 Xiao Chen
Xiao Chen Chundi Wang1†
Chundi Wang1† Bo Pan
Bo Pan Borong Lu
Borong Lu Zhuo Shen
Zhuo Shen Lifang Li
Lifang Li- 1Marine College, Shandong University, Weihai, China
- 2Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao, China
- 3Institute of Microbial Ecology and Matter Cycle, School of Marine Sciences, Sun Yat-sen University, Zhuhai, China
- 4Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai, China
- 5Department of Life Sciences, Natural History Museum, London, United Kingdom
Peritrichs are one of the largest groups of ciliates with over 1,000 species described so far. However, their genomic features are largely unknown. By single-cell genomic sequencing, we acquired the genomic data of three sessilid peritrichs (Cothurnia ceramicola, Vaginicola sp., and Zoothamnium sp. 2). Using genomic data from another 53 ciliates including 14 peritrichs, we reconstructed their evolutionary relationships and confirmed genome skimming as an efficient approach for expanding sampling. In addition, we profiled the stop codon usage and programmed ribosomal frameshifting (PRF) events in peritrichs for the first time. Our analysis reveals no evidence of stop codon reassignment for peritrichs, but they have prevalent +1 or -1 PRF events. These genomic features are distinguishable from other ciliates, and our observations suggest a unique evolutionary strategy for peritrichs.
Introduction
Peritrich ciliates have ubiquitous distribution and occupy a broad array of freshwater, brackish water, marine, and terrestrial ecosystems (Lynn, 2008; Zhuang et al., 2016; Lu et al., 2019, 2020). They are also one of the largest groups within the phylum Ciliophora Doflein, 1901 with over 1,000 nominal species. Historically, the subclass Peritrichia Stein, 1859 was considered to be a well-defined, monophyletic group comprising two orders, the Sessilida Stein, 1859, and the Mobilida Kahl, 1933 (Corliss, 1979; Lynn and Small, 2002; Figure 1). In recent years, however, the monophyly of the subclass Peritrichia has been questioned due to discordance between the molecular and morphological evidence (Miao et al., 2001; Utz and Eizirik, 2007; Williams and Clamp, 2007; Zhan et al., 2009; Sun et al., 2013). In particular, analyses based on sequences of ribosomal DNA (rDNA; 18S, 5.8S, and 28S) and alpha-tubulin, either individually or in combination, show that the order Sessilida clusters with the subclass Hymenostomatia rather than with the order Mobilida, rendering the subclass Peritrichia non-monophyletic (Utz and Eizirik, 2007; Zhan et al., 2009; Gao et al., 2016). Following the development of next-generation sequencing techniques, two studies using phylogenomic analysis of sequences from more than 100 genomic loci concluded that the orders Sessilida and Mobilida are sister groups, and the subclass Peritrichia is monophyletic (Gentekaki et al., 2017; Jiang et al., 2019). However, genomic data are available for only about 2% of known peritrich species and are lacking for several families (Jiang et al., 2019), so it is probably too early to draw solid conclusions on high-level peritrich systematics based on phylogenomic analyses.
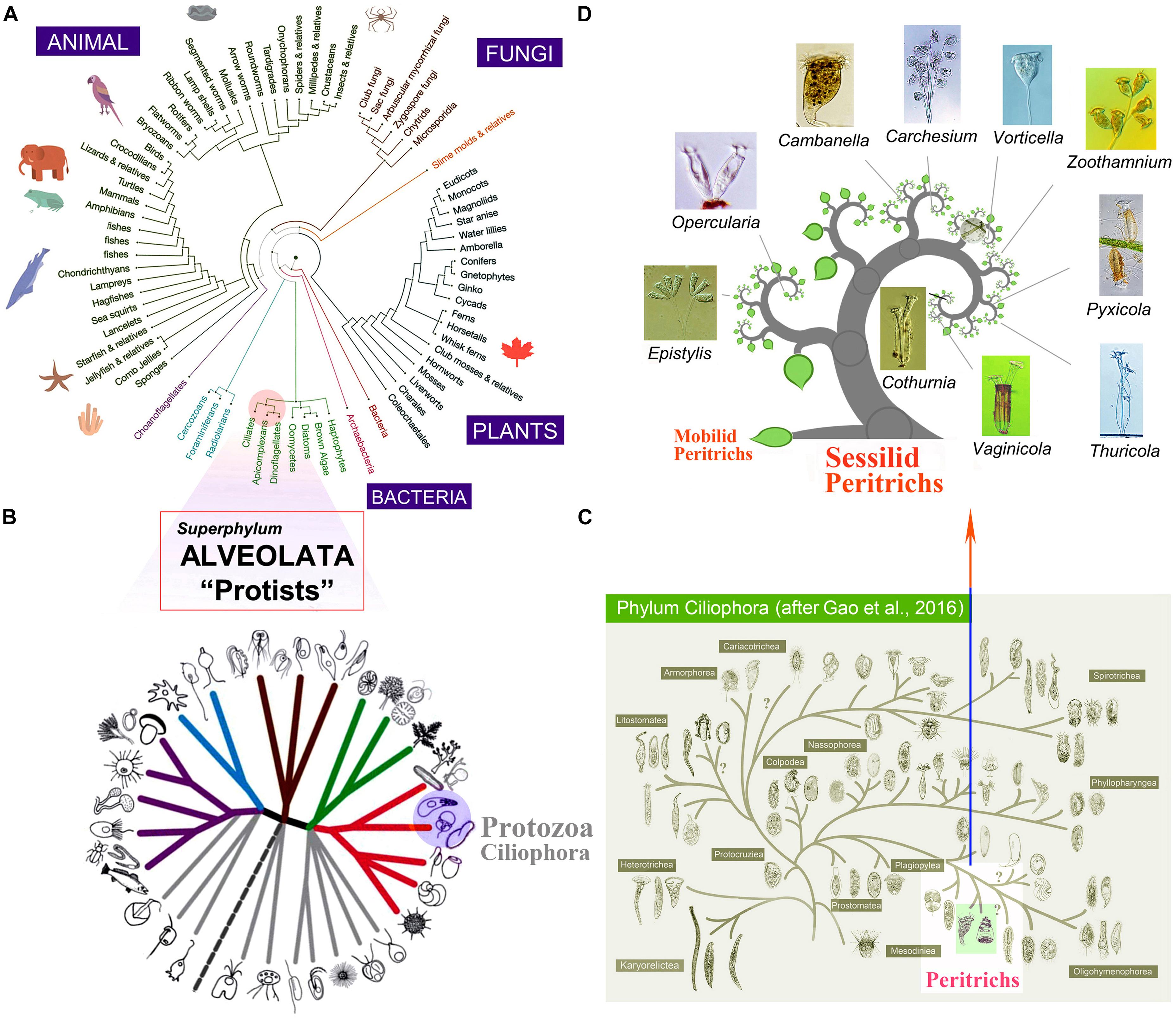
Figure 1. Systematic arrangement of peritrichs in the evolutionary history and current taxonomy of phylum Ciliophora. (A) Tree of life, adapted from Pennisi (2003). (B) Classification of eukaryotes, adapted from Adl et al. (2019). (C) Classification of ciliates, adapted from Gao et al. (2016). (D) The phylogeny of peritrichs based on 18S rDNA gene sequences, generated by OneZoom (Rosindell and Harmon, 2012).
Previous studies have reported the large flexibility of the nuclear genetic code in ciliates by demonstrating that standard stop codons are reassigned to amino acids (Lozupone et al., 2001; Swart et al., 2016). To address the question of how ambiguous genetic codes enabled species to thrive during their evolutionary history, evolutionary biologists have intensively investigated stop codon preference in both bacteria and eukaryotes (Alff-Steinberger and Epstein, 1994; Belinky et al., 2018). The stop codon reassignment in euplotid ciliates has also been linked to programmed ribosomal frameshifting (PRF) events (Wang et al., 2016; Lobanov et al., 2017; Chen et al., 2019). For peritrichs, such genomic features are still far from clear since many are not culturable and thus cannot satisfy the requirement of sufficient amounts of DNA for whole-genome sequencing.
To access the genomic profiles of unexplored peritrich lineages, we applied the single-cell genomic sequencing technique to three sessilid peritrichs, i.e., the marine species Cothurnia ceramicola and Zoothamnium sp. 2 and a freshwater species of Vaginicola. Based on genomic data of peritrichs and other ciliates, both from the current work and previous studies, we systematically reconstructed their phylogenomic relationships and, for the first time, profiled the stop codon usage and PRF events in peritrich genomes. Understanding the genomic features of peritrichs may help uncover their evolutionary history and could improve our understanding of their unique advantages for environmental adaptation.
Materials and Methods
Single-Cell Sample Preparation
Cothurnia ceramicola and Zoothamnium sp. 2 were collected from seawater along the coast of the Yellow Sea at Qingdao (35°56′18″ N, 120°12′44″ E and 36°03′03″ N, 120°21′01″ E, respectively), China. Vaginicola sp. was collected from a freshwater pond in Baihuayuan Park at Qingdao (36°03′58″ N, 120°20′24″ E), China. Ciliates were detached from their substrate (aquatic plants) using a glass micropipette under a stereomicroscope (40×). Specimens were observed in vivo with differential interference contrast microscopy (40× to 1,000×) and following silver staining, i.e., protargol staining to reveal their infraciliature and wet silver nitrate staining to reveal their silverline system (Song and Wilbert, 1995). They were identified based on morphological characteristics, such as body size, contractile vacuole, spasmoneme, lorica, branching pattern of the stalk patterns of infracilature, and numbers of silverlines using published keys and guides (Song et al., 2009; Lu et al., 2019). C. ceramicola is characterized by its cylindroid-shaped and annulated lorica, with a striated stalk, one-fourth to one-third of the body projecting outside the lorica, and marine habitat. Zoothamnium sp. 2 has the typical characters of its genus, i.e., colonial, with a continuous spasmoneme that runs throughout the entire colony causing the stalk to contract in a “zig-zag” fashion, and with transverse silverlines. Vaginicola sp. has a lorica that adheres directly to the substrate by the posterior end without a stalk. A single cell of each species was washed in phosphate-buffered saline (PBS) buffer (without Mg2+ or Ca2+), and genomic DNA amplification was carried out by MALBAC (Lu et al., 2012) using the Single-Cell WGA Kit (Yikon, YK001A) according to the manufacturer’s guidelines.
Illumina Sequencing and Genome Assembly
Illumina libraries were prepared from amplified single-cell genomic DNA according to manufacturer’s instructions and paired-end sequencing (150 bp read length) was performed using an Illumina HiSeq4000 sequencer. The sequencing adapter was trimmed, and low-quality reads (reads containing more than 10% Ns or 50% bases with Q value < 5) were filtered out. The single-cell genome of each species was assembled using SPAdes v3.7.1 (-k 21,33,55,77; Bankevich et al., 2012; Nurk et al., 2013). Oxytricha trifallax mitochondrial genomic peptides and bacterial genomes were downloaded from GenBank as BLAST databases to remove contamination caused by mitochondria or bacteria (BLAST E < 1.0e-5). CD-HIT v4.6.1 (CD-HIT-EST, -c 0.98 -n 8 -r 1) was employed to eliminate the redundancy of contigs (with sequence identity threshold = 98%; Fu et al., 2012). Poorly supported contigs (coverage < 1 or >20 and length < 400 bp) were discarded, considering the inherent characteristics of single-cell genomic sequencing technique (Zong et al., 2012).
Ortholog Detection and Phylogenomic Analysis
Genome-wide gene predictions were performed using AUGUSTUS v3.2.2 (—species = peritrichia, trained by transcriptomic data of the sessilid Vorticella microstoma; Stanke et al., 2006). Orthologs were detected from the top hits of homolog sequence alignment between predicted protein sequences and the ciliate protein library from National Center for Biotechnology Information (NCBI) GenBank using BLASTP version 2.3.0 (E value < 1e-5; Camacho et al., 2009). The orthologs detected from predicted protein sequences were aligned with 157 well-defined ciliate proteins from another 53 ciliates and from 13 apicomplexans/dinoflagellates that were used as outgroups taxa. Phylogenomic analysis was carried out using GPSit (relaxed masking mode, Supplementary Table 1 and Supplementary Figure 1; Chen et al., 2018). The gene recovery rate (GRR) of a taxon was equivalent to its coverage of a total of 157 multiple sequence alignments (MSAs) and calculated as previously described (Chen et al., 2018). The concatenated dataset output from GPSit was used for phylogenomic analysis on CIPRES Science Gateway server v3.3 (phylo.org; Miller et al., 2010). RAxML-HPC2 v8.2.9 under LG model of amino acid substitution (Γ distribution + F, four rate categories, 500 bootstrap replicates) was used to perform maximum likelihood (ML) analysis (Stamatakis, 2014). PhyloBayes MPI 1.5a (CAT-GTR model + Γ distribution, four independent chains, 10,000 generations with 10% burn-in, convergence Maxdiff < 0.3) was used to perform Bayesian inference (BI) analysis (Lartillot et al., 2009). The phylogenetic tree was visualized using MEGA v7.0.20 (Kumar et al., 2016).
Stop Codon Usage and PRF Events Detection
Usage of the stop codons (TAA, TGA, and TAG) was measured from the homolog sequence alignment between the predicted gene sequences, or transcripts from each species and the ciliate protein library, using BLASTX version 2.3.0 (E value cutoff = 1e-5) and a Perl script (Camacho et al., 2009; Chen et al., 2019). Stop codon usage bias was examined by the chi-squared test. PRF events were detected from the homolog sequence alignment using the R package FScanR1. To identify high-confidence PRF events and avoid false-positive frameshifting introduced by introns, the fidelity of homolog sequence alignments and the distance between two hits with changed frames were strictly controlled with rigorous cutoffs (evalue_cutoff = 1e-10, frameDist_cutoff = 10 nt).
Results and Discussion
Genomic Profiles of Peritrichs by Single-Cell Sequencing
We collected the genomic data from single cells and assembled the genomes for three peritrich species, C. ceramicola, Vaginicola sp., and Zoothamnium sp. 2 (Figure 2A). The mean size of genome assemblies is 104 Mb (Table 1). The large contig numbers (94,558 on average) and small N50 (1,539 on average) indicate that the genome assembly is not integral, probably due to the bias of whole-genome amplification, which is a necessary procedure during the library preparation in single-cell genomic sequencing (Lu et al., 2012). However, genome skimming is a suitable technique for generating data that can be used in several downstream analyses such as ortholog detection and phylogenetic analyses.
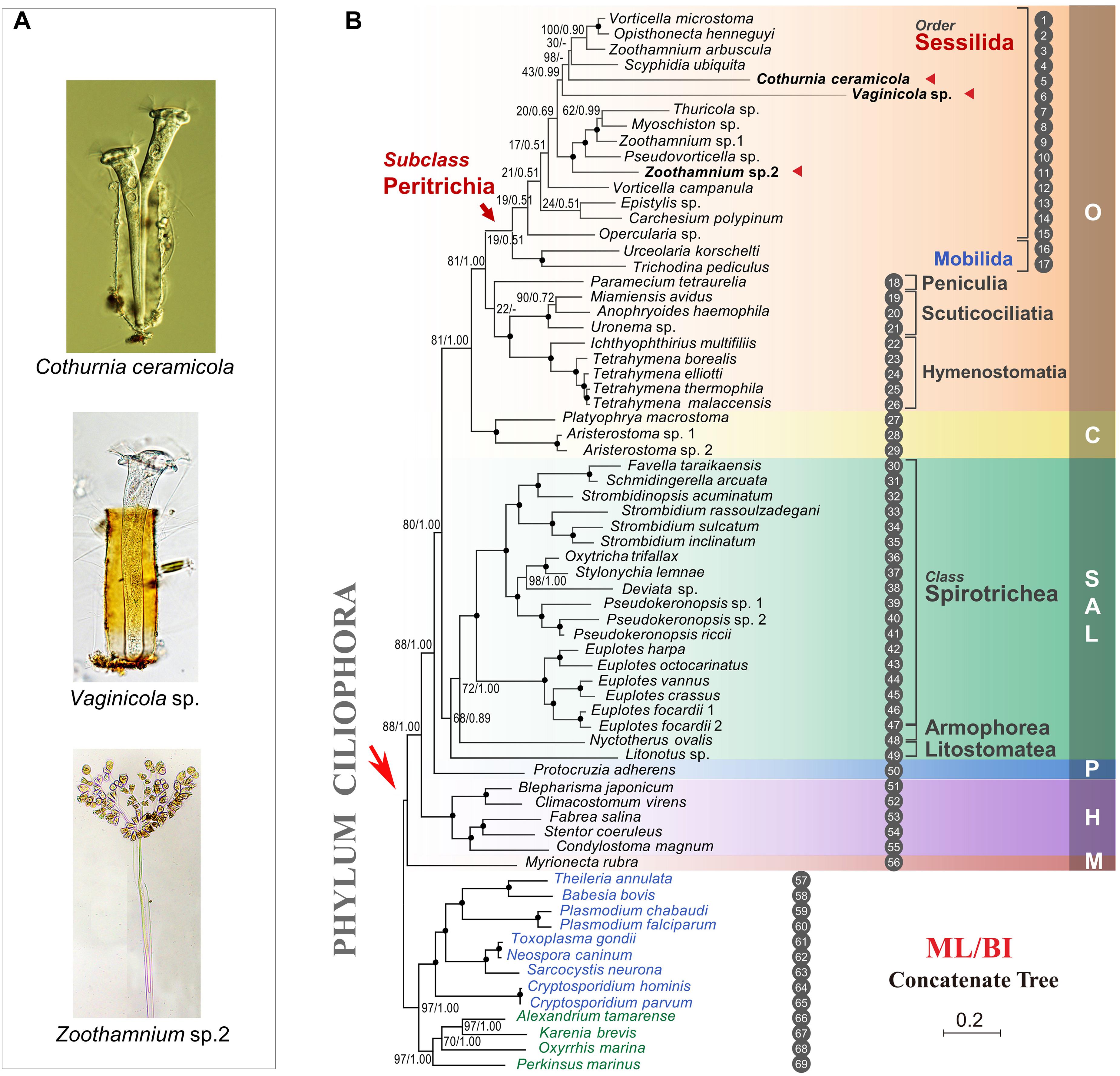
Figure 2. Phylogenomic analysis based on genome-wide homolog protein sequences. (A) Photomicrographs of Cothurnia ceramicola, Vaginicola sp., and Zoothamnium sp. 2 (B) Phylogenomic trees estimated from a 157-gene concatenated dataset by maximum likelihood (ML) and Bayesian inference (BI). Black dots denote full support (ML 100%/BI 1.0). Hyphens denote topological disagreement between BI and ML analyses. The three sessilid peritrichs sequenced in the current work are in bold and are marked by red triangles. The outgroup taxa, nine apicomplexans, and four dinoflagellates, are in blue and green, respectively. SAL, classes Spirotrichea, Armophorea, and Litostomatea; O, class Oligohymenophorea; C, class Colpodea; P, class Protocruziea; H, class Heterotrichea; and M, class Mesodiniea. Scale bar corresponds to 20 substitutions per 100 nucleotide positions. The numbers in the circles adjacent to species names correspond to those in Supplementary Table 1 and Supplementary Figure 1.
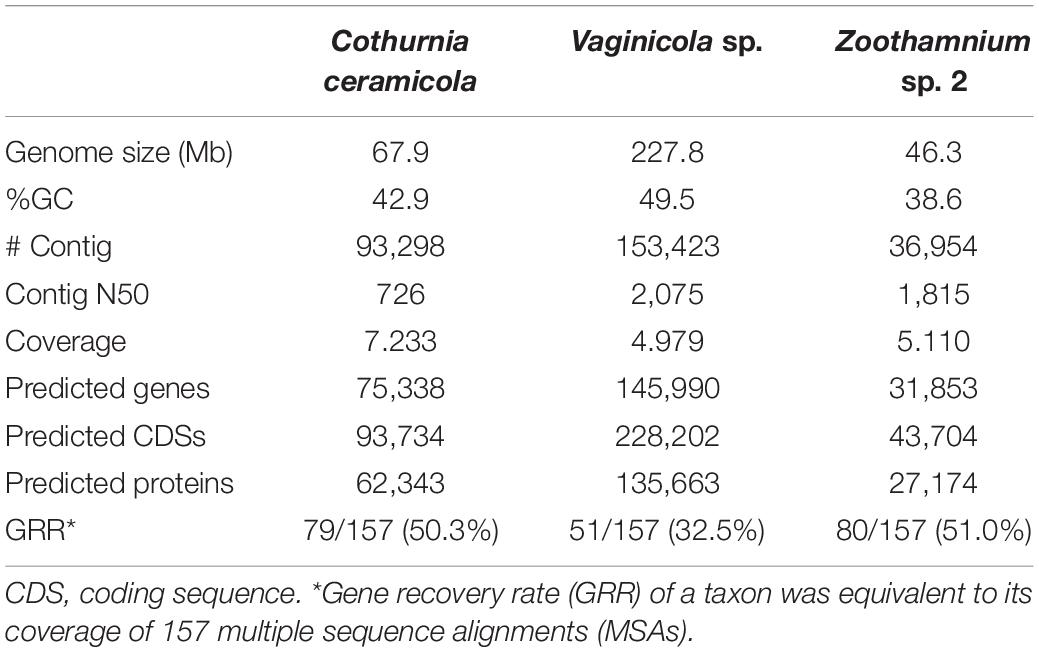
Table 1. Information of single-cell genome assemblies.
After gene annotation and ortholog detection, the GRR, which is equivalent to the coverage of 157 common orthologous genes in each ciliate taxon (Gentekaki et al., 2014; Chen et al., 2018), is 50.3% for C. ceramicola, 32.5% for Vaginicola sp., and 51.0% for Zoothamnium sp. 2. These values are comparable to those for other ciliates from public datasets (Table 1 and Supplementary Table 1). Although the coverage of gene sequences in these datasets is not as high as the bulk genomic or transcriptomic sequencing, single-cell whole-genome sequencing provides the opportunity to expand the phylogenetic scope to more peritrich lineages and to those species that are non-culturable.
Expanded Sampling by Single-Cell Sequencing Partially Supports the Monophyly of the Subclass Peritrichia
With expanded sampling and new single-cell genomic sequencing profiles from three sessilid peritrichs, we reconstructed their phylogenomic relationships using both ML and Bayesian inference (BI) methods (Figure 2B). The reconstructed phylogenomic trees show that the three species cluster with the 14 other peritrich species from previous studies using bulk genome/transcriptome sequencing. Furthermore, Zoothamnium species do not cluster together (see Zoothamnium sp. 1 and Z. sp. 2 in Figure 2B), as reported in previous studies based on either genomic-scale data or single locus sequences (Li et al., 2008; Zhuang et al., 2018; Jiang et al., 2019; Wu et al., 2020). These findings suggest that genome skimming from single cells is an efficient approach for expanding the sampling of genomic data to more ciliate species and resolving their phylogenomic positions. Our analysis supports the monophyly of the subclass Peritrichia, although the support values are low (ML, 19%; BI, 0.51). However, the application of genome skimming from single cells should enable more peritrich lineages to be included in future investigations, thereby facilitating more reliable conclusions to be drawn concerning the systematics of the Peritrichia.
Stop Codons Are Not Reassigned in Peritrichs
A recent study profiling stop codon usage in 33 ciliates across nine classes revealed that all 33 show stop codon reassignment (Pan et al., 2019). Most (30 out of 33) of the species investigated use either TAA or TGA as the biased stop codon and reassign the other two stop codons to code amino acids. In each of the five species representing the class Oligohymenophorea, TAA and TAG were reassigned to code the amino acid glutamine leaving TGA as the only stop codon. In the current study, we depict the stop codon usage in peritrichs for the first time (Figure 3). In contrast to other oligohymenophorean species, 10 out of 12 peritrichs show no sign of significant bias in stop codon usage, which suggests a unique evolutionary strategy of peritrichs.

Figure 3. Stop codon usage analysis shows most peritrichs have no significant bias in the usage frequency of three stop codons in their genomes. The color scale denotes the percentage of the usage frequency for each stop codon in each species. ***Significant biased stop codon usage (p < 0.001; Fisher’s exact test).
Previous studies in bacteria show that three stop codons are decoded by two release factors (RF1 for UAA and UAG and RF2 for UAA and UGA) and that UGA is used much more frequently than UAG in Escherichia coli, as RF2 is consistently more abundant than RF1 (Scolnick et al., 1968; Milman et al., 1969; Scolnick and Caskey, 1969; Korkmaz et al., 2014). This association between the frequency of a stop codon and its decoder concentration has also been documented in eukaryotes (Chavancy et al., 1979). Although a recent study suggests that GC content has a major impact on stop codon frequencies (Belinky et al., 2018), the translation termination mechanism in ciliates remains poorly understood and should be addressed in future studies.
+1 and −1 PRF Events Are Prevalent in Peritrichs
Based on the available ciliate genomic or transcriptomic data, we systematically investigated PRF events in ciliates (Figure 4). As the shortest spliceosomal introns currently known are reported in Stentor (15–16 nt; Slabodnick et al., 2017), unrecognized tiny introns may cause false-positive PRF events in those species for which transcriptomic data are lacking. To avoid this potential issue and identify high-confidence PRF events from genomic data, we searched for the adjacent homolog sequence alignment between ciliate genomic DNA and ciliate protein sequences with frames changed within a narrow window of 10 nt. PRF events (14 on average) were detected in 93% (42 out of 45) of the species examined. Furthermore, we identified a large number of +1 PRF events (34 on average) in all five species of Euplotes included in the analysis. This is consistent with previous studies that indicated that +1 PRF events are highly abundant in Euplotes (Wang et al., 2016; Chen et al., 2019). The present study revealed that, of 12 the peritrich species included in the analysis, 11 have prevalent +1 and -1 PRF events (12 and four events on average, respectively) in their genomes, which is not common to many ciliate groups. Our systematic investigation also revealed abundant +1 PRF and -1 PRF events in the genomes of Paramecium tetraurelia (58 events) and Ichthyophthirius multifiliis (37 events), respectively. The two representatives of the class Colpodea included in this study showed an intermediate frequency of PRF events of all four types (−2, −1, +1, +2) without an obvious bias toward any one in particular. These observations may help us understand their evolutionary history but need further validation by expanded sampling of more species.
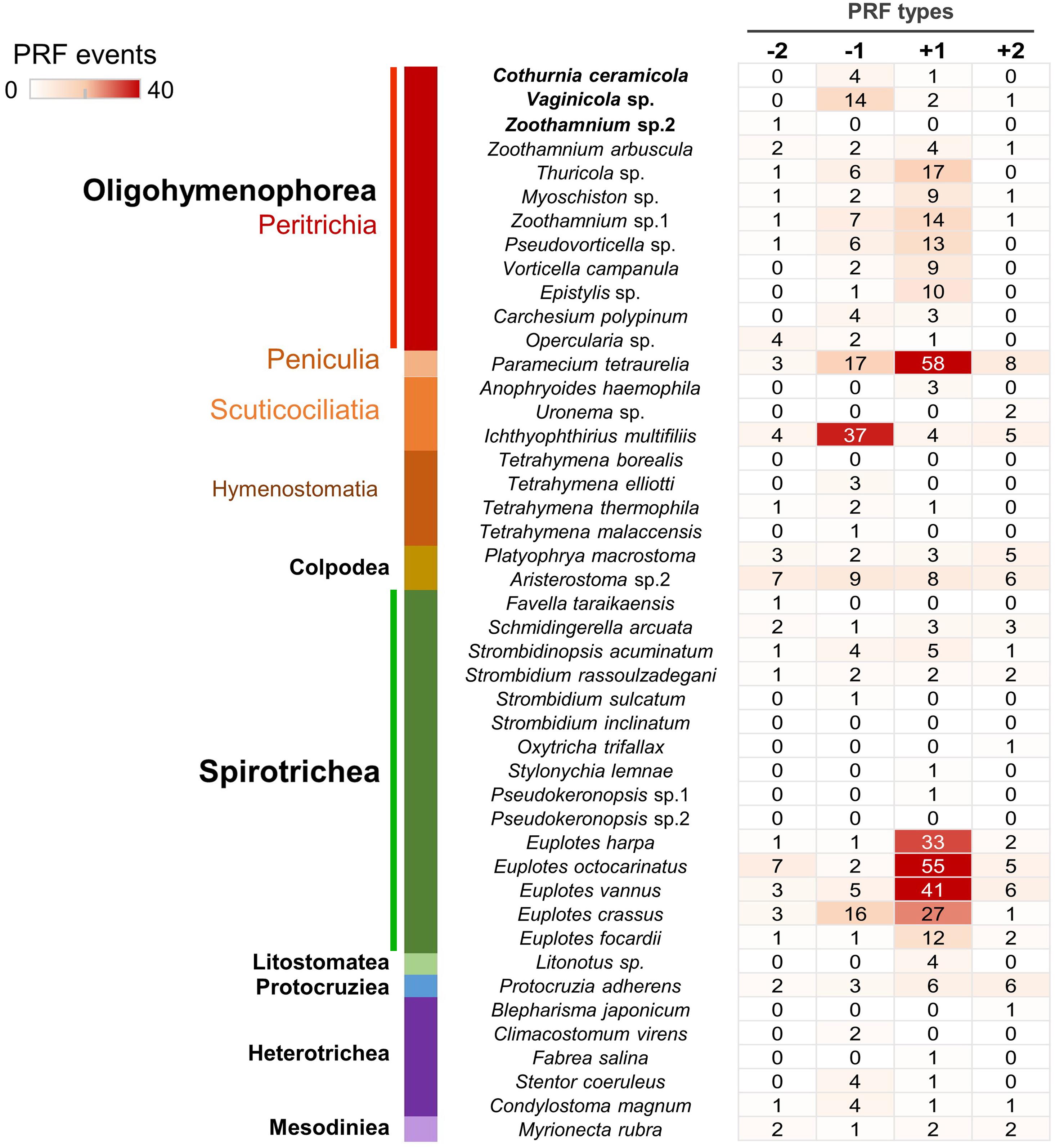
Figure 4. Systematic investigation of programmed ribosomal frameshifting (PRF) events in ciliate genomes reveals that +1 and -1 PRF events are prevalent in peritrichs. The color scale denotes the number of four types of PRF events in each species. The class names are labeled in bold, and the subclass names are labeled in color.
Concluding Remarks
In the current work, we applied single-cell genomic sequencing to representatives of three well-known sessilid peritrich genera. The results of our phylogenomic analysis partially supports the monophyly of the subclass Peritrichia. We also systematically investigated the stop codon reassignment and PRF events in peritrichs and other ciliates. Our findings reveal that peritrichs have less biased stop codon usage and more prevalent +1 and -1 PRF than other ciliates. Together, these findings suggest that peritrichs have a unique evolutionary strategy among ciliates.
Data Availability Statement
All Illumina sequencing data are deposited in the NCBI Short Read Archive (SRA), under the BioProject PRJNA609448.
Author Contributions
XC performed the conceptualization (lead), data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), resources (equal), software (lead), and validation (equal), visualization (lead), and wrote the original draft (lead). CW, BP, BL, and CL performed the data curation (equal), formal analysis (supporting), investigation (equal), and validation (supporting). ZS and AW reviewed and edited the manuscript (equal). LL performed funding acquisition (lead), project administration (lead), resources (equal), and supervision (lead), and reviewed and edited the manuscript (lead). All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (project numbers 31772431, 31801984, 31970486, and 32030015).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Weibo Song (Ocean University of China, China) for his advice during the preparation of the manuscript and Dr. Chao Lu (Columbia University Medical Center, United States) for institutional support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.602323/full#supplementary-material
Supplementary Figure 1 | The matrix of updated multiple sequence alignments showing the presence of 157 homologous genes in each species. The numbering of species corresponds to that in Supplementary Table 1.
Supplementary Table 1 | The data sets analyzed in the current work, including the information on taxonomy, numbers of homologous genes and genomic data source.
Footnotes
References
Adl, S. M., Bass, D., Lane, C. E., Lukeš, J., Schoch, C. L., Smirnov, A., et al. (2019). Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 66, 4–119. doi: 10.1111/jeu.12691
Alff-Steinberger, C., and Epstein, R. (1994). Codon preference in the terminal region of E. coli genes and evolution of stop codon usage. J. Theor. Biol. 168, 461–463. doi: 10.1006/jtbi.1994.1124
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Belinky, F., Babenko, V. N., Rogozin, I. B., and Koonin, E. V. (2018). Purifying and positive selection in the evolution of stop codons. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-27570-3
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chavancy, G., Chevallier, A., Fournier, A., and Garel, J.-P. (1979). Adaptation of iso-tRNA concentration to mRNA codon frequency in the eukaryote cell. Biochimie 61, 71–78. doi: 10.1016/S0300-9084(79)80314-4
Chen, X., Jiang, Y., Gao, F., Zheng, W., Krock, T. J., Stover, N. A., et al. (2019). Genome analyses of the new model protist Euplotes vannus focusing on genome rearrangement and resistance to environmental stressors. Mol. Ecol. Res. 19, 1292–1308. doi: 10.1111/1755-0998.13023
Chen, X., Wang, Y., Sheng, Y., Warren, A., and Gao, S. (2018). GPS it: an automated method for evolutionary analysis of nonculturable ciliated microeukaryotes. Mol. Ecol. Res. 18, 700–713. doi: 10.1111/1755-0998.12750
Corliss, J. O. (1979). The Ciliated Protozoa: Characterization, Classification and Guide to the Literature. Oxford: Pergamon Press.
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, F., Warren, A., Zhang, Q., Gong, J., Miao, M., Sun, P., et al. (2016). The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the phylum Ciliophora (Eukaryota, Alveolata). Sci. Rep. 6:24874. doi: 10.1038/srep24874
Gentekaki, E., Kolisko, M., Boscaro, V., Bright, K., Dini, F., Di Giuseppe, G., et al. (2014). Large-scale phylogenomic analysis reveals the phylogenetic position of the problematic taxon Protocruzia and unravels the deep phylogenetic affinities of the ciliate lineages. Mol. Phylogenet. Evol. 78, 36–42. doi: 10.1016/j.ympev.2014.04.020
Gentekaki, E., Kolisko, M., Gong, Y., and Lynn, D. (2017). Phylogenomics solves a long-standing evolutionary puzzle in the ciliate world: the subclass Peritrichia is monophyletic. Mol. Phylogenet. Evol. 106, 1–5. doi: 10.1016/j.ympev.2016.09.016
Jiang, C., Wang, G., Xiong, J., Yang, W., Sun, Z., Feng, J., et al. (2019). Insights into the origin and evolution of Peritrichia (Oligohymenophorea, Ciliophora) based on analyses of morphology and phylogenomics. Mol. Phylogenet. Evol. 132, 25–35. doi: 10.1016/j.ympev.2018.11.018
Korkmaz, G., Holm, M., Wiens, T., and Sanyal, S. (2014). Comprehensive analysis of stop codon usage in bacteria and its correlation with release factor abundance. J. Biol. Chem. 289, 30334–30342. doi: 10.1074/jbc.M114.606632
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lartillot, N., Lepage, T., and Blanquart, S. (2009). PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288. doi: 10.1093/bioinformatics/btp368
Li, L., Song, W., Warren, A., Shin, M. K., Chen, Z., Ji, D., et al. (2008). Reconsideration of the phylogenetic positions of five peritrich genera, Vorticella, Pseudovorticella, Zoothamnopsis, Zoothamnium, and Epicarchesium (Ciliophora, Peritrichia, Sessilida), based on small subunit rRNA gene sequences. J. Eukaryot. Microbiol. 55, 448–456. doi: 10.1111/j.1550-7408.2008.00351.x
Lobanov, A. V., Heaphy, S. M., Turanov, A. A., Gerashchenko, M. V., Pucciarelli, S., Devaraj, R. R., et al. (2017). Position-dependent termination and widespread obligatory frameshifting in Euplotes translation. Nat. Struct. Mol. Biol. 24:61. doi: 10.1038/nsmb.3330
Lozupone, C. A., Knight, R. D., and Landweber, L. F. (2001). The molecular basis of nuclear genetic code change in ciliates. Curr. Biol. 11, 65–74. doi: 10.1016/S0960-9822(01)00028-8
Lu, B., Li, L., Hu, X., Ji, D., Al-Rasheid, K. A., and Song, W. (2019). Novel contributions to the peritrich family Vaginicolidae (Protista: Ciliophora), with morphological and phylogenetic analyses of poorly known species of Pyxicola, Cothurnia and Vaginicola. Zool. J. Linn. Soc. 187, 1–30. doi: 10.1093/zoolinnean/zlz009
Lu, B., Shen, Z., Zhang, Q., Hu, X., Warren, A., and Song, W. (2020). Morphology and molecular analyses of four epibiotic peritrichs on crustacean and polychaete hosts, including descriptions of two new species (Ciliophora, Peritrichia). Eur. J. Protistol. 73:125670. doi: 10.1016/j.ejop.2019.125670
Lu, S., Zong, C., Fan, W., Yang, M., Li, J., Chapman, A. R., et al. (2012). Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 338, 1627–1630. doi: 10.1126/science.1229112
Lynn, D. (2008). The Ciliated Protozoa: Characterization, Classification, and Guide to the Literature. Berlin: Springer Science & Business Media.
Lynn, D. H., and Small, E. B. (2002). An Illustrated Guide to the Protozoa. Kansas: Allen Press Inc.
Miao, W., Yu, Y., and Shen, Y. (2001). Phylogenetic relationships of the subclass Peritrichia (Oligohymenophorea, Ciliophora) with emphasis on the genus Epistylis, inferred from small subunit rRNA gene sequences. J. Eukaryot. Microbiol. 48, 583–587. doi: 10.1111/j.1550-7408.2001.tb00194.x
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). Creating the CIPRES science gateway for inference of large phylogenetic trees. Gateway Comput. Environ. Workshop (GCE) 1, 1–8. doi: 10.1109/GCE.2010.5676129
Milman, G., Goldstein, J., Scolnick, E., and Caskey, T. (1969). Peptide chain termination, III. Stimulation of in vitro termination. Proc. Natl. Acad. Sci. U.S.A. 63, 183–190. doi: 10.1073/pnas.63.1.183
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
Pan, B., Chen, X., Hou, L., Zhang, Q., Qu, Z., Warren, A., et al. (2019). Comparative genomics analysis of ciliates provides insights on the evolutionary history within “Nassophorea–Synhymenia–Phyllopharyngea” assemblage. Front. Microbiol. 10:2819. doi: 10.3389/fmicb.2019.02819
Pennisi, E. (2003). Modernizing the tree of life. Science 300, 1692–1697. doi: 10.1126/science.300.5626.1692
Rosindell, J., and Harmon, L. J. (2012). OneZoom: a fractal explorer for the tree of life. PLoS Biol. 10:e1001406. doi: 10.1371/journal.pbio.1001406
Scolnick, E., and Caskey, C. (1969). Peptide chain termination, V. The role of release factors in mRNA terminator codon recognition. Proc. Natl. Acad. Sci. U.S.A. 64, 1235–1241. doi: 10.1073/pnas.64.4.1235
Scolnick, E., Tompkins, R., Caskey, T., and Nirenberg, M. (1968). Release factors differing in specificity for terminator codons. Proc. Natl. Acad. Sci. U.S.A. 61:768. doi: 10.1073/pnas.61.2.768
Slabodnick, M. M., Ruby, J. G., Reiff, S. B., Swart, E. C., Gosai, S., Prabakaran, S., et al. (2017). The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Curr. Biol. 27, 569–575. doi: 10.1016/j.cub.2016.12.057
Song, W., Warren, A., and Hu, X. (2009). Free-living Ciliates in the Bohai and Yellow Seas. Beijing: Science Press.
Song, W., and Wilbert, N. (1995). “Benthische ciliaten des Süßwassers,” in Praktikum der Protozoologie, ed. R. Röttger (Stuttgart: Gustav Fischer Verlag), 156–168.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Tzvetkova, A., and Morgenstern, B. (2006). AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7:1. doi: 10.1186/gb-2006-7-s1-s11
Sun, P., Clamp, J. C., Xu, D., Huang, B., Shin, M. K., and Turner, F. (2013). An ITS-based phylogenetic framework for the genus Vorticella: finding the molecular and morphological gaps in a taxonomically difficult group. P. Roy. Soc. Lond. B Bio. 280:20131177. doi: 10.1098/rspb.2013.1177
Swart, E. C., Serra, V., Petroni, G., and Nowacki, M. (2016). Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell 166, 691–702. doi: 10.1016/j.cell.2016.06.020
Utz, L. R., and Eizirik, E. (2007). Molecular phylogenetics of subclass Peritrichia (Ciliophora: Oligohymenophorea) based on expanded analyses of 18S rRNA sequences. J. Eukaryot. Microbiol. 54, 303–305. doi: 10.1111/j.1550-7408.2007.00260.x
Wang, R., Xiong, J., Wang, W., Miao, W., and Liang, A. (2016). High frequency of +1 programmed ribosomal frameshifting in Euplotes octocarinatus. Sci. Rep. 6:21139. doi: 10.1038/srep21139
Williams, D., and Clamp, J. C. (2007). A molecular phylogenetic investigation of Opisthonecta and related genera (Ciliophora, Peritrichia, Sessilida). J. Eukaryot. Microbiol. 54, 317–323. doi: 10.1111/j.1550-7408.2007.00262.x
Wu, T., Li, Y., Lu, B., Shen, Z., Song, W., and Warren, A. (2020). Morphology, taxonomy and molecular phylogeny of three marine peritrich ciliates, including two new species: Zoothamnium apoarbuscula n. sp. and Z. apohentscheli n. sp.(Protozoa, Ciliophora, Peritrichia). Mar. Life. Sci. Technol. 2, 334–348. doi: 10.1007/s42995-020-00046-y
Zhan, Z., Xu, K., Warren, A., and Gong, Y. (2009). Reconsideration of phylogenetic relationships of the subclass Peritrichia (Ciliophora, Oligohymenophorea) based on small subunit ribosomal RNA gene sequences, with the establishment of a new subclass Mobilia Kahl, 1933. J. Eukaryot. Microbiol. 56, 552–558. doi: 10.1111/j.1550-7408.2009.00435.x
Zhuang, Y., Clamp, J. C., Yi, Z., and Ji, D. (2016). A new peritrich ciliate from a hypersaline habitat in northern China. Zootaxa 4169, 179–186. doi: 10.11646/zootaxa.4169.1.10
Zhuang, Y., Clamp, J. C., Yi, Z., and Ji, D. (2018). Phylogeny of the families Zoothamniidae and Epistylididae (Protozoa: Ciliophora: Peritrichia) based on analyses of three rRNA-coding regions. Mol. Phylogenet. Evol. 118, 99–107. doi: 10.1016/j.ympev.2017.09.023
Keywords: Cothurnia, evolution, phylogenomics, Vaginicola, Zoothamnium
Citation: Chen X, Wang C, Pan B, Lu B, Li C, Shen Z, Warren A and Li L (2020) Single-Cell Genomic Sequencing of Three Peritrichs (Protista, Ciliophora) Reveals Less Biased Stop Codon Usage and More Prevalent Programmed Ribosomal Frameshifting Than in Other Ciliates. Front. Mar. Sci. 7:602323. doi: 10.3389/fmars.2020.602323
Received: 03 September 2020; Accepted: 04 November 2020;
Published: 02 December 2020.
Edited by:
Zhijun Dong, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Carolina Bastidas, Massachusetts Institute of Technology, United StatesAndreas Altenburger, Arctic University of Norway, Norway
Copyright © 2020 Chen, Wang, Pan, Lu, Li, Shen, Warren and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifang Li, cWRfbGlsaXlAc2luYS5jb20=
†These authors have contributed equally to this work