 Wei Yan
Wei Yan Xuejin Feng
Xuejin Feng Wei Zhang
Wei Zhang Muhammad Zohaib Nawaz2
Muhammad Zohaib Nawaz2 Tingwei Luo
Tingwei Luo Rui Zhang
Rui Zhang Nianzhi Jiao
Nianzhi Jiao- 1College of Marine Science and Technology, China University of Geosciences, Wuhan, China
- 2State Key Laboratory of Marine Environmental Science, Fujian Key Laboratory of Marine Carbon Sequestration, College of Ocean and Earth Sciences, Xiamen University, Xiamen, China
- 3Southern Marine Science and Engineering Guangdong Laboratory, Zhuhai, China
Introduction
Members of the cyanobacterium genus Prochlorococcus are the most abundant photosynthetic organisms in global oceans (Chisholm et al., 1992; Partensky et al., 1999), and contributes ~10% of ocean primary productivity (Flombaum et al., 2013). Prochlorococcus clades (ecotypes) are generally divided into viz. high-light-adapted (HL) clades and low-light-adapted clades based on the physiological characteristics, ecological distribution, and phylogeny (Moore and Chisholm, 1999; Rocap et al., 2002; Johnson et al., 2006). With diversified ecotypes, Prochlorococcus maintains a high genomic diversity, and has evolved continuously during the process of adapting to the marine environment (Biller et al., 2015). At least Fifty-two genomes of the Prochlorococcus genus were published (Biller et al., 2014; Yan et al., 2018a,b; Becker et al., 2019).
To date the Prochlorococcus HL clade II (HLII) is regarded as the dominant ecotype in global oceans and accounts for more than 90% of all Prochlorococcus in the upper layer of tropical waters (Johnson et al., 2006), exhibiting a fairly large repertoire of genomic and functional diversity (Partensky and Garczarek, 2010; Kashtan et al., 2014; Biller et al., 2015). HLII has gained significant research interests due to its streamlined genome, making it a model to study genome reduction (Kettler et al., 2007; Partensky and Garczarek, 2010; Biller et al., 2015). The western Pacific Ocean, having both a local and global climate impact (McPhaden et al., 2006), is well-known for low nutrient levels, low primary production, and strong light radiation (Schneider and Zhu, 1998). In addition, as part of the warmest ocean waters, the western Pacific Ocean represents an ideal site to study the effect of rising temperatures on the marine ecosystem (Rowe et al., 2012). However, only five Prochlorococcus genomes are reported in this region to date (Biller et al., 2014; Yan et al., 2018a,b). The present study reports 15 HLII Prochlorococcus and 101 co-cultured heterotrophic bacterial genomes in the western Pacific Ocean and the South China Sea. The genomes discussed here have been deposited in the National Center for Biotechnology Information, and require further analysis to explore the fine-scale diversity of Prochlorococcus and their future applications in marine microbiology and ecology.
Materials and Methods
Isolation of the Prochlorococcus HLII Strains
The Prochlorococcus HLII strains discussed in the present study were isolated from a depth of 50–150 m at seven different stations in the western Pacific Ocean and the South China Sea in 2014 (Supplementary Figure 1; Table 1). The isolation process was performed as previously described (Yan et al., 2021). Briefly, seawater collected by a Niskin bottle was subjected to gravity filtration through double polycarbonate filters (Millipore, USA) with a pore size of 0.6 μm (Chisholm et al., 1992). Then, a Pro2 medium nutrient stock solution was added to the filtrate (Moore et al., 2007). The filtrate was placed in an incubator onboard for initial enrichment for 4–8 weeks. After confirmation by a flow cytometer, the Prochlorococcus strains were maintained at a constant temperature of 22°C and a continuous light intensity of 10–20 μmol photons m−2s−1.
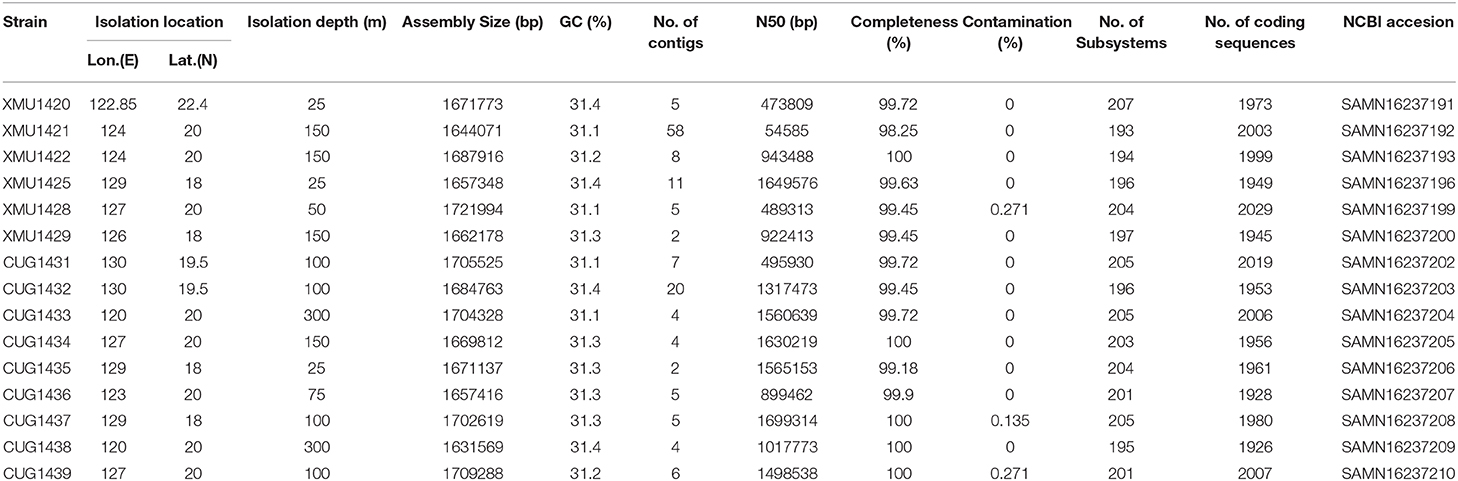
Table 1. Genome characteristics and assembly statistics of the Prochlorococcus HLII strains sequenced in the present study.
DNA Isolation, Library Preparation, and DNA Sequencing
DNA isolation, library preparation, and DNA sequencing were performed as previously described (Yan et al., 2021). Briefly, genomic DNA was collected from 25 ml laboratory cultures by centrifugation (10,000×g for 30 min) and extracted using a QIAamp DNA mini kit (Qiagen, Germany). One μg of extracted DNA was fragmented by a Covaris ME220 Focused-ultrasonicator (Covaris, USA). DNA library was constructed using a NEBNext® Ultra™ DNA Library Prep Kit for Illumina® in accordance with the manufacturer's instructions (NEB, USA). Ten ng of library DNA was taken and subjected to bidirectional sequencing using an Illumina NovaSeq 6000 instrument with a read length of 150 bp. All library construction and sequencing were performed at Shanghai Majorbio Bio-pharm Technology Co., Ltd (Shanghai, China).
Assembly and Annotation
To recover Prochlorococcus and heterotrophic bacterial genomes from non-axenic cultures, genome assembly was performed using the MetaWRAP v1.2.1 pipeline on a Linux cluster with 96 cores and 512 GB of RAM (Uritskiy et al., 2018). Briefly, the reads from all samples were trimmed using the metaWRAP-Read_qc module and then individually assembled with the metaWRAP assembly module using MEGAHIT as a metagenomic assembler (Li et al., 2015). Bins were calculated using three binning modules, including CONCOCT (Alneberg et al., 2014), MetaBat (Kang et al., 2015), and MaxBin2 (Wu et al., 2016). Then, the bin_refinement module was used to combine and improve the results generated by the three binners. Finally, the reassemble_bins module was used to attain better bins. The quality cutoffs of all steps were set as: completeness > 90% and contamination <5% using CheckM (Parks et al., 2015). The assembled genomes were annotated using the Rapid Annotation using Subsystem Technology online server (FIGfam version Release 70) (Aziz et al., 2008).
Phylogenomic Analysis
Phylogenetic relationships of HLII Prochlorococcus strains were reconstructed by using concatenated protein sequences. Briefly, protein sequences of 1102 core genes defined at 70% similarity level were aligned with MUSCLE (Edgar, 2004) and concatenated using Bacterial Pangenome Analysis Pipeline v1.3.0 (Chaudhari et al., 2016). Gblocks software v0.91b was used for degaping in the divergent regions (Castresana, 2000). The neighbor joining phylogenetic tree with 1000 bootstraps was constructed using MEGA X (Kumar et al., 2018), and visualized using iTOL version 4 (Letunic and Bork, 2019).
Evolutionary relationships and taxonomic classification of co-cultured heterotrophic bacteria were reconstructed based on concatenated amino acid sequences of 120 bacterial ortholog genes using GTDB-tk v1.3.0 (Chaumeil et al., 2019). Briefly, the 120 ortholog genes were identified and aligned with HMMER (Finn et al., 2011), concatenated into a single multiple sequence alignment, and trimmed with the 5,000-column bacterial mask (Chaumeil et al., 2019). The maximum likelihood phylogenetic tree was constructed using FastTree v2.1.10 (Price et al., 2009). The phylogenetic tree presented in the present study was visualized using iTOL version 4 (Letunic and Bork, 2019).
Pan-Genome Analysis
Gene-based pan-genome analysis of the HLII Prochlorococcus strains was conducted using the Bacterial Pangenome Analysis Pipeline v1.3.0 (Chaudhari et al., 2016), which uses the USEARCH algorithms to identify core genes. The similarity cutoff for amino acid sequences of the core gene was set as 50%.
Interpretation of the Data
Genomic Data of the Prochlorococcus HLII Strains
In the present study, The DNA of 15 Prochlorococcus HLII cultures were sequenced, and the raw data were quilty controlled and assemblied. Genome sizes of the 15 HLII isolates ranged from 1,631,569 bp to 1,721,994 bp, with an average GC content of 31.26% (s.d. = 0.11) and an average of 1,976 coding sequences per genome (s.d. = 28) (Table 1). Their phylogenetic position was confirmed through phylogenomic tree construction using core genome amino acid sequences at a similarity cutoff of 70% (Figure 1). In this study, 101 co-cultured heterotrophic bacterial genomes were obtained from the non-axenic HLII cultures using the binning method. The completeness of these genomes ranged from 90.2 to 100%, with an average of 97.7%. The completeness, contamination, genome size, GC content, and number of coding sequences of each genome are shown in Supplementary Table 1. Their phylogenetic positions were confirmed through phylogenetic trees using 120 bacterial orthologous genes (Figure 2). The phylogenetic tree comprised four bacterial clades, including Alphaproteobacteria, Rhodothermia, Gammaproteobacteria, and Bacteroidia, and was further sperated into six classes, 22 families, and 37 genera (Figure 2; Supplementary Table 1).

Figure 1. Phylogenetic tree of the Prochlorococcus strains sequenced in the present study. A neighbor joining phylogenetic tree was reconstructed using the protein sequences of 1102 core genes of 46 high-light-adapted clade II strains, with a high-light-adapted clade I strain (MED4) as an outgroup. Bootstrap values for 1,000 resamplings are indicated by numbers at the nodes (at least 50% support).
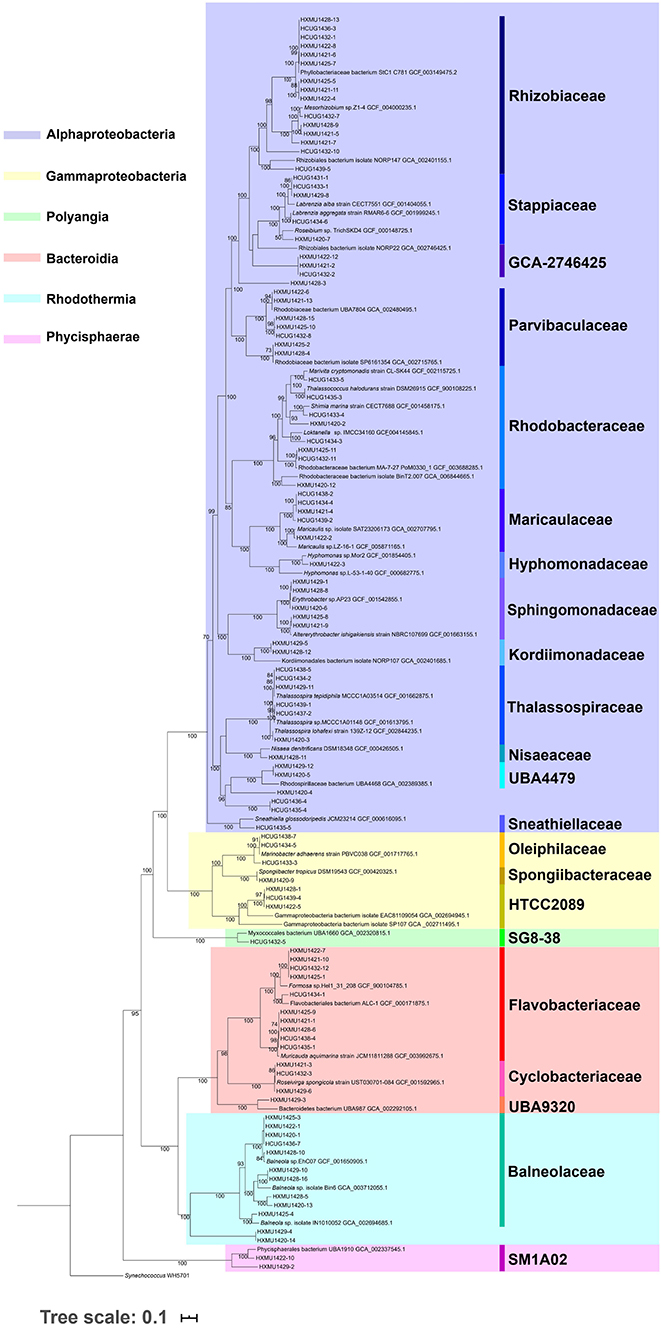
Figure 2. Phylogenetic tree of 101 co-cultured heterotrophic bacteria genomes, with the Synechococcus WH5701 strain as an outgroup. A maximum likelihood phylogenetic tree was reconstructed using the concatenated amino acid sequences of 120 bacterial ortholog genes. Bootstrap values for 1,000 resamplings are indicated by numbers at the nodes (at least 50% support).
Reuse Potential
The genomes of the Prochlorococcus HLII strains and their co-cultured heterotrophic bacteria discussed here warrant further analysis to explore the fine-scale diversity of Prochlorococcus and co-cultured heterotrophic bacteria, and their future applications in marine microbiology and ecology.
Data Availability Statement
The raw sequence data and assemblied genome data of the 15 HL II Prochlorococcus strains and 101 co-cultured heterotrophic bacteria reported in the present study have been deposited in the National Center for Biotechnology Information GenBank under the BioProject number PRJNA664924. Accession numbers for the individual genomes used in this study are listed in Table 1 and Supplementary Tables 1, 2.
Author Contributions
WY, RZ, and NJ designed the study. WY conducted the isolation of Prochlorococcus strains. XF, WZ, and TL participated in genome analysis. WY, MN, and RZ wrote the manuscript. All authors contributed to the final version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (41861144018 to WY, 91751207 to TL), China Ocean Mineral Resources R&D Association (DY135-E2-1-04 to NJ), the Fundamental Research Funds for the Central Universities (20720190141 to WY, 20720170107 to NJ). The Senior User Project of RV KEXUE (KEXUE2020G10 to RZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.619826/full#supplementary-material
References
Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Aziz, R. K., Bartels D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Becker, J. W., Hogle, S. L., Rosendo, K., and Chisholm, S. W. (2019). Co-culture and biogeography of Prochlorococcus and SAR11. ISME J.13, 1–14. doi: 10.1038/s41396-019-0365-4
Biller, S. J., Berube, P. M., Berta-Thompson, J. W., Kelly, L., Roggensack, S. E., Awad, L., et al. (2014). Genomes of diverse isolates of the marine cyanobacterium Prochlorococcus. Sci. Data 1:140034. doi: 10.1038/sdata.2014.34
Biller, S. J., Berube, P. M., Lindell, D., and Chisholm, S. W. (2015). Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Micro. 13, 13–27. doi: 10.1038/nrmicro3378
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chisholm, S. W., Frankel, S. L., Goericke, R., Olson, R. J., Palenik, B., Waterbury, J. B., et al. (1992). Prochlorococcus marinus nov. gen. nov. sp.: an oxyphototrophic marine prokaryote containing divinyl chlorophyll a and b. Arch. Microbiol. 157, 297–300. doi: 10.1007/BF00245165
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Flombaum, P., Gallegos, J. L., Gordillo, R. A., Rincón, J., Zabala, L. L., Jiao, N., et al. (2013). Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. U.S.A. 110, 9824–9829. doi: 10.1073/pnas.1307701110
Johnson, Z. I., Zinser, E. R., Coe, A., McNulty, N. P., Woodward, E. M. S., and Chisholm, S. W. (2006). Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311, 1737–1740. doi: 10.1126/science.1118052
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165–e1115. doi: 10.7717/peerj.1165
Kashtan, N., Roggensack, S. E., Rodrigue, S., Thompson, J. W., Biller, S. J., Coe, A., et al. (2014). Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344, 416–420. doi: 10.1126/science.1248575
Kettler, G. C., Martiny, A. C., Huang, K., Zucker, J., Coleman, M. L., Rodrigue, S., et al. (2007). Patterns and implications of gene gain and loss in the evolution of Prochlorococcus. PLoS Genet. 3:e231. doi: 10.1371/journal.pgen.0030231
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
McPhaden, M. J., Zebiak, S. E., and Glantz, M. H. (2006). ENSO as an integrating concept in earth science. Science 314, 1740–1745. doi: 10.1126/science.1132588
Moore, L. R., and Chisholm, S. W. (1999). Photophysiology of the marine cyanobacterium Prochlorococcus: ecotypic differences among cultured isolates. Limnol. Oceangr. 44, 628–638. doi: 10.4319/lo.1999.44.3.0628
Moore, L. R., Coe, A., Zinser, E. R., Saito, M. A., Sullivan, M. B., Lindell, D., et al. (2007). Culturing the marine cyanobacterium Prochlorococcus. Limnol. Oceanogr. Methods 5, 353–362. doi: 10.4319/lom.2007.5.353
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Partensky, F., and Garczarek, L. (2010). Prochlorococcus: advantages and limits of minimalism. Ann. Rev. Mar. Sci. 2, 305–331. doi: 10.1146/annurev-marine-120308-081034
Partensky, F., Hess, W. R., and Vaulot, D. (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63, 106–127. doi: 10.1128/MMBR.63.1.106-127.1999
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Rocap, G., Distel, D. L., Waterbury, J. B., and Chisholm, S. W. (2002). Resolution of Prochlorococcus and Synechococcus ecotypes by using 16S-23S ribosomal DNA internal transcribed spacer sequences. Appl. Environ. Microbiol. 68, 1180–1191. doi: 10.1128/AEM.68.3.1180-1191.2002
Rowe, J. M., DeBruyn, J. M., Poorvin, L., LeCleir, G. R., Johnson, Z. I., Zinser, E. R., et al. (2012). Viral and bacterial abundance and production in the Western Pacific Ocean and the relation to other oceanic realms. FEMS Microbiol. Ecol. 79, 359–370. doi: 10.1111/j.1574-6941.2011.01223.x
Schneider, E. K., and Zhu, Z. (1998). Sensitivity of the simulated annual cycle of sea surface temperature in the equatorial Pacific to sunlight penetration. J. Climate 11, 1932–1950. doi: 10.1175/1520-0442-11.8.1932
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 1–13. doi: 10.1186/s40168-018-0541-1
Wu, Y.-W., Simmons, B. A., and Singer, S. W. (2016). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Yan, W., Feng, X., Lin, T.-H., Huang, X., Le, X.ie, Wei, S., et al. (2021). Diverse subclade differentiation attribute to the ubiquity of Prochlorococcus high-light adapted clade II in marine environments. Submited.
Yan, W., Wei, S., Wang, Q., Xiao, X., Zeng, Q., Jiao, N., et al. (2018a). Genome rearrangement shapes Prochlorococcus ecological adaptation. Appl. Environ. Microb. 84, e01178–e01118. doi: 10.1128/AEM.01178-18
Keywords: Prochlorococcus, high-light-adapted clade II, genomes, the western Pacific Ocean, co-cultured heterotrophic bacteria
Citation: Yan W, Feng X, Zhang W, Nawaz MZ, Luo T, Zhang R and Jiao N (2021) Genomes of Diverse Isolates of Prochlorococcus High-Light-Adapted Clade II in the Western Pacific Ocean. Front. Mar. Sci. 7:619826. doi: 10.3389/fmars.2020.619826
Received: 09 November 2020; Accepted: 31 December 2020;
Published: 16 February 2021.
Edited by:
Zhijun Dong, Yantai Institute of Coastal Zone Research (CAS), ChinaReviewed by:
Zhenming Lv, Zhejiang Ocean University, ChinaJian-Wen Qiu, Hong Kong Baptist University, Hong Kong
Copyright © 2021 Yan, Feng, Zhang, Nawaz, Luo, Zhang and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Yan, eWFud2VpQGN1Zy5lZHUuY24=; Rui Zhang, cnVpemhhbmdAeG11LmVkdS5jbg==; Nianzhi Jiao, amlhb0B4bXUuZWR1LmNu