 Yunying Cheng
Yunying Cheng Jinlin Chen
Jinlin Chen Irum Mukhtar
Irum Mukhtar Jianming Chen
Jianming Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 14 June 2021
Sec. Marine Molecular Biology and Ecology
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.653447
This article is part of the Research Topic Molecular Evolution and Genomics of Marine Megafauna: Methodological Solutions and Key Advances View all 8 articles
Nuclear receptors (NRs), a series of key transcription factors that are mostly activated by endogenous ligands or environmental xenobiotics, are reportedly good phylogenetic markers of animal genome evolution. As the early diverging class of bilaterians, however, a comprehensive view of the NR family in a marine free-living flatworm Macrostomum lignano and comparative information in flatworms are still lacking, which is of significance to address the evolutionary diversification of the NR family and imply the adaptive evolution in the early diverging Bilateria. Herein, a total of 51, 26, and 23 putative NR genes were identified in M. lignano, Sparganum proliferum, and Clonorchis sinensis, respectively, which were classified into eight subfamilies, implying an extensive expansion of the NR family in M. lignano. It is presumed that the extensive expansion was mainly attributed to the M. lignano-specific hidden polyploidy, segmental, and tandem duplication events. The duplicated NR pairs in M. lignano and the NR orthologs in flatworms all experienced the purifying selection. Phylogenetic analyses indicated the presence of NR3-like genes in M. lignano, which is first reported in flatworms. Intron loss and reduced intron size were mainly contributed to the structural divergence of NR genes in flatworms. The combined data provide indispensable information for a better understanding of the complexity and the adaptive evolution of the NR gene family in metazoans.
Nuclear receptor (NR) gene superfamily comprises a large group of ligand-regulated transcription factors which are involved in various functions such as reproduction, differentiation, development, homeostasis, metabolism, and metamorphosis (Bain et al., 2007; Bodofsky et al., 2017; Lazar, 2017). The corresponding ligands of NRs contain a variety of endogenous molecules (e.g., estrogen, androgen, and secondary bile acids) and environmental xenobiotics (e.g., pharmaceutical agents, synthetic hormones, biocides, plastics, and personal care products) (Weatherman et al., 1999; Escriva et al., 2000). Consequently, NRs act as a conduit between the internal and external environments, closely linking to the function of endocrine systems (Fonseca et al., 2020). Although NRs are responsible for a tremendous diversity of functions, they share a common structural modular. A canonical NR possesses five to six functional regions including the N-terminal A/B domain, a highly conserved C domain (DNA binding domain, DBD), D domain (hinge), a moderately conserved E domain (ligand-binding domain, LBD), and F domain (Olefsky, 2001; Mazaira et al., 2018). Characterized by the most conserved domains DBD and LBD, the typical NRs are clustered into seven subfamilies, NR1–NR6 and NR8 (Auwerx et al., 1999; Huang et al., 2015). However, atypical NRs are grouped into NR0 with either DBD (NR0A) or LBD (NR0B) and NR7 with two DBD and an LBD in some animal lineages (Wu et al., 2007; Alvite et al., 2019). Additionally, all ctenophore NRs were atypical without a DBD domain (Reitzel et al., 2011).
Nuclear receptors are considered strong phylogenetic markers of animal genome evolution due to their common ancestral origin, distribution in all metazoan genomes, and highly conserved throughout the whole animal taxa (Escriva et al., 2003). Therefore, the genome-wide identification of NR genes can provide a path to link evolutionary and functional genomics, and trace the dynamic evolutionary route in metazoans. Recently, the rapid developments in the genome sequencing have facilitated a genome-wide identification of NR family members in many species. The NR gene repertoires have been characterized in different lineages with a clear species-dependent pattern from invertebrates to vertebrates, such as 2 members in sponge, 4 in placozoan, ≥250 in nematodes, ∼21 in arthropods, ∼48 in mammals, and 66–137 in teleost (Bertrand et al., 2004; Bridgham et al., 2010; Cheng et al., 2015; Yang et al., 2020). These data suggest the evolutionary complexity of the NR family, which is likely driven by gene duplication and loss (Fonseca et al., 2020). The early diverging bilaterians offer an exceptional opportunity to address the evolution and diversification of the NR family in bilaterians.
Flatworms occupy an assumed central position in the evolution of the Bilateria for that they have been widely reported as the early diverging bilaterians (Martín-Durán and Egger, 2012; Collins, 2017). Moreover, the flatworms have a characteristic of a simple bilateral body plan with the development of complex endocrine systems, which is crucial to coordinate the reaction of organisms to the environment (Noreña et al., 2015). They are conventionally divided into the free-living turbellarians and parasitic tapeworms and flake, and consequently exposed to different environments, including terrestrial, freshwater, and marine environments for the former and within the host tissues for the latter (Collins, 2017). Some parasitic flatworms (such as Schistosoma mansoni and Echinococcus multilocularis) and free-living fresh turbellarians (Schmidtea mediterranea) have been surveyed for NR homologs (Wu et al., 2006; Tharp et al., 2014; Wu and LoVerde, 2019); however, our knowledge regarding the repertoires of NRs in marine groups and the early branching flatworm lineages remains to be filled.
Macrostomum lignano from the high-tide interstitial sand fauna of the coast of the Mediterranean Sea is a marine free-living regenerative flatworm, which is an affiliate to the Macrostomorpha that is an earlier diverging Bilateria than the other often-studied free-living and parasitic flatworms (Wasik et al., 2015). Thus, M. lignano is an exceptional model organism for addressing the diversification of the NR family and implying the evolution of the endocrine system in the early diverging Bilateria. Moreover, a comparative study of NR genes in M. lignano and the other representative flatworms may provide important clues for understanding the adaptative evolution of early bilaterians. Herein, the comprehensive analyses based on the genome-wide identification, genomic distribution, and gene expansions of the NR family were performed in M. lignano. Furthermore, systematic investigations were also conducted to uncover the phylogenetic relationship, exon-intron organization, and selection pressures of this gene family in the representative flatworms.
All possible NR homologs were identified in the representative flatworms, including M. lignano (PRJNA371498), S. proliferum (PRJEB35374), and C. sinensis (PRJNA386618), by a four-step genome search strategy: (1) the reported NR homologs of parasitic flatworms were searched in the predicted proteomes of the representative flatworms by the BLASTP program with the default settings and similarity thresholds of ≥40% in the NCBI web1; (2) all NR sequences were aligned using the Bioedit software to retrieve the conserved domain DBD and LBD sequences; (3) to avoid omissions in the repertoires of NR genes, the DBD and LBD domain sequences were used as a query to search against the target genome sequences by iterative TBLASTN (E = 2e–5) until no novel sequence was retrieved; and (4) all of the positive hits were then identified and confirmed for the presence of NR genes by comparing them to the NCBI database using BLASTP, and redundancy was removed by sequence alignments and genomic location. Allowing for the incorrect genome assembly caused by the high repeated sequences content in the M. lignano genome, we artificially removed the NR genes with sequences that are the same in the middle of a nucleotide sequence but slightly different at the ends to minimize the number of potential redundant genes. To avoid the interference caused by the allelic variants, an analysis testing on the nucleotide sequence similarity starting from 90% with an increment of 1% was performed to choose an optimal cut-off value. Afterward, a cut-off value of 95% was chosen to eliminate the redundancy. Eventually, 51 potential NR proteins were identified and renamed based on their phylogenetic relationships.
Multiple sequence alignments of NR proteins from M. lignano, Amphimedon queenslandica, Trichoplax adhaeren, Nematostella vectensi, Crassostrea gigas, Bactrocera dorsalis, Branchiostoma floridae, and Homo sapiens were performed using the Bioedit software to obtain the aligned DBD and LBD amino acid sequences. Based on the alignments, Maximum-likelihood (ML) phylogenetic trees were built by the IQTREE v1.6.8 (Nguyen et al., 2015) with the automatic selection of an optimal model for protein substitution and rate heterogeneity. For branch support analysis, the SH-aLRT test and ultrafast bootstrapping were conducted with 1,000 replicates. Bayesian phylogenetic tree was constructed by MrBayes3.2 (Ronquist et al., 2012) with the following parameters: generations = 2,000,000, number of runs = 2, burnin fraction = 0.25, and temp = 0.15. The ML and Bayesian trees were visualized and polished by the iTOL v6 website2.
Chromosome size and genomic distribution of NR genes in M. lignano were obtained from the NCBI3 and displayed by the TBtools software with the Amazing Gene Location plugin. The information of NR gene structure was from the genomic annotations downloaded from the NCBI “(see text footnote 3).” The gene structure of NRs combined with the phylogenetic relationship were displayed by the TBtools software with the Amazing Optional Gene Viewer plugin (Chen et al., 2020).
Duplicated NR genes were analyzed by e-value cutoff BLAST-searching (e-value < 10–10) against each other, and identified as duplicated genes when the nucleic acid sequence identity of two or more NR genes reaches at least 80% and aligned region between the two genes were covered >80% of the longer genes (Kong et al., 2013). The NR genes that satisfied these two conditions are considered duplicates. To further explore the potential expansion mechanisms of the NR gene family in M. lignano, we performed all-vs.-all comparisons by the local BLASTP program to identify synteny blocks. SD events were identified according to two conditions as follows: (1) the NR genes derived from gene duplication were located on different scaffolds but clustered into the same group; and (2) the duplicated NRs were found in two or more syntenic regions which were defined to be ≥10 kb in size and ≥80% in sequence identity (Cao et al., 2011). These regions were identified as SDs. The genes adjacent to the NR genes were analyzed to determine whether tandem duplication had occurred. Tandem duplication events were characterized when a pair of duplicated NRs is nearby or separated by few genes in a 100-kb region of a chromosome (Wang et al., 2010).
The identification of positive selection and purifying selection at each site was performed by the SELECTON Server website4 with the ratio of non-synonymous (Ka) to synonymous substitutions (Ks), namely the Ka/Ks ratio (Doron-Faigenboim et al., 2005). If the Ka/Ks ratio of the sites is significantly greater than 1, it means that these sites is under positive pressure; that is, these sites in a sequence varied rapidly during evolution. If the Ka/Ks ratio of the sites is less than 1, it is interpreted as an evidence for negative selection. If the Ka/Ks ratio is 1, the sites is under natural selection, that means the absence of natural selection (Lequime et al., 2016). Based on these, the CDS sequences in each group of flatworms NR genes were submitted together to the SELECTON Server. The evolutionary model is M8, and other parameters are set as the default values.
To identify NR members in M. lignano, the NR proteins of parasitic flatworms and the consensus DBD or LBD domain sequences were used as a query to search against the M. lignano genome database. A total of 140 candidate NR genes with at least one DBD or LBD were originally obtained from the M. lignano genome. By removing 21 redundant sequences, 119 NR genes were obtained based on the presence of an apparently complete DBD or LBD domain. Since the remarkably complex genome structure with ∼75% of the sequence containing repeats and transposon sequences (Wasik et al., 2015), it is difficult to confirm the exact NR gene repertoires in M. lignano. To get more accurate repertoires of NR genes, an analysis testing on the nucleotide sequence similarity starting from 90% with an increment of 1% was performed to choose an optimal cut-off value. The total numbers of NR genes in M. lignano were 90, 62, 55, 52, and 51 with a cut off value of 99, 98, 97, 96, and 95%, respectively, and remained unchanged until to 90% (Supplementary Table 1 and Extended Table). Given the incorrect genome assembly caused by the high repeated sequences content and allelic variants in M. lignano, a cut-off value of 95% was chosen to filter the potential redundancy. Eventually, 51 NR genes were identified and renamed according to the phylogenetic relationships. The identified NR genes ranged from 654 to 2,958 bp, and the corresponding proteins ranged from 217 to 985 amino acids in length. The characteristic information of these NRs was listed in Supplementary Table 2, including the accession number, CDS length, and chromosomal locations.
For a comprehensive understanding of the phylogenetic relationships, an unrooted phylogenetic tree containing 198 NR protein sequences from Amphimedon queenslandica, Trichoplax adhaeren, Nematostella vectensis, Crassostrea gigas, Bactrocera dorsalis, Branchiostoma floridae, Homo sapiens, and M. lignano was constructed using the DBD plus LBD amino acid sequences by the IQTREE procedure with the ML method (Figure 1). All NR genes were clearly divided into nine major groups, as previously reported, referred to as subfamilies NR0–NR8. M. lignano possesses NR members belonging to eight of the nine NR subfamilies. There are two atypical NRs (NR0A1 and NR0A2) containing only one DBD from the NR0 subfamily, which is grouped with NR0A proteins from B. dorsalis rather than C. gigas, B. floridae, and H. sapiens. The largest family is the NR1 subfamily with 27 members, including 1 NR1As, 4 NR1Cs, 2 NR1Fs, and 20 NR1Js. The NR2 subfamily is made up of 7 genes. Interestingly, we have identified 10 members in the NR3 subfamily. Additionally, the rest contained 1 NR4A, 1 NR5B, 2 NR7 (2DBD), and 1 NR8 members. We did not identify any NR6 gene in the M. lignano genome.
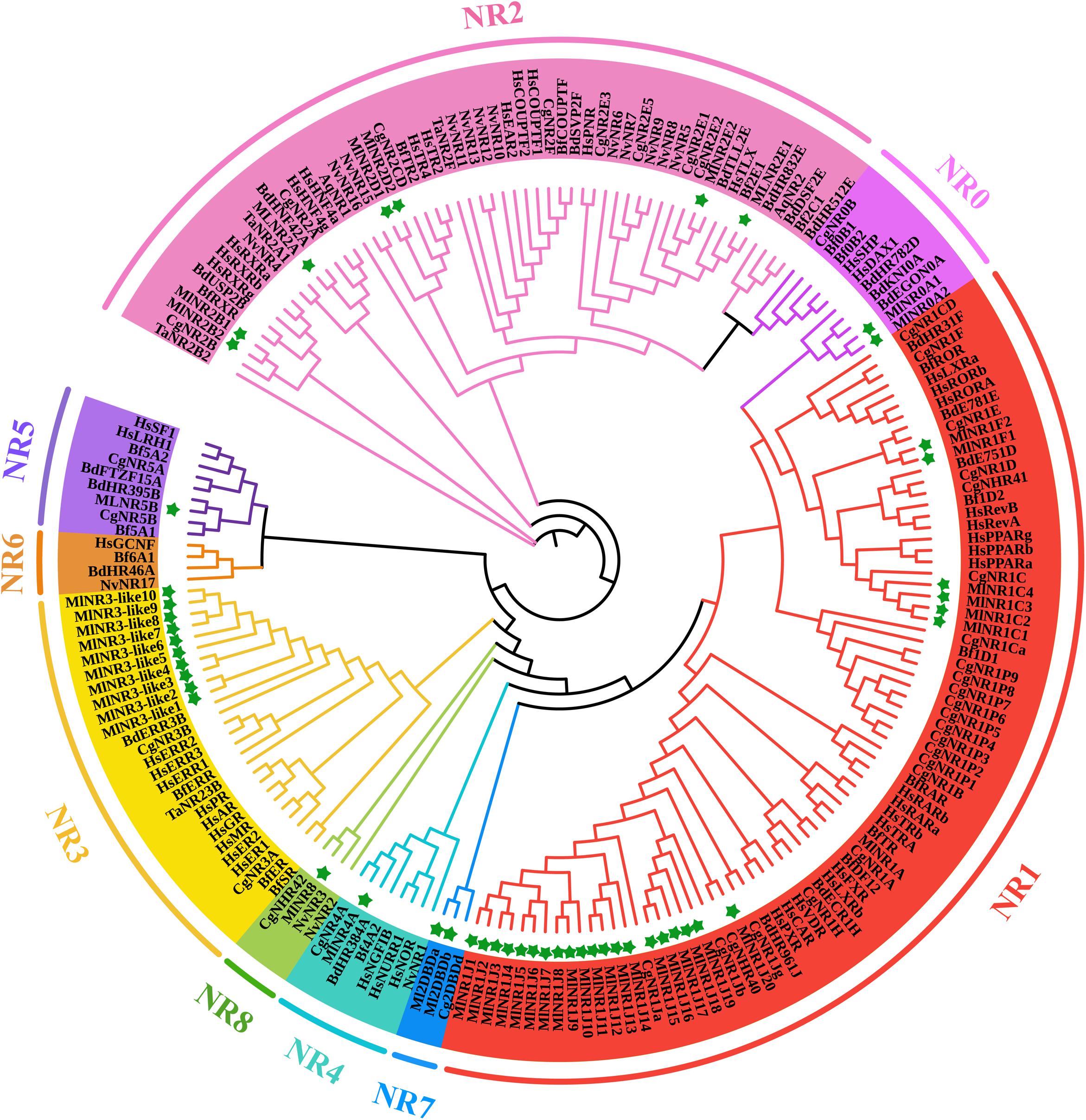
Figure 1. ML phylogenetic tree of NR proteins from M. lignano (Ml), Amphimedon queenslandica (Aq), Trichoplax adhaeren (Ta), Nematostella vectensis (Nv), Crassostrea gigas (Cg), Bactrocera dorsalis (Bd), Branchiostoma floridae (Bf), and Homo sapiens (Hs). The ML method was used to construct the unrooted tree by the program IQTREE. The amino acid sequences of the DBD and LBD domains that belong to NR proteins were aligned using the software Bioedit. Green stars indicate the NR proteins from M. lignano.
The above phylogenetic tree suggested that 10 NR3 proteins from M. lignano all clustered into the NR3B group with the other species. To further confirm the classification of these 10 NR genes, ML and Bayesian phylogenetic trees of the NR3 subfamily were constructed using software IQTREE1.6 and MrBayes 3.2, respectively, based on the multiple alignments of DBD plus LBD sequences (Figure 2). Comparative results showed that the topological structures of the three phylogenic trees were generally similar. However, the NR3 members from M. lignano (MlNR3s) were grouped with the vertebrate NR3C genes in the Bayesian phylogenetic trees, which is different from the result of the ML tree. Normally, the Bayesian Inference method shows a higher accuracy than the other tree-building methods (Hall, 2005) and, therefore, results were considered more credible. Nevertheless, the bootstrap value is moderate (0.532).
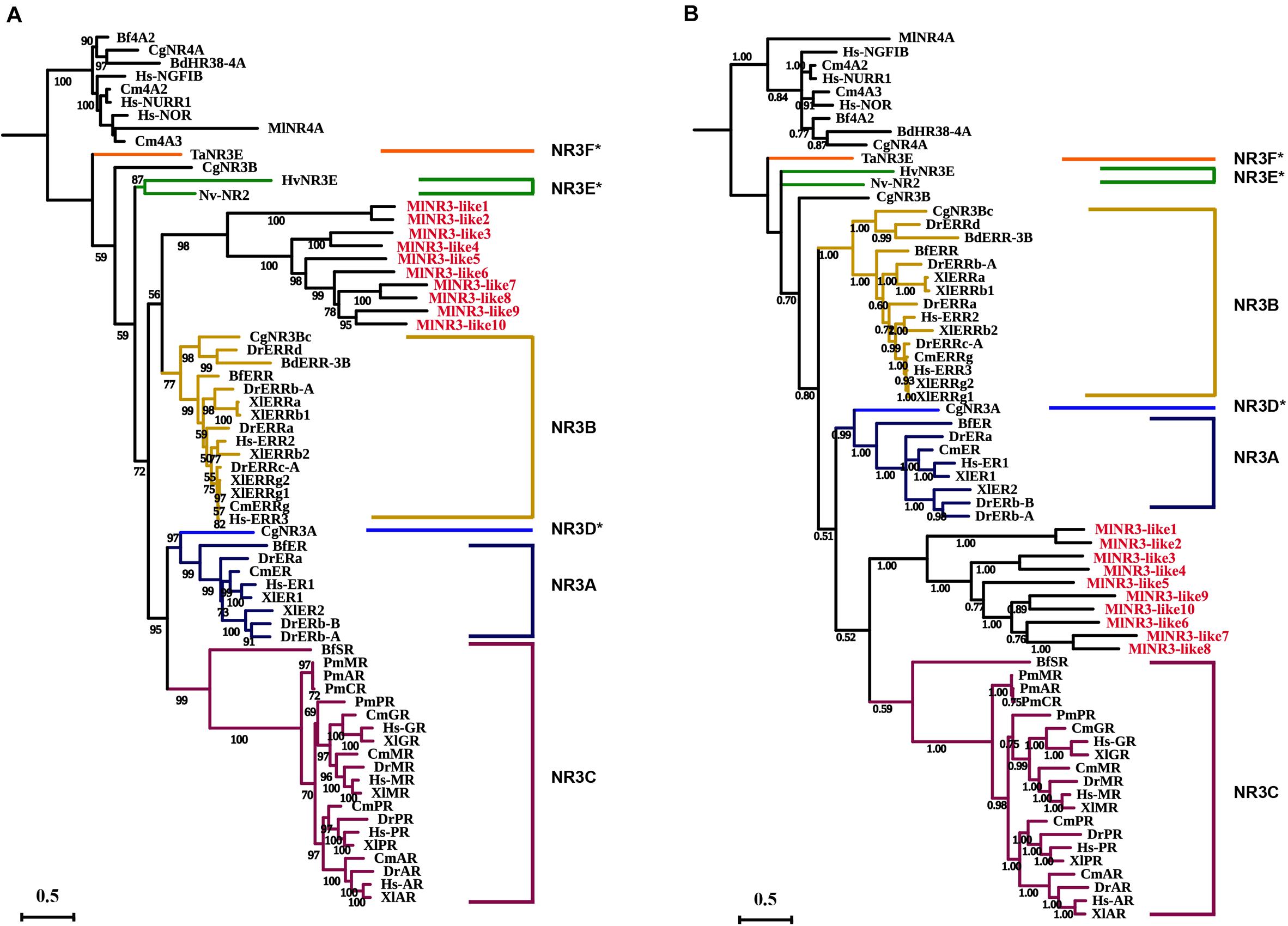
Figure 2. Phylogenetic analyses of the NR3 subfamily. (A) ML phylogenetic tree constructed by the IQTREE procedure. (B) Bayesian phylogenetic tree by the software MrBayes 3.2. The numbers on the branch indicate the likelihood ratio test branch support values. Aq, Amphimedon queenslandica; Ta, Trichoplax adhaerens; Hv, Hydra vulgaris; Nv, Nematostella vectensis; Cg, Crassostrea gigas, Bd, Bactrocera dorsalis; Bf, Branchiostoma floridae; Cm, Callorhinchus milii; Xt, Xenopus tropicalis; Dr, Danio rerio; Hs, Homo sapiens; Ml, M. lignano. NR3D*, NR3E*, and NR3F* represent the ER-like genes from the protostomia, cnidarian, and placozoa, respectively (Miglioli et al., 2021). All NR3-like genes from M. lignano were in red.
The analyses on the sequence identity of DBD and LBD in NR3s between M. lignano and the other species were also performed (Supplementary Tables 3, 4). The results suggested that the DBDs of MlNR3s have 39.3–55%, 44.9–64.4%, and 37.2–55% shared identity compared to the NR3A (ERs), NR3Bs (ERRs), and NR3Cs of the other species, respectively. Similarly, the LBDs of MlNR3s showed a low shared identity with 8.3–31.5%, 13.7–37.8%, and 9.5–28.7% when compared to the NR3A (ERs), NR3Bs (ERRs), and NR3Cs of the other species, respectively. This indirectly supported the result of the ML phylogenetic tree. The DBD and LBD sequences of MlNR3s were compared with the ERs, ERRs, and NR3Cs from the other species (Supplementary Figures 1, 2). The DBDs showed a high level of conservation for all sequences, and were more similar to the ERRs than the NR3Cs. For example, the remarkable conservation of four residues (21E, A22, 30T, and 32Q) in the ERRs was also observed in some of the MlNR3s, while two residues (78P and 79A) displayed conservation in Amphioxus SR, human AR, GR, MR, PR, and MlNR3B8-15. The LBD that constitutes the ligand-binding pocket (LBP) was less conserved. Similar to the other NR3 proteins, the LBDs of the MlNR3s also contained 12 α-helices (H1–H12). The detailed comparison of the LBD sequences suggested that the conserved residues involving ligand binding in the LBP of the ERs and ERRs were also found in the H3, H4–5, H7, H10–11, and H12 of the MlNR3s (Supplementary Figure 2), whereas only one conserved residue of human NR3Cs for oxosteroid binding was observed in the MlNR3s.
Phylogenetic results in this study revealed a massive expansion of the NR gene family in the M. lignano genome, especially in the NR1 and NR3 subfamilies. Generally, gene duplication events, including whole genome duplication (WGD), segmental duplication (SD), and tandem duplication (TD), has been recognized as the main mechanisms to drive the evolution and expansion of gene families. To explore the reason of NRs expansion in M. lignano, the genomic location was mapped. A complete view of the NR genes localization in the M. lignano genome clearly showed that NR genes displayed a highly dispersed distribution of 51 NR members across the 50 scaffolds (Figure 3 and Supplementary Table 5). Only two NR3-like genes located on Sc-73 formed a cluster, namely NR3-like12 and NR3-like13. As tandem duplicated genes represent an array of at least two homologous genes with a high sequence similarity of the encoded proteins separated by a chromosomal region within 100-kb, these 2 NR genes were clearly clustered into one tandem repeat event region. Therefore, they were identified as TD genes. Additionally, the genomic regions containing the NR members which are potential synteny were searched to locate the segmentally duplicated pairs. The searches at the whole-genome scale confirmed the widespread occurrence of SDs. Only one syntenic blocks containing 2 NR genes were detected in the M. lignano genome (Supplementary Figure 3), indicating that they were involved in SD. However, no TD or SD events were found in the NR gene family of the other flatworms.
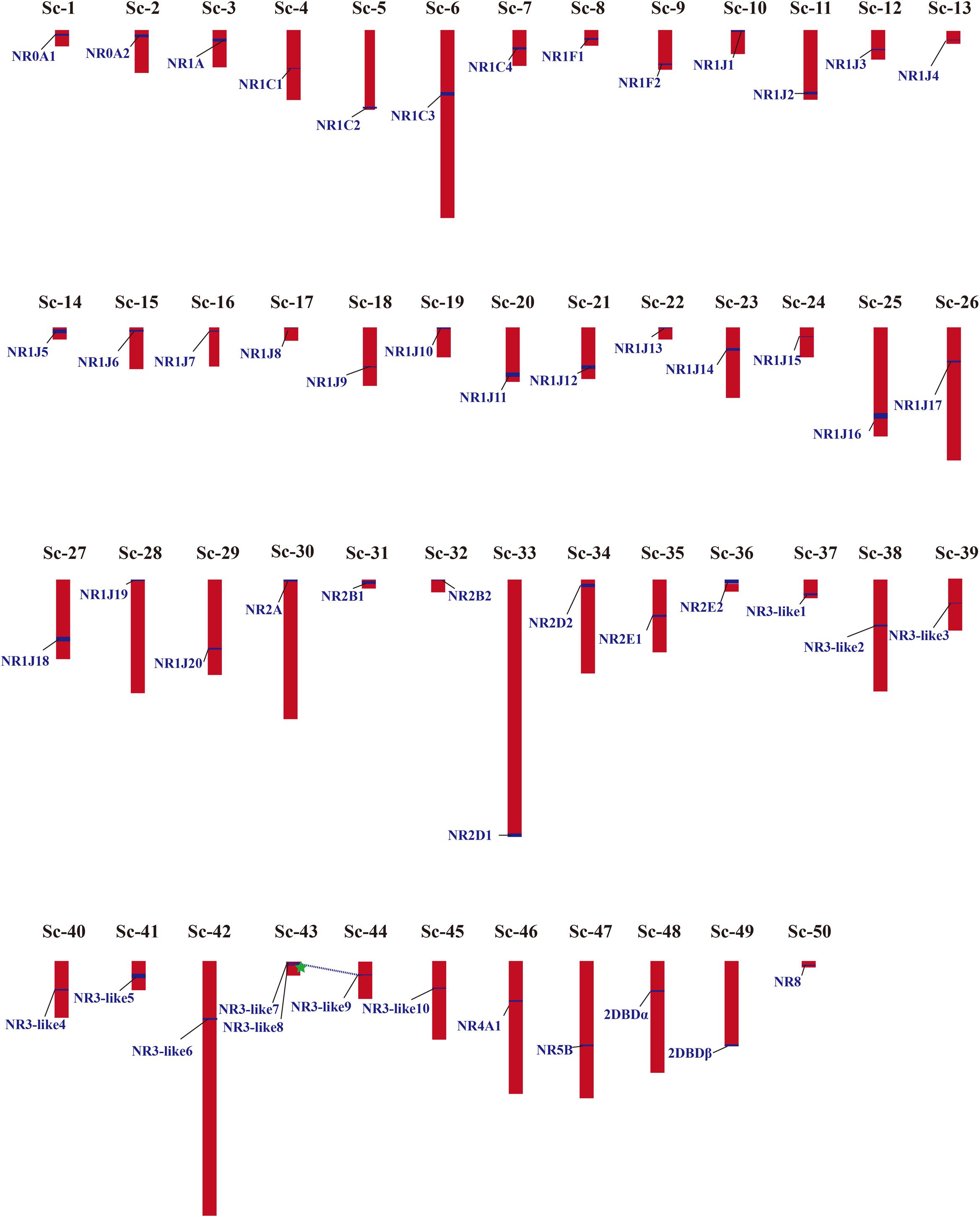
Figure 3. Chromosomal locations and gene duplication events of NR genes in M. lignano. Their detailed scaffold IDs are recorded in Supplementary Table 5. The NR genes from SD are linked by blue dashed lines, and from TD are marked by green star. The detailed map of the TD and SD is provided in Supplementary Figure S3.
Given that the comparative analyses of gene family can provide insights into interpreting the biological variation among flatworms in the light of evolution, we also identified the NR genes in other representative flatworms with 26 NR members in Sparganum proliferum (tapeworm) and 23 in C. sinensis (fluke) using the same methods (Supplementary Table 6). By combining the previously reported data of S. mansoni and S. mediterranea NR genes, comparison analyses on the scaffold number, genome size, coverage, total gene number, and identified NR gene number of six representative flatworms, including two free-living species, two tapeworms, and two flukes, were conducted (Table 1). The results revealed that the number of identified NR genes varies with the genome size of parasitic flatworm species in the range of 17--26 with similar genome sequencing quality and bioinformatic methods, but the free-living species. Among these species, the M. lignano genome contains a maximum of 51 NR genes, while E. granulosus has 17 NRs. The number of M. lignano NRs is 0.96--2 times more than that of the other flatworms, while the genome size was approximate. By searching the Genome database of NCBI5, we found that the total gene number of M. lignano is 49,027, which approximately is 1, 3.3, 1.2, 2.3, and 3.2 times larger than that of S. mediterranea (23,657), E. granulosus (11,325), S. proliferum (22,739), C. sinensis (14,936), and S. mansoni (11,713), respectively.
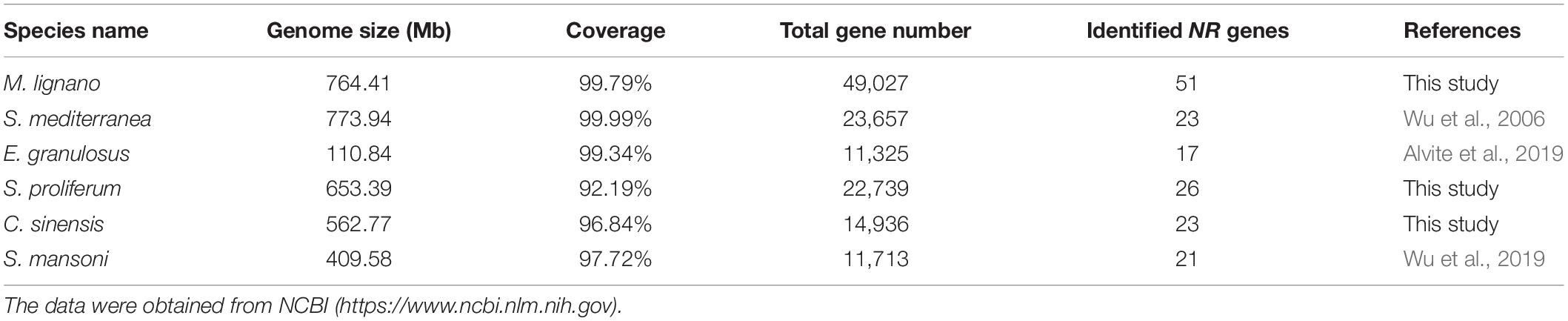
Table 1. Genomic information and identified NR gene numbers in representative species of flatworms.
The numbers of NR genes in certain groups were distinct among these species (Figure 4). We found that the number of genes in NR1 and NR3 were larger as compared to the other subfamilies in M. lignano. For instance, the NR1 subfamily was composed of 27 genes from M. lignano, but only 5 genes from S. mansoni, and 6 from S. proliferum, C. sinensis, and S. mediterranea. Additionally, 10 NR genes from M. lignano were classified under the NR3 subfamily, while no member in the NR3 subfamily was observed in the other flatworms. These results suggested that these groups have undergone an extensive expansion in M. lignano. Additionally, the largest clade is NR1, followed by NR3, NR2, NR0, NR7, NR5, and NR8 in M. lignano, while the NR2 subfamily is the largest group followed by the NR1, NR7, NR5, NR0, and NR4 subfamilies in the other flatworms. As compared to the other flatworms, NR1B and NR2F homologs were absent in the M. lignano genome. Conversely, the NR8 subfamily is unique to M. lignano. In addition, no homolog to the NR6 subfamily was identified in the flatworms.
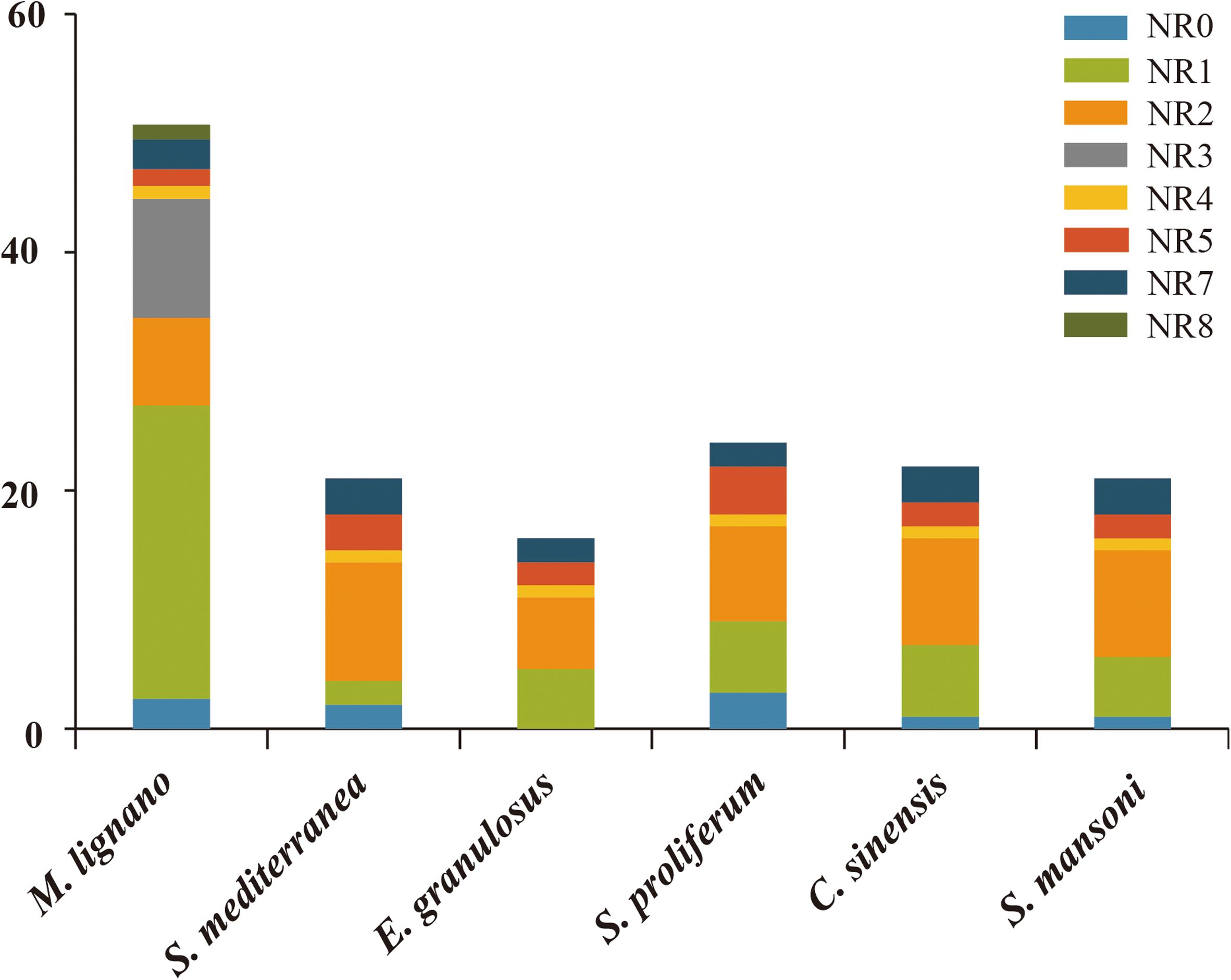
Figure 4. Different distribution patterns of NR genes in flatworms. The x-axis represents the species, and the y-axis represents the gene numbers in different subfamilies.
The structural stability of a gene is a prerequisite to maintain their biological functions, while divergence in the structures of a gene is essential for studying evolutionary divergence within members of gene families. Therefore, the evolutionary dynamics of an exon-intron organization are indicators for the evolutionary history of a gene family (Hajiahmadi et al., 2020). To inspect the correlation between the exon-intron organization and phylogenetic classification, a neighbor-joining phylogenetic tree was also constructed (Figure 5A). By comparing the CDS sequences and complete gene sequences of NR genes, we obtained the exon-intron distribution of NR genes to further understand the mechanisms of the structural evolution of NR paralogs in flatworms (Figure 5B). The results showed that the M. lignano NR members in the same subfamily had similar structures, and most of them had the approximate number of introns (Supplementary Table 7). For instance, the exon-intron organization of members in the NR0A, NR1F, NR2E, NR2D, and NR7 subfamilies is highly conserved with the approximate number of introns within each subfamily. The exceptions are the NR1J and NR3 subgroups, in which the number of introns differed most significantly with 1–6 and 1–4, respectively. In addition, the exon-intron distribution patterns and introns lengths of NR genes were usually diverse in different clades. The 2DBD and NR8 genes contained more introns than the other members, indicating that the structure of these genes were relatively complex. For the orthologs in these three flatworms, both the introns number and average length of NR genes in M. lignano were mostly less than their orthologs in S. proliferum and S. mansoni, while their genome sizes of were similar (Figure 6).
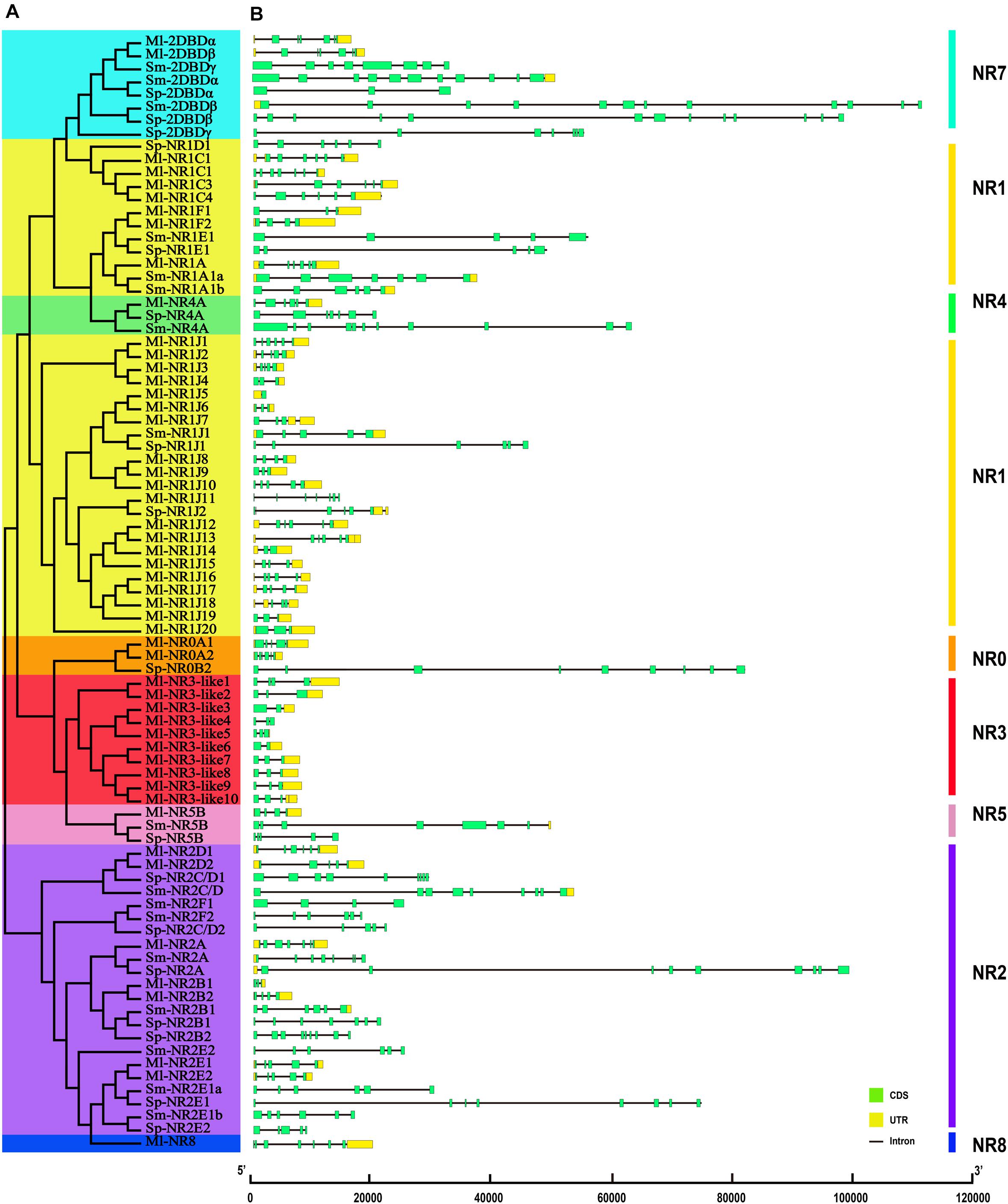
Figure 5. Phylogenetic relationships and exon-intron organizations of NR genes in M. lignano, S. proliferum, and S. mansoni. (A) Phylogenetic relationships. The phylogenic tree was constructed using full length NR protein sequences from the three species. The eight major groups were marked with different color backgrounds. (B) Exon-intron organizations of NR genes. Box represent exons, of which, green boxes signify coding sequence (CDS), and yellow boxes represent untranslated region introns (UTR). Black lines represent introns.
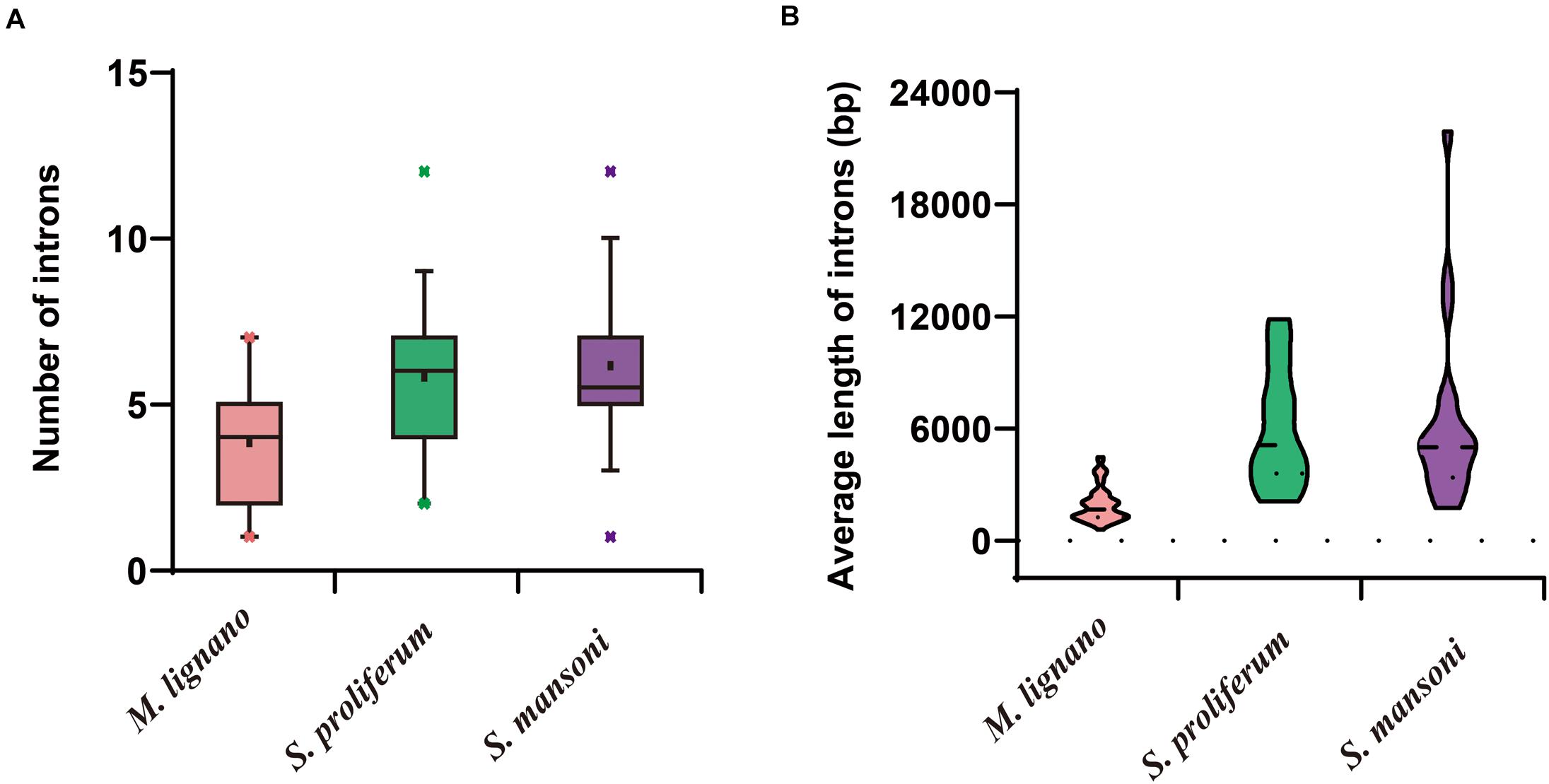
Figure 6. Differences of introns in M. lignano, S. proliferum, and S. mansoni. (A) Distribution of intron numbers in flatworm species. (B) Distribution of intron length in flatworm species.
Performing selection pressure analysis at the genetic level helps to understand not only the evolutionary history of organisms, but also the structural and functional variations of genes. According to the phylogenetic analysis, we found that the linage-specific gene duplication events happened in the NR family in M. lignano. For the sake of investigations on the selection pattern of these NR paralogs after duplication events, which might also contribute to the overall divergence of the NR gene family, the non-synonymous substitutions (Ka), synonymous substitutions (Ks), and their ratio Ka/Ks values of two pairs of paralogous NRs were obtained, respectively. As shown in Supplementary Table 8, the Ks value of NR genes derived from TD (1.7553) is far greater than that from SD (0.0449). The Ka/Ks ratios of the duplicated NR pairs in M. lignano were all lower than 1, implying that the duplicated NR genes has evolved under the purifying selection. To further assess which selection patterns drove the evolution of the NR gene family in flatworms, we also calculated the Ka/Ks ratio of orthologous NR genes among flatworms (Supplementary Table 9). The Ka/Ks ratios for all groups of NR genes in flatworms were <1, ranging from 0.2215 to 0.6785 with an average of 0.4405. These observations suggested that the evolution of the NR gene family in flatworms is under an intensive purifying selection pressure.
The in-depth investigation of well-chosen gene families appears as the most promising path for linking evolution to functional adaptations (García et al., 2003). The NR gene family, which originated in the common ancestor of metazoans, is just one of these families for several distinct characteristics, including highly conservation, dispersion in the metazoan genome as an unbiased sample of sequences, and the consistency between its duplication history with that of the whole gene population (García et al., 2003; Cheng et al., 2015). As one of the largest classes of transcriptional regulators, NRs are activated by endogenous or exogenous specific ligands and play crucial roles in maintaining the intra- and inter-cellular functions in metazoans (Lazar, 2017). Thus, their expansion events and evolution are inextricably linked to the function of endocrine systems.
Among the extant animals, the flatworms are close to one of the earlier groups of bilaterians, occupying the important evolutionary position in the tree of life. Importantly, the marine free-living M. lignano is the early diverging sub-taxon of the Platyhelminthes–Rhabditophora (Egger et al., 2015). However, the complete sets of NR family in this key species and their evolutionary relationships in flatworms are still unexplored. The availability of the sequenced genomes of M. lignano and the other flatworms provides a great genetic resource for the comprehensive investigation and comparison of this gene family. Here, we conducted an integrated analysis of the NR gene family in M. lignano containing the phylogenetic relationships, chromosomal distributions, duplication events, selective pressure, and comparison of exon-intron structures in flatworms.
A total of 119 NR genes were observed in M. lignano. After removing the potential redundancy caused by incorrected assembly and allelic variants, 51 NRs were eventually identified with a cut-off value of 95% sequence identity. Most of the removed NR genes with a high sequence similarity (>97%) were located in the duplicated segments as reported by Wasik et al. (2015). It has been reported that the segmentally duplicated genes, especially those that occurred recently, have a high sequence similarity with a range of 90–99.5% (Samonte and Eichler, 2002). Even the segmental duplication has been defined as “long stretches of duplicated sequences that can span between 1–200 kb and that share a sequence identity higher than 95%” by Lallemand et al. (2020). Given that a more recent large segmental duplications may occur in the M. lignano genome (Wasik et al., 2015), the possibility that the above-mentioned removed NR genes are caused by fragmental duplication rather than allelic variants or the result of misassembly cannot be ruled out. Therefore, the total number of NR genes may change when a completer and more continuous genome is subsequently released.
Based on the phylogenetic grouping relationships, a total of 198 NR proteins were organized into nine major groups, from NR0–NR8 in selected species (Figure 1), which largely coincided with the previous reports (Kim et al., 2017; Hyde et al., 2019). The genes which are grouped into the same subclades always possessed similar functions, which provides a valuable reference to predict the function of homologous genes (Zhang et al., 2014). Remarkably, 10 NRs were clustered with the NR3 genes from other species well. However, the results of the ML and Bayesians phylogenetic analyses are inconsistent (Figure 2). The former indicated that they belong to the NR3B subfamily, while the latter suggested that they should be assigned to the NR3C subfamily. The DBD and LBD sequence identity and alignments supported the results of the ML phylogenetic analyses (Supplementary Figures 1, 2). Although we could not confirm which subfamily they belong to, this is true for these 10 sequences identified as NR3 genes. To avoid misleading inferences, we named them as NR3-like genes. Notably, this is the first report of the flatworm NR3 members.
The NR gene repertoire is significantly larger than those in S. mediterranea (21), E. granulosus (17), S. proliferum (26), C. sinensis (23), and S. mansoni (21) with similar genome sequencing quality and bioinformatic methods (Table 1 and Supplementary Table 4), clearly showing the size multiplicity of this gene family among different flatworms. The genome size varied in different flatworms, such as M. lignano (764.41 Mb), E. granulosus (110.84 Mb), S. proliferum (653.39 Mb), C. sinensis (562.77 Mb), and S. mansoni (409.58 Mb) (Table 1), suggesting that the number of NR family members has an absolute correlation with the genome size in parasitic flatworms. However, the genome size of free-living species S. mediterranea (773.94 Mb) is much larger than that of the other parasitic flatworms, while it bears less NR genes, implying that the different evolutionary histories for each species is another reason except for the genome size. This was assumedly attributed to the relatively similar environment surrounding the parasitic flatworms, which is one of the evolutionary driving forces that contribute to the final shape of the organism genome (Cases et al., 2003).
Comparative analyses suggested that 12 of 17 subfamilies were shared by M. lignano and the other flatworms; this means that 12 subfamilies of M. lignano contained NR orthologs to the other flatworms (Supplementary Table 6). Remarkably, the NR3 and NR8 subgroups were exclusive to M. lignano. As the second-oldest subfamily, a member of the NR3 subfamily (ERR) has been identified in the placozoan T. adhaerens (Novotný et al., 2017). In addition, NR3 members were also found in the Cnidaria, Lophotrochozoa, Ecdysozoa, and Deuterostomia (Kim et al., 2017; Baker, 2019), whereas, they were absent in C. elegans and flatworms except in M. lignano (Eick and Thornton, 2011; Wu and LoVerde, 2019), indicating that NR3 genes possibly appeared as secondary loss in the flatworm species diverged from the Macrostomorpha. The NR8 gene, which could not be assigned to the NR0–NR7 subfamilies, was first identified and named in C. gigas, and found in most invertebrate phyla, including the cnidaria, rotifera, mollusks, annelids, echinoderm, hemichordate, and cephalochordate (Huang et al., 2015; Kim et al., 2017). Nevertheless, no homolog of the NR8 gene was found in the vertebrate, ecdysozoans, and the other flatworms up to date. Thus, NR8 is likely to have undergone lineage-specific gene loss events in the flatworm species diverged from the taxon Macrostomorpha, the ancestors of ecdysozoans and vertebrates. Apart from the subfamilies exclusive to M. lignano, there are two subfamilies that include members in the other flatworms except M. lignano, namely, the NR1B and NR2F subfamilies. It can be assumed that these members were lost in M. lignano. The flatworm genomes did not contain a NR6 subfamily homolog which was also not observed in C. gigas and four of the monogonont rotifer species. Besides, the homologs have been discussed both in the other protostomes and deuterostomes (Robinson-Rechavi et al., 2001; Zhang et al., 2004; Hwang et al., 2014; Vogeler et al., 2014; Kaur et al., 2015). Therefore, gene loss events occurred independently during the ecdysozoans-lophotrochozoans split.
The number of NR genes identified in M. lignano was much more than the other flatworms, which is nearly three times, showing an extensive expansion in the M. lignano genome. The independent species-duplications were also detected in the homeobox (Hox) gene family in M. lignano, leaving multiple copies of Hox genes (Wasik et al., 2015). Generally, gene duplication was considered a major mechanism for gene expansion, including whole WGD, SD, and TD (Fernández and Gabaldón, 2020; Herath and Verchot, 2021). For example, the expansion of the NR and TGF-β signaling pathway members is primarily driven by WGD in teleost (Cheng et al., 2015; Zheng et al., 2018), and SD and TD promote the expansion of the Expansins family in Moso bamboo (Jin et al., 2020). As proven by the phylogenetic analyses, the NR1C, NR1J, and NR3 subfamilies had experienced a species-specific expansion in M. lignano. The genomic location and the syntenic analysis of NR genes in M. lignano revealed that only one SD and TD (∼8%) were observed (Figure 3 and Supplementary Figure 3), implying that they have a less contribution in the expansion of the NR gene family. These SD and TD events, even the expansion of these gene subfamilies, were specific to M. lignano and lacking in the other flatworms. Besides the SD and TD, the M. lignano-specific hidden tetraploidy may be the main mechanism of this species-specific gene duplication. Recent studies have showed that hidden tetraploidy may occur in the M. lignano genome with ∼75% of the sequence containing repeats and transposon sequences, potentially suggesting whole-genome duplication or more recent large segmental duplications (Wasik et al., 2015; Zadesenets et al., 2017, 2020). Thus, we presume that polyploidy, SD, and TD are the main mechanisms driving the extensive expansion of the NR gene family in M. lignano. The species-specific expansion in the NR gene family and hidden tetraploidy indicated the independent evolution of the M. lignano genome.
The extensive expansion was mainly attributed to the impressive number of NR1J, NR3, and NR1C genes with 20, 10, and 4 duplicates in M. lignano, respectively. Seemingly, species- or lineage-specific gene duplications appeared to be relatively frequent in invertebrates. For instance, lineage-specific gene duplication of 10 NR1H has been observed in Branchiostoma floridae (Lecroisey et al., 2012). The analogous duplications have been also detected in NR1L of Tigriopus japonicus (12 duplicates), NR1P of Crassostrea gigas (11 duplicates), and NR1O of the monogonont rotifer Brachionus spp. (19 duplicates) (Thomson et al., 2009; Hwang et al., 2014; Kim et al., 2017). These species- or lineage-specific gene duplications may be interpreted as undergoing sporadic evolutionary processes for adaptation to their different environments. Remarkably, these duplications in previous and recent reports almost all occurred in the NR1 subfamily, implying the crucial roles of the NR1 genes in environmental adaptation for invertebrates. It was previously suggested that Scrobicularia plana NR1J genes respond to a natural toxin (okadaic acid) in the way that was previously observed in tunicates and vertebrates (Cruzeiro et al., 2016). Additionally, NR1C of the pacific oyster reportedly bound to the environmental chemicals, and potentially offered pathways for the disruptive function of these chemicals (Vogeler et al., 2017). M. lignano typically inhabits the interstitial spaces between sand grains in the upper intertidal zone, which is exposed to variable environments and brimming with bioactive compounds such as toxins (Wudarski et al., 2020). Consequently, the expansion of these NR genes in M. lignano may help cope with a wide array of toxins, temperatures, salinities, and oxygen concentrations. However, the specific function of these NR genes in M. lignano is unclear.
The structure of a specific gene regarding the intron numbers and length is typically conserved in the closely related species (Parvathaneni et al., 2017). Unexpectedly, the NR genes of M. lignano had an incongruent exon-intron structure as compared to S. mediterranea and S. proliferum. The incongruence was due to the dynamic changes in the intron composition among flatworms. As shown in Figure 5B and Supplementary Table 7, the intron numbers and average lengths of NR genes in M. lignano are generally less or shorter than that of S. mediterranea and S. proliferum. This observation may suggest that the intron loss occurred in the evolution of aquatic species (free-living) to land species (parasitism), which was also detected in plants (Wang S. et al., 2017). In particular, M. lignano is the first species in which prevailing intron loss events and a reduction in intron size have been reported in the NR family. Previous studies have suggested that gene duplication is a key force driving the dynamic changes in exon-intron organizations, and the segmental duplication can even cause the frequent intron losses (Cao et al., 2011; Xu et al., 2012; Wang Y. et al., 2017). Considering whole-genome duplication or more recent large segmental duplications caused by the M. lignano-specific hidden tetraploidy, therefore, our study in the high frequency of intron loss also suggested the association between the intron losses and duplications. Additionally, this hypothesis was also supported by the fact that the most expanded subfamily, such as NR1J and NR3, possessed the most diverse intron numbers in M. lignano. This mechanism may also underlie the evolution of the NR family in M. lignano. It is worth to mention that the average full-lengths of NR genes in S. mediterranea and S. proliferum were also far longer than that in M. lignano, implying that the M. lignano genome is extraordinarily compact. It explains why M. lignano genome bears 2–3 times the total number of genes compared to the other flatworms.
During the evolutionary process, the conservation or divergence must occur in the NR gene family of flatworms, especially in M. lignano, for extensive duplication. In general, duplicated genes reportedly undergo different evolutionary fates after duplication events, including functional conservation, neo-functionalization, sub-functionalization, and pseudogenization (Moore and Purugganan, 2005). The genes with neo-functionalization undergo a positive selection, and those with sub-functionalization undergo a purifying selection pressure. Statistically, significant evidence suggested that the duplicated NR genes were subjected to a purifying selection in M. lignano since the Ka/Ks values of them were all <1, making their functions trend toward a relative conservation. Furthermore, the Ka/Ks values of NR orthologs were lower than 1 in flatworms, and we presumed that the NR gene family underwent strong purifying selection pressures, leading to a limited functional divergence during the long-term evolutionary process of flatworms. The Ks values of NR genes derived from tandem duplication (1.755) were far larger than those from SD (0.045). The approximate time of the duplication events can be calculated using a synonymous substitution rate Ks based on the formula T = Ks/2λ (Cheng et al., 2009). Consequently, we concluded that the tandem duplication occurred earlier than the SD in the NR family of M. lignano.
The present study displays a genome-wide comprehensive identification and comparative analyses of NR family in three flatworms and provides insight for understanding the mode of evolution of the NR family in flatworms. Our analyses suggest an extensive expansion of the NR gene family with 51 members in M. lignano as compared to the other flatworm species (17–26 members). Remarkably, the NR3-like genes were identified in M. lignano, which is not presented in the other flatworms. Based on the genomic location and syntenic analysis, the expansion of NR family seems to be attributed to the M. lignano-specific hidden tetraploidy, SDs and TD. Furthermore, the evolutionary conservation and diversity of NR genes in flatworms is denoted by the comparative analyses on the exon-intron organizations and selective pressure. Intron loss and a reduction in intron size have been observed in M. lignano, which may mainly attribute to the massive gene duplications. The purifying selection is the primary force driving the evolution of the NR gene family in flatworms. This study provides important cues for addressing the evolutionary diversification of NR family, and understanding the adaptative evolution in the early diverging Bilateria.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
YYC contributed to the conceptualization, analyses, interpretation, writing – original draft, and writing – review and editing. JLC contributed to the software and data analyses. IM contributed to the writing – review and editing. JMC contributed to the project administration and funding acquisition. All authors have read and agreed to the submitted version of the manuscript.
This research was funded by China Ocean Mineral Resources Research and Development Association Program (Grant No. DY135-B2-13), Program for Innovative Research Team in Science and Technology of Education Department of Fujian Province, Project of Fujian Provincial Department of Science and Technology (Grant No. 2020J01868), Project of Minjiang University (Grant No. MYK20004), and National Natural Science Foundation of China (Grant No. 31902352).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.653447/full#supplementary-material
Alvite, G., Riera, X., Cancela, S., Paulino, M., and Esteves, A. (2019). Bioinformatic analysis of a novel Echinococcus granulosus nuclear receptor with two DNA binding domains. PLoS One 14:e0224703. doi: 10.1371/journal.pone.0224703
Auwerx, J., Baulieu, E., Beato, M., Becker-Andre, M., Burbach, P. H., Camerino, G., et al. (1999). A unified nomenclature system for the nuclear receptor superfamily. Cell 97, 161–163. doi: 10.1016/s0092-8674(00)80726-6
Bain, D. L., Heneghan, A. F., Connaghan-Jones, K. D., and Miura, M. T. (2007). Nuclear receptor structure: implications for function. Annu. Rev. Physiol. 69, 201–220. doi: 10.1146/annurev.physiol.69.031905.160308
Baker, M. E. (2019). Steroid receptors and vertebrate evolution. Mol. Cell Endocrinol. 496:110526. doi: 10.1016/j.mce.2019.110526
Bertrand, S., Brunet, F. G., Escriva, H., Parmentier, G., Laudet, V., and Robinson-Rechavi, M. (2004). Evolutionary genomics of nuclear receptors: from twenty-five ancestral genes to derived endocrine systems. Mol. Biol. Evol. 21, 1923–1937. doi: 10.1093/molbev/msh200
Bodofsky, S., Koitz, F., and Wightman, B. (2017). Conserved and exapted functions of nuclear receptors in animal development. Nucl. Receptor. Res. 4:101305. doi: 10.11131/2017/101305
Bridgham, J. T., Eick, G. N., Larroux, C., Deshpande, K., Harms, M. J., Gauthier, M. E. A., et al. (2010). Protein evolution by molecular tinkering: diversification of the nuclear receptor superfamily from a ligand-dependent ancestor. PLoS Biol. 8:e1000497. doi: 10.1371/journal.pbio.1000497
Cao, J., Huang, J., Yang, Y., and Hu, X. (2011). Analyses of the oligopeptide transporter gene family in poplar and grape. BMC Genomics 12:465. doi: 10.1186/1471-2164-12-465
Cases, I., de Lorenzo, V., and Ouzounis, C. A. (2003). Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 11, 248–253. doi: 10.1016/S0966-842X(03)00103-3
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, X., Zhang, D., Cheng, Z., Keller, B., and Ling, H. Q. (2009). A new family of ty1-copia-like retrotransposons originated in the tomato genome by a recent horizontal transfer event. Genetics 181:1183. doi: 10.1534/genetics.108.099150
Cheng, Y. Y., Tao, W. J., Chen, J. L., Sun, L. N., Zhou, L. Y., Song, Q., et al. (2015). Genome-wide identification, evolution and expression analysis of nuclear receptor superfamily in Nile tilapia, Oreochromis niloticus. Gene 569, 141–152. doi: 10.1016/j.gene.2015.05.057
Cruzeiro, C., Lopes-Marques, M., Ruivo, R., Rodrigues-Oliveira, N., Santos, M. M., Rocha, M. J., et al. (2016). A mollusk VDR/PXR/CAR-like (NR1J) nuclear receptor provides insight into ancient detoxification mechanisms. Aquat. Toxicol. 174, 61–69. doi: 10.1016/j.aquatox.2016.02.007
Doron-Faigenboim, A., Stern, A., Mayrose, I., Bacharach, E., and Pupko, T. (2005). Selecton: a server for detecting evolutionary forces at a single amino-acid site. Bioinformatics 21, 2101–2103. doi: 10.1093/bioinformatics/bti259
Egger, B., Lapraz, F., Tomiczek, B., Müller, S., Dessimoz, C., Girstmair, J., et al. (2015). A transcriptomic-phylogenomic analysis of the evolutionary relationships of flatworms. Curr. Biol. 25, 1347–1353. doi: 10.1016/j.cub.2015.03.034
Eick, G. N., and Thornton, J. W. (2011). Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol. Cell Endocrinol. 334, 31–38. doi: 10.1016/j.mce.2010.09.003
Escriva, H., Delaunay, F., and Laudet, V. (2000). Ligand binding and nuclear receptor evolution. Bioessays 22, 717–727. doi: 10.1002/1521-1878(200008)22:8<717::aid-bies5>3.0.co;2-i
Escriva, H. G., Laudet, V., and Robinson-Rechavi, M. (2003). Nuclear receptors are markers of animal genome evolution. J. Struct. Funct. Genomics 3, 177–184. doi: 10.1007/978-94-010-0263-9_17
Fernández, R., and Gabaldón, T. (2020). Gene gain and loss across the metazoan tree of life. Nat. Ecol. Evol. 4, 524–533. doi: 10.1038/s41559-019-1069-x
Fonseca, E., Machado, A. M., Vilas-Arrondo, N., Gomes-dos-Santos, A., Veríssimo, A., Esteves, P., et al. (2020). Cartilaginous fishes offer unique insights into the evolution of the nuclear receptor gene repertoire in gnathostomes. Gen. Comp. Endocr. 295:113527. doi: 10.1016/j.ygcen.2020.113527
García, H. E., Laudet, V., and Robinson-Rechavi, M. (2003). Nuclear receptors are markers of animal genome evolution. J. Struct. Funct. Genomics 3, 177–184.
Hajiahmadi, Z., Abedi, A., Wei, H., Sun, W., Ruan, H., Zhuge, Q., et al. (2020). Identification, evolution, expression, and docking studies of fatty acid desaturase genes in wheat (Triticum aestivum L.). BMC Genomics 21:778. doi: 10.1186/s12864-020-07199-1
Hall, B. G. (2005). Comparison of the accuracies of several phylogenetic methods using protein and DNA sequences. Mol. Biol. Evol. 22:792. doi: 10.1093/molbev/msi066
Herath, V., and Verchot, J. (2021). Insight into the bZIP gene family in solanum tuberosum: genome and transcriptome analysis to understand the roles of gene diversification in spatiotemporal gene expression and function. Int. J. Mol. Sci. 22:253. doi: 10.3390/ijms22010253
Huang, W., Xu, F., Li, J., Li, L., Que, H., and Zhang, G. (2015). Evolution of a novel nuclear receptor subfamily with emphasis on the member from the Pacific oyster Crassostrea gigas. Gene 567, 164–172. doi: 10.1016/j.gene.2015.04.082
Hwang, D. S., Lee, B. Y., Kim, H. S., Lee, M. C., Kyung, D. H., Om, A. S., et al. (2014). Genome-wide identification of nuclear receptor (NR) superfamily genes in the copepod Tigriopus japonicus. BMC Genomics 15:993. doi: 10.1186/1471-2164-15-993
Hyde, C. J., Fitzgibbon, Q. P., Elizur, A., Smith, G. G., and Ventura, T. (2019). Transcriptional profiling of spiny lobster metamorphosis reveals three new additions to the nuclear receptor superfamily. BMC Genomics 20:531. doi: 10.1186/s12864-019-5925-5
Jin, K. M., Zhuo, R. Y., Xu, D., Wang, Y. J., Fan, H. J., Huang, B. Y., et al. (2020). Genome-wide identification of the expansin gene family and its potential association with drought stress in Moso bamboo. Int. J. Mol. Sci. 21:9491. doi: 10.3390/ijms21249491
Kaur, S., Jobling, S., Jones, C. S., Noble, L. R., Routledge, E. J., and Lockyer, A. E. (2015). The nuclear receptors of biomphalaria glabrata and lottia gigantea: implications for developing new model organisms. PLoS One 10:e0121259. doi: 10.1371/journal.pone.0121259
Kim, D. H., Kim, H. S., Hwang, D. S., Kim, H. J., Hagiwara, A., Lee, J. S., et al. (2017). Genome-wide identification of nuclear receptor (NR) genes and the evolutionary significance of the NR1O subfamily in the monogonont rotifer Brachionus spp. Gen. Comp. Endocr. 252, 219–225. doi: 10.1016/j.ygcen.2017.06.030
Kong, X., Lv, W., Jiang, S., Zhang, D., Cai, G., Pan, J., et al. (2013). Genome-wide identification and expression analysis of calcium-dependent protein kinase in maize. BMC Genomics 14:433. doi: 10.1186/1471-2164-14-433
Lallemand, T., Leduc, M., Landès, C., Rizzon, C., and Lerat, E. (2020). An overview of duplicated gene detection methods: why the duplication mechanism has to be accounted for in their choice. Genes 11:1046. doi: 10.3390/genes11091046
Lazar, M. A. (2017). Maturing of the nuclear receptor family. J. Clin. Invest. 127, 1123–1125. doi: 10.1172/JCI92949
Lecroisey, C., Laudet, V., and Schubert, M. (2012). The cephalochordate amphioxus: a key to reveal the secrets of nuclear receptor evolution. Brief. Funct. Genomics 11, 156–166. doi: 10.1093/bfgp/els008
Lequime, S., Fontaine, A., Ar Gouilh, M., Moltini-Conclois, I., and Lambrechts, L. (2016). Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes. Plos Genet. 12:e1006111. doi: 10.1371/journal.pgen.1006111
Martín-Durán, J. M., and Egger, B. (2012). Developmental diversity in free-living flatworms. Evodevo 3:7. doi: 10.1186/2041-9139-3-7
Mazaira, G. I., Zgajnar, N. R., Lotufo, C. M., Daneri-Becerra, C., Sivils, J. C., Soto, O. B., et al. (2018). The nuclear receptor field: a historical overview and future challenges. Nucl. Receptor. Res. 5:101320.
Miglioli, A., Canesi, L., Gomes, I. D. L., Schubert, M., and Dumollard, R. (2021). Nuclear receptors and development of marine invertebrates. Genes 12:83. doi: 10.3390/genes12010083
Moore, R. C., and Purugganan, M. D. (2005). The evolutionary dynamics of plant duplicate genes. Curr. Opin. Plant Biol. 8, 122–128. doi: 10.1016/j.pbi.2004.12.001
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Noreña, C., Damborenea, C., and Brusa, F. (2015). “Chapter 10 – phylum platyhelminthes,” in Thorp and Covich’s Freshwater Invertebrates, 4th Edn, eds J. H. Thorp and D. C. Rogers (Boston: Academic Press), 181–203.
Novotný, J. P., Chughtai, A. A., Kostrouchová, M., Kostrouchová, V., Kostrouch, D., Kaššák, F., et al. (2017). Trichoplax adhaerens reveals a network of nuclear receptors sensitive to 9-cis-retinoic acid at the base of metazoan evolution. PeerJ. 5:e3789. doi: 10.7717/peerj.3789
Olefsky, J. M. (2001). Nuclear receptor minireview series. J. Biol. Chem. 276, 36863–36864. doi: 10.1074/jbc.r100047200
Parvathaneni, R. K., DeLeo, V. L., Spiekerman, J. J., Chakraborty, D., and Devos, K. M. (2017). Parallel loss of introns in the ABCB1 gene in angiosperms. BMC Evol. Biol. 17:238. doi: 10.1186/s12862-017-1077-x
Reitzel, A. M., Pang, K., Ryan, J. F., Mullikin, J. C., Martindale, M. Q., Baxevanis, A. D., et al. (2011). Nuclear receptors from the ctenophore Mnemiopsis leidyi lack a zinc-finger DNA-binding domain: lineage-specific loss or ancestral condition in the emergence of the nuclear receptor superfamily? Evodevo 2:3. doi: 10.1186/2041-9139-2-3
Robinson-Rechavi, M., Carpentier, A.-S., Duffraisse, M., and Laudet, V. (2001). How many nuclear hormone receptors are there in the human genome? Trends Genet. 17, 554–556. doi: 10.1016/s0168-9525(01)02417-9
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Samonte, R. V., and Eichler, E. E. (2002). Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 3, 65–72. doi: 10.1038/nrg705
Thomson, S. A., Baldwin, W. S., Wang, Y. H., Kwon, G., and LeBlanc, G. A. (2009). Annotation, phylogenetics, and expression of the nuclear receptors in Daphnia pulex. BMC Genomics 10:500. doi: 10.1186/1471-2164-10-500
Tharp, M. E., Collins, J. J., and Newmark, P. A. (2014). A lophotrochozoan-specific nuclear hormone receptor is required for reproductive system development in the planarian. Dev. Biol. 396, 150–157. doi: 10.1016/j.ydbio.2014.09.024
Vogeler, S., Galloway, T. S., Isupov, M., and Bean, T. P. (2017). Cloning retinoid and peroxisome proliferator-activated nuclear receptors of the Pacific oyster and in silico binding to environmental chemicals. PLoS One 12:e0176024. doi: 10.1371/journal.pone.0176024
Vogeler, S., Galloway, T. S., Lyons, B. P., and Bean, T. P. (2014). The nuclear receptor gene family in the Pacific oyster, Crassostrea gigas, contains a novel subfamily group. BMC Genomics 15:369. doi: 10.1186/1471-2164-15-369
Wang, L., Guo, K., Li, Y., Tu, Y., Hu, H., Wang, B., et al. (2010). Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 10:282. doi: 10.1186/1471-2229-10-282
Wang, S., Zhu, X. q, and Cai, X. (2017). Gene duplication analysis reveals no ancient whole genome duplication but extensive small-scale duplications during genome evolution and adaptation of Schistosoma mansoni. Front. Cell Infect. Microbiol. 7:412. doi: 10.3389/fcimb.2017.00412
Wang, Y., Xu, L., Thilmony, R., You, F. M., Gu, Y. Q., and Coleman-Derr, D. (2017). PIECE 2.0: an update for the plant gene structure comparison and evolution database. Nucleic Acids Res. 45(D1), D1015–D1020. doi: 10.1093/nar/gkw935
Wasik, K., Gurtowski, J., Zhou, X., Ramos, O. M., Delás, M. J., Battistoni, G., et al. (2015). Genome and transcriptome of the regeneration-competent flatworm, Macrostomum lignano. Proc. Natl. Acad. Sci. U.S.A. 112, 12462–12467. doi: 10.1073/pnas.1516718112
Weatherman, R. V., Fletterick, R. J., and Scanlan, T. S. (1999). Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem. 68, 559–581. doi: 10.1146/annurev.biochem.68.1.559
Wu, A., Hao, P., Wei, H., Sun, H., Cheng, S., Chen, P., et al. (2019). Genome-wide identification and characterization of glycosyltransferase family 47 in cotton. Front. Genet. 10:824. doi: 10.3389/fgene.2019.00824
Wu, W., and LoVerde, P. T. (2019). Nuclear hormone receptors in parasitic Platyhelminths. Mol. Biochem. Parasit. 233:111218. doi: 10.1016/j.molbiopara.2019.111218
Wu, W., Niles, E. G., El-Sayed, N., Berriman, M., and LoVerde, P. T. (2006). Schistosoma mansoni (Platyhelminthes, Trematoda) nuclear receptors: sixteen new members and a novel subfamily. Gene 366, 303–315. doi: 10.1016/j.gene.2005.09.013
Wu, W., Niles, E. G., Hirai, H., and LoVerde, P. T. (2007). Evolution of a novel subfamily of nuclear receptors with members that each contain two DNA binding domains. BMC Evol. Biol. 7:27.
Wudarski, J., Egger, B., Ramm, S. A., Schärer, L., Ladurner, P., Zadesenets, K. S., et al. (2020). The free-living flatworm Macrostomum lignano. Evodevo 11:5. doi: 10.1186/s13227-020-00150-1
Xu, G., Guo, C., Shan, H., and Kong, H. (2012). Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. U.S.A. 109, 1187–1192. doi: 10.1073/pnas.1109047109
Yang, P. J., Chen, E. H., Song, Z. H., He, W., Liu, S. H., Dou, W., et al. (2020). Molecular characterization and expression profiling of nuclear receptor gene families in oriental fruit fly, Bactrocera Dorsalis (Hendel). Insects 11:126. doi: 10.3390/insects11020126
Zadesenets, K. S., Jetybayev, I. Y., Schärer, L., and Rubtsov, N. B. (2020). Genome and karyotype reorganization after whole genome duplication in free-living flatworms of the genus Macrostomum. Int. J. Mol. Sci. 21:680. doi: 10.3390/ijms21020680
Zadesenets, K. S., Schärer, L., and Rubtsov, N. B. (2017). New insights into the karyotype evolution of the free-living flatworm Macrostomum lignano (Platyhelminthes, Turbellaria). Sci. Rep. 7, 6066.
Zhang, Y. M., Zhang, S. Z., and Zheng, C. C. (2014). Genomewide analysis of LATERAL ORGAN BOUNDARIES Domain gene family in Zea mays. J. Genet. 93, 79–91. doi: 10.1007/s12041-014-0342-7
Zhang, Z., Burch, P. E., Cooney, A. J., Lanz, R. B., Pereira, F. A., Wu, J., et al. (2004). Genomic analysis of the nuclear receptor family: new insights into structure, regulation, and evolution from the rat genome. Genome Res. 14, 580–590. doi: 10.1101/gr.2160004
Keywords: nuclear receptor, Macrostomum lignano, flatworms, gene family expansion, intron loss, reduced intron size, evolution
Citation: Cheng Y, Chen J, Mukhtar I and Chen J (2021) Genome-Wide Characterization of the Nuclear Receptor Gene Family in Macrostomum lignano Imply Its Evolutionary Diversification. Front. Mar. Sci. 8:653447. doi: 10.3389/fmars.2021.653447
Received: 14 January 2021; Accepted: 07 May 2021;
Published: 14 June 2021.
Edited by:
Oleg Simakov, University of Vienna, AustriaReviewed by:
Adam Michael Reitzel, University of North Carolina at Charlotte, United StatesCopyright © 2021 Cheng, Chen, Mukhtar and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianming Chen, Y2hlbmptQG1qdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.