 Kazuaki Yamaguchi
Kazuaki Yamaguchi Shigehiro Kuraku
Shigehiro Kuraku
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Mar. Sci. , 10 March 2021
Sec. Marine Megafauna
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.573853
This article is part of the Research Topic Molecular Evolution and Genomics of Marine Megafauna: Methodological Solutions and Key Advances View all 8 articles
While mostly confined to marine environments, species in the taxon Chondrichthyes (cartilaginous fishes) exhibit a wide variety of body size, longevity, and habitat depth. Despite their diversity, genome analysis has concentrated on species with high public visibility (Read et al., 2017; Marra et al., 2019). Previously, a study reporting whole genome sequencing of the white shark, Carcharodon carcharias (Marra et al., 2019) suggested unique molecular evolution accounted for the maintenance of genome stability and the elongated lifespan of the whale shark, Rhincodon typus as well as the white shark. The results reported by Marra et al. (2019) included positive selection of dozens of protein-coding genes potentially involved in genome stability and wound healing. We performed a reanalysis of some of the data presented in Marra et al. (2019) using the genome resources of other shark species and report our opinion based the results of the reanalysis.
Marra et al. (2019) reported positive selection in the gene encoding Mdm4 in the whale shark lineage. Mdm4 is a key regulator of the tumor suppressor gene, p53 (Toledo and Wahl, 2007). In our view, the whale shark Mdm4 ortholog sequence used in their analysis (XP_020377040.1 in NCBI; presented in Figure 3 by Marra et al., 2019) seems to harbor an incorrect open reading frame (ORF), when it is curated by transcript sequencing (Figure 1A). The sequence used by Marra et al. (2019) shows a remarkable dissimilarity to their orthologs, as well as the curated sequence of this whale shark gene (Figure 1A).
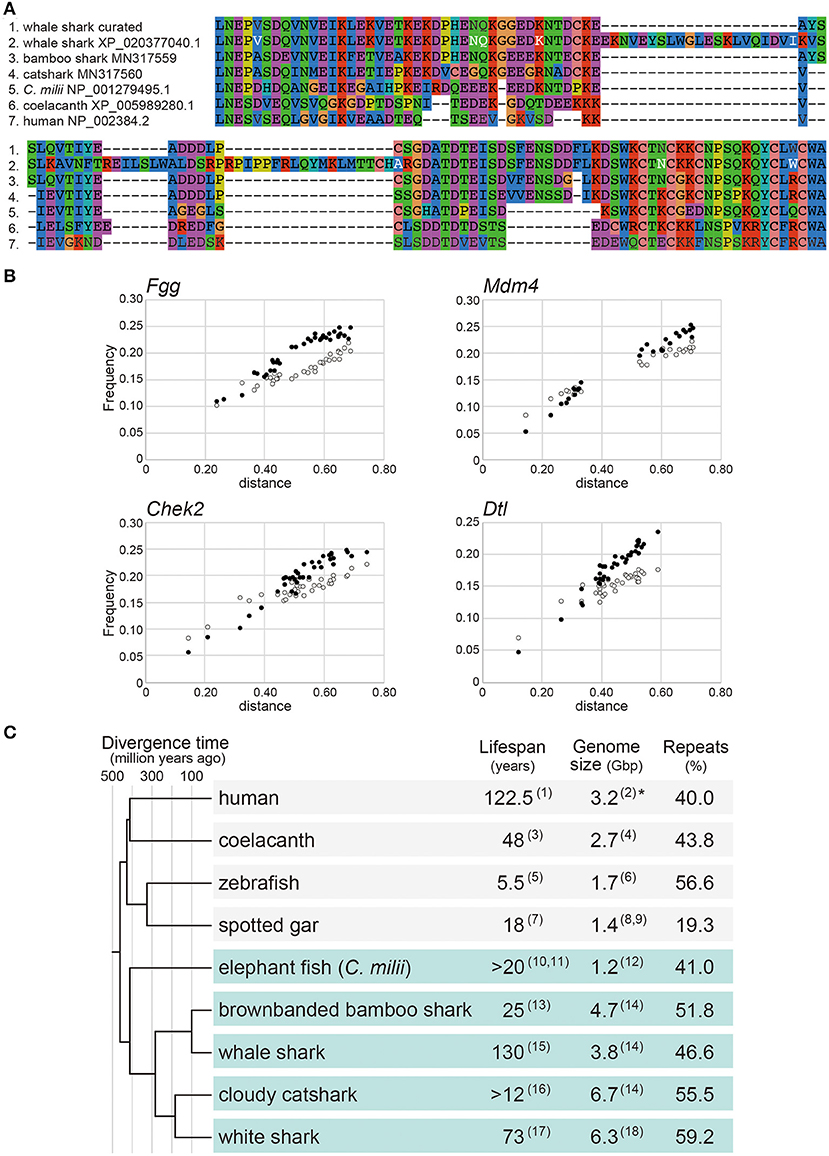
Figure 1. Reanalysis of shark molecular sequences. (A) Multiple alignment of Mdm4 for the amino acid sequence stretch corresponding to the residue 241 to 325 of human Mdm4 (NP_002384.2 in NCBI). The residues in white letters indicate positively selected residues in the whale shark sequence identified previously (Marra et al., 2019), but most of them are neither unique to the whale shark nor included in the curated whale shark sequence. The GenBank accession ID for the curated whale shark sequence (top) supported by transcript sequencing is MN317558.1. Details of the transcript sequencing will be reported elsewhere. (B) Substitution saturation plots for transition and transversion of the coding region of the Fgg, Mdm4, Chek2, and Dtl genes chosen from those previously regarded as positively selected (Marra et al., 2019). Each dot indicates a pair of species in the dataset. The white and black dots indicate transitions and transversions, respectively. The horizontal axis indicates the distance based on the TN93 substitution model (Tamura and Nei, 1993). The vertical axis indicates the observed proportion of transition and transversion. The amino acid sequences were aligned with MAFFT v7.299b (Katoh and Standley, 2013) using the L-INS-i option. Nucleotide sequences were aligned based on the amino acid sequence alignment using the emboss tranalign tool. Unreliably aligned regions were removed using Gblocks v0.91b (Talavera and Castresana, 2007) based on the default parameters. Transversion and transition frequencies were calculated with the TN93 substitution model using the DAMBE program (Xia, 2018). The details of the sequences used for the analysis are included in Supplementary Data (https://doi.org/10.6084/m9.figshare.13521329). (C) Phylogenetic overview of maximum recorded lifespan, genome size, and repeat content in a uniform presentation. The branch lengths are proportional to the geological times based on information retrieved from the Timetree of Life website (http://www.timetree.org/). The lifespan of the coelacanth was suggested to be over 100 years in other literature (Froese and Palomares, 2000). Repetitiveness in the genomes was quantified uniformly as previously described (Hara et al., 2018). The genome sizes are based on flow cytometry except for the human for which a total length of the genome sequences (3.2 Gbp) is conventionally referred to as its genome size (*). Because the documented lifespan of the brownbanded bamboo shark, Chiloscyllium punctatum is unavailable, that of a different species in the same genus (C. plagiosum) is included. The numbers in parentheses correspond to the following publications: (1) Allard et al. (1998); (2) Morton (1991); (3) Fricke et al. (2011); (4) Noonan et al. (2004); (5) Gerhard et al. (2002); (6) Postlethwait et al. (2009); (7) Hugg (1996); (8) Hardie and Hebert (2004); (9) Ojima (1990); (10) Francis (1997); (11) Francis and Ó Maolagáin (2019); (12) Venkatesh et al. (2005); (13) Chen et al. (2007); (14) Hara et al. (2018); (15) Perry et al. (2018); (16) Michael (2005); (17) Hamady et al. (2014); (18) Schwartz and Maddock (1986).
Because one of the main findings by Marra et al. (2019) is challenged by our finding of this simple ORF misidentification, we investigated the other genes that were judged to be positively selected in their study. Here, we focused on Denticleless E3 ubiquitin protein ligase homolog (Dtl), Coenzyme Q3, methyltransferase (Coq3), and Sirtuin 7 (Sirt7) genes. We could not find orthologous coding sequences of these genes in the white shark protein-coding sequences supplied by Marra et al. (2019) but were identified in the genomic sequences, guided by the record of their gene prediction in the general feature format (GFF) file described the exact coordinates and attributes of genes and transcripts. Specifically, of these genes, the coding sequences of Coq3 and Sirt7 seem to be erroneously predicted and possibly used without curation (Supplementary Data, https://doi.org/10.6084/m9.figshare.13521329), as shown above for the whale shark Mdm4 sequence.
Dataset S1 in the publication by Marra et al. (2019) frequently exhibits inflated values (e.g., 999) for the ratio of non-synonymous to synonymous substitutions (ω). The inflation may be caused by ORF misidentification as shown above for Mdm4 but could also be due to the inclusion of phylogenetically distant sequences (e.g., paralogs) or species (e.g., teleost fishes that diverged from chondrichthyan species more than 400 million years ago; Irisarri et al., 2017). The three chondrichthyan species included by Marra et al. (2019) diverged more than 150 million years ago (Irisarri et al., 2017), leaving long branches connecting taxa that would ideally be broken by including more closely related shark species with the genome sequences made available earlier (such as the brownbanded bamboo shark and the cloudy catshark; Hara et al., 2018).
To examine the appropriateness of interspecific comparisons in the previous study (Marra et al., 2019), we estimated the frequencies of transversion and transition in the Mdm4, Dtl, Checkpoint kinase 2 (Chek2), and Fibrinogen gamma chain (Fgg) genes shown as positively selected by Marra et al. (2019). For this analysis, we employed the sequence sets used in the previous analysis (Marra et al., 2019). In particular, the Fgg gene associated with wound healing was reported by Marra et al. (2019) as a positively selected gene in both the white shark and whale shark lineages. The inclusion of distantly related species likely resulted in inflated ω values, indicated by the fact that the number of transversions in the Fgg gene exceeds the number of transitions (Figure 1B). Similarly, the other genes, Mdm4, Chek2, and Dtl, exhibited a larger number of transversions than transitions together with increasing evolutionary distance (Figure 1B).
To investigate possible substitution saturation, we further analyzed the Fgg, Mdm4, Chek2, and Dtl genes. Indices of substitution saturation (Iss), introduced by Xia (2009), were computed for the first, second, and third codon positions of ortholog sequences using the DAMBE program (Xia, 2018). As a result, Iss exceeded the index of substitution saturation for asymmetric topologies (Iss.cAsym) at the third codon positions of these genes (for Fgg, Iss = 0.6858 and Iss.cAsym = 0.5958; for Mdm4, Iss = 0.6988 and Iss.cAsym = 0.6299; for Chek2, Iss = 0.7438 and Iss.cAsym = 0.5935; for Dtl, Iss = 0.7004 and Iss.cAsym = 0.6041). These results indicate the saturation of substitutions at the third codon positions of these genes.
Ortholog sequences of multiple closely related species and accurate alignments are essential for the proper detection of positively selected genes. It was previously cautioned that imprecise alignments containing erroneous ORFs likely induce ω value inflation in the detection of positively selected genes (Jordan and Goldman, 2012). Our reanalysis illustrates the use of mis-predicted ORF sequences and excessively distant sequences by Marra et al. (2019). The saturation of substitutions at the third codon positions, caused by the use of distant sequences, increases the possibility of falsely detecting positive selection (Weber et al., 2014). Overall, for the genes we examined, at least, no evidence of positive selection was obtained.
In Figure 1C, we reconstructed the schematic summary presented as Figure 1 originally by Marra et al. (2019). Therein, we included genome sizes and repeat element abundance presented in a uniform style. Marra et al. (2019), argued for the association of larger genome size and higher repeat abundance of the white shark and whale shark with their high genome stability. However, the comparison in Figure 1C, involving other shark species, shows that some shark species with relatively small body sizes and short lifespans have comparable or even larger genome sizes than the white shark and the whale shark (Hara et al., 2018). The current dataset does not support any association of genome stability with genome size or repeat abundance, which remains to be further explored when whole genomes of more species are sequenced.
In our opinion, the findings reported by Marra et al. (2019) need to be reassessed without any bias that genome analysis of those long-lived shark species will readily account for their capacities of genome stability maintenance and wound healing. This reassessment could be facilitated by the inclusion of emerging sequence information for other cartilaginous fishes (reviewed in Yamaguchi et al., 2021).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
KY performed data analysis. KY and SK wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Allard, M., Lèbre, V., Robine, J.-M., and Calment, J. (1998). Jeanne Calment: From Van Gogh's Time To Ours, 122 Extraordinary Years. New York, NY: WH Freeman & Company.
Chen, W. K., Chen, P. C., Liu, K. M., and Wang, S. B. (2007). Age and growth estimates of the whitespotted bamboo shark, Chiloscyllium plagiosum, in the northern waters of Taiwan. Zool. Stud. 46, 92–102.
Francis, M. P. (1997). Spatial and temporal variation in the growth rate of elephantfish (Callorhinchus milii). New Zeal. J. Mar. Fresh. 31, 9–23. doi: 10.1080/00288330.1997.9516741
Francis, M. P., and Ó Maolagáin, C. (2019). Growth-band counts from elephantfish Callorhinchus milii fin spines do not correspond with independently estimated ages. J. Fish Biol. 95, 743–752. doi: 10.1111/jfb.14060
Fricke, H., Hissmann, K., Froese, R., Schauer, J., Plante, R., and Fricke, S. (2011). The population biology of the living coelacanth studied over 21 years. Mar. Biol. 158, 1511–1522. doi: 10.1007/s00227-011-1667-x
Froese, R., and Palomares, M. L. D. (2000). Growth, natural mortality, length-weight relationship, maximum length and length-at-first-maturity of the coelacanth Latimeria chalumnae. Environ. Biol. Fishes 58, 45–52. doi: 10.1023/A:1007602613607
Gerhard, G. S., Kauffman, E. J., Wang, X., Stewart, R., Moore, J. L., Kasales, C. J., et al. (2002). Life spans and senescent phenotypes in two strains of Zebrafish (Danio rerio). Exp. Gerontol. 37, 1055–1068. doi: 10.1016/S0531-5565(02)00088-8
Hamady, L. L., Natanson, L. J., Skomal, G. B., and Thorrold, S. R. (2014). Vertebral bomb radiocarbon suggests extreme longevity in white sharks. PLoS ONE 9:e84006. doi: 10.1371/journal.pone.0084006
Hara, Y., Yamaguchi, K., Onimaru, K., Kadota, M., Koyanagi, M., Keeley, S. D., et al. (2018). Shark genomes provide insights into elasmobranch evolution and the origin of vertebrates. Nat. Ecol. Evol. 2, 1761–1771. doi: 10.1038/s41559-018-0673-5
Hardie, D. C., and Hebert, P. D. (2004). Genome-size evolution in fishes. Can. J. Fish. Aquat. Sci. 61, 1636–1646. doi: 10.1139/f04-106
Hugg, D. O. (1996). “MAPFISH georeferenced mapping database,” in Freshwater and Estuarine Fishes of North America. Life Science Software, eds O. Dennis and Steven Hugg (Edgewater, MD).
Irisarri, I., Baurain, D., Brinkmann, H., Delsuc, F., Sire, J.-Y., Kupfer, A., et al. (2017). Phylotranscriptomic consolidation of the jawed vertebrate timetree. Nat. Ecol. Evol. 1, 1370–1378. doi: 10.1038/s41559-017-0240-5
Jordan, G., and Goldman, N. (2012). The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol. Biol. Evol. 29, 1125–1139. doi: 10.1093/molbev/msr272
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Marra, N. J., Stanhope, M. J., Jue, N. K., Wang, M., Sun, Q., Bitar, P. P., et al. (2019). White shark genome reveals ancient elasmobranch adaptations associated with wound healing and the maintenance of genome stability. Proc. Natl. Acad. Sci. U.S.A. 116, 4446–4455. doi: 10.1073/pnas.1819778116
Michael, S. W. (2005). Reef Sharks and Rays of the World: A Guide to their Identification, Behavior and Ecology. Inglewood, CA: ProStar Publications, 54.
Morton, N. E. (1991). Parameters of the human genome. Proc. Natl. Acad. Sci. U.S.A. 88, 7474–7476. doi: 10.1073/pnas.88.17.7474
Noonan, J. P., Grimwood, J., Danke, J., Schmutz, J., Dickson, M., Amemiya, C. T., et al. (2004). Coelacanth genome sequence reveals the evolutionary history of vertebrate genes. Genome Res. 14, 2397–2405. doi: 10.1101/gr.2972804
Ojima, Y. (1990). Cellular DNA contents of fishes determined by flow cytometry. La Kromosomo II 57, 1871–1888.
Perry, C. T., Figueiredo, J., Vaudo, J. J., Hancock, J., Rees, R., and Shivji, M. (2018). Comparing length-measurement methods and estimating growth parameters of free-swimming whale sharks (Rhincodon typus) near the South Ari Atoll, Maldives. Mar. Freshwater Res. 69, 1487–1495. doi: 10.1071/MF17393
Postlethwait, J., Amores, A., Force, A., and Yan, Y. L. (2009). “The zebrafish genome,” in Essential Zebrafish Methods: Genetics and Genomics, eds H. W. Detrich III, M. Westerfield, and L. Zon (Cambridge, MA: Academic Press), 47–60.
Read, T. D., Petit, R. A., Joseph, S. J., Alam, M. T., Weil, M. R., Ahmad, M., et al. (2017). Draft sequencing and assembly of the genome of the world's largest fish, the whale shark: Rhincodon typus Smith 1828. BMC Genom. 18:532. doi: 10.1186/s12864-017-4138-z
Schwartz, F. J., and Maddock, M. B. (1986). “Comparisons of karyotypes and cellular DNA contents within and between major lines of elasmobranchs,” in Indo-Pacific Fish Biology: Proceedings of the Second International Conference on Indo-Pacific Fishes (Tokyo: Ichthyol. Soc. of Japan), 148–157.
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Toledo, F., and Wahl, G. M. (2007). MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 39, 1476–1482. doi: 10.1016/j.biocel.2007.03.022
Venkatesh, B., Tay, A., Dandona, N., Patil, J. G., and Brenner, S. (2005). A compact cartilaginous fish model genome. Curr. Biol. 15, R82–R83. doi: 10.1016/j.cub.2005.01.021
Weber, C. C., Nabholz, B., Romiguier, J., and Ellegren, H. (2014). Kr/Kc but not dN/dS correlates positively with body mass in birds, raising implications for inferring lineage-specific selection. Genome Biol. 15:542. doi: 10.1186/s13059-014-0542-8
Xia, X. (2009). “Assessing substitution saturation with DAMBE,” in The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny, eds M. Salemi and A. M. Vandamme (Cambridge, MA: Cambridge University Press), 615–630. doi: 10.1017/CBO9780511819049.022
Xia, X. (2018). DAMBE7: new and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 35, 1550–1552. doi: 10.1093/molbev/msy073
Keywords: Mdm4, positive selection, genome size, longevity, genome, shark
Citation: Yamaguchi K and Kuraku S (2021) Unbiasing Genome-Based Analyses of Selection: An Example Using Iconic Shark Species. Front. Mar. Sci. 8:573853. doi: 10.3389/fmars.2021.573853
Received: 13 October 2020; Accepted: 16 February 2021;
Published: 10 March 2021.
Edited by:
Rob Harcourt, Macquarie University, AustraliaReviewed by:
Gail Schofield, Queen Mary University of London, United KingdomCopyright © 2021 Yamaguchi and Kuraku. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shigehiro Kuraku, c2hpZ2VoaXJvLmt1cmFrdUByaWtlbi5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.