 Yunbang Zhang
Yunbang Zhang Jian Gao
Jian Gao Yunhai Zhang
Yunhai Zhang Yuanchao Zou
Yuanchao Zou Xiaojuan Cao
Xiaojuan Cao- 1Engineering Research Center of Green Development for Conventional Aquatic Biological Industry in the Yangtze River Economic Belt, Ministry of Education, College of Fisheries, Huazhong Agricultural University, Wuhan, China
- 2Hubei Provincial Engineering Laboratory for Pond Aquaculture, Hubei, China
- 3Wuhan Sinoeco Ecological Science and Technology Co., Ltd., Wuhan, China
- 4Conservation and Utilization of Fishes Resources in the Upper Reaches of the Yangtze River Key Laboratory of Sichuan Province, College of Life Sciences, Neijiang Normal University, Neijiang, China
Elongate loach (Leptobotia elongata) is endemic to middle and upper reaches of the Yangtze River in China. Due to overfishing and habitat destruction, this loach has become an endangered species. So far, lack of reliable genetic information and molecular markers has hindered the conservation and utilization of elongate loach resources. Therefore, we here performed an Illumina sequencing and de novo transcriptome assembly in elongate loach, and then developed polymorphic simple sequence repeat markers (SSRs). After assembly, 51,185 unigenes were obtained, with an average length of 1,496 bp. A total of 23,901 expressed sequence tag-simple sequence repeats (EST-SSRs) were identified, distributing in 14,422 unigenes, with a distribution frequency of 28.18%. Out of 16,885 designed EST-SSR primers, 150 primers (3 or 4 base repetition-dominated) were synthesized for polymorphic EST-SSR development. Then, 52 polymorphic EST-SSRs were identified, with polymorphism information contents (PIC) ranging from 0.03 to 0.88 (average 0.54). In conclusion, this was the first report of transcriptome sequencing of elongate loach. Meanwhile, we developed a set of polymorphic EST-SSRs for the loach. This study will provide an important basis, namely genetic information and polymorphic SSRs, for further population genetics and breeding studies of this endangered and economic loach in China.
Introduction
Elongate loach (Leptobotia elongata), also known as Queen loach, is the largest species in the Cobitidae family (Yuan et al., 2010). This loach is recorded in the middle and lower reaches of the Jinsha River, the upper reaches of the Yangtze River, the Minjiang River, the Jialing River, the Qujiang River and the Lancang River. It is endemic to the middle and upper reaches of the Yangtze River in China (Ding, 1994). Elongate loach contains rich nutrients (protein, fat and essential amino acids) and is of diuretic, nourishing Yin and other medicinal values, and ornamental value as well (Liang et al., 2009), making it with high market price. Meanwhile, the loach grows fast. These advantages bring a bright future for elongate loach aquaculture. Due to over-fishing, pollution of the water environment and the construction of water conservancy projects, the resources and distribution area of elongate loach have been seriously reduced (Liang et al., 2009; Yuan et al., 2010). This loach is currently listed as a highly endangered fish in the “China Species Red List” (Yue and Chen, 1998). Therefore, protection and sustainable utilization of elongate loach resource are imperative, imminent and of great significance.
Nowadays, molecular methods and means, e.g., transcriptomics and molecular markers, are often applied in the conservation and utilization of fish resources. The RNA-seq technology combined with bioinformatics can generate extensive data in a very cost-effective way (Parchman et al., 2010; Ekblom and Galindo, 2011; Harkess et al., 2017; Mercati et al., 2019). It not only facilitates the acquisition of a large number of gene sequences, but is also conducive to develop expressed sequence tag (EST)-derived markers (Qiao et al., 2014; Weisberg et al., 2017; Penin et al., 2019). Simple sequence repeats (SSRs) are sequences formed by short tandem repeating of 1–6 nucleotide bases, also known as microsatellites, widely found in the nuclear genome of eukaryotes (Jarne and Lagoda, 1996). They were first discovered in the genomes of plant chloroplasts and mitochondria (Powell et al., 1995; Provan et al., 1996; Whitton et al., 1997; Procaccini and Mazzella, 1998). As a molecular marker, SSR has the advantages of high polymorphism, co-dominance, quantity and easy experimentation (Zhang et al., 2016). It is a powerful tool in researches on resource conservation, phylogeny and population genetics, and genetic breeding. Different from genomic SSRs, EST-SSRs are located in the coding regions and highly transferable to related taxa (Zhao et al., 2013). Thus, EST-SSRs are considered as effective functional markers (Li et al., 2004; Mercati et al., 2019).
There have been a few reports on SSR development of elongate loach. Liu et al. (2012) developed a total of 22 polymorphic SSRs in the loach by using the fast isolation by AFLP of sequences containing repeats protocol (FIASCO). 18 polymorphic SSRs were developed in the loach based on the method of triplex affinity capture (Lian et al., 2013). In addition, Liu et al. (2014) developed 14 polymorphic SSRs through 454 sequencing technology of elongate loach genomic DNA. In this study, RNA-seq analysis was conducted in elongate loach for the first time to enrich its genomic resources. Then, we mined sequences containing SSRs from the transcriptome dataset and 52 polymorphic EST-SSRs were validated. This study will benefit for studies of population genetics, gene functions and genetic breeding in elongate loach and other related species.
Materials and Methods
Ethics Statement
This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of Huazhong Agricultural University. All efforts were made to minimize suffering of the loach.
Fish Samples
In October 2019, thirty elongate loach adults used here were collected from the waters of the Minjiang River (104°37′12′′ N; 28°46′12′′ E) in China. The heart, liver, brain and muscle tissues from each elongate loach adult (n = 4) were dissected. The samples were immediately frozen in liquid nitrogen for 24 h, and then stored at −80°C for RNA sequencing.
RNA Isolation, Library Preparation, and Sequencing
Total RNA was isolated from each sample by using TRIzol Reagent (TaKaRa, Dalian, China) according to the manufacturer’s protocol. RNA purity and concentration were determined by gel electrophoresis and Nanodrop 2,000 (Thermo Fisher Scientific, Waltham, MA, United States). The RNA integrity number (RIN) was examined using Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States). The isolated RNAs from the four different tissues of each loach, with RIN values greater than 9.0, were mixed with equivalent concentration as one biological sample. Since a RIN value of a live tissue was less than 9.0, three biological samples were finally obtained. The mixed RNA samples were then used for cDNA synthesis (Invitrogen, Carlsbad, CA, United States). Then the synthesized cDNA was subjected to end-repair, phosphorylation and “A” base addition according to Illumina’s library construction protocol. Three cDNA libraries were constructed (namely LE1, LE2, and LE3) in this study. The cDNA libraries were sequenced on the Illumina sequencing platform (Illumina Hiseq xten/NovaSeq 6,000 sequencer Illumina, San Diego, CA, United States) by using paired-end technology. The Illumina GA pipeline (version 1.8.2, San Diego, CA, United States) was used to analyze the raw image and for base calling.
De novo Assembly and Annotations
The sequencing data was filtered with the Sickle1 and SeqPrep2 software to remove the linker and low-quality sequences. Then, all clean reads were de novo assembled into transcripts by Trinity (Version v2.8.5) software, and the details were described by Zhang et al. (2017). After assembly, the TGICL clustering software (Version 2.1, J. Craig Venter Institute, Rockville, MD, United States) was used to cluster and remove redundant transcripts, and then the remaining sequences were defined as unigenes. TransRate3 and CD-HIT4 were then used to filter and optimize the sequences obtained. Assembly results were evaluated using BUSCO (Version 3.0.2) software. Unigenes were functionally annotated by BLASTX alignment with an E-value threshold of 10–5 against the following databases: NCBI non-redundant protein database (Nr), swiss-prot database, protein family database (Pfam), clusters of orthologous groups of proteins database (COG), gene ontology database (GO), and kyoto encyclopedia of genes and genomes database (KEGG). The GO annotations were performed using Blast2GO based on the NCBI Nr database. The KEGG automatic annotation server was used to map these unigenes to the KEGG pathway database. The expression levels of unigenes were calculated using transcripts per million (TPM) with RSEM (Version 1.3.1) software.
Detection of EST-SSRs
The software MISA v1.05 was used to detect SSRs in the unigenes with the following parameters: the motif sizes ranged from one to six nucleotide repeat units (six for mono- and di-, five for tri-, tetra-, penta-, and hexa-nucleotides), and the maximum interruption distance between two SSRs was 100 bp. We used the program Premier 5.0 software (Premier Biosoft Inc., Palo Alto, CA, United States) to design SSR primers and the methods in details referred to Feng et al. (2017). In this study, out of 16,885 designed EST-SSR primers, 150 pairs of primers were randomly selected and synthesized for polymorphic EST-SSR (3 or 4 base repetition-dominated) development.
EST-SSRs Amplification and Genotyping
Thirty elongate loaches were used for DNA extractions. The genomic DNA was extracted from caudal fin by using ammonium acetate method (Richardson et al., 2001), diluted with deionized water, and then subjected to agarose gel electrophoresis to detect the quality.
Amplification and polymorphism of the 150 EST-SSRs were tested through polymerase chain reaction (PCR) and electrophoresis, respectively. DNA amplification was performed by using a T100 Gradient Thermal cycler (Bio-Rad Laboratories, Inc., Pleasanton, CA, United States) within a 10 μL final reaction volume: 3.5 μL of double distilled water, 0.5 μL of each primer (100 μmol/L), 0.5 μL of genomic DNA (70 ng/μL), and 5 μL of 2X DreamTaq Green PCR Master Mix. PCR amplification program was listed as following: an initial denaturation at 98°C for 5 min, then 35 cycles of 30 s at 96°C, 10–16 s at 60°C, 1 min at 72°C, and final extension at 72°C for 10 min. 2% agarose gel electrophoresis was used to evaluate the success of the PCR reaction and the polymorphisms of EST-SSRs.
Simple sequence repeat genotyping was determined by capillary electrophoresis on a 96-capillary Applied Biosystem 3730 Genetic Analyzer. Three different dyes including 6-carboxyfluorescein (FAM), hexachloro-6-carboxy-fluorescein (HEX) and 5-carboxy-X-rhodamine (ROX), were used to label forward primers. GeneScan 500 ROX size standard (Applied Biosystem, United States) was the internal lane size standard. The genotyping data were obtained by using GeneMapper v4.0.
The POPGENE32 (Yeh et al., 1999) software was used to assess the number of observed alleles (Na), observed heterozygosity (Ho), expected heterozygosity (He), and polymorphic information content (PIC) of each EST-SSR.
Results and Discussion
De novo Assembly and Functional Annotation of Elongate Loach Transcriptome
Before the 2000s, the annual catch of elongate loach was about 10,000 kg. By 2008, the annual catch had reduced to 2,000–3,000 kg (Duan et al., 2008). At present, elongate loach is an endangered fish species endemic to Yangtze River in China (Yuan et al., 2010). Like elongate loach, populations of many fish species in the Yangtze River are getting smaller (Zhang et al., 2020). That’s why Chinese government has planned to carry out a ten-year forbidden fishing in the Yangtze River, which will be fully implemented since January 1, 2021 (Huang, 2020). Along with the obviously continuous reduction in wild resources, elongate loach is getting more and more attention. Many studies of the loach have been done. However, they mainly focus on its ecological habits, morphological characteristics, artificial reproduction, etc (Ku and Wen, 1997; Li, 2014; Zhang et al., 2018). Although the sequencing technologies have developed rapidly, different from other endangered animal species (Magalhães et al., 2019; Chen et al., 2020; Grandjean et al., 2020; Wen et al., 2020), there were few researches on genetic information of elongate loach. In this study, we developed a reference transcriptome in elongate loach. This was the first report of transcriptome sequencing in elongate loach, which could provide useful information for its protection and effective utilization.
Sequencing details of each library of elongate loach are shown in Table 1. All of the sequenced libraries were mixed for de novo assembling. After cleaning and quality checks, 202,296,650 clean reads with a total length of 29.98 Gb were obtained, which assembled into 51,185 unigenes with a mean length of 1,495.84 bp and a N50 of 2,785 bp (Table 2). The assembled unigene lengths ranged from 201 to 48,765 bp. In addition, the TransRate score and BUSCO score were 0.36 and 84.9%, respectively. Supplementary Table 1 showed the comparison of sequencing data and assembly results by using Transdecoder, indicating that the reference transcriptome had a good quality. The length distribution of the assembled unigenes is shown in Figure 1.
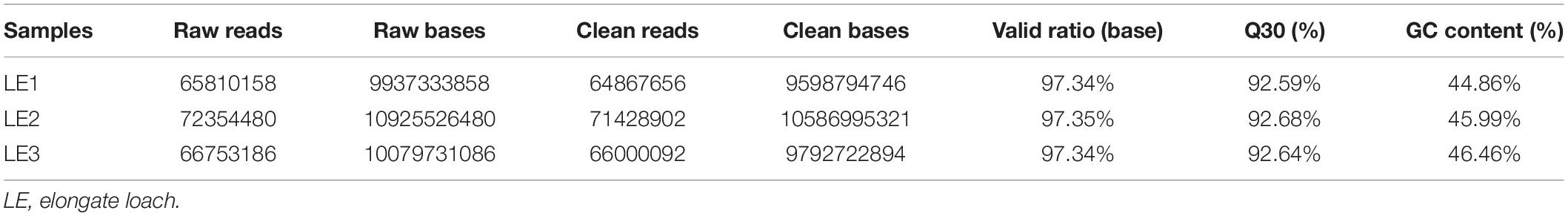
Table 1. Sequencing details of each library for mixed-assembly in elongate loach.

Table 2. Summary of mixed-assembling of elongate loach.
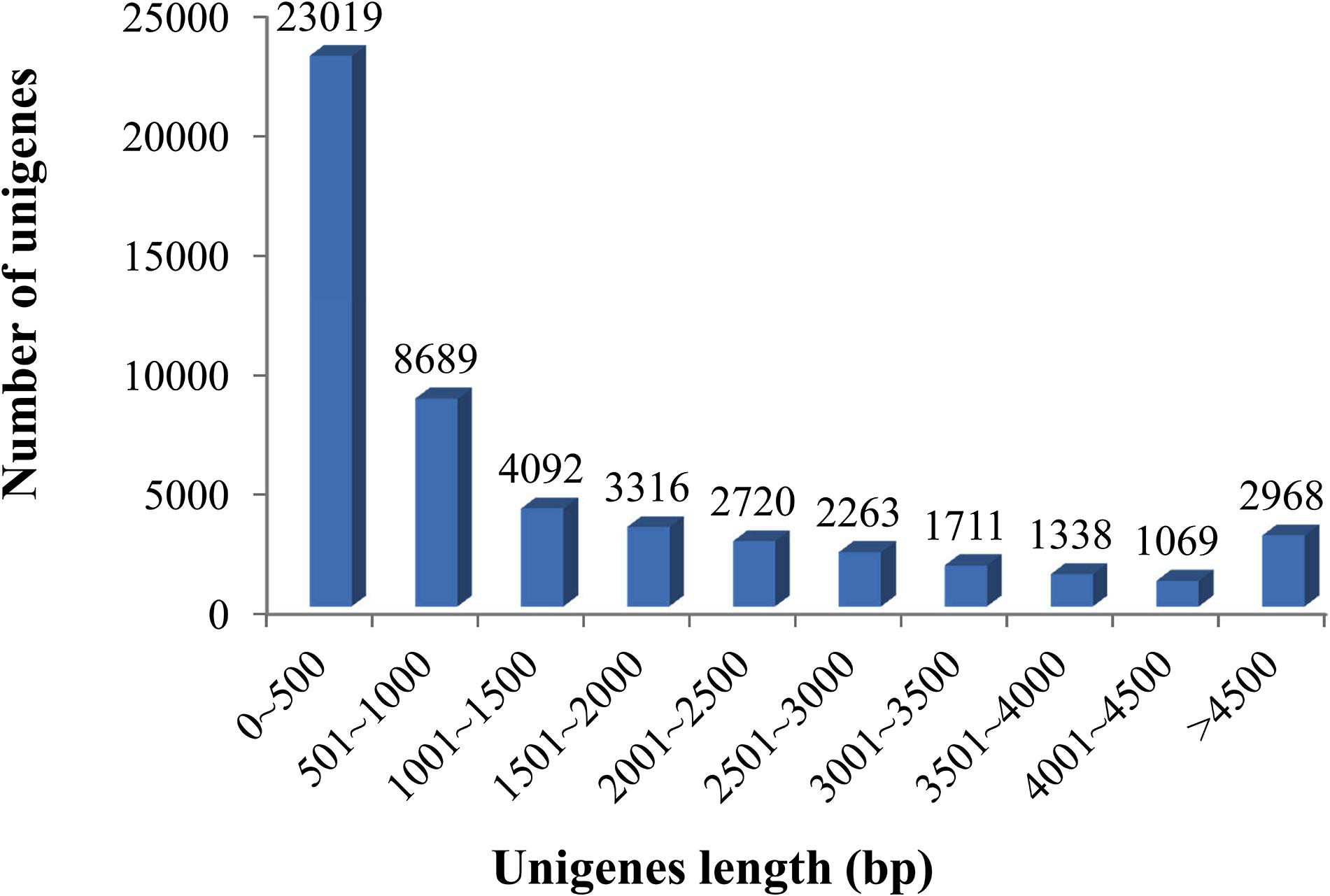
Figure 1. Length distributions of the assembled unigenes in elongate loach. Of all unigenes (51,185), 44.97% (23,019) were shorter than 500 bp, 16.98% (8,689) ranged from 501 to 1,000 bp, 14.47% (7,408) from 1,001 to 2,000 bp, and 23.58% (12,069) longer than 2,000 bp.
All unigenes were annotated against the six functional databases: NCBI Nr, Swiss-Prot, Pfam, COG, GO, and KEGG. As indicated in Figure 2, 49.43% unigenes (25,303) were significantly matched with known genes in the Nr database, while 42.45% unigenes (21,726) matched with entries in the Swiss-Prot database, and 38.39% unigenes (19,651) matched with proteins in the Pfam database. Based on the E-value distribution, 17.21% of annotated sequences showed high scores for homology (E-value <1 × 10–30), and 82.80% showed homology with E-values ranging from 0 to 1 × 10–30 (Figure 3A). Figure 3B showed that 71.57% of sequences had similarities over 80%. These results reflected the high identities of the mapped sequences with known sequences, suggesting good assembly quality of the reference transcriptome in elongate loach. Species-based distribution analysis indicated that Sinocyclocheilus rhinocerous (22.37%) provided most of the BLASTx hits for the elongate loach unigenes (Figure 3C).
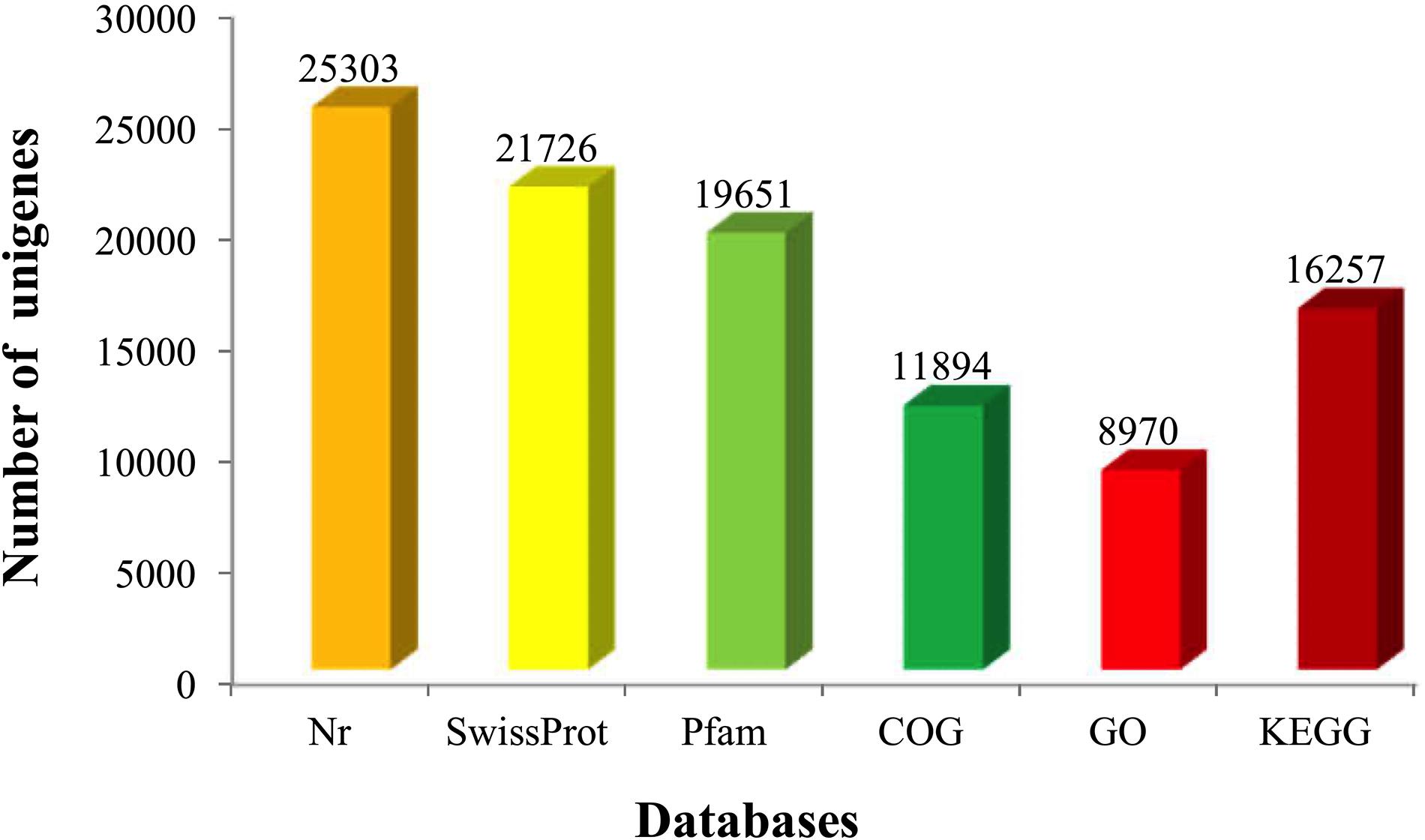
Figure 2. A summary of numbers of annotated unigenes against by six databases.
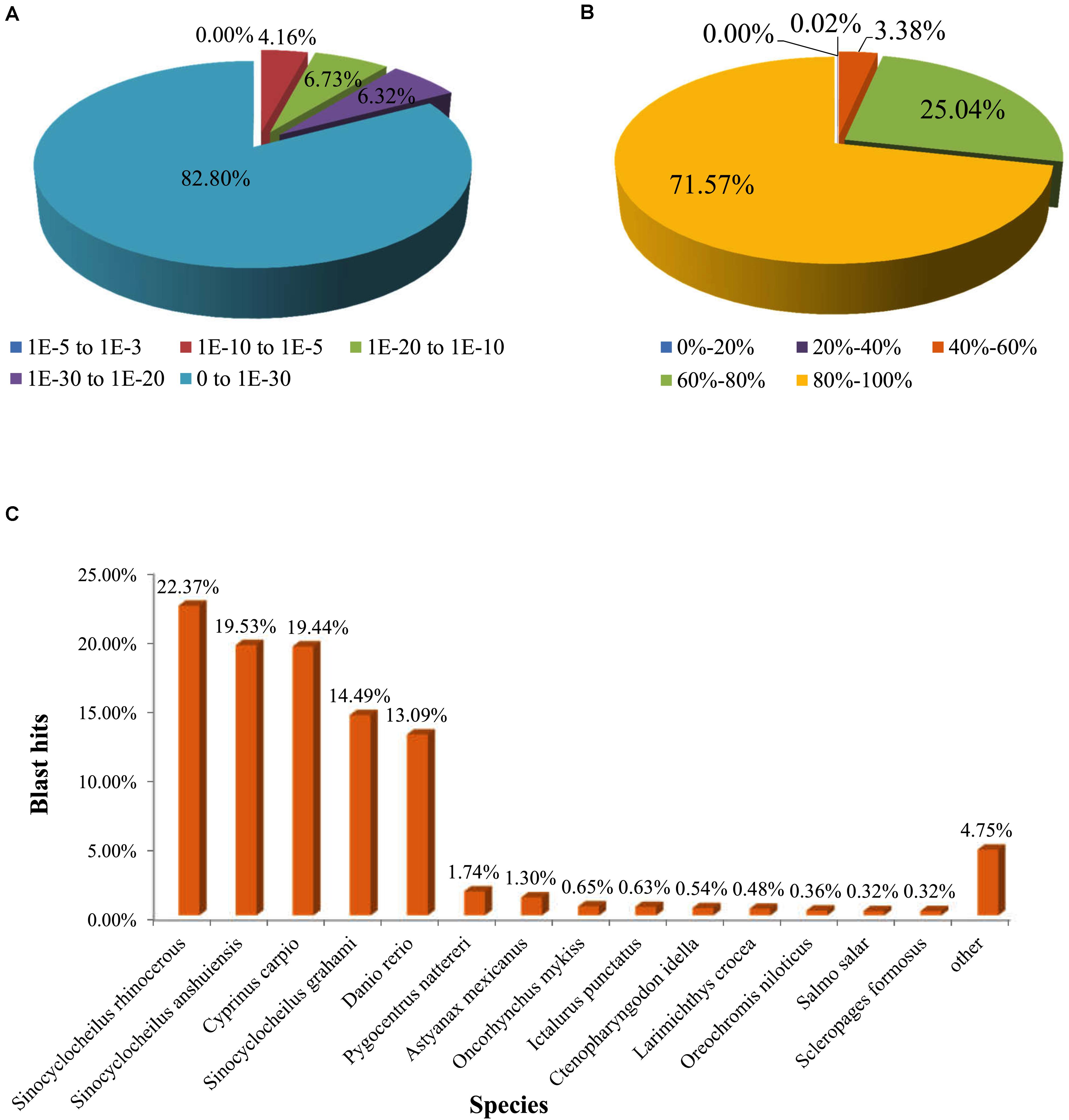
Figure 3. Characteristics of homology analysis for elongate loach unigenes against the non-redundant protein database (Nr). (A) The E-value distribution of BLASTx hits for each assembled unigene. (B) The similarity distribution of BLASTx hits for each assembled unigene. (C) Species-based distribution of the top BLASTx hits.
Functional prediction and classification of all unigenes were performed using their annotations with COG, GO, and KEGG databases. A total of 11, 894 unigenes (23.24%) in elongate loach were annotated in COG and grouped into 24 COG classifications (Supplementary Figure 1A). Based on Nr annotation and 60,229 functional terms, 8,970 (14.89%) elongate loach unigenes were assigned to GO term (Supplementary Figure 1B). It is classified into 59 functional groups from three aspects: biological process, cellular component and molecular function. All unigenes were analyzed with the KEGG pathway database as well. Carrying significant matches in this present study, 16,257 (31.76%) unigenes were assigned to 43 predicted metabolic pathways (Supplementary Figure 1C).
Through the reference transcriptome, we calculated the relative abundance of the assembled unigenes from four tissues of elongate loach. Cytochrome c oxidase subunit genes (COX1/3) were the highest expressed, followed by some genes related to muscle growth, including GAPDH, TPM4, and ACTC1 (Supplementary Table 2). Moreover, many ribosomal genes including RPL37, RPL23, RPS2, RPL32, RPS19, RPS23, RPL27A, and RPL15, were highly expressed in elongate loach.
Identification and Classification of EST-SSRs
A total of 23,901 EST-SSRs in elongate loach were identified in elongate loach (Table 3), which provided foundation for further investigations of population genetics and breeding of this species. Among all the unigenes, 28.18% (14,422) of them contained SSRs. Meanwhile, 4,450 unigenes had more than one SSR. The EST-SSR type in elongate loach was abundant, with a total of 54 repeat types from mononucleotide to hexanucleotides (Supplementary Table 3). 96.36% of mononucleotide repeats was A/T motif, accounting for 49.99% of the total EST-SSRs. Dinucleotide was dominated by AC/GT and AG/CT, accounting for 22.73% and 11.76%, respectively. Trinucleotide was mainly AAT/ATT and ATC/ATG, accounting for 1.58 and 1.21% respectively. The proportion of the remaining motifs did not exceed 1%. AAAC/GTTT and AAAG/CTTT motif accounted for the highest proportion in tetranucleotide. Pentanucleotide and hexanucleotide were the least of all motifs, and their proportions were 0.04% (Supplementary Table 3). Among the perfect SSRs, mononucleotide repeats were the most abundant (51.87%), followed by dinucleotide (40.28%), tri- (6.06%), tetra- (1.69%), penta- (0.084%), and hexa- (0.013%) nucleotide repeats (Table 4). In addition, we analyzed the numbers of repeat units and found that most EST-SSRs motifs had fewer than 15 repeats and accounted for 89.57% of all SSRs. 6–10 and 11–15 repeats harbored the most SSRs. In this study, we found that mononucleotide-repeat SSRs were the most abundant in elongate loach. This was consistent with the findings in Acipenser dabryanus (Chen et al., 2020). However, dinucleotide-repeat SSR was the main type in other fishes (Magalhães et al., 2019).

Table 3. Occurrence of SSRs in the surveyed elongate loach transcriptome.
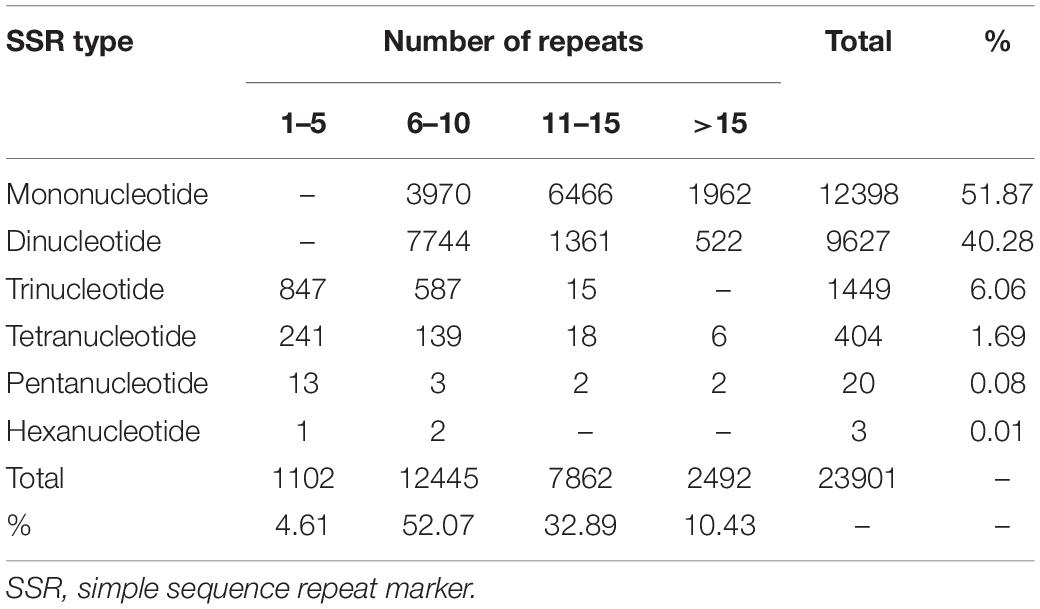
Table 4. Frequency distributions of SSRs in elongate loach transcriptome based on the number of repeat units.
Development of Polymorphic EST-SSRs
Out of 23,901 EST-SSRs, 16,885 primer pairs were successfully designed, and 150 primers (3 or 4 base repetition-dominated) were randomly selected for amplification of genomic DNA from elongate loach. Among the 150 primer pairs, 96 pairs (64%) were able to generate clear bands by PCR, which implied a decent EST-SSR primer dataset. Considering the amplification and polymorphic results, 52 SSRs (including 4 dinucleotide-repeat SSRs, 16 trinucleotide-repeat SSRs, and 32 tetranucleotide-repeat SSRs) were selected to evaluate polymorphism levels within the elongate loach population (Table 5). Previously, a total of 54 polymorphic SSRs were developed in elongate loach (Liu et al., 2012, 2014; Lian et al., 2013). Among them, 29 were dinucleotide-repeat SSRs. Compared with mono- and dinucleotide-repeat SSRs, tri- and tetranucleotide-repeat SSRs were more efficient in genotyping researches (Tibihika et al., 2020). Therefore, it could be told that this study developed many efficient SSR marks to conserve and utilize the threaten and economic loach.
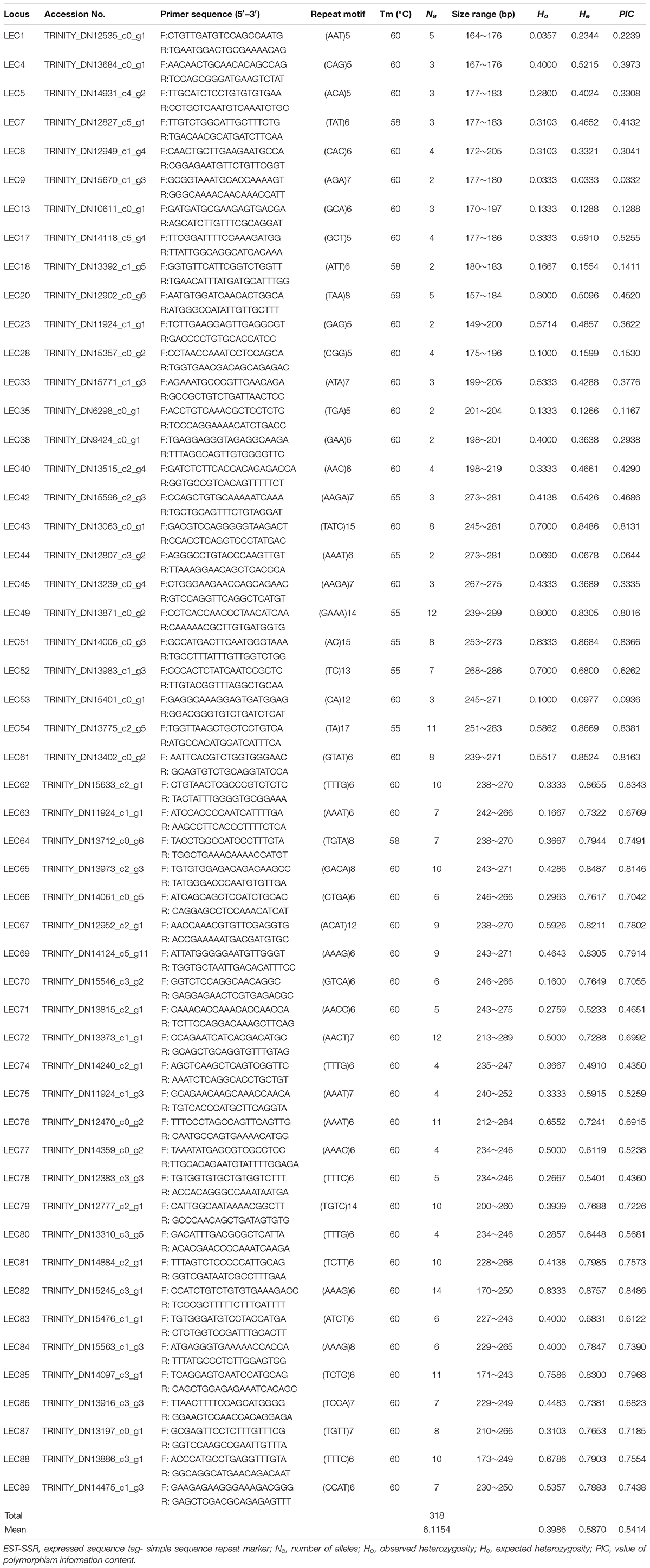
Table 5. Characteristics of 52 EST-SSRs that amplified successfully and exhibited polymorphism in elongate loach.
In October 2019, we collected an elongate loach population from the Yangtze River and found that a total of 318 alleles were detected in all the 52 polymorphic SSRs developed here. Meanwhile, the average number of alleles was 6.1. Ho varied from 0.0333 to 0.8333 with an average of 0.3986, while He from 0.3333 to 0.8757 with an average of 0.5870, were found. The value of PIC ranged from 0.0332 to 0.8381 with a mean value of 0.5414. Among the 52 SSRs obtained here, 30 SSRs were highly polymorphic (PIC > 0.5), and 14 were moderately polymorphic (0.25 < PIC < 0.5) (Botstein et al., 1980). Liu et al. (2017) reported that the average number of alleles and Ho for seven populations of elongate loach collected from July 2011 to December 2013, were respectively 11.5 and 0.74, which were obviously higher than those in our study. To a certain extent, it indicated an obvious reduction in population genetic diversity of elongate loach in the Yangtze River recently, which would be caused by over-fishing and habitat destruction. Therefore, it needs to pay more attention to the protection of elongate loach.
In summary, this study provides a reference transcriptome of multiple tissues and 52 polymorphic SSRs (3 or 4 base repetition-dominated) in elongate loach, a highly threatened fish of the Yangtze River in China. These will benefit for the protection and sustainable utilization of elongate loach.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Ethics Committee and the Use of Laboratory Animals of Huazhong Agricultural University.
Author Contributions
XC and YCZ designed and supervised the experiments. YBZ, JG, and YHZ carried out the experiments. YBZ analyzed the data and wrote the manuscript. XC revised the manuscript. All authors reviewed and approved the manuscript.
Funding
This study was financially supported by Ministry of Agriculture and Rural Affairs of the People’s Republic of China (Project Title: Investigations of the Wild Resources and Genetic Diversity of Elongate Loach in Yangtze River. Project Number: 81), Fundamental Research Funds for the Central Universities (Project Number: 2662020SCPY002), and Open Project Program of Fishery Resources and Environmental Science Experimental Station of The Upper-Middle Reaches of Yangtze River Ministry of Agriculture (Project Number: FRESES201901).
Conflict of Interest
YHZ was employed by Wuhan Sinoeco Ecological Science and Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Many thanks to the two reviewers for their comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.616727/full#supplementary-material
Footnotes
- ^ https://github.com/najoshi/sickle
- ^ https://github.com/jstjohn/SeqPrep
- ^ http://hibberdlab.com/transrate/
- ^ http://weizhongli-lab.org/cd-hit/
- ^ http://pgrc.ipkgatersleben.de/misa/
References
Botstein, D., White, R. L., Skolnick, M., and Davis, R. W. (1980). Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331. doi: 10.1016/0165-1161(81)90274-0
Chen, Y. Y., Liu, Y., Du, J., Song, M. J., Lai, J. S., and Gong, Q. (2020). Isolation and characterization of 47 SNP markers in the critically endangered Acipenser dabryanus. Consery Genet Resour. 12, 9–12. doi: 10.1007/s12686-018-1052-x
Duan, X. B., Liu, S. P., Xiong, F., Chen, D. Q., Yang, R. H., Chi, C. G., et al. (2008). Analysis of fishing structure and biodiversity in the upper mainstream of the Yangzte River before and after 3 years spring fishing off. Resour. Environ. Yangtze Basin 17, 878–884.
Ekblom, R., and Galindo, J. (2011). Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107, 1–15. doi: 10.1038/hdy.2010.152
Feng, S. J., Zhao, L. L., Liu, Z. S., Liu, Y. L., and Yang, T. X. (2017). De novo transcriptome assembly of Zanthoxylum bungeanum using Illumina sequencing for evolutionary analysis and simple sequence repeat marker development. Sci. Rep. 7:16754. doi: 10.1038/s41598-017-15911-7
Grandjean, F., Gan, H. M., Moumen, B., Giraud, I., Hatira, S., Cordaux, R., et al. (2020). Dataset for sequencing and de novo assembly of the european endangered white-clawed crayfish (Austropotamobius pallipes) abdominal muscle transcriptome. Data Brief. 29:105166. doi: 10.1016/j.dib.2020.105166
Harkess, A., Zhou, J. S., Xu, C. Y., Bowers, J. E., Hulst, R. V., Ayyampalayam, S., et al. (2017). The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 8, 1279–1286. doi: 10.1038/s41467-017-01064-8
Huang, Y. J. (2020). Forbidden fishing for ten years: the measures of promoting the development of Yangtze River economic zone. Study Times 7, 1–3.
Jarne, P., and Lagoda, P. J. L. (1996). Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 11, 424–429. doi: 10.1016/0169-5347(96)10049-5
Ku, Y. M., and Wen, X. B. (1997). A preliminary study on the biological characteristics of Leptobatia elongata (Bleeker). J. Hubei Agric. College 17, 40–42.
Li, L. (2014). Studies on the Feeding Ecology of Dominant Fishes and Food Web Structure of Fishes in the Yibin Reach of Yangtze River. Doctoral thesis, Huazhong Agricultural University, Hongshan.
Li, Y. C., Korol, A. B., Fahima, T., and Nevo, E. (2004). Microsatellites within genes: structure, function, and evolution. Mol. Biol. Evol. 21, 991–1007. doi: 10.1093/molbev/msh073
Lian, H., Que, Y. F., Xu, D. M., Liang, Y. Q., and Zhu, B. (2013). Development and characterization of 18 microsatellite loci from the endemic elongate loach (Leptobotia elongata). Conserv Genet Resour. 5, 371–373. doi: 10.1007/s12686-012-9806-3
Liang, H. L., Zhang, X. Z., and Liang, Y. Q. (2009). Threatened fishes of the world: Leptobotia elongata (Bleeker, 1870) (Botiidae). Environ. Biol. Fish. 85, 287–288. doi: 10.1007/s10641-009-9500-9
Liu, D. Q., Wu, J. Y., Deng, L. J., Gan, W. X., Du, L. M., and Song, Z. B. (2014). Development of microsatellite markers for Leptobotia elongata (Cypriniformes: Cobitidae) using 454 sequencing and cross-species amplification. Pak. J. Zool. 46, 1147–1151.
Liu, H. Y., Xiong, F., Duan, X. B., and Chen, D. Q. (2012). Isolation and characterization of polymorphic microsatellite loci from elongate loach (Leptobotia elongate), a threatened fish species endemic to the Yangtze River. Conserv. Genet. Resour. 4, 129–131. doi: 10.1007/s12686-011-9491-7
Liu, H. Y., Xiong, F., Duan, X. B., Tian, H. W., Liu, S. P., and Chen, D. Q. (2017). Low population differentiation revealed in the highly threatened elongate loach (Leptobotia elongata, Bleeker), a species endemic to the fragmented upper reaches of the Yangtze River. Biochem. Syst. Ecol. 70, 22–28. doi: 10.1016/j.bse.2016.10.015
Magalhães, M. G. P., Moreira, D. A., Furtado, C., and Parente, T. E. (2019). Development of SNVs and indels markers mined out of the first multi-organ transcriptomes from Hypancistrus zebra (Loricariidae), an endangered Amazonian catfish. Genomics 112, 971–980. doi: 10.1016/j.ygeno.2019.06.013
Mercati, F., Fontana, I., Gristina, A. S., Martorana, A., Nagar, M. E., and Michele, R. D. (2019). Transcriptome analysis and codominant markers development in caper, a drought tolerant orphan crop with medicinal value. Sci. Rep. U.K. 9;10411. doi: 10.1038/s41598-019-46613-x
Parchman, T. L., Geist, K. S., Grahnen, J. A., Benkman, C. W., and Buerkle, C. A. (2010). Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11:180. doi: 10.1186/1471-2164-11-180
Penin, A. A., Klepikova, A. V., Kasianov, A. S., Gerasimov, E. S., and Logacheva, M. D. (2019). Comparative analysis of developmental transcriptome maps of Arabidopsis thaliana and Solanum lycopersicum. Genes 10, 50–63. doi: 10.3390/genes10010050
Powell, W., Morgante, M., Andre, C., Mcnicol, J. W., Machray, G. C., Doyle, J. J., et al. (1995). Hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr. Biol. 5, 1023–1029. doi: 10.1016/S0960-9822(95)00206-5
Procaccini, G., and Mazzella, L. (1998). Population genetic structure and gene flow in the seagrass Posidonia oceanica assessed using microsatellite analysis. Mar. Ecol. Prog. Ser. 169, 133–141. doi: 10.3354/meps169133
Provan, J., Corbett, G., Waugh, R., Mcnicol, J. W., Morgante, M., and Powell, W. (1996). DNA fingerprints of rice (Oryza sativa) obtained from hypervariable chloroplast simple sequence repeats. Proc. R. Soc. Lond. B 263, 1275–1281. doi: 10.1098/rspb.1996.0187
Qiao, F., Cong, H. Q., Jiang, X. F., Wang, R. X., Yin, J. M., Qian, D., et al. (2014). De novo characterization of a Cephalotaxus hainanensis transcriptome and genes related to paclitaxel biosynthesis. PLoS One 9:e106900. doi: 10.1371/journal.pone.0106900
Richardson, D. S., Jury, F. L., Blaakmeer, K., Komdeur, J., and Burke, T. (2001). Parentage assignment and extra-group paternity in a cooperative breeder: the Seychelles warbler (Acrocephalus sechellensis). Mol. Ecol. 10, 2263–2273. doi: 10.1046/j.0962-1083.2001.01355.x
Tibihika, P. D., Curto, M., Alemayehu, E., Waidbacher, H., Masembe, C., Akoll, P., et al. (2020). Molecular genetic diversity and differentiation of Nile tilapia (Oreochromis niloticus, L. 1758) in East African natural and stocked populations. BMC Evol. Biol. 20:16. doi: 10.21203/rs.2.11849/v4
Weisberg, A. J., Kim, G., Westwood, J. H., and Jelesko, J. G. (2017). Sequencing and de novo assembly of the Toxicodendron radicans (Poison Ivy) transcriptome. Genes 8, 317–342. doi: 10.3390/genes8110317
Wen, S. N., Li, P., Wang, F., Li, J. L., Liu, H. X., and Li, N. (2020). De novo assembly and microsatellite marker development of the transcriptome of the endangered Brachymystax lenok tsinlingensis. Genes Genom. 42, 727–734. doi: 10.1007/s13258-020-00939-3
Whitton, J., Rieseberg, L. H., and Ungerer, M. C. (1997). Microsatellite loci are not conserved across the asteraceae. Mol. Biol. Evol. 14, 204–209. doi: 10.1093/oxfordjournals.molbev.a025755
Yeh, F. C., Yang, R. C., and Boyle, T. B. J. (1999). POPGENE. Ver.1.31. Microsoft Windows-Based Free Ware for Population Genetic Analysis. Alberta: University of Alberta and Centre for International Forestry Research.
Yuan, Y. C., Yang, H. J., Gong, S. Y., and Liang, Y. Q. (2010). Threatened fishes of the world: Leptobotia elongata Bleeker, 1870 (Bottiinae). Environ. Biol. Fish. 87, 295–296. doi: 10.1007/s10641-010-9582-4
Yue, P., and Chen, Y. (1998). China Red Data Book of Endangered Animals: Pisces. Beijing, China: Science Press, 240–243.
Zhang, L. L., Ren, Q. Q., Liu, M., Xu, Q., Bin Kang, B., and Jiang, X. B. (2020). Fishery stock assessments in the min river estuary and its adjacent waters in southern china using the length-based bayesian estimation (LBB) method. Front. Mar. Sci. 7:507. doi: 10.3389/fmars.2020.00507
Zhang, P. F., Zhou, X. R., Pang, B. P., Tan, Y., Chang, J., and Gao, L. J. (2016). High-throughput discovery of microsatellite markers in Galeruca daurica (Coleoptera: Chrysomelidae) from a transcriptome database. J. Appl. Entomol. 53, 1058–1064. doi: 10.7679/j.issn.2095-1353.2016.130
Zhang, Y., Li, Y., Liang, X., Cao, X. J., Huang, L. F., Yan, J., et al. (2017). Hepatic transcriptome analysis and identification of differentially expressed genes response to dietary oxidized fish oil in loach Misgurnus anguillicaudatus. PLoS One 12:e0172386. doi: 10.1371/journal.pone.0172386
Zhang, Y. H., Ding, Y., Gu, Z. X., Huang, X. X., Yu, Z. W., and Shu, M. (2018). Artificial propagation and embryonic development observation of Leptobotia elongata from Jinsha River and Yangtze River. Hub Agric. Sci. 57, 104–107.
Keywords: Leptobotia elongate, transcriptome, SSRs, polymorphism, endangered species
Citation: Zhang Y, Gao J, Zhang Y, Zou Y and Cao X (2021) Transcriptome Sequencing of the Endangered Species Elongate Loach (Leptobotia elongata) From the Yangtze River: De novo Transcriptome Assembly, Annotation, Identification and Validation of EST-SSR Markers. Front. Mar. Sci. 8:616727. doi: 10.3389/fmars.2021.616727
Received: 13 October 2020; Accepted: 26 January 2021;
Published: 04 March 2021.
Edited by:
Andrew Stanley Mount, Clemson University, United StatesReviewed by:
Vittoria Roncalli, University of Naples Federico II, ItalySushil Kumar, Anand Agricultural University, India
Copyright © 2021 Zhang, Gao, Zhang, Zou and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanchao Zou, MzgxNDE3MDg3QHFxLmNvbQ==; Xiaojuan Cao, Y2FveGlhb2p1YW5AbWFpbC5oemF1LmVkdS5jbg==