
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 03 May 2021
Sec. Marine Biotechnology and Bioproducts
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.644730
This article is part of the Research Topic Marine Biotechnology, Revealing an Ocean of Opportunities View all 26 articles
 Luciana C. Furtado1
Luciana C. Furtado1 Anelize Bauermeister1,2
Anelize Bauermeister1,2 Rafael de Felicio3
Rafael de Felicio3 Raquel Ortega3
Raquel Ortega3 Francisco das Chagas L. Pinto4
Francisco das Chagas L. Pinto4 João Agostinho Machado-Neto1
João Agostinho Machado-Neto1 Daniela B. B. Trivella3
Daniela B. B. Trivella3 Otilia D. L. Pessoa4
Otilia D. L. Pessoa4 Diego V. Wilke5
Diego V. Wilke5 Norberto P. Lopes2
Norberto P. Lopes2 Paula C. Jimenez6
Paula C. Jimenez6 Leticia V. Costa-Lotufo1*
Leticia V. Costa-Lotufo1*Marine natural products have emerged as an important source for drug development, notably in the field of anticancer therapy. Still, the limited effectiveness of current therapies for central nervous system tumors indicates the need to identify new therapeutic targets and also novel pharmacological agents. In this context, proteasome inhibitors are appearing as a promising new treatment for these diseases. Herein, cytotoxic extracts produced by four marine bacteria recovered from the Brazilian endemic ascidian Euherdmania sp. were screened to evaluate their potential as proteasome inhibitors. The extract from marine Streptomyces sp. BRA-346 was selected for further investigation due to the potent proteasome inhibitory activity it displayed. Bioassay-guided fractionation led to an enriched fraction (proteasome inhibition IC50 = 45 ng/mL), in which the presence of dihydroeponemycin (DHE), known for its proteasome inhibitory effect, and related compounds were annotated by mass spectrometry and further confirmed by comparison with DHE standard. Both DHE and the epoxyketone-containing fraction were evaluated in glioma cell lines, displaying high cytotoxicity in HOG and T98G cells (GI50 of 1.6 and 1.7 ng/mL for DHE, and 17.6 and 28.2 ng/mL for the BRA-346 fraction, respectively). Additional studies showed that the epoxyketone-containing fraction (at GI50 levels) led to an accumulation of ubiquitinated proteins and up-regulation of genes related to ER-stress response, suggesting treated cells are under proteasome inhibition. DHE induced similar effects in treated cells but at concentrations 25 times its GI50, suggesting that the other epoxyketone compounds in the bacteria extract derived fraction may contribute to enhance proteasome inhibition and further cellular effects in glioma cells. These findings revealed the molecular pathways modulated by this class of compounds in glioma cells and, moreover, reinforced the potential of this marine bacteria in producing a cocktail of structurally-related compounds that affect the viability of glioma cells.
The marine environment encompasses more than 90% of the volume of the biosphere, housing an enormous and still majorly unknown number of species. The most optimistic estimate from the Census of Marine Life points out to barely 10% of properly described species in the oceans1 (Snelgrove, 2010; National Oceanic and Atmospheric Administration, 2020). However, if a broader concept of diversity is considered, one that includes genetic and chemical diversities, it becomes even more evident that this surface has hardly been scratched (Mora et al., 2011; Snelgrove, 2016). Indeed, from a chemical diversity perspective, the possibilities of exploring this multitude of molecules are virtually limitless, once a marine organism, such as an invertebrate, may be contemplated as a complex ecosystem in itself, with billions of associated microbial cells interacting and responding to biotic and abiotic factors through the production of their metabolites to warrant the holobiont’s homeostasis (Cheng et al., 2020; McCauley et al., 2020).
The uniqueness of the metabolites produced by marine species gathered interest from the scientific community for their potent biological activities, resulting in the development of thirteen drugs, of which most are being used to treat cancer2 (Jimenez et al., 2020; Midwestern University, 2020). In such realm, tunicates – filter-feeding animals distributed across the ocean – are among the most resourceful organisms considering the return of pharmacological capital (Palanisamy et al., 2017; Bauermeister et al., 2019), being recognized for the ability to produce nitrogen-containing compounds, like alkaloids, peptides and their derivatives (Watters, 2018). Didemnin B, a depsipeptide isolated from Trididemnum solidum, was the first marine natural product to enter clinical trials, back in 1986, having been later discharged due to marked cardio and muscular toxicity and lack of efficacy (Rinehart et al., 1981; Vera and Joullié, 2002). Nonetheless, plitidepsin (Aplidin®), also known as dehydrodidemnin B, isolated from Aplidium albicans, has been recently approved by the ATGA (Australian Therapeutics Goods Administration) for treatment of hematological cancers in association with dexamethasone3 (Sakai et al., 1996; Alonso-Álvarez et al., 2017; Therapeutic Goods Administration, 2020), and is now under clinical trials for COVID-19 (White et al., 2021). Additionally, trabectedin, an alkaloid isolated from Ecteinascidia turbinata, and its synthetic derivative, lurbinectedin, make up for the three tunicate-sourced anticancer drugs approved for clinical use4 (Rinehart et al., 1990; Markham, 2020). Both didemnin B and trabectedin are recognized as products of the metabolism of symbiotic bacteria, emphasizing the importance of the associated microbiota on the chemical versatility previously attributed to tunicates (Tsukimoto et al., 2011; Xu et al., 2012; Schofield et al., 2015; Bauermeister et al., 2019).
Although the majority of current anticancer drugs have been obtained from natural sources (Newman and Cragg, 2020), only a few marine compounds are included in this arsenal. However, marine natural products have already distinguished themselves from their outnumbered terrestrial counterparts in terms of their mode of action (Pereira et al., 2019). Plitidepsin, for example, is a first in the class drug that impairs de novo protein synthesis by blocking the function of eEF1A2 (eukaryotic elongation factor 1A2), a protagonist protein in delivering amino-acylated tRNA to ribosomes and thereby promoting polypeptide elongation (Mateyak and Kinzy, 2010; Losada et al., 2016). However, a secondary role to eEF1A2, which favors cancer cells, involves the elimination of misfolded proteins through clustering them for lysosome degradation or tagging them for proteasome degradation. Therefore, inhibition of this elongation factor causes the accumulation of toxic misfolded proteins in the cytoplasm, which then leads to cell death (Hotokezaka et al., 2002; Losada et al., 2020). In fact, provoking cell buildup of misfolded proteins revealed to be a successful anticancer strategy, and one such approach involves hampering the catalytic function of the proteasome itself.
The proteasome is a multicatalytic enzyme complex responsible for most non-lysosomal cellular proteolysis. In fact, the proteasome can be considered the headquarter of protein turnover, influencing many cellular processes such as cell cycle, angiogenesis, immune response, apoptosis, and gene expression. The altered proteostasis of cell cycle regulators (cyclins, tumor suppressors, and oncogenes) and apoptosis-related proteins influence cancer development. For example, non-degradation of cyclin-dependent kinase inhibitors and Ik-Bα prevents cell cycle progression and activation of NF-kB, which switches on prosurvival genes (BCL-2) (Catalgol, 2012; Thibaudeau and Smith, 2019). In light of this, inhibition of the 20S proteasome core particle (20S-CP) is a validated strategy for cancer therapy, where impairment of the chymotrypsin-like activity (ChTL, beta 5) is sufficient to achieve cancer cell apoptosis with a favorable therapeutic window (Kisselev et al., 2012).
In this context, microorganisms have been proven as a promising source of proteasome inhibitors, including the α′-β′-epoxyketones epoxomicin, eponemycin and related compounds (Sugawara et al., 1990; Hanada et al., 1992), and the β-lactone-γ-lactam marizomib (Feling et al., 2003; Potts et al., 2011). Among them, epoxomicin led to the development of carfilzomib (kyprolis®, Onyx Pharmaceuticals), approved by the United States Food and Drug Administration (2020) in 2012 for the treatment of patients with relapsed or refractory myeloma (Muchtar et al., 2016). Additionally, marizomib, produced by the marine free-living actinobacteria Salinispora tropica, is undergoing phase III clinical trials for the treatment of hematological and central nervous system tumors (Jimenez et al., 2020). The α′-β′-epoxyketone inhibitors bind to the β5-subunit of the proteasome, exhibiting a potent and highly specific inhibitory effect on the chymotrypsin-like activity (Kim et al., 1999). Marizomib, in turn, inhibits three catalytic subunits of the proteasome, with, generally, a greater affinity toward the β5 subunit (Potts et al., 2011).
Herein, we evaluated the presence of proteasome inhibitors in cytotoxic extracts of bacteria isolated from the Brazilian endemic tunicate Euherdmania sp. The genus Euherdmania comprises merely 13 species and none have been so far characterized for their secondary metabolism nor associated microbiota (Monniot, 1983; Lotufo and Silva, 2006). Still, a previous effort to access the biological activity of crude extracts obtained from tunicates collected on the coast of Ceará State (Jimenez et al., 2003) highlighted the extract from Euherdmania sp. for a prominent cytotoxic activity against leukemia, melanoma, and colorectal tumor cell lines. Using a HPLC-MS/MS analysis and comparison with standard compounds, the presence of a series of α′-β′-epoxyketones related to eponemycin, including dihydroeponemycin (DHE), were verified in Streptomyces sp. BRA-346 extract along with a potent inhibitory activity of the proteasome. Then, the antiglioma activities of DHE and BRA-346 fraction-containing a cocktail of α′-β′-epoxyketones were evaluated focusing on the modulation of pathways related to the unfolded protein response (UPR).
Four bacteria isolated from the ascidian Euherdmania sp., BRA-339, BRA-341, BRA-346, and BRA-386, collected at Taíba beach, Brazilian northeast coast (3°34.125′S; 38°54.469′W), were used for this work and the procedure used was previously described in Velasco-Alzate et al. (2019). Briefly, ascidian samples were sorted for epibionts and debris, dipped in ethanol 70% for superficial decontamination and kept under sterile conditions. Samples were suspended in sterile seawater (1:1 w/v), macerated, and separated into two aliquots: the first was heated to 55°C over 10 min to minimize the growth of Gram-negative bacteria prior to inoculation; the second was directly inoculated onto 90 mm Petri dishes filled with starch-casein agar (SCA) prepared with reconstituted seawater. Cycloheximide (0.1 mg/mL) was added to isolation plates to reduce fungal contamination. Colonies owning an actinomycete-phenotype (color, presence of spores, texture, etc.) were picked out, transferred to fresh A1 (soluble starch, peptone, yeast extract, reconstituted seawater, and agar) medium dishes and subjected to subsequent replatings onto A1 dishes for purification.
Purified bacteria strains were cryopreserved in the microorganism strain bank of the Laboratory of Marine Bioprospecting and Biotechnology at the Federal University of Ceara. The selection of strains was based on the high cytotoxic activity observed against colon carcinoma cell lines (Supplementary Table 1). A license for the collection of tunicates was granted by Biodiversity Authorization and Information System (SISBIO, authorization number 48522-2). A license for genetic access was granted by the National System for the Management of Genetic Heritage and Associated Traditional Knowledge (SISGen, authorization number AC0781C).
The extracts were obtained from the growth of bacteria strains in Erlenmeyer flasks (250 mL) containing 100 mL of A1 liquid medium (peptone, soluble starch, yeast extract, and reconstituted seawater) for 7 days at 140 rpm maintained at 26°C. To recover the extracts, 100 mL of ethyl acetate was added to the bacteria culture for 1 h under 60 rpm at 26°C. The organic phases were separated from culture bacteria media and collected. The solvent was then removed under reduced pressure and extracts were recovered.
The colon carcinoma cell line [HCT-116 (ATCC CCL-247)] was used for the MTT assay. This assay measures cellular metabolic function based on reduction of tetrazolium salt, MTT 3-(4,5-dimethylthia-zol-2-yl)-2,5-diphenyltetrazolium bromide, to the formazan by mitochondrial dehydrogenases of viable cells. An increase in the formazan concentration indicates greater cell proliferation (Mosmann, 1983).
Cells were cultured in RPMI media supplemented with 10% SBF and 1% penicillin-streptomycin and seeded in 96-well plates (1 × 104 cells per well) 24 h before the test. The extracts and fractions were dissolved in DMSO and tested at increasing concentrations (0.0032–50 μg/mL) for 72 h of incubation at 37°C, 5% of CO2. DMSO 0.5% was used as a negative control, reflecting 100% cell viability. After the incubation period, the media was removed and an MTT solution (5 mg/mL) was added to plates for an additional incubation (3 h). Next, the supernatant was discarded and the precipitate (formazan) was dissolved in DMSO to measure absorbance at 570 nm. Three independent experiments were performed and absorbance values were normalized using blank as 0%, and negative control (DMSO, 0.5%) as 100% of cell proliferation. Inhibitory concentration mean (IC50) values and their 95% confidence intervals (CI95%) were calculated using non-linear regression by GraphPad Prism 7.0. (GraphPad Software, Inc., San Diego, CA, United States).
The EtOAc bacteria crude extracts, obtained as described above, were assayed against the purified 20S-CP from Saccharomyces cerevisiae cells. Yeast 20S proteasome complex was purified in-house from S. cerevisiae using affinity, ion exchange, and size exclusion chromatography, as previously described (Souza et al., 2018). The assay procedures were carried out according to Souza et al. (2018) with minor modifications. Briefly, bacteria crude extracts were diluted in DMSO and assayed by pipetting 1 μL in 384-well black v-shape microplates containing 19 μL of the yeast proteasome at 1 μg/mL, in Tris 10 mM pH 7.5, SDS 0.01%. The yeast proteasome was exposed to the extracts for 2 h before addition of 5 μL of the fluorogenic proteasome substrate Suc-LLVY-Amc at 200 μM (final substrate concentration was 40 μM). The fluorescence was monitored in each well for 1 h, every 2 min, using the Clario (BMG Labtech) plate reader. Liquid handling was assisted by the Versette (Thermo Scientific) automated pipetting system. Each experiment was conducted in triplicates. Remaining proteasome activity (%) for test samples was calculated relative to the DMSO control group, carried out in the same assay plate, however, in the absence of the tested samples. The control group corresponds to 100% proteasome activity and was obtained by incubating the enzyme in the reaction buffer with 5% DMSO before adding the substrate. The intrinsic fluorescence of the samples was also accounted for in the measurement of the fluorescence in each well before the addition of the substrate. Initial experiments were carried out at single concentrations (50 μg/μL) of the crude extracts. Inhibition curves were performed at ten inhibitor concentrations, with final concentrations ranging from 0.003 to 50 μg/mL, in a dilution factor of threefold. The data were plotted as average ± SD of enzyme activity (%) as a function of logarithmic concentrations of the inhibitor. Curves were fitted using the normalized four-parameter concentration-response equation with variable slope implemented in Prism (Graph Pad, San Diego, v. 7.0).
To assess the inhibitory potential of dihydroeponemycin (DHE) (ApexBio, A8172), epoxomicin (ABCAM, ab144598), and epoxyketone-containing fraction obtained from the fractionation of the BRA-346 extract (BRA-346 fraction) on human purified proteasome catalytic subunit, the 20S Proteasome Activity Assay (Merck Millipore, Cat. No. APT280) was used. DMSO (0.3%) was used as the negative control. The entire test was performed according to manufacturer protocol (Zhang et al., 2011). To calculate the half-maximum inhibitory concentration (IC50) of epoxyketone-containing fraction, the sample was evaluated in graded concentrations (0.001, 0.01, 0.1, 1, and 10 μg/mL). The emission fluorescence converted values (%) were compared to the negative control. Assays were analyzed using GraphPad Prism 7.0 software.
For molecular identification, genomic DNA of the isolated BRA-346 strain was obtained from pure culture as previously described by Pinto et al. (2020) using phenol:chloroform:isoamyl alcohol-based protocol. The gene 16S rRNA was amplified by polymerase chain reaction (PCR) using primers 27F (5′- AGAGTTTGATCCTGGCTCAG-3′) e 1494R (5′-ACGGCTACCTTGTTACGACTT-3′) (Heuer et al., 1997). The PCR reaction was also performed as previously described by Pinto et al. (2020). The PCR product was stained using SYBRTM Safe DNA Gel (Life Technologies), analyzed by electrophoresis on agarose gel 1%, and purified with ExoSAP-IT (Affymetrix). Sequencing of amplicons was conducted at the Laboratory of Human Molecular Genetics (LGMH), Department of Genetics of Federal University of Pernambuco (UFPE) using Bigdye TM terminator v3.1 cycle sequencing kit (Thermo Fisher). Sequences forward and reverse were analyzed and edited using Geneious Prime 2020.2 software5. The obtained sequence was deposited in Genbank6 under the number MW342808 and compared with sequences from EzBioCloud database7.
BRA-346 strain was grown in Erlenmeyer flasks (30 × 1 L) containing 250 mL of A1 medium at 26°C, 180 rpm. The crude extract (1.35 g) recovered with ethyl acetate was dissolved in 100 mL of MeOH/H2O (8:2, v/v) and partitioned with n-hexane and EtOAc (2 mL × 100 mL), followed by treatment with anhydrous Na2SO4. The organic solvents were dried by distillation under reduced pressure and, subsequently, by compressed airflow, providing their respective fractions: BRA-346 hexane (506.3 mg) and BRA-346 ethyl acetate (416.5 mg). The resultant MeOH/H2O mixture was dried by solvent distillation under reduced pressure and lyophilized, yielding BRA-346 MeOH/H2O (375.6 mg).
The BRA-346 EtOAc fraction (400.5 mg) was fractionated on a Sephadex LH-20 column (50 mm × 30 mm i.d.) using a mixture of MeOH/CH2Cl2 (8:2, v/v) to provide 20 fractions of approximately 10 mL each, which were monitored by thin layer chromatography (TLC) to yield five fractions, F1-4 (5.6 mg), F5-8 (25.0 mg), F9-11 (65.1 mg), F12-14 (217.5 mg), and F15-20 (86.5 mg), which were screened for cytotoxicity. F12-14 showed the most significant activity, therefore, an aliquot of this fraction (20.0 mg) was subjected to solid phase extraction (SPE) eluted with A (H2O), B (MeOH/H2O 1:1, v/v), C (MeCN/H2O 7:3, v/v), D (MeCN), and E (CHCl3/MeOH 9:1, v/v) to provide their respective fractions, A (0.7 mg), B (5.0 mg), C (9.0 mg), D (0.9 mg), and E (0.3 mg) as described by Cutignano et al. (2015). All obtained fractions were evaporated under reduced pressure to remove solvents and diluted in DMSO before screening for cytotoxicity. Fraction C was selected for sequential studies based on its proteasome inhibitory activity and HPLC-MS/MS analysis. Supplementary Table 2 shows the yield and biological activity data of extract and fractions.
BRA-346 crude extract and fractions A, B, C, D, and E were analyzed by HPLC-MS/MS for metabolic profiling. Dihydroeponemycin (DHE) (ApexBio, A8172), epoxomicin (ABCAM, ab144598) were used as standards for comparison. The analyses were acquired on an HPLC System (Shimadzu) coupled to a micrOTOF QII mass spectrometer (Bruker Daltonics), equipped with an electrospray ionization source (operating in positive ionization mode), and a quadrupole and a time of flight (TOF) analyzers. Chromatographic separation was performed on a Supelco Ascentis Express C18 column (5 μm, 150 mm × 3.0 mm), equipped with a pre-column. The column temperature was maintained at 40°C. The elution was carried out with a linear gradient of the mobile phase, starting with 5–100% acetonitrile for 25 min with a flow rate of 0.7 mL/min. Both solvents (H2O and ACN) contained 0.1% formic acid. The injection volume was 15 μL (1.0 mg/mL). The parameters used in the ionization source were as follows: voltage: 3.5 kV, cone: 4.5 kV, drying gas: 9.0 L/min, gas temperature: 220°C, m/z range: 50–1,500. A data-dependent acquisition (DDA) method was used, which selects to fragment, separately, all ions above 1.0E3 of intensity, in this method a collision energy ramp of 20–75 eV was used to fragment the ions.
The HPLC-MS/MS data were converted to the .mzXLM format, a file type compatible with the GNPS platform (Wang et al., 2016), using the MSConvert software. The molecular network was constructed following the workflow on the GNPS platform. GNPS is an online platform8, which deposits and shares a mass spectrometry data library (MS-MS), allowing the identification of natural products, mainly of marine origin, and which has been widely used in metabolomics works. Briefly, the data (.mzXML) was transferred to the GNPS server (ucsd.edu). The MS/MS spectra were grouped by spectral similarity using cosine score, and the parameters used are described in Supplementary Table 3. After the construction of the molecular network, the file was viewed and analyzed in the Cytoscape software (v. 3.2).
Due to the presence of proteasome inhibitors in the BRA-346 fraction, which can interfere on the turnover protein, and possible over and underestimation of cell viability by the MTT assay in glioblastoma cell lines (Stepanenko and Dmitrenko, 2015), SRB assay was used to evaluate initial cytotoxicity studies of the BRA-346 epoxyketone-containing fraction and dihydroeponemycin in glioma cell lines.
This is a colorimetric assay developed in 1990 which quantifies the total proteins and can be used for investigating cell proliferation and screening of the anticancer drugs. It entails the binding of sulforhodamine B dye to basic amino-acid residues of cell proteins, allowing measure increase or decrease of cellular mass (Skehan et al., 1990). SRB assay setup permits the identification of three main values: mean growth inhibitory concentration (GI50) when the treated cells growth 50% in relation to “time-zero” plate cells; total growth inhibitory concentration (TGI), when treated cells do not growth in relation to cells seeded in “time-zero” plate; and mean lethal concentration (LC50), when 50% cells died in relation observed cells concentration in “time-zero” plate. Cell proliferation was calculated as: % of control cell growth = 100 × (absorbance sample value − absorbance “time-zero” plate)/(absorbance negative control − absorbance “time-zero” plate) for 50% of cell proliferation; and absorbance sample value = absorbance “time-zero” plate for 0% of cell proliferation (Vichai and Kirtikara, 2006; NCI, 20219), and each drug concentration was related. The percentage of proliferation was set range to −100% to +100% to facilitate finding GI50 and LC50 values. These values can be observed in the screening methodology applied by USA-NCI (National Cancer Institute, 2021).
Glioma cell lines [HOG (CVCL_D354) and T98G (ATCC CRL-1690)], certified by Rede PREMIUM FMUSP, São Paulo, Brazil (ATCC – STR Profile Report method), were used. Both cell lines were cultured in RPMI media supplemented with 10% SBF and 1% penicillin-streptomycin and seeded in 96-well plates (6 × 103 cells per well). 24 h later, a plate designated by “time-zero,” containing only cells, was fixed with 100 μL/well of 10% trichloroacetic acid (TCA) and stored at 4°C for 1 h. Meanwhile, DHE and the epoxyketone-containing fraction, both dissolved in DMSO, were tested on the test plates at increasing concentrations (0.0032–50 μM or μg/mL) for 24, 48, and 72 h of incubation at 37°C and under a 5% CO2 atmosphere. As a negative control, i.e., 100% cell growth, 0.5% DMSO was used. After each incubation period, the medium of test plates was removed, cells were fixed with 100 μL/well of 10% TCA and plates were stored for 1 h at 4°C. This solution was removed and all plates were washed three times with deionized water. After that, 100 μL of 0.4% SRB solution was added to each well and kept at 37°C and 5% CO2 for 30 min. The SRB solution was removed by washing three times with 1% acetic acid and, to solubilize the bound SRB, fixed cells were dissolved in 200 μL/well of 10 mM Tris Base before the absorbance of each well was measured at 570 nm. Three independent experiments were performed. Mean growth inhibitory concentration (GI50), total growth inhibitory concentration (TGI), and mean lethal concentration (LC50) were calculated by interpolation of normalized data by GraphPad Prism 7.0. (GraphPad Software, Inc., San Diego, CA, United States).
Accumulation of ubiquitinated proteins and expression of HSP70 were evaluated by Western blotting as molecular markers of proteasome inhibition. With this purpose, HOG and T98G cells were seeded in 100 mm dishes at 1 × 106 cells/mL. After 24 h, the cells were treated with DHE or BRA-346 epoxyketone-containing fraction at concentrations corresponding to GI50 values obtained after 48 h incubation (1.6 and 1.7 ng/mL for DHE, and 17.6 and 28.2 ng/mL for the BRA-346 fraction for HOG and T98G, respectively). Incubation time was set at 36 h to evaluate the onset of the effects. DHE was further tested in both cell lines at 40 ng/mL, approximately 25 times the GI50 values obtained after 48 h incubation. Proteins of treated and untreated cells were extracted using a buffer containing 100 mM Tris (pH 7.6), 1% Triton X-100, 150 mM NaCl, 2 mM PMSF, 10 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, and 4 mM EDTA and their quantification was performed by the Bradford assay (Bradford, 1976) with a BSA curve (1 mg/mL) as standard. The proteins were submitted to SDS-PAGE then transferred to a nitrocellulose membrane for immunodetection. Antibodies against ubiquitin P4D1 (#3936) and α-tubulin (#2144) were obtained from Cell Signaling, while that against HSP70 (#648002) from BioLegend. Three independent experiments were performed. Band intensities were quantified by the UN-SCAN-IT gel 6.1 program that captures and quantifies the pixels in each band. Values associated to test proteins were normalized by standard α-tubulin for the relative expression measure.
HOG and T98G cells were seeded in 100 mm dishes at 1 × 106 cells/mL and 24 h later treated with DHE or BRA-346 epoxyketone-containing fraction at concentrations corresponding to GI50 values obtained after 48 h incubation (1.6 and 1.7 ng/mL for DHE, and 17.6 and 28.2 ng/mL for the BRA-346 fraction for HOG and T98G, respectively). Incubation time was set at 36 h to evaluate the onset of the effects. DHE was further tested in both cell lines at 40 ng/mL, approximately 25 times GI50 values obtained after 48 h incubation. For the quantitative PCR analysis, total RNA was extracted from cells using Trizol (as recommended by the manufacturer). Sixteen genes related to UPR and to the ubiquitin-proteasome system (UPS) were analyzed in control and treated cells. The primer sequences are described in Supplementary Table 4. RNA samples were quantified and used for the production of cDNA that was performed according to the High-Capacity cDNA Reverse Transcription Kit protocol (Thermo Fisher Scientific) with the addition of ribonuclease inhibitor RNaseOUTTM (Thermo Fisher Scientific), according to the following steps: 10′ at 25°C; 2 cycles from 60′ to 37°C and one cycle from 5′to 85°C. Sybr Green PCR Master Mix and solution of the forward and reverse primers were added to the cDNA. Hypoxanthine phosphoribosyltransferase 1 (HPRT1) and beta-actin (ACTB) were used as reference genes. Quantitative PCR (qPCR) was performed using an ABI 7500 Sequence Detector System and the qPCR reaction volume was 12 μL, which steps were 2′ at 50°C and 10′ at 95°C for the maintenance stage, 40 cycles of 15′ at 95°C and 1′ at 60°C for the PCR stage; and 15′ at 95°C, 1′ at 60°C and 15′ at 95°C for the melting curve stage. Three independent experiments were performed. For the analysis of relative expression, the values from the Cycle Threshold (CT) average of each gene were used and the values were normalized by the average reference genes values by the 2–ΔΔCT method (Livak and Schmittgen, 2001). The software MeV (Multiple Experiment Viewer10) was used to construct the heatmap. Network construction was performed using modulated genes in both glioma cell lines (1.4-fold-change) and the GeneMANIA database11.
Statistical analyses were performed using GraphPad InStat 7.0. (GraphPad Software, Inc., San Diego, CA, United States). For comparisons, ANOVA and Tukey multiple post-test were used. A p-value < 0.05 was considered statistically significant.
Since our prior study showed the cytotoxic potential of an extract obtained from the Brazilian endemic ascidian Euherdmania sp. (Figure 1A) (Jimenez et al., 2003), we proceeded with the isolation of four bacteria strains (Figure 1B and Supplementary Table 1) associated with the ascidian and evaluation of the cytotoxicity of their respective extracts in the colorectal carcinoma cells HCT-116. All tested extracts presented cytotoxicity with IC50 values varying from 0.03 (BRA-346) to 5.15 μg/mL (BRA-341) (Figure 1C and Supplementary Table 1). The extracts were further evaluated for their inhibitory potential against the proteasome chymotrypsin-like (ChTL) catalytic subunit (Figure 1C) for which only the extract from BRA-346 significantly inhibited this enzymatic activity, with IC50 of 0.42 μg/mL (Figure 1D).
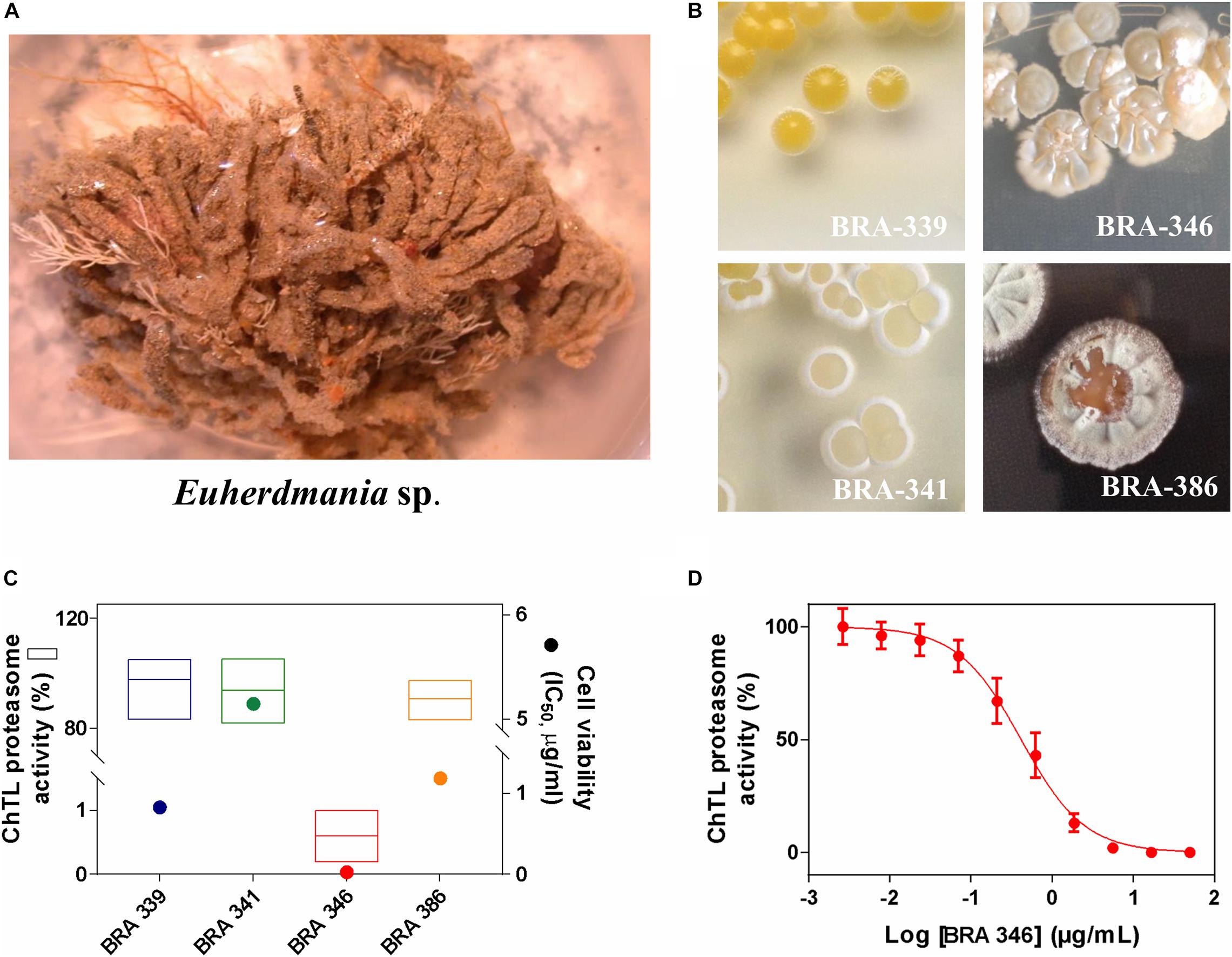
Figure 1. (A) Photo of the ascidian Euherdmania sp. collected on the northeast coast of Brazil. (B) Photographs of Euherdmania sp. associated bacteria: BRA-339, BRA-341, BRA-346 and BRA-386. (C) Graph showing the chymotrypsin-like (ChTL) enzymatic activity of proteasome purified from Saccharomyces cerevisiae (1 μg/mL) after 2 h of incubation with bacterial extracts at 50 μg/mL (left axis; n = 3) and IC50 values of the bacterial extracts against HCT-116 cell line assessed by MTT assay after 72 h of incubation (right axis; n = 3, in concentrations ranging from 0.0032 to 50 μg/mL). (D) Concentration-response curve of BRA-346 extract on the ChTL enzymatic (IC50 = 0.42 μg/mL) activity. The ChTL substrate (LLVY-AMC) was set at a concentration of 40 μM.
Therefore, BRA-346 was selected for further studies, including molecular identification of the bacteria, bioassay-guided fractionation of the extract and mechanism of action (MoA). Through 16S rRNA sequencing and comparison with the EzBioCloud database, BRA-346 was identified as Streptomyces sp. (MW342808), with close similarity (above 99%) to Streptomyces amphotericinicus (KX777593) (National Center for Biotechnology Information, 2020).
The BRA-346 strain was grown in liquid media and extracted with ethyl acetate. The crude extract was then subjected to a fractionation process guided by cytotoxicity against HCT-116 cell line, assessed through the MTT assay (Supplementary Figure 1 and Supplementary Table 2). Ultimately, fraction C, obtained after a SPE step applied to a mid-polarity fraction eluted from the ethyl acetate partition subjected to a Sephadex LH-20 column, was shown to produce the highest cytotoxicity, with IC50 of 30 ng/mL, being 20 times more potent than the second most active one, fraction B (Supplementary Figure 1), and was selected to the MoA studies.
Studies with BRA-346 fraction C were conducted to confirm the proteasome inhibitory activity and to access its chemical profile. Known proteasome inhibitors, including DHE and epoxomicin, were included in this analysis as positive controls. BRA-346 fraction inhibited 92.6% of the ChTL enzymatic activity at 1 μg/mL, while comparable inhibition of this enzyme by DHE and epoxomicin, by 94.6 and 93.9%, were reached at 12 and 16.6 μg/mL, respectively (Figure 2A), which is equivalent to nearly 30 μM of either compound. BRA-346 fraction showed an IC50 of 0.04 μg/mL (Figure 2B), a potency 10 times greater than that of the crude extract (IC50 = 0.42 μg/mL) when tested against the proteasome ChTL subunit.
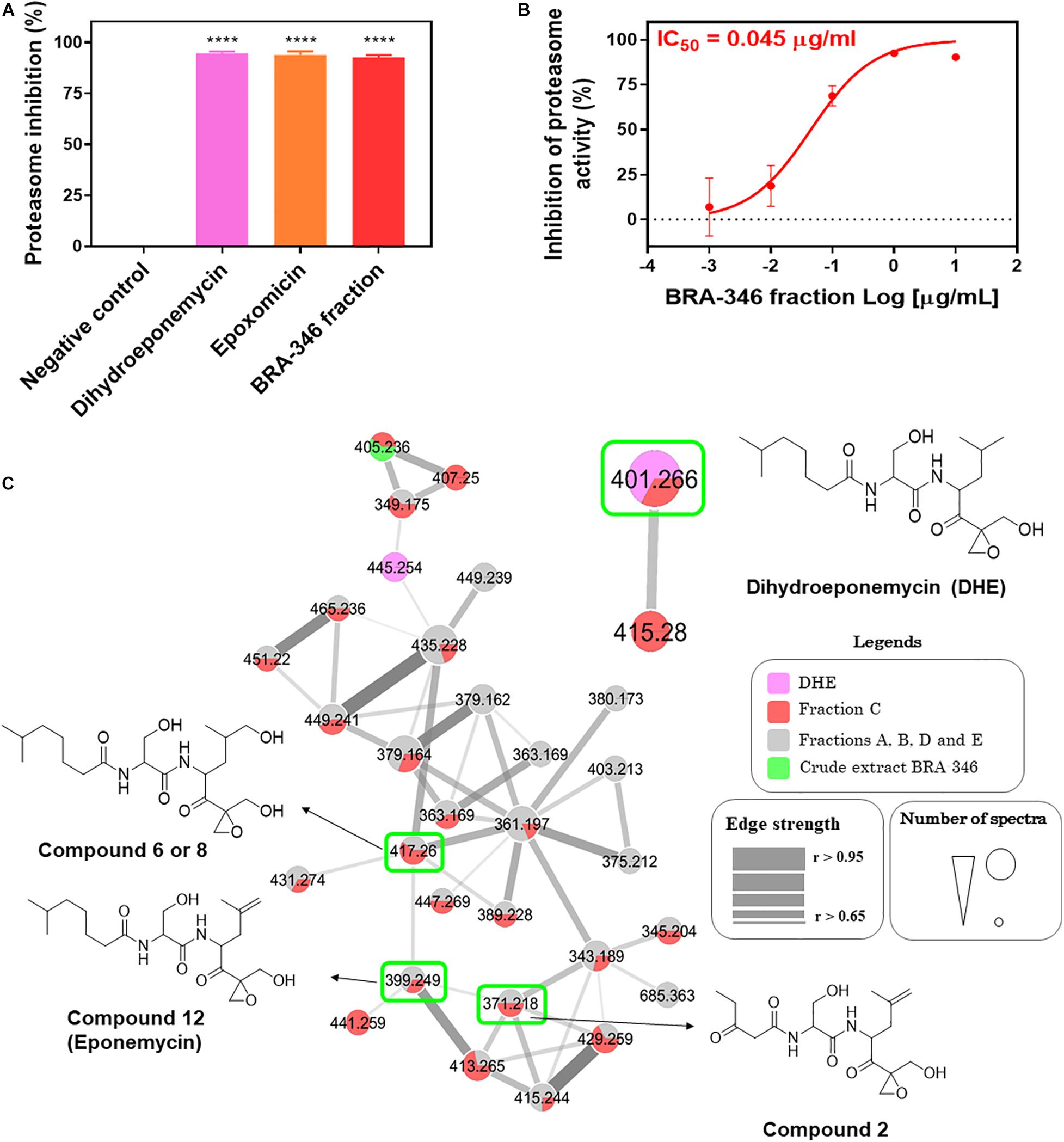
Figure 2. Evaluation of proteasome inhibition by dihydroeponemycin, epoxomicin, and BRA-346 epoxyketone-containing fraction, and molecular networking profile of proteasome inhibitors, BRA-346 crude extract, and fractions. (A) Comparative graph of inhibition of the catalytic activity of the Chymotrypsin-like (ChTL) subunit of the proteasome by dihydroeponemycin and epoxomicin (at 12 and 16.6 μg/mL, respectively), and BRA-346 fraction (fraction C Supplementary Figure 1) (at 1 μg/mL) through the enzymatic assay. Data were analyzed using GraphPad Prism 7.0 software by ANOVA followed by Tukey multiple comparison test, ****p-value < 0.0001. (B) Dose-response curve of the proteasome inhibition test by BRA-346 fraction, with IC50 value = 0.04 μg/mL, confidence interval (CI 95%) = 0.02 to 0.07. All values were obtained from three independent experiments. (C) Molecular network profile of dihydroeponemycin, BRA-346 crude extract and fractions A, B, C, D, and E. Compounds were analyzed by grouping from the fragmentation profile data of the compounds obtained by the ESI positive mode (ESI+). Only clusters related to dihydroeponemycin are shown. Nodes represent the ions present in the samples, which are indicated by related colors. Highlighted nodes refer to dihydroeponemycin (m/z 401.2) and analog ions present in the BRA-346 fractions.
HPLC-MS/MS analysis of the extract and fractions A, B, C, D, and E were conducted to investigate the chemical nature of the proteasome inhibitor present in the BRA-346 extract. Data was used to build a molecular network using the GNPS platform (Figure 2C). Two known proteasome inhibitors, DHE and epoxomicin, were included in the HPLC-MS/MS analysis for comparison. The fractionation was efficient and allowed the detection and identification of several compounds that were not observed in crude extract. The ion of m/z 401.26, compatible with DHE, was observed in DHE (Figure 2C) and exclusively in fraction C (Figure 2C and Supplementary Figure 2), while epoxomicin compatible signals were not detected in the extract or fractions. The molecular networking actually highlighted the presence of a large family of epoxyketone derivatives mainly in BRA-346 fraction C, but also in other fractions. Therefore, aiming at the identification of these compounds we decided to investigate their fragmentation pathway.
Present MS/MS data and that of Schorn et al. (2014) were used to propose the fragmentation pathways for DHE (Figure 3), and to guide the dereplication of compounds from the same family. As expected, the elimination of water makes up the picture of major ions, since there is an OH group in the central part of the molecule that can be easily eliminated due to its proximity to a more acidic alpha-carbonyl hydrogen. This route is represented in pathway C (Figure 3). The second water elimination could occur directly by a mechanism suggested in pathway E, however, a primary carbocation would have been formed, which does not occur due to its large instability. Thus, the alternative route goes through opening of the epoxide by an initial hydrogen rearrangement, affording an enol ion and presumably balancing the keto form (Li et al., 2007). Through this path, the elimination of a second water (m/z 365) can occur expending low energy. An additional loss of H2CO confirms the actuality of this pathway. Furthermore, elimination of an amide from the same ion at m/z 365, resulting in ion m/z 153, and the subsequent elimination of the side chain, affording ion at m/z 109, which is thermodynamically favored due to conjugations extended (Figure 3), will be useful to recognize additional derivatives. The ion at m/z 109 can also be produced directly after loss of water from the epoxide moiety (fragmentation mechanism is present at Supplementary Material, Supplementary Figure 4 and Supplementary Scheme 2).
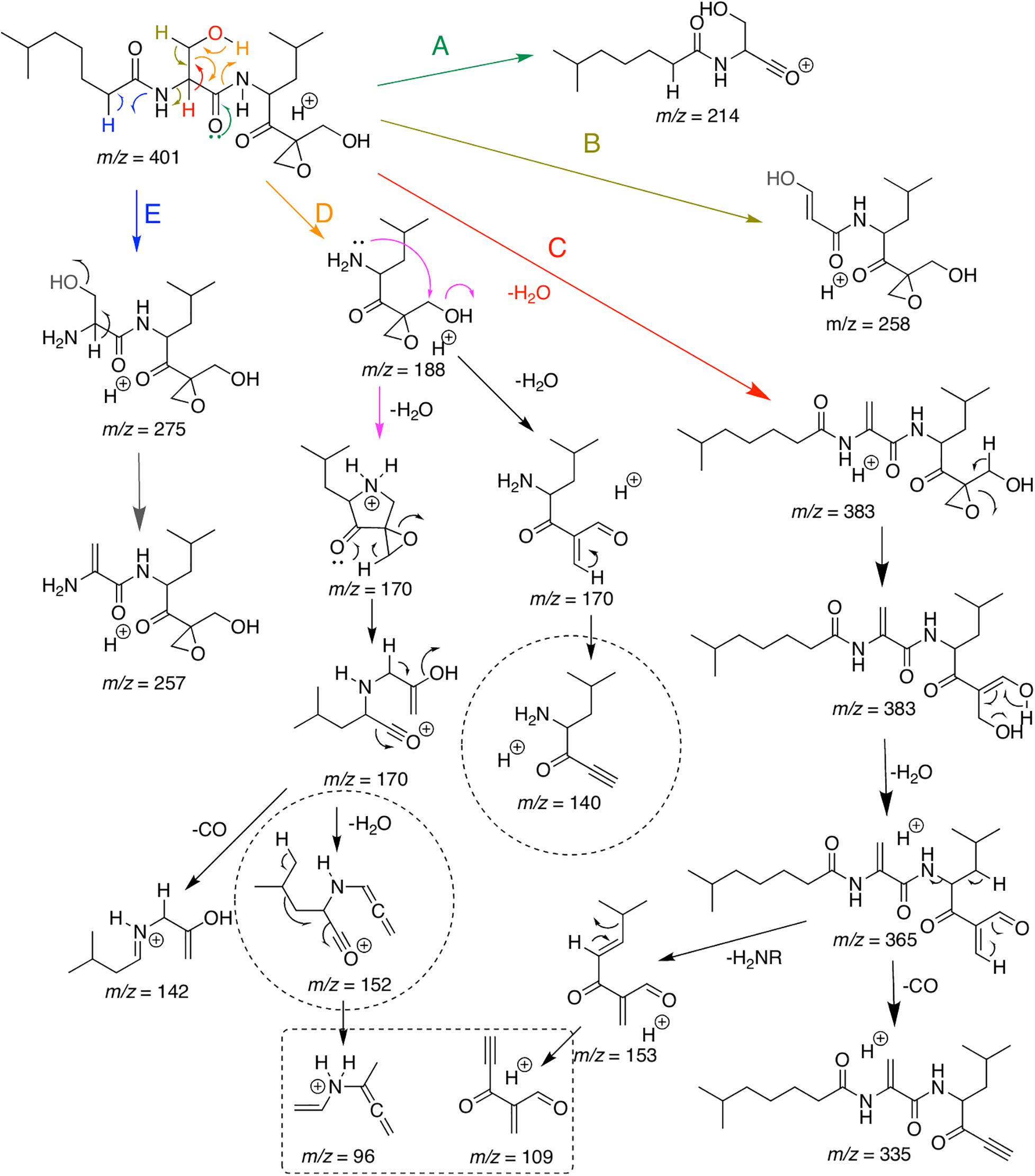
Figure 3. Major putative ions detected in ESI-MS/MS analysis of a commercial standard dihydroeponemycin and the ion at (retention time) tR 12.0 min (m/z 401). Analyses were carried out on Bruker Daltonic machines and the isolated protonated molecule was excited at a CID chamber applying N2 as the collision gas.
Fragmentation pathways A, B, D, and E also start as previously described (Schorn et al., 2014) and some important fragments with m/z 214 (A), m/z 258 (B), m/z 188 (D) m/z 275, and m/z 257 (E) can be useful to define possible modifications at the molecule moiety. Additional important information can be assessed by fragmentation of ion m/z 188. After elimination of the side chain through a retro-heteroene reaction (Crotti et al., 2005), the amine has freedom to undergo a long hand displacement reaction (McLafferty and Tureek, 1993), thus affording a water elimination product (ion at m/z 170, Figure 3). Now, two mechanisms for water elimination can occur from the same part of the molecule. The open-ring form of the ion at m/z 170 can lose H2CO affording a final ion at m/z 140, confirming, once again, opening of the epoxide ring. The cyclic ion at m/z 170 can be opened by an anchimeric assistance (Lopes et al., 2002) and, at the same concerted process, the epoxide is also opened. Sequential loss of water and of CO affords the ion at m/z 96, that can be helpful to find new derivatives in combination with the previously described ions.
Once the main fragmentation mechanisms were defined, we started to analyze the LC-UV-MS/MS data for the structural determination of possible derivatives (Figure 4 and Supplementary Figures 3–13 in the Supplementary Material shows de MS/MS data of all identified compounds). The first step was to confirm the spectrum of DHE (compound 13) detected in BRA-346 fraction C at retention time (tR) 12.0 min. The perfect spectral match between the commercial DHE and the ion at m/z 401 present in BRA-346 fraction C undoubtedly confirmed its occurrence (see spectra in Supplementary Figure 2). A substance with m/z 401 with two losses of water and the ion at m/z 109 with significant intensity (see spectra in Supplementary Figure 3) were observed at tR 10.6 min. This initial analysis suggests that it could be a slightly more polar isomer. Additionally, the second H2O elimination is the most intense ion in the spectrum (which does not occur in the DHE spectrum) and there is a lower intensity ion from a third H2O elimination. These information suggest an expected reduction of the carbonyl group in the side chain and a double-bond in the same position as eponemycin 12. MS/MS analysis also shows an ion at m/z 151, which confirms the double-bond, as proposed for compound 9. As expected, eponemycin (m/z 399) was detected at a lower tR (11.4 min) than DHE. Only the initial pathway A shown in Figure 3 could not be observed in the MS/MS analysis for eponemycin (Scheme 2 in Supplementary Material). Also, the ion at m/z 109 was formed directly from the ion at m/z 363 (losses of two molecules of water). The majority of other ions have two mass units less than the observed for DHE, confirming the chemical structure.
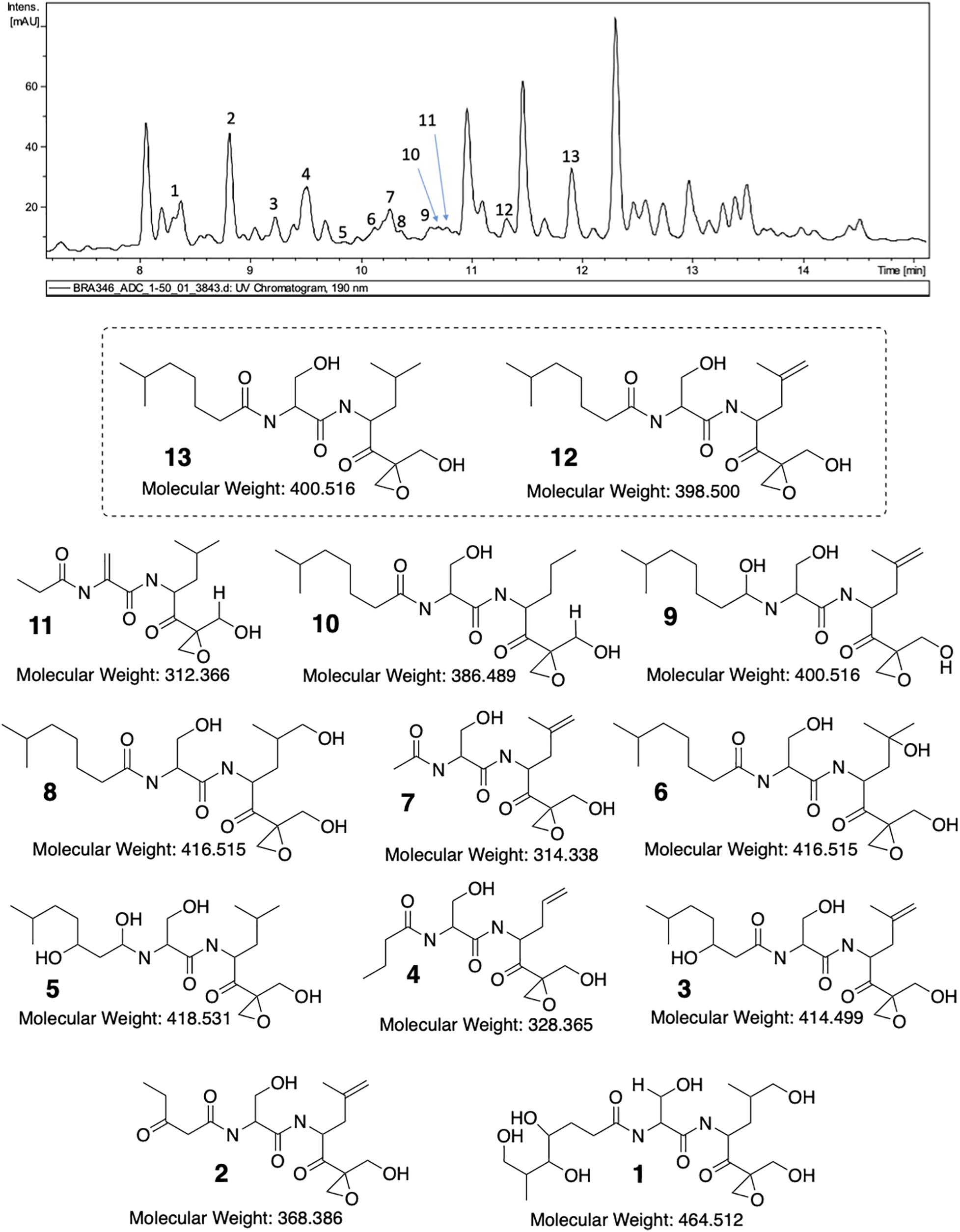
Figure 4. LC-MS/MS analysis of BRA-346 fraction C (BRA-346 epoxyketone-containing fraction) and proposed structures for eponemycin derivatives.
Previous work from Schorn et al. (2014) showed several analogs of eponemycin with lower side chains. In the present work compounds with tR at 9.0 min (compound 2, m/z 371), 9.6 min (compound 4, m/z 329), 10.2 min (compound 7, m/z 315), 10.7 min (compound 10, m/z 387), and 10.8 min (compound 11, m/z 313) showed spectral feature consistent with this family of compounds. For all compounds, the major fragmentation pathway starts with the loss of water and ends with the diagnostic ion at m/z 109 (see Supplementary Figure 10). Pathway A (green) presented in Scheme 3 (Supplementary Material) was important to confirm the size of the side chain and a double-bond replacing the hydroxyl group present in DHE. These information suggest N-(1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-yl)-2-propionamidoacrylamide as the structure for compound 11 (Figure 4). For compound 10, the fragment related to the side chain elimination was key for structure elucidation. Therein, demetallation may occur only at the isobutane section and the ion at m/z 109 also confirms that there are no modifications in the epoxide chain. Based on this, we propose compound 10 as N-(3-hydroxy-1-((1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-yl)amino)-1-oxopropan-2-yl)-6-methylheptanamide (Figure 4). Compound 7 has two more mass units than compound 11, but in this case, the hydroxyl group remains in the structure and the side chain has only one alpha carbonyl carbon. Therefore, it was not possible to observe elimination from the side chain and all ions were afforded from the initial pathway C (Scheme 6 in the Supplementary Material). In this case, the ion at m/z 109 was a minor signal, but all ions collectively allowed confirmation of compound 7 as 2-acetamido-3-hydroxy-N-(1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopent-4-en-2-yl)propanamide (Figure 4). Like 10, compound 4 also owns a demetallation at the same site, probably by an oxidation of the metabolites followed by CO elimination, resulting in a loss of carbon for the molecule moiety. Once again, pathway C is not held as the most important, but pathway D (Figure 3 and Supplementary Scheme 8) contributed to the final elucidation. The initial ion from pathway D has less restrictions and an additional ion can be observed at m/z 130. The mechanism of all other ions formation are in agreement with the proposition displayed in Figure 3 and we confirm the compound as N-(3-hydroxy-1-((1-(2-(hydroxymethyl)oxiran-2-yl)-1-oxopent-4-en-2-yl)amino)-1-oxopropan-2-yl)butyramide. Compound 2 is another compound with an eponemycin core, owning a small side chain and a second carbonyl group. The presence of a second carbonyl group was already described in some analogs annotated by Schorn et al. (2014). The ions observed in the spectra and the major fragmentation pathways (Supplementary Figure 12 and Supplementary Scheme 10, respectively, in Supplementary Material) are fully in agreement with a protonated ion of N-(3-hydroxy-1-((1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopent-4-en-2-yl)amino)-1-oxopropan-2-yl)-3-oxopentanamide.
The last group of compounds had higher mass values than eponemycin. In all cases, we observed the increased number of oxygen atoms and no modifications in the carbon skeleton. We observed two ions with m/z 417 with identical spectra and their only difference is the tR. Comparison to a previously published spectrum (Schorn et al., 2014) confirms the presence of the same major ions. Detailed analysis in Supplementary Figure 7 and Supplementary Scheme 5 indicate no other possibility than the expected addition of water to the double-bond. In this case, we can assume that one compound is similar to that previously published and the other is an isomer of position. Therefore, we believe that compound 6 can be the first to be eluted, but keeping in mind that this order can be reversed with compound 8. Compound 3 (3-hydroxy-N-(3-hydroxy-1-((1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopent-4-en-2-yl)amino)-1-oxopropan-2-yl)-6-methylheptanamide) has been previously identified (Schorn et al., 2014) and the ions observed herein are in full agreement (Supplementary Figure 11 and Supplementary Scheme 9). Compound 5, a minor signal in the HPLC-MS/MS analysis, has an additional hydroxyl group and an expected reduction of the carbonyl group in the side chain. Therefore, we verified the occurrence of pathway E (Supplementary Figure 9 and Supplementary Scheme 7) and the ions at m/z 275 and 255 confirmed we had an unmodified residue of 3-hydroxy-N-(1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-yl)propanamide. Moreover, the ion at m/z 109 reinforces this information. Hence, we propose compound 5 as 2-((1,3-dihydroxy-6-methylheptyl)amino)-3-hydroxy-N-(1-(2-(hydroxymethyl)oxiran-2-yl)-4-methyl-1-oxopentan-2-yl)propanamide. Finally, compound 1 showed the highest oxidation level for all series. In this case, the core is still the same as eponemycin and a new hydroxyl group was observed in the terminal isobutil group and in the side chain. While it is not possible to define the position of all groups, it is indeed possible to confirm the presence of a terminal hydroxyl group, which afforded the fragment ion from the heteroene reaction. So, a putative structure is presented in Figure 4 and details on spectrum and fragmentation pathways are offered in the Supplementary Figure 13 and Supplementary Scheme 11.
The activities of DHE and Streptomyces sp. BRA-346 epoxyketone-containing fraction were further evaluated in glioma cell lines HOG and T98G, since proteasome inhibitors are emerging as promising compounds in the treatment of glioblastoma multiforme, which remains a poorly manageable disease with high mortality rates (World Health Organization, 2020). The analysis was based on the assessment of cytotoxicity and the evaluation of molecular markers related to proteasome inhibition. Table 1 and Supplementary Figure 14 show the results of the SRB analysis after 24, 48, and 72 h incubation. Generally, growth inhibitory activity was amplified 10-fold when incubation time increased from 24 to 48 h for both DHE and BRA-346 epoxyketone-containing fraction but remained constant when the incubation time was further increased to 72 h. The sensitivity of both cell lines was similar for DHE, with 48 h incubation GI50 values of 1.6 and 1.7 ng/mL for HOG and T98G cells, respectively, while T98G cells were slightly less sensitive to BRA-346 epoxyketone-containing fraction, with GI50 of 28.2 ng/mL when compared to 17.6 ng/mL in HOG cells.
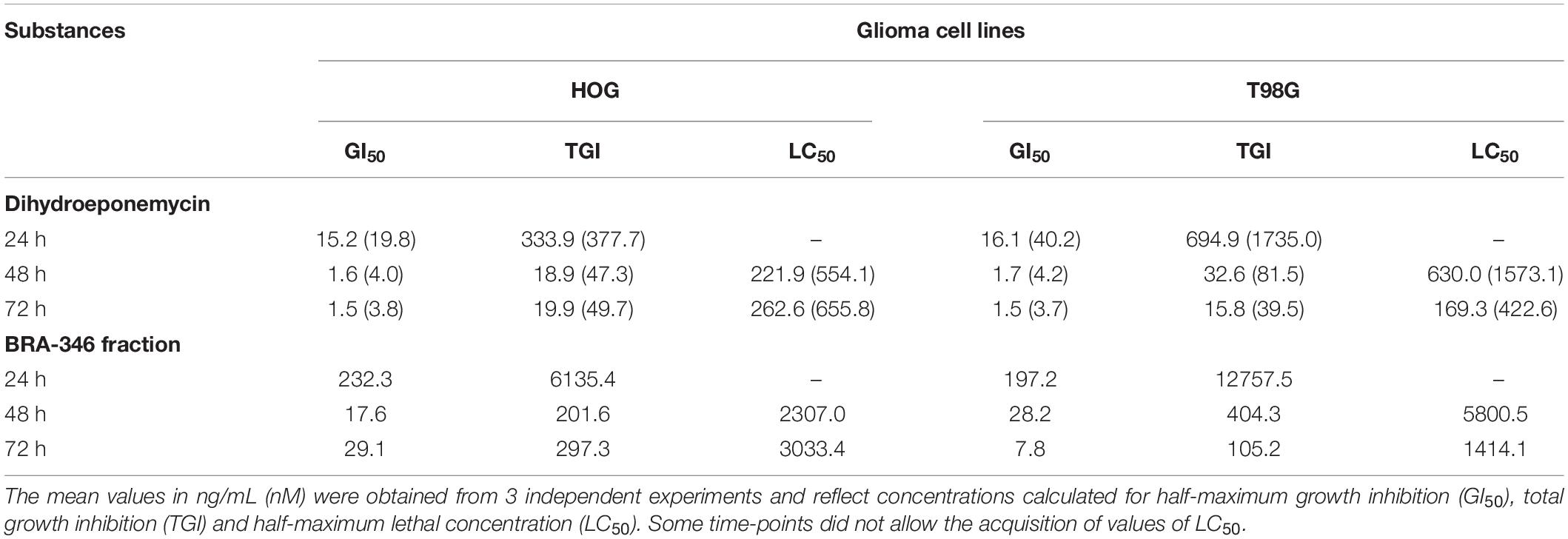
Table 1. Evaluation of cell growth inhibition of HOG and T98G cell lines exposed to dihydroeponemycin (DHE) and BRA-346 epoxyketone-containing fraction through SRB assay after 24, 48, and 72 h treatment.
To investigate the molecular changes incited by DHE and BRA-346 epoxyketone-containing fraction, glioma cells were exposed to the samples and incubated for 24, 36, or 48 h. Protein expression analysis demonstrated an increase in HSP70 and ubiquitinated proteins levels in both HOG and T98G cells treated with the BRA-346 epoxyketone-containing fraction, but no significant changes were observed when cells were treated with DHE at GI50 levels (Figures 5A,B and Supplementary Figure 15). Nonetheless, at concentration 25 times higher (40 ng/mL or 100 nM, Figure 5C) and 36 h of incubation, DHE induced comparable cellular effects with those of BRA-346 epoxyketone-containing fraction at 17.6 and 28.2 ng/mL in HOG and T98G cells, respectively (Figures 5A,C and Supplementary Figure 15).
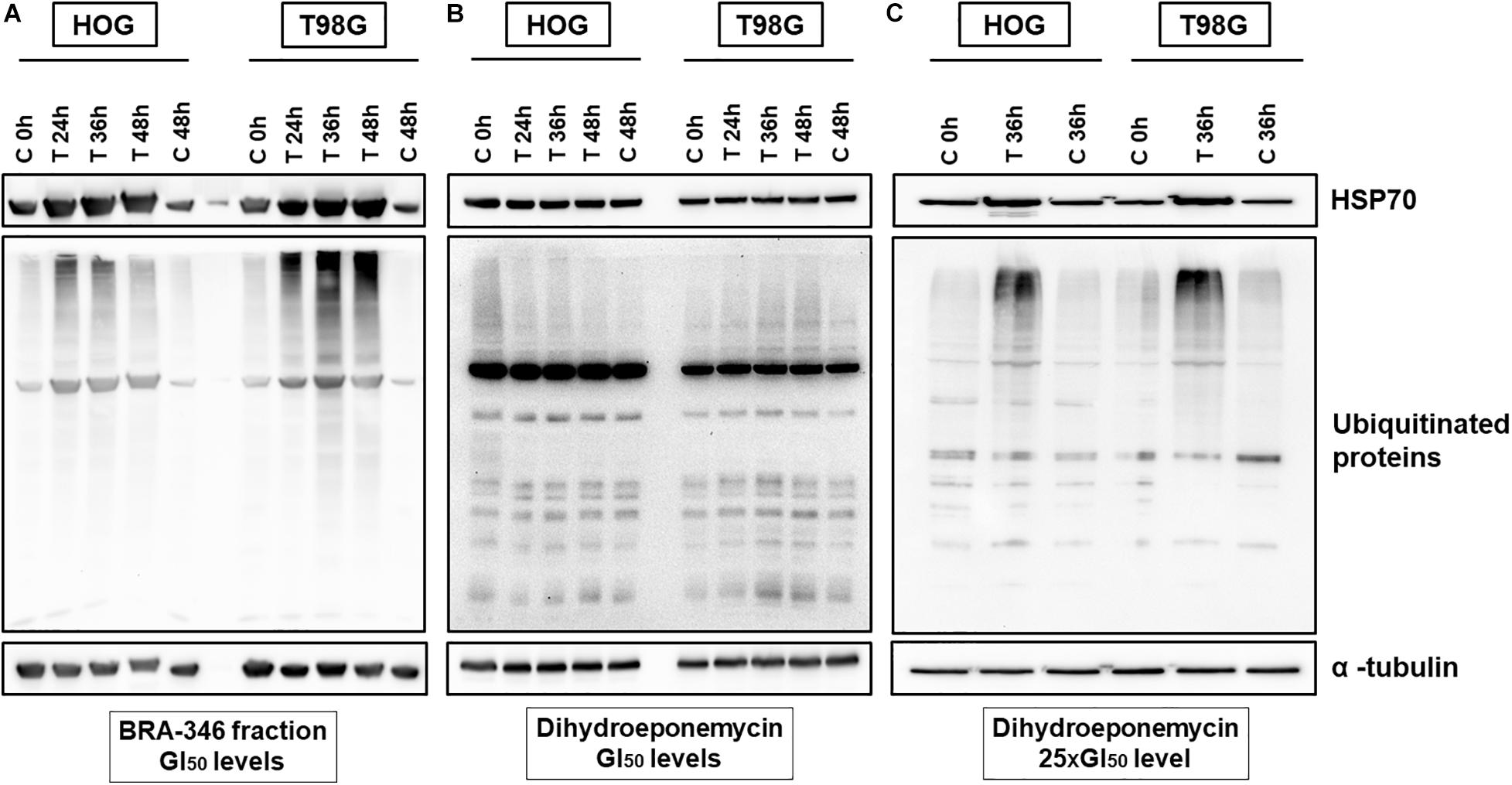
Figure 5. Dihydroeponemycin (DHE) and BRA-346 epoxyketone-containing fraction led to the accumulation of ubiquitinated proteins and HSP70 in glioma cell lines HOG and T98G. (A) Glioma cells treated with BRA-346 fraction at GI50 levels (17.6 and 28.2 ng/mL for HOG and T98G, respectively) for 3 different times of exposition (24, 36, and 48 h). (B) Glioma cells treated with DHE at GI50 levels (1.6 and 1.7 ng/mL for HOG and T98G, respectively) for 3 different times of exposition (24, 36, and 48 h). (C) Glioma cells treated with DHE at 40 ng/mL (100 nM) for 36 h of exposure. Negative controls (DMSO 0.1%) were set at 0, 36, and 48 h. Experiments were repeated three times.
We then turn to evaluate gene expression of treated cells, revealing a similar profile for both DHE at 25 times GI50 and BRA-346 epoxyketone-containing fraction (Figures 6A,C). DHE, at GI50 levels, had limited effects on the genes analyzed as also observed for protein alterations. Genes with a 1.4-fold-change were included in the network by GeneMANIA (see text footnote 11), which highlighted UPR as the main modulated mechanism for both DHE and BRA-346 fraction (Figures 6B,D). At 40 ng/mL, among the sixteen genes analyzed, DHE increased the expression of 9 and 13 genes in HOG and T98G cells, respectively (Figures 6B,D and Supplementary Table 5). BRA-346 epoxyketone-containing fraction, on the other hand, upregulated 12 and 5 genes at 17.6 and 28.2 ng/mL in HOG and T98G cell lines, respectively. Six genes were upregulated for both treatments in HOG – ATF6, CALR, DDIT3, HSPA4, HSPA5, and PPP1R15A – and five genes – CANX, DDIT3, HSPA5, MAPK8, and PPP1R15A – in T98G cells (Figure 7). The analysis of functions related to the modulated genes showed that both DHE and BRA-346-fraction are inducing a UPR phenotype, as expected by the inhibition of proteasome function, and ultimately leading treated cells to apoptosis (Figure 7).
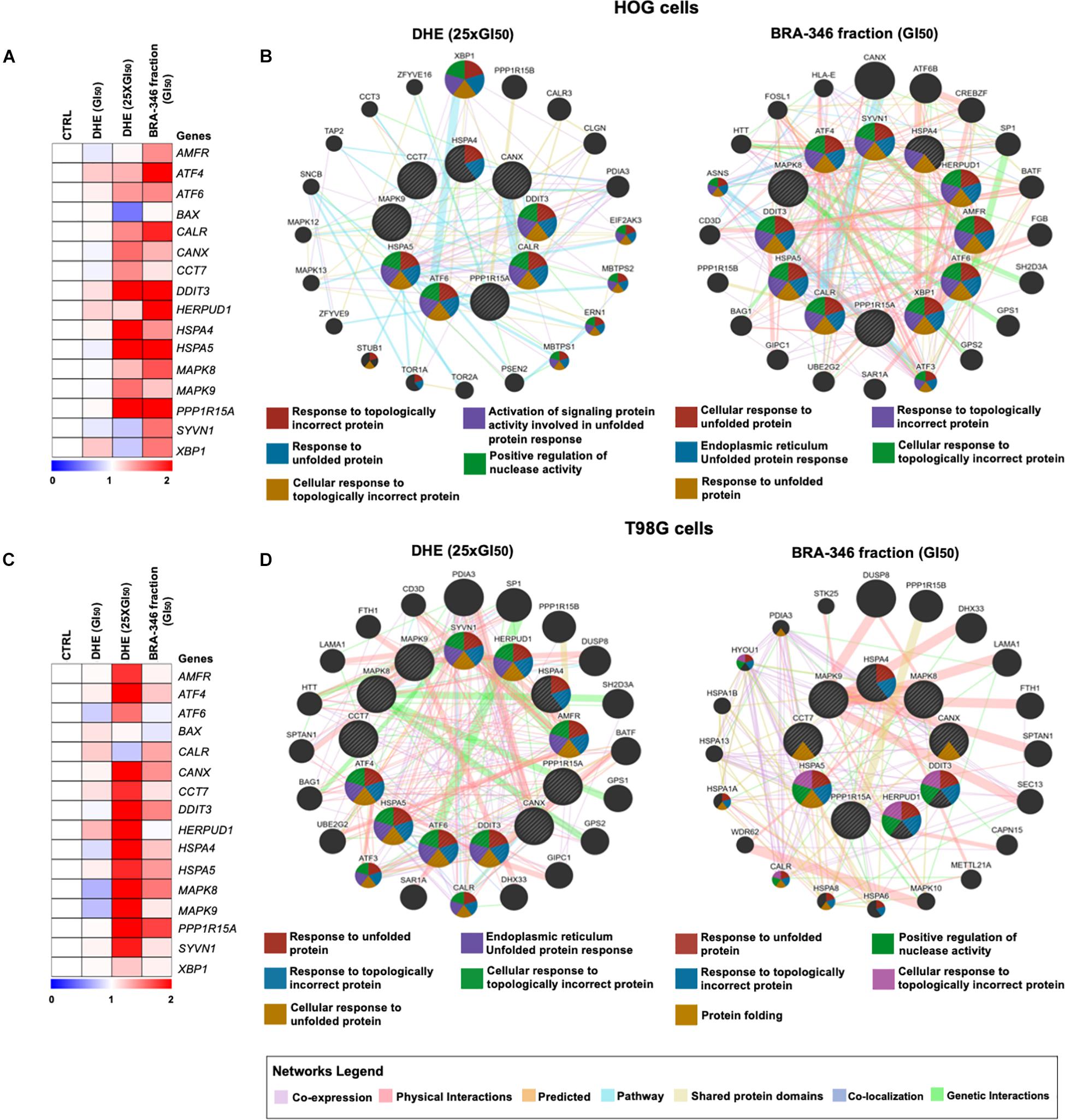
Figure 6. Molecular changes promoted by dihydroeponemycin (DHE) and BRA-346 epoxyketone-containing fraction in glioma cells. (A,C) Heatmap of gene expression of HOG and T98G cells treated with DHE and BRA-346 epoxyketone-containing fraction for 36h. DHE was evaluated at GI50 (n = 1) and 25xGI50 (40 ng/mL) (n = 3), while the BRA-346 fraction was tested at GI50 levels (17.6 and 28.2 ng/mL) (n = 3). Values for relative expression were established in comparison to control values and the heatmap was constructed by MeV 4.9.0 software (http://mev.tm4.org). All genes analyzed are related to ER-stress, except for BAX, which mostly relates to apoptosis. (B,D) The networks illustrate interactions of 1.4-fold-change in genes modulated by DHE at 40 ng/mL and BRA-346 epoxyketone-containing fraction (17.6 and 28.2 ng/mL) treatment in HOG and T98G. The up-regulated genes in the quantitative PCR analysis are illustrated as crosshatched circles and the interacting genes included by software modeling are indicated by non-crosshatched circles. The main networks and function of genes are indicated by colored lines and color in circles are described in the legend.
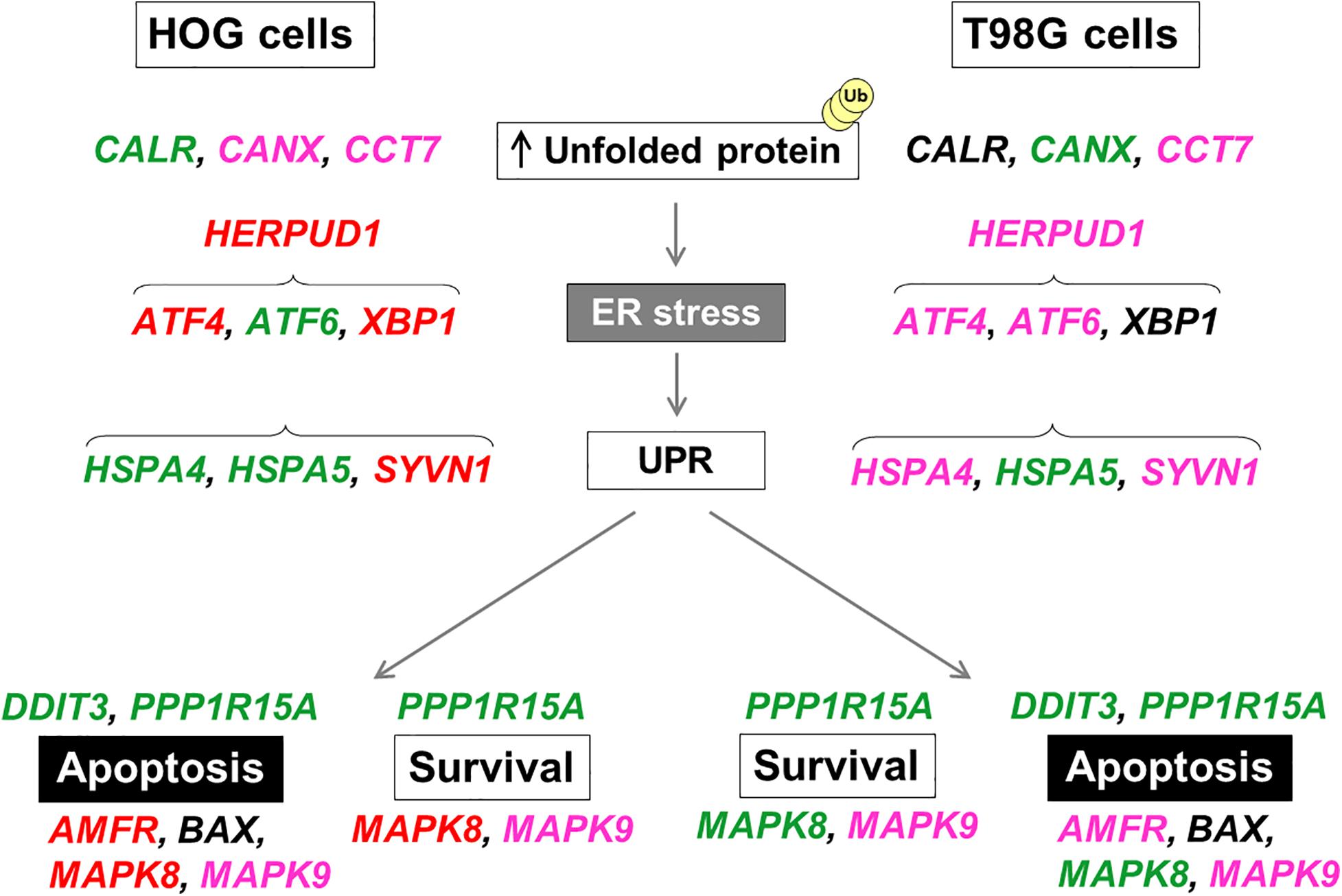
Figure 7. Schematic representation of upregulated UPR-related genes in HOG and T98G cells by DHE or BRA-346 epoxyketone-containing fraction. Each gene selected for validation acts on the unfolded protein response (UPR) and endoplasmic reticulum stress (ER-stress) and shows different regulation in HOG and T98G cells for each treatment. Treatments were performed with DHE at 40 ng/mL (both cell lines) and BRA-346 epoxyketone-containing fraction at 17.6 and 28.2 ng/mL for HOG and T98G, respectively. Genes in green were found to be upregulated following exposure to both DHE and BRA-346 epoxyketone-containing fraction; genes in red were upregulated only by BRA-346 epoxyketone-containing fraction; genes in pink were upregulated only by DHE; and genes in black were not upregulated. Data of relative expression were obtained from average values for each gene normalized by the average values for reference genes by the 2− ΔΔCT method from three experiments (Supplementary Table 5).
Marine bacteria have been studied in the last years due to their large potential to produce molecules with interesting biological activities, including antibacterial, antifungal, antitumor, anti-inflammatory, antimalaria, and antiviral actions (Manivasagan et al., 2014). The chemical diversity of such bioactive marine natural products range from peptides, alkaloids, terpenes, lactones, and steroids (Hu et al., 2015) to complex secondary metabolites that have offered new mechanisms of action to be untangled by pharmacological sciences (Reen et al., 2015). Of the four extracts tested, three displayed a relevant cytotoxicity against HCT-116 cells and may be promising sources of anticancer compounds. However, the extract derived from BRA-346, besides its high cytotoxicity, also presented a potent inhibition of proteasome ChTL activity. Proteasome inhibitors have been shown to be important pharmacological resources for the treatment of central nervous system tumors. Therefore, this extract was selected herein for a further investigation of its chemical composition and antiglioma potential.
Herein, we present a Streptomyces bacterium recovered from an ascidian collected in the northeast coast of Brazil, Euherdmania sp., produces antiproteasome compounds. Evaluation of the most potent fraction obtained through a bioassay-guided fractionation protocol of this extract revealed potent proteasome inhibitory properties along with the presence of a family of eponemycin related compounds, including dihydroeponemycin (DHE). Both DHE and the bacteria epoxyketone-containing fraction showed potent cytotoxicity against human oligodendroglioma and glioblastoma cell lines (HOG and T98G, respectively), resulting in significant changes in expression levels of genes and proteins related to ER-stress, unfolded protein response (UPR) and the ubiquitin-proteasome system (UPS).
The proteasome is a multicatalytic enzyme complex dedicated to recycling intracellular proteins. The main function of such organelle is to degrade damaged or unnecessary proteins, thus maintaining cell proteostasis (Voges et al., 1999; Bard et al., 2018). In the context of cancer, a malady inflicted by intense protein synthesis at the cellular level, the proteasome has been established as a valuable target, as its downmodulation implies poor handling of unwanted and unfolded proteins, along with further cell perturbations leading to cell-apoptosis (Manasanch and Orlowski, 2017; Jang, 2018). Importantly, proteasome inhibitors display a favorable therapeutic window, selectively killing cancer cells in humans, and are within the most tolerated chemotherapeutic options for multiple myeloma, for example. Proteasome inhibitors that reached the clinic are small peptides with an electrophilic warhead that can bind to the catalytic subunits of the proteasome and impair the proper function of the enzymatic complex (Micale et al., 2014; Park et al., 2018). Examples are bortezomib and carfilzomib, the latter belonging to the epoxyketone class, which was inspired by the actinobacteria-derived natural product epoxomicin. Many other bacterial natural products have been reported with these features, including carmaphycin, eponemycin, and other non-ribosomal peptides. These compounds form covalent bonds with threonine residues in the N-terminal region present in the catalytic active β subunits of the proteasome, leading to irreversible binding and, thus, inhibition of proteasome activity (Borissenko and Groll, 2007). Proteasome inhibitors use, including bortezomib and carfilzomib, is indicated for multiple myeloma patients, but many studies have highlighted its potential in the treatment of gliomas (Areeb et al., 2016; Wang et al., 2018; Tang et al., 2019). Nonetheless, the proper functioning of the ubiquitin-proteasome system is also relevant for other nervous system diseases, including neurodegenerative ones, where the accumulation of unfolded or mutated proteins generating protein aggregates and inclusion bodies trigger the death of neurons (Cacabelos, 2017; Momtaz et al., 2020). Thus, proteasome inhibitors may impact the therapy of many diseases, highlighting the importance of better understanding their mechanism of action and the pathways modulated by these compounds in treated cells.
The occurrence of the epoxyketone proteasome inhibitor, dihydroeponemycin (DHE), in fractions obtained from the Streptomyces sp. BRA-346 extract was suggested by comparison of the HPLC-MS/MS spectra with the commercially available DHE. In fact, detailed dereplication studies showed the presence of several ions related to eponemycin in BRA-346 fraction. Eponemycin was first isolated from the extract of the bacterium Streptomyces hygroscopicus, recovered from a soil sample from the Philippines, showing cytotoxicity toward HCT116 cells with IC50 value of 9.7 ng/mL (Sugawara et al., 1990). Herein, BRA-346 epoxyketone-containing fraction restrains proliferation of HCT116 cells, displaying IC50 value of 30 ng/mL. The activity of this fraction against the proteasome ChTL catalytic subunit was also confirmed by enzymatic assay, revealing an IC50 value of 45 ng/mL. DHE and tetrahydroeponemycin was first described as the product of hydrogenation of eponemycin over palladium carbon (Sugawara et al., 1990), and while DHE showed IC50 of 13 ng/mL in HCT116, tetrahydroeponemycin was not active at 1 μg/mL, reinforcing the importance of the epoxy group in the biological activity. DHE was also shown to inhibit the proteasome activity in micromolar concentrations (Meng et al., 1999). More recently, Schorn et al. (2014) described the biosynthetic gene cluster responsible for the production of eponemycin and, through heterologous expression in Streptomyces albus J1046, described several putative epoxyketone derivatives, including DHE. Herein, we compare the obtained MS/MS data of BRA-346 fraction C with that described by Schorn et al. (2014). As expected, a comparison of MS/MS data with previously published spectra shows there are slight differences in the initial fragmentation pathways (Schorn et al., 2014). Distinct ESI-MS/MS equipments can apply a variety of collision gases which results in a non-linear transfer of collisional energy in the CID chamber, therefore resulting in different numbers of initiating pathways since the protonated molecule (Demarque et al., 2016). All ions observed have their fragmentation pathways rationalized through the ESI-CID-MS/MS gas-phase chemistry concepts (Demarque et al., 2016). Thus, we started our analysis by proposing the expected fragmentation pathways from DHE and compared MS/MS data for the ions grouped in the same cluster, leading to the identification of thirteen eponemycin analogs in BRA-346 fraction C (designated, from now on, as BRA-346 epoxyketone-containing fraction), including DHE and eponemycin itself.
Meng et al. (1999) described that DHE antitumor activity, as also observed for structurally related compounds, is explained through inhibition of the proteasome function, but epoxyketone-containing antitumor natural products may differ in their antiproliferative activity, proteasome subunit binding specificity, and rates of proteasome inhibition. Considering the intriguing structure-activity relationship for this class of compounds (Kim et al., 1999; Groll et al., 2000) and the emerging knowledge on the antiglioma effects of the proteasome inhibitors (Di et al., 2016; Zhang et al., 2018; Veggiani et al., 2019), we further assayed DHE and BRA-346 epoxyketone-containing fraction in glioma cells HOG and T98G, revealing their potent antiproliferative effects in the ng/mL-range and the modulation of ER-stress related and pro-apoptotic related genes, along with unfolded protein response and the ubiquitin-proteasome system itself.
BRA-346 epoxyketone-containing fraction at GI50 levels promoted an increased expression of HSP70 and ubiquitinated proteins, while DHE required higher concentrations, corresponding to 25 times the GI50, to achieve a comparable effect. Accumulation of ubiquitinated proteins is regarded as one hallmark feature of proteasome inhibition, since misfolded proteins are labeled by multiple ubiquitin molecules to be degraded by the proteasome (Myung et al., 2001). Therefore, proteasome inhibitors prevent the degradation of ubiquitinated proteins, promoting an accumulation of these in the cytoplasm, as demonstrated in HUVECs and neuronal cells (Canu et al., 2000; Myeku and Figueiredo-Pereira, 2011). Furthermore, HSP70 is a family of chaperones related to proper functioning and maintenance of the cell as it supports the folding of new proteins, improving the transport of proteins out of the ER. The high expression levels of such chaperones in both cell lines promoted by proteasome inhibitors, as observed herein for both DHE and BRA-346 epoxyketone-containing fraction, can be interpreted as the ultimate attempt to restore homeostasis by handling excessive unwanted proteins, which, in turn, may be a consequence of proteasome inhibition (Bush et al., 1997; Mayer and Bukau, 2005). The difference in the potency of DHE and the BRA-346 fraction, considering the relation of GI50 levels and the accumulation of ubiquitinated proteins, may be explained by the presence of several epoxyketone in the fraction, some owning, supposably, higher potencies that enhance the observed effects. The presence of other compounds not related to eponemycin cannot be excluded at this moment, since the studied fraction is indeed very complex.
That said, we then turned to evaluate the effects of DHE and BRA-346 epoxyketone-containing fraction on genes related to pathways that showed modulation of ER-stress response in gliomas (Markouli et al., 2020). While notable differences were observed between responses elicited by each cell line, a similar molecular profile, in which most of the assessed genes were found to be upregulated by both DHE and the BRA-346 epoxyketone-containing fraction was revealed, reflecting a compensatory mechanism undergoing in cells exposed to either treatment to overcome ER disturbances (Kadowaki and Nishitoh, 2013). Figure 7 illustrated the genes upregulated for both treatments (in green) or individually (DHE: pink; BRA-346 fraction: red) in both cell lines. Despite slight differences, the main functions impaired by either treatment are related to unfolded protein response (UPR) in glioma cell lines. Indeed, UPR is triggered by an overload of unfolded proteins in the ER lumen and accounts, in principle, for a transitory state aimed at re-establishing normal ER function, mainly characterized by synthesis, maturation, and export of proteins (Walter and Ron, 2011). Still, as many natural compounds have been shown to induce transitory or sustained UPR, we wish to highlight that to the best of our knowledge, this is the first report on the effects of DHE on glioma cells assessed to this extent and, moreover, in comparison to a complex natural fraction containing a myriad of structurally related epoxyketones.
HSPA5 refers to the gene that encodes GRP78 or BiP protein, a key chaperone for newly synthesized proteins, supporting co-translational modifications, which is an essential component of the UPR and an important target to cancer therapy (Wang et al., 2009; Lu et al., 2020). As a matter of fact, GRP78, a member of the HSP70 family of proteins, is the common initial response that detaches and activates the downstream ER-stress sensors, therefore a typical marker to UPR (Kopp et al., 2019). However, different cells may, indeed, launch then handle UPR in distinct manners, even if subjected to similar stressors, as the intrinsic capacity for protein synthesis varies among cell types (Bertolotti et al., 2000; Carrara et al., 2013). In thyroid cancer cells, proteasome inhibitors such as PSI, epoxomicin and MG132 induced upregulation of GRP78 and CHOP at mRNA and protein levels (Wang et al., 2007). HSPA4, in turn, encodes a large heat-shock chaperone from the HSP110 family, HSPA4 (also referred to as Apg-2), which works in concert with HSP70s to restore proper protein folding activity and counter ER-stress (Zuo et al., 2016). While HSPA4 has not been as broadly assessed as their closely related HSP70 assemblage, it has been associated to maintenance of neuronal functions under stress conditions (Okui et al., 2000) and a significant correlation was observed between the expression of this chaperone and increased levels of the antiapoptotic marker Bcl-2 in inflammatory cells, further suggesting an interplay of the protein in cell survival pathways (Adachi et al., 2015). Herein, both genes, HSPA4 and HSPA5, which respond to misfolded or unfolded proteins, as shown in the networks in Figure 6, were found upregulated in HOG cells exposed to either DHE or BRA-346 epoxyketone-rich fraction, indicating these cells are under proteotoxic stress due to proteasome malfunction and signalizing an attempt to neutralize misfolded proteins. In T98G cells, however, expression of HSPA4 was not affected by the treatments. Thus, it may be suggested that T98G cells represent a more resistant cell line to ER-stress elicited by compounds of this chemical class. Indeed, the product of the XBP1 gene, a typical downstream response of the dimerization and activation of the ER transmembrane protein IRE-1 UPR sensor further stimulating the UPR (Ron and Walter, 2007; Kadowaki and Nishitoh, 2013), also remained mostly unaltered in T98G cells.
PERK is a second transmembrane protein that acts as a sensor for ER-stress and, mainly, blocks general protein translation and cell cycle while also stimulating antioxidant systems to compensate for ER saturation. ATF4, a transcription factor encoded by the gene ATF4, is a key downstream marker for activation of this pathway which can contribute to survival or cell death (Okada et al., 2002; Iurlaro and Muñoz-Pinedo, 2016; Rozpedek et al., 2016). Studies highlighted the modulation of PERK-eIF2a-ATF4 pathway by proteasome inhibitors like bortezomib in human myeloma and squamous carcinoma cell lines, evidencing that this class of compounds may activate UPR-related molecular markers (Fribley et al., 2004; Obeng et al., 2006). Still, prolonged ER-stress can lead to apoptosis in cancer cells, for these overturn the survival process into a cell death trigger (Jimbo et al., 2003; Kim and Kim, 2018). In this sense, the PPP1R15A gene, found to be upregulated in both cell lines following exposure to either treatment, encodes the GADD34 protein. Its expression is stress-dependent and elicited following phosphorylation of the initiation factor eIF2a, a key event to activate UPR. Once GADD34 levels rise, it binds protein phosphatase 1 (PP1) to dephosphorylate eIF2a, generating a feedback loop that reverses eIF2a signalization (Reid et al., 2016). GADD34 is essential for progression of UPR as it has been shown that sustained activation of eIF2a drives cells out of UPR and, therefore, out of survival mode. Therefore, its increase in cellular levels is related to both cell survival and death, as its transcription is amplified in an attempt to normalize protein synthesis, prevented by the PERK pathway. The reversal of the inhibition of the translation process leads not only to the production of apoptotic proteins, but also to those related to cell continuity (Ma and Hendershot, 2003; Adams et al., 2019; Féral et al., 2021). The activation of this pathway reinforces the importance of proteasome inhibitors as an interesting option for cancer therapy.
At treatment level, the distinction between the expression of genes CALR and CANX, can be noted; while the BRA-346 fraction mostly increased CARL, DHE enhanced CANX. These genes encode calreticulin and calnexin, respectively, which are ER-resident Ca2+-binding chaperones involved in protein folding (Wang et al., 2012). Inhibition of the proteasome can modulate these proteins, promoting an increase in apoptosis mediated by exposure to calreticulin, as observed in glioma and multiple myeloma cell lines treated with bortezomib and carfilzomib, respectively (Jarauta et al., 2016; Comba et al., 2019). In fact, increased expression of calreticulin is associated, mostly, to the third arm for eliciting UPR, that is the pathway initiated by ATF6, an ER transmembrane protein which is translocated to be cleaved in the Golgi apparatus into a functional transcription factor (Hetz, 2012). Therefore, ATF4, ATF6, and XPB1 are transcription factors in close relation within UPR pathways and all three branches may be activated in a situation of ER-stress like these presented herein. Indeed, throughout treatments and cells lines, an evident and consistent upregulation of DDIT3, a gene that encodes the homologous protein CHOP, a pro-apoptotic factor connected majorly to the PERK pathway is as well, even if to a lower extent, downstream to the ATF6 and to IRE-1 branches (Fribley et al., 2009; Iurlaro and Muñoz-Pinedo, 2016). Furthermore, the increase in CHOP, by bortezomib, amplifies cytotoxicity in multiple myeloma and breast cancer cells when combined with inhibitors of early and intermediate autophagy processes, modulating apoptosis and the autophagic pathway (Kawaguchi et al., 2011; Komatsu et al., 2012). Actually, autophagy, the lysosome-mediated degradation of defective proteins, protein aggregate and even damaged organelles, is also included among cell survival strategies during ER stress, and is actively interconnected to UPR (Rashid et al., 2015). The changeover signal between pathways, however, is largely unknown. Still, besides CHOP, MAPK8 and closely related MAPK9, also known by JNK-1 and JNK-2, respectively, may be partly to blame in such a process and the duration of the signaling may be determinant cell fate (Wei et al., 2008). Interestingly, the treatments employed herein have distinguished between the expression of both these genes, as only DHE notably increased expression of MAPK9, but MAPK 8 increased under either treatment.
Nonetheless, unrepaired or aggravation of ER-stress promotes cell death for the benefit of the organism and, in such a situation, UPR is no longer portrayed as a survival mechanism (Tabas and Ron, 2011; Hetz, 2012; Urra et al., 2013). Alike launching and handling, there is also variability considering the stress levels a particular cell type can endure before veering UPR to apoptosis (Luo and Lee, 2013; Peñaranda Fajardo et al., 2016). In this sense, overactivation of the UPR, either through proteasome inhibition or by other mechanisms, has been a recurring subject of investigation and also used therapeutically to treat cancers. Moreover, many natural products have been shown to induce ER-stress-mediated apoptosis (Kim and Kim, 2018), highlighting these compounds for their remarkable anticancer potential.
Herein, we described the recovery of the ascidian-associated bacterium Streptomyces BRA-346 that produces dihydroeponemycin and related compounds with proteasome inhibitory activity. Furthermore, we described the potent cytotoxicity and the pathways modulated by DHE and BRA-346 epoxyketone-containing fraction in glioma cell lines. While the present study advances the knowledge on the mode of action of DHE in gliomas cells, reinforcing that the antiproteasome effect is indeed in play thereof, then leading to ER-stress reflected by manifestation of UPR, it also reveals comparable signaling pathways are activated following exposure of cells to the epoxyketone-rich fraction. Nonetheless, at this point, we cannot exclude the participation of other minor compounds, including dihydroeponemycin derivatives, also present in the fraction in the displayed cellular effects.
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW342808.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This research was funded by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Finance code 001), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, 306913/2017-8; 465637/2014-0; and 443281/2019-0), and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, grant numbers 2015/17177-6; 2017/18235-5; 2017/17648-4; and 2018/24865-4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Helori Vanny Domingos and Simone Aparecida Teixeira for technical assistance. The license for the collection of tunicates was granted by Biodiversity Authorization and Information System (SISBIO, authorization number 48522-2). The license for genetic access was granted by the National System for the Management of Genetic Heritage and Associated Traditional Knowledge (SISGen, authorization number AC0781C). We are grateful to Professor Tito Monteiro da Cruz Lotufo from Oceanography Institute, University of São Paulo for providing the photo of ascidian Euherdmania sp. (Figure 1A).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.644730/full#supplementary-material
Adams, C. J., Kopp, M. C., Larburu, N., Nowak, P. R., and Ali, M. (2019). Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mole. Biosci. 6:11.
Adachi, T., Sakurai, T., Kashida, H., Mine, H., Hagiwara, S., Matsui, S., et al. (2015). Involvement of heat shock protein a4/apg-2 in refractory inflammatory bowel disease. Inflamm. Bowel Dis. 21, 31–39. doi: 10.1097/MIB.0000000000000244
Alonso-Álvarez, S., Pardal, E., Sánchez-Nieto, D., Navarro, M., Caballero, M. D., Mateos, M. V., et al. (2017). Plitidepsin: design, development, and potential place in therapy. Drug Design Dev. Ther. 11, 253–264.
Areeb, Z., Stylli, S. S., Ware, T. M., Harris, N. C., Shukla, L., Shayan, R., et al. (2016). Inhibition of glioblastoma cell proliferation, migration and invasion by the proteasome antagonist carfilzomib. Medical Oncol. 33:53. doi: 10.1007/s12032-016-0767-3
Bard, J., Goodall, E. A., Greene, E. R., Jonsson, E., Dong, K. C., and Martin, A. (2018). Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 87, 697–724.
Bauermeister, A., Branco, P. C., Furtado, L. C., Jimenez, P. C., Costa-Lotufo, L. V., and Lotufo, T. M. C. (2019). Tunicates: A model organism to investigate the effects of associated-microbiota on the production of pharmaceuticals. Drug Dis. Today: Dis. Models 28, 13–20. doi: 10.1016/j.ddmod.2019.08.008
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Borissenko, L., and Groll, M. (2007). 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 107, 687–717. doi: 10.1021/cr0502504
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Bush, K. T., Goldberg, A. L., and Nigam, S. K. (1997). Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J. Biol. Chem. 272, 9086–9092. doi: 10.1074/jbc.272.14.9086
Cacabelos, R. (2017). Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mole. Sci. 18:551. doi: 10.3390/ijms18030551
Canu, N., Barbato, C., Ciotti, M. T., Serafino, A., Dus, L., and Calissano, P. (2000). Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J. Neurosci. Off. J. Soc. Neurosci. 20, 589–599. doi: 10.1523/JNEUROSCI.20-02-00589.2000
Carrara, M., Prischi, F., and Ali, M. M. (2013). UPR Signal Activation by Luminal Sensor Domains. Int. J. Mole. Sci. 14, 6454–6466. doi: 10.3390/ijms14036454
Catalgol, B. (2012). Proteasome and cancer. Prog. Mole. Biol. Transl. Sci. 109, 277–293. doi: 10.1016/B978-0-12-397863-9.00008-0
Cheng, M. M., Tang, X. L., Sun, Y. T., Song, D. Y., Cheng, Y. J., Liu, H., et al. (2020). Biological and Chemical Diversity of Marine Sponge-Derived Microorganisms over the Last Two Decades from 1998 to 2017. Molecules 25:853. doi: 10.3390/molecules25040853
Comba, A., Bonnet, L. V., Goitea, V. E., Hallak, M. E., and Galiano, M. R. (2019). Arginylated Calreticulin Increases Apoptotic Response Induced by Bortezomib in Glioma Cells. Mole. Neurobiol. 56, 1653–1664. doi: 10.1007/s12035-018-1182-x
Crotti, A. E., Lopes, J. L., and Lopes, N. P. (2005). Triple quadrupole tandem mass spectrometry of sesquiterpene lactones: a study of goyazensolide and its congeners. J. Mass Spectr. 40, 1030–1034. doi: 10.1002/jms.877
Cutignano, A., Nuzzo, G., Ianora, A., Luongo, E., Romano, G., Gallo, C., et al. (2015). Development and Application of a Novel SPE-Method for Bioassay-Guided Fractionation of Marine Extracts. Mar. Drugs 13, 5736–5749. doi: 10.3390/md13095736
Demarque, D. P., Crotti, A. E., Vessecchi, R., Lopes, J. L., and Lopes, N. P. (2016). Fragmentation reactions using electrospray ionization mass spectrometry: an important tool for the structural elucidation and characterization of synthetic and natural products. Nat. Product Rep. 33, 432–455. doi: 10.1039/c5np00073d
Di, K., Lloyd, G. K., Abraham, V., MacLaren, A., Burrows, F. J., Desjardins, A., et al. (2016). Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro-Oncol. 18, 840–848. doi: 10.1093/neuonc/nov299
Feling, R. H., Buchanan, G. O., Mincer, T. J., Kauffman, C. A., Jensen, P. R., and Fenical, W. (2003). Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angewandte Chemie 42, 355–357. doi: 10.1002/anie.200390115
Féral, K., Jaud, M., Philippe, C., Di Bella, D., Pyronnet, S., Rouault-Pierre, K., et al. (2021). Stress and Unfolded Protein Response in Leukemia: Friend, Foe, or Both? Biomolecules 11:199. doi: 10.3390/biom11020199
Food and Drug Administration (2020). Novel Drug Approvals for 2020. Available online at: https://www.fda.gov/ [accessed September 10, 2020]
Fribley, A., Zeng, Q., and Wang, C. Y. (2004). Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mole. Cell. Biol. 24, 9695–9704. doi: 10.1128/MCB.24.22.9695-9704.2004
Fribley, A., Zhang, K., and Kaufman, R. J. (2009). Regulation of apoptosis by the unfolded protein response. Methods Mole. Biol. 559, 191–204.
Groll, M., Bajorek, M., Köhler, A., Moroder, L., Rubin, D. M., Huber, R., et al. (2000). A gated channel into the proteasome core particle. Nat. Struct. Biol. 7, 1062–1067. doi: 10.1038/80992
Hanada, M., Sugawara, K., Kaneta, K., Toda, S., Nishiyama, Y., Tomita, K., et al. (1992). Epoxomicin, a new antitumor agent of microbial origin. J. Antib. 45, 1746–1752. doi: 10.7164/antibiotics.45.1746
Heuer, H., Krsek, M., Baker, P., Smalla, K., and Wellington, E. M. (1997). Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microb. 63, 3233–3241. doi: 10.1128/AEM.63.8.3233-3241.1997
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mole. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hotokezaka, Y., Tobben, U., Hotokezaka, H., Van Leyen, K., Beatrix, B., Smith, D. H., et al. (2002). Interaction of the eukaryotic elongation factor 1A with newly synthesized polypeptides. J. Biol. Chem. 277, 18545–18551. doi: 10.1074/jbc.M201022200
Hu, Y., Chen, J., Hu, G., Yu, J., Zhu, X., Lin, Y., et al. (2015). Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 13, 202–221. doi: 10.3390/md13010202
Iurlaro, R., and Muñoz-Pinedo, C. (2016). Cell death induced by endoplasmic reticulum stress. FEBS J. 283, 2640–2652. doi: 10.1111/febs.13598
Jang, H. H. (2018). Regulation of Protein Degradation by Proteasomes in Cancer. J. Cancer Prevent. 23, 153–161. doi: 10.15430/JCP.2018.23.4.153
Jarauta, V., Jaime, P., Gonzalo, O., de Miguel, D., Ramírez-Labrada, A., Martínez-Lostao, L., et al. (2016). Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 382, 1–10. doi: 10.1016/j.canlet.2016.08.019
Jimbo, A., Fujita, E., Kouroku, Y., Ohnishi, J., Inohara, N., Kuida, K., et al. (2003). ER stress induces caspase-8 activation, stimulating cytochrome c release and caspase-9 activation. Exp. Cell Res. 283, 156–166. doi: 10.1016/s0014-4827(02)00033-2
Jimenez, P. C., Fortier, S. C., Lotufo, T. M. C., Pessoa, C., Moraes, M. E. A., de Moraes, M. O., et al. (2003). Biological activity in extracts of ascidians (Tunicata, Ascidiacea) from the northeastern Brazilian coast. J. Exp. Mar. Biol. Ecol. 287, 93–101. doi: 10.1016/S0022-0981(02)00499-9
Jimenez, P. C., Wilke, D. V., Branco, P. C., Bauermeister, A., Rezende-Teixeira, P., Gaudêncio, S. P., et al. (2020). Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharm. 177, 3–27. doi: 10.1111/bph.14876
Kadowaki, H., and Nishitoh, H. (2013). Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 4, 306–333. doi: 10.3390/genes4030306
Kawaguchi, T., Miyazawa, K., Moriya, S., Ohtomo, T., Che, X. F., Naito, M., et al. (2011). Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int. J. Oncol. 38, 643–654. doi: 10.3892/ijo.2010.882
Kim, C., and Kim, B. (2018). Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 10:1021. doi: 10.3390/nu10081021
Kim, K. B., Myung, J., Sin, N., and Crews, C. M. (1999). Proteasome inhibition by the natural products epoxomicin and dihydroeponemycin: insights into specificity and potency. Bioorganic Med. Chem. Lett. 9, 3335–3340. doi: 10.1016/s0960-894x(99)00612-5
Kisselev, A. F., van der Linden, W. A., and Overkleeft, H. S. (2012). Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol. 19, 99–115. doi: 10.1016/j.chembiol.2012.01.003
Komatsu, S., Miyazawa, K., Moriya, S., Takase, A., Naito, M., Inazu, M., et al. (2012). Clarithromycin enhances bortezomib-induced cytotoxicity via endoplasmic reticulum stress-mediated CHOP (GADD153) induction and autophagy in breast cancer cells. Int. J. Oncol. 40, 1029–1039. doi: 10.3892/ijo.2011.1317
Kopp, M. C., Larburu, N., Durairaj, V., Adams, C. J., and Ali, M. (2019). UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mole. Biol. 26, 1053–1062. doi: 10.1038/s41594-019-0324-9
Li, H., Wan, L., Hashi, Y., and Chen, S. (2007). Fragmentation study of a 8-C-glycosyl isoflavone, puerarin, using electrospray ion trap time-of-flight mass spectrometry at high resolution. Rapid Commun. Mass Spectr. Rcm 21, 2497–2504. doi: 10.1002/rcm.3087
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lopes, N. P., Stark, C. B., Gates, P. J., and Staunton, J. (2002). Fragmentation studies on monensin A by sequential electrospray mass spectrometry. Analyst 127, 503–506. doi: 10.1039/b110412h
Losada, A., Muñoz-Alonso, M. J., García, C., Sánchez-Murcia, P. A., Martínez-Leal, J. F., Domínguez, J. M., et al. (2016). Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 6:35100.
Losada, A., Berlanga, J. J., Molina-Guijarro, J. M., Jiménez-Ruiz, A., Gago, F., Avilés, P., et al. (2020). Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells. Biochem. Pharm. 172:113744.
Lotufo, T. M. C., and Silva, A. M. B. (2006). “Ascidiacea,” in Biota Marinha da Costa Oeste do Ceará, eds H. Matthews-Cascon and T. M. C. Lotufo (Brasília, DF: Ministério do Meio Ambiente), 221–247.
Lu, G., Luo, H., and Zhu, X. (2020). Targeting the GRP78 Pathway for Cancer Therapy. Front. Med. 7:351.
Luo, B., and Lee, A. S. (2013). The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 32, 805–818. doi: 10.1038/onc.2012.130
Ma, Y., and Hendershot, L. M. (2003). Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278, 34864–34873. doi: 10.1074/jbc.M301107200
Manasanch, E. E., and Orlowski, R. Z. (2017). Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 14, 417–433. doi: 10.1038/nrclinonc.2016.206
Manivasagan, P., Venkatesan, J., Sivakumar, K., and Kim, S. K. (2014). Pharmaceutically active secondary metabolites of marine actinobacteria. Microbiol. Res. 169, 262–278. doi: 10.1016/j.micres.2013.07.014
Markham, A. (2020). Lurbinectedin: First Approval. Drugs 80, 1345–1353. doi: 10.1007/s40265-020-01374-0
Markouli, M., Strepkos, D., Papavassiliou, A. G., and Piperi, C. (2020). Targeting of endoplasmic reticulum (ER) stress in gliomas. Pharmacol. Res. 157:104823.
Mateyak, M. K., and Kinzy, T. G. (2010). eEF1A: thinking outside the ribosome. J. Biol. Chem. 285, 21209–21213. doi: 10.1074/jbc.R110.113795
Mayer, M. P., and Bukau, B. (2005). Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mole. Life Sci. 62, 670–684. doi: 10.1007/s00018-004-4464-6
McCauley, E. P., Piña, I. C., Thompson, A. D., Bashir, K., Weinberg, M., Kurz, S. L., et al. (2020). Highlights of marine natural products having parallel scaffolds found from marine-derived bacteria, sponges, and tunicates. J. Antib. 73, 504–525. doi: 10.1038/s41429-020-0330-5
McLafferty, F. W., and Tureek, F. (1993). Interpretation of Mass Spectra, 4th Edn. Sausalito: University Science Books.
Meng, L., Kwok, B. H., Sin, N., and Crews, C. M. (1999). Eponemycin exerts its antitumor effect through the inhibition of proteasome function. Cancer Res. 59, 2798–2801.
Micale, N., Scarbaci, K., Troiano, V., Ettari, R., Grasso, S., and Zappalà, M. (2014). Peptide-based proteasome inhibitors in anticancer drug design. Med. Res. Rev. 34, 1001–1069. doi: 10.1002/med.21312
Midwestern University (2020). Marine Pharmacology. Available online at: https://www.midwestern.edu/departments/marinepharmacology.xml [accessed July 16, 2020]
Momtaz, S., Memariani, Z., El-Senduny, F. F., Sanadgol, N., Golab, F., Katebi, M., et al. (2020). Targeting Ubiquitin-Proteasome Pathway by Natural Products: Novel Therapeutic Strategy for Treatment of Neurodegenerative Diseases. Front. Physiol. 11:361. doi: 10.3389/fphys.2020.00361
Mora, C., Tittensor, D. P., Adl, S., Simpson, A. G. B., and Worm, B. (2011). How Many Species Are There on Earth and in the Ocean? PLoS Biol. 2011:1001127. doi: 10.1371/journal.pbio.1001127
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63. doi: 10.1016/0022-1759(83)90303-4
Muchtar, E., Gertz, M. A., and Magen, H. (2016). A practical review on carfilzomib in multiple myeloma. Eur. J. Haematol. 96, 564–577. doi: 10.1111/ejh.12749
Myeku, N., and Figueiredo-Pereira, M. E. (2011). Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J. Biol. Chem. 286, 22426–22440. doi: 10.1074/jbc.M110.149252
Myung, J., Kim, K. B., and Crews, C. M. (2001). The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 21, 245–273. doi: 10.1002/med.1009
National Cancer Institute (2021). NCI-60 Screening Methodology. Available online at: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm. [accessed March 19, 2021].
National Center for Biotechnology Information (2020). Available online at: https://www.ncbi.nlm.nih.gov/ [accessed November 11, 2020].
National Oceanic and Atmospheric Administration (2020). How many species live in the ocean?. Available online at: https://oceanservice.noaa.gov/facts/ocean-species.html [accessed November 20, 2020].
Newman, D. J., and Cragg, G. M. (2020). Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803. doi: 10.1021/acs.jnatprod.9b01285
Obeng, E. A., Carlson, L. M., Gutman, D. M., Harrington, W. J. Jr., Lee, K. P., and Boise, L. H. (2006). Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107, 4907–4916. doi: 10.1182/blood-2005-08-3531
Okada, T., Yoshida, H., Akazawa, R., Negishi, M., and Mori, K. (2002). Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 366, 585–594. doi: 10.1042/BJ20020391
Okui, M., Ito, F., Ogita, K., Kuramoto, N., Kudoh, J., Shimizu, N., et al. (2000). Expression of APG-2 protein, a member of the heat shock protein 110 family, in developing rat brain. Neurochem. Int. 36, 35–43. doi: 10.1016/s0197-0186(99)00095-9
Palanisamy, S. K., Rajendran, N. M., and Marino, A. (2017). Natural Products Diversity of Marine Ascidians (Tunicates; Ascidiacea) and Successful Drugs in Clinical Development. Nat. Prod. Biopros. 7:115. doi: 10.1007/s13659-016-0115-5
Park, J. E., Miller, Z., Jun, Y., Lee, W., and Kim, K. B. (2018). Next-generation proteasome inhibitors for cancer therapy. Transl. Res. 198, 1–16.
Peñaranda Fajardo, N. M., Meijer, C., and Kruyt, F. A. (2016). The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharm. 118, 1–8. https://doi.org/10.1016/j.bcp.2016.04.008,Google Scholar
Pereira, R. B., Evdokimov, N. M., Lefranc, F., Valentão, P., Kornienko, A., Pereira, D. M., et al. (2019). Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets. Mar. Drugs 17:329. doi: 10.3390/Md17060329
Pinto, Francisco, C. L., Silveira, Edilberto, R., Vasconcelos, Ana Caroline, L., et al. (2020). Dextrorotatory Chromomycins from the Marine Streptomyces sp. Associated to Palythoa caribaeorum. J. Brazilian Chem. Soc. 31, 143–152.
Potts, B. C., Albitar, M. X., Anderson, K. C., Baritaki, S., Berkers, C., Bonavida, B., et al. (2011). Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets 11, 254–284. doi: 10.2174/156800911794519716
Rashid, H. O., Yadav, R. K., Kim, H. R., and Chae, H. J. (2015). ER stress: Autophagy induction, inhibition and selection. Autophagy 11, 1956–1977. doi: 10.1080/15548627.2015.1091141
Reen, F. J., Gutiérrez-Barranquero, José, A., Dobson, Alan, D. W., and Adams, et al. (2015). Emerging Concepts Promising New Horizons for Marine Biodiscovery and Synthetic Biology. Mar. Drugs 5, 2924–2954.
Reid, D. W., Tay, A. S., Sundaram, J. R., Lee, I. C., Chen, Q., George, S. E., et al. (2016). Complementary Roles of GADD34- and CReP-Containing Eukaryotic Initiation Factor 2α Phosphatases during the Unfolded Protein Response. Mole. Cell. Biol. 36, 1868–1880. doi: 10.1128/MCB.00190-16
Rinehart, K. L. Jr., Gloer, J. B., Hughes, R. G. Jr., Renis, H. E., McGovren, J. P., Swynenberg, E. B., et al. (1981). Didemnins: antiviral and antitumor depsipeptides from a caribbean tunicate. Science 212, 933–935. doi: 10.1126/science.7233187
Rinehart, K. L., Holt, T. G., Fregeau, N. L., Stroh, J. G., Keifer, P. A., Sun, F., et al. (1990). Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinate. J. Organ. Chem. 55, 4512–4515. doi: 10.1021/jo00302a007
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews. Mole. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Rozpedek, W., Pytel, D., Mucha, B., Leszczynska, H., Diehl, J. A., and Majsterek, I. (2016). The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mole. Med. 16, doi: 10.2174/1566524016666160523143937
Sakai, R., Rinehart, K. L., Kishore, V., Kundu, B., Faircloth, G., Gloer, J. B., et al. (1996). Structure–activity relationships of the didemnins. J. Med. Chem. 39, 2819–2834. doi: 10.1021/jm960048g
Schofield, M. M., Jain, S., Porat, D., Dick, G. J., and Sherman, D. H. (2015). Identification and analysis of the bacterial endosymbiont specialized for production of the chemotherapeutic natural product ET-743. Env. Microb. 17, 3964–3975. doi: 10.1111/1462-2920.12908
Schorn, M., Zettler, J., Noel, J. P., Dorrestein, P. C., Moore, B. S., and Kaysser, L. (2014). Genetic basis for the biosynthesis of the pharmaceutically important class of epoxyketone proteasome inhibitors. ACS Chem. Biol. 9, 301–309. doi: 10.1021/cb400699p
Skehan, P., Storeng, R., Scudiero, D., Monks, A., McMahon, J., Vistica, D., et al. (1990). New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82, 1107–1112. doi: 10.1093/jnci/82.13.1107
Snelgrove, P. V. R. (2010). Discoveries of the Census of Marine Life: making ocean life count. Cambridge: Cambridge University Press.
Snelgrove, P. V. (2016). An Ocean of Discovery: Biodiversity Beyond the Census of Marine Life. Planta Med. 82, 790–799. doi: 10.1055/s-0042-103934
Souza, R., Freire, V. F., Gubiani, J. R., Ferreira, R. O., Trivella, D., Moraes, F. C., et al. (2018). Bromopyrrole Alkaloid Inhibitors of the Proteasome Isolated from a Dictyonella sp. Marine Sponge Collected at the Amazon River Mouth. J. Nat. Prod. 81, 2296–2300. doi: 10.1021/acs.jnatprod.8b00533
Stepanenko, A. A., and Dmitrenko, V. V. (2015). Pitfalls of the MTT assay: Direct and off-target effects of inhibitors can result in over/underestimation of cell viability. Gene 574, 193–203. doi: 10.1016/j.gene.2015.08.009
Sugawara, K., Hatori, M., Nishiyama, Y., Tomita, K., Kamei, H., Konishi, M., et al. (1990). Eponemycin, a new antibiotic active against B16 melanoma. I. Production, isolation, structure and biological activity. J. Antib. 43, 8–18. doi: 10.7164/antibiotics.43.8
Tabas, I., and Ron, D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190. doi: 10.1038/ncb0311-184
Tang, J. H., Yang, L., Chen, J. X., Li, Q. R., Zhu, L. R., Xu, Q. F., et al. (2019). Bortezomib inhibits growth and sensitizes glioma to temozolomide (TMZ) via down-regulating the FOXM1-Survivin axis. Cancer Commun. 39, 81. doi: 10.1186/s40880-019-0424-2
Therapeutic Goods Administration (2020). Available online at: https://www.tga.gov.au/. [accessed September 10, 2020].
Thibaudeau, T. A., and Smith, D. M. (2019). A Practical Review of Proteasome Pharmacology. Pharm. Rev. 71, 170–197. doi: 10.1124/pr.117.015370
Tsukimoto, M., Nagaoka, M., Shishido, Y., Fujimoto, J., Nishisaka, F., Matsumoto, S., et al. (2011). Bacterial production of the tunicate-derived antitumor cyclic depsipeptide didemnin B. J. Nat. Prod. 74, 2329–2331. doi: 10.1021/np200543z
Urra, H., Dufey, E., Lisbona, F., Rojas-Rivera, D., and Hetz, C. (2013). When ER stress reaches a dead end. Biochim. Biophys. Acta 1833, 3507–3517. doi: 10.1016/j.bbamcr.2013.07.024
Veggiani, G., Gerpe, M., Sidhu, S. S., and Zhang, W. (2019). Emerging drug development technologies targeting ubiquitination for cancer therapeutics. Pharm. Therapeut. 199, 139–154.
Velasco-Alzate, K. Y., Bauermeister, A., Tangerina, M., Lotufo, T., Ferreira, M., Jimenez, P. C., et al. (2019). Marine Bacteria from Rocas Atoll as a Rich Source of Pharmacologically Active Compounds. Mar. Drugs 17, 671. doi: 10.3390/md17120671
Vera, M. D., and Joullié, M. M. (2002). Natural products as probes of cell biology: 20 years of didemnin research. Med. Res. Rev. 22, 102–145. doi: 10.1002/med.10003
Vichai, V., and Kirtikara, K. (2006). Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Prot. 1, 1112–1116. doi: 10.1038/nprot.2006.179
Voges, D., Zwickl, P., and Baumeister, W. (1999). The 26S proteasome: a molecular machine designed for controlled proteolysis. Ann. Rev. Biochem. 68, 1015–1068.
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, H. Q., Du, Z. X., Zhang, H. Y., and Gao, D. X. (2007). Different induction of GRP78 and CHOP as a predictor of sensitivity to proteasome inhibitors in thyroid cancer cells. Endocrinology 148, 3258–3270. doi: 10.1210/en.2006-1564
Wang, M., Carver, J. J., Phelan, V. V., Sanchez, L. M., Garg, N., Peng, Y., et al. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837. doi: 10.1038/nbt.3597
Wang, M., Wey, S., Zhang, Y., Ye, R., and Lee, A. S. (2009). Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 11, 2307–2316. doi: 10.1089/ars.2009.2485
Wang, W. A., Groenendyk, J., and Michalak, M. (2012). Calreticulin signaling in health and disease. Int. J. Biochem. Cell Biol. 44, 842–846. doi: 10.1016/j.biocel.2012.02.009
Wang, W., Cho, H. Y., Rosenstein-Sisson, R., Marín Ramos, N. I., Price, R., Hurth, K., et al. (2018). Intratumoral delivery of bortezomib: impact on survival in an intracranial glioma tumor model. J. Neurosurg. 128, 695–700. doi: 10.3171/2016.11.JNS161212
Watters, D. J. (2018). Ascidian Toxins with Potential for Drug Development. Mar. Drugs 16:162. doi: 10.3390/md16050162
Wei, Y., Sinha, S., and Levine, B. (2008). Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951. doi: 10.4161/auto.6788
White, K. M., Rosales, R., Yildiz, S., Kehrer, T., Miorin, L., Moreno, E., et al. (2021). Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021:eabf4058. doi: 10.1126/science.abf4058
World Health Organization (2020). Available online at: https://www.who.int/ [accessed November 10, 2020].
Xu, Y., Kersten, R. D., Nam, S. J., Lu, L., Al-Suwailem, A. M., Zheng, H., et al. (2012). Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 134, 8625–8632. doi: 10.1021/ja301735a
Zhang, Q. G., Han, D., Wang, R. M., Dong, Y., Yang, F., Vadlamudi, R. K., et al. (2011). C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proc. Natl. Acad. Sci. U S A 108, E617–E624. doi: 10.1073/pnas.1104391108
Zhang, M., Lu, L., Ying, M., Ruan, H., Wang, X., Wang, H., et al. (2018). Enhanced Glioblastoma Targeting Ability of Carfilzomib Enabled by a DA7R-Modified Lipid Nanodisk. Mole. Pharm. 15, 2437–2447. doi: 10.1021/acs.molpharmaceut.8b00270
Keywords: marine natural products, cytotoxicity, proteasome inhibitors, epoxyketones, ER-stress response
Citation: Furtado LC, Bauermeister A, de Felicio R, Ortega R, Pinto FCL, Machado-Neto JA, Trivella DBB, Pessoa ODL, Wilke DV, Lopes NP, Jimenez PC and Costa-Lotufo LV (2021) Marine Streptomyces sp. Isolated From the Brazilian Endemic Tunicate Euherdmania sp. Produces Dihydroeponemycin and Analogs With Potent Antiglioma Activity. Front. Mar. Sci. 8:644730. doi: 10.3389/fmars.2021.644730
Received: 21 December 2020; Accepted: 01 April 2021;
Published: 03 May 2021.
Edited by:
Marlen Ines Vasquez, Cyprus University of Technology, CyprusReviewed by:
Maria Luisa Marques Serralheiro, University of Lisbon, PortugalCopyright © 2021 Furtado, Bauermeister, de Felicio, Ortega, Pinto, Machado-Neto, Trivella, Pessoa, Wilke, Lopes, Jimenez and Costa-Lotufo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leticia V. Costa-Lotufo, Y29zdGFsb3R1Zm9AdXNwLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.