 Zhe Zheng1,2,3,4
Zhe Zheng1,2,3,4 Yongshan Liao1,2Jianming Ye1
Yongshan Liao1,2Jianming Ye1 Chuangye Yang1,2,3,4
Chuangye Yang1,2,3,4 Linda Adzigbli1,5
Linda Adzigbli1,5 Qingheng Wang1,2,3,4*
Qingheng Wang1,2,3,4* Xiaodong Du1,2,3,4
Xiaodong Du1,2,3,4 Yuewen Deng1,2,3,4
Yuewen Deng1,2,3,4- 1Fisheries College, Guangdong Ocean University, Zhanjiang, China
- 2Pearl Breeding and Processing Engineering Technology Research Centre of Guangdong Province, Zhanjiang, China
- 3Guangdong Provincial Engineering Laboratory for Mariculture Organism Breeding, Zhanjiang, China
- 4Guangdong Provincial Key Laboratory of Pathogenic Biology and Epidemiology for Aquatic Economic Animals, Zhanjiang, China
- 5Department of Marine Biology, University of Rostock, Rostock, Germany
Environmental microbiota plays a vital role in the intestinal microbiota of aquatic organisms. However, data concerning the association between the intestinal microbiota of pearl oyster Pinctada fucata martensii and the surrounding seawater are limited. The existing bacterial communities in pearl oyster intestine and surrounding water from two sites (D and H, within Liusha Bay in Guangdong, China) were investigated using 16S rRNA-based sequencing to explore the relationship among the two. D located in the inner bay, and H located in the open sea area outside bay. Results revealed the richness and diversity of pearl oyster intestinal microbiota to be less than those of the surrounding water, with 38 phyla and 272 genera observed as a result of the classifiable sequence. The microbiota compositions in the intestine and the surrounding water were diversified at the phylum and genus levels, with the sequencing data being statistically significant. However, the functional prediction of microbiota emphasized the overall similarity in the functional profile of the surrounding seawater and intestinal microbiomes. This profile was associated with metabolism of cofactors and vitamin, carbohydrates metabolism, amino acids metabolism, metabolism of terpenoids, and polyketides, metabolism of other amino acids, lipids metabolism, and energy metabolism. Seven common operational taxonomic units (OTUs), which belonged to phyla Tenericutes, Cyanobacteria, and Planctomycetes, were noted in the intestines of pearl oysters from two different sites. These OTUs may be affiliates to the core microbiome of pearl oyster. Significantly different bacterial taxa in the intestines of pearl oysters from two different sites were found at the phylum and genus levels. This finding suggested that the bacterial communities in pearl oyster intestines may exhibit some plasticity to adapt to changes in the surrounding water-cultured environment. This study generally offers constructive discoveries associated with pearl oyster intestinal microbiota and provides guidance for sustainable aquaculture.
Introduction
Symbiotic associations observed between macroorganisms and bacteria (among other microbes) are ubiquitous and extensively researched. These microbial symbionts provide a number of probiotic functions (i.e., immunological regulation, defense against pathogens, and enhanced nutritional efficiency) to the host aiding homeostasis and health (O’Brien et al., 2019; Rausch et al., 2019; Simon et al., 2019). Therefore, microbiota is considered a vital part of the host physiology, and balanced intestinal microbiota is critical to the health of the host (Belkaid and Hand, 2014; Fan and Li, 2019). Previous studies revealed that the composition of the microbial community associated with the host is not random nor probabilistic but usually determined by the host phylogeny and living environment (Cárdenas et al., 2014; Brooks et al., 2016; Carrier and Reitzel, 2018). Swift progresses in sequencing technology and a reduction in its associated cost has prompted an upsurge in microbiome research, aiming at multiple organisms and environments (Caporaso et al., 2011). Aquatic animals, such as sponges (Fan et al., 2013), oysters (Laroche et al., 2018), crustaceans (Zhang et al., 2016; Gao et al., 2019), and fish (Shi et al., 2019), have been extensively investigated in recent years. Unlike terrestrial organisms, aquatic organisms are directly prone to water-cultured environment, wherein microorganisms are the vital constituents of nutrient cycling, productivity, and water quality and play an imperative function in defining the fitness of aquaculture organisms (Blancheton et al., 2013; Carbone and Faggio, 2016; Li et al., 2017). Therefore, establishing effective microbial and ecological strategies is essential for a sustainable aquaculture production and an in-depth and complete understanding of the characteristics of microflora in aquatic environments (Xiong et al., 2016). Numerous studies have examined microbial community variations that resulted from cultured environment variances (Zurel et al., 2011; King et al., 2012; Trabal et al., 2012; Fernández et al., 2014).
As a usual filter-feeder, bivalves ingest water-suspended particles, such as organic debris, bacteria, microzooplankton, and microalgae, as food sources (Yang et al., 2019b), and numerous bacteria predominant in seawater are first harbored in the algal culture. Thus, the bivalve larval microbiota comprise bacteria growing in seawater (Asmani et al., 2016), but some could come from the broodstock. Thus, understanding the relationship between the intestinal microbiota of bivalves and their cultured environment is crucial in determining the formation of intestinal microbial community and the adaptation of microorganisms in the intestine of the host to the environment. Although the relationships of bacterial communities in bivalve intestine and its aquaculture environment have been considered (Asmani et al., 2016; Lokmer et al., 2016; Laroche et al., 2018; Sun et al., 2019; Musella et al., 2020), those of pearl oyster intestine and its aquaculture environment have not been discussed. Pearl oyster (Pinctada fucata martensii) is a well-known filter feeder worldwide due to its capability to produce high-quality pearls (Yang et al., 2019a; Zhang et al., 2021). This species of oyster produces valuable pearls through biomineralization, succeeding the insertion of a mantle graft from a donor into the gonad of a recipient oyster together with a nucleus (He et al., 2020). Meanwhile, current studies have explored the genome (Du et al., 2017) and transcriptome (Hao et al., 2019; Zheng et al., 2019) to ascertain possible links to cultured pearl quality traits in P. f. martensii. However, data related to the peal oyster microbiome are currently unavailable. Therefore, characterizing the microbial composition of symbiont assemblages in P. f. martensii is critical because these microbial communities offer an important function in sustaining oyster fitness (Lokmer et al., 2016). This fitness, by extension, may consequently influence pearl quality (Cuif et al., 2018).
To address these issues, bacterial communities in pearl oyster P. f. martensii intestine and its aquaculture environment were investigated and analyzed in this study. The present work could also help accumulate basic data for the development of healthy breeding, disease prevention and control, and micro-ecological preparations and the optimization of feed formulations.
Materials and Methods
Sample Collection
In September 2017, samples, including pearl oysters which were selected from our breed of the black shell-colored line, and surrounding water, were collected from two commercial farms H (20°48′N, 109°53′E) and D (20°25′N, 109°57′E) within the Liusha Bay (Supplementary Figure 1) in Guangdong, China. Liusha Bay is a semi-closed bay with a gourd shape, which is the largest marine pearl breeding base in China. D commercial farm located in the inner bay, the wind and waves are small, the water flow is gentle. H commercial farm located in the open sea area outside bay, the current is fast, the water is exchanged well. Forty healthy 2 year-old pearl oysters (P) with total weight of 42.91 ± 7.81 g were randomly selected from D and H. The intestines were dissected and rinsed with sterilized seawater. Each replicate included four intestines, and was put into a 2.0 mL sterile centrifuge tube, froze in liquid nitrogen and immediately stored at −80°C. Five water samples (W) were simultaneously selected from the two farms, 2 L seawater was prefiltered to remove large particles, then refiltered using a polycarbonate membrane with a pore size of 0.22 μm. Following filtration, the membrane was placed in a 2.0 mL sterile centrifuge tube, froze in liquid nitrogen and then stored at −80°C. In accordance with the grouping, the samples obtained from different locations were recorded as DP, DW, HP, and HW.
DNA Extraction, PCR Amplification, and Illumina MiSeq Sequencing
The total DNA of the intestinal and water samples was extracted using the OMEGA Stool DNA Kit (D4015-01). The quality of the DNA was verified via electrophoresis on 1.0% agarose gels and ethidium bromide staining. The purity and concentration of the extracted DNA was detected using NanoDrop, and it was stored at -20°C until further use. The extracted DNA from the individual samples was diluted to 2 ng/μL as a PCR amplification DNA template. Each sample DNA template was amplified using a bacterial 16S rDNA V4 region primer. The PCR reaction system and amplification conditions were conducted following Zhao et al. (2016).
Extracted amplicons from 2% agarose gels were purified with AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) in accordance with the manufacturer’s instructions and quantified using QuantiFluor-ST (Promega, United States). The purified amplicons were then pooled in equimolar and paired-end sequences (2 × 250 bp) on an Illumina MiSeq platform in accordance with the standard protocols.
Data Analysis
Raw FASTA files were de-multiplexed, quality filtered, and analyzed on QIIME 1.80. The 250 bp reads were truncated at sites of more than three sequential bases receiving a Phred quality score of < Q20. Any reads comprising ambiguous base calls or barcode/primer errors were rejected. Operational taxonomic units (OTUs) were clustered with 97% similarity cutoff on UPARSE (version 7.1)1. Chimeric sequences were recognized and eliminated using UCHIME. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed using the RDP Classifier2 against the Silva 16S rRNA database under the confidence threshold of 70%. Transcriptomics raw data were deposited at NCBI, and the accession numbers were in Supplementary Table 1.
Alpha diversity was applied to analyze the species diversity complexity for a sample through numerous indexes, such as observed species, Chao1, Ace, Shannon, and Simpson. The sample complexity is proportional to the first four values, with a negative correlation with the Simpson value. Principal coordinate analysis (PCoA) was used to display the variances between the samples in accordance to the matrix of beta diversity distance. The close distance represents the similar species composition of the samples. Unweighted pair group method with arithmetic mean (UPGMA), a type of hierarchical clustering method that uses average linkage, was employed to deduce the distance matrix produced by beta diversity. Jackknifing analysis was performed to ascertain the robustness of the results to sequencing effort. In this analysis, 75% of the lowest sample sequences from individual samples were chosen haphazardly; the resultant UPGMA tree from this subset of data and the tree representative of the entire available data set were then compared on QIIME (v1.80). This comparison was repetitive with 100 arbitrary subsets of data and tree nodes to prove consistency across jackknifed datasets, which were considered robust. In addition, the figure was illustrated using R software (v3.1.1). PICRUSt recaptured the vital discoveries from the Human Microbiome Project and precisely predicted the richness of gene families in host-associated and environmental communities with quantifiable uncertainty using 16S information. PICRUSt 2 was used to perform the functional classification scheme of KEGG orthology and Clusters of Orthologous Groups (COGs).
Statistical Analysis
The statistical method was used to attain the abundant differences in microbial communities between samples, and false discovery rate (FDR) was obtained to assess the significance of the difference. The samples that caused diversity in species composition between the two groups were recognized on basis of the results. Statistical significance was analyzed at the genus and phylum levels among the groups using t-test. Metastats3 and R (v3.1.1) were used to decide the significance among samples in various taxonomies. The resultant p-value was adjusted using the Benjamini–Hochberg FDR correction.
Results
Overview of 16S rRNA Gene Sequencing
Illumina MiSeq platform was used to sequence the bacterial 16S rRNA gene V4 regions and profile the microbiota between P. f. martensii intestine and the surrounding water. The raw data after quality check and chimera filtration produced a total of 1,879,032 high-quality sequencing reads from 30 samples, pertaining to four groups, with an average of 62,634 reads (Supplementary Table 2). The high-quality sequences with a sequence identity of 97% were grouped into a total of 2,222 OTUs with individual library containing diverse phylogenetic OTUs within a range of 628–1,042. The Good’s coverage of individual sample to approximate the wholeness of sequencing was 0.995 (from 0.994 to 0.996). This value shows that the identified sequences are representative of most bacteria identified in individual samples.
Overall Microbiota Structures
The 16S rRNA gene sequences presented the OTUs of all sample microbiota being classified into 38 prokaryotic phyla. A group annotated as “others” denoted sequences that did not fit into any category. As shown in Figure 1A, the relatively abundant phyla in all samples were Cyanobacteria (38.93%), Tenericutes (22.84%), Proteobacteria (18.41%), Bacteroidetes (8.97%), Planctomycetes (4.87%), Actinobacteria (3.34%), Verrucomicrobia (1.43%), and others (1.22%).
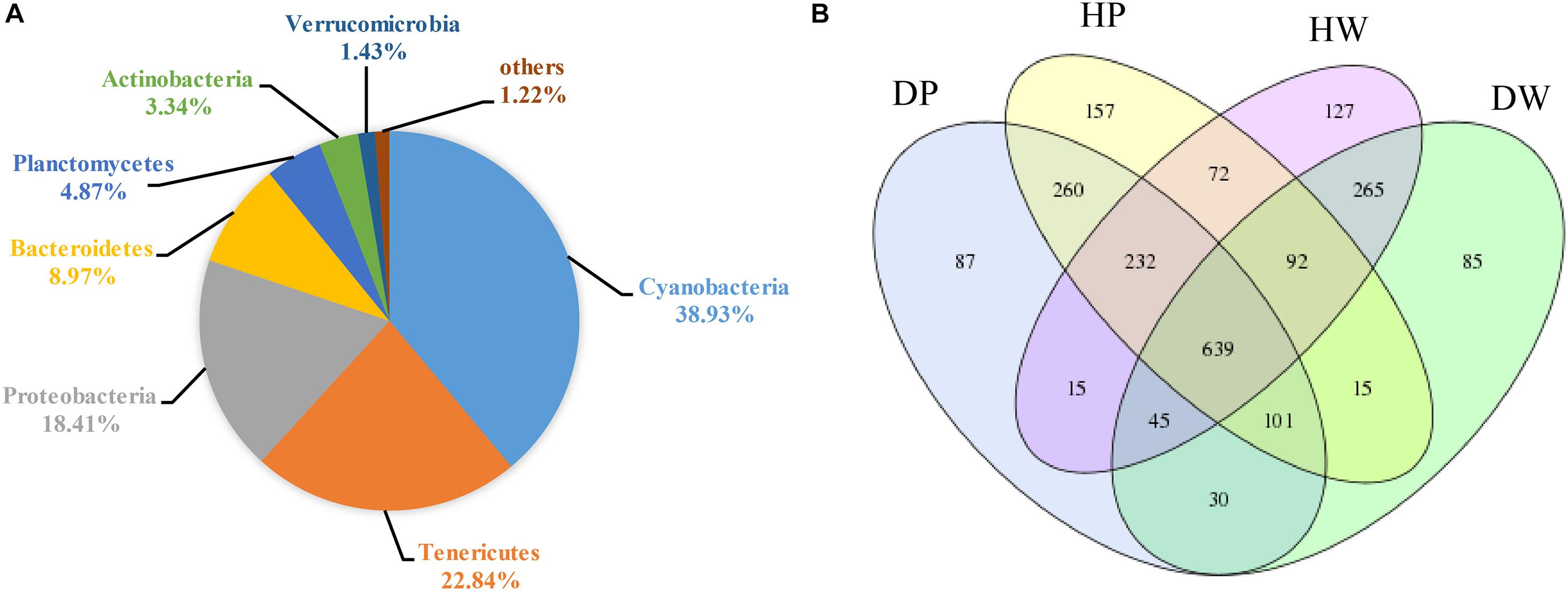
Figure 1. (A) Bacterial community in all samples at the phylum level. Less than 1% abundance of the phyla was merged into others. (B) Comparison of OTUs in DP, DW, HP, and HW by Venn diagram.
A Venn diagram was created to categorize the predominant OTUs in the four groups and further investigate the predominant microbiota between the intestines and the cultured environments in all samples (Figure 1B). A total of 639 OTUs were shared among DP, DW, HP, and HW, representing 28.76% of the total reads. A total of 1,232 and 1,041 OTUs were shared between DP and HP and between DW and HW, representing 55.45 and 46.85% of the total reads, respectively. A total of 815 and 1,035 OTUs were shared between DP and DW and between HP and HW, representing 36.68 and 46.58% of the total reads, respectively.
The first 10 abundant OTUs in the four groups (DP, DW, HP, and HW) are presented in Table 1. In group DP, the relative abundance of the first 10 OTUs accounted for 78.87%, which belonged to Tenericutes (47.88%), Cyanobacteria (28.98%), Planctomycetes (4.52%), and Proteobacteria (1.28%). In group DW, the relative abundance of the first 10 OTUs accounted for 44.67%, which belonged to Proteobacteria (19.72%), Cyanobacteria (19.50%), Bacteroidetes (2.26%), and Actinobacteria (3.19%). In group HP, the relative abundance of the first 10 OTUs accounted for 56.35%, which belonged to Cyanobacteria (33.65%), Tenericutes (18.08%), Proteobacteria (2.19%), and Planctomycetes (2.44%). In group HW, the relative abundance of the first 10 OTUs accounted for 52.53%, which belonged to Cyanobacteria (34.69%), Proteobacteria (6.90%), Actinobacteria (6.21%), and Bacteroidetes (4.71%). Seven OTUs (OTU1, OTU2, OTU6, OTU7, OTU5, OTU8, and OTU3) with relative abundances ranked among the first 10 OTUs in the DP and HP groups. Among the top 10 OTUs, only one (OTU2) was shared by the DP and DW groups, and two (OTU2 and OTU274) were shared by the HP and HW groups.
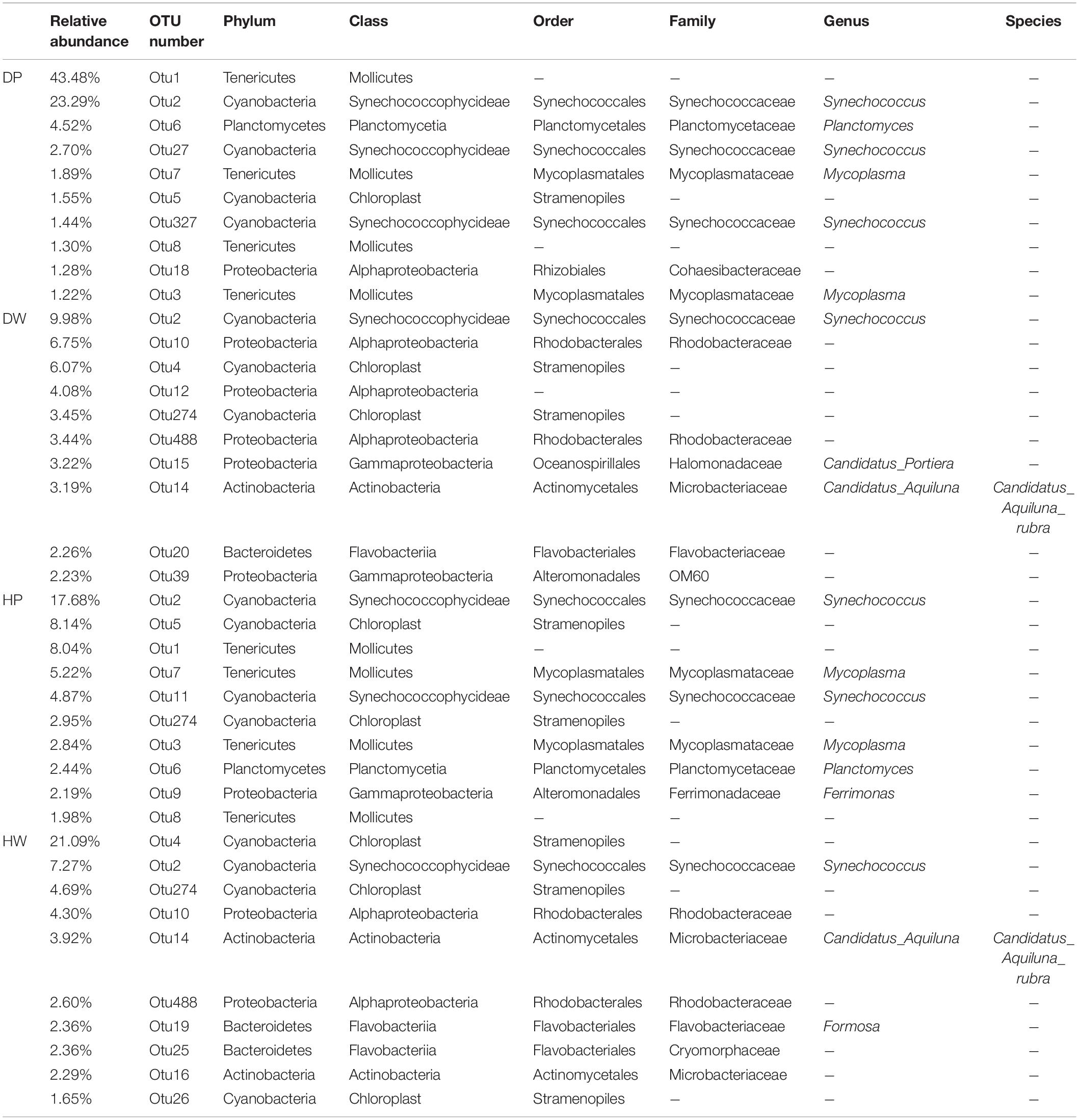
Table 1. Top 10 abundant OTUs in DP, DW, HP, and HW.
Bacterial Community Diversity
The richness and diversity indexes of the samples were calculated to demonstrate the complexity of individual sample (Supplementary Table 2). The diversity of samples was quantified using Shannon and Simpson indices. The Shannon index ranged from 2.3471 ± 0.5041 to 4.4266 ± 0.0605, whereas the Simpson index was from 0.0297 ± 0.0021 to 0.2935 ± 0.1175. The Sobs, Chao, and Ace indices were used to calculate the richness. The Sobs index ranged from 736.00 ± 93.47 to 952.40 ± 57.30, the Chao index ranged from 988.36 ± 108.37 to 1218.10 ± 38.73, and the Ace index ranged from 1012.05 ± 93.92 to 1250.08 ± 52.44. The richness and diversity of the bacterial species in the samples were arranged as follows: pearl oyster intestine < surrounding water, pearl oyster intestine in D < pearl oyster intestine in H, and surrounding water in D < surrounding water in H (Figure 2 and Supplementary Table 2).
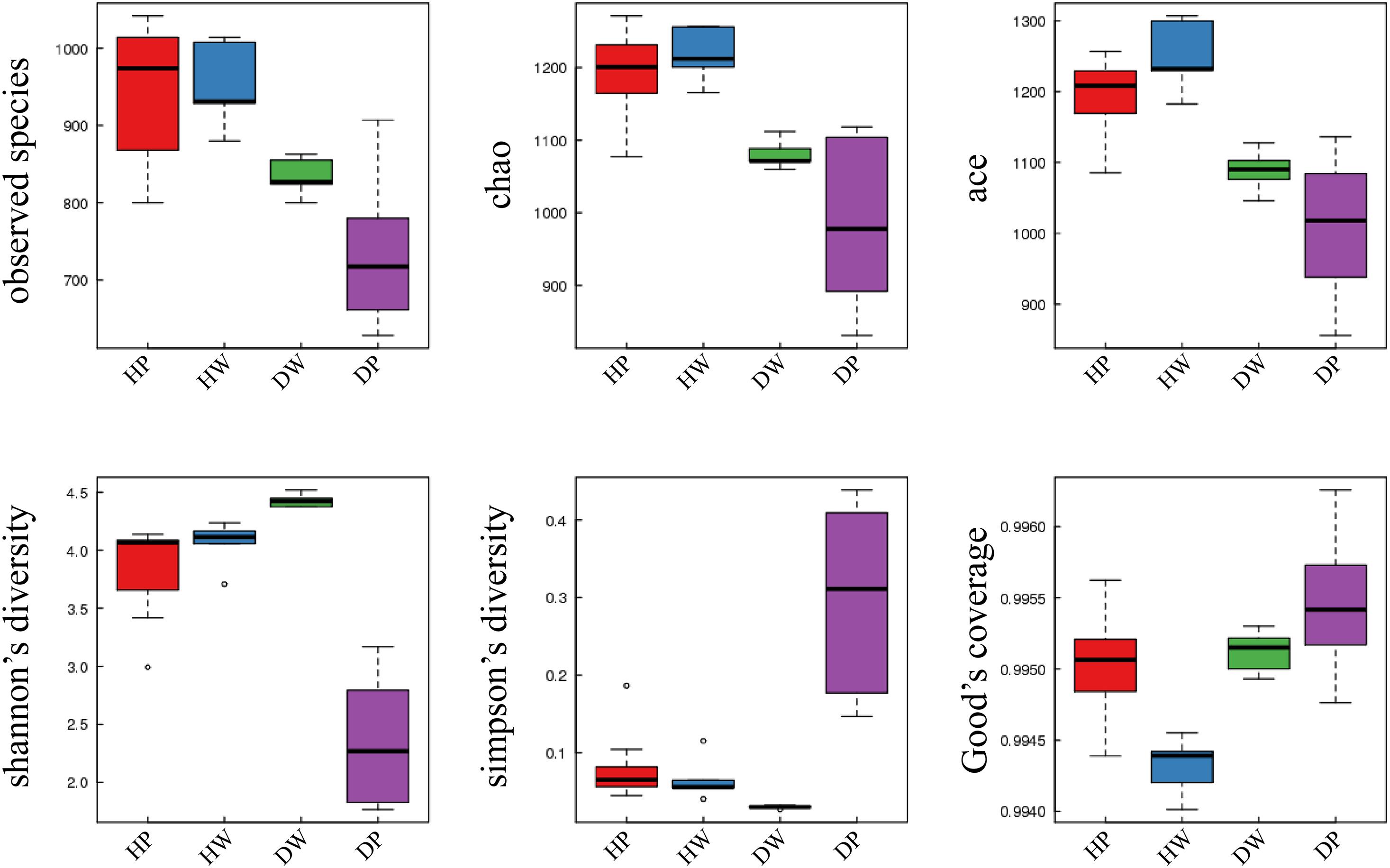
Figure 2. Richness and diversities of bacterial species in DP, DW, HP, and HW.
The difference and similarity in microbial communities of the samples were analyzed using Beta diversity. Figure 3A represents the hierarchical clustering tree of the samples, with those of the same group clustered together. PCoA was also used to analyze the similarity matrix of the samples with PC1 = 31.32% and PC2 = 17.18% of the total variations (Figure 3B). The samples within the same group were more closely clustered than those in the intergroup. Furthermore, two P. f. martensii intestine groups (DP vs. HP) tended to cluster closer than the P. f. martensii intestines and related surrounding water samples (DP vs. DW and HP vs. HW). Two surrounding water groups displayed a similar status.
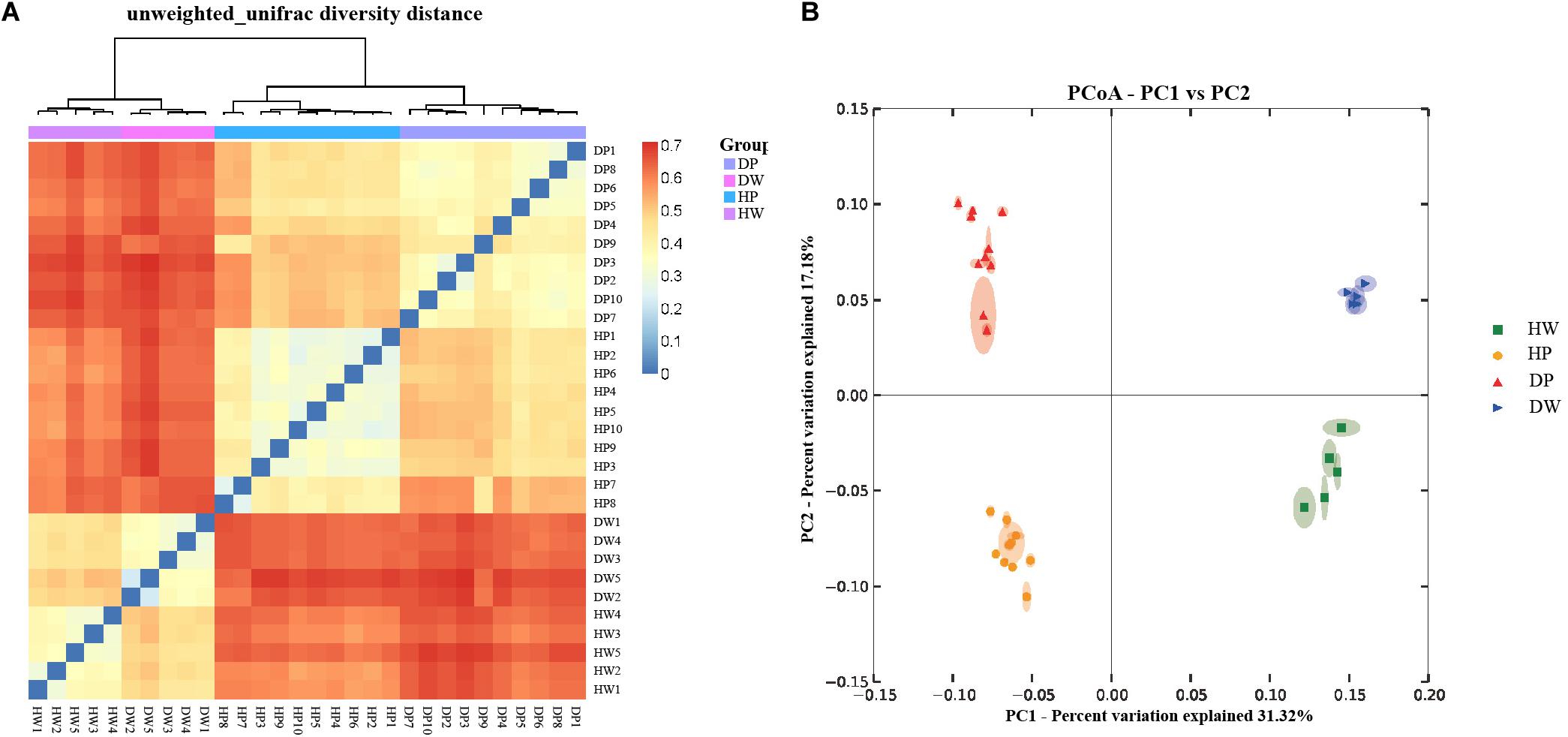
Figure 3. Hierarchical clustering tree (A) and principal coordinate analysis (PCoA) of the bacterial community (B) on the OTU level. The hierarchical clustering tree was calculated using the unweighted pair-group method with arithmetic mean (UPGMA) method, and the relationship between samples was determined using Bray distance and the average clustering method.
Among the four groups, the relative abundance of bacterial taxa was significantly different at the phylum and genus levels (Figures 4A,B for phylum and genus levels, respectively). In the 38 phyla identified, 33 from the four groups had p < 0.05, and the predominant phyla (with relative abundance of >5% in at least one sample) were Cyanobacteria, Proteobacteria, Bacteroidetes, Actinobacteria, Planctomycetes, and Tenericutes (Figure 4A and Supplementary Table 3). In the 272 genera identified, 208 had p < 0.05 in the four groups. The predominant genera (with relative abundance of > 1% in at least one sample) were Synechococcus, Mycoplasma, Planctomyces, Ferrimon, Persicirhabdus, Tenacibaculum, Marivita, Ruegeria, Formosa, Candidatus_Portiera, Nautella, Candidatus_Aquiluna, and Sediminicola (Figure 4B and Supplementary Table 4). All these genera were statistically significant among the four groups. These results proposed that the microbial composition was significantly different among the pearl oyster P. f. martensii intestine and the surrounding water-cultured environment per the relative abundance of sequences.
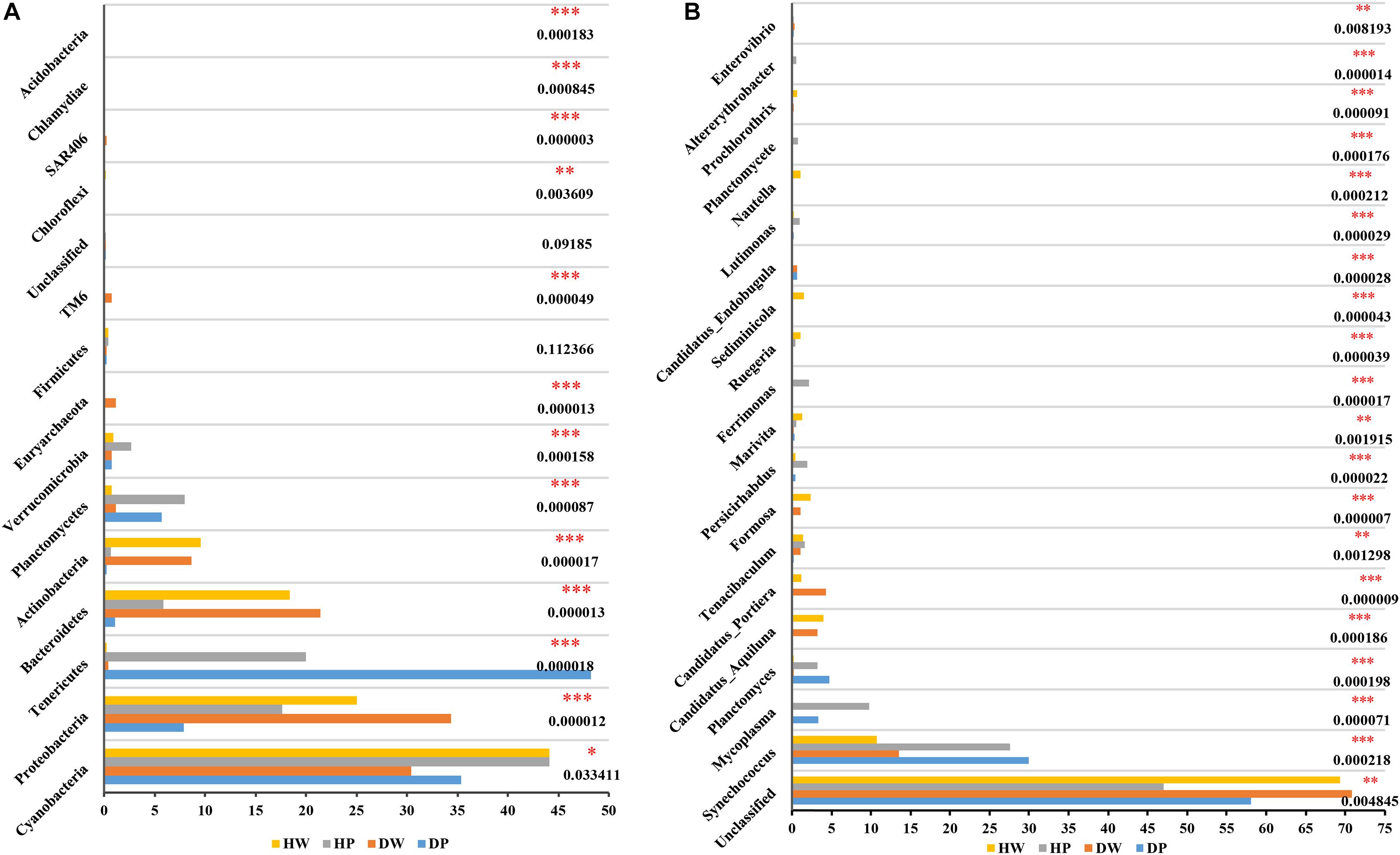
Figure 4. Comparison of bacterial abundances among HW, HP, DW, and DP at the phylum level (A) and genus level (B). ∗0.01 < p ≤ 0.05, ∗∗0.001 < p ≤ 0.01, and ∗∗∗p ≤ 0.001.
The relative abundances of bacterial taxa showed a statistical significance between two groups (DW vs. DP, HP vs. DP, HW vs. DW, and HW vs. HP) at the genus and phylum levels (Figures 5, 6 indicate the first 15 and 20 abundant phyla and genera, respectively). At the phylum level, the statistically significant bacterial taxa between DW and DP were Tenericutes, Proteobacteria, Bacteroidetes, Actinobacteria, Planctomycetes, Euryarchaeota, TM6, SAR406, Chlamydiae, Chloroflexi, and Acidobacteria. The bacterial taxa between HP and DP were Tenericutes, Proteobacteria, Bacteroidetes, Verrucomicrobia, Actinobacteria, Firmicutes, Acidobacteria, NKB19, Crenarchaeota, and Gemmatimonadetes. The bacterial taxa between HW and DW were Cyanobacteria, Proteobacteria, Planctomycetes, Euryarchaeota, TM6, Tenericutes, SAR406, Chloroflexi, GN02, and Chlamydiae. The bacterial taxa between HW and HP were Proteobacteria, Bacteroidetes, Tenericutes, Actinobacteria, Planctomycetes, Verrucomicrobia, Chloroflexi, Acidobacteria, NKB19, GN02, Chlamydiae, and Euryarchaeota.
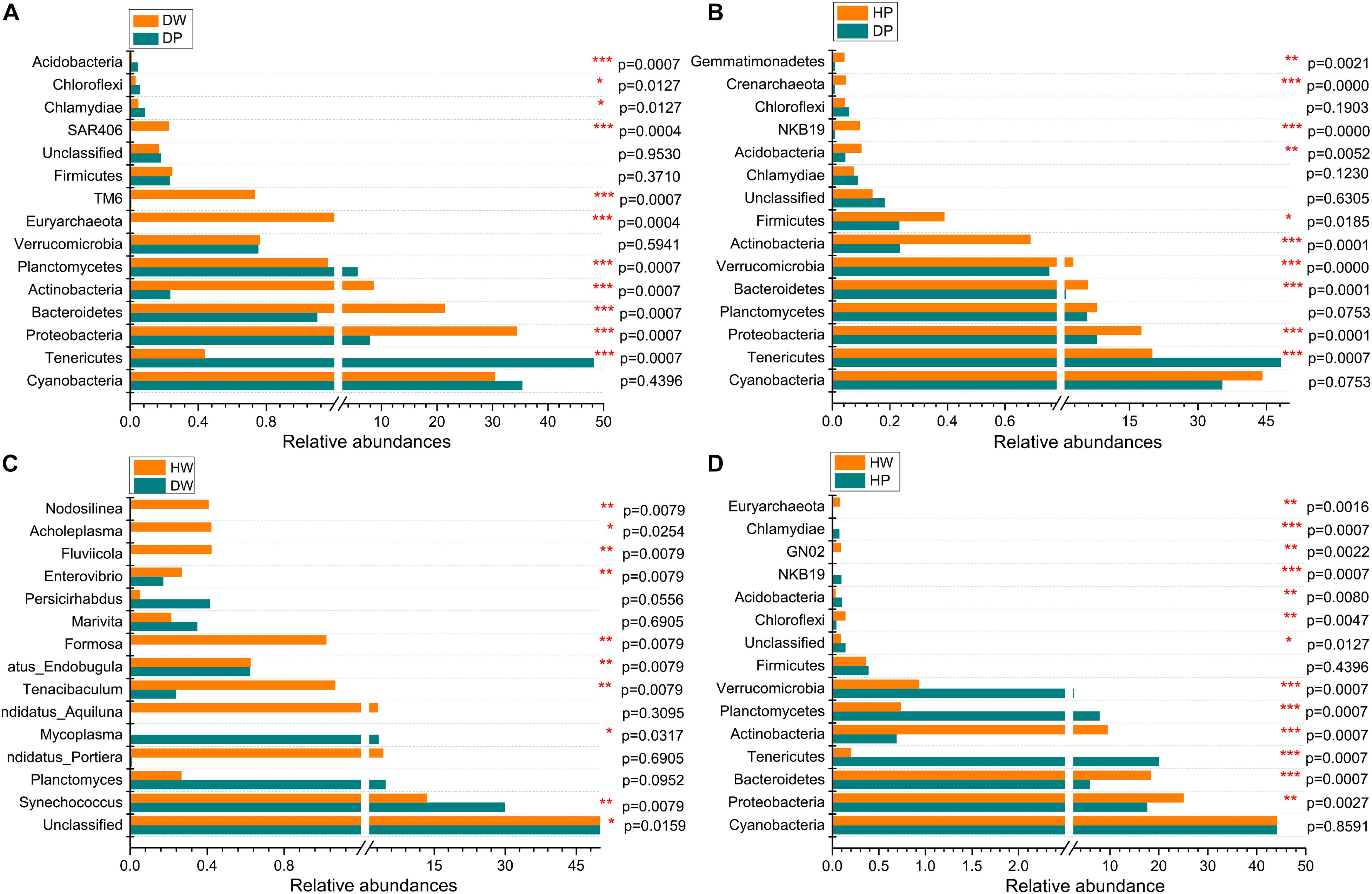
Figure 5. Comparison of microbial community between DW and DP (A), HP and DP (B), HW and DW (C), and HW and HP (D) at the phylum level. *0.01 < p ≤ 0.05, **0.001 < p ≤ 0.01, and ***p ≤ 0.001.
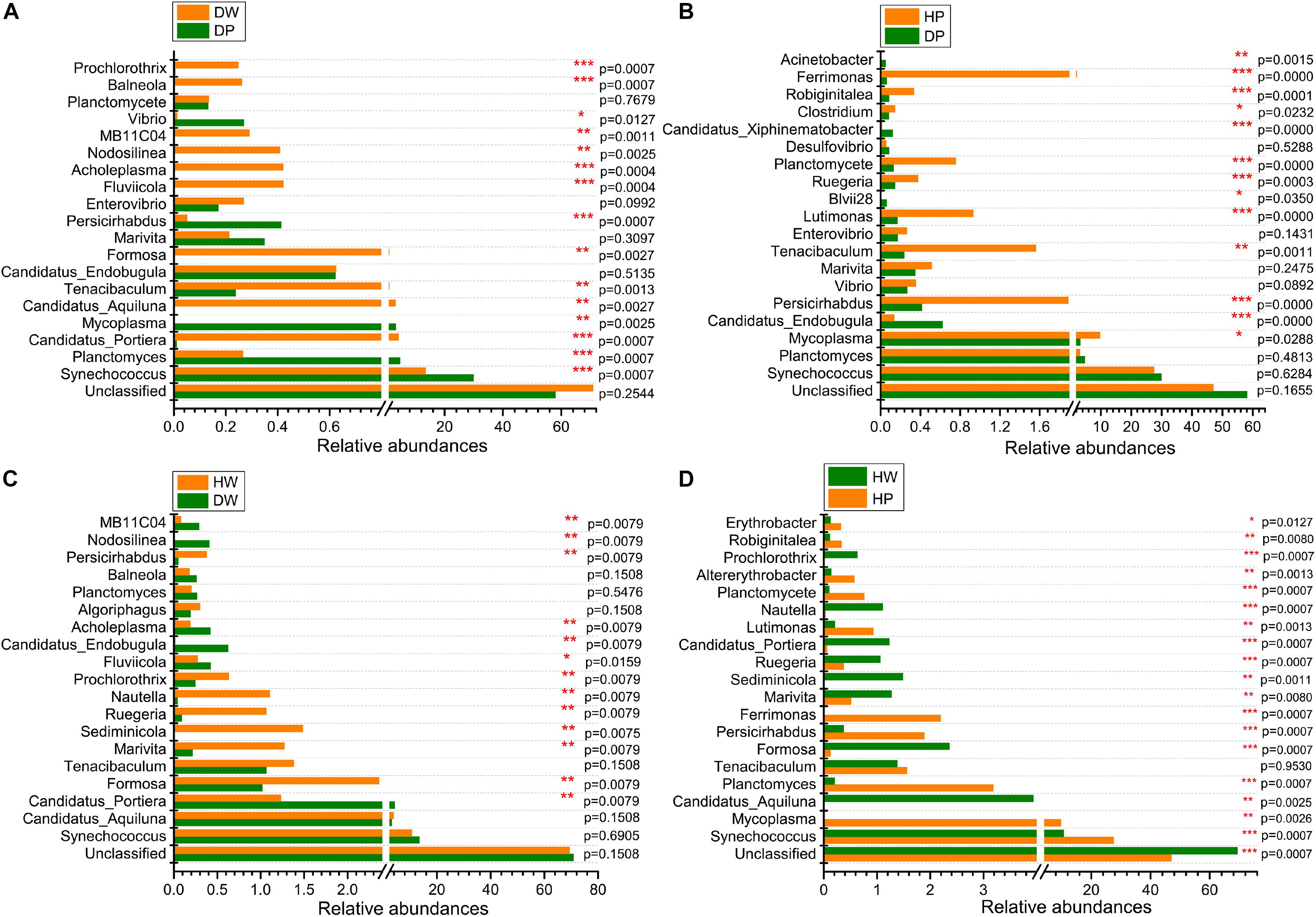
Figure 6. Comparison of microbial community between DW and DP (A), HP and DP (B), HW and DW (C), and HW and HP (D) at the genus level. *0.01 < p ≤ 0.05, **0.001 < p ≤ 0.01, and ***p ≤ 0.001.
At the genus level, the statistically significant microbiota between DW and DP were Synechococcus, Planctomyces, Candidatus_Portiera, Mycoplasma, Formosa, Candidatus_Aquiluna, Tenacibaculum, Persicirhabdus, Fluviicola, Acholeplasma, Nodosilinea, MB11C04, Vibrio, Balneola, and Prochlorothrix. The microbiota between HP and DP were Mycoplasma, Candidatus_Endobugula, Persicirhabdus, Tenacibaculum, Lutimonas, Blvii28, Ruegeria, Planctomycete, Clostridium, Candidatus_Xiphinematobacter, Robiginitalea, Ferrimonas, and Acinetobacter. The microbiota between HW and DW were Candidatus_Portiera, Formosa, Marivita, Sediminicola, Ruegeria, Nautella, Prochlorothrix, Fluviicola, Candidatus_Endobugula, Acholeplasma, Persicirhabdus, Nodosilinea, and MB11C04. The microbiota between HW and HP were Synechococcus, Mycoplasma, Candidatus_Aquiluna, Planctomyces, Formosa, Persicirhabdus, Ferrimonas, Marivita, Sediminicola, Ruegeria, Candidatus_Portiera, Lutimonas, Planctomycete, Altererythrobacter, Robiginitalea, Erythrobacter, Nautella, and Prochlorothrix. These observations suggested that the relative abundance of the predominant genera varied between two groups. Taken together, the abundance and composition of the dominant genera specifically differed among the four groups.
Functional Prediction of Microbiota
The presumptive roles of the microbiota of pearl oyster intestine and the surrounding water-cultured environment were demonstrated on PICRUSt 2. A substantial similarity was observed between the functional profiles of all groups compared with their taxonomic profiles (Figure 7). The functions of these groups were predominantly related to the metabolism of cofactors and vitamin (relative abundance of 10.19–15.62%), carbohydrates metabolism (relative abundance of 11.73–12.73%), amino acids metabolism (relative abundance of 10.40–13.61%), metabolism of terpenoids and polyketides (relative abundance of 8.70–12.13%), metabolism of other amino acids (relative abundance from 8.20 to 9.00%), lipids metabolism (relative abundance of 5.41–8.06%), energy metabolism (relative abundance of 5.83–7.38%), replication and repair (relative abundance of 4.76–7.55%), xenobiotic biodegradation and metabolism (relative abundance of 4.26–6.30%), and folding, sorting, and degradation (relative abundance of 3.27–4.01%). The results from the COG function suggested a maintenance and similarity in biological functions of pearl oyster intestinal microbial taxa to that observed in the groups at different locations or the surrounding water-cultured environment.
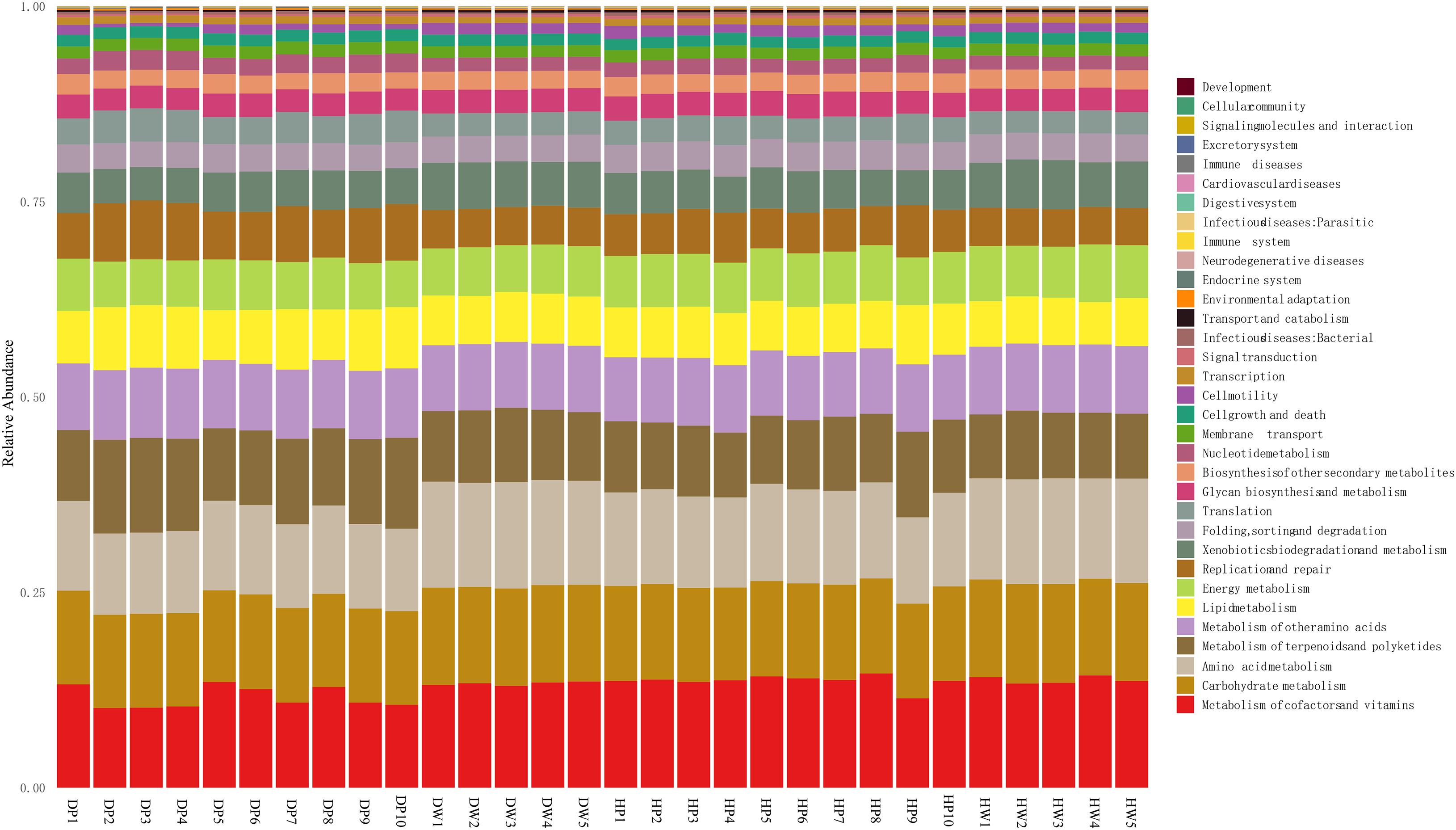
Figure 7. COG function classification of DP, DW, HP, and HW.
Discussion
High-throughput sequencing technology delivers a good visual of microbiota related to the cultivation of pearl oyster (Dubé et al., 2019; Liao et al., 2020). The current study explored the relationship between the intestinal microbiota of pearl oyster and its association with the microbial ecosystem of the surrounding seawater. The findings statistically showed that the varied microbiota compositions exhibited a significance at the phylum and genus levels in the pearl oyster P. f. martensii intestines and its association with the surrounding water. Other works also established that many microbial communities of marine invertebrates, such as crustaceans (Harris et al., 1991), sponges (Fan et al., 2012), and corals (Ravindran et al., 2013), differ from those of the environment. Meanwhile, by analyzing the diversity index of pearl oyster intestines and the surrounding water-cultured environments in different sea areas, the richness and diversity of the intestinal flora of P. f. martensii was found to be less than those of the surrounding water. This finding agreed with the result from other aquatic animals and bacterial flora, such as Sinonovacula constricta (Wang et al., 2019), Scylla paramamosain (Wang et al., 2017), and Litopenaeus vannamei (Sun et al., 2016).
Some studies have demonstrated that the gut bacterial communities of matured bivalves often comprise resident bacteria/specific indigenous/core microbiome with permanent types of organisms that persist over time, irrespective of the ingested particles by the host, that is, the bacteria spontaneously recover despite disturbance (King et al., 2012; Trabal et al., 2012; Lokmer and Wegner, 2015; Dubé et al., 2019). For example, King et al. (2012) speculated the existence of core gut microbiome in eastern oyster (Crassostrea virginica), which contained 44 OTUs in 12 phyla. Pierce et al. (2016) also suggested the existence of a core microbiome in eastern oyster C. virginica. Although the bacteria in the surrounding seawater were not analyzed, Trabal et al. (2012) proposed a relative stability of oyster microbiota from hatchery to open sea in determining the relationship among microbiota of both compartments. Cleary et al. (2015) also reported the similarity of mussel microbial communities in Indonesian marine lakes and open coastal mangroves, whereas their environments are considerably different. In terms of pearl oyster, Dubé et al. (2019) reported that Bacteroidetes, Proteobacteria, and Spirochaetes were the core members of the microbiome of the black-lipped pearl oyster P. margaritifera despite their tissue affiliation. In the present study, 1232 OTUs were shared by DP and HP, representing 55.45% of the total reads. Seven OTUs with relative abundances ranked among the first 10 OTUs in the DP and HP groups, which indicated that the composition of the predominant bacteria in the intestine of pearl oyster P. f. martensii is similar despite differences in cultured environments. This result proposed that these OTUs may be a part of the core gut microbiome. These bacterial phyla are commonly associated with bivalve guts, such as in pearl oysters (King et al., 2012; Lokmer et al., 2016; Dubé et al., 2019; Liao et al., 2020). For instance, phyla Planctomycetes and Tenericutes are dominant microbials of P. margaritifera gut (Dubé et al., 2019). Mollicutes (Mycoplasma) have high abundance in the gut, whereas mantle and gill microbiota demonstrates a slight abundance (Lokmer et al., 2016).
Another category of regular bacterial microbiota could be defined in bivalves as suspension feeders: transient or non-indigenous microbiota, which consist of microbes ingested with food that survived en-route through the gut (possibly proliferating in the gut); that is, many members of the gut microbiomes appear to be transients or opportunists (King et al., 2012). Previous scientific evidence also indicated that bacterial communities in oyster intestines may exhibit some plasticity to survive within a host in different environments (Pierce et al., 2016). In the current study, the varied trends of bacterial species richness and diversity in pearl oyster intestine were consistent with those of the surrounding water-cultured environment (DP < HP, DW < HW). Laroche et al. (2018) also observed that the diversity of bacterial communities in Pacific oyster larval changes is caused by seasonal and temporal changes in the arriving seawater. A total of 266 OTUs in the intestine of DP were observed but remained undetected in HP, and 477 OTUs in the intestine of HP did not appear in DP. Meanwhile, some of the shared OTUs between HP and DP exist statistically significant on the phylum and genus level. These differences may be due to the changes in the external environment. Liao et al. (2020) reported that protein sources in formulated diets change the diversity of intestinal microflora in pearl oyster P. f. martensii. The diverse or variable microbiome could be linked to oyster clearance rates (Pierce et al., 2016). On basis of these results, the present study proposed that pearl oyster could regulate the intestinal bacterial communities through certain mechanisms to adapt to changes in the external environment; that is, bacterial communities in pearl oyster intestine may exhibit some plasticity.
These altered bacteria possess functions to respond to a changing environment. Proteobacteria, a gram-negative bacteria, are one of the phyla profusely found in sediments, wastewater treatment reactor, and activated sludge, with a key function in degrading carbon complex and eradicating nitrogen (Shu et al., 2015). Proteobacteria participate in diverse biogeochemical processes (such as sulfur, nitrogen, and carbon cycling) in aquatic ecosystems (Klase et al., 2019). Actinobacteria, a gram-positive bacteria, are found in aquatic and terrestrial environments (Servin et al., 2008) and believed to degrade glucose dominantly (Ito et al., 2012). In addition, Actinobacteria are well-known producers of bioactive natural product utilized in isolating potential probiotics (Bernal et al., 2015). They are essential in maintaining gut homeostasis (Binda et al., 2018). The gram-negative (Proteobacteria) and gram-positive (Actinobacteria) bacteria from the intestine of P. f. martensii were stored in balance to preserve the organic homeostasis compared with the sediment data. In the gut microbiota of humans, the ratio of Bacteroidetes to Firmicutes is substantially important. In the microbiota of obese subjects, the Firmicutes-to-Bacteroidetes ratio exhibited an increasing trend (Zhou et al., 2018), and this ratio evolved at various life stages (Mariat et al., 2009). An increasing number of Firmicutes could cause an increase in the quantity of lipid droplets, hence intensifying the proportionate absorption of fatty acid (Semova et al., 2012). The present findings indicated that the Firmicutes-to-Bacteroidetes ratio of DP was 0.23:1.10 compared with that of HP at 0.39:5.90. This result may indicate that D-cultured pearl oysters were fatter than the H-cultured ones. The phylum Chloroflexi, an aerobic facultative bacterium with a photosynthetic ability under anaerobic conditions, is profuse in freshwater, intertidal, marine, and surface and subsurface sediments (Lv et al., 2018). Chloroflexi existed in the intestine of P. f. martensii and the surrounding water in the present study. The predominant genus Synechococcus (Cyanobacteria) at the genus level is abundant in ocean regions (Flombaum et al., 2013). Thus, the present study identified that Synechococcus was substantially abundant in the pearl oyster intestine and the surrounding water-cultured environment. A previous study stated the possibility of a compromised health and susceptibility to diseases in aquatic animals due to the superfluity of Vibrio (Fan et al., 2019). In the present study, Vibrio obtained a low abundance (mean 0.17%) in the pearl oyster intestine and the surrounding water, and its abundance in D (pearl oyster intestine and surrounding water) was higher than that in H. This finding may be attributed to the location of H, which is in the open sea area outside the Liusha Bay, wherein the water exchange is good, and the cage culture in the adjacent waters was fewer than that in H. Therefore, the changes in the pearl oyster intestine bacteria were recorded to adapt to changes in the surrounding water-cultured environment.
Conclusion
This work ascertained the relationship between the intestinal microbes of pearl oyster P. f. martensii and the surrounding water-cultured environment. Even though the existing intestinal microbial community of pearl oysters from the two sites differed, some similar characteristics were observed in the intestine samples. These characteristics varied among water samples. Seven OTUs, which belonged to the phyla Tenericutes, Cyanobacteria, and Planctomycetes, may be the core microbiome of pearl oyster P. f. martensii. The bacterial communities in pearl oyster intestines may exhibit some plasticity to adapt to changes in the surrounding water-cultured environment. With the limited research available and the importance of intestinal microbial communities and those in cultured environment, this study presented pivotal background information to prevent and control diseases caused by aquatic microorganisms.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA639226, SRR12008329–SRR12008353, SRR12165640–SRR12165644, and SAMN15065811–SAMN15065815).
Ethics Statement
The pearl oyster Pinctada fucata martensii is a lower invertebrate, and therefore, the study was not subject to ethical approval.
Author Contributions
QW, ZZ, YD, and XD designed the research. ZZ, CY, YL, and YD conducted the research. ZZ, YL, and CY analyzed the data. ZZ, YL, LA, CY, JY, QW, XD, and YD contributed to the final writing of the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by the Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams, Department of Agriculture and Rurual Affairs of Guangdong Province (Grant no. 2020KJ146), Young Innovative Talents Project (Grant no. 2019KQNCX043), Innovation Team Project (Grant no. 2017KCXTD016) from the Department of Education of Guangdong Province, Guangdong Basic and Applied Basic Research Foundation (Grant nos. 2019A1515111026 and 2020A1515010691), the China Agriculture Research System (Grant no. CARS-049), and Students Innovation and Entrepreneurship Training Program (Grant no. CXXL2020045).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.655698/full#supplementary-material
Supplementary Figure 1 | Study area map showing the position of study locations sampled in the Liusha Bay in Guangdong, China.
Footnotes
References
Asmani, K., Petton, B., Grand, J. L., Mounier, J., Robert, R., and Nicolas, J. L. (2016). Establishment of microbiota in larval culture of Pacific oyster, Crassostrea gigas. Aquaculture 464, 434–444. doi: 10.1016/j.aquaculture.2016.07.020
Belkaid, Y., and Hand, T. W. (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. doi: 10.1016/j.cell.2014.03.011
Bernal, M. G., Campa-Córdova, ÁI., Saucedo, P. E., González, M. C., Marrero, R. M., and Mazón-Suástegui, J. M. (2015). Isolation and in vitro selection of actinomycetes strains as potential probiotics for aquaculture. Vet. World 8, 170–176. doi: 10.14202/vetworld.2015.170-176
Binda, C., Lopetuso, L. R., Rizzatti, G., Gibiino, G., Cennamo, V., and Gasbarrini, A. (2018). Actinobacteria: a relevant minority for the maintenance of gut homeostasis. Dig. Liver Dis. 50, 421–428. doi: 10.1016/j.dld.2018.02.012
Blancheton, J. P., Attramadal, K. J. K., Michaud, L., Roqued’Orbcastel, E., and Vadstein, O. (2013). Insight into bacterial population in aquaculture systems and its implication. Aquacult. Eng. 53, 30–39. doi: 10.1016/j.aquaeng.2012.11.009
Brooks, A. W., Kohl, K. D., Brucker, R. M., van Opstal, E. J., and Bordenstein, S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14:e2000225. doi: 10.1371/journal.pbio.2000225
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Carbone, D., and Faggio, C. (2016). Importance of prebiotics in aquaculture as immunostimulants. Effects on immune system of Sparus aurata and Dicentrarchus labrax. Fish Shellfish Immunol. 54, 172–178. doi: 10.1016/j.fsi.2016.04.011
Cárdenas, C. A., Bell, J. J., Davy, S. K., Hoggard, M., and Taylor, M. W. (2014). Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiol. Ecol. 88, 516–527. doi: 10.1111/1574-6941.12317
Carrier, T. J., and Reitzel, A. M. (2018). Convergent shifts in host-associated microbial communities across environmentally elicited phenotypes. Nat. Commun. 9:952.
Cleary, D. F. R., Becking, L. E., Polonia, A. R. M., Freitas, R. M., and Gomes, N. C. M. (2015). Composition and predicted functional ecology of mussel-associated bacteria in Indonesian marine lakes. Antonie Van Leeuwenhoek 107, 821–834. doi: 10.1007/s10482-014-0375-1
Cuif, J. P., Perez-Huerta, A., Lo, C., Oulfa Belhadj, O., and Dauphin, Y. (2018). On the deep origin of the depressed rings on pearl surface illustrated from Polynesian Pinctada margaritifera (Linnaeus 1758). Aquac. Res. 49, 1834–1847. doi: 10.1111/are.13638
Du, X. D., Fan, G. Y., Jiao, Y., Zhang, H., Guo, X. M., Huang, R. L., et al. (2017). The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. Gigascience 6, 1–12. doi: 10.1007/978-981-13-1459-9_1
Dubé, C. E., Ky, C. L., and Planes, S. (2019). Microbiome of the black-lipped pearl oyster Pinctada margaritifera, a multi-tissue description with functional profiling. Front. Microbiol. 10:1548. doi: 10.3389/fmicb.2019.01548
Fan, L., Liu, M., Simister, R., Webster, N. S., and Thomas, T. (2013). Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 7, 991–1002. doi: 10.1038/ismej.2012.165
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, 1878–1887.
Fan, L. F., and Li, Q. X. (2019). Characteristics of intestinal microbiota in the Pacific white shrimp Litopenaeus vannamei differing growth performances in the marine cultured environment. Aquaculture 505, 450–461. doi: 10.1016/j.aquaculture.2019.02.075
Fan, L. F., Wang, Z. L., Chen, M. S., Qu, Y. X., Li, J. Y., Zhou, A. G., et al. (2019). Microbiota comparison of Pacific white shrimp intestine and sediment at freshwater and marine cultured environment. Sci. Total Environ. 657, 1194–1204. doi: 10.1016/j.scitotenv.2018.12.069
Fernández, N. T., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Ascencio-Valle, F., and Romero, J. (2014). Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. Fems. Microbiol. Eco. 88, 69–83. doi: 10.1111/1574-6941.12270
Flombaum, P., Gallegos, J. L., Gordillo, R. A., Rincón, J., Zabala, L. L., Jiao, N., et al. (2013). Present and future global distributions of the marine cyanobacteria prochlorococcus and synechococcus. Proc. Natl. Acad. Sci. U.S.A. 110, 9824–9829. doi: 10.1073/pnas.1307701110
Gao, S., Pan, L. Q., Huang, F., Song, M. S., Tian, C. C., and Zhang, M. Y. (2019). Metagenomic insights into the structure and function of intestinal microbiota of the farmed Pacific white shrimp (Litopenaeus vannamei). Aquaculture 499, 109–118. doi: 10.1016/j.aquaculture.2018.09.026
Hao, R. J., Du, X. D., Yang, C. Y., Deng, Y. W., Zheng, Z., and Wang, Q. H. (2019). Integrated application of transcriptomics and metabolomics provides insights into unsynchronized growth in pearl oyster Pinctada fucata martensii. Sci. Total Environ. 666, 46–56. doi: 10.1016/j.scitotenv.2019.02.221
Harris, J. M., Seiderer, L. J., and Lucas, M. I. (1991). Gut microflora of two saltmarsh detritivore thalassinid prawns, Upogebia africana and Callianassa kraussi. Microb. Ecol. 21, 277–296. doi: 10.1007/bf02539159
He, C. Z., Hao, R. J., Deng, Y. W., Yang, C. Y., and Du, X. D. (2020). Response of pearl oyster Pinctada fucata martensii to allograft-induced stress from lipid metabolism. Fish Shellfish Immunol. 98, 1001–1007. doi: 10.1016/j.fsi.2019.11.028
Ito, T., Yoshiguchi, K., Ariesyady, H. D., and Okabe, S. (2012). Identification and quantification of key microbial trophic groups of methanogenic glucose degradation in an anaerobic digester sludge. Bioresour. Technol. 123, 599–607. doi: 10.1016/j.biortech.2012.07.108
King, G. M., Judd, C., Kuske, C. R., and Smith, C. (2012). Analysis of stomach and gut microbiomes of the eastern oyster (Crassostrea virginica) from coastal louisiana, USA. PLoS One 7:e51475. doi: 10.1371/journal.pone.0051475
Klase, G., Lee, S. J., Liang, S., Kim, J. N., Zo, Y. G., and Lee, J. Y. (2019). The microbiome and antibiotic resistance in integrated fishfarm water: implications of environmental public health. Sci. Total Environ. 649, 1491–1501. doi: 10.1016/j.scitotenv.2018.08.288
Laroche, O., Symonds, J. E., Smith, K. F., Banks, J. C., Mae, H., Bowman, J. P., et al. (2018). Understanding bacterial communities for informed biosecurity and improved larval survival in Pacific oysters. Aquaculture 497, 164–173. doi: 10.1016/j.aquaculture.2018.07.052
Li, T. T., Li, H., Gatesoupe, F. J., She, R., Lin, Q., Yan, X. F., et al. (2017). Bacterial signatures of “red-operculum” disease in the gut of crucian carp (Carassius auratus). Microb. Ecol. 74, 510–521. doi: 10.1007/s00248-017-0967-1
Liao, Y. S., Cai, C. X., Yang, C. Y., Zheng, Z., Wang, Q. H., Du, X. D., et al. (2020). Effect of protein sources in formulated diets on the growth, immune response, and intestinal microflora of pearl oyster Pinctada fucata martensii. Aquacult. Rep. 16:100253. doi: 10.1016/j.aqrep.2019.100253
Lokmer, A., Kuenzel, S., Baines, J. F., and Wegner, K. M. (2016). The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ. Microbiol. 18, 970–987. doi: 10.1111/1462-2920.13163
Lokmer, A., and Wegner, K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9, 670–682. doi: 10.1038/ismej.2014.160
Lv, X. F., Yu, P., Mao, W. T., and Li, Y. C. (2018). Vertical variations in bacterial community composition and environmental factors in the culture pond sediment of sea cucumber Apostichopus japonicus. J. Coastal Res. 84, 69–76. doi: 10.2112/si84-010.1
Mariat, D., Firmesse, O., Levenez, F., Guimarăes, V. D., Sokol, H., Doré, J., et al. (2009). The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 9:123. doi: 10.1186/1471-2180-9-123
Musella, M., Wathsala, R., Tavella, T., Rampelli, S., Barone, M., Palladino, G., et al. (2020). Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 717:137209. doi: 10.1016/j.scitotenv.2020.137209
O’Brien, P. A., Webster, N. S., Miller, D. J., and Bourne, D. G. (2019). Host-microbe coevolution: applying evidence from model systems to complex marine invertebrate holobionts. mBio 10:e2241–18.
Pierce, M. L., Ward, J. E., Holohan, B. A., Zhao, X. W., and Hicks, R. E. (2016). The influence of site and season on the gut and pallial fluid microbial communities of the eastern oyster, Crassostrea virginica (Bivalvia, Ostreidae): community-level physiological profiling and genetic structure. Hydrobiologia 765, 97–113. doi: 10.1007/s10750-015-2405-z
Rausch, P., Rühlemann, M., Hermes, B. M., Doms, S., Dagan, T., Dierking, K., et al. (2019). Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 7:133.
Ravindran, J., Kannapiran, E., Manikandan, B., Francis, K., Arora, S., Karunya, E., et al. (2013). UV-absorbing bacteria in coral mucus and their response to simulated temperature elevations. Coral Reefs 32, 1043–1050. doi: 10.1007/s00338-013-1053-x
Semova, I., Carten, J. D., Stombaugh, J., Mackey, L. C., Knight, R., Farber, S. A., et al. (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the Zebrafish. Cell Host Microbe 12, 277–288. doi: 10.1016/j.chom.2012.08.003
Servin, J. A., Herbold, C. W., Skophammer, R. G., and Lake, J. A. (2008). Evidence excluding the root of the tree of life from the actinobacteria. Mol. Biol. Evol. 25, 1–4. doi: 10.1093/molbev/msm249
Shi, Y., Zhong, L., Ma, X. K., Liu, Y. L., Tang, T., and Hu, Y. (2019). Effect of replacing fishmeal with stickwater hydrolysate on the growth, serum biochemical indexes, immune indexes, intestinal histology and microbiota of rice field eel (Monopterus albus). Aquacult. Rep. 15:100223. doi: 10.1016/j.aqrep.2019.100223
Shu, D. T., He, Y. L., Yue, H., and Wang, Q. Y. (2015). Microbial structures and community functions of anaerobic sludge in six full-scale wastewater treatment plants as revealed by 454 high-throughput pyrosequencing. Bioresour. Technol. 186, 163–172. doi: 10.1016/j.biortech.2015.03.072
Simon, J. C., Marchesi, J. R., Mougel, C., and Selosse, M. A. (2019). Host-microbiota interactions: from holobiont theory to analysis. Microbiome 7:5.
Sun, F. L., Wang, Y. S., Wang, C. Z., Zhang, L., Tu, K., and Zheng, Z. P. (2019). Insights into the intestinal microbiota of several aquatic organisms and association with the surrounding environment. Aquaculture 507, 196–202.
Sun, Z. L., Xuan, Y. M., Zhang, H., Jiang, M., Pan, Y. S., Zhang, Y. M., et al. (2016). Bacterial diversity in the Penaeus vannamei Boone intestine and aquaculture environment. J. Fish. Sci. China 23, 594–605. (in Chinese with English abstract)
Trabal, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Asencio-Valle, F., Morales-Bojórquez, E., and Romero, J. (2012). Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microb. Ecol. 64, 555–569.
Wang, X. F., Zhao, Y. F., Song, Z. F., Zhong, S. P., Huang, G. Q., Tong, T., et al. (2017). Application of high-throughput sequencing techniques for analyzing bacterial communities in pond-raised mud crab (Scylla paramamosain) intestine and its aquaculture environment. J. Fish. Sci. China 24, 1245–1253. (in Chinese with English abstract) doi: 10.3724/sp.j.1118.2017.16386
Wang, X. Y., Xie, X., Jin, S., Zhu, J. Q., Zhao, Q. S., and Zhou, S. M. (2019). Seasonal variation of microflora in Sinonovacula constricta and its aquacultural pond based on high-throughput sequencing. Chin. J. Appl. Ecol. 30, 4267–4276. (in Chinese with English abstract)
Xiong, J. B., Dai, W. F., and Li, C. H. (2016). Advances, challenges, and directions in shrimp disease control: the guidelines from an ecological perspective. Appl. Microbiol. Biot. 100, 6947–6954. doi: 10.1007/s00253-016-7679-1
Yang, C. Y., Du, X. D., Hao, R. J., Wang, Q. H., Deng, Y. W., and Sun, R. J. (2019a). Effect of vitamin D3 on immunity and antioxidant capacity of pearl oyster Pinctada fucata martensii after transplantation: insights from LC–MS-based metabolomics analysis. Fish Shellfish Immunol. 94, 271–279.
Yang, C. Y., Hao, R. J., Du, X. D., Wang, Q. H., Deng, Y. W., and Sun, R. J. (2019b). Response to different dietary carbohydrate and protein levels of pearl oysters (Pinctada fucata martensii) as revealed by GC–TOF/MS based metabolomics. Sci. Total Environ. 650, 2614–2623. doi: 10.1016/j.scitotenv.2018.10.023
Zhang, J. B., Xiong, X. W., Deng, Y. W., Zheng, Z., Yang, C. Y., and Du, X. D. (2021). Integrated application of transcriptomics and metabolomics provides insights into the larval metamorphosis of pearl oyster (Pinctada fucata martensii). Aquaculture 532:736067. doi: 10.1016/j.aquaculture.2020.736067
Zhang, M. L., Sun, Y. H., Chen, L. Q., Cai, C. F., Qiao, F., Du, Z. Y., et al. (2016). Symbiotic bacteria in gills and guts of Chinese mitten crab (Eriocheir sinensis) differ from the free-living bacteria in water. PLoS One 11:0148135. doi: 10.1371/journal.pone.0148135
Zhao, Y., Yuan, L., Wan, J., Sun, Z., Wang, Y., and Sun, H. (2016). Effects of potential probiotic Bacillus cereus EN25 on growth, immunity and disease resistance of juvenile sea cucumber Apostichopus japonicus. Fish Shellfish Immunol. 49, 237–242. doi: 10.1016/j.fsi.2015.12.035
Zheng, Z., Hao, R. J., Xiong, X. W., Jiao, Y., Deng, Y. W., and Du, X. D. (2019). Developmental characteristics of pearl oyster Pinctada fucata martensii: insight into key molecular events related to shell formation, settlement and metamorphosis. BMC Genom. 20:122. doi: 10.1186/s12864-019-5505-8
Zhou, M., Liang, R. L., Mo, J. F., Yang, S., Gu, N., Wu, Z. H., et al. (2018). Effects of brewer’s yeast hydrolysate on the growth performance and the intestinal bacterial diversity of largemouth bass (Micropterus salmoides). Aquaculture 484, 139–144. doi: 10.1016/j.aquaculture.2017.11.006
Keywords: Pinctada fucata martensii, bacterial community, intestine, environmental microbiota, 16S rRNA-based sequencing
Citation: Zheng Z, Liao Y, Ye J, Yang C, Adzigbli L, Wang Q, Du X and Deng Y (2021) Microbiota Diversity in Pearl Oyster Pinctada fucata martensii Intestine and Its Aquaculture Environment. Front. Mar. Sci. 8:655698. doi: 10.3389/fmars.2021.655698
Received: 19 January 2021; Accepted: 03 March 2021;
Published: 23 March 2021.
Edited by:
Xiaotong Wang, Ludong University, ChinaReviewed by:
Wenguang Liu, Chinese Academy of Sciences, ChinaZhenhua Ma, Chinese Academy of Fishery Sciences (CAFS), China
Emmanuel Abarike, University for Development Studies, Ghana
Copyright © 2021 Zheng, Liao, Ye, Yang, Adzigbli, Wang, Du and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingheng Wang, d2FuZ3FpbmdoZW5nX2hhaWRhQDE2My5jb20=