
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 31 May 2021
Sec. Microbial Symbioses
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.659180
 Golam Rabbani1
Golam Rabbani1 Bertrand Chengxiang Yan1
Bertrand Chengxiang Yan1 Nicole Li Ying Lee2
Nicole Li Ying Lee2 Jillian Lean Sim Ooi3
Jillian Lean Sim Ooi3 Jen Nie Lee4
Jen Nie Lee4 Danwei Huang2,5,6
Danwei Huang2,5,6 Benjamin J. Wainwright1*
Benjamin J. Wainwright1*Plant-microbe relationships play critical roles in the functioning and health of terrestrial plants, but little is known about this relationship in marine angiosperms such as seagrasses. Here, we investigated the microbial communities associated with the seagrass Enhalus acoroides throughout Singapore and Peninsular Malaysia. At each sampling location we collected 10 individual and unconnected plants. Each plant was subsequently broken down into leaves, roots, and rhizomes. In addition to living plant parts a sediment sample was taken in close proximity to each. Using high throughput 16S rRNA gene amplicon sequencing we characterised the bacterial communities associated with each plant part and the associated sediment sample. Results indicate geographic structuring of bacterial communities, with a significant pattern of distance decay suggesting dispersal limitation is a contributing factor to the differences we see in bacterial community structure. Bacterial communities can be further differentiated by the function of the collected sample (leaf, root, and rhizome), and we identified a number of microbial indicator species that are associated with each plant part. Further analysis revealed the presence of several microbial taxa that have previously been identified as indicators of “unhealthy” or “stressed” seagrass meadows. This study addresses a current scientific gap related to the characterisation of seagrass microbiomes, and provides a foundation on which future studies can build, particularly those in the Southeast Asian seagrass biodiversity hotspot.
Seagrasses are marine angiosperms whose ancestors made the transition from land back to marine environments between 70 and 100 million years ago (Olsen et al., 2016). Forming large meadows they provide numerous important ecological services, they act as nursery habitats for many marine species, help prevent shoreline erosion through the dissipation of wave energy, reduce human contact with bacterial pathogens and play crucial roles in nutrient cycling and carbon sequestration (Harborne et al., 2006; Waycott et al., 2009; Kannan et al., 2010; Fourqurean et al., 2012; Christianen et al., 2013; Dewsbury et al., 2016; Lamb et al., 2017). It is estimated that seagrasses are responsible for 10% of total global carbon sequestration (Fourqurean et al., 2012). Yet, despite this recognised importance, seagrasses are increasingly threatened by anthropogenic stresses such as coastal development, overfishing, pollution from land based runoff, and climate change (Waycott et al., 2009; Unsworth et al., 2018), and such effects are especially apparent in Southeast Asia (Fortes et al., 2018). Globally, annual seagrass loss is estimated to be approximately 7% per year (Waycott et al., 2009) with declines of approximately 45% reported in localised cases (Yaakub et al., 2014, 2018).
Plant-microbe associations are critical to the health and functioning of terrestrial plants (Vandenkoornhuyse et al., 2015; Crump et al., 2018), but little is known of the role, diversity and structure of microbial communities in seagrasses and other marine plants (Cúcio et al., 2016; Fahimipour et al., 2017; Crump et al., 2018). However, this is beginning to change as researchers start to examine the microbiomes of marine and coastal plants (Fahimipour et al., 2017; Wainwright et al., 2018, 2019b; Hurtado-McCormick et al., 2019; Lee et al., 2019; Ettinger and Eisen, 2020). Marine plant-associated microbes are involved in sulphide oxidation and reduction, nitrogen fixation, and nutrient cycling. They promote host nutrient uptake and play vital roles in the decomposition of plant matter (Harlin, 1973; Hemminga et al., 1991; Stapel and Hemminga, 1997; Hansen et al., 2000; Crump et al., 2018).
Terrestrial plants show discrete microbial assemblages and communities dependent upon whether leaves, fruits, or roots are examined (Amend et al., 2019). Similarly, seagrass associated microbes occupy a number of distinct microenvironments across the plant (e.g., the phyllosphere, and the rhizosphere) (Hurtado-McCormick et al., 2019; Wainwright et al., 2019c). Oxygen levels and the density of inorganic carbon and dissolved organic compounds exuded vary across the seagrass phyllosphere and rhizosphere (Wetzel and Penhale, 1979; Moriarty and Iverson, 1986; Rubio et al., 2017; Ugarelli et al., 2017). Similarly, the roots and rhizomes influence the availability of nutrients in the rhizosphere and create oxygen gradients, and different parts of the seagrass can be submerged, or exposed for differing durations. These differences can promote and lead to the development of bacterial communities specific to each environment or plant part (Ugarelli et al., 2017; Hurtado-McCormick et al., 2019; Wang et al., 2020).
Plants of the same species generally require similar environmental conditions to sustain life, for example, species that are adapted to tropical conditions would not be expected to survive in the tundra, and vice versa. Relatedly, if members of the same species require similar environmental conditions to sustain life, it could be expected that over time plants will select a species-specific consortium of bacteria, and this consortia would likely reflect their requirements for survival and reproduction. Consequently, microbial communities could become homogenised between plant members of the same species, especially when taking into account the high dispersal potential microbes have, or, “everything is everywhere: but the environment selects” (O’Malley, 2008), or in this case the plant selects. Yet, research is frequently showing discrete microbial (fungi and bacteria) communities do exist in different sampling locations within the same species (e.g., seagrasses and mangorves) (Ettinger et al., 2017; Fahimipour et al., 2017; Crump et al., 2018; Wainwright et al., 2019a; Lee et al., 2020).
Southeast Asia is the global epicentre of seagrass biodiversity (Short et al., 2007), while knowledge of seagrass ecosystems is increasing throughout the region, a paucity of research centred on seagrass remains, and considerable knowledge gaps persist (Yaakub et al., 2018). This is especially true of research examining seagrass-associated bacterial communities and microbiomes, which is unfortunate given the increasingly acknowledged vital roles that microbes play in promoting seagrass fitness (Brodersen et al., 2018; Ugarelli et al., 2019, 2017). Understanding these host and microbe relationships is especially pressing in Southeast Asia where seagrass loss is particularly acute, with meadows here suffering from the combined anthropogenic impacts associated with rapid coastal urbanisation and climate change.
With this work we examine the bacterial communities associated with the seagrass Enhalus acoroides throughout Singapore and Peninsular Malaysia and provide foundations on which to build and monitor change. We investigate site-specific associations in an attempt to reveal any patterns of bacterial biogeography, and we profiled above and below ground plant parts to determine whether specific bacterial communities reside in particular structures.
Ten individual and unconnected Enhalus acoroides plants free of any visible epiphytes were collected at low tide from each location studied (Figure 1 and Supplementary Table 1). To minimise the possible collection of clones, each plant was sampled from a location at least 10 m apart from the last. Collected plants were separated into leaves, roots and rhizomes with a sterile razor blade. Additionally, one sediment sample was collected in close proximity (<1 m) to each plant, with sediment samples taken from approximately 4 cm below the surface. Samples were immediately placed in individual, sealed tubes containing salt saturated CTAB (Hammer et al., 2015), transported on ice to a −20°C freezer (usually in a guest house). Samples were then transported to the laboratory on ice where they were stored at −80°C until DNA extraction was performed (Cobian et al., 2019).

Figure 1. Location of sampling sites throughout Singapore and Peninsular Malaysia.
All tissues and sediment samples were disrupted in an Omni Bead Ruptor 24 (Omni International) at 8 m/s for 2 min, and DNA was extracted with a Qiagen DNeasy Powersoil kit following the manufacturer’s instructions. PCR amplification targeting the V4 region of the 16S rRNA gene was performed using the bacterial and archaeal primers 515F and 806R (515F – GTG CCA GCM GCC GCG GTA A; 806R – GGA CTA CHV GGG TWT CTA AT). Forward and reverse primers were modified to include Illumina adaptors, a linker and a unique barcode (Caporaso et al., 2011). Each reaction was performed in a total volume of 25 μl, containing 1 μl of undiluted template, 0.1 μl of KAPA 3G Enzyme (Kapa Biosystems, Inc., Wilmington, MA, United States), 0.75 μl of each primer at 10 μM, 12.5 μl KAPA PCR Buffer and water to 25 μl. PCR cycling protocol was 94∘C for 180 s, followed by 35 cycles of 94∘C for 45 s, 50∘C for 60 s and 72∘C for 90 s, with a final extension at 72∘C for 10 min. Negative extraction and PCR controls were included to identify possible contamination issues.
PCR products were visualised on a 1% TBE buffer agarose gel. Normalisation and cleaning of PCR products were performed in SequalPrep normalisation plates (Invitrogen, Frederick, MD, United States) and submitted for sequencing on the Illumina MiSeq platform (600 cycles, V3 chemistry, 300-bp paired end reads) with a 30% PhiX spike (Macrogen Korea).
Sequences were demultiplexed by Macrogen, barcodes and adaptors were removed with Cutadapt (Martin, 2011). Reads were filtered based on quality scores and trimmed using the DADA2 package version 1.14.1 (Callahan et al., 2016) in R version 3.6.2 (R Core Team, 2017). Forward reads were truncated at 250 nucleotides, and reverse reads were truncated at 200 nucleotides. Both forward and reverse reads were filtered to remove any reads with a max EE (expected error) of 2, and reads were additionally truncated at the end of ‘‘a good quality sequence’’ with the parameter truncQ = 2 (see1 for a detailed explanation of filtering parameters).
The DADA2 algorithm was next used to estimate error rates from all quality-filtered reads and then to merge forward and reverse reads and infer amplicon sequence variants (ASVs). Chimeras were removed with de novo detection. Sequenced extraction negatives were used to identify possible contaminants using the prevalence method implemented in the decontam R package (Davis et al., 2018), and remaining ASVs were assigned taxonomy with the RDP classifier (Cole et al., 2007) against a training set based on the Silva v138 16S database (Quast et al., 2013).
Any ASVs assigned to mitochondrial or chloroplast genomes, and those not present in at least 5% of samples were removed. Rarefaction curves were produced using the rarecurve() function implemented in the vegan R package version 2.5-6 (Oksanen et al., 2019). Raw sequence counts were then converted to relative abundance. The Shannon diversity for each sample was calculated, and non-metric multi-dimensional scaling (NMDS) was performed on the Bray–Curtis dissimilarity matrix of samples using the phyloseq R package version 1.30.0 (McMurdie and Holmes, 2013). NMDS plots were generated for all sampled compartments together, and then for each compartment individually (leaf, rhizome, root, sediment). PCoA and network plots were constructed using the phyloseq package. Bar plots of relative abundance were made using ggplot2 version 3.3.1 (Wickham, 2011).
Permutational multivariate ANOVA was performed using the adonis() function of the vegan R package version 2.5-6 (Oksanen et al., 2019). Venn diagrams were created using the VennDiagram R package (Chen and Boutros, 2011). Tests for distance decay of similarity (Mantel test and multiple regression on distance matrices) were performed using the ecodist package. Indicator species were identified for each compartment and each site using the indicspecies R package version 1.7.9 (Cáceres et al., 2012).
All sequences associated with this work have been deposited at the National Center for Biotechnology Information under BioProject ID: PRJNA649070.
After quality control and filtering, 5,539,364 reads were retained for downstream analysis, see Supplementary Table 2 for sequencing statistics of individual samples. Rarefaction curves indicate that sufficient sequencing depth was achieved (Supplementary Figure 1). Bacterial communities appear to be structured by the function of sampled compartments (leaf, rhizome, root, and sediment; Supplementary Figure 2), and these communities can be further differentiated by geographic location (Figure 2). This community structuring is further confirmed by network plots and PCoA plots that show similar patterns of clustering by seagrass part and location (Supplementary Figures 3, 4). Permutational multivariate analysis of variance (PERMANOVA) indicates significant differences in bacterial community according to structure and location (R2 = 0.183; p = 0.001 and R2 = 0.092; p = 0.001, respectively; Supplementary Table 3).

Figure 2. Non-metric multidimensional scaling (NMDS) of bacterial communities based on Bray–Curtis dissimilarity, coloured by location with shapes indicating structure sampled. Plots (A–D), show the leaf, rhizome, root, and sediment samples respectively.
Structures that are below ground (i.e., rhizome and root) share more similar bacterial communities than those that are above ground, and biological structures have similar communities and diversity in comparison to non-biological structures (e.g., sediment) (Figure 3 and Supplementary Figure 2). Bacterial communities show a significant pattern of distance decay, with those closer together more similar in community structure (Mantel test: R = 0.128, p = 0.001). This relationship is further supported by multiple regression on distance matrices (MRM) for all samples combined (R2 = 0.038, p = 0.001) (Table 1). Bacterial diversity is highest in sediment samples and lowest in the leaves (Figure 3). Samples from Merambong Shoal have the highest median diversity (Shannon), and all locations generally have similar levels of diversity (Supplementary Figure 5). All samples are dominated by members from the phylum Proteobacteria, irrespective of structure and location, and the phylum Fusobacteria has the highest relative abundance in sediment samples from Perhentian (Figure 4). The class Gammaproteobacteria has been found in all compartments, at all locations but tends to be more abundant in living structures (leaf, rhizome, and root samples) (Supplementary Figure 6). Along with Gammaproteobacteria, heatmaps show that Alphaproteobacteria, and Acidimicrobiia and Bacteroidia are common in all compartments. Desulfobacteria and Anaerolineae are more frequently observed in the sediment and below ground structures, and Planctomycetes tends to be most frequently encountered in sediment samples (Figure 5; Supplementary Figures 6–11).
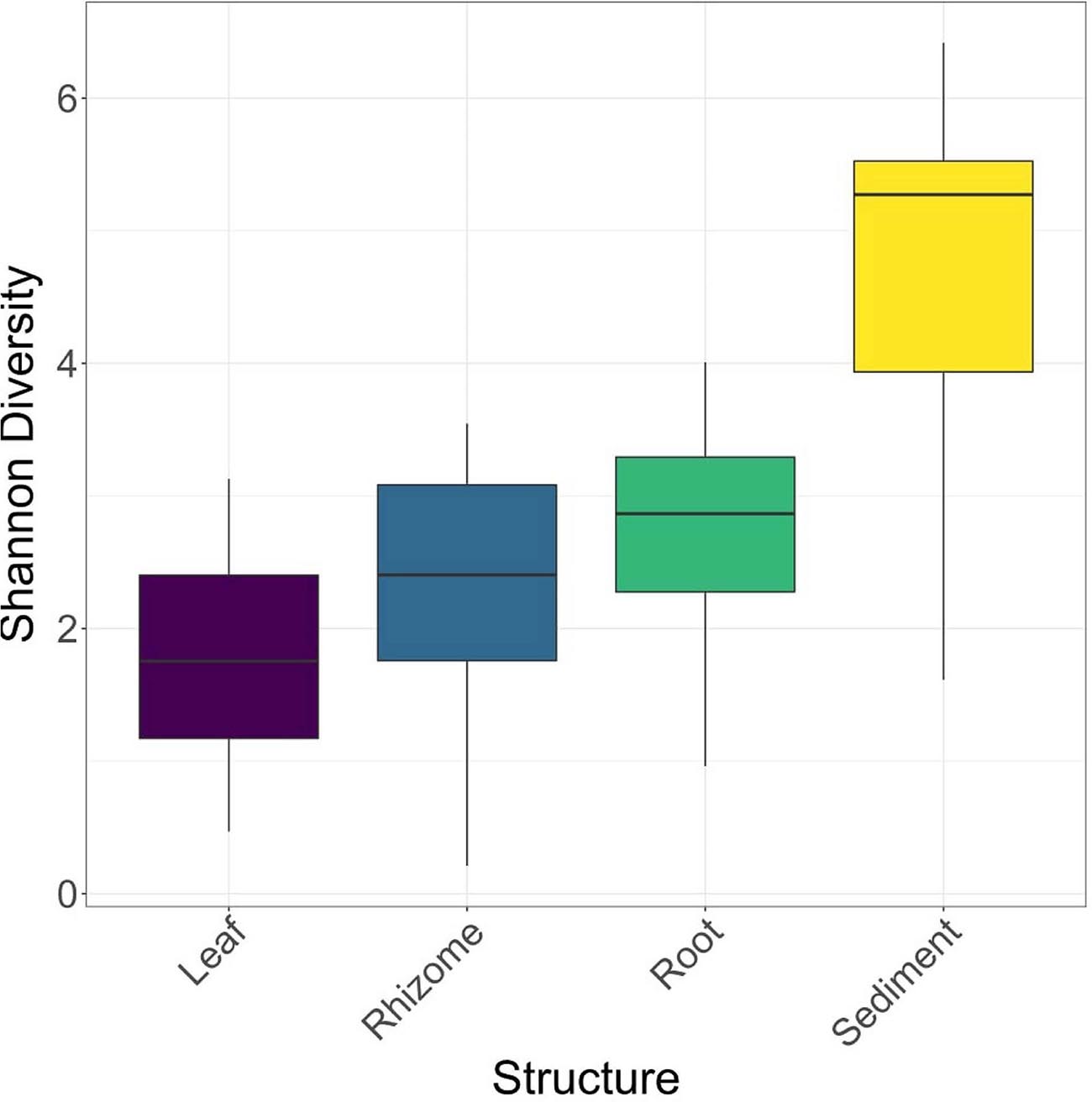
Figure 3. Box plots of Shannon diversity for each location.
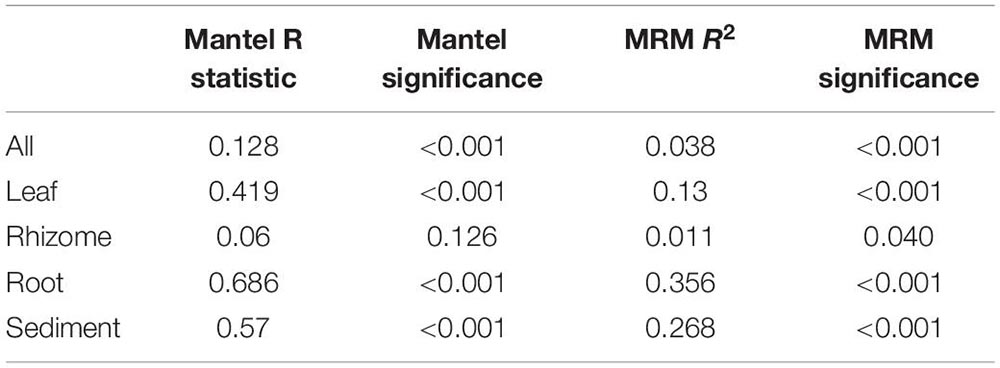
Table 1. Mantel test and multiple regression on distance matrices (MRM) results for all compartments combined, and each individual compartment.
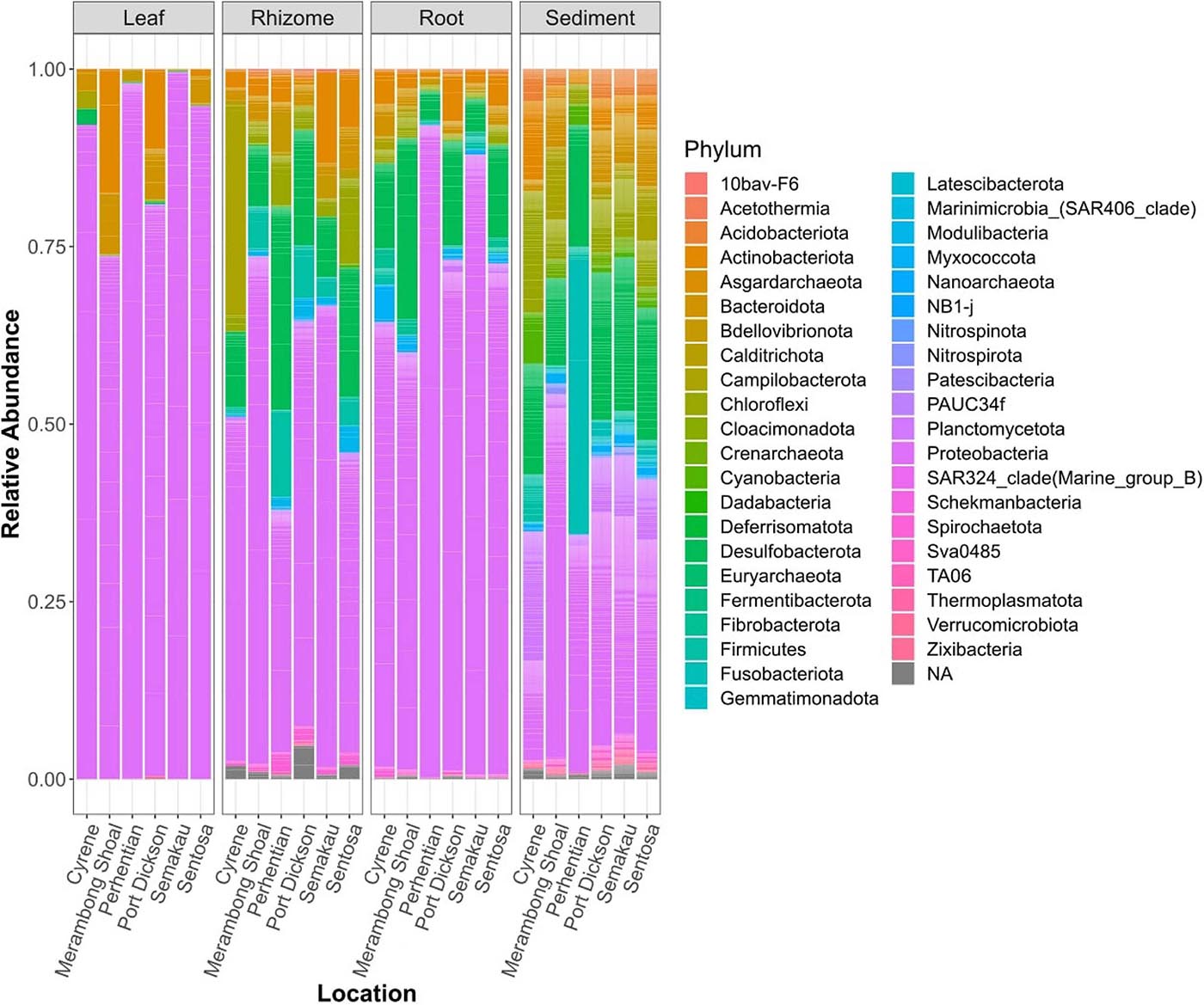
Figure 4. Stacked bar plots of relative bacterial abundance. All samples of a specific type, from one location combined.
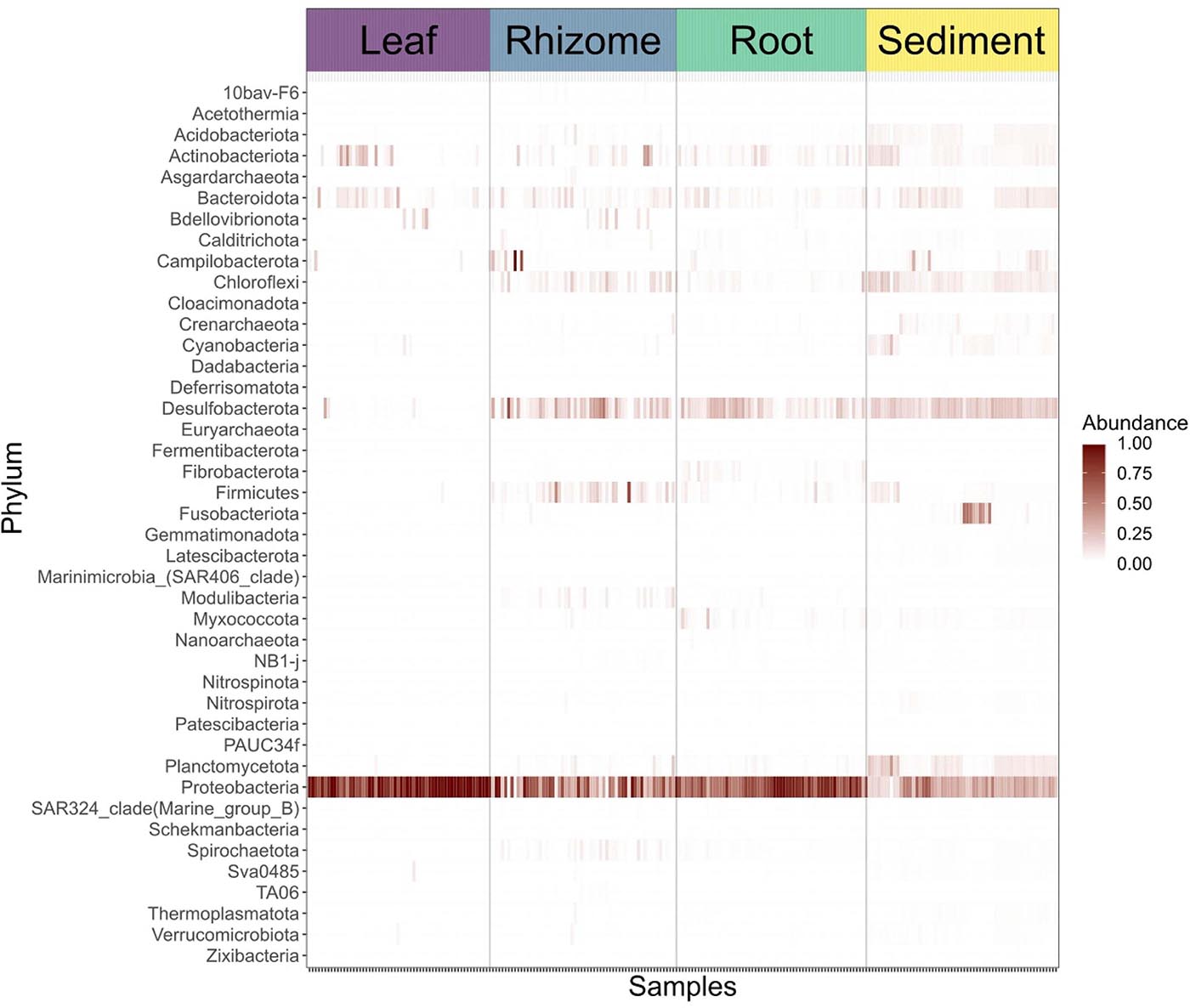
Figure 5. Heat map showing the abundances of bacterial phyla in sediment samples and specific plant parts.
Amplicon sequence variant (ASV) richness was highest in sediment, followed by root, rhizome, and leaf (1,426, 844, 659, 140, respectively). 64 ASVs are shared between all structures including sediment, and the highest number of shared ASVs (321) are found between rhizome, root, and sediment samples (Figure 6). Indicator species analysis shows leaves are significantly associated with 19 ASVs, rhizomes 16 ASVs, root 48 ASVs and 425 are associated with sediment samples. See Table 2 for the top 5 associations with each structure and Supplementary Table 4 for a complete list. Roots and rhizomes tend to associate with sulphate reducing and oxidising bacteria. The top 5 genera associated with half of the sampling locations have ASVs from the genus Vibrio significantly associated with them (Table 3), see Supplementary Table 5 for full details of all significant associations.
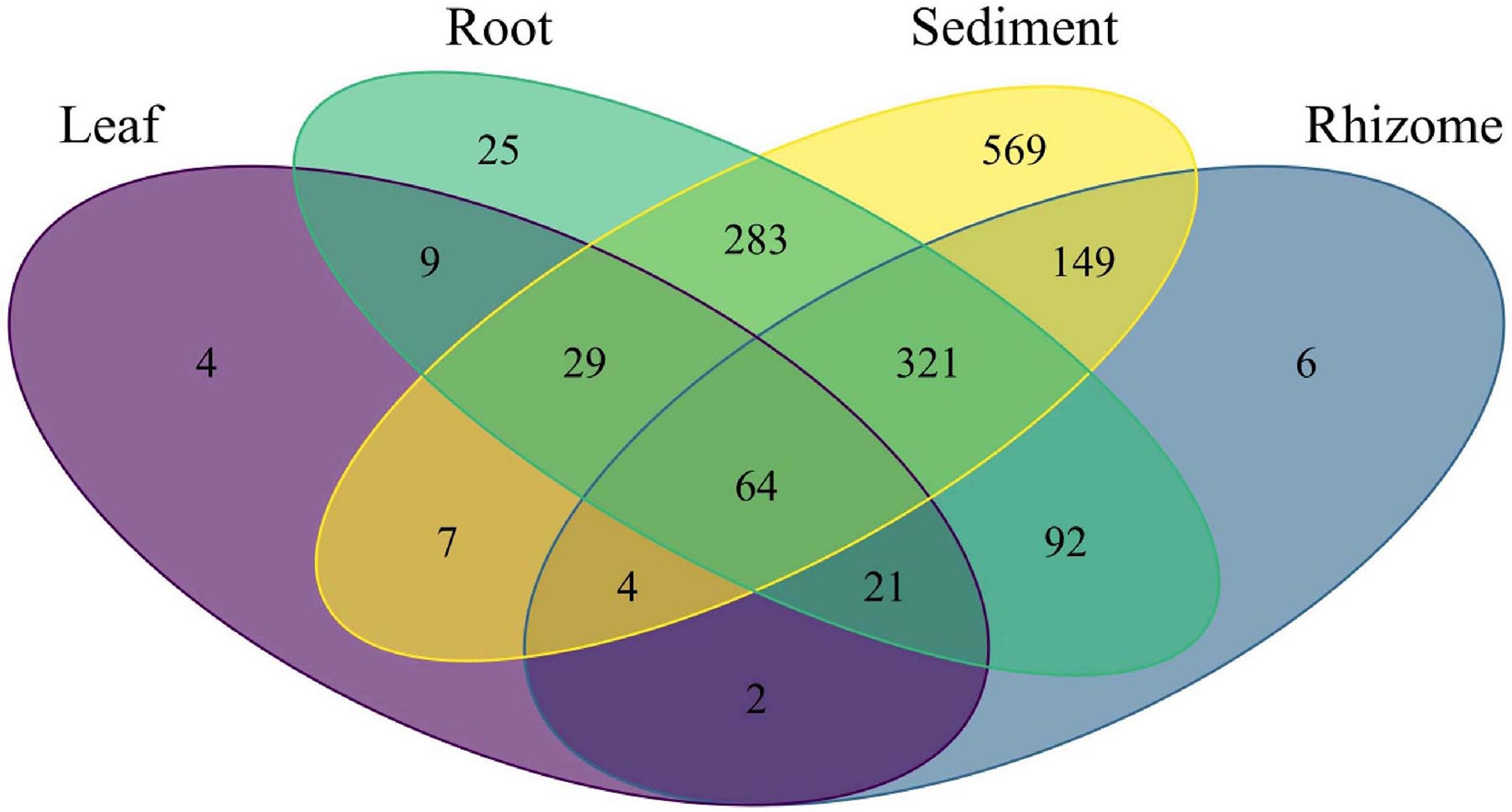
Figure 6. Venn diagram showing the number of bacterial ASVs unique to each part and shared between parts.

Table 2. Top 5 genera significantly associated with each structure see Supplementary Table 2 for a complete list of significant associations.

Table 3. Top 5 genera significantly associated with the seagrass, Enhalus acoroides, at each location, see Supplementary Table 3 for a complete list of significant associations.
Stemming from their high dispersal ability and high abundance, it is a commonly held belief that bacterial communities will show lower levels of spatial variation than larger multicellular organisms (Finlay, 2002; Horner-Devine et al., 2004; Meyer et al., 2018; Bay et al., 2020). Facilitating this expected low spatial variance, microbes are capable of entering extremely low-energy requiring states where they can persist in dormancy for up to 101.5 Ma. These microbes, when presented with suitable conditions are then able to readily incorporate carbon and nitrogen and go through cell division to rapidly increase in numbers (Bradley et al., 2020; Morono et al., 2020). The ability to enter a dormant state allows bacteria to overcome high environmental heterogeneity, or unfavourable conditions and emerge when conditions are suitable (Jones and Lennon, 2010). This idea suggests that while microbes could be everywhere, they might not actually be playing biological or ecological roles in the ecosystem at the particular point in time sampling took place. Molecular methods would still detect their presence irrespective of whether they are active or dormant, likewise, relic DNA (that from dead microorganisms) can persist for years (Cangelosi and Meschke, 2014; Carini et al., 2017). The presence of dormant microbes and relic DNA can obscure biologically relevant patterns and phenomena and suggest an absence of geographically localised community structure (Finlay, 2002; Carini et al., 2017).
However, work is now challenging the notion that variance is low in microbial communities, and congruent with other research examining marine microbiomes (Cúcio et al., 2016; Crump et al., 2018; Bay et al., 2020; Osman et al., 2020; Tan et al., 2020; Wainwright et al., 2020) we show that bacterial communities can be significantly different between sampling location and structure examined, and this spatial variance is particularly strong in sediment samples in comparison to living seagrass structures. This is consistent with previously proposed hypotheses suggesting that habitats offered by living plant organs (i.e., leaf, fruit, etc.) act as a biological filter allowing the plant to exert some degree of control over the constituents of their microbial communities (Goldmann et al., 2016; Jones et al., 2019; Wang et al., 2020). Consequently, the microbial communities associated with living compartments should be more similar to each other over wider geographic scales, whereas soil and sediment associated communities are not subjected to any filtering, therefore we see higher community variance between sample sites in these non-living sample types, and this could allow the development of more unique communities. This idea is supported by the results of our ordinations, MRM and Mantel tests. Ordinations show tight and distinct clustering in sediment samples, and a greater degree of community overlap in living structures, but there are still significant differences in community structure between living parts. MRM and Mantel tests confirm this structuring and the strongest patterns of distance decay of similarity in bacterial communities is found in sediment samples. Further supporting this idea of filtering we see the highest microbial diversity in sediment samples and much lower, but similar level of diversity in all living samples. This likely reflects the specific requirements that each living structure requires for correct functioning, and similar to our work, numerous other studies report highly diverse microbial communities in soil and sediment samples (Serna-Chavez et al., 2013; Fierer, 2017).
Our analysis of indicator species shows a number of microbes significantly associated with each structure and location, for example, rhizome and root tissues both associate with bacteria that are involved in sulphide-oxidation. Sulphur containing compounds are highly toxic to seagrasses and cause a reduction in photosynthetic performance and block aerobic respiration by inhibition of the mitochondrial cytochrome c oxidase enzyme (Lee et al., 1999). Under anoxic conditions sulphide compounds (H2S, HS–, and S2–), especially hydrogen sulphide, a common metabolic poison can accumulate to levels that are toxic (Goodman et al., 1995). These sulphide containing compounds enter root tissue where they inhibit enzyme performance and functioning (Lee et al., 1999), and field experiments show that sulphide exposure results in stunted growth (King et al., 1982). The presence, and significant associations of microbes that can reduce the availability of toxic compounds in these structures is a likely adaption to the anoxic conditions that exist in water logged sediment environments. Leaves were significantly associated with bacteria from the genus Marinomonas. This genus has previously been found associated with the leaves of the seagrass Posidonia oceanica (Garcias-Bonet et al., 2012; Tarquinio et al., 2019), and plays a role in the production of oxidative enzymes such as melanin (Sanchez-Amat et al., 2010). Melanin is a free radical scavenger that helps prevent cellular damage that can be caused by oxidative stress at high light intensities and the resultant excess excitation energy (Mittler, 2002; Costa et al., 2015; Tarquinio et al., 2019). Microbes that minimise oxidative stress are particularly useful in the leaves of seagrasses from tropical regions where exposure to direct and intense sunlight at low tide is frequent. Taxa from the genus Marinomonas are also recognised as Plant Growth Promoting Bacteria (PGPB), which assist and increase growth and development in macroalgae (Singh et al., 2011), and it is suggested that the same PGBPs have profound effects and enhance the development rate of seagrass leaves (Celdrán et al., 2012). Similarly, leaves were significantly associated with the genera Labrenzia which also helps promote plant growth and has antimicrobial properties (Martin et al., 2020).
Microbial indicators have been proposed as sensitive, and effective markers of environmental perturbation and stress, and the development/identification of these markers and their associations can aid in ecosystem management and restoration programmes (Glasl et al., 2019; Wainwright et al., 2019a; Martin et al., 2020). Previous studies have identified a number of bacterial genera that are indicative of “healthy” and “stressed” seagrass states. For example, methylotrophic bacteria, iron cycling bacteria, and N2 fixing bacteria are generally more abundant and associated with seagrass meadows identified as “healthy.” Conversely, sulphur-cycling bacteria (sulphide-oxidising and sulphate-reducing) are more frequently observed in “stressed” meadows (Martin et al., 2020), and the genus Vibrio has been implicated as a causative agent of disease in many marine species (Colwell and Grimes, 1984; Jayasree et al., 2006; Vezzulli et al., 2016; Weynberg et al., 2016; Rubio-Portillo et al., 2018). Several studies have putatively identified Vibrio spp. as bacterial pathogens in seagrass meadows, and Vibrio spp. tend to show increased abundance in more impacted sites (Liu et al., 2018; Tarquinio et al., 2019). Indicative of the anthropogenic stresses associated with coastal urbanisation and increasing coastal development throughout the region, several of our sample sites have significant associations with Vibrio spp. and sulphur cycling bacteria. We did not detect any of the bacterial genera that have been identified as indicators of “healthy” seagrass meadows, and given the conservation challenges that seagrasses in the region face, this is not entirely surprising. However, caution should be exercised when interpreting what bacteria are indicative of “healthy” seagrass meadows, especially when comparing study results from different countries, particularly given the complexities and dynamics of microbial systems (Archer et al., 2019). This study is the first of this nature from the region, and the composition of the microbial constituents in a healthy seagrass meadow here could be very different to those in other regions. Nevertheless, a number of the bacteria we identified are associated with degraded seagrass meadows and it would be wise to consider these findings in seagrass conservation schemes. Doing so could facilitate the taking of steps to mitigate known stressors such as increased sedimentation resulting from deforestation, particularly as these stresses have already been documented to increase the prevalence of potential seagrass pathogens (Liu et al., 2018).
As has previously been suggested (Santos et al., 2011; O’Callaghan, 2016; Zahn and Amend, 2017; Lee et al., 2019; Tarquinio et al., 2019; Vanwonterghem and Webster, 2020), understanding microbial community dynamics and structure is likely a key determinant of restoration and conservation success. Unfortunately, the majority of seagrass restoration projects end in failure (Bayraktarov et al., 2016; van Katwijk et al., 2016) and a variety of reasons are thought to contribute to these unfavourable outcomes, amongst them – poor site location, techniques used, or ongoing human stressors (Fonseca, 2011). To our knowledge, and despite the widespread manipulation of microbiomes in terrestrial systems to achieve positive outcomes (O’Callaghan, 2016; Zahn and Amend, 2017), this has not been attempted in seagrass restoration projects. Considering the microbial constituents in seagrass restoration is likely even more important when using transplants from other regions, or transplants grown in ex situ nurseries, especially, as we show that microbial communities can differ over comparatively small spatial scales (<10 km). If microbes are not considered it is possible that transplants could be maladapted to their new environments, or suffer from transplantation shock (Wang et al., 2021) which could be a contributing factors leading to restoration failure (Vogel et al., 2021).
With this work we characterise the bacterial communities associated with the seagrass Enhalus acoroides throughout Singapore and Peninsular Malaysia, providing a framework on which future studies can build and monitor bacterial community change and composition.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA649070.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was funded by the National Research Foundation, Prime Minister’s Office, Singapore under its Marine Science R&D Programme (MSRDP-P03) and the Mandai Nature Fund. Student support was provided by the Summer Research Programme, administered by the Yale-NUS College Centre for International & Professional Experience (CIPE) and co-funded by the Yale-NUS Dean of Faculty Office. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All samples were collected under permit number NP/RP16-156 issued by the National Parks Board of Singapore. Collections from Malaysia were made under permit JTLM 630-7Jld.9(9).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.659180/full#supplementary-material
Amend, A. S., Cobian, G. M., Laruson, A. J., Remple, K., Tucker, S. J., Poff, K. E., et al. (2019). Phytobiomes are compositionally nested from the ground up. PeerJ 7:e6609. doi: 10.7717/peerj.6609
Archer, S. D. J., Lee, K. C., Caruso, T., King-Miaow, K., Harvey, M., Huang, D., et al. (2019). Air mass source determines airborne microbial diversity at the ocean–atmosphere interface of the Great Barrier Reef marine ecosystem. ISME J. 14, 871–876. doi: 10.1038/s41396-019-0555-0
Bay, S. K., McGeoch, M. A., Gillor, O., Wieler, N., Palmer, D. J., Baker, D. J., et al. (2020). Soil bacterial communities exhibit strong biogeographic patterns at fine taxonomic resolution. mSystems 5:e00540–20. doi: 10.1128/mSystems.00540-20
Bayraktarov, E., Saunders, M. I., Abdullah, S., Mills, M., Beher, J., Possingham, H. P., et al. (2016). The cost and feasibility of marine coastal restoration. Ecol. Appl. 26, 1055–1074. doi: 10.1890/15-1077
Bradley, J. A., Arndt, S., Amend, J. P., Burwicz, E., Dale, A. W., Egger, M., et al. (2020). Widespread energy limitation to life in global subseafloor sediments. Sci. Adv. 6:eaba0697. doi: 10.1126/sciadv.aba0697
Brodersen, K. E., Siboni, N., Nielsen, D. A., Pernice, M., Ralph, P. J., Seymour, J., et al. (2018). Seagrass rhizosphere microenvironment alters plant-associated microbial community composition. Environ. Microbiol. 20, 2854–2864. doi: 10.1111/1462-2920.14245
Cáceres, M. D., Legendre, P., Wiser, S. K., and Brotons, L. (2012). Using species combinations in indicator value analyses. Methods Ecol. Evol. 3, 973–982. doi: 10.1111/j.2041-210X.2012.00246.x
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Cangelosi, G. A., and Meschke, J. S. (2014). Dead or alive: molecular assessment of microbial viability. Appl. Environ. Microbiol. 80, 5884–5891. doi: 10.1128/AEM.01763-14
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Carini, P., Marsden, P. J., Leff, J. W., Morgan, E. E., Strickland, M. S., and Fierer, N. (2017). Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2:16242. doi: 10.1038/nmicrobiol.2016.242
Celdrán, D., Espinosa, E., Sánchez-Amat, A., and Marín, A. (2012). Effects of epibiotic bacteria on leaf growth and epiphytes of the seagrass Posidonia oceanica. Mar. Ecol. Prog. Ser. 456, 21–27. doi: 10.3354/meps09672
Chen, H., and Boutros, P. C. (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12:35. doi: 10.1186/1471-2105-12-35
Christianen, M. J. A., van Belzen, J., Herman, P. M. J., van Katwijk, M. M., Lamers, L. P. M., van Leent, P. J. M., et al. (2013). Low-canopy seagrass beds still provide important coastal protection services. PLoS One 8:e62413. doi: 10.1371/journal.pone.0062413
Cobian, G. M., Egan, C. P., and Amend, A. S. (2019). Plant–microbe specificity varies as a function of elevation. ISME J. 1:470. doi: 10.1038/s41396-019-0470-4
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., et al. (2007). The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 35, D169–D172. doi: 10.1093/nar/gkl889
Colwell, R. R., and Grimes, D. J. (1984). Vibrio diseases of marine fish populations. Helgolander Meeresunters 37, 265–287. doi: 10.1007/BF01989311
Costa, M. M., Barrote, I., Silva, J., Olivé, I., Alexandre, A., Albano, S., et al. (2015). Epiphytes modulate posidonia oceanica photosynthetic production, energetic balance, antioxidant mechanisms, and oxidative damage. Front. Mar. Sci. 2:111. doi: 10.3389/fmars.2015.00111
Crump, B. C., Wojahn, J. M., Tomas, F., and Mueller, R. S. (2018). Metatranscriptomics and amplicon sequencing reveal mutualisms in seagrass microbiomes. Front. Microbiol. 9:388. doi: 10.3389/fmicb.2018.00388
Cúcio, C., Engelen, A. H., Costa, R., and Muyzer, G. (2016). Rhizosphere microbiomes of European + seagrasses are selected by the plant, but are not species specific. Front. Microbiol. 7:440. doi: 10.3389/fmicb.2016.00440
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., and Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226. doi: 10.1186/s40168-018-0605-2
Dewsbury, B. M., Bhat, M., and Fourqurean, J. W. (2016). A review of seagrass economic valuations: gaps and progress in valuation approaches. Ecosyst. Serv. 18, 68–77. doi: 10.1016/j.ecoser.2016.02.010
Ettinger, C. L., and Eisen, J. A. (2020). Fungi, bacteria and oomycota opportunistically isolated from the seagrass, Zostera marina. PLoS One 15:e0236135. doi: 10.1371/journal.pone.0236135
Ettinger, C. L., Voerman, S. E., Lang, J. M., Stachowicz, J. J., and Eisen, J. A. (2017). Microbial communities in sediment from Zostera marina patches, but not the Z. marina leaf or root microbiomes, vary in relation to distance from patch edge. PeerJ 5:e3246. doi: 10.7717/peerj.3246
Fahimipour, A. K., Kardish, M. R., Lang, J. M., Green, J. L., Eisen, J. A., and Stachowicz, J. J. (2017). Global-scale structure of the eelgrass microbiome. Appl. Environ. Microbiol. 83, e03391–16. doi: 10.1128/AEM.03391-16
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Finlay, B. J. (2002). Global dispersal of free-living microbial eukaryote species. Science 296, 1061–1063. doi: 10.1126/science.1070710
Fonseca, M. S. (2011). Addy revisited: what has changed with seagrass restoration in 64 years? Ecol. Restor. 29, 73–81.
Fortes, M. D., Ooi, J. L. S., Tan, Y. M., Prathep, A., Bujang, J. S., and Yaakub, S. M. (2018). Seagrass in Southeast Asia: a review of status and knowledge gaps, and a road map for conservation. Bot. Mar. 61, 269–288. doi: 10.1515/bot-2018-0008
Fourqurean, J. W., Duarte, C. M., Kennedy, H., Marbà, N., Holmer, M., Mateo, M. A., et al. (2012). Seagrass ecosystems as a globally significant carbon stock. Nat. Geosci. 5, 505–509. doi: 10.1038/ngeo1477
Garcias-Bonet, N., Arrieta, J. M., de Santana, C. N., Duarte, C. M., and Marbà, N. (2012). Endophytic bacterial community of a Mediterranean marine angiosperm (Posidonia oceanica). Front. Microbiol. 3:342. doi: 10.3389/fmicb.2012.00342
Glasl, B., Bourne, D. G., Frade, P. R., Thomas, T., Schaffelke, B., and Webster, N. S. (2019). Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome 7:94. doi: 10.1186/s40168-019-0705-7
Goldmann, K., Schröter, K., Pena, R., Schöning, I., Schrumpf, M., Buscot, F., et al. (2016). Divergent habitat filtering of root and soil fungal communities in temperate beech forests. Sci. Rep. 6:31439. doi: 10.1038/srep31439
Goodman, J. L., Moore, K. A., and Dennison, W. C. (1995). Photosynthetic responses of eelgrass (Zostera marina L.) to light and sediment sulfide in a shallow barrier island lagoon. Aquat. Bot. 50, 37–47. doi: 10.1016/0304-3770(94)00444-Q
Hammer, T. J., Dickerson, J. C., and Fierer, N. (2015). Evidence-based recommendations on storing and handling specimens for analyses of insect microbiota. PeerJ 3:e1190. doi: 10.7717/peerj.1190
Hansen, J., Udy, J., Perry, C., Dennison, W., and Lomstein, B. A. (2000). Effect of the seagrass Zostera capricorni on sediment microbial processes. Mar. Ecol. Prog. Ser. 199, 83–96. doi: 10.3354/meps199083
Harborne, A. R., Mumby, P. J., Micheli, F., Perry, C. T., Dahlgren, C. P., Holmes, K. E., et al. (2006). The functional value of Caribbean coral reef, seagrass and mangrove habitats to ecosystem processes. Adv. Mar. Biol. 50, 57–189. doi: 10.1016/S0065-2881(05)50002-6
Harlin, M. M. (1973). Transfer of products between epiphytic marine algae and host plants1,2. J. Phycol. 9, 243–248. doi: 10.1111/j.1529-8817.1973.tb04088.x
Hemminga, M. A., Harrison, P. G., and van Lent, F. (1991). The balance of nutrient losses and gains in seagrass meadows. Mar. Ecol. Prog. Ser. 71, 85–96. doi: 10.3354/meps071085
Horner-Devine, M. C., Lage, M., Hughes, J. B., and Bohannan, B. J. M. (2004). A taxa–area relationship for bacteria. Nature 432, 750–753. doi: 10.1038/nature03073
Hurtado-McCormick, V., Kahlke, T., Petrou, K., Jeffries, T., Ralph, P. J., and Seymour, J. R. (2019). Regional and microenvironmental scale characterization of the Zostera muelleri seagrass microbiome. Front. Microbiol. 10:1011. doi: 10.3389/fmicb.2019.01011
Jayasree, L., Janakiram, P., and Madhavi, R. (2006). Characterization of Vibrio spp. associated with diseased shrimp from culture ponds of Andhra Pradesh (India). J. World Aquac. Soc. 37, 523–532. doi: 10.1111/j.1749-7345.2006.00066.x
Jones, P., Garcia, B. J., Furches, A., Tuskan, G. A., and Jacobson, D. (2019). Plant host-associated mechanisms for microbial selection. Front. Plant Sci. 10:862. doi: 10.3389/fpls.2019.00862
Jones, S. E., and Lennon, J. T. (2010). Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U. S. A. 107, 5881–5886. doi: 10.1073/pnas.0912765107
Kannan, R. R. R., Arumugam, R., and Anantharaman, P. (2010). Antibacterial potential of three seagrasses against human pathogens. Asian Pac. J. Trop. Med. 3, 890–893. doi: 10.1016/S1995-7645(10)60214-3
King, G. M., Klug, M. J., Wiegert, R. G., and Chalmers, A. G. (1982). Relation of soil water movement and sulfide concentration to Spartina alterniflora production in a Georgia Salt Marsh. Science 218, 61–63. doi: 10.1126/science.218.4567.61
Lamb, J. B., Water, J. A. J. M., van de Bourne, D. G., Altier, C., Hein, M. Y., Fiorenza, E. A., et al. (2017). Seagrass ecosystems reduce exposure to bacterial pathogens of humans, fishes, and invertebrates. Science 355, 731–733. doi: 10.1126/science.aal1956
Lee, N. L. Y., Huang, D., Quek, Z. B. R., Lee, J. N., and Wainwright, B. J. (2019). Mangrove-associated fungal communities are differentiated by geographic location and host structure. Front. Microbiol. 10:2456. doi: 10.3389/fmicb.2019.02456
Lee, N. L. Y., Huang, D., Quek, Z. B. R., Lee, J. N., and Wainwright, B. J. (2020). Distinct fungal communities associated with different organs of the mangrove Sonneratia alba in the Malay Peninsula. IMA Fungus 11:17. doi: 10.1186/s43008-020-00042-y
Lee, R. W., Kraus, D. W., and Doeller, J. E. (1999). Oxidation of sulfide by Spartina alterniflora roots. Limnol. Oceanogr. 44, 1155–1159. doi: 10.4319/lo.1999.44.4.1155
Liu, S., Jiang, Z., Deng, Y., Wu, Y., Zhang, J., Zhao, C., et al. (2018). Effects of nutrient loading on sediment bacterial and pathogen communities within seagrass meadows. MicrobiologyOpen 7:e00600. doi: 10.1002/mbo3.600
Martin, B. C., Alarcon, M. S., Gleeson, D., Middleton, J. A., Fraser, M. W., Ryan, M. H., et al. (2020). Root microbiomes as indicators of seagrass health. FEMS Microbiol. Ecol. 96:fiz201. doi: 10.1093/femsec/fiz201
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an r package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meyer, K. M., Memiaghe, H., Korte, L., Kenfack, D., Alonso, A., and Bohannan, B. J. M. (2018). Why do microbes exhibit weak biogeographic patterns? ISME J. 12, 1404–1413. doi: 10.1038/s41396-018-0103-3
Mittler, R. (2002). Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 7, 405–410. doi: 10.1016/S1360-1385(02)02312-9
Moriarty, D. J. W., and Iverson, R. L. (1986). Exudation of organic carbon by the seagrass halodule wrzghtzz aschers. And its effect on bacterial growth in the sediment. J. Exp. Mar. Biol. Ecol. 96:12. doi: 10.1016/0022-0981(86)90237-6
Morono, Y., Ito, M., Hoshino, T., Terada, T., Hori, T., Ikehara, M., et al. (2020). Aerobic microbial life persists in oxic marine sediment as old as 101.5 million years. Nat. Commun. 11:3626. doi: 10.1038/s41467-020-17330-1
O’Callaghan, M. (2016). Microbial inoculation of seed for improved crop performance: issues and opportunities. Appl. Microbiol. Biotechnol. 100, 5729–5746. doi: 10.1007/s00253-016-7590-9
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R., et al. (2019). Package ‘vegan.’ Community Ecology Package, version 2.5-6. 1–295.
Olsen, J. L., Rouzé, P., Verhelst, B., Lin, Y.-C., Bayer, T., Collen, J., et al. (2016). The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335. doi: 10.1038/nature16548
O’Malley, M. A. (2008). ‘Everything is everywhere: but the environment selects’: ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Philos. Sci. Biol. Biomed. Sci. 39, 314–325. doi: 10.1016/j.shpsc.2008.06.005
Osman, E. O., Suggett, D. J., Voolstra, C. R., Pettay, D. T., Clark, D. R., Pogoreutz, C., et al. (2020). Coral microbiome composition along the northern Red Sea suggests high plasticity of bacterial and specificity of endosymbiotic dinoflagellate communities. Microbiome 8:8. doi: 10.1186/s40168-019-0776-5
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rubio, L., García, D., García-Sánchez, M. J., Niell, F. X., Felle, H. H., and Fernández, J. A. (2017). Direct uptake of HCO 3 - in the marine angiosperm Posidonia oceanica (L.) Delile driven by a plasma membrane H + economy. Plant Cell Environ. 40, 2820–2830. doi: 10.1111/pce.13057
Rubio-Portillo, E., Gago, J. F., Martínez-García, M., Vezzulli, L., Rosselló-Móra, R., Antón, J., et al. (2018). Vibrio communities in scleractinian corals differ according to health status and geographic location in the Mediterranean Sea. Syst. Appl. Microbiol. 41, 131–138. doi: 10.1016/j.syapm.2017.11.007
Sanchez-Amat, A., Solano, F., and Lucas-Elío, P. (2010). Finding new enzymes from bacterial physiology: a successful approach illustrated by the detection of novel oxidases in marinomonas mediterranea. Mar. Drugs 8, 519–541. doi: 10.3390/md8030519
Santos, H. F., Carmo, F. L., Paes, J. E. S., Rosado, A. S., and Peixoto, R. S. (2011). Bioremediation of mangroves impacted by petroleum. Water Air Soil Pollut. 216, 329–350. doi: 10.1007/s11270-010-0536-4
Serna-Chavez, H. M., Fierer, N., and van Bodegom, P. M. (2013). Global drivers and patterns of microbial abundance in soil. Glob. Ecol. Biogeogr. 22, 1162–1172. doi: 10.1111/geb.12070
Short, F., Carruthers, T., Dennison, W., and Waycott, M. (2007). Global seagrass distribution and diversity: a bioregional model. J. Exp. Mar. Biol. Ecol. 350, 3–20. doi: 10.1016/j.jembe.2007.06.012
Singh, R. P., Mantri, V. A., Reddy, C. R. K., and Jha, B. (2011). Isolation of seaweed-associated bacteria and their morphogenesis-inducing capability in axenic cultures of the green alga Ulva fasciata. Aquat. Biol. 12, 13–21. doi: 10.3354/ab00312
Stapel, J., and Hemminga, M. A. (1997). Nutrient resorption from seagrass leaves. Mar. Biol. 128, 197–206. doi: 10.1007/s002270050083
Tan, Y. T. R., Wainwright, B. J., Afiq-Rosli, L., Ip, Y. C. A., Lee, J. N., Nguyen, N. T. H., et al. (2020). Endosymbiont diversity and community structure in Porites lutea from Southeast Asia are driven by a suite of environmental variables. Symbiosis 80, 269–277. doi: 10.1007/s13199-020-00671-2
Tarquinio, F., Hyndes, G. A., Laverock, B., Koenders, A., and Säwström, C. (2019). The seagrass holobiont: understanding seagrass-bacteria interactions and their role in seagrass ecosystem functioning. FEMS Microbiol. Lett. 366:fnz057. doi: 10.1093/femsle/fnz057
Ugarelli, K., Chakrabarti, S., Laas, P., and Stingl, U. (2017). The seagrass holobiont and its microbiome. Microorganisms 5:81. doi: 10.3390/microorganisms5040081
Ugarelli, K., Laas, P., and Stingl, U. (2019). The microbial communities of leaves and roots associated with turtle grass (Thalassia testudinum) and manatee grass (Syringodium filliforme) are distinct from seawater and sediment communities, but are similar between species and sampling sites. Microorganisms 7:4. doi: 10.3390/microorganisms7010004
Unsworth, R. K. F., Ambo-Rappe, R., Jones, B. L., La Nafie, Y. A., Irawan, A., Hernawan, U. E., et al. (2018). Indonesia’s globally significant seagrass meadows are under widespread threat. Sci. Total Environ. 634, 279–286. doi: 10.1016/j.scitotenv.2018.03.315
van Katwijk, M. M., Thorhaug, A., Marbà, N., Orth, R. J., Duarte, C. M., Kendrick, G. A., et al. (2016). Global analysis of seagrass restoration: the importance of large-scale planting. J. Appl. Ecol. 53, 567–578. doi: 10.1111/1365-2664.12562
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Van, A. L., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Vanwonterghem, I., and Webster, N. S. (2020). Coral reef microorganisms in a changing climate. iScience 23:100972. doi: 10.1016/j.isci.2020.100972
Vezzulli, L., Grande, C., Reid, P. C., Hélaouët, P., Edwards, M., Höfle, M. G., et al. (2016). Climate influence on vibrio and associated human diseases during the past half-century in the coastal North Atlantic. Proc. Natl. Acad. Sci. U. S. A. 113, E5062–E5071. doi: 10.1073/pnas.1609157113
Vogel, M. A., Mason, O. U., and Miller, T. E. (2021). Composition of seagrass phyllosphere microbial communities suggests rapid environmental regulation of community structure. FEMS Microbiol. Ecol. 97:fiab013. doi: 10.1093/femsec/fiab013
Wainwright, B. J., Afiq-Rosli, L., Zahn, G. L., and Huang, D. (2019a). Characterisation of coral-associated bacterial communities in an urbanised marine environment shows strong divergence over small geographic scales. Coral Reefs 38, 1097–1106. doi: 10.1007/s00338-019-01837-1
Wainwright, B. J., Bauman, A. G., Zahn, G. L., Todd, P. A., and Huang, D. (2019b). Characterization of fungal biodiversity and communities associated with the reef macroalga Sargassum ilicifolium reveals fungal community differentiation according to geographic locality and algal structure. Mar. Biodiv. 49, 2601–2608. doi: 10.1007/s12526-019-00992-6
Wainwright, B. J., Zahn, G. L., Afiq-Rosli, L., Tanzil, J. T. I., and Huang, D. (2020). Host age is not a consistent predictor of microbial diversity in the coral Porites lutea. Sci. Rep. 10:14376. doi: 10.1038/s41598-020-71117-4
Wainwright, B. J., Zahn, G. L., Arlyza, I. S., and Amend, A. S. (2018). Seagrass-associated fungal communities follow Wallace’s line, but host genotype does not structure fungal community. J. Biogeogr. 45, 762–770. doi: 10.1111/jbi.13168
Wainwright, B. J., Zahn, G. L., Zushi, J., Lee, N. L. Y., Ooi, J. L. S., Lee, J. N., et al. (2019c). Seagrass-associated fungal communities show distance decay of similarity that has implications for seagrass management and restoration. Ecol. Evol. 9, 11288–11297. doi: 10.1002/ece3.5631
Wang, L., English, M. K., Tomas, F., and Mueller, R. S. (2021). Recovery and Community succession of the Zostera marina rhizobiome after transplantation. Appl. Environ. Microbiol. 87:16. doi: 10.1128/AEM.02326-20
Wang, L., Tomas, F., and Mueller, R. S. (2020). Nutrient enrichment increases size of Zostera marina shoots and enriches for sulfur and nitrogen cycling bacteria in root-associated microbiomes. FEMS Microbiol. Ecol. 96:fiaa129. doi: 10.1093/femsec/fiaa129
Waycott, M., Duarte, C. M., Carruthers, T. J. B., Orth, R. J., Dennison, W. C., Olyarnik, S., et al. (2009). Accelerating loss of seagrasses across the globe threatens coastal ecosystems. Proc. Natl. Acad. Sci. U. S. A. 106, 12377–12381. doi: 10.1073/pnas.0905620106
Wetzel, R. G., and Penhale, P. A. (1979). Transport of carbon and excretion of dissolved organic carbon by leaves and roots/rhizomes in seagrasses and their epiphytes. Aquat. Bot. 6, 149–158. doi: 10.1016/0304-3770(79)90058-5
Weynberg, K. D., Voolstra, C. R., Neave, M. J., Buerger, P., and van Oppen, M. J. H. (2016). From cholera to corals: viruses as drivers of virulence in a major coral bacterial pathogen. Sci. Rep. 5:17889. doi: 10.1038/srep17889
Yaakub, S. M., McKenzie, L. J., Erftemeijer, P. L. A., Bouma, T., and Todd, P. A. (2014). Courage under fire: seagrass persistence adjacent to a highly urbanised city–state. Mar. Pollut. Bull. 83, 417–424. doi: 10.1016/j.marpolbul.2014.01.012
Yaakub, S. M., Ooi, J. L. S., Buapet, P., and Unsworth, R. K. F. (2018). Seagrass research in Southeast Asia. Bot. Mar. 61, 177–179. doi: 10.1515/bot-2018-0046
Keywords: indicator species, microbial ecology, plant-microbe interactions, seagrass microbiome, Southeast Asia, South China Sea, Strait of Malacca
Citation: Rabbani G, Yan BC, Lee NLY, Ooi JLS, Lee JN, Huang D and Wainwright BJ (2021) Spatial and Structural Factors Shape Seagrass-Associated Bacterial Communities in Singapore and Peninsular Malaysia. Front. Mar. Sci. 8:659180. doi: 10.3389/fmars.2021.659180
Received: 27 January 2021; Accepted: 28 April 2021;
Published: 31 May 2021.
Edited by:
Hirokazu Toju, Kyoto University, JapanReviewed by:
Cassie L. Ettinger, University of California, Riverside, United StatesCopyright © 2021 Rabbani, Yan, Lee, Ooi, Lee, Huang and Wainwright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin J. Wainwright, QmVuLldhaW53cmlnaHRAWWFsZS1OVVMuZWR1LnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.