 Tomoko Hamabata1*
Tomoko Hamabata1* Ayumi Matsuo2
Ayumi Matsuo2 Mitsuhiko P. Sato2Satomi Kondo3
Mitsuhiko P. Sato2Satomi Kondo3 Kazunari Kameda4Isao Kawazu5,6
Kazunari Kameda4Isao Kawazu5,6 Takuya Fukuoka7,8
Takuya Fukuoka7,8 Katsufumi Sato7Yoshihisa Suyama2Masakado Kawata1
Katsufumi Sato7Yoshihisa Suyama2Masakado Kawata1- 1Laboratory of Evolutionary Biology, Graduate School of Life Sciences, Tohoku University, Sendai, Japan
- 2Kawatabi Field Science Center, Graduate School of Agricultural Science, Tohoku University, Osaki, Japan
- 3Ogasawara Marine Center, Everlasting Nature of Asia, Ogasawara, Japan
- 4Kuroshima Research Station, Sea Turtle Association of Japan, Yaeyama, Japan
- 5Okinawa Churashima Research Center, Okinawa Churashima Foundation, Motobu, Japan
- 6Okinawa Churaumi Aquarium, Okinawa Churashima Foundation, Motobu, Japan
- 7Atmosphere and Ocean Research Institute, The University of Tokyo, Kashiwa, Japan
- 8International Coastal Research Center, Atmosphere and Ocean Research Institute, The University of Tokyo, Otsuchi, Japan
Although studies using genome-wide single nucleotide polymorphisms (SNPs) are growing in non-model species, it is still difficult to prepare sufficient high-quality genomic DNA (gDNA) for population genomic analyses in many wild species. In this study, we first analyzed the population structure of green turtles in the North Pacific; four regions in Japan (the Yaeyama, Okinawa, Amami, and Ogasawara Islands), the Southeast Asia, Taiwan, the Federated States of Micronesia, the Republic Marshall Islands, and the Central or Eastern Pacific (C/E Pacific) using ≃1 ng of gDNA from green turtle field samples – including ones from dead carcasses – and multiplexed inter-simple sequence repeat (ISSR) genotyping by sequencing (MIG-seq). In addition, we explored whether the genome-wide SNP data narrowed down the natal origins of foraging turtles that were difficult to identify by mitochondrial DNA (mtDNA) haplotype alone. The overall nesting population structure observed from genome-wide SNPs was consistent with results from mtDNA, and the population isolation of the C/E Pacific and Ogasawara Islands from the other North Pacific populations was highlighted. However, the population boundaries among the Northwestern Pacific, other than those of the Ogasawaras, were not clear. The uniqueness of the Ogasawara population in genome-wide SNP data enabled the identification of turtles that were more likely to have been born on the Ogasawara Islands. Our results show that genome-wide SNP data are more practical in identifying the natal origins of turtles that were difficult determine by the conventional mtDNA-basis mixed stock analysis. This study also showed that MIG-seq can be expected to meet the potential demand to utilize many preserved or fragmented gDNA samples for population genomics in many marine megafaunas for which sample collections are difficult.
Introduction
Marine turtles are one of the most actively studied marine megafauna. Six of the seven extant species, except for the flatback turtle (Natator depressus), which has the most restricted migratory range in the tropical waters of the Australian continental shelf, exhibit long-distance migrations from hundreds to thousands of kilometers (Bolten, 2003). To date, studies with genetic markers have clarified various life history traits and evolutionary histories of marine turtles. Notably, since studies on maternally inherited mitochondrial DNA (mtDNA) have revealed that nesting populations are regionally differentiated by the philopatric behavior of female marine turtles to the natal site (i.e., natal homing) (Meylan et al., 1990), mtDNA haplotype data of nesting populations have been collected globally to elucidate the population structures and boundaries. Haplotype data on regional nesting populations have been subsequently applied to estimate the natal origin of turtles in foraging grounds using mixed stock analysis (MSA) (Lahanas et al., 1998). These studies have revealed that marine turtles at a foraging ground are usually composed of individuals from various nesting sites (i.e., mixed stock) (reviewed in Jensen et al., 2013). To date, many studies using MSA have clarified the connections between nesting populations and foraging grounds. Like other marine megafauna, marine turtles have longevity, and thus it is difficult to track their movements during their lifetime. The data on identification of the natal origins of individuals in foraging sites are applied to estimate the patterns of developmental migration (Hamabata et al., 2018), and those on habitat connections are utilized to define the distinct population segments (DPS) for conservation and management (Seminoff et al., 2015).
The green turtle (Chelonia mydas) is the largest hard-shell marine turtle and has a cosmopolitan distribution, centered on warm tropical and subtropical waters. The northernmost nesting sites of the green turtle in the North Pacific are situated on the Ryukyu Archipelago (Ryukyus) and Ogasawara Islands, Japan (Figure 1). Previous studies based on mtDNA have suggested that there are multiple nesting populations in Japan (two in the southern Ryukyus, several in the central Ryukyus, and at least one in the Ogasawara Islands). These populations all comprise a mixture of three divergent mtDNA lineages: one highly endemic lineage (clade VII in Jensen et al., 2019), suggesting the existence of a refugial population around the Ogasawara Islands (Hamabata et al., 2014), and two other lineages (clade III and VIII in Jensen et al., 2019), reflecting the historical colonization from lower latitudes (Nishizawa et al., 2011). In addition to rookery distribution, the coastal waters of the Ryukyus and the Japanese main islands provide foraging grounds for green turtles (Nishizawa et al., 2013, 2014; Fukuoka et al., 2015, 2019; Hamabata et al., 2015; Kameda et al., 2017). Previous studies using MSA have revealed that the coasts of the Ryukyus provide foraging habitats to turtles from the southern tropical rookeries, such as islands in the Federated States of Micronesia (FSM), the Republic Marshall Islands (RMI), and Southeast Asia, as well as those from Japan (Nishizawa et al., 2013; Hamabata et al., 2018). However, the accuracy of MSA in the Western Pacific is low, because some haplotypes are often detected from multiple geographically remote nesting populations (called widespread haplotypes), impeding the identification of the origins of the turtles. In addition to the widespread haplotypes, several haplotypes that are endemic to Japan are also shared by populations in the Ryukyu and Ogasawara islands (Nishizawa et al., 2011; Hamabata et al., 2014). These shared haplotypes among nesting populations within Japan especially decrease the accuracy of MSA estimation for the contribution of nesting populations to foraging aggregation around Japan. The development of a new method to estimate the candidate natal origins of turtles with widespread or common Japanese haplotypes more accurately remains important. Although nuclear microsatellite markers have been developed for all marine turtles, the application of microsatellite data to MSA is inefficient; it is a time- and labor-intensive process in the laboratory (Komoroske et al., 2017).
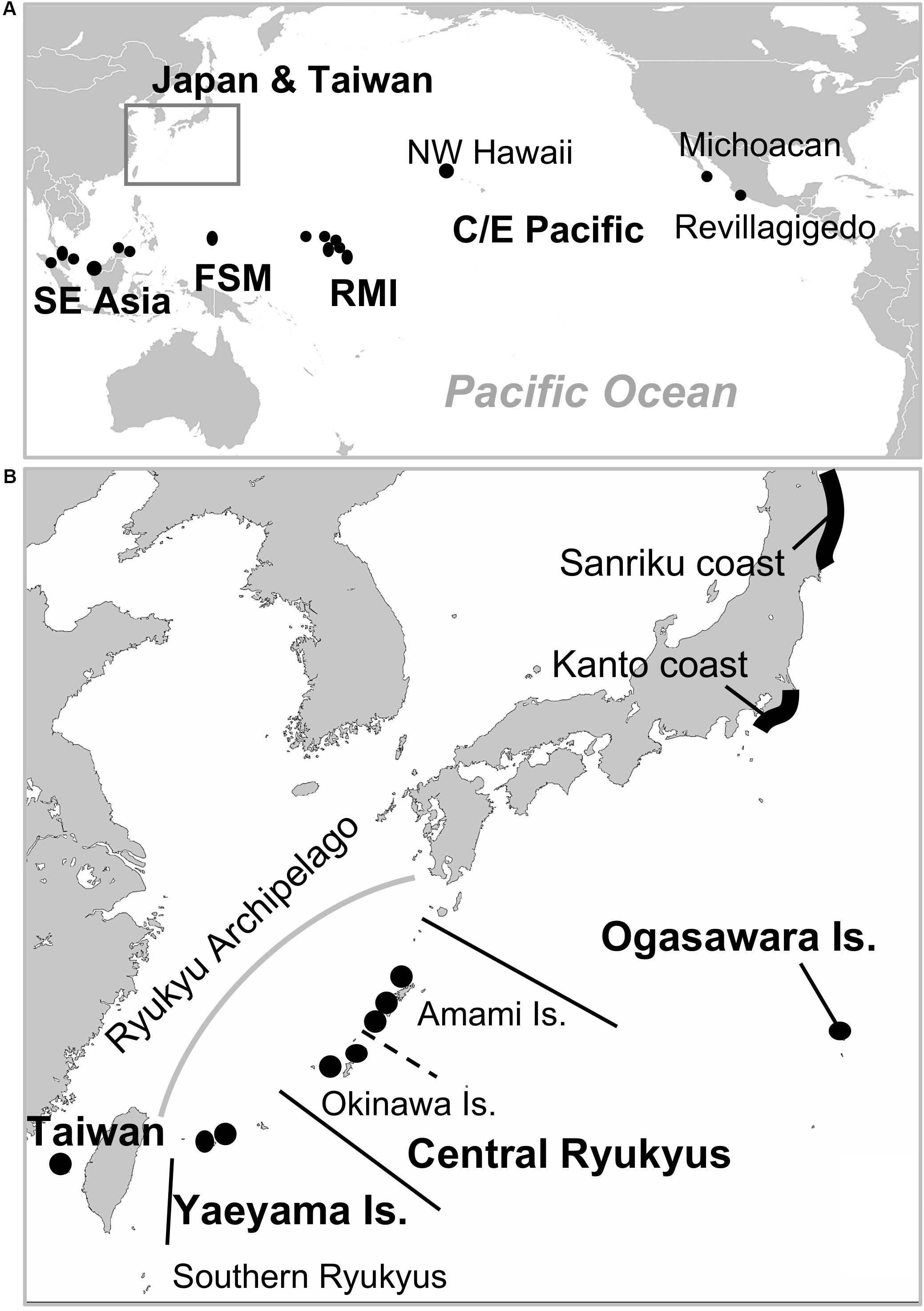
Figure 1. Map of study area (A) and enlarged map around Japan (B). Black dots show the sampling nesting sites (in Japan) and the candidate natal origins of turtles that were included as samples from the nesting populations outside of Japan in this study. Abbreviations of locations are as follows: SE Asia, Southeast Asia; FSM, Federated States of Micronesia; RMI, Republic Marshall Islands; NW Hawaii, Northwestern Hawaii; C/E Pacific, Central/Eastern Pacific.
Genome-wide population studies based on single nucleotide polymorphisms (SNPs) obtained by high-throughput sequencing (HTS) platforms are now growing, even for non-model organisms. Methods using the HTS platform generally require certain amounts of high-quality genomic DNA (gDNA). However, samples collected in the field, especially those collected as non-invasively as possible from endangered marine turtles, often contain very small amounts of gDNA. Therefore, many methods are not applicable to population studies of marine turtles. Thus, it is important to select suitable methods according to the gDNA condition. Among the various genotyping methods using HTS, a multiplexed inter-simple sequence repeats (ISSR) genotyping by sequencing (MIG-seq) approach was developed to adapt to low-quality field samples using a PCR-based procedure (Suyama and Matsuki, 2015). Studies using the MIG-seq method have shown that this approach is highly applicable to field samples with small amounts of gDNA in various taxonomic groups (e.g., plants, Gutiérrez-Ortega et al., 2018; Park et al., 2019 and animals, Hirai, 2019; Takata et al., 2019). It is expected that this method can be used to solve the issues in analysis of marine turtle population genomics.
In this study, we first applied MIG-seq to the population analysis of green turtles in the North Pacific, focusing on Japanese nesting populations. Based on the nesting population structure from genome-wide analysis, we explored whether the natal origins of turtles with widespread or common Japanese mtDNA haplotypes could be narrowed down. More than half of the samples used in this study were preserved samples that had been collected in the field under the assumption of conducting mtDNA haplotype analysis. Samples included those from dead stranded carcasses. Thus, they initially had large variations in the quality or fragmentation of gDNA (Supplementary Figure S1A). We tested whether these non-ideal samples for general HTS platform methods could be made available by MIG-seq for the future utility of the preserved samples.
Materials and Methods
Samples for Nesting Population Structure
For nesting population samples, we collected samples of turtles whose breeding sites or natal origins were identified in the North Pacific. From the Japanese nesting populations, we collected samples from the Ogasawara Islands (n = 55), and Yaeyama (n = 8), Okinawa (n = 19), and Amami Islands (n = 41) of the Ryukyus (Table 1 and Supplementary Table S1). The samples from the Ogasawara Islands included not only those from nesting females (n = 15), but also females (n = 23) and males (n = 17) that were mating (14 females and 14 males) or resting (nine females and three males). These turtles were legally captured by local fishers during the breeding season under the fishery adjustment regulation of the Tokyo metropolitan area. According to the mating behavior or examination of the gonads, the individuals were estimated to be migrating to the Ogasawaras to breed. Of the samples collected from the Ryukyus, only five samples were newly collected from dead hatchlings on the Yaeyama Islands in this study. The remaining samples were the same specimens used in the previous study (Hamabata et al., 2014). Regarding the nesting population samples included from outside of Japan, 22 were from foraging turtles that were probably born in Southeast Asia (SE Asia) (n = 4), Taiwan (n = 3), the FSM/RMI (n = 10), or the Central/Eastern Pacific (C/E Pacific) (n = 5). The natal origins of these 22 turtles were identified from regionally unique mtDNA haplotypes according to previous studies (Dutton et al., 2014a,b; Nishizawa et al., 2014, 2018; Jensen et al., 2016b; Joseph et al., 2016). These turtles were captured in set nets around the Yaeyama or Okinawa Islands, or on the Sanriku coast (Figure 1). All of the tissue samples from turtles caught in set nets were obtained during tagging for mark-recapture research. Tagging was performed ethically with the permissions of each prefecture or the Animal Ethic Committee of the University of Tokyo. They were released near the captured location after a physical health evaluation. Detailed information on the haplotypes and the candidate natal origins within each region are described in Supplementary Table S2 for the 20 specimens that were included in the final analyses.
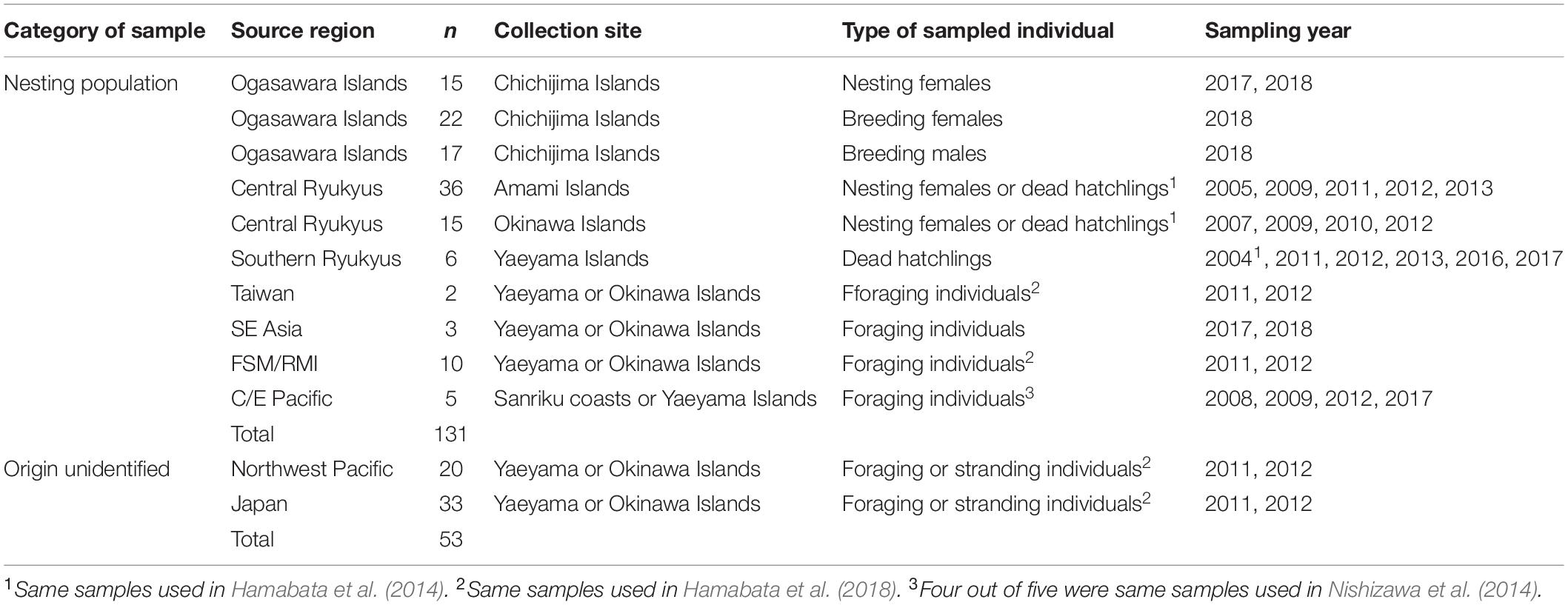
Table 1. Information on samples analyzed in this study.
Samples for Natal Origin Identification
In the Indo-Pacific region, the mtDNA haplotypes CmP20.1 and CmP49.1 are known as widespread haplotypes detected from females in a wide range of nesting sites, including Japan (Supplementary Table S3). In addition, within Japanese nesting sites, the haplotype CmP39.1, CmP50.1, CmP53.1, CmP54.1, and CmP121.1 (and their shorter haplotypes) were widely detected in females nesting on the Ryukyus and in Ogasawara Islands (Supplementary Table S3). Turtles with these seven mtDNA haplotypes are often captured in coastal set nets or stranded along the Ryukyus (Nishizawa et al., 2013; Hamabata et al., 2018). To explore whether the candidate natal region of foraging turtles with those haplotypes can be narrowed down, we included samples of foraging turtles with these widespread (n = 20) and common Japanese haplotypes (n = 36). These 56 samples were the same specimens used in the previous MSA study by Hamabata et al. (2018).
DNA Extraction
Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (QIAGEN) or phenol chloroform extraction method. The concentration of gDNA was measured using a NanoDrop (Thermo Scientific, Waltham, United States), and was adjusted to 1 ng/μL for further analysis.
Mitochondrial DNA Haplotype Identification
To identify the mtDNA haplotypes of the newly collected samples, 820-bp fragments of the mtDNA control region were amplified by PCR and sequenced using an Applied Biosystems 3730xl DNA Analyzer using the primers LCM15382 and H950g (Abreu-Grobois et al., 2006) and the same procedure as described by Hamabata et al. (2018). All sequences were confirmed to have been registered on GenBank by a nucleotide– nucleotide basic local alignment search tool (BLASTn) search on the NCBI website. The mtDNA haplotypes of green turtles in the Pacific region are now termed with standardized nomenclature (“CmP” prefix) based on the alignments of sequences >770-bp. When the sequence was unregistered or registered only as a shorter haplotype (e.g., “CMJ” prefix series of <500-bp sequences), a new sequence designation was assigned according to the nomenclature for the Pacific green turtle with the assistance of the Southwest Fisheries Science Center, National Oceanic and Atmospheric Administration (NOAA).
MIG-seq
The MIG-seq library was prepared according to the method described by Suyama and Matsuki (2015), except for a modification of the thermal cycle conditions in the first PCR. First, a large number of anonymous genome-wide regions were amplified using multiplex ISSR primers that were designed to anneal to the 3′ ends of repetitive motifs, including two bases of anchor sequences, and to amplify multiple non-repetitive regions between various SSRs. PCR was performed using 1.0 μL of the template gDNA and the primer set-1 reported in the protocol by Suyama and Matsuki (2015) using the Multiplex PCR Assay Kit Ver.2 (Takara Bio, Kusatsu, Japan). The original PCR thermal cycling conditions were modified as follows: initial denaturation step of 1 min at 94°C, and 25 cycles of PCR denaturation at 94°C for 30 s, annealing at 38°C for 1 min, and extension at 72°C for 1 min, followed by a final extension at 72°C for 10 min. Successful amplification of the ISSR regions in the first PCR was confirmed by smeared bands in agarose gel electrophoresis and microchip electrophoresis system (MultiNA; Shimadzu, Kyoto, Japan) analysis with the DNA-2500 Reagent kit. PCR products with smeared bands are expected to have a large number of amplified loci, and thus, many shared SNPs with other samples after filtering are expected. Specific amplification bands were highlighted in electrophoresis profiles for the first PCR products of some samples (Supplementary Figure S1B). Although genome-wide loci were not likely to be amplified in these samples, we proceeded with subsequent library preparation without excluding. In the subsequent steps for a second round of PCR, the complementary sequences for the binding sites of Illumina sequencing flow cell and indices (barcodes) for each sample were added to the first PCR products of the first round, and the libraries were purified and sequenced on Illumina MiSeq sequencers (MiSeq Control Software v2.0.12; Illumina, San Diego, United States) with a MiSeq Reagent kit v3 (150 cycle; Illumina) at 80-bp paired-end reads.
Data Analysis
Low-quality reads, which occur when over 20% of the bases have a quality score <30, were discarded by the FASTQ Quality Filter implemented in the FASTX-Toolkit1. SNP discovery and genotyping were carried out using Stacks version 2.2.0. The draft genome of the green turtle is available. However, it is not recommended to apply the reference-aligned pipeline for draft quality reference in Stacks because a draft genome can act as a filter by excluding loci not contained in the assembly and may fail to align reads that belong to loci captured in the reference more than once (Paris et al., 2017; Rochette and Catchen, 2017). Thus, we used a de novo approach to build the loci. While the minimum depth in the de novo assembly of identical raw reads into stacks (putative alleles) was set to three (default value for the m option in ustacks), the optimal values of other Stacks parameters were explored by repeating the entire analysis process and determined based on the highest value of the proportion of overall correct assignment by discriminant analysis of principal components (DAPC) using the adegenet package (v.2.1.1) in R (Jombart et al., 2010). In the ustacks program, the number of mismatches allowed between stacks to merge them into a putative locus (-M option in ustacks) was changed from two to six bases, according to the method described by Paris et al. (2017). In the subsequent cstacks program, the number of mismatches allowed between stacks (putative loci) during the construction of a population-wide catalog of loci (-n option) was changed from M − 1 to M + 1 depending on the parameter sets in the previous ustacks process. The data on each individual could then be matched to the data contained in the catalog with sstacks. After data converted to locus orientation were stored as standard BAM files in the tsv2bam module, variants in each individual were genotyped based on the SNPs identified within all populations for each locus in the gstacks process. SNP data that were shared by at least 80% of the individuals in all eight regional populations were outputted with the populations program in Stacks. Finally, we set the parameter values to three for the number of mismatches allowed for a putative locus (-M option in ustacks) and population-wide catalog (-n option in cstacks), and seventeen samples (14 nesting population samples and three unidentified natal origin samples) with missing call rates exceeding 0.5 were filtered out using PLINK 1.9 (Purcell et al., 2007). Finally, all analyses were carried out for 131 nesting samples and 53 unidentified natal origin samples. Among the 184 samples analyzed, 24 were derived from carcasses (Supplementary Table S2). The final filtering and calculation of the significance of deviation from the Hardy–Weinberg equilibrium (HWE) were performed again using the populations program in Stacks. SNP pairs in linkage disequilibrium (LD) were identified using PLINK 1.9 (r2 > 0.8).
Phylogenetic and Population Differentiation Analysis
To infer phylogenetic relationships among individuals and regions, maximum likelihood estimation was conducted using RAxML (version 8.2.10) with several variations in the proportion of shared SNPs among individuals of a population (10–50%, -r option). The minimum number of populations (-p option) with a locus was set at two, and 17,462–36,185 SNPs were extracted. Differentiation among regional populations was examined by population pairwise FST values, with the significance determined by permutation tests using Arlequin 3.5 (1,000 permutations, Excoffier and Lischer, 2010), and SNPs that were not significantly deviated from the HWE (p > 0.01) and were shared by at least 80% of the individuals in all eight regional populations. Although the values for minimum minor allele frequency threshold were tested 0.02 and 0.05, the results were almost the same for both values. Then, we set it at 0.02. Only the first SNP per locus was used, and SNPs that were likely to be in LD were excluded. Finally, 387 SNPs matched to these criteria were used for the pairwise FST calculations and determination of nucleotide diversity (π). Prior to the analysis, differentiation within Ogasawara individuals (among nesting females and breeding females and males) was examined using mtDNA haplotype compositions and the same SNPs from the regional population tests derived from MIG-seq; neither supported significant population differentiation within the Ogasawara individuals. Thus, samples from the Ogasawara populations were pooled for analysis.
Discriminant Analysis of Principal Components (DAPC)
Discriminant analysis of principal component was used to investigate the structural patterns of nesting populations and for the group assignment of individuals of unidentified natal origin in foraging grounds. All DAPCs were run with SNPs that were shared by all candidate nesting populations with more than 2% of the minimum minor allele frequency and more than 80% of individuals in a population. Because DAPC is free of assumptions about the HWE or LD, all SNPs on each locus were used without filtering. DAPC for nesting population structure was carried out with given regional nesting groups and 629 SNPs satisfied the screening criteria in 131 individuals. The number of retained principal components was determined by calculating the a-score. The group assignment of individuals of unidentified natal origin was carried out without prior groups. The C/E Pacific population was excluded from candidate natal origin in the group assignment because they did not share any mtDNA haplotypes. To identify the optimal number of clusters, the sequential k-means algorithm was run sequentially with increasing numbers of actual regional clusters (nesting populations), and the optimal clustering was determined as that corresponding to the lowest Bayesian information criterion. Clustering of individuals with widespread and common Japanese haplotypes was independently carried out to maximize the number of available SNPs. Group assignments of individuals with widespread and common Japanese haplotypes were carried out using 1,093 SNPs in 146 individuals (20 individuals with widespread haplotypes and 126 individuals from 7 western Pacific nesting populations) and 2,326 SNPs in 144 individuals (33 individuals with common Japanese haplotypes and 111 individuals from Japanese populations), respectively.
Results
Newly Identified mtDNA Haplotypes
Of the newly collected samples, the sequences of four samples were unregistered in GenBank as longer sequences. The first sequence, identified from a female nesting on the Ogasawara Islands, matched with a 384-bp haplotype named CMJ40 (AB661782.1), and was termed CmP247.1 (GenBank accession no. LC492975.1). CMJ40 was previously identified in a turtle foraging in the Kanto coastal area (Figure 1B) but was not detected in nesting turtles. Thus, it was treated as an “orphan haplotype,” whose candidate natal origins were unknown. The second sequence, identified from another nesting female on the Ogasawara Islands, had a 1-bp substitution of CmP53.1 and was termed CmP248.1 (GenBank accession no. LC492976.1). The third sequence, identified from a breeding female captured around the Ogasawaras, matched with a 385-bp haplotype named CMJ38 and has a 1-bp substitution of CmP39.1 and was termed CmP209.1 (GenBank accession no. LC492977.1). The fourth sequence, identified from hatchlings born on Iriomote Island in the Yaeyama Islands, had a 1-bp substitution of CmP54.1 and CMJ6, and was termed CmP54.2 (GenBank accession no. LC522564). In addition, another orphan haplotype, CmP208.1, was identified from two individuals captured during mating behavior off Ogasawara Islands.
Population Structure and Genetic Analysis
Discriminant analysis of principal component based on the given regional nesting populations indicated that the C/E Pacific population was isolated from the seven Western Pacific populations (Figures 2A,B). Within the Western Pacific populations, the Ogasawara population was relatively isolated from the other six, although it slightly overlapped with the Amami population. It was difficult to determine the boundaries among Western Pacific populations, except for the Ogasawara population as an independent cluster in the scatterplot of the DAPC. In particular, the scatterplot of the Okinawa and Amami populations largely overlapped (Figure 2A). Although the nucleotide diversity was slightly high in the Japanese and SE Asia populations, the level of genetic diversity was similar for all populations (Figure 3). The differentiation of the C/E Pacific and Ogasawara populations from most other western populations was also supported by pairwise FST (Figure 3). The population structure among the Western Pacific populations, except for the Ogasawara population, was relatively weak, and no differentiation was supported by pairwise FST among the nesting populations in the Ryukyus (Yaeyama, Okinawa, and Amami) (Figure 3). There were no significant differentiations among the SE Asian, Taiwan, FSM/RMI, and Yaeyama populations. The Taiwanese population showed relatively higher pairwise FST values than did the other populations, and significant differences from the populations other than Japanese ones were not found.
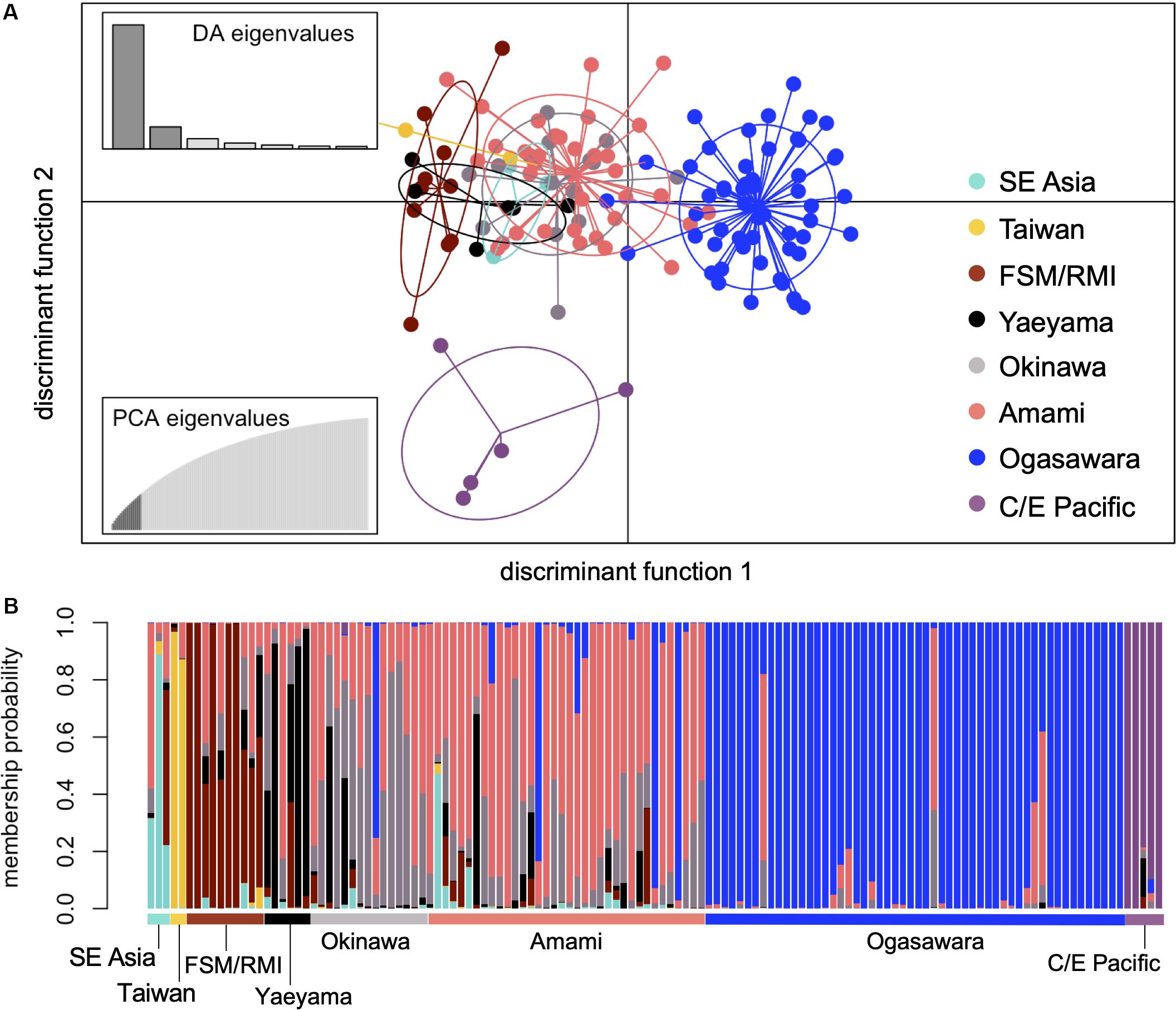
Figure 2. Scatterplot showing the structure patterns of nesting populations (A) and compoplot showing the admixed pattern of each individual (B). Both structures were generated from discriminant analysis of principal components (DAPC) based on the eight regional nesting groups and 629 SNPs.
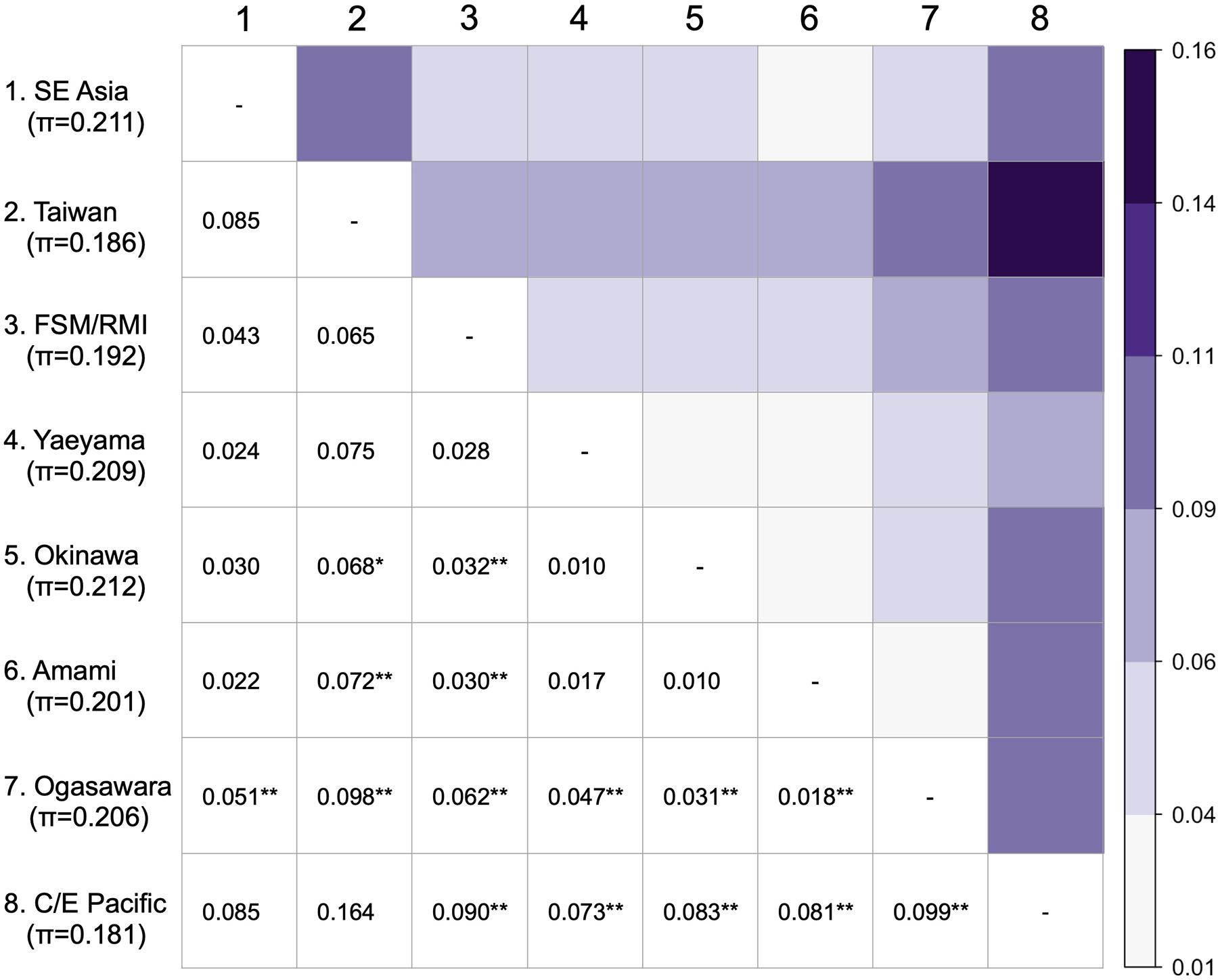
Figure 3. Population pairwise FST values (below the diagonal) and heatmap of populations drawn by the FST values (above the diagonal). The superscripts * and ** on the FST values denote significance levels of <0.05 and <0.01, respectively. The average nucleotide diversity (π) of each population is indicated below the population.
Maximum likelihood phylogenetic trees were constructed using various numbers of SNPs. Although no regional nesting populations as a whole were distinguished with high bootstrap values (>75), the branches of four out of five individuals from the C/E Pacific were supported with high bootstrap values (>90) in two trees constructed by 24,406 SNPs and 17,462 SNPs that were shared by at least two populations and more than 30 and 50% of the individuals in a population, respectively. Only the tree constructed with 24,406 SNPs is shown in Supplementary Figure S3. The mixed structure in the Ryukyu populations was also supported by their mixed phylogenetic relationships. Based on these results and the geographic proximity, the samples from Okinawa and Amami were treated as one central Ryukyu population in the estimation of group assignment to identify the natal origins of foraging turtles.
Identification of the Natal Origin of Foraging Turtles
The number of optimal genetic clusters across the Western Pacific and within Japanese populations was suggested to be two. One out of 20 individuals with widespread haplotypes was assigned to Cluster 2, where individuals of the Ogasawara population were dominant. The remaining 19 individuals were assigned to Cluster 1, which comprised the majority of individuals from nesting populations other than those of Ogasawara (Figure 4A). Nine of the 33 individuals with common Japanese haplotypes were assigned to Cluster 2, where individuals of the Ogasawara population dominated, and all the rest were assigned to Cluster 1, together with most nesting individuals from the Ryukyus (Figure 4B).
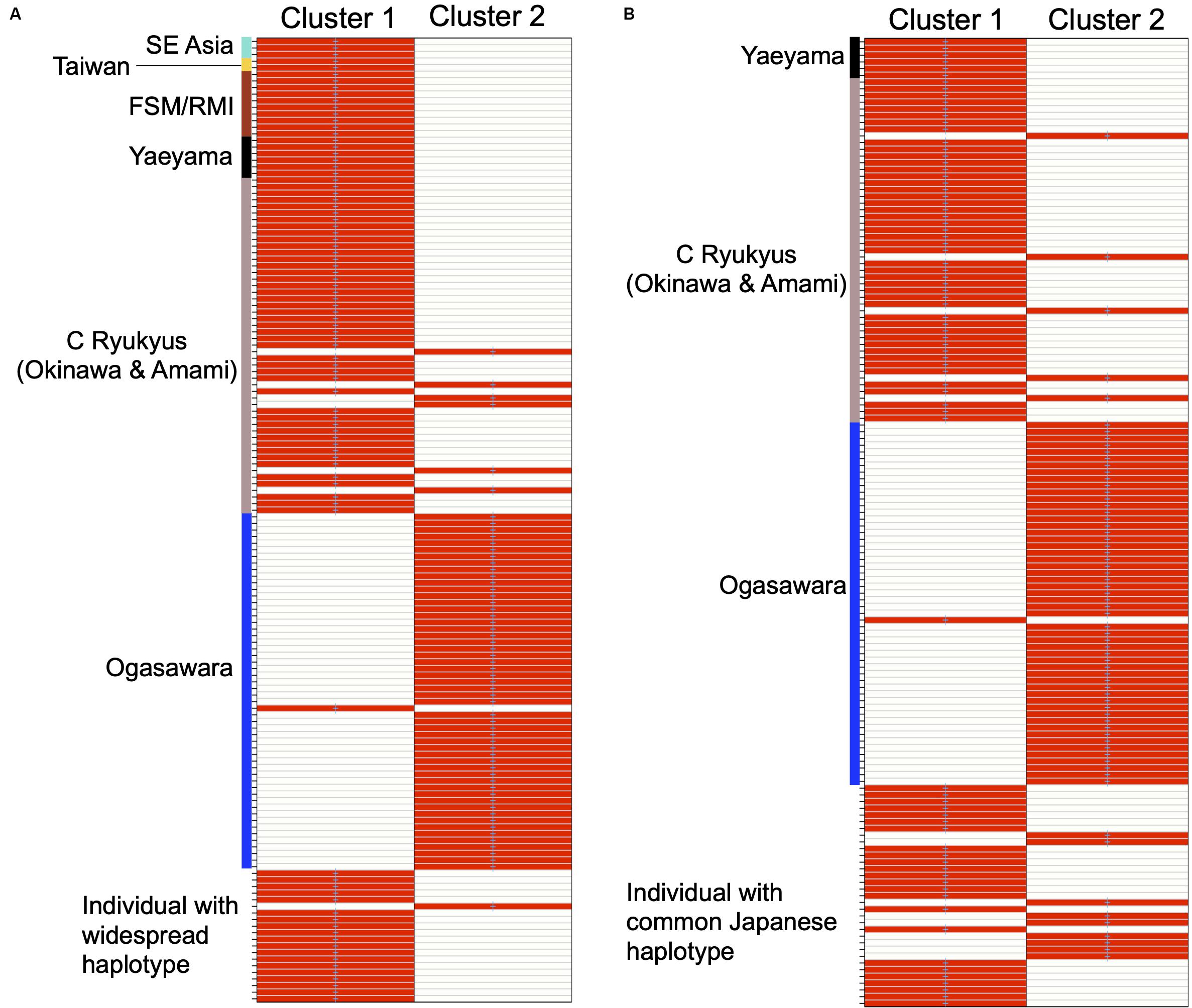
Figure 4. Results of the group assignments of the turtles carried out with 20 individuals with widespread haplotypes and 126 origin identified individuals [SE Asia, Taiwan, Yaeyama, C Ryukyus (Okinawa and Amami combined), and Ogasawara] based on 1,093 SNPs (A) and carried out with 33 individuals with common Japanese haplotypes and 111 origin identified individuals [Yaeyama, C Ryukyus (Okinawa and Amami combined), and Ogasawara] based on 2,326 SNPs (B), respectively. Heat colors represent membership probabilities (red = 1, white = 0); blue crosses represent the prior cluster provided to DAPC by the find.clusters function. Blue crosses on red rectangles show that the individuals successfully reassigned to the prior clusters based on the discriminant functions.
Discussion
Population Structure of Genome-Wide SNPs
In the present study, we conducted a genome-wide population analysis that showed that the population structure of green turtles in the North Pacific was overall partly inconsistent with the structure observed by mtDNA analysis. Our results showed that the C/E Pacific populations were isolated from western populations within the North Pacific, and that the Ogasawara population was relatively isolated from other Western Pacific populations. These observations agree with the population genealogies based on mtDNA. In the mtDNA genealogies, the C/E Pacific populations were composed of an endemic monophyletic lineage (clade IX in Jensen et al., 2019), and the Ogasawara population was considered to have maintained an endemic lineage (clade VII in in Jensen et al., 2019) from the central distribution pattern of its haplotypes (Hamabata et al., 2014). Furthermore, there was an overall agreement between results from the mtDNA and genome-wide SNP data for the Japanese population structure. The present analysis in Japanese populations elucidated the admixed population structure in the Ryukyus at the genomic level. The results of DAPC and phylogeny analysis showed that the population in the southern Ryukyus (i.e., Yaeyama) seemed to be closer to southern populations in SE Asia and FSM/RMI, while the northern population (i.e., Amami) partly overlapped with the Ogasawara population. The mtDNA studies showed that the proportion of the haplotypes in clade VII, which is highly endemic to Ogasawara, increased from south to north in the Ryukyu populations. In contrast, the proportion of the haplotypes in two lineages, which suggest historical colonization from the SE Asia (clade VIII in Jensen et al., 2019) and from the Western Pacific Equatorial Islands around Micronesia (clade III in Jensen et al., 2019), decreased from south to north (Supplementary Figure S4). It is notable that these observations were consistent with the mtDNA haplotype compositions of these populations based on the substantial sample sizes. In contrast, while population differentiation among the regional populations at lower latitudes (SE Asia, FSM/RMI, and Taiwan) was also strongly supported in mtDNA, population boundaries seemed to be ambiguous in genome-wide SNPs. A similar admixed population structure among these regions was also suggested from the PCA and phylogeny by restriction-site associated DNA (RAD)-Capture data (Komoroske et al., 2019). It was difficult to determine the structure of these populations at lower latitudes owing to the small sample sizes in both this study and previous studies (Komoroske et al., 2019). Thresholds during SNP detection might filter out specific SNPs from populations outside of Japan because of their small sample sizes. Although further research focused on these regions is needed, the genetic distance among these regional populations is probably smaller than that previously estimated from mtDNA.
Global analysis of mtDNA suggested that there was generally little to no genetic differentiation between green turtle rookeries located within 500 km of each other and a significant difference between more distant rookeries (Jensen et al., 2019). The distance between sampling sites in the Yaeyama and central Ryukyu Islands (Okinawa and Amami) is 400 km at its nearest point and over 700 km at its furthest. The Ryukyus are 1,200 km away from the Ogasawaras. The mixed structure within the Ryukyus and the population isolation of the Ogasawaras are consistent with the prediction of population differentiation according to the geographical distance calculated based on mtDNA. The entire Ryukyus lay along the Kuroshio Current. Thus, not only the geographical distance, but also the ease of access or migration facilitated by the currents and other factors, may be reflected in the admixed population structure within the Ryukyus. Factors other than geographical distance are especially necessary to explain the larger genetic distance between Ogasawara and FSM/RMI populations than among the Ryukyus, SE Asia, and FSM/RMI populations. While female marine turtles have strong natal philopatry to the nesting sites, the phylogeographic structure of marine turtle populations is affected by various other factors such as historical colonization, incidental nest site sifts by gravid females, and male-mediated gene flow among populations. In this study, it was difficult to reliably estimate the migration rates among populations due to unequal sample sizes and small genetic distances among proximate populations. Population genomic analysis, including the comparison with results from mtDNA analysis, will clarify the history and ecology of populations in more detail, by elucidating the important factors that affect population structures in future studies.
Identification of Natal Origin
In conventional MSA, the mtDNA haplotype compositions of nesting populations is the baseline data to estimate their contribution to the foraging grounds. As some widespread haplotypes are detected from multiple geographically remote nesting populations, there is a limitation in the accuracy of MSA (reviewed in Jensen et al., 2013 and Komoroske et al., 2017). This is an especially serious problem in estimation of the origins of foraging green turtles in Japanese waters, not only because either widespread haplotypes of CmP20.1 and CmP49.1 have been detected in almost all areas of the population in the Western Pacific (Dutton et al., 2014b; Jensen et al., 2016a; Nishizawa et al., 2018), but also because several common haplotypes were shared within the Japanese nesting populations. Our genome-wide population analysis demonstrated that the distinctiveness of the Ogasawara population from other Western Pacific populations was highly practical for narrowing down the natal origins of foraging turtles with widespread or common Japanese haplotypes. We examined the origins of foraging turtles around Yaeyama in the Ryukyus. This method is especially useful for examining the candidate natal origins of turtles foraging around the Japanese main islands where the turtles are almost exclusively from Japan (Nishizawa et al., 2013; Hamabata et al., 2015, 2018). Elucidation of differences in the distribution and migration ranges between the Ryukyus and the Ogasawaras will contribute to further studies on the biology and evolution of this northernmost population of this species, which usually prefers warm waters. In contrast, it was unfortunately difficult to narrow down the candidate natal origins of the turtles at lower latitudes because of the ambiguous population structures in the present data. When the population structures around the SE Asia and FSM/RMI improved by a substantial sample size growth and genome-wide SNPs, group assignment of turtles from these regions will also be realized in future studies.
In addition to the improved resolution of natal origins, the present group assignment has another advantage: unlike the conventional mtDNA-basis MSA, which predicts the contributions from the nesting population as probabilities for an entire foraging aggregation, group assignment by genome-wide SNPs can determine the cluster to which each individual may be assigned (Figures 4A,B). Furthermore, in the foraging aggregations, there are some turtles with orphan haplotypes, which were observed only in the foraging grounds and not in any of the nesting rookeries, as well as turtles with widespread haplotypes. Although the proportion of turtles with orphan haplotypes varies depending on the foraging grounds, it sometimes reaches more than 5% in the foraging grounds of the Northwestern Pacific (e.g., Hamabata et al., 2018). The data on the individuals with orphan haplotypes must be excluded in the conventional MSA because they are absent in the haplotype compositions of candidate nesting populations. In the estimation by DAPC, they do not have to be excluded, as long as the data of turtles in candidate nesting populations were sufficiently included. The challenges in resolution of MSA caused by widespread haplotypes and the presence of orphan haplotypes are common in marine turtle populations [e.g., loggerhead turtles (Caretta caretta) in the North Atlantic (Shamblin et al., 2012), and green turtles in the Mediterranean Sea (Tikochinski et al., 2012)]. In conclusion, DAPC estimation of the natal origins of turtles ensures a more efficient use of samples than conventional MSAs worldwide.
Availability of MIG-seq and Caveats
Recently, RAD-Capture was presented as a versatile genotyping platform using over 1,000 marine turtle samples (Komoroske et al., 2019), and improved 2b−RAD protocols on non−model organisms were introduced using samples of loggerhead turtle (Barbanti et al., 2020). These studies create expectations for a deeper understanding of the population structures, evolution, and ecology of marine turtles. Meanwhile, the low initial concentrations of gDNA in field samples remains as an issue (Komoroske et al., 2019). In this study, we first examined the marine turtle population structure by MIG-seq, using 1 ng of gDNA. Hundreds to a few thousands of SNPs were available according to the analyses, even in fragmented gDNA from dead carcasses, despite the relatively high filtering thresholds to avoid the missing data. We confirmed that excessive fragmentation of the original gDNA tended to result in a lower number of detected SNPs. However, fragmentation was sometimes observed not only in samples from carcasses but also in samples from fresh tissues. The actual success of the MIG-seq analysis largely depends on the number of sites that are amplified by ISSR primers in the first PCR because such samples can share many loci with other samples after filtering. Thus, the smeared bands in the electrophoresis profile of the first PCR products more reliably predicted the success in MIG-seq than the observed degree of fragmentation or type of sample (carcass or fresh tissue). In addition, we confirmed that a high concentration of template DNA amplified specific regions in the first PCR. Thus, PCR attempts with a gradient concentration of template DNA can improve the results of the first PCR in the MIG-seq process.
It is difficult to compare the number of SNPs obtained by MIG-seq and RAD-Capture or 2b-RAD because, unlike the previous studies using the Illumina HiSeq series, the present data were obtained using Illumina MiSeq, which outputs less data than the HiSeq series. In addition, the number of available SNPs can vary according to the filtering thresholds, and it was also decreased when we compared remote populations (i.e., including the C/E Pacific). The intra-specific differences in ISSR and the adjacent non-repetitive regions, as well as the small sample sizes in nesting populations outside of Japan may cause lower numbers of shared SNPs among remote populations. Therefore, when comparing geographically remote populations that are expected to result in data sets with larger amounts of missing data, the sequencing method should be carefully considered (sequencing platform and data size per sample). Furthermore, it should be reexamined whether more SNPs that are shared among populations can improve the resolution of phylogeny of green turtles in the North Pacific in future studies. However, the significant point in our results is that MIG-seq using fragmented gDNA clarifies the genomic differences of turtles whose mtDNA haplotypes are the same, and can demonstrate a more reliable estimation of natal origin of foraging turtles than the estimation obtained with a conventional MSA. Studying migratory marine megafauna presents a common challenge in sample collection due to the inaccessibility of animals. Additionally, from the perspective of animal welfare, skin biopsies and fecal samples in the field (e.g., Valqui et al., 2010) have played important roles in population genetic studies. Researchers often need to investigate dead carcasses that are stranded on the coasts (e.g., Rankin-Baransky et al., 2001; Cassens et al., 2005; Natoli et al., 2005); it is not expected that high-quality gDNA is obtained from such field samples. Our results indicated that MIG-seq, using low concentration of gDNA or even fragmented gDNA, makes it possible to perform genome-wide population analyses with preserved field samples. It is expected that the potential demand is not limited to marine turtles and that this method may be used to perform genome-wide population analyses using samples preserved worldwide.
Data Availability Statement
The datasets generated for this study can be found in the NCBI Sequence Read Archive (Bioproject PRJDB9358, BioSample Accession SAMD00207387–SAMD00207570).
Ethics Statement
Samplings in this study were conducted with permissions from the government of the Okinawa Prefecture, the governor of the Tokyo Metropolitan area, and the Animal Ethic Committee of the University of Tokyo.
Author Contributions
TH conducted the field sampling, DNA extractions, laboratory experiments, data analysis, and wrote the manuscript. AM, MS, and YS conduct the laboratory experiments, MIG-seq sequencing, helped the design of experiment, data analysis and interpretation, and commented on the manuscript. SK, KK, IK, TF, and KS conducted the filed sampling and commented on the manuscript. MK helped with the data interpretation and discussion and commented on the manuscript. All authors read and approved the manuscript.
Funding
This research was supported by Grant-in-Aid for JSPS Fellows Grant Number 18J00353.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the staffs of Ogasawara Marine Research Center to help our field sampling. Sampling at Sanriku, Iwate, was performed under the Cooperative Program of Atmosphere and Ocean Research Institute, the University of Tokyo. We also thank Michael P. Jensen and Erin LaCasella for their assistance for the haplotype nomenclature of unregistered sequences. Computations in this study were partially performed on the NIG supercomputer at ROIS National Institute of Genetics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2020.00658/full#supplementary-material
Footnotes
References
Abreu-Grobois, F. A., Horrocks, J. A., Krueger, B., Formia, A., and Beggs, J. (2006). “New mtDNA dloop primers which work for a variety of marine turtle species may increase the resolution of mixed stock analyses,” in Book of Abstracts from the 26th Annual Symposium on Sea Turtle Biology and Conservation. International Sea Turtle Society, eds M. Frick, A. Panagopoulou, A. F. Rees, and K. Williams, (Athens: International Sea Turtle Society), 179.
Barbanti, A., Torrado, H., Macpherson, E., Bargelloni, L., Franch, R., Carreras, C., et al. (2020). Helping decision making for reliable and cost-effective 2b-RAD sequencing and genotyping analyses in non-model species. Mol. Ecol. Resour. 20, 756–806. doi: 10.1111/1755-0998.13144
Bolten, A. B. (2003). “Chapter 9: variation in sea turtle life history patterns: neritic vs. oceanic developmental stages,” in The Biology of Sea Turtles, Vol. II, eds P. L. Lutz, J. A. Musick, and J. Wyneken, (Boca Raton, FL: CRC Press), 243–257. doi: 10.1201/9781420040807.ch9
Cassens, I., Van Waerebeek, K., Best, P. B., Tzika, A., Van Helden, A. L., Crespo, E. A., et al. (2005). Evidence for male dispersal along the coasts but no migration in pelagic waters in dusky dolphins (Lagenorhynchus obscurus). Mol. Ecol. 14, 107–121. doi: 10.1111/j.1365-294X.2004.02407.x
Dutton, P. H., Jensen, M. P., Frey, A., LaCasella, E., Balazs, G. H., Zárate, P., et al. (2014a). Population structure and phylogeography reveal pathways of colonization by a migratory marine reptile (Chelonia mydas) in the central and eastern Pacific. Ecol. Evol. 4, 4317–4331. doi: 10.1002/ece3.1269
Dutton, P. H., Jensen, M. P., Frutchey, K., Frey, A., LaCasella, E., Balazs, G. H., et al. (2014b). Genetic stock structure of green turtle (Chelonia mydas) nesting populations across the Pacific islands. Pac. Sci. 68, 451–464. doi: 10.2984/68.4.1
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fukuoka, T., Narazaki, T., Kinoshita, C., and Sato, K. (2019). Diverse foraging habits of juvenile green turtles (Chelonia mydas) in a summer-restricted foraging habitat in the northwest Pacific Ocean. Mar. Biol. 166:25. doi: 10.1007/s00227-019-3481-9
Fukuoka, T., Narazaki, T., and Sato, K. (2015). Summer-restricted migration of green turtles Chelonia mydas to a temperate habitat of the northwest Pacific Ocean. Endanger. Species Res. 28, 1–10. doi: 10.3354/esr00671
Gutiérrez-Ortega, J. S., Jiménez-Cedillo, K., Pérez-Farrera, M. A., Vovides, A. P., Martínez, J. F., Molina-Freaner, F., et al. (2018). Considering evolutionary processes in cycad conservation: identification of evolutionarily significant units within Dioon sonorense (Zamiaceae) in northwestern Mexico. Conserv. Genet. 19, 1069–1081. doi: 10.1007/s10592-018-1079-2
Hamabata, T., Hikida, T., Okamoto, K., Watanabe, S., and Kamezaki, N. (2015). Ontogenetic habitat shifts of green turtles (Chelonia mydas) suggested by the size modality in foraging aggregations along the coasts of the western Japanese main islands. J. Exp. Mar. Bio. Ecol. 463, 181–188. doi: 10.1016/j.jembe.2014.12.007
Hamabata, T., Kamezaki, N., and Hikida, T. (2014). Genetic structure of green turtle (Chelonia mydas) peripheral populations nesting in the northwestern Pacific rookeries: evidence for northern refugia and postglacial colonization. Mar. Biol. 161, 495–507. doi: 10.1007/s00227-013-2352-z
Hamabata, T., Nishizawa, H., Kawazu, I., Kameda, K., Kamezaki, N., and Hikida, T. (2018). Stock composition of green turtles Chelonia mydas foraging in the Ryukyu Archipelago differs with size class. Mar. Ecol. Prog. Ser. 600, 151–163. doi: 10.3354/meps12657
Hirai, J. (2019). Insights into reproductive isolation within the pelagic copepod Pleuromamma abdominalis with high genetic diversity using genome-wide SNP data. Mar. Biol. 167:1. doi: 10.1007/s00227-019-3618-x
Jensen, M. P., FitzSimmons, N. N., Bourjea, J., Hamabata, T., Reece, J., and Dutton, P. H. (2019). The evolutionary history and global phylogeography of the green turtle (Chelonia mydas). J. Biogeogr. 46, 860–870. doi: 10.1111/jbi.13483
Jensen, M. P., FitzSimmons, N. N., and Dutton, P. H. (2013). “Chapter 6: molecular genetics of sea turtles,” in The Biology of Sea Turtles, Vol. III, eds J. Wyneken, K. J. Lohmann, and J. A. Musick, (Boca Raton, FL: CRC Press), 135–161.
Jensen, M. P., Ian, B., Limpus, C. J., Hamann, M., Amber, S., Whap, T., et al. (2016a). Spatial and temporal genetic variation among size classes of green turtles (Chelonia mydas) provides information on oceanic dispersal and population dynamics. Mar. Ecol. Prog. Ser. 543, 241–256. doi: 10.3354/meps11521
Jensen, M. P., Pilcher, N., and FitzSimmons, N. N. (2016b). Genetic markers provide insight on origins of immature green turtles Chelonia mydas with biased sex ratios at foraging grounds in Sabah, Malaysia. Endanger. Species Res. 31, 191–201. doi: 10.3354/esr00763
Jombart, T., Devillard, S., and Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94. doi: 10.1186/1471-2156-11-94
Joseph, J., Nishizawa, H., Arshaad, W. M., Kadird, S. A. S., Jaaman, S. A., Bali, J., et al. (2016). Genetic stock compositions and natal origin of green turtle (Chelonia mydas) foraging at Brunei Bay. Glob. Ecol. Conserv. 6, 16–24. doi: 10.1016/j.gecco.2016.01.003
Kameda, K., Wakatsuki, M., Kuroyanagi, K., Iwase, F., Shima, T., Kondo, T., et al. (2017). Change in population structure, growth and mortality rate of juvenile green turtle (Chelonia mydas) after the decline of the sea turtle fishery in Yaeyama Islands, Ryukyu Archipelago. Mar. Biol. 164:143. doi: 10.1007/s00227-017-3171-4
Komoroske, L. M., Jensen, M. P., Stewart, K. R., Shamblin, B. M., and Dutton, P. H. (2017). Advances in the application of genetics in marine turtle biology and conservation. Front. Mar. Sci. 4:156. doi: 10.3389/fmars.2017.00156
Komoroske, L. M., Miller, M. R., O’Rourke, S. M., Stewart, K. R., Jensen, M. P., and Dutton, P. H. (2019). A versatile Rapture (RAD-Capture) platform for genotyping marine turtles. Mol. Ecol. Resour. 19, 497–511. doi: 10.1111/1755-0998.12980
Lahanas, P. N., Bjorndal, K. A., Bolten, A. B., Encalada, S. E., Miyamoto, M. M., Valverde, R. A., et al. (1998). Genetic composition of a green turtle (Chelonia mydas) feeding ground population: evidence for multiple origins. Mar. Biol. 130, 345–352. doi: 10.1007/s002270050254
Meylan, A. B., Bowen, B. W., and Avise, J. C. (1990). A genetic test of the natal homing versus social facilitation models for Green turtle migration. Science 248, 724–727. doi: 10.1126/science.2333522
Natoli, A., Birkun, A., Aguilar, A., Lopez, A., and Hoelzel, A. R. (2005). Habitat structure and the dispersal of male and female bottlenose dolphins (Tursiops truncatus). Proc. R. Soc. B Biol. Sci. 272, 1217–1226. doi: 10.1098/rspb.2005.3076
Nishizawa, H., Abe, O., Okuyama, J., Kobayashi, M., and Arai, N. (2011). Population genetic structure and implications for natal philopatry of nesting green turtles Chelonia mydas in the Yaeyama Islands, Japan. Endanger. Species Res. 14, 141–148. doi: 10.3354/esr00355
Nishizawa, H., Joseph, J., Chong, Y. K., Kadir, S. A. S., Isnain, I., Ganyai, T. A., et al. (2018). Comparison of the rookery connectivity and migratory connectivity: insight into movement and colonization of the green turtle (Chelonia mydas) in Pacific–Southeast Asia. Mar. Biol. 165:77.
Nishizawa, H., Naito, Y., Suganuma, H., Abe, O., Okuyama, J., Hirate, K., et al. (2013). Composition of green turtle feeding aggregations along the Japanese archipelago: implications for changes in composition with current flow. Mar. Biol. 160, 2671–2685. doi: 10.1007/s00227-013-2261-1
Nishizawa, H., Narazaki, T., Fukuoka, T., Sato, K., Hamabata, T., Kinoshita, M., et al. (2014). Juvenile green turtles on the northern edge of their range: mtDNA evidence of long-distance westward dispersals in the northern Pacific Ocean. Endanger. Species Res. 24, 171–179. doi: 10.3354/esr00592
Paris, J. R., Stevens, J. R., and Catchen, J. M. (2017). Lost in parameter space: a road map for stacks. Methods Ecol. Evol. 8, 1360–1373. doi: 10.1111/2041-210X.12775
Park, J.-S., Takayama, K., Suyama, Y., and Choi, B.-H. (2019). Distinct phylogeographic structure of the halophyte Suaeda malacosperma (Chenopodiaceae/Amaranthaceae), endemic to Korea–Japan region, influenced by historical range shift dynamics. Plant Syst. Evol. 305, 193–203. doi: 10.1007/s00606-018-1562-8
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Rankin-Baransky, K., Williams, C. J., Bass, A. L., Bowen, B. W., and Spotila, J. R. (2001). Origin of loggerhead turtles stranded in the northeastern United States as determined by mitochondrial DNA analysis. J. Herpetol. 35, 638–646. doi: 10.2307/1565903
Rochette, N. C., and Catchen, J. M. (2017). Deriving genotypes from RAD-seq short-read data using Stacks. Nat. Protoc. 12, 2640–2659. doi: 10.1038/nprot.2017.123
Seminoff, J. A., Allen, C. D., Balazs, G. H., Dutton, P. H., Eguchi, T., Haas, H. L., et al. (2015). Status review of the green turtle (Chelonia mydas) under the Endangered Species Act. NOAA Tech. Memo. NOAANMFS-SWFSC- 539:571.
Shamblin, B. M., Bolten, A. B., Bjorndal, K. A., Dutton, P. H., Nielsen, J. T., Abreu-Grobois, F. A., et al. (2012). Expanded mitochondrial control region sequences increase resolution of stock structure among North Atlantic loggerhead turtle rookeries. Mar. Ecol. Prog. Ser. 469, 145–160. doi: 10.3354/meps09980
Suyama, Y., and Matsuki, Y. (2015). MIG-seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 5:16963. doi: 10.1038/srep16963
Takata, K., Taninaka, H., Nonaka, M., Iwase, F., Kikuchi, T., Suyama, Y., et al. (2019). Multiplexed ISSR genotyping by sequencing distinguishes two precious coral species (Anthozoa: Octocorallia: Coralliidae) that share a mitochondrial haplotype. PeerJ 7:e7769. doi: 10.7717/peerj.7769
Tikochinski, Y., Bendelac, R., Barash, A., Daya, A., Levy, Y., and Friedmann, A. (2012). Mitochondrial DNA STR analysis as a tool for studying the green sea turtle (Chelonia mydas) populations: the Mediterranean Sea case study. Mar. Genomics 6, 17–24. doi: 10.1016/j.margen.2012.01.002
Keywords: marine turtle, population genomics, field sample, MIG-seq, natal origin identification
Citation: Hamabata T, Matsuo A, Sato MP, Kondo S, Kameda K, Kawazu I, Fukuoka T, Sato K, Suyama Y and Kawata M (2020) Natal Origin Identification of Green Turtles in the North Pacific by Genome-Wide Population Analysis With Limited DNA Samples. Front. Mar. Sci. 7:658. doi: 10.3389/fmars.2020.00658
Received: 18 March 2020; Accepted: 20 July 2020;
Published: 07 August 2020.
Edited by:
Shigehiro Kuraku, RIKEN Center for Biosystems Dynamics Research (BDR), JapanReviewed by:
Hans Recknagel, University of Glasgow, United KingdomTetsuya Akita, Japan Fisheries Research and Education Agency, Japan
Copyright © 2020 Hamabata, Matsuo, Sato, Kondo, Kameda, Kawazu, Fukuoka, Sato, Suyama and Kawata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoko Hamabata, dG1rLmhhbWFiYXRhQGdtYWlsLmNvbQ==