 Hailong Ge1,2†
Hailong Ge1,2† Haoyu Zhang3†
Haoyu Zhang3† Qingyuan Zhao1,2Fang Li1,2Haoran Gu1,2Siying Liu1,2Haibo Yang1,2
Qingyuan Zhao1,2Fang Li1,2Haoran Gu1,2Siying Liu1,2Haibo Yang1,2 Yun Li1,2*
Yun Li1,2* Zhijian Wang1,2*
Zhijian Wang1,2*- 1Key Laboratory of Freshwater Fish Reproduction and Development, Ministry of Education, Southwest University, Chongqing, China
- 2Key Laboratory of Aquatic Science of Chongqing, Southwest University, Chongqing, China
- 3Animal Disease Prevention and Food Safety Key Laboratory of Sichuan Province, Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China
Background
The Chinese sucker (Myxocyprinus asiaticus), which belongs to the genus Myxocyprinus in the family Catostomidae, is the only Catostomidae species in China and the only endemic Catostomidae species in Asia. It is distributed mainly in the Yangtze River. The Chinese sucker, a unique and rare freshwater species in China, has been listed as a second-class national key protected animal and is nicknamed the Asian Mermaid. The Chinese sucker is brightly colored and has a unique body shape and delicious, nutritious flesh, giving it certain value as an ornamental and edible fish (Huang et al., 2015). It is a valuable aquaculture fish (Yuan et al., 2011; Xu et al., 2013).
To date, most studies on the Chinese sucker have focused on several topics, including its resource status and artificial breeding (Songliang et al., 2002; Adeoba et al., 2019), its non-specific immunity (Zhang et al., 2009, 2021), its genetic diversity and mitochondrial genome (Chen, 2013; Liu et al., 2018), and its feed application (Guo et al., 2016; Jianhua et al., 2016; Li et al., 2018, 2019; Lu et al., 2020). As the conservation value of this species has become increasingly prominent, related research has gradually expanded. Due to the importance of germplasm resource protection, extensive research and development plans have been launched. The goals of these plans are to better understand Chinese sucker biology and to use basic knowledge to improve resource conservation and artificial breeding programs in order to achieve long-term maintenance and utilization of the germplasm resource. The ultimate goal is to realize the sustainable development of Chinese suckers.
In this study, we applied the isoform sequencing (Iso-Seq) technique of Pacific Biosciences (PacBio) for long reads to generate the first full-length transcriptome assembly of the Chinese sucker. The PacBio sequencing platform is an ideal method for constructing reference transcriptome components without reference genome sequences (Dong et al., 2015; Kuo et al., 2017; Workman et al., 2018). Full-length mRNA sequences from five major organs were obtained using PacBio's circular consensus sequencing. Due to the lack of high-quality genome sequence data for the Chinese sucker, the high-quality reference transcriptome assembly obtained in this study will be an important resource for further transcriptome analyses of Chinese suckers under various conditions.
Data Description
Sample Collection
Five organs were collected from healthy 1-year-old Chinese suckers. The animal welfare and laboratory procedures for this study were in accordance with the relevant ethical requirements of the Key Laboratory of Freshwater Fish Reproduction and Development (Ministry of Education). The following organs were dissected and immediately frozen in liquid nitrogen: the brain, gills, liver, skin, and muscles. We mixed the frozen ground tissue samples together, then extracted RNA from that. All samples were stored at −80°C before RNA extraction.
Library Construction and Single-Molecule Real-Time (SMRT) Sequencing
Total RNA was extracted by grinding tissue in TRIzol reagent (Life Technologies) on dry ice and processed following the protocol provided by the manufacturer. The integrity of the RNA was determined with an Agilent 2100 Bioanalyzer and agarose gel electrophoresis. The purity and concentration of the RNA were determined with a NanoDrop microspectrophotometer (Thermo Fisher). mRNA was enriched with oligo (dT) magnetic beads. Then, the enriched mRNA was reverse-transcribed into cDNA using a Clontech SMARTer PCR cDNA Synthesis Kit. The PCR cycling parameters were optimized to determine the best number of amplification cycles for the downstream large-scale PCR procedures. Then, double-stranded cDNA was generated with the determined number of cycles. In addition, cDNA molecules >5 kb in size were selected using a BluePippinTM Size-Selection System and mixed equally with non-size-selected cDNA. Then, large-scale PCR was performed for the next step: SMRTbell library construction. The cDNA was subjected to DNA damage repair and end repair and was ligated to sequencing adapters. The SMRTbell template was annealed to a sequencing primer, bound to polymerase, and sequenced on the PacBio Sequel II platform by Gene Denovo Biotechnology Co. (Guangzhou, China).
Data Processing
The raw sequencing reads of the cDNA libraries were analyzed by using the SMRT Link (V9.0.0) pipeline. First, high-quality circular consensus sequences (CCSs, HiFi reads) were extracted from the BAM subread file using the CCS function with the following parameters: min_predicted_accuracy 0.8, min_length 50, min_passes 1. We analyzed the integrity of transcripts on the basis of by whether the CCS reads contained 5′ primers, 3′ primers and polyA structures. Sequences containing all three structures were considered full-length sequences (full-length reads, FL reads). Afterwards, the primers, barcodes, polyA tails and concatemers of full passes were removed to obtain the full-length nonchimeric (FLNC) reads. The FLNC reads were clustered to generate the entire isoform. The Cluster function (which use minimap2 for clustering, and use transcripts as reference) was used to cluster the consensus sequences for similar FLNC reads with the following parameters: hq_quiver_min_accuracy 0.99, qv_trim_5p 100, and qv_trim_3p 30, use the Quiver algorithm for correction. Then, we used the Cluster Database at High Identity with Tolerance (CD-HIT) program (v4.6.7) to further correct the consensus sequences with the following parameters: -c 0.99, -T 6, -G 0, -aL 0.90, -AL 100, -aS 0.99, and -AS 30. We used BUSCO for assembly access According to the results, high-quality isoforms (prediction accuracy ≥ 0.99) were used for further analysis (Supplementary Figure 1).
Gene Annotation
For annotation, the isoforms were BLAST-analyzed against the NCBI non-redundant protein (Nr) database (http://www.ncbi.nlm.nih.gov, updated 04/19/2020), the Swiss-Prot protein database (http://www.expasy.ch/sprot), the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg, release 94.0), and the Clusters of Orthologous Groups (COG)/EuKaryotic Orthologous Groups of proteins (KOG) database (http://www.ncbi.nlm.nih.gov/COG) with the BLASTx program at an E-value threshold of 1e−5 to evaluate the sequence similarity with genes of other species. Gene Ontology (GO) (updated 01/05/2019) annotation was performed with Blast2GO software (v2.3.5) with the default parameters against the Nr results (Conesa et al., 2005). The 20 highest-scoring isoforms with no fewer than 33 high-scoring segment pair (HSP) hits were selected for Blast2GO analysis. Then, functional classification of the isoforms was performed using WEGO software (v2.0) with the default parameters (Ye et al., 2006).
The open reading frames (ORFs) of the isoform sequences were detected by using ANGEL software (version 2.4) with the default parameters to obtain the coding sequences (CDSs), protein sequences, and UTR sequences (Shimizu et al., 2006). Protein domain prediction was performed by aligning the protein sequences of the isoforms to the Pfam database (v26.0) with the Pfam_Scan program (v1.3) and to the SMART database (updated 06/08/2012) with HMMER (v3.0). The parameters -E 0.00001 and –domE 0.00001 were used to obtain protein domain annotations (Eddy, 2011; Letunic et al., 2012; Finn et al., 2015). The protein-coding sequences of the isoforms were aligned with hmmscan (v3.1b2) with the default parameters against TFdb (v2.0) to predict the transcription factor (TF) families (Zhang et al., 2015).
Potential transmembrane helices in the proteins were predicted with TMHMM Server 2.0 (Möller et al., 2001).
The presence and locations of signal peptide cleavage sites in the amino acid sequences were predicted with SignalP 4.1 Server (Nielsen, 2017). Mucin-type GalNAc O-glycosylation sites in mammalian proteins were predicted with the NetOGlyc 4.0 Server (Steentoft et al., 2013). Arginine and lysine propeptide cleavage sites in eukaryotic protein sequences were predicted with ProP 1.0 Server (Duckert et al., 2004).
Gene Structure Prediction
MIcroSAtellite (MISA, v1.0) was employed for microsatellite mining of the whole transcriptome. The parameters were as follows:
definition (n,m): 2,6; 3,5; 4,4; 5,4; and 6,4
interruptions (max_difference_between_2_SSRs): 100
where n is the length of repeat units and m is the minimum number of repeat units.
The interruptions parameter indicates that if the distance between two simple sequence repeats (SSRs) is shorter than 100 bp, the SSRs will be considered as one SSR.
Based on the MISA results, Primer 1.1.4 was used to design primer pairs in the flanking regions of the SSRs for subsequent validation (Rozen and Skaletsky, 2000).
CPC (v0.92r2) was used to assess the protein-coding potential of transcripts without annotations according to the default parameters in order to identify potential long non-coding RNAs (lncRNAs), CPC reference database is made of uniport sequences which come from Swiss-prot database. LncRNA analysis was performed on the full-length transcript sequences that were not annotated to the four major databases. The RNAs predicted as “non-coding” by both software programs were considered the lncRNAs. To better annotate lncRNAs at the evolutionary level, the software Infernal (v1.1.2) (http://eddylab.org/infernal/) was used for sequence alignment with the default parameters (Nawrocki and Eddy, 2013). The lncRNAs were classified by their secondary structures and sequence conservation.
To analyze the alternative splicing (AS) events of the transcript isoforms, the COding GENome reconstruction Tool (Cogent, v3.3) was initially used with the default parameters to divide the transcripts into gene families based on k-mer similarity and to reconstruct each family into a coding reference genome based on a De Bruijn graph (Li et al., 2017). Then, the SUPPA (2.2) tool was used to analyze AS events of isoforms using the default parameters (Alamancos et al., 2015).
Results
Data Summary
A total of 71.58 Gb of nucleotide data was obtained from Iso-Seq using the PacBio SMRT Sequencing method. The raw sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) database with the accession number PRJNA718002. After initial quality control involving removal of the adaptor reads and subreads <50 bp in length, a total of 33,957,771 reads (71.58 Gb of nucleotides) were generated (Supplementary Figure 2a), with an average length and N50 of 2,263 and 2,684 bp, respectively. Thereafter, all subreads were used for CCS analysis. A total of 997,309 CCSs were produced (Supplementary Figure 2b), with an average length of 2,805 bp. FLNC read clustering was performed using Minimap2 for error correction of the third-generation sequencing data; 58,375 high-quality (HQ) isoforms (prediction accuracy ≥ 0.99) were obtained for subsequent analysis, and 1,731 low-quality (LQ) isoforms were obtained (prediction accuracy < 0.99) (Supplementary Figure 2c). CD-HIT was used to cluster redundant sequences with >99% similarity. Ultimately, 54,470 full-length non-redundant transcripts were obtained (Supplementary Figure 2d), with a maximum length of 14,296 bp and an N50 length of 3,406 bp (Table 1). Using the SMRT Sequencing pipeline, the number of non-redundant full-length transcripts was significantly reduced. BUSCO evaluation indicates suitable sequence completeness (Supplementary Figure 2e).
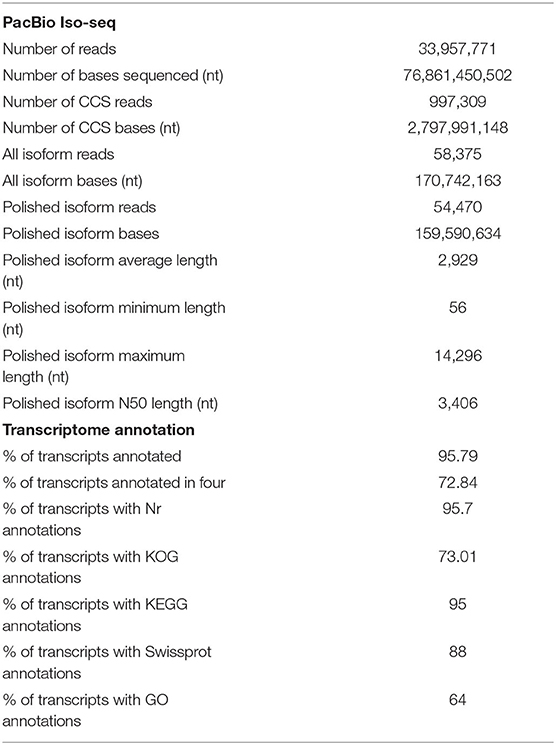
Table 1. Assembly statistics for transcriptome.
Gene Annotations and Taxonomy
The full-length non-redundant transcripts were annotated from four databases. A total of 52,178 (95.79%) transcripts were annotated, among which 39,675 (72.84%) transcripts were annotated from all four databases. Prediction and functional annotation of the coded transcripts showed that 52,129 (95.7%), 51,894 (95.27%), 48,054 (88.22%), and 39,765 (73.01%) were annotated in the Nr database, KEGG, Swiss-Prot database and KOG database, respectively (Table 1 and Supplementary Figure 2f).
In the Nr database, most transcripts were annotated to the Cyprinidae family. The top 10 species classifications were Danio rerio (6,546, 12.56%), Cyprinus carpio (6,416, 12.31%), Triplophysa tibetana (5,926, 11.37%), Sinocyclocheilus graham (3,640, 6.98%), Carassius auratus (2,890, 5.54%), Labeo rohita (2,728, 5.23%), Anabarilius graham (2,565, 4.92%), Sinocyclocheilus anshuiensis (2,562, 4.91%), Danionella translucida (2,285, 4.38%), and Pangasianodon hypophthalmus (1,663, 3.19%) (Figure 1B).
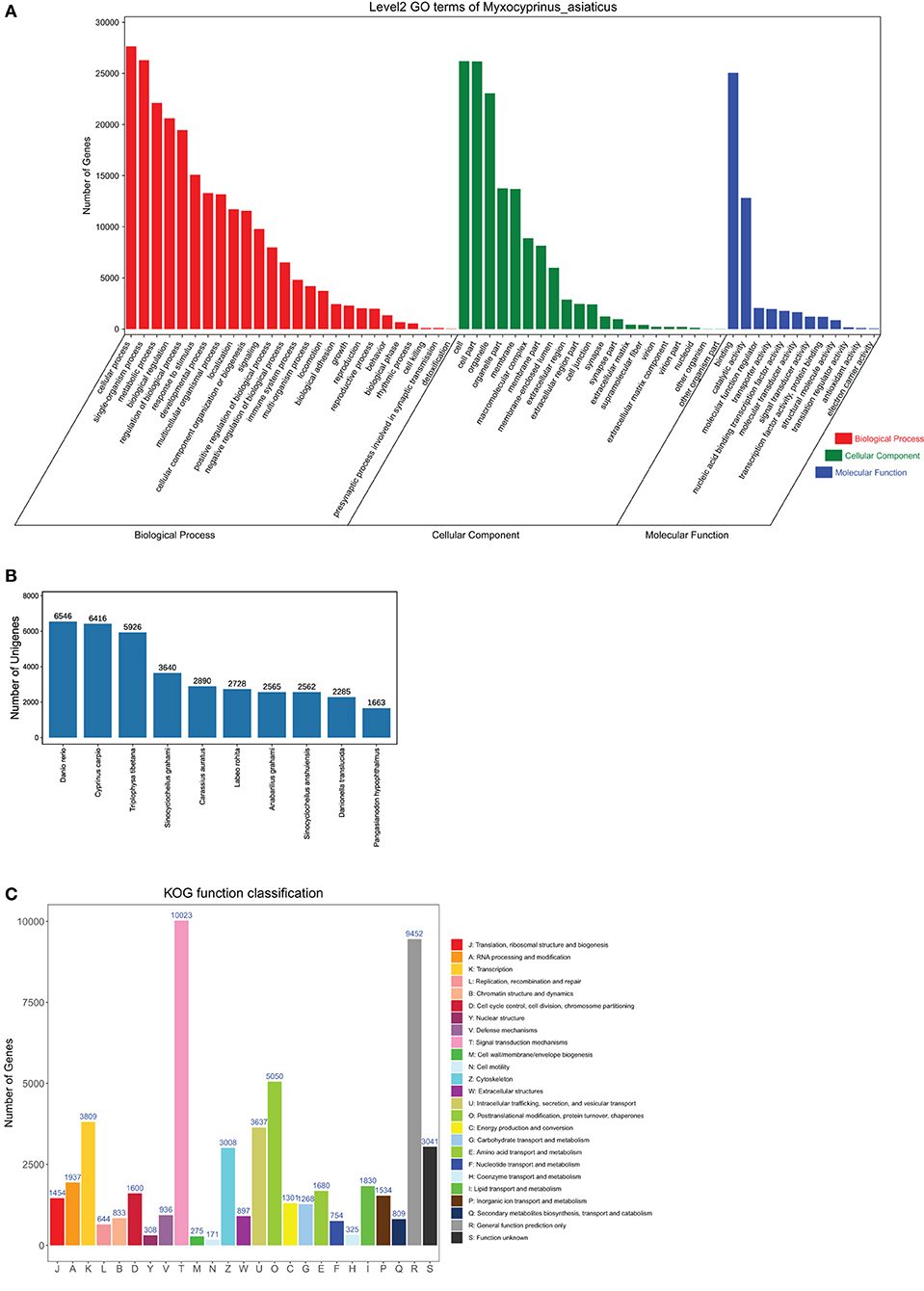
Figure 1. Functional classification. (A) 2 level of GO functional annotations of full-length transcripts. (B) Protein taxonomic distribution annotated by non-redundant protein database. (C) Function classification annotated in KOG database.
With regard to KOG annotation, 39,764 (73.01%) transcripts were divided into 24 subcategories. The subcategory with the highest percentage was the signal transduction mechanisms subcategory (Figure 1A). Regarding functional annotations, 34,870 (64.02%) transcripts were associated with 59 terms in the GO database. The most enriched terms in the biological processes category (55.19%) were the cellular process (6.65%), single-tissue process (6.32%), metabolic process (5.32%) and biological regulation (4.96%) terms. Within the cellular component category (33.06%), the most enriched GO terms were the cell (6.3%), cellular component (6.29%) and organelle (5.55%) terms. Within the molecular function category (11.75%), the most abundant GO terms were the binding (6.03%), and catalytic activity (3.08%) terms (Figure 1C).
A total of 357 pathways were derived from the KEGG database, and a total of 51,894 transcripts were annotated. These pathways include 6 primary categories with 46 secondary subcategories. The most prominent subcategory was “metabolism.”
Gene Structure
AS of RNA is widespread in biology. AS occurs after mRNA transcription but before template DNA is formed and produces multiple proteins from a single protein-coding gene. In total, we identified 9,499 AS events (Supplementary Figure 3a). Since no reference genome was available, for these data, only 1,855 events were classified into 6 types of AS events (Supplementary Figure 3b). The most prominent type of AS was intron retention (RI, 1382).
To study genetic diversity, assess quality and promote genetic research, the SSRs identified in the Iso-Seq library were analyzed. In addition, a total of 21,428 SSRs were detected in 13,714 transcripts (Supplementary Figure 3c). Of these SSRs, 14,028 (65.14%) were dinucleotide repeats, most of which consisted of 4–7 repeated sequences; 3,977 SSRs were dinucleotide repeats with 8–11 repeats. In addition, 3,919 SSRs were trinucleotide repeats with 4–7 repeats (Supplementary Figure 3d).
LncRNA Prediction, Coding Sequence, and TF Analyses
In this study, Iso-Seq was also used to sequence lncRNAs with polyA ends. Due to the lack of a genome, the exons of each lncRNA were not evaluated, and the expression densities of lncRNAs and protein-coding RNAs were analyzed at the same time (Supplementary Figure 4b).
The protein-encoding transcripts identified from the mRNA transcripts were submitted to ANGEL to predict the CDSs. A total of 52,465 (96.32%) protein-coding transcripts were predicted (Supplementary Figure 4a). Most of the CDSs were shorter than 2,500 bp.
Hmmsearch was used to predict the TFs among all single genes, and 3,316 TF transcripts were identified. The four most common families were the zf-C2H2 (858), TF_bZIP (266), bHLH (254), and homeobox (186) families (Supplementary Figure 4c). The results provide convenient data for further elucidation of transcriptional regulation.
In this study, we used the SMRT Sequencing method to sequence the full-length transcriptome and obtained 54,470 non-redundant full-length transcripts. A total of 52,178 (95.79%) transcripts were annotated into four databases. In addition, 2,155 (3.96%) transcripts were predicted to be lncRNAs, and 3,316 (6.09%) were predicted to encode TFs. A total of 9,499 AS events and 21,428 SSRs were detected. Thus, we obtained the full-length transcriptome of Myxocyprinus asiaticus and genetic information via analysis of known data. Our findings significantly increase the genetic information on Myxocyprinus asiaticus and will aid future research on the functions of genes related to Chinese sucker development and reproduction as well as the evolution of the Chinese sucker.
Reuse Potential
Here, we report the first full-length transcriptome of the Chinese sucker, which was obtained using PacBio long-read SMRT Sequencing technology. The Iso-Seq-based full-length transcriptome assembly reported here will greatly benefit future transcriptome analysis and genome sequencing of the Chinese sucker and closely related species.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI [accession: PRJNA718002].
Ethics Statement
The animal study was reviewed and approved by Key laboratory of Freshwater Fish Reproduction and Development (Ministry of Education), Key Laboratory of Aquatic Science of Chongqing, Southwest University, Chongqing, China. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
HGe and HZ conceived and designed the experiment and drafted the manuscript. SL and HY raised the fish. HGe and HGu dissected the fish and collected fish tissue samples. HZ and QZ completed the bioinformatics analysis. HGe and FL provided the tables and figures. ZW and YL revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Financial Program of Ministry of Agriculture and Rural Affairs of China (Grant No. YYJZHC201921301350063) and National Special Research Fund for Non-Profit Sector (Agriculture) (201203086).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.699504/full#supplementary-material
References
Adeoba, M. I., Tesfamichael, S., and Yessoufou, K. (2019). Preserving the tree of life of the fish family Cyprinidae in Africa in the face of the ongoing extinction crisis. Genome. 62, 170–182. doi: 10.1139/gen-2018-0023
Alamancos, G. P., Pagès, A., Trincado, J. L., Bellora, N., and Eyras, E. (2015). Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 21, 1521–1531. doi: 10.1261/rna.051557.115
Chen, I.-S. (2013). The complete mitochondrial genome of Chinese sucker Myxocyprinus asiaticus (Cypriniformes, Catostomidae). Mitochondrial DNA 24, 680–682. doi: 10.3109/19401736.2013.773985
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dong, L., Liu, H., Zhang, J., Yang, S., Kong, G., and Chu, J. S. (2015). Single-molecule real-time transcript sequencing facilitates common wheat genome annotation and grain transcriptome research. BMC Genomics 16:1039. doi: 10.1186/s12864-015-2257-y
Duckert, P., Brunak, S., and Blom, N. (2004). Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 17, 107–112. doi: 10.1093/protein/gzh013
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2015). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Guo, Z., Liu, Y., and Wang, Z. J. (2016). Effects of different weaning strategies on the growth and survival rate in larvae and juvenile of Chinese sucker, Myxocyprinus asiaticus. 40, 64–72. doi: 10.11964/jfc.20150609923
Huang, F., Wu, F., Zhang, S., Jiang, M., Liu, W., Tian, J., et al. (2015). Dietary vitamin C requirement of juvenile Chinese sucker (Myxocyprinus asiaticus). Aquac. Res. 48, 37–46. doi: 10.1111/are.12858
Jianhua, Y., Zhongdi, G., Benxiang, L., Lijuan, Y., Deyong, P., Zhong, Y., et al. (2016). Effects of early weaning and co-feeding on growth and digestive capacity in larvae of Myxocyprinus asiaticus. J. Fish. China 40, 225–235. doi: 10.11964/jfc.20150409853
Kuo, R. I., Tseng, E., Eory, L., Paton, I. R., Archibald, A. L., and Burt, D. W. (2017). Normalized long read RNA sequencing in chicken reveals transcriptome complexity similar to human. BMC Genomics 18:323. doi: 10.1186/s12864-017-3691-9
Letunic, I., Doerks, T., and Bork, P. (2012). SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40:gkr931. doi: 10.1093/nar/gkr931
Li, F., Liu, X.-H., Ge, H.-L., Xie, C.-Y., Cai, R.-Y., Hu, Z.-C., et al. (2018). The discovery of Clinostomum complanatum metacercariae in farmed Chinese sucker, Myxocyprinus asiaticus. Aquaculture 495, 273–280. doi: 10.1016/j.aquaculture.2018.05.052
Li, F., Wu, D., Gu, H., Yin, M., Ge, H., Liu, X., et al. (2019). Aeromonas hydrophila and Aeromonas veronii cause motile Aeromonas septicaemia in the cultured Chinese sucker, Myxocyprinus asiaticus. Aquac. Res. 5, 1515–1526. doi: 10.1111/are.14028
Li, J., Harata-Lee, Y., Denton, M. D., Feng, Q., Rathjen, J. R., Qu, Z., et al. (2017). Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 3:17031. doi: 10.1038/celldisc.2017.31
Liu, D., Zhou, Y., Yang, K., Zhang, X., Chen, Y., Li, C., et al. (2018). Low genetic diversity in broodstocks of endangered Chinese sucker, Myxocyprinusasiaticus: implications for artificial propagation and conservation. Zookeys. 792, 117–132. doi: 10.3897/zookeys.792.23785
Lu, X., Zhang, Z.-Q., Wen, H., Jiang, M., and Du, H. (2020). Dietary choline requirement of juvenile Chinese sucker (Myxocyprinus asiaticus). Aquacult. Rep. 18:100484. doi: 10.1016/j.aqrep.2020.100484
Möller, S., Croning, M. D., and Apweiler, R. (2001). Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17, 646–653. doi: 10.1093/bioinformatics/17.7.646
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nielsen, H. (2017). Predicting secretory proteins with SignalP. Methods Mol. Biol. 1611, 59–73. doi: 10.1007/978-1-4939-7015-5_6
Rozen, S., and Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386. doi: 10.1385/1-59259-192-2:365
Shimizu, K., Adachi, J., and Muraoka, Y. (2006). Angle: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 4, 649–664. doi: 10.1142/S0219720006002260
Songliang, W., Jitian, P., and Xingguo, L. (2002). Preliminary study on artificial propagation and fry cultivation of Chinese sucker. J. Hydroecol. 22, 1–2. doi: 10.3969/j.issn.1003-1278.2002.02.001
Steentoft, C., Vakhrushev, S. Y., Joshi, H. J., Kong, Y., Vester-Christensen, M. B., Schjoldager, K. T., et al. (2013). Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488. doi: 10.1038/emboj.2013.79
Workman, R. E., Myrka, A. M., Wong, G. W., Tseng, E., Welch, K. C. Jr., and Timp, W. (2018). Single-molecule, full-length transcript sequencing provides insight into the extreme metabolism of the ruby-throated hummingbird Archilochus colubris. GigaScience 7:giy009. doi: 10.1093/gigascience/giy009
Xu, N., Xu, D. M., Que, Y. F., Shi, F., Xiong, M. H., and Shao, K. (2013). Identification of novel polymorphic microsatellite loci in the endangered Chinese sucker (Myxocyprinus asiaticus). J. Genet. 93, 96–98. doi: 10.1007/s12041-013-0276-5
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Yuan, Y., Gong, S., Yang, H., Lin, Y., Yu, D., and Luo, Z. (2011). Effects of supplementation of crystalline or coated lysine and/or methionine on growth performance and feed utilization of the Chinese sucker, Myxocyprinus asiaticus. Aquaculture 316, 31–36. doi: 10.1016/j.aquaculture.2011.03.015
Zhang, G., Gong, S., Yu, D., and Yuan, H. (2009). Propolis and Herba Epimedii extracts enhance the non-specific immune response and disease resistance of Chinese sucker, Myxocyprinus asiaticus. Fish Shellfish Immunol. 26, 467–472. doi: 10.1016/j.fsi.2009.01.011
Zhang, H. M., Liu, T., Liu, C. J., Song, S., Zhang, X., Liu, W., et al. (2015). AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 43, D76–D81. doi: 10.1093/nar/gku887
Keywords: Chinese sucker, Myxocyprinus asiaticus, transcriptome, Iso-Seq, PacBio sequencing, reference resource
Citation: Ge H, Zhang H, Zhao Q, Li F, Gu H, Liu S, Yang H, Li Y and Wang Z (2021) Construction of a Full-Length Transcriptome Resource for the Chinese Sucker (Myxocyprinus asiaticus), a Rare Protected Fish, Based on Isoform Sequencing (Iso-Seq). Front. Mar. Sci. 8:699504. doi: 10.3389/fmars.2021.699504
Received: 23 April 2021; Accepted: 17 May 2021;
Published: 14 June 2021.
Edited by:
Manuel Aranda, King Abdullah University of Science and Technology, Saudi ArabiaReviewed by:
Arun Prasanna, King Abdullah University of Science and Technology, Saudi ArabiaNam V. Hoang, Seoul National University, South Korea
Copyright © 2021 Ge, Zhang, Zhao, Li, Gu, Liu, Yang, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Li, YXF1YXRpY3NAc3d1LmVkdS5jbg==; eXVubGljbkAxMjYuY29t; Zhijian Wang, d2FuZ3pqQHN3dS5lZHUuY24=; d2FuZ3pqMTk2OUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship