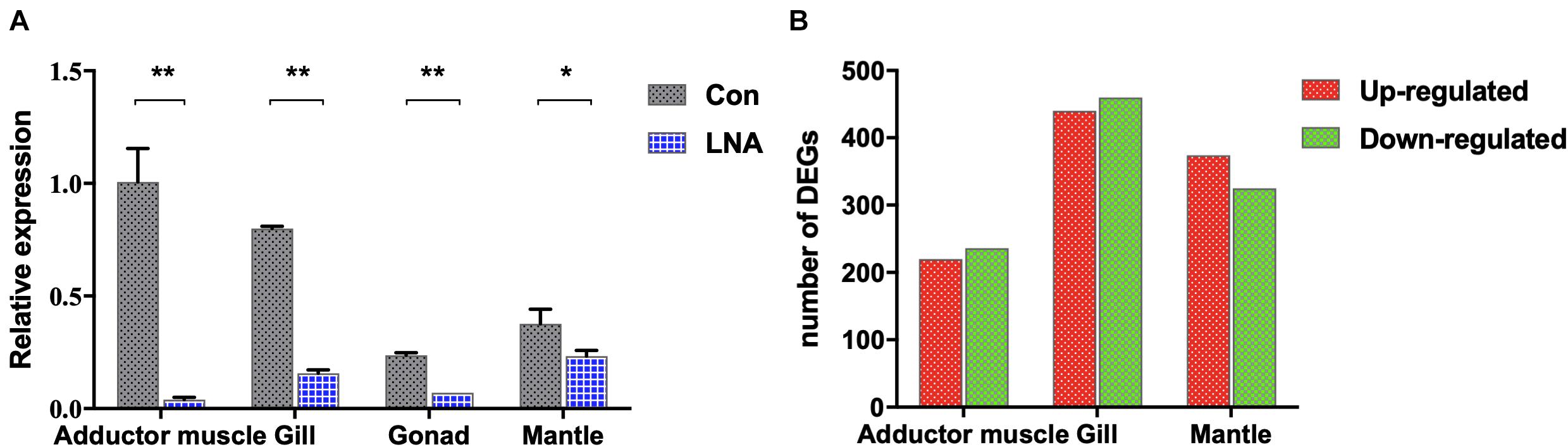
Figure 6. piRNA0001 silencing in P. fucata. (A) piRNA0001 expression profiles in P. fucata treated with LNA-antagonist. U6 was used as the reference gene. The relative expression of piRNA0001 was normalized by piRNA0001 expression following LNA-antagonist treatment. Differences were statistically analyzed between Con and LNA groups using one-way analysis of variance (ANOVA). Significant differences are marked with * (p < 0.05) and ** (p < 0.01). (B) Number of differentially expressed genes (DEGs) between LNA and Con groups below the following threshold: P-value < 0.05 and folds > 2.
Twenty-four libraries from the Con and LNA groups, including adductor muscle, gill, gonad, and mantle tissues were constructed for RNA sequencing, with three replicates. Following data cleaning and quality checks, RNA sequencing produced 265.71 million clean reads (Supplementary Table 4), that is, a total length of 26.57 Gb, further assembled into 213,323 transcripts by Trinity (Table 1, RNA_Seq2). The Trinotate pipeline annotated 48.63% of these transcripts, based on five public databases (NT, Swiss-Prot, Pfam, GO, and KEGG), and identified 31.06, 63.37, 80.60, 60.88, and 38.14% transcripts in the NT, Swiss-Prot, Pfam, GO, and KEGG databases, respectively. In addition, most transcripts were blasted with P. fucata and other mollusk species (Supplementary Figure 6).
Analysis of DEGs in somatic tissues between Con and LNA groups was performed to understand further the molecular events involved in the functions of piRNA0001 in P. fucata. We selected the genes from at least one group with a mean TPM value > 5 for analysis using edgeR (P-value < 0.05 and fold > 2). A total of 2,904 transcripts showed significant differential expression between the Con and LNA groups (Supplementary Table 5). Pairwise comparison of the adductor muscle revealed 456 differentially expressed transcripts, including 220 upregulated and 236 downregulated genes after LNA-antagonist treatment (in comparison with the Con group). In addition, analysis of 900 transcripts revealed 440 upregulated and 460 downregulated DEGs in the gill tissue and 374 upregulated and 325 downregulated DEGs in the mantle tissues, following LNA-antagonist treatment (Figure 6B). Among these, 16 and 15 DEGs were typically upregulated and downregulated, respectively, in the three examined LNA-antagonist-treated tissue types.
All DEGs were used for piRNA0001 target prediction, with genes containing target sites in the 3′UTR considered as potential target genes. Four, nine, and eleven DEGs were predicted to be targeted by piRNA0001 in the adductor muscle, gill, and mantle tissues, respectively, with an average alignment score of 162.17 and average energy value of −14.00 kcal mol–1 (Figure 7). Target prediction was anticipated for FHOD3, SRS10, VINC, and ZNF622 in the adductor muscle, for ART2, EF1A, EMC7, ESI1L, NAC2, PA2HB, SYWC, TM87A, and ZNF622 in the gill tissue, and for CAH14, CBF, CDC42, FRRS1, GOLI4, KAT3, NBR1, PA2HB, TYR1, TYR2, and ZNF622 in the mantle tissue. The predicted piRNA-mRNA interaction is shown in Supplementary Table 6, including two sites on both the ART2 and CBF 3′UTR. No polyA signal was observed in the assembly transcripts of ART2, EF1A, ESI1L, FHOD3, PA2HB, SRS10, and TM87A, and predicted interaction sites on CAH14, NBR1, SYWC, and VINC were located after the polyA signal (Supplementary Sequences). The second piRNA:mRNA interaction site on ART2, NAC2, and VINC, were located far from the stop codon (Supplementary Table 6). Alignment pairing from the second to eighth nucleotides, considered to be the piRNA seed region, showed perfect complementarity. Furthermore, base-pairing outside the seed region is also important for piRNA target prediction. Following piRNA0001 silencing in P. fucata, FHOD3, SRS10, and ZNF622 were upregulated, whereas VINC were downregulated in the adductor muscle tissue. In the gill tissue, ART2, EF1A, EMC7, ESI1L, SYWC, TM87A, and ZNF622 were upregulated, whereas NAC2 and PA2HB were downregulated. In the mantle tissue, CBF, CDC42, NBR1, and ZNF622 were upregulated, while CAH14, FRRS1, GOLI4, KAT3, PA2HB, YTR1, and TYR2 were downregulated. ZNF622 was upregulated in all the examined tissues. Both upregulated and downregulated genes were predicted to be targeted by piRNA0001, demonstrating the diverse ways of piRNA-mediated gene regulation in P. fucata.
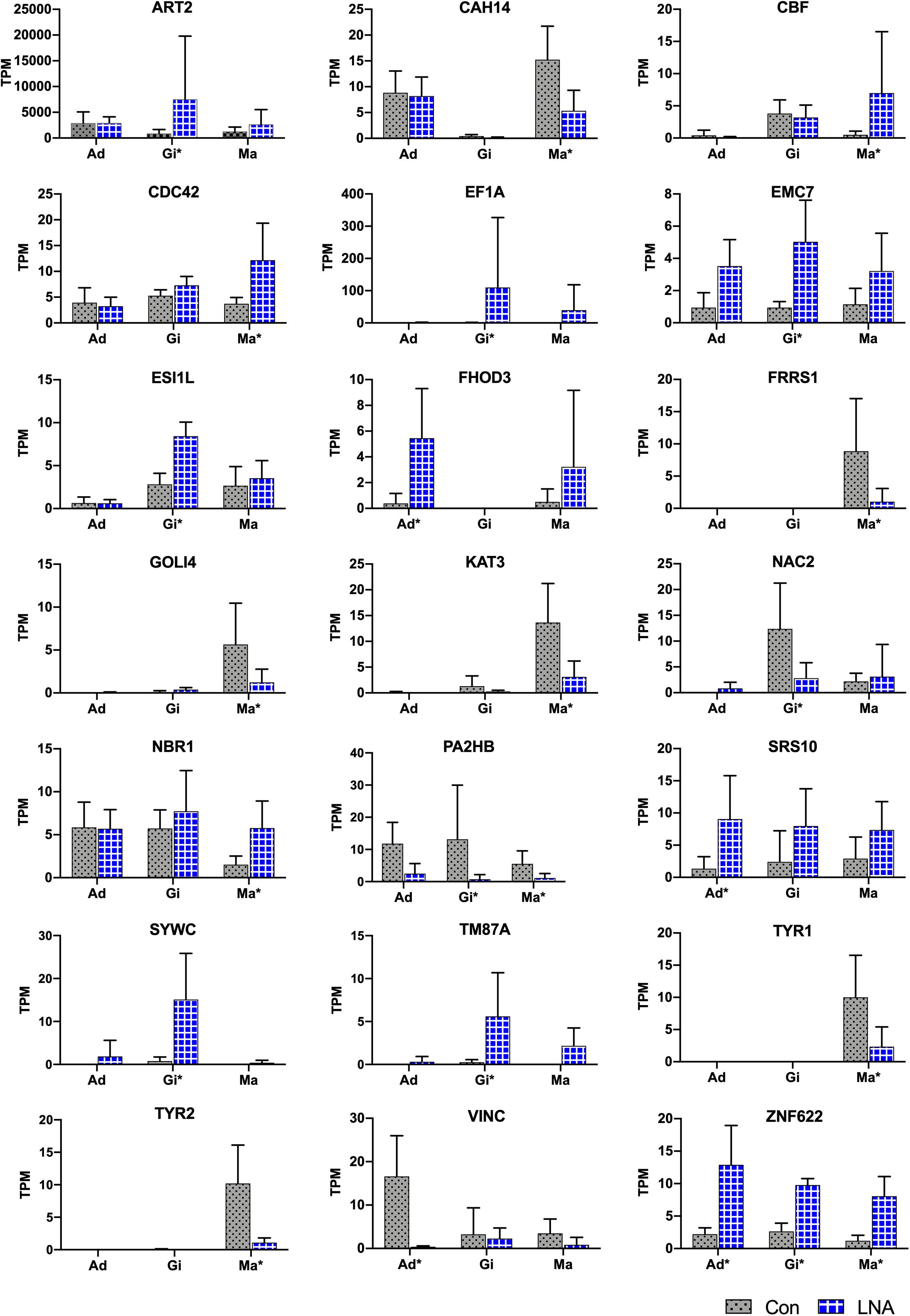
Figure 7. Target predictions of DEGs in P. fucata. TPM shows the relative expression level of target genes analyzed by RNA-seq. Tissue: adductor muscle (Ad), gill (Gi), and mantle (Ma). Large difference in standard deviations of relative expression level of genes analyzed by RNA-seq might cause errors during the complex sequencing protocols or differences among individual samples. Tissue marked with an asterisk (*) represents differentially expressed gene under the following threshold: P-value < 0.05 and folds > 2 in the present tissue.
Validation of Gene Expression by RT-PCR
Total RNA extracted from LNA and Con P. fucata somatic tissues was analyzed using RT-PCR, to determine the authenticity of mRNA expression calculated by RNA sequencing. Inconsistencies were detected in the adductor muscle VINC expression obtained from RNA sequencing and RT-PCR analysis (Figure 8), possibly caused by ineffective RT-PCR primers or by sequencing errors that occurred during the complex process, with the predicted products located after the polyA signal on the 3′UTR, far from the stop codon (Supplementary Sequences: VINC). However, the remaining eight predicted target genes demonstrated consistent levels of relative expression levels through RT-PCR, compared with the results obtained from RNA sequencing. Among these genes, ZNF622 was upregulated in all examined somatic tissues, following piRNA0001 silencing in P. fucata. In contrast, PA2HB and TYR1 were downregulated in the gill and mantle tissues, based on RNA sequencing and RT-PCR data.
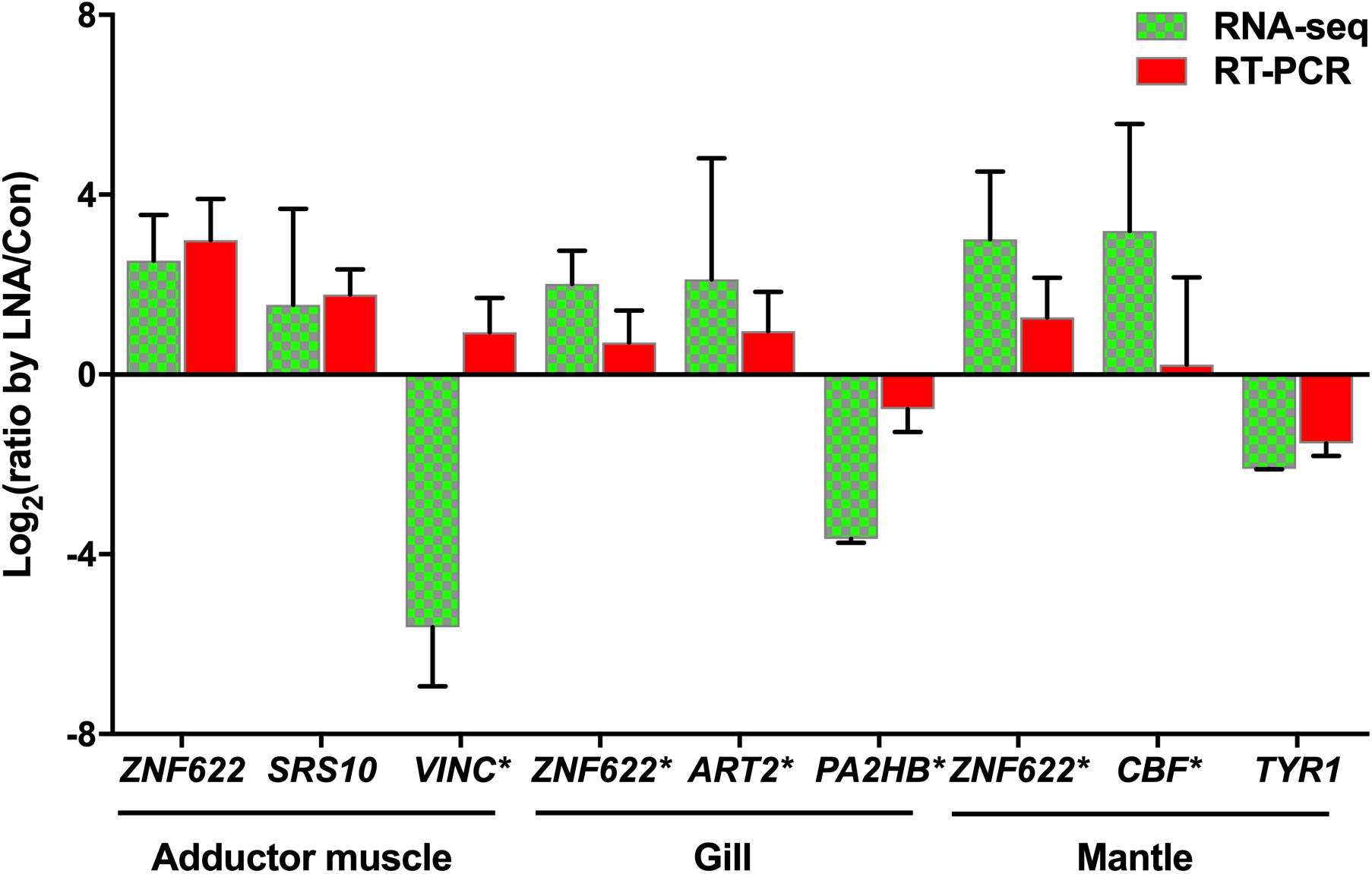
Figure 8. Validation of potential piRNA0001 predicted target genes in P. fucata. t-test was used to compare the differences in relative expressions of LP/LC for the eight DEGs found common between RNA-seq and qPCR. Gene marked with an asterisk (*) represents significant differences in the gene expression trend observed between RNA-seq and RT-PCR analysis using a t-test (P < 0.05).
Discussion
Analysis of the PIWI/piRNA pathway representative of several animals revealed an extensive diversity of lineage-specific adaptations, challenging the universal validity of data obtained from model organisms. PIWI proteins are characterized by two protein domains, namely the PAZ domain, an RNA-binding motif that binds the 3′ end of short RNAs, and the PIWI domain, which is structurally similar to the RNaseH catalytic domain (Parker and Barford, 2006). Numerous PIWI homologs have been identified in various organisms, such as four homologous Hiwi, Hili, Hiwi2, and Hiwi3 have been identified in Homo sapiens, and play a crucial role in human cancer and male germline cell development (Qiao et al., 2002; Sasaki et al., 2003; Liu et al., 2006); homologous Miwi, Mili, and Miwi2 were identified in Mus musculus, and their knockdown led to male sterility (Deng and Lin, 2002; Aravin et al., 2006; Lau et al., 2006); and two homolog, Ziwi and Zili, were identified in Danio rerio, and were both were crucial for germ cell differentiation and meiosis (Houwing et al., 2007, 2008). These findings indicate that PIWI may play an important role in germline cell development in organisms.
PIWI was first discovered in Drosophila, where it functions in germline cell maintenance and self-renewal (Cox et al., 1998). PIWI/piRNA research in germline cells has rapidly advanced. PIWI mutations lead to a profound infertility phenotype in mice (Deng and Lin, 2002; Carmell et al., 2007). In addition to PIWI function in germline cells, recent studies have demonstrated the expression and function of PIWI in the soma, from low eukaryotes to mammals. PIWI expression has been detected in human cancer cells (Ross et al., 2014; Krishnan et al., 2016) and mammalian somatic tissues (Yan et al., 2011). Although somatic PIWI was previously detected in Drosophila fat bodies and ovarian somatic tissues (Malone et al., 2009; Jones et al., 2016), further evidence of somatic PIWI/piRNA expression was observed in 16 out of 20 surveyed arthropod species (Lewis et al., 2018). In mollusks, PIWI proteins are widely expressed in Lymnaea stagnalis and C. gigas somatic tissues, and homologous PIWIs were also detected in 11 mollusk species, suggesting the occurrence of somatic PIWI/piRNA expression in early bilaterian ancestors (Jehn et al., 2018). In this study, Piwil1 and Piwil2, assembled by RNA sequencing in P. fucata, were ubiquitously expressed in all of the examined somatic tissues and gonadal tissues. These results provide further evidence of somatic PIWI/piRNA expression as an ancestral bilaterian trait (Lewis et al., 2018).
piRNA biogenesis in metazoa involves synthesizing primary piRNA from a piRNA cluster, assisted by several factors in the cytoplasm and nucleus. Thereafter, the long, single-stranded piRNA precursor was exported from the nucleus to the cytoplasm, where it was shortened to piRNA-like small RNAs by an undetermined endonuclease (Ishizu et al., 2012; Ross et al., 2014). Recent studies have indicated that Zuc may be an endonuclease that forms the 5′ end of piRNAs in Drosophila and mice (Ipsaro et al., 2012; Inoue et al., 2017; Izumi et al., 2020). The 3′ end was 2-O-methylated by HEN1 (Saito et al., 2007), whereas an uncharacterized 3′-5′ exonuclease has been shown to trim the 3′ end of piRNAs (Kawaoka et al., 2011; Izumi et al., 2016). In this study, several crucial piRNA biogenesis factors, including PIWI, Zuc, and HEN1, were assembled by RNA sequencing. These were ubiquitously expressed in all examined tissues, particularly in gonads, which previously showed a higher percentage of piRNA expression than somatic tissues (Huang et al., 2019a).
The ping-pong amplification loop is responsible for post-transcriptional silencing of transposable elements (Jehn et al., 2018). In Drosophila and mice, this process normally involves two PIWI proteins (heterotypic ping-pong), one loaded with antisense piRNAs targeting piRNA cluster transcripts, which contain transposon sequences in an antisense orientation (Aravin et al., 2008). The homotypic Aub:Aub ping-pong process occurs in Drosophila (Huang et al., 2014) and wild-type prenatal mouse testes (Miwi2:Miwi2 and Mili:Mili) (Aravin et al., 2008). In P. fucata, we also determined the amplification system for a secondary piRNA biogenesis pathway using a comprehensive computational analysis. A Piwil1-Piwil1-dependent homotypic ping-pong amplification loop was observed in the somatic and gonadal tissues, with a Piwil1-Piwil2 dependent heterotypic ping-pong amplification loop, simultaneously occurring in the gonadal tissues; however, we cannot determine whether the binding preferences of PIWI proteins have changed in P. fucata. Both PIWI proteins may bind to the entire range of piRNAs. However, based on the presence of piRNA populations with length profiles and their representation in ping-pong pairs, together with the differences in their number of 1U and 10A reads, we believe the above explanation is a reasonable and parsimonious interpretation of the data while acknowledging the possibility of others.
The expression of piRNA biogenesis factors in somatic tissues implies the potential existence of somatic piRNAs. Recent studies have revealed somatic piRNAs from sponges to humans (Yan et al., 2011; Ross et al., 2014). A functional non-gonadal somatic piRNA pathway in Drosophila fat bodies affects normal metabolism and overall organismal health (Jones et al., 2016). Somatic piRNAs are considered an ancestral trait of arthropods, which predominantly target transposable elements, suggesting that the piRNA pathway was active in the soma of the last common ancestor of arthropods to maintain mobile genetic elements in check (Lewis et al., 2018). Abundant putative piNRAs were observed in the somatic and gonadal tissues of P. fucata in our previous study (Huang et al., 2019a). In this study, we analyzed the expression of piRNAs and piRNA clusters in the somatic and gonadal tissues of P. fucata. The PCA analysis of piRNAs and heatmap of expression patterns of piRNA clusters clustered the somatic tissues together, therefore, the piRNA and piRNA clusters expressed in somatic tissues were not substantially different, but varied significantly between the somatic and gonadal tissues. The transposon annotation results of piRNA clusters also indicated that the gonadal piRNAs are more likely related to transposon silencing. These results imply different functions of piRNAs between the somatic and gonadal tissues in P. fucata.
To explore the function of gene regulation of piRNAs in P. fucata, an LNA-antagonist was used to silence single piRNA (piRNA0001) expression. LNA-antagonists have been used for mouse and non-human primate small RNA silencing without affecting health (Elmén et al., 2008). In this study, we used an LNA-antagonist to silence piRNA0001expression, which was the most highly expressed piRNA in P. fucata somatic tissues, followed by a stem-loop RT-PCR for piRNA0001 quantification analysis, as in previous studies (Hong et al., 2016; Wang et al., 2018; Zhang et al., 2018). Stem-loop RT-PCR is a powerful and reliable tool for quantitatively analyzing and monitoring dynamic changes in piRNAs, specifically at the cellular and tissue levels in invertebrates. In this study, piRNA0001 was effectively downregulated in somatic tissues by a specific LNA-antagonist.
Recent studies have shown that piRNAs may regulate endogenous mRNA expression in Drosophila and mice (Gou et al., 2014; Wu et al., 2018). Thousands of genes were differentially expressed after piRNA0001 silencing in P. fucata, which might be caused by piRNA0001 silencing, or changes in other biological processes, cellular components or molecular functions caused by LNA injection. Although targeting rules have been determined in Drosophila (Wu et al., 2018; Zhang et al., 2018), the available tools cannot be used for piRNA targeting site prediction in other species. An RNA-RNA interacting prediction tool, such as miRanda (Gou et al., 2014), was alternatively used to predict potential targeting sites between piRNAs and endogenous genes. In this study, we used miRanda to predict potential piRNA-mRNA interaction sites on DEGs in P. fucata. piRNA target recognition was strikingly similar to that of miRNA pairing in the seed sequence (positions 2–8 relative to the 5′ end of the piRNA), which is the primary determinant of target recognition. Therefore, seed pairing is not sufficient, and additional base-pairing outside of the seed sequence is also important for piRNA target sites.
Among the predicted piRNA0001 target genes in P. fucata, the majority were related to metabolism and biological processes. ZNF622 was up-regulated in all examined tissues following piRNA0001 silencing, and was predicted to be targeted by piRNA0001 with an alignment score of 154.00 and energy value of −10.64 kcalmol–1. Zinc finger proteins (ZNFs) are one of the most abundant groups of proteins, with a wide range of molecular functions, such as transcriptional regulation, ubiquitin-mediated protein degradation, signal transduction, actin targeting, DNA repair, cell migration, and numerous other processes (Cassandri et al., 2017). A single piRNA (fem piRNA) from a sex chromosome downregulates Masc mRNA, which encodes a C3H-type ZNF that induces masculinization and plays an important role in sex determination in silkworms (Kiuchi et al., 2014). CAH14, NBR1, SYWC, and VINC were predicted targets of piRNA0001, whereas interaction sites were located after the polyA signal on the 3′ UTR, suggesting that these interaction sites may be false positive results. This may be due to the insufficient assembly result of RNA sequencing, with the polyA signal not appearing in the assembling gene sequences, as observed in ART2, EF1A, ESI1L, FHOD3, PA2HB, SRS10, and TM87A. Single and double target sites were both observed in these target genes, indicating that piRNA may regulate endogenous genes at multiple sites on the 3′ UTR. Except for upregulated genes predicted to be targeted by piRNA0001, nine down-regulated genes may also have been targeted by piRNA0001, extending the uncertainty surrounding piRNAs in P. fucata. The lowest piRNA silencing efficiency was observed in the mantle tissue, possibly explaining the majority of downregulated genes. Although, we cannot finalize the target rules or genes of piRNAs, we have provided new insights into the gene regulatory function of piRNAs in P. fucata. The PIWI/piRNA might have a great diversity of functions in P. fucata, including but not limited to gene regulation. Further studies are needed to fully understand somatic piRNAs in mollusks.
In summary, this study reported a PIWI/piRNA pathway, including piRNA biogenesis and piRNA-mediated transposon silencing and gene regulation, in the pearl oyster P. fucata (Mollusca). These findings have successfully contributed to our understanding of the role of piRNAs in mollusks and will also help to gain further insights into the PIWI/piRNA pathway function outside of germline cells.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ddbj.nig.ac.jp/, DRA008674 and https://www.ddbj.nig.ac.jp/, DRA007432.
Author Contributions
SA and SK designed the study. FO, KM, and KN provided the experimental animal samples. MA and SW performed the transcriptomic library construction and sequencing with assistance from SK. YI did the LNA-mediated piRNA silencing. SH and KY performed all bioinformatic analyses. SH collected and analyzed the data. SA and SH wrote the manuscript. All authors reviewed and commented on the manuscript.
Funding
This research was supported by the Japan Society for the Promotion of Science (Grant No. JP24248034) and the Research Fellowship of the Japan Society for the Promotion of Science Postdoctoral Fellowship for Overseas Researchers (Grant No. P20395).
Conflict of Interest
KM was employed by company Mikimoto Pharmaceutical Co., Ltd. KN was employed by company Pearl Research Laboratory, K. MIKIMOTO & Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Yukihide Tomari and Natsuko Izumi for technical support in the piRNA analysis. We also thank the two reviewers for their valuable comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.730556/full#supplementary-material
Footnotes
- ^ https://trinotate.github.io/
- ^ https://www.ncbi.nlm.nih.gov/orffinder/
- ^ http://smart.embl-heidelberg.de/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ http://www.microrna.org/microrna/home.do
References
Aravin, A. A., Gaidatzis, D., Pfeffer, S., Lagos-Quintana, M., Landgraf, P., Iovino, N., et al. (2006). A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207. doi: 10.1038/nature04916
Aravin, A. A., Hannon, G. J., and Brennecke, J. (2007). The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 318, 761–764. doi: 10.1126/science.1146484
Aravin, A. A., Naumova, N. M., Tulin, A. V., Vagin, V. V., Rozovsky, Y. M., and Gvozdev, V. A. (2001). Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. 11, 1017–1027. doi: 10.1016/S0960-9822(01)00299-8
Aravin, A. A., Sachidanandam, R., Bourc’his, D., Schaefer, C., Pezic, D., Toth, K. F., et al. (2008). A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 31, 785–799. doi: 10.1016/j.molcel.2008.09.003
Bryant, D. M., Johnson, K., DiTommaso, T., Tickle, T., Couger, M. B., Payzin-Dogru, D., et al. (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776. doi: 10.1016/j.celrep.2016.12.063
Carmell, M. A., Girard, A., Kant, H. J., Bourc’his, D., Bestor, T. H., Rooij, D. G., et al. (2007). MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev. Cell 12, 503–514. doi: 10.1016/j.devcel.2007.03.001
Cassandri, M., Smirnov, A., Novelli, F., Pitolli, C., Agostini, M., Malewicz, M., et al. (2017). Zinc-finger proteins in health and disease. Cell Death Discov. 3:17071.
Cerutti, L., Mian, N., and Bateman, A. (2000). Domains in gene silencing and cell differentiation proteins: the novel PAZ domain and redefinition of the Piwi domain. Trends Biochem. Sci. 25, 481–482. doi: 10.1016/S0968-0004(00)01641-8
Chen, G., Zhang, C., Jiang, F., Wang, Y., Xu, Z., and Wang, C. (2014). Bioinformatics analysis of hemocyte miRNAs of scallop Chlamys farreri against acute viral necrobiotic virus (AVNV). Fish Shellfish Immunol. 37, 75–86. doi: 10.1016/j.fsi.2014.01.002
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 1–14. doi: 10.1002/0471250953.bi0410s05
Cox, D. N., Chao, A., Baker, J., Chang, L., Qian, D., and Lin, H. (1998). A novel class of evolutionarily conserved genes defined by piwi are essential for stem cell self-renewal. Genes Dev. 12, 3715–3727. doi: 10.1101/gad.12.23.3715
Deng, W., and Lin, H. F. (2002). Miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev. Cell 2, 819–830. doi: 10.1016/S1534-5807(02)00165-X
Elmén, J., Lindow, M., Schütz, S., Lawrence, M., Petri, A., Obad, S., et al. (2008). LNA-mediated microRNA silencing in non-human primates. Nature 452, 896–900. doi: 10.1038/nature06783
Enright, A. J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D. S. (2003). MicroRNA targets in Drosophila. Genome Biol. 5:R1. doi: 10.1186/gb-2003-5-1-r1
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). Protein Identification and Analysis Tools on the ExPASy Server; The Proteomics Protocols Handbook. Totowa, NJ: Humana Press, 571–607.
Gebert, D., Hewel, C., and Rosenkranz, D. (2017). unitas: the universal tool for annotation of small RNAs. BMC Genomics 18:644. doi: 10.1186/s12864-017-4031-9
Gou, L. T., Dai, P., Yang, J. H., Wang, E. D., and Liu, M. F. (2014). Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 24, 680–700. doi: 10.1038/cr.2014.41
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 9, 95–98.
Hong, Y., Wang, C., Fu, Z., Liang, H., Zhang, S., Lu, M., et al. (2016). Systematic characterization of seminal plasma piRNAs as molecular biomarkers for male infertility. Sci. Rep. 6:24229. doi: 10.1038/srep24229
Houwing, S., Berezikov, E., and Ketting, R. F. (2008). Zili is required for germ cell differentiation and meiosis in zebrafish. EMBO J. 27, 2702–2711. doi: 10.1038/emboj.2008.204
Houwing, S., Kamminga, L. M., Berezikov, E., Cronembold, D., Girard, A., Elst, H., et al. (2007). A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 129, 69–82. doi: 10.1016/j.cell.2007.03.026
Huang, H. D., Li, Y. J., Szulwach, K. E., Zhang, G. Q., Jin, P., and Chen, D. H. (2014). AGO3 slicer activity regulates mitochondria-nuage localization of armitage and piRNA amplification. J. Cell Biol. 206, 217–230. doi: 10.1083/jcb.201401002
Huang, S. Q., Ichikawa, Y., Igarashi, Y., Yoshitake, K., Kinoshita, S., Omori, F., et al. (2019a). Piwi-interacting RNA (piRNA) expression patterns in pearl oyster (Pinctada fucata) somatic tissues. Sci. Rep. 9:247. doi: 10.1038/s41598-018-36726-0
Huang, S. Q., Ichikawa, Y., Yoshitake, K., Kinoshita, S., Igarashi, Y., Omori, F., et al. (2019b). Identification and characterization of microRNAs and their predicted functions in biomineralization in the pearl oyster (Pinctada fucata). Biology 8:47.
Inoue, K., Ichiyanagi, K., and Fukuda, K. (2017). Switching of dominant retrotransposon silencing strategies from posttranscriptional to transcriptional mechanisms during male germ-cell development in mice. PLoS Genet. 13:e1006926. doi: 10.1371/journal.pgen.1006926
Ipsaro, J. J., Haase, A., Knott, S. R., Joshua-Tor, L., and Hannon, G. J. (2012). The structural biochemistry of Zucchini implicates it as a nuclease in piRNA biogenesis. Nature 491, 279–283. doi: 10.1038/nature11502
Ishizu, H., Siomi, H., and Siomi, M. C. (2012). Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 26, 2361–2373. doi: 10.1101/gad.203786.112
Izumi, N., Shoji, K., Sakaguchi, Y., Honda, S., Kirino, Y., Suzuki, T., et al. (2016). Identification and functional analysis of the pre-piRNA 3’ trimmer in silkworms. Cell 164, 962–973. doi: 10.1016/j.cell.2016.01.008
Izumi, N., Shoji, K., Suzuki, Y., Katsuma, S., and Tomari, Y. (2020). Zucchini consensus motifs determine the mechanism of pre-piRNA production. Nature 578, 311–316. doi: 10.1038/s41586-020-1966-9
Izumi, N., and Tomari, Y. (2014). Diversity of the piRNA pathway for nonself silencing: worm-specific piRNA biogenesis factors. Genes Dev. 28, 665–671. doi: 10.1101/gad.241323.114
Jehn, J., Gebert, D., Pipilescu, F., Stern, S., Kiefer, J., Hewel, C., et al. (2018). PIWI genes and piRNAs are ubiquitously expressed in mollusks and show patterns of lineage-specific adaptation. Commun. Biol. 1, 1–11. doi: 10.1038/s42003-018-0141-4
Jiao, Y., Zheng, Z., Du, X. D., Wang, Q. H., Huang, R. L., Deng, Y. W., et al. (2014). Identification and characterization of miRNAs in pearl oyster Pinctada martensii by solexa sequencing. Mar. Biotechnol. 16, 54–62.
Jones, B. C., Wood, J. G., Chang, C., Tam, A. D., Franklin, M. J., Siegel, E. R., et al. (2016). A somatic piRNA pathway in the Drosophila fat body ensures metabolic homeostasis and normal lifespan. Nat. Commun. 7:13856. doi: 10.1038/ncomms13856
Kawaoka, S., Izumi, N., Katsuma, S., and Tomari, Y. (2011). 3’ end formation of PIWI-interacting RNAs in vitro. Mol. Cell 43, 1015–1022. doi: 10.1016/j.molcel.2011.07.029
Kim, V. N., Han, J., and Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139. doi: 10.1038/nrm2632
Kiuchi, T., Koga, H., Kawamoto, M., Shoji, K., Sakai, H., Arai, Y., et al. (2014). A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature 509, 633–638.
Krishnan, P., Ghosh, S., Graham, K., Mackey, J. R., Kovalchuk, O., and Damaraju, S. (2016). Piwi-interacting RNAs and PIWI genes as novel prognostic markers for breast cancer. Oncotarget 7, 37944–37956. doi: 10.18632/oncotarget.9272
Lau, N. C., Seto, A. G., Kim, J., Kuramochi-Miyagawa, S., Nakano, T., Bartel, D. P., et al. (2006). Characterization of the piRNA complex from rat testes. Science 313, 363–367. doi: 10.1126/science.1130164
Letunic, I., and Bork, P. (2017). 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496. doi: 10.1093/nar/gkx922
Lewis, S. H., Quarles, K. A., Yang, Y., Tanguy, M., Frézal, L., Smith, S. A., et al. (2018). Pan-arthropod analysis reveals somatic piRNAs as an ancestral defence against transposable elements. Nat. Ecol. Evol. 2, 174–181. doi: 10.1038/s41559-017-0403-4
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Liu, J., Carmell, M. A., Rivas, F. V., Marsden, C. G., Thomson, J. M., Song, J. J., et al. (2004). Argonaute2 is the catalytic engine of mammalian RNAi. Science 305, 1437–1441. doi: 10.1126/science.1102513
Liu, X., Sun, Y., Guo, J., Ma, H., Dong, B., Jin, G., et al. (2006). Expression of hiwi gene in human gastric cancer was associated with proliferation of cancer cells. Int. J. Cancer 118, 1922–1929. doi: 10.1002/ijc.21575
Ma, X. S., Ji, A. C., Zhang, Z. F., Yang, D. D., Liang, S. S., Wang, Y. H., et al. (2017). Piwi1 is essential for gametogenesis in mollusk Chlamys farreri. PeerJ 5:e3412. doi: 10.7717/peerj.3412
Malone, C. D., Brennecke, J., Dus, M., Stark, A., McCombie, W. R., Sachidanandam, R., et al. (2009). Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137, 522–535. doi: 10.1016/j.cell.2009.03.040
Parker, J. S., and Barford, D. (2006). Argonaute: a scaffold for the function of short regulatory RNAs. Trends Biochem. Sci. 31, 622–630. doi: 10.1016/j.tibs.2006.09.010
Peters, L., and Meister, G. (2007). Argonaute proteins: mediators of RNA silencing. Mol. Cell 26, 611–623. doi: 10.1016/j.molcel.2007.05.001
Pfaffl, M. W. (2011). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. doi: 10.1093/nar/29.9.e45
Qiao, D., Zeeman, A. M., Deng, W., Looijenga, L. H., and Lin, H. F. (2002). Molecular characterization of hiwi, a human member of the piwi gene family whose over expression is correlated to seminomas. Oncogene 21, 3988–3999. doi: 10.1038/sj.onc.1205505
Rajasethupathy, P., Antonov, I., Sheridan, R., Frey, S., Sander, C., Tuschi, T., et al. (2012). A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149, 693–707. doi: 10.1016/j.cell.2012.02.057
Robine, N., Lau, N. C., Balla, S., Jin, Z., Okamura, K., Kuramochi-Miyagawa, S., et al. (2009). A broadly conserved pathway generates 3’UTR-directed primary piRNAs. Curr. Biol. 19, 2066–2076. doi: 10.1016/j.cub.2009.11.064
Robinson, M. D., Mccarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139. doi: 10.1093/bioinformatics/btp616
Ross, R. J., Weiner, M. M., and Lin, H. F. (2014). PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505, 353–359. doi: 10.1038/nature12987
Saito, K., Inagaki, S., Mituyama, T., Kawamura, Y., Ono, Y., Sakota, E., et al. (2009). A regulatory circuit for piwi by the large Maf gene traffic jam in Drosophila. Nature 461, 1296–1299. doi: 10.1038/nature08501
Saito, K., Sakaguchi, Y., Suzuki, T., Suzuki, T., Siomi, H., and Siomi, M. C. (2007). Pimet, the Drosophila homolog of HEN1, mediates 2’-O-methylation of Piwi-interacting RNAs at their 3’ ends. Genes Dev. 21, 1603–1608. doi: 10.1101/gad.1563607
Sasaki, T., Shiohama, A., Minoshima, S., and Shimizu, N. (2003). Identification of eight members of the Argonaute family in the human genome. Genomics 82, 323–330. doi: 10.1016/s0888-7543(03)00129-120
Shen, E. Z., Chen, H., Ozturk, A. R., Tu, S. K., Shirayama, M., Tang, W., et al. (2018). Identification of piRNA binding sites reveals the argonaute regulatory landscape of the, C. elegans, germline. Cell 172, 937.e18–951.e18. doi: 10.1016/j.cell.2018.02.002
Song, J. J., Smith, S. K., Hannon, G. J., and Joshua-Tor, L. (2004). Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305, 1434–1437.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2014). MEGA5: molecular evolutionary genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tolia, N. H., and Joshua-Tor, L. (2007). Slicer and the argonautes. Nat. Chem. Biol. 3, 36–43. doi: 10.1038/nchembio848
Vourekas, A., Zheng, Q., Alexiou, P., Maragkakis, M., Kirino, Y., Gregory, B. D., et al. (2012). Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat. Struct. Mol. Biol. 19, 773–781. doi: 10.1038/nsmb.2347
Wang, Y., Jin, B., Liu, P., Li, J., Chen, X., and Gu, J. (2018). piRNA profiling of dengue virus type 2-infected Asian tiger mosquito and midgut tissues. Viruses 10:213. doi: 10.3390/v10040213
Weick, E. M., and Miska, E. A. (2014). piRNAs: from biogenesis to function. Development 141, 3458–3471. doi: 10.1242/dev.094037
Wu, W. S., Huang, W. C., Brown, J. S., Zhang, D. L., Song, X. Y., Chen, H., et al. (2018). pirScan: a webserver to predict piRNA targeting sites and to avoid transgene silencing in C. elegans. Nucleic Acids Res. 46, W43–W48. doi: 10.1093/nar/gky277
Xu, F., Wang, X. T., Feng, Y., Huang, W., Wang, W., Li, L., et al. (2014). Identification of conserved and novel microRNAs in the Pacific oyster Crassostrea gigas by deep sequencing. PLoS One 9:e104371. doi: 10.1371/journal.pone.0104371
Yan, Z., Hu, H. Y., Jiang, X., Maierhofer, V., Neb, E., He, L., et al. (2011). Widespread expression of piRNA-like molecules in somatic tissues. Nucleic Acids Res. 39, 6596–6607. doi: 10.1093/nar/gkr298
Yang, F., and Xi, R. W. (2017). Silencing transposable elements in the Drosophila germline. Cell. Mol. Life Sci. 74, 435–448. doi: 10.1007/s00018-016-2353-4
Zhang, D. L., Tu, S. K., Stubna, M., Wu, W. S., Huang, W. C., Weng, Z. P., et al. (2018). The piRNA targeting rules and the resistance to piRNA silencing in endogenous genes. Science 359:587. doi: 10.1126/science.aao2840
Keywords: piRNA, PIWI genes, ping-pong amplification, transposon silencing, gene regulation, Pinctada fucata
Citation: Huang S, Ichikawa Y, Yoshitake K, Kinoshita S, Asaduzzaman M, Omori F, Maeyama K, Nagai K, Watabe S and Asakawa S (2021) Conserved and Widespread Expression of piRNA-Like Molecules and PIWI-Like Genes Reveal Dual Functions of Transposon Silencing and Gene Regulation in Pinctada fucata (Mollusca). Front. Mar. Sci. 8:730556. doi: 10.3389/fmars.2021.730556
Received: 28 June 2021; Accepted: 13 August 2021;
Published: 10 September 2021.
Edited by:
Jian-Wen Qiu, Hong Kong Baptist University, Hong Kong, SAR ChinaReviewed by:
Shiguo Li, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences (CAS), ChinaYang Zhang, South China Sea Institute of Oceanology, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Huang, Ichikawa, Yoshitake, Kinoshita, Asaduzzaman, Omori, Maeyama, Nagai, Watabe and Asakawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songqian Huang, aHVhbmdzb25ncWlhbjAxMTVAZy5lY2MudS10b2t5by5hYy5qcA==; Shuichi Asakawa, YXNha2F3YUBtYWlsLmVjYy51LXRva3lvLmFjLmpw