 April N. Abbott
April N. Abbott Stefan Löhr
Stefan Löhr Megan Trethewy
Megan Trethewy
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 20 August 2019
Sec. Marine Biogeochemistry
Volume 6 - 2019 | https://doi.org/10.3389/fmars.2019.00504
This article is part of the Research Topic REE Marine Geochemistry in the 21st Century: A Tribute to the Pioneering Research of Henry Elderfield (1943–2016) View all 11 articles
The rare earth elements (REEs) are an important tool for understanding biogeochemical cycling and sedimentary processes in the global ocean. However, ambiguities in the marine REE budgets, including questions around the dominant source of REEs to the ocean, hinder the application of this tool. A bottom-up model for REE release into the ocean has recently been proposed, driven by early diagenetic processes such as sediment dissolution, with potentially significant implications for the interpretation of marine REE and Nd isotope paleo-records. Here, our goal is to identify the phase or phases that interact with the pore waters to drive such a benthic flux. We use new pore water REE, microbeam imaging and mineralogical data in combination with published pore water REE data to evaluate potential sedimentary REE host phases. Mineralogical and direct imaging observations suggest that authigenic Fe or Mn oxyhydroxides, which are widely considered a dominant REE host phase, are not sufficiently abundant sediment components to account for the high Nd concentrations recovered in reductive leaches, and are unlikely to be the primary source of pore water REEs. Pore water REE signatures similar to river sourced clays indicate a detrital clay dissolution source, while the spread in heavy to light REE enrichment in pore waters and bottom waters relative to this clay source is best explained by fractionation during authigenic clay uptake of REEs. We therefore conclude that clay mineral dissolution and authigenesis are likely the primary influences on the REE cycling near the seafloor. We propose that the balance between dissolution and authigenesis controls the concentration, ratio of heavy and light REE abundances, and the isotopic composition of the pore waters. We discuss the implications of this hypothesis on an oceanic REE budget controlled by a benthic flux from a sedimentary REE source, and the use of authigenic neodymium isotopes as a paleoproxy for shifts in ocean circulation.
The rare earth elements (REEs) are widely used in paleoceanographic studies. Also known as the lanthanides, the REEs are a series of 14 elements with largely coherent chemical properties (Elderfield and Greaves, 1982) used for applications ranging from reconstructing circulation and oxygen content to examining the influence of diagenesis. These applications rely on examining fractionation within the REEs or the behavior of individual REEs. The fractionation of REEs is visualized through normalization to a reference such as shale or chondrite and can reveal information on oceanic and sedimentary processes (e.g., Elderfield and Greaves, 1982; Sholkovitz et al., 1994; Nozaki and Alibo, 2003; Alibo and Nozaki, 2004; Haley et al., 2004; Akagi, 2013; Hathorne et al., 2015; Abbott et al., 2015b; Skinner et al., 2019). For example, an increased stability of heavy rare earths (HREEs) in aqueous complexes results in the HREEs complexing more readily and remaining in solution (e.g., Goldberg et al., 1963; Turner et al., 1981; Wood, 1990; Akagi, 2013) whereas the light rare earths (LREEs) will more readily adsorb on particle surfaces (e.g., Byrne and Kim, 1990; Sholkovitz et al., 1994). Neodymium (Nd) is one REE that is commonly used individually as the Nd isotopic composition of seawater is considered a useful tracer of water mass circulation (e.g., von Blanckenburg, 1999). The isotopic signature (εNd) of ferromanganese coatings, fossil fish teeth, and foraminifera recovered from the marine sedimentary record is a widely used tool for reconstructing past ocean circulation. Changes in global ocean circulation are inferred from the changes in the εNd of these authigenic records under the assumption that εNd is quasi-conservative in the global oceans (e.g., Vance and Burton, 1999; Frank, 2002; Haley et al., 2008; Böhm et al., 2015; Abbott et al., 2016b; Deaney et al., 2017).
The usefulness of REEs to our understanding of the ocean is hampered by ambiguities in the modern cycling and oceanic budget of these REEs. Despite extensive progress in field and laboratory research on the utility of REE proxies (e.g., Amakawa et al., 2009; Carter et al., 2012; Grasse et al., 2012; Singh et al., 2012; Grenier et al., 2013; Pearce et al., 2013; Du et al., 2016; Skinner et al., 2019) and REE cycling in the ocean (e.g., Elderfield and Greaves, 1982; de Baar et al., 1985; Byrne and Kim, 1990; Bertram and Elderfield, 1993; Greaves et al., 1994; Zhang and Nozaki, 1996; Douville et al., 1999; Nozaki and Alibo, 2003; Haley et al., 2004, 2017; Lacan and Jeandel, 2005; Zhang et al., 2008; Johannesson et al., 2011; Jeandel et al., 2013; Hathorne et al., 2015; Rousseau et al., 2015; Abbott et al., 2015b; Grenier et al., 2018), the processes governing the modern budget remain surprisingly poorly constrained. The riverine dissolved flux was long considered the primary source of neodymium to the ocean (e.g., Bertram and Elderfield, 1993; Sholkovitz et al., 1999; Frank, 2002) but oceanic budgets that consider dissolved riverine and dust Nd sources still cannot balance the oceanic budget for either Nd concentrations or Nd isotopes (e.g., Bertram and Elderfield, 1993; Tachikawa et al., 2003). Recent estimates suggest that upward of 90% of oceanic neodymium inputs are “missing from the budget” (Tachikawa et al., 2003; Jones et al., 2008; Arsouze et al., 2009) with sediment dissolution, submarine groundwater discharge, dust, hydrothermal input, fluid-particle interaction, and reversible scavenging among the potential sources investigated to date (e.g., Piper, 1974; Bertram and Elderfield, 1993; Douville et al., 1999; Lacan and Jeandel, 2005; Jones et al., 2008; Arsouze et al., 2009; Freslon et al., 2014; Rickli et al., 2014; Rousseau et al., 2015; Stichel et al., 2015; Abbott et al., 2015b; Howe et al., 2016; Stewart et al., 2016; van de Flierdt et al., 2016; Zheng et al., 2016). Similar gaps are predicted for the budgets of the other lanthanides. Since the particulate flux dominates fluvial REE transport to the ocean, dissolution of less than 3% of the particulate input could represent the largest oceanic REE source term (Jeandel and Oelkers, 2015), consistent with recent estimates that a benthic flux may account for near 90% of oceanic REE budget (Arsouze et al., 2009; Abbott et al., 2015b). Furthermore, the apparent decoupling between the non-conservative behavior of Nd concentrations and the quasi-conservative behavior of Nd isotopes, coined the “neodymium paradox” (e.g., Goldstein and Hemming, 2003; Lacan and Jeandel, 2005; Arsouze et al., 2009) raises questions around the validity of εNd as a quasi-conservative tracer of ocean circulation.
The uncertainties surrounding oceanic REE cycling are at least in part due to gaps in our understanding of the sedimentary host phase or phases acting as either REE sources or REE sinks. The relevance of these host phases to both the oceanic REE budget and the application of REE paleoproxies centers around (i) whether and under what conditions each potential phase acts as a sink of REE from the fluid phase (e.g., scavenging), a source of REE to the fluid phase (e.g., dissolution, remineralization), or exchanges with the fluid phase and (ii) over what time scale these solid-fluid interactions are occurring. A particulate phase acting as a sink when in contact with seawater will provide the best record of changes in ocean conditions through time. However, a phase that continues to act as a sink during burial will have any original seawater signature diagenetically overprinted as has already been proposed for biogenic phosphates and foraminifera (Palmer and Elderfield, 1985; Toyoda and Tokonami, 1990; Takahashi et al., 2015; Trotter et al., 2016; Zhang et al., 2016; Skinner et al., 2019).
A wide range of sediment components have been identified as potentially important REE host phases. These could either contribute REEs to the dissolved phase and drive a benthic source of REEs to the ocean or act as a sink of REEs from the fluid phase and thus potentially record environmental conditions. However, to date little robust, direct evidence of the role of these phases has been found. Potential host phases include Fe-Mn oxyhydroxides (e.g., Sholkovitz et al., 1994; Bayon et al., 2004; Haley et al., 2004; Ren et al., 2015; Takahashi et al., 2015), cerium oxides (e.g., Haley et al., 2004), phosphates (e.g., Kon et al., 2014; Takahashi et al., 2015; Zhang et al., 2016), biogenic silicates (e.g., Akagi et al., 2014), organic matter (e.g., Duncan and Shaw, 2003; Haley et al., 2004; Schacht et al., 2010; Kim et al., 2012; Freslon et al., 2014), volcanic ash (e.g., Vance et al., 2004; Elmore et al., 2011; Wilson et al., 2013), and clays (e.g., Cullers et al., 1975; Grandjean et al., 1987; Zhang et al., 2016). These sediment components may not contribute equally to REE cycling, and their contributions will vary in space and time as the amount of REEs remobilized from each phase is likely a function of mineralogy, mineral chemistry, reactivity, surface area, and other factors such as local pore water chemistry and redox state (e.g., Elderfield and Sholkovitz, 1987; Wilson et al., 2012). The distinct conditions of formation, alteration or dissolution of each potential host phase makes the identification of this phase important to paleo-interpretations. Currently, Fe-Mn oxyhydroxides are widely considered a sink for seawater REEs found ubiquitously throughout the global ocean and thus an important reactive pool of REEs in the sediments (e.g., Bayon et al., 2004), but clays are likely a larger total reservoir in the sediments (e.g., Cullers et al., 1975) and the overall impact of silicate dissolution is potentially dominant (e.g., Jeandel and Oelkers, 2015). Uncertainty as to the major REE host phases can be attributed to limitations in the operationally defined techniques used to quantify their abundance and REE signature. The standard protocols used to target the Fe-Mn oxyhydroxide phases, for instance, use prescribed leaching procedures and are often assumed successful based on chemical characterization of the recovered leachate (e.g., Fe/Al and Nd/Fe ratios), while lacking direct observation of the targeted phase. The limitations of this purely geochemical approach is illustrated in recent work which showed that the “oxide coating” in foraminifera consists of Fe, Mn, and REEs absorbed directly from the surrounding pore fluids rather than a discrete oxyhydroxide phase (Skinner et al., 2019).
Here, we combine geochemical, mineralogical, and sedimentological data from sites in the North Pacific and Tasman Sea to identify the sedimentary phase or phases that control benthic cycling of REEs. We use standard mineralogical techniques (i.e., X-ray diffraction) coupled to novel microbeam imaging and mineral mapping for direct, unambiguous identification of potential REE host phases in marine sediments, allowing us to examine the relative abundance of these phases and determine their likely origin. We employ REE fingerprinting to identify the processes influencing the REEs in marine sediments and in the ocean via a pore water controlled benthic flux and examine the implications on the oceanic budget of REEs as well as the use of REEs as proxies in paleoceanographic studies. We argue that a significant role of surface sediments in the REE cycle is consistent with observations that corals, Fe-Mn nodules, and co-located sediment phases have the same εNd as bottom waters, with sediment diagenesis providing the Nd to bottom waters as well as the nodules and corals (e.g., Elderfield and Greaves, 1982; Abbott et al., 2015a; Roberts and Piotrowski, 2015).
We focus on sediment cores collected from three sites in the eastern North Pacific (2012–2013, R/V Oceanus) and three sites in the western Tasman Sea (2016, R/V Investigator) at water depths of approximately 200, 1500, and 3000 m (Figure 1 and Table 1). For simplicity, we will refer to the 3 North Pacific sites as NP200, NP1500, and NP3000 and the Tasman Sea sites as TS200, TS1500, and TS3000, respectively. Exact water depths, locations, and site names from previous literature are available in Table 1. Detailed descriptions of core and pore water collection are available elsewhere (Abbott et al., 2015b; Abbott, 2019). Briefly, a multi-corer was deployed and cores with a visibly intact sediment-water interface were sectioned on board the ship into HCl cleaned centrifuge tubes in a glove bag filled with an inert (N2) atmosphere. After centrifugation, pore water was pulled off using an HCl cleaned syringe, filtered with a 0.45 μm syringe filter, and acidified to pH ≤ 2 with distilled HCl. Sediments were frozen and later freeze dried prior to analyses.
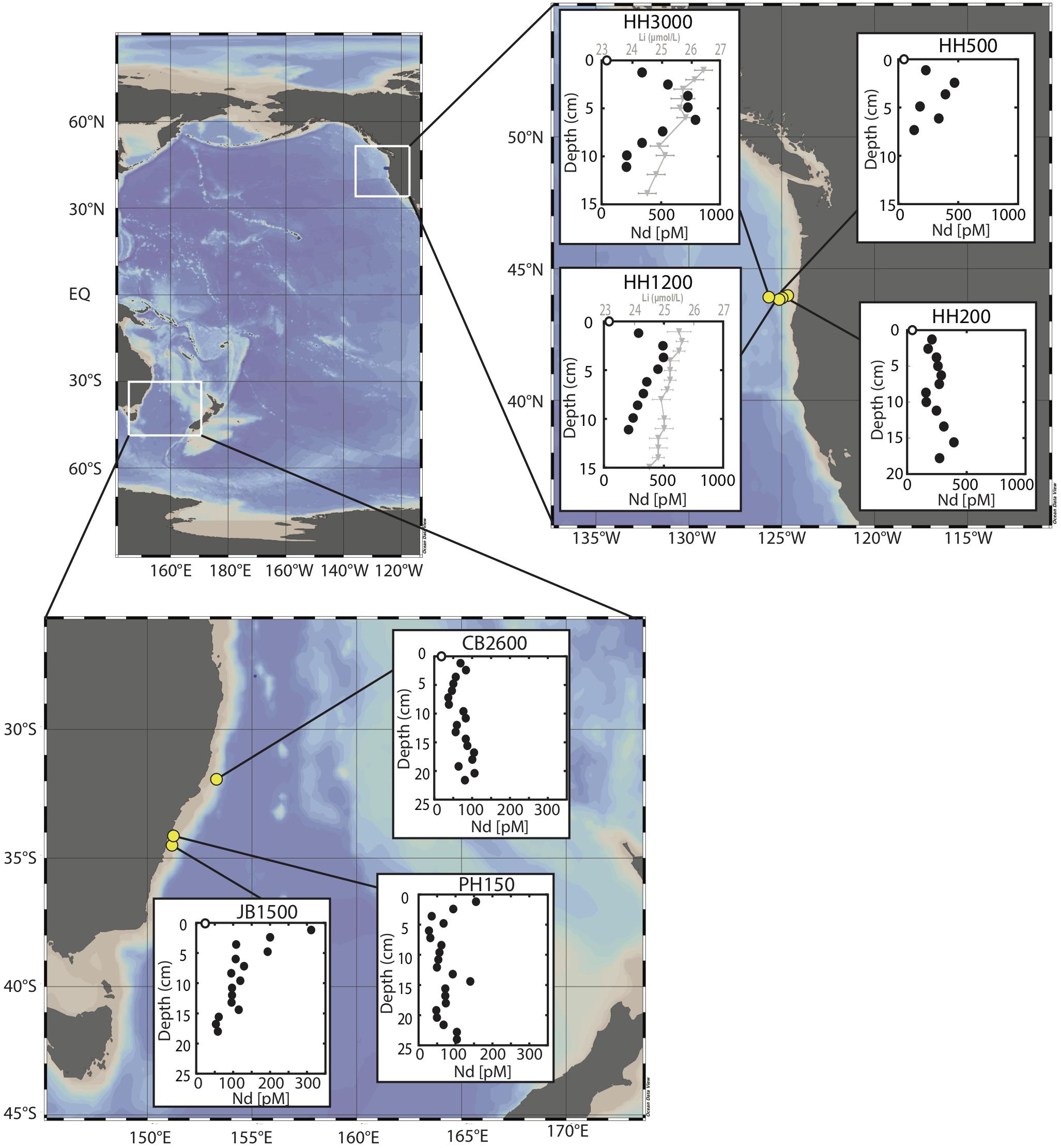
Figure 1. Site locations in the North Pacific and Tasman Sea. Pore water neodymium profiles shown for each site (neodymium concentrations from Abbott et al., 2015b; Abbott, 2019). Dissolved lithium pore water profiles are provided for NP1500 and NP3000 from Abbott et al., 2016a.
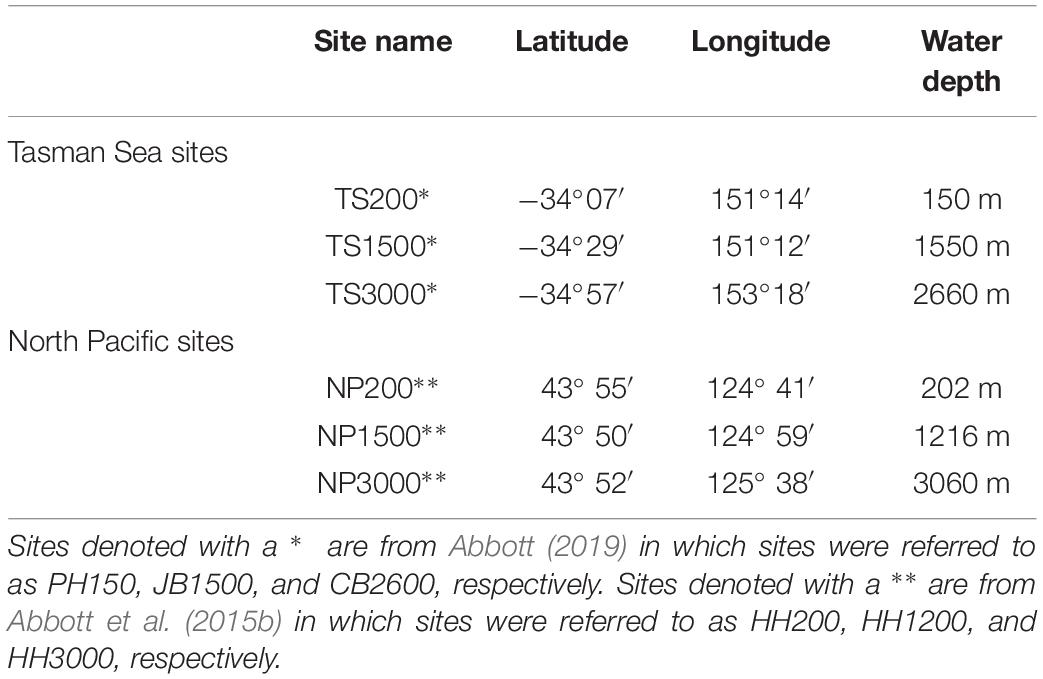
Table 1. Site locations and water depths.
Rare earth element analyses for the North Pacific pore waters have been previously reported (Abbott et al., 2015b, 2016a). Tasman Sea pore waters were analyzed using a SeaFAST II inline with a Thermo X-series II Inductively Coupled Plasma-Mass Spectrometer at the Keck Collaboratory for Plasma Mass Spectrometry (as described by Yang and Haley, 2016; Abbott, 2019). To compare the REE patterns of pore waters to potential host phases, we considered all pore water samples collected from the six sites (three North Pacific and three Tasman Sea) and other published marine pore water values (Haley et al., 2004; Abbott et al., 2015b). We did not include pore waters collected using Rhizons© because of the REE fractionation associated with this technique (Abbott et al., 2015b). We also excluded pore water data with less than ten of the REEs reported because these data did not allow for consistent comparison of REE signatures. The included data were grouped into quartiles using the neodymium concentration in each sample. Neodymium was chosen for its applicability to paleoceanographic reconstructions. All concentration data was normalized to PAAS (Taylor and McLennan, 1985) and then to praseodymium (Pr). The normalization to PAAS facilitates inter-REE comparisons despite large differences in natural abundance and the normalization to Pr facilitates direct inter-sample pattern comparison despite differences in concentration.
Grain size was measured on the Tasman Sea samples using a MasterSizer 2000 laser granulometer at Macquarie University. Samples were analyzed pre-leach (freeze-dried) and post a buffered acetic acid decarbonization step (leach procedure after Abbott et al., 2016a, b). Two subsamples were loaded into the MasterSizer for each pre- and post- leach steps, which completed 2 measurement cycles 5 s apart per load for a total of four analyses per sample. All samples were analyzed using deionized water with the sample refractive index set to 1.544 and absorption set to 1. Samples were measured for 20 s (20,000 snaps) and the background was measured between each load for 10 s (10,000 snaps). The ultrasonic probe was set at 50% and remained on while the sample was loaded to a laser obscuration between 5 and 20% and then the probe was turned off at the start of each analysis. The stirrer was set at 500 rpm and the pump was set at 1250 rpm and remained on throughout loading and analyses. Grain size data for the North Pacific sites reported for comparison is from Abbott et al. (2016a).
Bulk mineralogy of representative samples from each site was determined by X-ray diffractometry (XRD) at Macquarie University. Freeze-dried but otherwise untreated samples were homogenized in an agate mortar and pestle, before back-loading into 25 mm internal diameter stainless steel sample holders. X-ray powder diffraction patterns spanning 5 to 90 ∘2θ were collected using a PANalytical Aeris benchtop XRD instrument (0.02∘ step size, Cu-radiation source with 40 kV generator voltage and 15 mA tube current, 1/8 inch divergence slits, and 23 mm beam mask). Diffraction patterns were interpreted using Panalytical HighscorePlus software with the ICSD database for phase identification. The detection limit for bulk XRD analyses depends on the density, Z number, and crystal structure of the compounds in the sample, but corresponds to approximately 1% by volume (Cullity and Stock, 2001).
The mineralogy of the clay fraction in representative samples from each site was determined on oriented preparations of <2 μm separates. The <2 μm fraction was obtained by settling after carbonate removal using sodium acetate buffered acetic acid (for Tasman Sea sites only because North Pacific sites are low carbonate) and ultrasonic dispersal. An aliquot was pipetted onto low background Si samples holders, air-dried and measured using the same instrument (Tasman Sea samples: 2–30 ∘2θ, 0.02∘ step size, 1/8 inch slits, 23 mm beam mask, beam knife in low position) or on a PANalytical Xpert-Pro MPD System (Oregon Margin samples: 2–65 ∘2θ, 0.0167∘ step size, Cu source with 45 kV generator voltage and 40 mA tube current, automatic slits). Clay diffraction patterns were collected both on air-dried samples and after treatment with ethylene glycol, with interpretation following Moore and Reynolds (1997). We did not attempt to directly geochemically characterize the detrital or authigenic clays in the sediments studied here because authigenic clays are typically small, interspersed grains and thus cannot be separated from detrital clays using physical separation techniques. Furthermore, our SEM-based petrographic work shows that even relatively large pellets of authigenic clays contain abundant detrital contaminants including igneous silicates and barite, preventing picked grains from representing a true authigenic signature. Since detrital and authigenic clay phases cannot be confidently separated from <2 μm, non-clay sedimentary fractions, the geochemical characterization of the clay size fraction would represent a mixed detrital clay, non clay, and authigenic clay signature. We therefore use available published characterizations of riverine clay to represent a detrital end member and authigenic phases (fish teeth, Fe-Mn crusts, glauconite) in our phase modeling.
Polished resin mounts of the freeze-dried but otherwise untreated sediments were prepared using standard methods. The samples were not homogenized prior to resin embedding in order to preserve the spatial associations present within the sediment. Resin mounts were subsequently ion polished using a Hitachi IM4000 Argon Ion Mill (30 min at 10° beam incident angle, 5 kV accelerating voltage and continuous sample rotation) to remove surface damage from mechanical polishing which can otherwise obscure phase relations and the presence of nm-scale coatings. Ion milled samples were carbon-coated prior to scanning electron microscope (SEM) analysis on an FEI Teneo LoVac field emission SEM equipped with dual Bruker XFlash Series 6 energy dispersive X-ray spectroscopy (EDS) detectors. High-resolution back-scatter electron (BSE) images and mineral maps of 1 to 3 regions of interest were collected for each sample (13 mm working distance, 15 kV accelerating voltage). BSE image tilesets (100 nm pixel resolution) and EDS spectra (1 μm step size, 8 ms acquisition time) for mineral mapping were collected sequentially using the FEI Maps Mineralogy software, followed by classification of the individual EDS spectra using the FEI Nanomin software (Haberlah et al., 2015). Mineral identification is achieved by comparing EDS spectra collected in the mapped area against reference spectra collected on known mineral standards, and is further constrained by independent XRD-based mineral identification. Unlike earlier SEM-based mineral mapping techniques (e.g., QEMSCAN), the Nanomin mineral classification system can de-convolve mixed X-ray spectra and assign up to three minerals per analyzed spot (Haberlah et al., 2015). This is a critical requirement for the correct interpretation of the mixed phase X-ray spectra characteristic of heterogeneous fine-grained sediments, where the X-ray generating electron interaction volume is commonly larger than the grain size. BSE photomicrographs, EDS elemental maps, and classified mineral maps provide direct visual evidence of grain relationships and common associations. While it is possible to convert these mineral maps into quantified percent compositions, the high heterogeneity of the samples coupled with the relatively small mapped areas results in a large uncertainty relative to bulk sample analyses such as XRD. Mineral mapping is employed here solely to test for the presence of, and establish the identity of trace phases that are too low for detection with the XRD.
We expect the REE pattern of the pore water to be influenced by both the source of REEs to the pore water and any sink of the REEs from the pore water. The fine-grained nature and intermingled distribution of most authigenic clays complicates an assessment of their REE content, and therefore of their potential influence on the pore water REE signature. However, glauconite is a widely occurring authigenic clay mineral which forms easily separated, macroscopic pellets (Baldermann et al., 2013, 2015) for which the REE signature is relatively well constrained (e.g., Huggett et al., 2017). Therefore, to test the feasibility of a clay driven pore water REE signature, we model the impact of glauconite formation on the pore water REE signature. For this calculation, we assume world river average clay (WRAC, Bayon et al., 2015) is the dominant source of pore water REEs (see section “Pore Water REE Patterns Support a Clay Dissolution Source” for a justification). Thus, we set the initial pore water value equivalent to WRAC (PW0 = WRAC) and we average the Shadwell, Abbey Mills, and top and bottom Victoria glauconites of Huggett et al. (2017) for our glauconite REE pattern. We estimate a partition coefficient, k, by taking the ratio of WRAC to average glauconite concentration of each REE. We model the alteration of the pore water REE pattern with continued uptake into glauconite as:
where Pc is the percentage of PW0 lost to glauconite (G) formation.
Pore water REE concentrations are up to two orders of magnitude enriched relative to ambient seawater with pore water concentrations ranging from 25 to 790 pmol Nd L–1 and 1 to 30 pmol Lu L–1 at these sites. In general, the highest pore water REE concentrations were observed in the North Pacific (Abbott et al., 2015b) with lower concentrations observed in the Tasman Sea (Abbott, 2019). The enrichment of REE in pore fluid at these six sites is not as high as has been observed at other North Pacific sites (e.g., Haley et al., 2004 – 1280 pmol Nd L–1 at Station 10; Abbott et al., 2015a – 2250 pmol Nd L–1 at Station 2; Deng et al., 2017 – 1654 pmol Nd L–1 at MABC25-1). Pore fluid concentrations show a shallow sub-surface maximum (<10 cm below the sediment water interface) across all sites, occurring in the upper 1–2 cm (Tasman Sea) and in the upper 4–7 cm (North Pacific) of the sediment column (Figure 1), which is consistent with an early diagenetic release of REEs to the pore water and a benthic source of REEs to the ocean (Abbott et al., 2015a, b; Abbott, 2019). The pore water REE maximum does not directly correlate with dissolved iron at the Tasman Sea sites nor the North Pacific sites, with the REE maximum occurring shallower in the sediment column than the appearance of dissolved iron (Abbott et al., 2015a; Abbott, 2019). The pore waters with the highest concentration of REEs have the flattest REE patterns with some middle REE (MREE) enrichment (Figure 2). These patterns typically become increasingly heavy REE (HREE) enriched as the amount of REEs in the pore water decreases (Figure 2). However, even the most HREE enriched pore waters do not match the HREE enrichment of seawater.
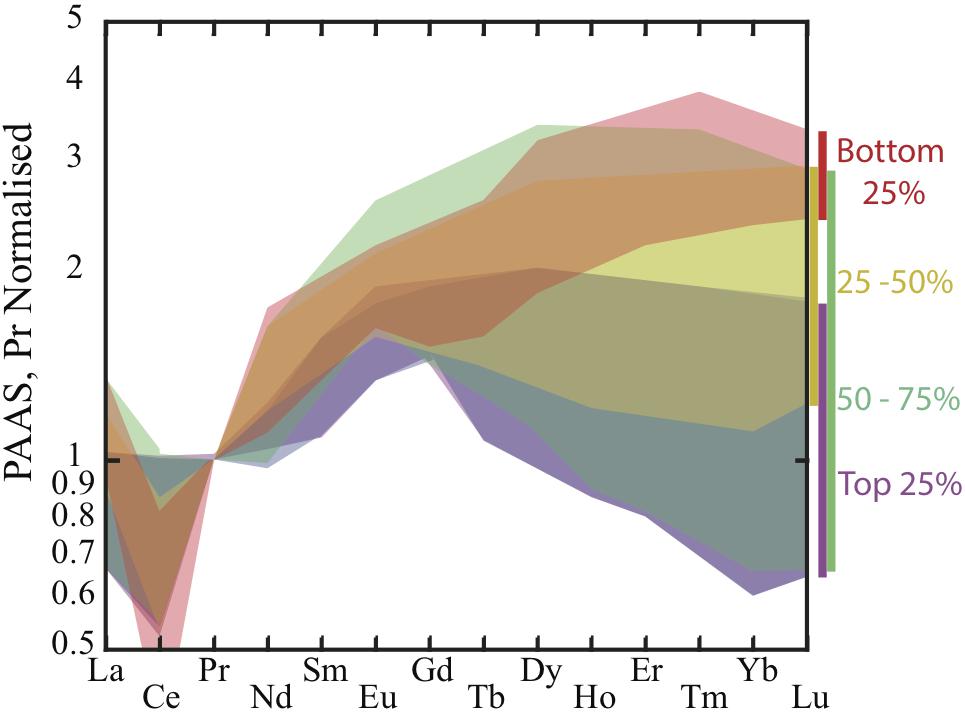
Figure 2. General trends in pore water shale-normalized REE pattern by concentration. REE concentrations from North Pacific and Tasman Sea sites (Figure 1) were ranked by neodymium concentration and split into quartiles, the range is displayed for each quartile. Normalized to PAAS values of Taylor and McLennan (1985). Europium data is excluded for samples with suspected oxide formation (e.g., Abbott et al., 2015b) and Samarium data is excluded for Tasman Sea samples due to a high blank.
Grain size distributions were consistent down-core at each site for both pre- and post-leach measurements. Tasman Sea grain size distributions are largely bimodal pre-leach with only a single peak in post-leach (Supplementary Material). The primary grain size in pre-leached samples was coarse silt to very fine sand at both the 200 and 1500 m Tasman Sea sites, and the two dominant pre-leach grain sizes at the 3000 m site are very fine clay and medium silt. The primary grain size in post-leached samples was very fine clay at the 1500 and 3000 m sites and medium silt at the 200 m site, reflecting dissolution of coarser biogenic carbonates during the acetic acid leach. The 200 m site in the North Pacific had a similar grain size distribution to the 200 m Tasman Sea site with medium silt the dominant grain size. The 3000 m site in the North Pacific was most similar to both the 1500 m and 3000 m Tasman Sea sites with a dominant grain size of very fine clay in the post-leach samples (Abbott et al., 2016a).
At all three Tasman Sea Sites clay fraction XRD reveals a consistent clay mineral assemblage comprised of kaolinite, illite, smectite and mixed-layer illite-smectite (Figure 3). However, two distinct sediment types can be distinguished on the basis of bulk XRD (Supplementary Material) and SEM-based mineral mapping (Figure 4). The shallow TS200 site features a poorly sorted, clay to fine sand (up to 150 μm) comprised of a mixture of terrestrially derived silt to fine sand grains (quartz, feldspar (albite), kaolinite, ankerite, lithics with quartz and feldspar ± ankerite ± rutile), biogenic calcite, aragonite and high-magnesium calcite (intact and fragmented foraminifera, bivalves, echinoid spines) and opal (sponge spicules) as well as a <5 μm fraction comprised of clay minerals and coccolithophore remains (Figure 4). Samples recovered at the deeper sites TS1500 and TS3000, by contrast, are calcareous oozes containing calcite, quartz, feldspar (albite), along with muscovite/illite, kaolinite, and smectite (Figures 3, 4). SEM analysis reveals potentially wind-blown silt size quartz, feldspar and kaolinite at TS1500, with abundant silt and sand size foraminiferal tests, as well as siliceous sponge spicules (Figure 4 and Supplementary Figure 2). The <5 μm fraction is predominantly clay minerals and coccolithophore remains, with trace abundances of Fe-oxide, rutile, barite and silt-size zircon. TS3000 is very similar to TS1500, but has terrestrially derived detrital grains up to 150 μm in size.
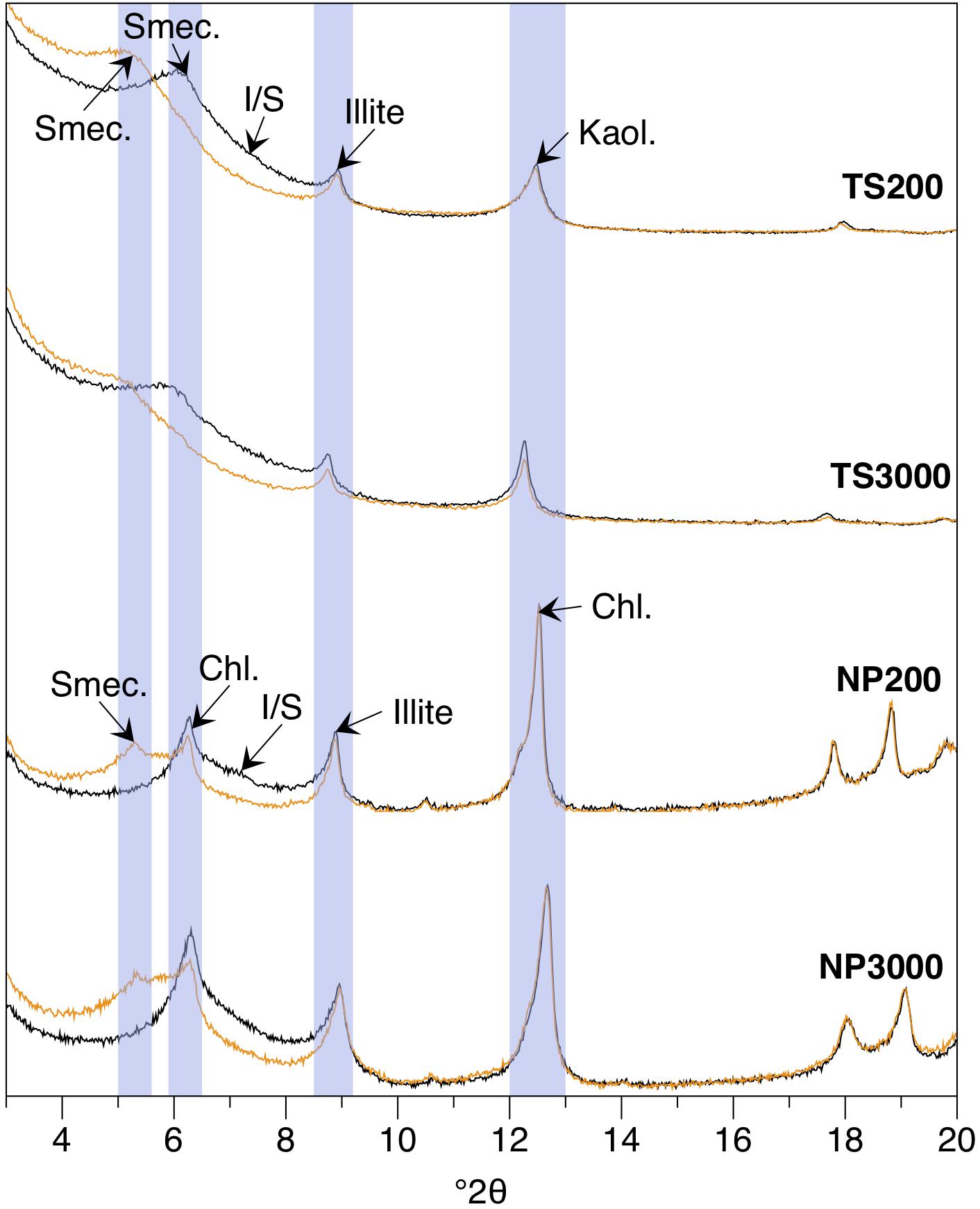
Figure 3. Representative X-ray diffractograms of the air dried (black) and ethylene glycol-treated (orange) <2 μm fraction from the shallow and deep North Pacific and Tasman Sea sites. Peaks corresponding to smec., smectite; chl., chlorite; I/S, mixed-layer illite-smectite; kaol., illite and kaolinite are indicated. Bulk XRD information is provided in the Supplementary Material.

Figure 4. SEM images BSE (left) and mineral map (right) for panel (A) TS200 1–2 cm, (B) TS1500 1–2 cm, (C) TS3000 1–2 cm, (D) NP200 2–3 cm, (E) NP1500 0–1 cm, and (F) NP3000 0–1 cm with selected features labeled as: L, lithic grain; cal, calcite; sps, sponge spicule; ank, ankerite; FeO, iron oxide; dol, dolomite; hmc, high Mg calcite; qtz, quartz; rtl, rutile; fsp, feldspar; afp, feldspar partially weathered to kaolinite; dtm, diatom; zrc, zircon; epi, epidote; olv, olivine; il-sm, illite and smectite; apt, apatite; chl, chlorite; brt, barite. Scale bars are 40 μm for all panels.
Bulk XRD analysis identifies a mineral assemblage consisting of quartz, feldspar (primarily albite), chlorite, mica (muscovite and/or illite), pyroxenes (augite or clinopyroxene), and amphibole (magnesian hornblende) at all three North Pacific sites (Supplementary Material). In addition, the two deeper sites show evidence for a significant X-ray amorphous component, identified as opaline silica (see below). Clay fraction XRD confirms the presence of illite, chlorite and smectite at all three sites (Figure 3). Mineral mapping confirms the presence of a similar mineral assemblage across all three North Pacific sites, but with a systematically decreased grain size, increased clay mineral, and increased biogenic component with depth (Figure 4). NP200 is a poorly sorted fine sand. The silt to sand size fraction is primarily comprised of quartz, Na-feldspar, pyroxene (augite), epidote, and a Mg-silicate phase identified as Mg-olivine. Lithic grains (quartz and feldspar ± rutile, mica/illite, chlorite, and illmenite) are abundant, while glass shards of likely volcanic origin are also observed (Figure 5). Abundant chlorite occurs both as discrete silt to fine sand-sized grains and within larger lithic grains. Siliceous biogenic remains (diatom frustules and sponge spicules) are present in both the silt and clay size fractions. NP1500 has a mineral assemblage similar to NP200, but with detrital mineral grains of silt size or finer (<60 μm diameter), fewer pyroxene, epidote and olivine, and no volcanic glass. Authigenic Fe smectite and glauconite pellets up to 1500 μm are abundant at this site (Figure 5), and there is an increased contribution of clay minerals and siliceous biogenic debris relative to the shallower site. The EDS elemental maps and subsequent Nanomin mineral maps reveal a wide range of textural maturities of the glauconite and Fe-smectite pellets present, ranging from fully formed mature pellets to fragile incipient pellets. While the mature pellets would be relatively resistant to physical breakdown if transported, the presence of incipient pellets that would not survive such transport makes in-situ clay authigenesis most likely. NP3000 is a well sorted siliceous ooze dominated by diatom remains. Detrital grains (feldspar, quartz, chlorite and muscovite, no lithic grains, volcanic glass, or glauconite) are up to 20 μm diameter, with trace abundances of zircon, rutile, and apatite (Figure 4). However, the bulk of the detrital sediment component is <5 μm in size, comprising clays, minor feldspar and quartz, and trace amounts of barite and Fe oxides.

Figure 5. Elemental iron (green) maps overlaid on SEM BSE images of (A) an incipient glauconite grain from NP200 2–3 cm, FeSmc, iron rich smectite (B) sediment grains from TS1500 1–2 cm, (C) an immature glauconite grain from the NP1500 0–1 cm that is low in K, high in iron, (D) volcanic glass shards surrounded by clay minerals from NP200 2–3 cm, and (E,F) iron rich clay aggregations from TS3000 1–2 cm. Small detrital iron oxide grains are visible in panels (B,E,F) as indicated by yellow arrows. All scale bars are 10 μm except for panel (C) (100 μm). Selected features labeled as: MgOl, magnesium olivine; apt, apatite; qtz, quartz; scm, smectite; tnt, titanate; chl, chlorite; dtm, diatom; ccl, coccolithophore; cal, calcite; fsp, feldspar; L, lithic grain; rtl, rutile.
Fe and Mn oxyhydroxides are not evident in bulk X-ray diffraction patterns of any site studied here but can be difficult to detect when present as poorly crystalline or amorphous forms. Mineral mapping does reveal trace quantities of Fe oxides present as discrete, μm size grains which are likely of detrital origin (Figure 5) at all sites. However, EDS elemental maps show that iron is primarily hosted in the clay size fraction (all sites, Figure 5), but is also present in silt/sand size chlorite grains, their unaltered igneous precursor minerals (NP sites only – Figure 4), as well as Fe-smectite and glauconite pellets (NP1500 only – Figure 5). Mineral mapping does not identify Mn oxyhydroxides, but Mn elemental maps show patches of Mn enrichment relative to background at Site NP3000, which may represent local enrichments of nm-scale Mn oxyhydroxide intermingled with the clay mineral fraction. Positive identification of this Mn enriched material is beyond the instrumental resolution. However, while the 1 μm step size and potentially several μm3 beam interaction volume during the mineral mapping and EDS analyses does not permit the direct identification of nm-scale coatings, the high Z-number of Fe and Mn oxyhydroxides relative to siliciclastic sediment components mean that aggregates of nanoscale Fe-Mn oxyhydroxides and coatings >100 nm should be visible as bright zones in the high resolution backscatter electron images. Higher resolution (∼30 nm) field emission reveals some high Z-number material intermingled with the clay fraction, which may be Fe-Mn oxyhydroxides, but could also be barite, rutile, and apatite minerals as identified in larger grains through mineral maps. While this material is present in all samples, the amount of the material is ≪1% in all cases. This leads us to conclude that authigenic Fe or Mn grain coatings are a minor component of the sediments studied here, although we cannot rule out the presence of oxyhydroxide nanoparticles (10 s of nm) intermingled with the clay size fraction.
Fe-Mn oxyhydroxides in marine sediments are a commonly targeted archive for reconstructing paleocirculation because they are assumed to precipitate directly from ambient seawater, preserving a record of seawater REE composition (Rutberg et al., 2000; Bayon et al., 2002, 2004; Frank, 2002; Roberts and Piotrowski, 2015). Although macroscale Fe-Mn crusts and nodules are known to be important REE host phases (e.g., Bau et al., 2014; Conrad et al., 2016), they have a limited spatial distribution in the modern ocean. However, it is widely thought that Fe-Mn oxyhydroxides are ubiquitously present as REE enriched, nanoscale authigenic grain coatings similar to the Fe oxyhydroxides coatings known from terrestrially derived detrital silt and sand size grains (Manceau et al., 2007) or the nano-scale particles and aggregates attached to clays in the suspended fraction of tropical rivers (Poulton and Raiswell, 2005). Crucially, the distribution, preservation, and formation of these coatings remains poorly understood (e.g., Bayon et al., 2004) with no direct, imaging-based identification of their systematic presence to date. This is an important gap because the partitioning of REEs into these phases as well as the susceptibility of these coatings to reductive dissolution means that, where present, they are likely important contributors to pore water REE (e.g., Haley et al., 2004).
Attempts to quantify the abundance of these coatings have mostly relied on indirect, operationally defined chemical extractions rather than direct crystallographic, microscopic, or microbeam identification. This is also true more broadly of our knowledge of sedimentary trace element and REE host phases, which is largely informed by operationally defined chemical extractions; procedures known to be hampered by incomplete dissolution of the target phase, redistribution of elemental species through dissolution and subsequent secondary mineral precipitation, and dissolution of non-target phases (e.g., Taylor and McKenzie, 1966; Schwertmann and Pfab, 1994; Koschinsky and Hein, 2003; Wilson et al., 2013; Homoky et al., 2016; Skinner et al., 2019). Although sediment leachate trace metal ratios are reasonably similar to those measured on macroscopic Fe-Mn crusts (e.g., Gutjahr et al., 2007; Du et al., 2016), the well-established presence of phyllosilicate and phosphate phases in many Fe-Mn crusts (e.g., Rao, 1987; Bau et al., 1996; Hein et al., 1997) and the large range in sediment reductive leachate trace element ratios (e.g., Du et al., 2016 and references therein) leaves significant uncertainty as to the identity of the REE host phases dissolved by commonly used operationally defined leaching procedures. Nd/Fe ratios reported for reductive sediment leachates, for example, range from 0.001 to 0.01 compared to a much tighter range of only 0.001 to 0.002 for Fe-Mn crusts and nodules, whereas Al/Nd ratios in reductive sediment leachates are reported to fall between 50 – 400 compared to only 40 – 250 for Fe-Mn nodules and crusts (Du et al., 2016 and references therein) and Nd recovery associated with high Fe high Al phases has been observed (e.g., Wilson et al., 2013).
In addition, the large proportion of REEs recoverable through the same operationally defined sediment leaching procedures discussed above suggest that the phase or phases driving REE cycling in early sediment diagenesis should be sufficiently abundant to be readily identified by XRD or microscopically. Specifically, mass balance calculations show that 55 to 70% of the bulk sediment REE content is recoverable through combined acetic acid and hydroxylamine hydrochloride sequential leaching, with up to 55% of those REEs recovered during the reductive hydroxylamine hydrochloride leach that is believed to target the oxyhydroxide phase (Abbott et al., 2016b). Based on an average concentration of 58 mg Nd kg–1 for authigenic Fe-Mn nodules (Bau et al., 2014), 5300 ng Nd cm–3 recovered through reductive leaching from NP3000 (Abbott et al., 2016a), and assuming a sediment density of 2.6 g cm–3, Fe-Mn phases would need to make up 3.5% of the bulk marine sediment to account for the Nd recovered in the reductive leaching procedure using:
where Sp is the calculated percent of sediment, R is the recovered Nd through reductive leaching (in mg Nd kg–1) and N is the measured concentration of Nd in the nodules (in mg Nd kg–1). This value ranges from 1.5 to 13% if we consider the entire spread of Fe-Mn nodule Nd concentrations reported by Bau et al. (2014), and the lower proportion of bulk sediment Nd concentrations recovered through reductive leaching at NP200 (Abbott et al., 2016a). Although this assessment of Sp is likely subject to a substantial margin of error due to the limited data available with which to constrain the proportion of leachable Nd relative to total Nd, in addition to differences in mineralogy between Fe-Mn nodules, crusts and authigenic Fe-Mn oxyhydroxide coatings, it nonetheless allows us to estimate the likely range of Fe-Mn oxyhydroxide abundance required to account for the Nd recovered by chemical leaching. While the lower limit of the Sp range we calculate is just sufficiently small that Fe-Mn oxyhydroxide phases may not be detected by powder X-ray diffraction (detection limit for Fe-Mn oxyhydroxide phases in siliciclastic matrix is approx. 2 wt%), careful microbeam mineralogical characterization of these samples also fails to identify a ubiquitous presence of authigenic Fe-Mn oxyhydroxides (Figures 4, 5) despite 5 to 9 wt% iron in the bulk sediment from each North Pacific site (Abbott et al., 2016a). This microbeam characterization revealed only trace quantities of 2–4 μm, detrital Fe oxyhydroxide grains (Figure 5). Instead, the bulk of the iron present at all sites appears to be hosted in the clay mineral fraction.
This is not to say that nanoscale Fe-Mn oxyhydroxides are not present. Recent work by Blaser et al. (2016), for example, repeatedly applied a modified “weak” reductive leach to sediments from over ten sites in the North Atlantic and consistently identified a rapid decline in Nd and Mn concentrations from the first leach to subsequent leaches. Leachate elemental ratios and Nd isotopic composition indicated only minimal contamination from non-target siliciclastic phases during the first leach step. Overall, this suggests that labile Nd-bearing Mn phases may be widespread but relatively low-abundance phase in marine sediments. Mössbauer spectroscopic analysis of a range of marine sediments further confirms the presence of widespread, low abundance reactive Fe oxyhydroxides in marine sediments, and identifies these to be dominantly nanoscale (<12 nm diameter) goethite (van der Zee et al., 2003). Interestingly, goethite is considered to be relatively resistant to reductive leaching (Poulton and Canfield, 2005). The imaging approach used here does not have the resolution to identify nanoscale Fe-Mn oxyhydroxide, identifying only aggregates >200 nm in size. It is therefore likely that the Fe-clay association we identify in our samples is partly due to nano-scale intermingling and coating of clays by Fe-Mn oxyhydroxides. However, the presence of iron-bearing clays such as chlorite and Fe-smectite (nontronite) and the failure to identify Fe-Mn oxyhydroxides by XRD suggests that the major portion of clay-associated Fe is hosted within the clays, rather than representing discrete nanoscale oxyhydroxide coatings. This interpretation is consistent with the findings of Homoky et al. (2011) that Fe-Mn bearing clays may be a thermodynamically favorable explanation to the presence of nano-scale iron and manganese phases in pore water. We therefore argue that while Fe-Mn oxyhydroxides maybe an ubiquitous trace component of marine sediments, they appear to be insufficiently abundant to represent the primary source of Nd in reductive leachates or pore fluids, consistent with recent observations that the REEs recovered by leaching procedures are not necessarily sourced from oxyhydroxide phases (e.g., Vance et al., 2004; Tachikawa et al., 2013; Osborne et al., 2017; Skinner et al., 2019).
The complex, but commonly observed relationship between Fe and Nd in marine pore waters and the range of observed Nd/Fe ratios (e.g., Haley et al., 2004; Gutjahr et al., 2007; Du et al., 2016; Abbott et al., 2016a) leads us to suggest that the REEs are remobilizing from iron containing phases other than Fe oxyhydroxides. This suggestion is consistent with recent research revealing the same sedimentary origin for REEs and iron in the Southern Ocean (Blain et al., 2008; Zhang et al., 2008; Grenier et al., 2018). Our EDS elemental mapping suggests the primary iron host in the sediments studied here to be the clay mineral fraction (Figure 5). Indicators more commonly interpreted as evidence for oxyhydroxide coating recovery during reductive leaches, including trace metal ratios within the leachate (e.g., Gutjahr et al., 2007; Du et al., 2016), REE patterns (e.g., Gutjahr et al., 2007), and the agreement between the recovered neodymium with the overlying seawater (e.g., Elderfield and Greaves, 1982; Palmer and Elderfield, 1985; Rutberg et al., 2000; Roberts and Piotrowski, 2015) are also consistent with a benthic control model of REEs (Haley et al., 2017) that could be driven by clay minerals as we propose here. Further supporting this hypothesis is the correlation between finer average grain sizes, higher pore water REE concentrations (e.g., Abbott et al., 2016a), and higher sediment REE concentrations (e.g., Sa et al., 2018); consistent with higher reactivities of fine-grained, high surface clays than of coarser sediment component. We propose that clay dissolution and authigenesis may provide the universal controls on the sedimentary release (e.g., reverse scavenging; Elderfield and Greaves, 1982; Jeandel et al., 1995) and uptake (e.g., authigenic clay formation; Michalopoulos and Aller, 1995; Rahman et al., 2017) of REEs, and may therefore be the primary driver of REE distribution in seawater, pore water, and marine authigenic phases.
Chemical transfer reactions among terrestrially derived particles and seawater include adsorption/desorption, ion exchange, and dissolution/precipitation reactions (e.g., Elderfield and Sholkovitz, 1987; Jeandel et al., 1995; Rousseau et al., 2015; Homoky et al., 2016). Desorption and ion exchange tend to be relatively rapid processes, driven by salinity increases and changes in fluid major ion composition occurring as soon as river-borne particles come into contact with higher salinity estuarine waters (e.g., Rousseau et al., 2015). The conservative nature of major seawater ions and uniform salinity of bottom water versus pore water therefore suggest that exchange reactions are unlikely to play a significant role in the transfer of REEs from clays to pore water at our study sites. Dissolution and precipitation reactions, on the other hand, are longer-term processes that continue as long as the minerals are out of equilibrium with the adjacent fluid. The rate of dissolution and precipitation will change through time as a function of the saturation of the surrounding pore waters, as seen in batch reactor experiments (Oelkers et al., 2011). While the rates and extent of dissolution are hard to quantify due to simultaneously occurring secondary precipitation reactions (Oelkers et al., 2011; Pearce et al., 2013; Rousseau et al., 2015; Homoky et al., 2016), these experiments and investigations of Nd isotope evolution along estuarine salinity gradients do show that dissolution progresses rapidly (days to weeks) and is of sufficient magnitude to impact on oceanic REE budgets. Closed-system dissolution experiments, for instance, show 0.4% of the Nd contained in natural basaltic particles is released after only one month, sufficient to alter the initial εNd composition of the seawater to that of the bulk basalt (Pearce et al., 2013). Modeling studies constrained by oceanic εNd distributions suggest up to 1–3% of the continentally derived sediments may dissolve into seawater (Lacan and Jeandel, 2005; Arsouze et al., 2009). This estimated dissolution is several orders of magnitude greater than the amount of dissolution required (<0.001%) to account for elevated (relative to seawater) pore water Nd concentrations at the North Pacific sites (Abbott et al., 2016a; we do not estimate this for Tasman Sea sites as bulk sediment REE data is currently unavailable).
A disproportional contribution to this 0.001 to 3% total sediment dissolution from clay minerals is likely because mineral dissolution rates are commonly a function of surface area (e.g., Schott et al., 2009, 2012). For comparison, the specific surface areas of common clay minerals [N2 sorption Brunauer–Emmett–Teller (BET) surface areas of illite and smectite are 76–91 m2g–1 and 32–97 m2g–1, respectively; Van Olphen and Fripiat, 1979] are up to two orders of magnitude greater than that of the ground mineral powders used in typical laboratory dissolution experiments (e.g., Chaïrat et al., 2007; Oelkers et al., 2008). In addition, ubiquitously distributed Fe3+ containing clays (including chlorite, illite, smectite, and mixed-layer clays) are susceptible to microbially mediated alteration via reductive dissolution (Kim et al., 2004; Vorhies and Gaines, 2009; Liu et al., 2012; Zhang et al., 2012; Ijiri et al., 2018). Given that microbially mediated clay mineral dissolution occurs because of the reduction of structural Fe3+ (Dong et al., 2009), it stands to reason that these same minerals would be targeted by the reductive leachates used to isolate Fe-Mn oxyhydroxide phases. Evidence for dissolution of silicate phases, including clays, by reductive hydroxylamine hydrochloride leaches can be found in a recent sequential leaching study of North Atlantic sediments, which documented substantial Al and Fe mobilization from the silicate fraction after repeated exposure to a “weak” reductive leach (Blaser et al., 2016). This could reconcile the operational defined leachate evidence for a significant Fe-Mn oxyhydroxide REE host phase in the absence of visible authigenic Fe-Mn oxyhydroxide coatings. Furthermore, clay mineral dissolution could at least partly explain the relationship between pore water dissolved iron and REEs in some sedimentary settings and a lack of a clear relationship in others (e.g., Haley et al., 2004; Abbott et al., 2015b), depending on the Fe content of the clays in question.
If we assume that a single process, or source, dominates the benthic REE cycle then the REE pattern and concentrations of the pore water should be a function of the source of REEs and any simultaneous sink, such that:
whereas PW is the pore water, S is the host phase of REE in the sediments acting as the source and P is the loss of REEs through adsorption or authigenic mineral precipitation. In our considerations of P, we ignore the diffusive flux out of the sediments as this flux is not considered a mechanism for fractionation and thus will not change the REE pattern or isotopic signature of the pore water. Using this basic framework, we can examine the range of REE patterns observed in pore waters and authigenic phases in order to test the hypothesis that clays are the primary drivers of benthic REE cycles.
We expect the pore water REE signature to be most similar to the source phase REE signature at depths that have the highest overall pore water REE concentrations, i.e., the depth at which P is most likely to approach 0 (“source” depth of Abbott et al., 2015b). We find that the most Nd enriched pore waters show REE signatures similar to the pattern of World River Average Clay (WRAC, Figure 6; Bayon et al., 2015), consistent with clay dissolution as the source of REEs. Pore waters with lower REE concentrations, by contrast, have more variable REE patterns that do not closely match the WRAC pattern, and are generally more HREE enriched than the higher concentration pore waters (Figure 7). This could reflect REE sourced from a host phase other than clays (i.e., a different S-term in framework above), but we argue that this is more likely caused by a modification of the pore water pattern by fractionation from REE sorption or incorporation in authigenic minerals (P term in framework above).
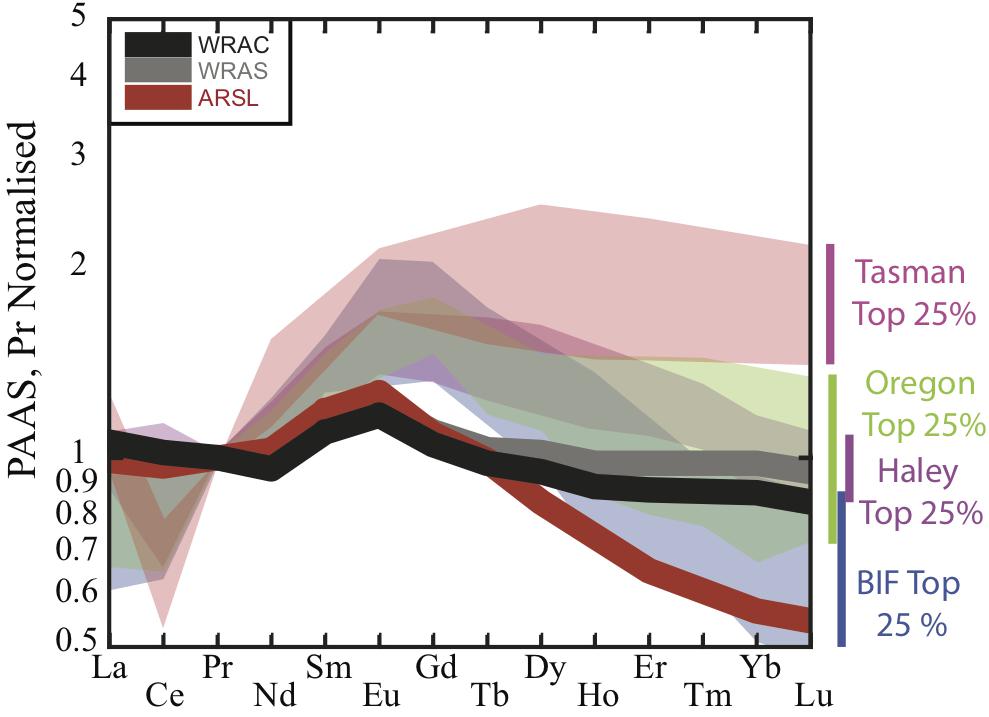
Figure 6. Range of shale-normalized REE patterns of pore water source depths from the Tasman Sea (this study) and available published pore water data (Haley et al., 2004; Abbott et al., 2015b). The most HREE enriched range is observed in the Tasman Sea, where REE concentrations were the lowest overall. Average river suspended load (ARSL, red), world river average clay (WRAC, gray), and world river average silt (WRAS, black) plotted for comparison (data from Bayon et al., 2015). Normalized to PAAS values of Taylor and McLennan (1985).
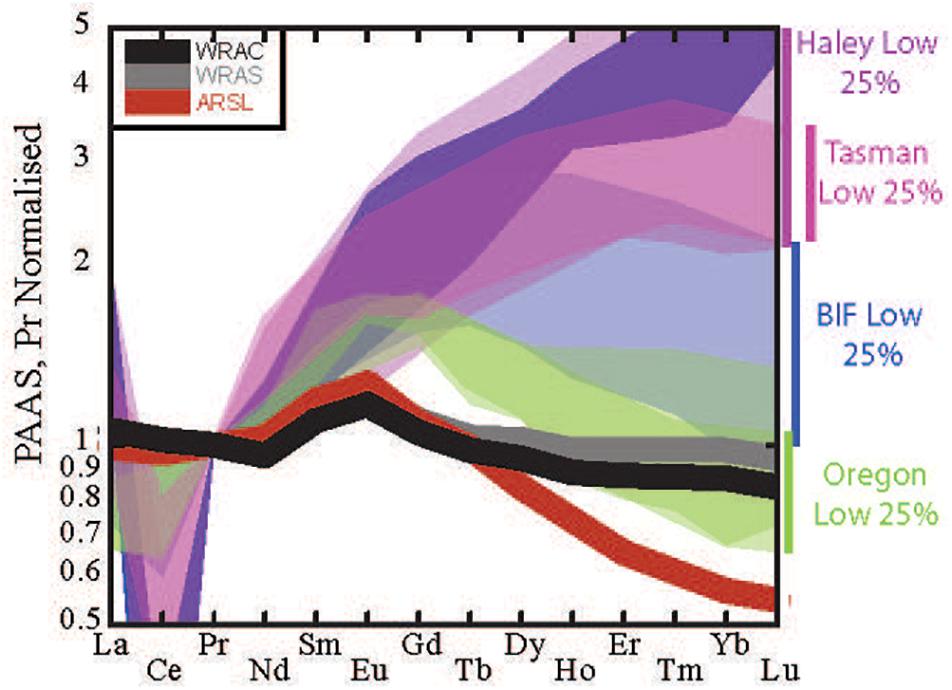
Figure 7. Range of shale-normalized REE patterns of pore water with the lowest REE concentrations from the Tasman Sea (this study) and available published pore water data (Haley et al., 2004; Abbott et al., 2015b) including the pore waters corresponding to the Oregon margin sediments analyzed for clay in this study. The most and least HREE enriched ranges were observed in North Pacific samples (Haley et al., 2004; Abbott et al., 2015b) highlighting the variability in these low concentration intervals. Average river suspended load (ARSL, red), world river average clay (WRAC, gray), and world river average silt (WRAS, black) plotted for comparison (data from Bayon et al., 2015). The range of REE patterns of bottom water from the two study regions is also provided (Abbott et al., 2015a; Abbott, 2019). Normalized to PAAS values of Taylor and McLennan (1985).
As noted in section “Mechanisms of REE Release From Clay Minerals,” experimental quantification of the rates and extent of dissolution of detrital sedimentary material is complicated by simultaneously occurring precipitation reactions producing a range of secondary minerals (e.g., Oelkers et al., 2011), mostly aluminosilicates. A wide range of secondary minerals including phyllosilicates, Fe-Mn oxyhydroxides and phosphates are known to form during early diagenesis, each of which are known to preferential scavenge LREEs, leading to HREE enrichment in the pore water (Cantrell and Byrne, 1987; Byrne and Kim, 1990; Byrne and Kim, 1993; Jeandel and Oelkers, 2015). Of these, authigenic clays are likely the most widespread and quantitatively significant authigenic phase. We explore the impact of secondary mineral precipitation on pore water REE patterns using a simple mass balance calculation, assuming that the starting REE pattern of pore water resembles WRAC (in agreement with section “Pore Water REE Patterns Support a Clay Dissolution Source”), and that the secondary phase responsible for REE uptake is an authigenic glauconite clay (Figure 8, see also section “Fractionation Feasibility Modeling”). We find that increased removal of REEs by glauconite produces an increasingly seawater-like pattern in the residual dissolved phase (Figure 9). Specifically, we observe an increase in HREE/LREE and a decrease in the MREE anomaly. Because experimental and modeling work suggests dissolution of detrital silicates releases up to 0.4 to 3% of the solid phase REEs, but pore water has several orders of magnitude less REEs per volume then the accompanying solid phase (<0.001%, Abbott et al., 2016a), we consider the high authigenic phase REE uptake (∼90%) required to produce a seawater-like HREE/LREE and MREE signal from a WRAC dissolution source to be reasonable. However, the resulting MREE anomaly is slightly lower than that observed in pore water, fish teeth, foraminifera, and Fe-Mn nodules. This discrepancy is likely because while WRAC is a useful starting point for our assessment of a clay source, as an average value WRAC masks the broad range of REE signature of clays sourced from different bedrocks (e.g., Bayon et al., 2015). The dissolution of clay size fraction during early diagenesis is unlikely to be congruous, with some clay phases dissolving more rapidly or more completely than others. For example, clays in suspended load of rivers draining volcanic (basaltic) terrains are known to have greater MREE anomalies than WRAC and this detrital volcanic material may preferentially influence pore water (e.g., Dessert et al., 2003; Vance et al., 2004; Wilson et al., 2013). Rivers draining basaltic terrains contribute heavily to clays throughout the Pacific Ocean, compatible with the MREE enrichment in pore water relative to WRAC even at source depths in the pore water profiles (Figure 9). The deep-sea clay fraction likely contains detrital clays sourced from a range of source areas, and disproportionate dissolution of a particular clay fraction, such as fine-grained, high surface smectite clay that is commonly of volcanic origin, may be what results in MREEs higher than WRAC. Equilibrium modeling identified Fe-bearing smectite clay as the most likely nano-colloidal phase present in pore water (Homoky et al., 2011) supporting the plausibility of this mechanism.
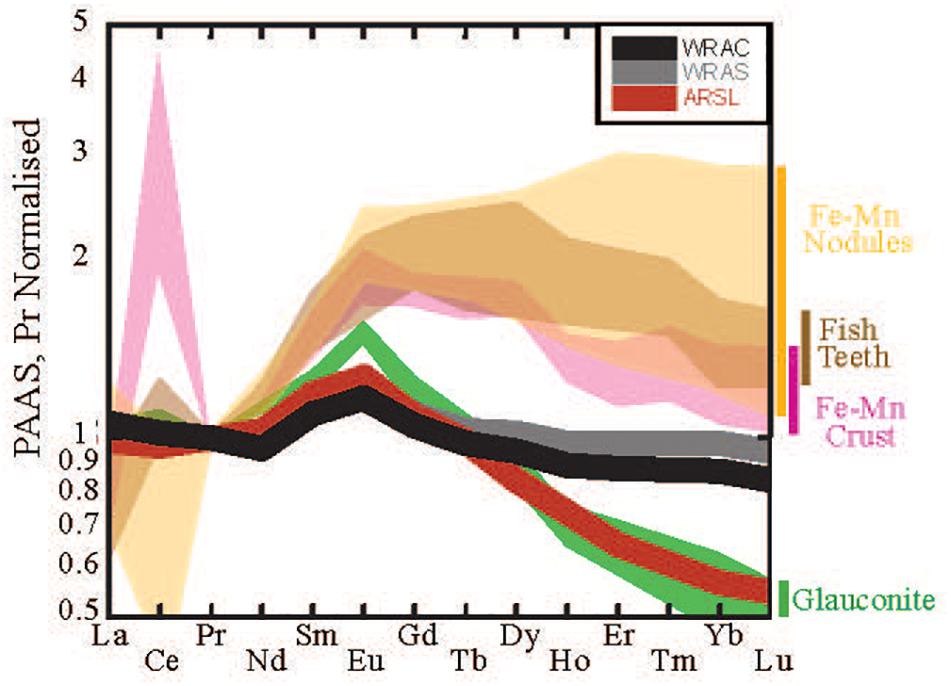
Figure 8. Range of shale-normalized REE patterns of potential authigenic host phases including glauconite (green, Huggett et al., 2017), Fe-Mn nodules (yellow, Bau et al., 2014), hydrogenetic Fe-Mn crusts, (pink, Azami et al., 2018), and fish teeth (brown, Huck et al., 2016). Average river suspended load (ARSL, red), world river average clay (WRAC, gray), and world river average silt (WRAS, black) plotted for comparison (data from Bayon et al., 2015). Normalized to PAAS values of Taylor and McLennan (1985).
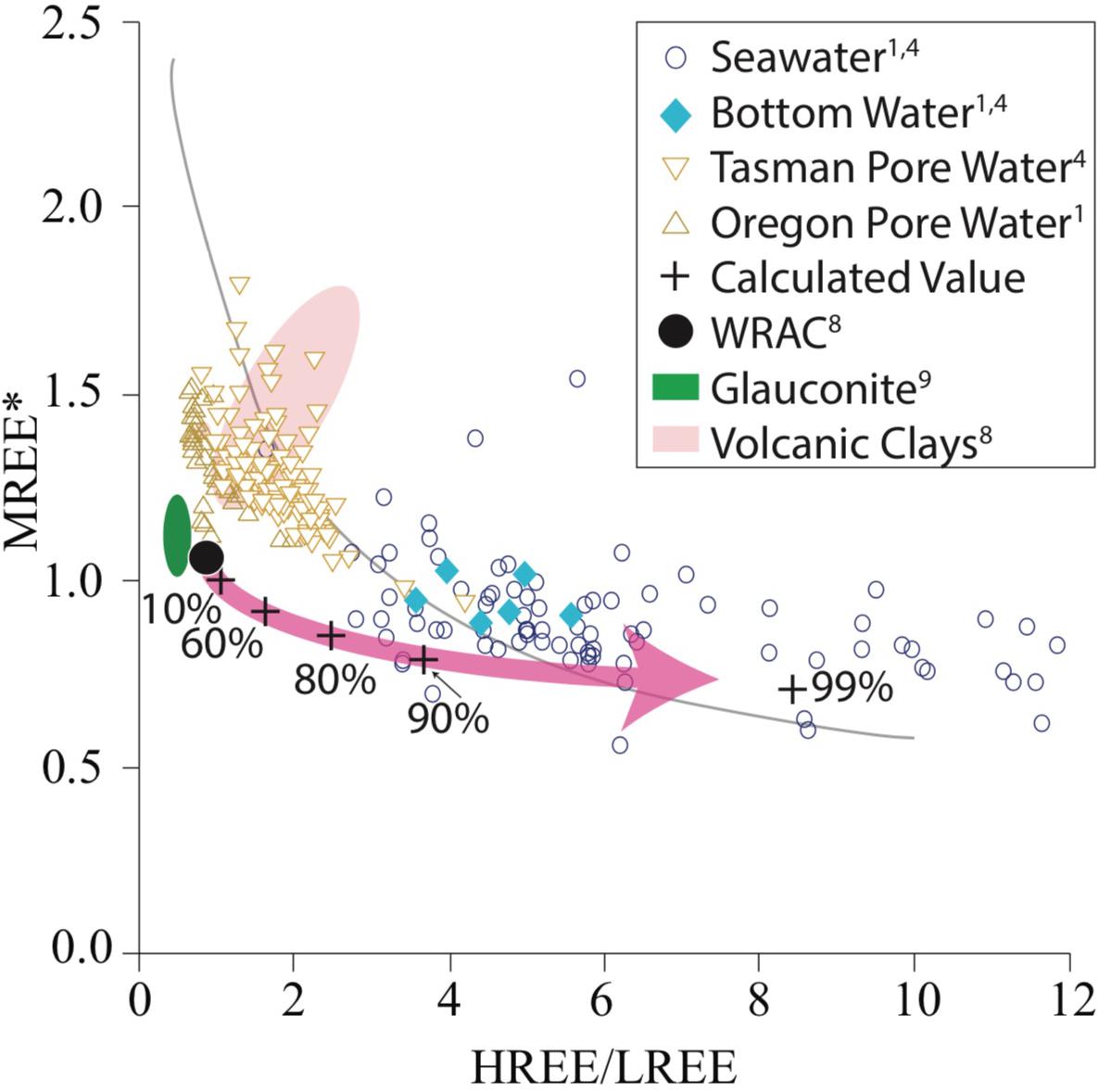
Figure 9. HREE/LREE versus MREE∗ of pore water and sedimentary phases including pore water (brown) and seawater (blue) from the six sites (Abbott et al., 2015a; Abbott, 2019), clay size fraction from volcanic rivers (red envelope, data from Bayon et al., 2015), and glauconites (green envelope, data from Huggett et al., 2017). World river average clay (WRAC, dark black dot, data from Bayon et al., 2015), and calculated resulting pore water patterns (+) from a glauconite sink as discussed in section “Authigenic Clay Formation May Modulate Pore Water REE Signature.”
Several other independent lines of evidence support our inference that the HREE enrichment of the lower concentration pore waters is largely due to fractionation from clay authigenesis rather than the formation of authigenic oxyhydroxide or phosphate phases. Firstly, while not conclusive, microbeam characterization of the samples reveals only trace quantities of Fe oxyhydroxide and calcium phosphates, neither of which are of authigenic origin suggesting that these phases are not abundant enough to drive the REE composition of pore water. Secondly, findings that authigenic clays are the primary sink for Li in the ocean (e.g., Vigier et al., 2008) support the importance of authigenic clay formation in marine elemental cycling. Pore water Li concentrations decrease with depth and with decreasing REE concentration at the same North Pacific sites (Abbott et al., 2015b). Together with the presence of Fe-smectite and glauconite pellets at NP1500, the pore water Li concentrations at sites with and without visible glauconite formation are evidence of authigenic clay formation (e.g., Stoffyn-Egli and Mackenzie, 1984). Furthermore, while we are unable to quantify authigenic clays at our sites, recent studies have proposed a significantly increased magnitude and spatial extent of authigenic clay formation/reverse weathering than previously believed (Michalopoulos and Aller, 1995; Rahman et al., 2017). Baldermann et al. (2015) for example, have shown that authigenic clays (glauconite) are a potentially more significant Fe sink in low-oxygen settings than authigenic Fe sulfides, whereas Rahman et al. (2017) use cosmogenic 32Si to estimate that authigenic clay formation is responsible for the burial of an estimated 4.5–4.9 × 1012 mol/yr Si, representing almost 50% of the total dissolved Si inputs to the ocean (Frings et al., 2016). Finally, our simple model demonstrates that clay authigenesis (reverse weathering) has the potential to push the pore water REE pattern away from the REE pattern of the primary host phase toward more seawater-like compositions.
Observations of the non-conservative behavior of neodymium isotopes in the ocean have largely been concentrated around the margins (e.g., Lacan and Jeandel, 2005; Arsouze et al., 2009; Grasse et al., 2012; Wilson et al., 2013). In line with these observations, several additional but geographically limited sources of Nd to the ocean have been proposed, including submarine groundwater discharge (e.g., Johannesson et al., 2011), hydrothermal vents (e.g., Douville et al., 1999), and even localized detrital inputs (e.g., Howe et al., 2016). The relatively strict geographic limits of these inputs and the non-conservative behavior of εNd associated with these has allowed proxy records collected from outside of these regions to be interpreted under the assumption of quasi-conservative behavior of εNd. However, the ubiquitous distribution of clays throughout the global ocean means that a benthic source driven by clay minerals would not be limited by proximity to land or as a function of water depth. This supports the proposal of Abbott et al. (2015b) that a widespread benthic source of REEs from the pore water could be the major source of REEs to the ocean, thus balancing the oceanic budget. While studies have noted the potential influence of reactive, REE-enriched sedimentary components with distinctive Nd isotopic composition (relative to the bulk sediment) on the oceanic REE budget (e.g., Tachikawa et al., 2004; Wilson et al., 2013; Abbott et al., 2015b; Du et al., 2016), particularly at continental margin sites (e.g., “Boundary Exchange,” Lacan and Jeandel, 2005; Jeandel et al., 2007; Rousseau et al., 2015; Jeandel, 2016), our results suggest the margins are not necessarily the regions with the largest flux. Indeed, the more distal sites with smaller grained sediments likely have larger fluxes (e.g., Holdren and Berner, 1979; Abbott et al., 2016a; Sa et al., 2018). A higher flux from distal sites could explain the isotopic agreement between modern core top leachates, pore water, and bottom water particularly if such a benthic flux is the dominant source of ocean Nd (e.g., Elderfield and Greaves, 1982; Abbott et al., 2015a; Haley et al., 2017). From this perspective, the absence of an anomalous isotopic signature in deep water regions is not indicative of a modest benthic flux, rather it is a predictable consequence of a long-term benthic flux from a major proportion of the seafloor. In other words, the absence of an anomalous signature in these regions is expected if sediment diagenesis is the dominant source of Nd to bottom waters (Elderfield and Greaves, 1982; Abbott et al., 2015b). On the other hand, regions near the margin are more prone to the additional influence of fresh reactive phases making the εNd of the flux in these areas more likely to be unique, and therefore noticeable.
Significant variation in εNd between suspended and dissolved riverine loads, dissolved riverine load and local bedrock, different sized sediment grains, as well as between organic matter and detrital material (VanLaningham et al., 2008; Viers et al., 2008; Freslon et al., 2014; Bayon et al., 2015; Hindshaw et al., 2018) all demonstrate the importance of host phase identification in balancing the oceanic REE budget and for the application of REEs as tools in biogeochemical studies. For instance, in some large river systems there is more than a 1 epsilon unit offset between silt and clay sized fraction, with the smaller clay sized particles being systematically more radiogenic (Bayon et al., 2015). Attributed to preferential weathering (Bayon et al., 2015), this suggests that some of the most labile components of the sediment flux to the ocean are not necessarily representative of the average composition of the corresponding drainage basin. Minor components of the bulk sediment are therefore able to shift pore water εNd away from the bulk sediment if these components are reactive and enriched in REEs, making for an εNd of the flux that is noticeably different to the bulk sediment and the expected bottom water value. This discrepancy is most likely to occur proximal to the source (e.g., continental margins), especially in regions with more recently eroded materials, consistent with existing observations of boundary exchange concentrated near the margins (e.g., Lacan and Jeandel, 2005; Arsouze et al., 2009; Jeandel, 2016). Generally, these considerations result in the actual source being more radiogenic than the bulk rock (e.g., Viers et al., 2008; Grenier et al., 2018; Hindshaw et al., 2018). This model could explain North Pacific pore water neodymium isotopes that were more radiogenic than the bulk sediment or leachable phases (Abbott et al., 2016a) and why near shore sites were more radiogenic off the Kerguelen Plateau (Grenier et al., 2018). If the glass shards at NP200 (Figure 5) are relatively easily dissolved after deposition, they may release a more radiogenic signature than that of the bulk sediments at that site.
The array of REE patterns associated with authigenic phases such as those measured in sediment leaches, fish teeth, and pore waters (Figure 9; the “authigenic array” of Du et al., 2016) can largely be explained as a balance between a WRAC REE source and the preferential uptake of LREEs by authigenic clays as demonstrated by the REE pattern of glauconite. While this combination of clay dissolution and clay authigenesis can explain the HREE/LREE variation observed in pore water and seawater, the low MREE anomaly of these HREE enriched waters may indicate the role of additional processes but the formation of a MREE enriched authigenic phase (Figures 8, 9; e.g., Kang et al., 2014; Bayon et al., 2016; Trotter et al., 2016) would further decrease, rather than increase the MREE anomaly in the fluid phases. However, the MREE anomaly may be a result of local variability in the clay source, preferential dissolution of more labile clay fractions (i.e., volcanic), or fractionation associated with clay authigenesis not captured by the glauconites considered above. WRAC only considered large rivers covering an estimated 30% of the area that drains into the ocean (Bayon et al., 2015) and does not allow for the locally dominant, chemically distinct contribution of smaller river systems as mentioned above. The potential importance of the local to regional variability in the clay supply to the ocean is illustrated by the range of REE patterns associated with clays coming from volcanic landscapes versus large rivers (Figure 8; Bayon et al., 2015). This sensitivity to clay supply also means that the magnitude and signature of the benthic source may vary as a function of continental weathering, ocean circulation, and sediment provenance influences on the overall sediment supply.
The association of iron and REEs with clay phases in the sediments emphasizes the need for caution in interpretations of the marine authigenic record. We suggest the leaching protocols typically employed to recover this authigenic record are not only targeting oxyhydroxide phases, but also attacking clay host phases. Conventional leaching techniques mobilize significant quantities of silicate hosted REEs particularly in carbonate poor sediments and can release up to 16% of sedimentary iron (e.g., Wilson et al., 2013; Blaser et al., 2016). Recent developments in leaching techniques using a gentler approach can minimize the “contamination” from a silicate fraction, but even these refined methods mobilize some of the silicate fraction in the absence of a carbonate buffer (Blaser et al., 2016, 2019). For instance, the recent sequential leaching experiments on a wide range of North Atlantic sediments that used a refined reductive leaching approach demonstrated progressively increased Al/Nd, accompanied by shifts in leachate εNd away from a foraminiferal/authigenic value toward a volcaniclastic/detrital silicate signature (Blaser et al., 2016). Several clay minerals contain structural Fe3+, which is prone to reductive dissolution especially in smectite (Vorhies and Gaines, 2009) and thus could be a source of the iron recovered during reductive leaching protocols. Even our simple model of the effect of glauconite formation on a WRAC-like REE pattern in the pore water demonstrated that uptake by glauconites can plausibly lead to the HREE enrichment commonly observed in pore waters, seawater, and other authigenic phases (Figure 9, e.g., fish teeth, Fe-Mn nodules). We argue that the similarity of the REE pattern of authigenic phases such as Fe-Mn nodules (e.g., Bau et al., 2014) and fish teeth (e.g., Huck et al., 2016) to lower concentration pore waters (Figures 8, 9) is likely due to continuing exchange (and homogenization) between these authigenic phases and the pore waters, and not an original Fe-Mn or phosphatic source of REEs to the pore water. This argument is consistent with diagenetic transfer of REE from clay minerals to biogenic phosphates in Ce anomaly mass balances (Grandjean et al., 1987) and would mean that the exchange between the sediments and pore water is continuing through early diagenesis. Thus, the authigenic phases in the sediments are going to record the balance of fluid-particle exchange in the pore water rather than a true bottom water signature, consistent with observed pore water-like REE patterns in Fe-Mn oxyhydroxide and biogenic phosphates (Figures 8, 9) with implications for our application of authigenic records to paleocirculation reconstructions.
The magnitude and character of the REE flux is likely to change in response to changing climate and the resulting changes in sediment supply. In this way, the apparent correlation between authigenic εNd records and major climatic events (e.g., Böhm et al., 2015; Howe et al., 2016; Abbott et al., 2016b; Deaney et al., 2017) makes sense. Ocean circulation is going to, at least in part, be responsible for sediment provenance at any given site with the climate and tectonic activity both exerting control over the sources of those sediments (e.g., continental weathering; VanLaningham et al., 2008; Viers et al., 2008); thus, we would still expect a correlation between the timing of εNd value excursions and intervals of climatic or oceanic change. This means that in a clay-driven system, the changes in REE and εNd in authigenic phases may reflect both changes in sediment provenance and changes in continental weathering regime. If widely applicable, clays as a source of pore water REEs may make clay type and distribution potentially useful factors for estimating the diagenetic overprinting of authigenic neodymium isotope records. The resulting authigenic signature in sediments would be a function of the REE content and reactivity of the solid phases present as well as the location, mineral association, and nature of the host phase as observed with strontium (McKinley et al., 2007). Furthermore, if reductive dissolution is an important component of overall sediment dissolution, then we may see a relationship between organic matter production and REE remobilization. If this is the case, a relationship between organic matter supply and the spatial and temporal variability of a benthic flux may exist which could link the benthic source of neodymium to the ocean and changes in continental weathering regimes through time.
This study combined new high-resolution SEM images, mineral maps, pore water REE data, and sedimentological measurements to identify the phases actively involved in REE cycling in the upper sediment column. We conclude that:
• Although the Fe-Mn oxyhydroxide phases commonly invoked as sedimentary REE host phases are likely present as nanoscale grains smaller than the resolution of the imaging techniques used here, our calculations suggest that they are not present at sufficiently high abundances to account for the REEs recovered using reductive leaching protocols.
• The REEs are likely associated with iron-bearing phases, but we argue that clay minerals are likely more significant REE host phases than Fe-Mn oxyhydroxides. The REE patterns associated with the pore water source depth are consistent with clay dissolution as the primary source of REEs.
• The fractionation associated with REE uptake during clay authigenesis provides a mechanism to explain the HREE/LREE spread of the authigenic array. An additional process or MREE depleted authigenic phase may be needed to completely explain the observed MREE anomalies depending on the variability of clay REE patterns not captured by WRAC and the glauconites included in this model.
• A clay driven benthic REE model also suggests that the sedimentary REE and εNd record will be subject to changes in sediment provenance and continental weathering, but potentially also sensitive to processes such as surface water export production which impact on pore-water pH and redox state and must be interpreted with caution.
A benthic control model of REE cycling that is driven by clay dissolution and clay authigenesis during early diagenesis would be widely applicable throughout the global ocean as a result of the ubiquitous distribution of clay minerals. This suggests sediments from the deep sea play an important role in the oceanic REE budget by supplying a significant benthic flux of REEs that has gone largely undetected due to the similarity between the isotopic value of the flux and the bottom water.
AA collected the samples on board R/V Oceanus and R/V Investigator including pore water separations and filtration, and completed the REE analyses on the SeaFAST II. SL and MT prepared the sediment samples and completed the XRD analyses and grain size measurements. SL and AA prepared the samples for SEM imaging, supervised MT during her undergraduate research experience, drafted the manuscript, and prepared the figures. SL performed the mineral classifications. All authors contributed to the data representation and interpretation.
All analyses were made possible by internal funding from Macquarie University to AA and SL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the Captain and Crew of the R/V Investigator and the R/V Oceanus as well as Mark Lewis, Jason Fazey, and Rebecca D’arcy for making sample collection possible; Benat Oliveira Bravo for help with data analyses; and Martin Kennedy, Damian Gore, Peter Wieland, Bruce Schaefer, Brian Haley, Chris Russo, and Russell Field for laboratory access and support. We also sincerely thank our reviewers and editor for improvements on previous versions of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2019.00504/full#supplementary-material
Abbott, A. N. (2019). A benthic flux from calcareous sediments results in non-conservative neodymium behavior during lateral transport: a study from the Tasman Sea. Geology 47, 363–366. doi: 10.1130/G45904.1
Abbott, A. N., Haley, B., and McManus, J. (2015a). Bottoms up: sedimentary control of the deep North Pacific Ocean’s εNd signature. Geology 43, 1035–1038. doi: 10.1130/G37114.1
Abbott, A. N., Haley, B., McManus, J., and Reimers, C. (2015b). The sedimentary source of dissolved rare earth elements to the ocean. Geochim. Cosmochim. Acta 154, 186–200. doi: 10.1016/j.gca.2015.01.010
Abbott, A. N., Haley, B., and McManus, J. (2016a). The impact of sedimentary coatings on the diagenetic Nd flux. Earth and Planet. Sci. Lett. 144, 217–227. doi: 10.1016/j.epsl.2016.06.001
Abbott, A. N., Haley, B. A., Tripati, A. K., and Frank, M. (2016b). Constraints on ocean circulation at the paleocene-eocene thermal maximum from neodymium isotopes. Clim. Past 12, 837–847. doi: 10.5194/cp-12-837-2016
Akagi, T. (2013). Rare earth element (REE) silicic acid complexes in seawater to explain the incorporation of REEs in opal and the “leftover” REEs in surface water: new interpretation of dissolved REE distribution profiles. Geochim. Cosmochim. Acta 113, 174–192. doi: 10.1016/j.gca.2013.03.014
Akagi, T., Yasuda, S., Asahara, Y., Emoto, M., and Takahashi, K. (2014). Diatoms spread a high εNd-signature in the North Pacific Ocean. Geochem. J. 48, 121–131. doi: 10.2343/geochemj.2.0292
Alibo, D. S., and Nozaki, Y. (2004). Dissolved rare earth elements in the eastern Indian Ocean: chemical tracers of the water masses. Deep Sea Res. 51, 559–576. doi: 10.1016/j.dsr.2003.11.004
Amakawa, H., Sasaki, K., and Ebihara, M. (2009). Nd isotopic composition in the central North Pacific. Geochim. Cosmochim. Acta 73, 4705–4719. doi: 10.1016/j.gca.2009.05.058
Arsouze, T., Dutay, J.-C., Lacan, F., and Jeandel, C. (2009). Reconstructing the Nd oceanic cycle using a coupled dynamical biogeochemical model. Biogeosciences 6, 2829–2846. doi: 10.5194/bg-6-2829-2009
Azami, K., Hirano, N., Machida, S., Yasukawa, K., and Kato, Y. (2018). Rare earth elements and yttrium (REY) variability with water depth in hydrogenetic ferromanganese crusts. Chem. Geol. 493, 224–233. doi: 10.1016/j.chemgeo.2018.05.045
Baldermann, A., Warr, L. N., Grathoff, G. H., and Dietzel, M. (2013). The Rate and mechanism of deep-sea glauconite formation at the ivory coast – ghana marginal ridge. Clays Clay Miner. 61, 258–276. doi: 10.1346/CCMN.2013.0610307
Baldermann, A., Warr, L. N., Letofsky-Papst, I., and Mavromatis, V. (2015). Substantial iron sequestration during green-clay authigenesis in modern deep-sea sediments. Nat. Geosci. 6, 885–889. doi: 10.1038/ngeo2542
Bau, M., Koschinsky, A., Dulski, P., and Hein, J. R. (1996). Comparison of the partitioning behaviors of yttrium, rare earth elements, and titanium between hydrogenetic marine ferromanganese crusts and seawater. Geochim. Cosmochim. Acta 60, 1709–1725. doi: 10.1016/0016-7037(96)00063-4
Bau, M., Schmidt, K., Koschinsky, A., Hein, J., Kuhn, T., and Usui, A. (2014). Discriminating between different genetic types of marine ferro-manganese crusts and nodules based on rare earth elements and yttrium. Chem. Geol. 381, 1–9. doi: 10.1016/j.chemigeo.2014.05.004
Bayon, G., German, C. R., Boella, R. M., Milton, J. A., Taylor, R. N., and Nesbitt, R. W. (2002). An improved method for extracting marine sediment fractions and its application to Sr and Nd isotopic analysis. Chem. Geol. 187, 179–199. doi: 10.1016/s0009-2541(01)00416-8
Bayon, G., German, C. R., Burton, K. W., Nesbitt, R. W., and Rogers, N. (2004). Sedimentary Fe-Mn oxyhydroxides as paleoceanographic archives and the role of Aeolian flux in regulating oceanic dissolved REE. Earth Planet. Sci. Lett. 224, 477–492. doi: 10.1016/j.epsl.2004.05.033
Bayon, G., Skonieczny, C., Delvigne, C., Toucanne, S., Bermell, S., Ponzevera, E., et al. (2016). Environmental Hf-Nd isotopic decoupling in World river clays. Earth Planet. Sci. Lett. 438, 25–36. doi: 10.1016/j.epsl.2016.01.010
Bayon, G., Toucanne, S., Skonieczny, C., André, L., Bermell, S., Cheron, S., et al. (2015). Rare earth elements and neodymium isotopes in world river sediments revisted. Geochim. Cosmochim. Acta 170, 17–38. doi: 10.1016/j.gca.2015.08.001
Bertram, C. J., and Elderfield, H. (1993). The geochemical balance of the rare earth elements and neodymium isotopes in the ocean. Geochim. Cosmochim. Acta 57, 1957–1986. doi: 10.1016/0016-7037(93)90087-d
Blain, S., Sarthou, G., and Laan, P. (2008). Distribution of dissolved iron during the natural iron-fertilization experiment KEOPS (Kerguelen Plateau, Southern Ocean). Deep Sea Res. II 55, 594–605. doi: 10.1016/j.dsr2.2007.12.028
Blaser, P., Lippold, J., Gutjahr, M., Frank, N., Link, J. M., and Frank, M. (2016). Extracting foraminiferal seawater Nd isotope signatures from bulk deep sea sediment by chemical leaching. Chem. Geol. 439, 189–204. doi: 10.1016/j.chemgeo.2016.06.024
Blaser, P., Pöppelmeier, F., Schulz, H., Gutjahr, M., Frank, M., Lippold, J., et al. (2019). The resilience and sensitivity of Northeast Atlantic deep water εNd to overprinting by detrital fluxes over the past 30,000 years. Geochim. Cosmochim. Acta 245, 79–97. doi: 10.1016/j.gca.2018.10.018
Böhm, E., Lippolf, J., Gutjahr, M., Frank, M., Blaser, P., Antz, B., et al. (2015). Strong and deep Atlantic meridional overturning circulation during the last glacial cycle. Nature 517, 73–79. doi: 10.10138/nature14059
Byrne, R. H., and Kim, K.-H. (1990). Rare earth element scavenging in seawater. Geochim. Cosmochim. Acta 54, 2645–2656. doi: 10.1016/j.chemosphere.2014.05.049
Byrne, R. H., and Kim, K. H. (1993). Rare earth precipitation and coprecipitation behavior: the limiting role of PO43- on dissolved rare earth concentrations in seawater. Geochim. Cosmochim. Acta 57, 519–526. doi: 10.1016/0016-7037(93)90364-3
Cantrell, K. J., and Byrne, R. H. (1987). Rare-Earth element complexation by carbonate and oxalate ions. Geochim. Cosmochim. Acta 54, 2645–2656.
Carter, P., Vance, D., Hillenbrand, C. D., Smith, J. A., and Shoosmith, D. R. (2012). The neodymium isotopic composition of water masses in the eastern Pacific sector of the Southern Ocean. Geochim. Cosmochim. Acta 79, 41–59. doi: 10.1016/j.gca.2011.11.034
Chaïrat, C., Schott, J., Oelkers, E. H., Lartigue, J.-E., and Harouiya, N. (2007). Kinetics and mechanism of natural fluorapatite dissolution at 25°C and pH from 3 to 12. Geochim. Cosmochim. Acta 71, 5901–5912. doi: 10.1016/j.gca.2007.08.031
Conrad, T., Hein, J. R., Paytan, A., and Clague, D. A. (2016). Formation of Fe-Mn crusts within a continental margin environment. Ore Geol. Rev. 87, 25–40. doi: 10.1016/j.oregorev.2016.09.010
Cullers, R. L., Chaudhuri, S., Arnold, B., Lee, M., and Wolf, C. W. Jr. (1975). Rare earth distributions in clay minerals and in the clay-sized fraction of the Lower Permian Havensville and Eskridge shales of Kansas and Oklahoma. Geochim. Cosmochim. Acta 39, 1691–1703. doi: 10.1016/0016-7037(75)90090-3
Cullity, B. D., and Stock, S. R. (2001). Elements of X-ray Diffraction, 3rd Edn. Boston, MA: Addison-Wesley Publishing Co.
de Baar, H. J. W., Bacon, M. P., Brewer, P. G., and Bruland, K. W. (1985). Rare earth elements in the Pacific and Atlantic Ocean. Geochim. Cosmochim. Acta 49, 1943–1959.
Deaney, E. L., Barker, S., and van de Flierdt, T. (2017). Timing and nature of AMOC recovery across Termination 2 and magnitude of deglacial CO2 change. Nat. Commun. 8:14595. doi: 10.1038/ncomms14595
Deng, Y., Ren, J., Guo, Q., Cao, J., Wang, H., and Liu, C. (2017). Rare earth element geochemistry characteristics of seawater and porewater from deep sea in western Pacific. Sci. Rep. 7:16539. doi: 10.1038/s41598-017-16379-1
Dessert, C., Dupre, B., Gaillardet, J., Louis, M., François, L. M., and Allegre, C. J. (2003). Basalt weathering laws and the impact of basalt weathering on the global carbon cycle. Chem. Geol. 202, 257–273. doi: 10.1016/j.chemgeo.2002.10.001
Dong, H., Jaisi, D. P., Kim, J., and Zhang, G. (2009). Microbe-clay mineral interactions. Am. Mineral. 94, 1505–1519. doi: 10.2138/am.2009.3246
Douville, E., Bienvenu, P., Charlou, J. L., Donval, J. P., Fouquet, Y., and Appriou, P. (1999). Yttrium and rare earth elements in fluids from various deep-sea hydrothermal systems. Geochim. Cosmochim. Acta 63, 627–643. doi: 10.1016/s0016-7037(99)00024-1
Du, J., Haley, B. A., and Mix, A. C. (2016). Neodymium isotopes in authigenic phases, bottom waters and detrital sediments in the Gulf of Alaska and their implications for paleo-circulation reconstruction. Geochim. Cosmochim. Acta 193, 14–35. doi: 10.1016/j.gca.2016.08.005
Duncan, T., and Shaw, T. J. (2003). The Mobility of rare earth elements and redox sensitive elements in the groundwater/seawater mixing zone of a shallow coastal aquifer. Aquat. Geochem. 9, 233–255. doi: 10.1023/b:aqua.0000022956.20338.26
Elderfield, H., and Greaves, M. J. (1982). The rare earth elements in seawater. Nature 296, 214–219. doi: 10.1038/296214a0
Elderfield, H., and Sholkovitz, E. R. (1987). Rare earth elements in the pore waters of reducing nearshore sediments. Earth Planet. Sci. Lett. 82, 280–288. doi: 10.1016/0012-821x(87)90202-0
Elmore, A. C., Piotrowski, A. M., Wright, J. D., and Scrivner, A. E. (2011). Testing the extraction of past seawater Nd isotopic composition from North Atlantic deep sea sediments and foraminifera. Geochem. Geophys. Geosyst. 12:Q09008. doi: 10.1029/2011gc003741
Frank, M. (2002). Radiogenic isotopes: tracers of past ocean circulation and erosional inputs. Rev.Geophys. 40, 1–1–1–38. doi: 10.1029/2000RG000094
Freslon, N., Bayon, G., Toucanne, S., Bermell, S., Bollinger, C., Chéron, S., et al. (2014). Rare earth elements and neodymium isotopes in sedimentary organic matter. Geochim. Cosmochim. Acta 140, 177–198. doi: 10.1016/j.gca.2014.05.016
Frings, P. J., Clymans, W., Fontorbe, G., La Rocha, De, C. L., and Conley, D. J. (2016). The continental Si cycle and its impact on the ocean Si isotope budget. Chem. Geol. 25, 12–36. doi: 10.1016/j.chemgeo.2016.01.020
Goldberg, E. D., Koide, M., Schmitt, R. A., and Smith, R. H. (1963). Rare earth distributions in the marine environment. J. Geophys. Res. 68, 4209–4217. doi: 10.1029/jz068i014p04209
Goldstein, S. L., and Hemming, S. R. (2003). “Long lived isotopic tracers in oceanography, paleoceanography, and ice sheet dynamics,” in Treatise on Geochemistry, ed. H. Elderfield (Amsterdam: Elsevier Pergamon Press).
Grandjean, P., Cappetta, H., Michard, A., and Albarede, F. (1987). The assessment of REE patterns and 143Nd/144Nd ratios in fish remains. Earth Planet. Sci. Lett. 84, 181–196. doi: 10.1016/0012-821x(87)90084-7
Grasse, P., Stichel, T., Stumpf, R., Stramma, L., and Frank, M. (2012). The distribution of neodymium isotopes and concentrations in the Eastern Equatorial Pacific: water mass advection versus particle exchange. Earth Planet. Sci. Lett. 35, 198–207. doi: 10.1016/j.epsl.2012.07.044
Greaves, M. J., Statham, P. J., and Elderfield, H. (1994). Rare earth element mobilization from marine atmospheric dust into seawater. Mar. Chem. 46, 255–260. doi: 10.1016/0304-4203(94)90081-7
Grenier, M., Garcia-Solsona, E., Lemaitre, N., Trull, T. W., Bouvier, V., Nonnotte, P., et al. (2018). Differentiating lithogenic supplies, water mass transport, and biological processes on and off the Kerguelen Plateau using rare earth element concentrations and neodymium isotopic compositions. Front. Mar. Sci. 5:426. doi: 10.3389/fmars.2018.00426
Grenier, M., Jeandel, C., Lacan, F., Vance, D., Venchiarutti, C., Cros, A., et al. (2013). From the subtropics to the central equatorial Pacific Ocean: neodymium isotopic composition and rare earth element concentration variations. J. Geophys. Res. Oceans 118, 592–618. doi: 10.1029/2012JC008239
Gutjahr, M., Frank, M., Stirling, C. H., Klemm, V., van de Flierdt, T., and Halliday, A. N. (2007). Reliable extraction of a deepwater trace metal isotope signal from Fe-Mn oxyhydroxide coatings of marine sediments. Chem. Geol. 242, 351–370. doi: 10.1016/j.chemgeo.2007.03.021
Haberlah, D., Löhr, S., Kennedy, M. J., Debenham, N., and Lattanzi, D. (2015). “Innovative sub-micron SEM-EDS mineral mapping and analysis applied to australian shale samples,” in Proceedings of the AAPG International Conference and Exhibition, Melbourne, Australia 13-16 September 2015: SEG Global Meeting Abstracts, (Tulsa, OK: Society of Exploration Geophysicists), 419. doi: 10.1190/ice2015-2211146
Haley, B. A., Du, J., Abbott, A. N., and McManus, J. (2017). The impact of benthic processes on rare earth element and neodymium isotope distributions in the oceans. Front. Mar. Sci. 4:426. doi: 10.3389/fmar.2017.00426
Haley, B. A., Frank, M., Spielhagen, R. F., and Eisenhauer, A. (2008). Influence of brine formation on Arctic Ocean circulation over the past 15 million years. Nat. Geosci. 1, 68–72. doi: 10.1038/ngeo.2007.5
Haley, B. A., Klinkhammer, G. P., and McManus, J. (2004). Rare earth elements in pore waters of marine sediments. Geochim. Cosmochim. Acta 68, 1265–1279. doi: 10.1016/j.gca.2003.09.012
Hathorne, E. C., Stichel, T., Brück, B., and Frank, M. (2015). Rare earth element distribution in the Atlantic sector of the Southern Ocean: the balance between particle scavenging and vertical supply. Mar. Chem. 177, 157–171. doi: 10.1016/j.marchem.2015.03.011
Hein, J. R., Koschinsky, A., Halbach, P., Manheim, F. T., Bau, M., Kanj, J.-K., et al. (1997). Iron and manages oxide mineralization in the Pacific. Geol. Soc. Lond. 119, 123–138. doi: 10.1144/GSL.Sp.1997.119.01.09
Hindshaw, R. S., Aciego, S. M., Piotrowski, A. M., and Tipper, E. T. (2018). Decoupling of dissolved and bedrock neodymium isotopes during sedimentary cycling. Geophys. Perspect. Lett. 8, 43–46. doi: 10.7185/geochemlet.1828
Holdren, G. R., and Berner, R. A. (1979). Mechanism of feldspar weathering – I. Experimental studies. Geochim. Cosmochim. Acta 43, 1161–1171. doi: 10.1016/0016-7037(79)90109-1
Homoky, W. B., Hembury, D., Hepburn, L. E., Mills, R. A., Statham, P. J., Fones, G. R., et al. (2011). Iron and manganese diagenesis in deep sea volcanogenic sediments and the origins of pore water colloids. Geochim. Cosmochim. Acta 75, 5032–5048. doi: 10.1016/j.gca.2011.06.019
Homoky, W. B., Weber, T., Berelson, W. M., Conway, T. M., Henderson, G. M., van Hulten, M., et al. (2016). Quantifying trace element and isotope fluxes at the ocean-sediment boundary: a review. Philos. Trans. A Math. Phys. Eng. Sci. 374:20160246. doi: 10.1098/rsta.2016.0246
Howe, J. N. W., Piotrowski, A. M., and Rennie, V. C. F. (2016). Abyssal origin for the early Holocene pulse of unradiogenic neodymium isotopes in Atlantic seawater. Geology 44, 831–834. doi: 10.1130/G38155.1
Huck, C. E., van de Flierdt, T., Jiménez-Espejo, F. J., Bohaty, S. M., Röhl, U., and Hammond, S. J. (2016). Robustness of fossil fish teeth for seawater neodymium isotope reconstructions under variable redox conditions in an ancient shallow marine setting. Geochem. Geophys. Geosyst. 17, 679–698. doi: 10.1002/2015GC006218
Huggett, J., Adetunji, J., Longstaffe, F., and Wray, D. (2017). Mineralogical and geochemical characterisation of warm-water, shallow-marine glaucony from the Tertiary of the London Basin. Clay Miner. 52, 25–50. doi: 10.1180/claymin.2017.052.1.02
Ijiri, A., Tomioka, N., Wakaki, S., Masuda, H., Shozugawa, K., Kim, S., et al. (2018). Low-temperature clay mineral dehydration contributes to porewater dilution in bering sea slope subseafloor. Front. Earth Sci. 6:36. doi: 10.3389/feart.2018.00036
Jeandel, C., Arsouze, T., Lacan, F., Téchiné, P., and Dutay, J.-C. (2007). Isotopic Nd compositions and concentrations of the lithogenic inputs into the ocean: a compilation, with an emphasis on the margins. Chem. Geol. 239, 156–164. doi: 10.1016/j.chemgeo.2006.11.013
Jeandel, C. (2016). Overview of the mechanisms that could explain the ‘Boundary Exchange’ at the land-ocean contact. Philos. Trans. R. Soc. 374:20150287. doi: 10.1098/rsta.2015.0287
Jeandel, C., Bishop, J. K., and Zindler, A. (1995). Exchange of neodymium and its isotopes between seawater and small and large particles in the Sargasso Sea. Geochim. Cosmochim. Acta 59, 535–547. doi: 10.1016/0016-7037(94)00367-u
Jeandel, C., Delattre, H., Grenier, M., Pradoux, C., and Lacan, F. (2013). Rare earth element concentrations and Nd isotopes in the Southeast Pacific Ocean. Geochem. Geophys. Geosyst. 14, 328–341. doi: 10.1029/2012GC004309
Jeandel, C., and Oelkers, E. H. (2015). The influence of terrigenous particulate material dissolution on ocean chemistry and global element cycles. Chem. Geol. 395, 50–66. doi: 10.1016/j.chemgeo.2014.12.001
Johannesson, K. H., Chevis, D. A., Burdige, D. J., Cable, J. E., Martin, J. B., and Roy, M. (2011). Submarine groundwater discharge is an important net source of light and middle REEs to coastal waters of the Indian River Lagoon, Florida, USA. Geochim. Cosmochim. Acta 75, 825–843. doi: 10.1016/j.gca.2010.11.005
Jones, K. M., Khatiwala, S. P., Goldstein, S. L., Hemming, S. R., and van de Flierdt, T. (2008). Modelling the distribution of Nd isotopes in the oceans using an ocean general circulation model. Earth Planet. Sci. Lett. 272, 610–619. doi: 10.1016/j.epsl.2008.05.027
Kang, J., Jeong, K. S., Cho, J. H., Lee, J. H., Jang, S., and Kim, S. R. (2014). Post-depositional redistribution processes and their effects on middle rare earth element precipitation and the cerium anomaly in sediments in the South Korea Plateau, East Sea. J. Asian Earth Sci. 82, 66–79. doi: 10.1016/j.jseaes.2013.11.019
Kim, J., Dong, H., Seabaugh, J., Newell, S. W., and Eberl, D. D. (2004). Role of microbes in the smectite-to-illite reaction. Science 303, 830–832. doi: 10.1126/science.1093245
Kim, J., Torres, M. E., Haley, B. A., Kastner, M., Pohlman, J. W., Riedel, M., et al. (2012). The effect of diagenesis and fluid migration on rare earth element distribution in pore fluids of the northern Cascadia accretionary margin. Chem. Geol. 291, 152–165. doi: 10.1016/j.chemgeo.2011.10.010
Kon, Y., Hoshino, M., Sanematsu, K., Morita, S., Tsunematsu, M., Okamoto, N., et al. (2014). Geochemical characteristics of apatite in heavy REE-rich deep-sea mud from the Minami-Torishima area, southeastern Japan. Resour. Geol. 64, 47–57. doi: 10.1111/rge.12026
Koschinsky, A., and Hein, J. R. (2003). Uptake of elements from seawater by ferromanganese crusts: solid-phase associations and seawater speciation. Mar. Geol. 198, 331–351. doi: 10.1016/S0025-3227(03)00122-1
Lacan, F., and Jeandel, C. (2005). Neodymium isotopes as a new tool for quantifying exchange fluxes at the continent-ocean interface. Earth Planet. Lett. 232, 245–257. doi: 10.1016/j.epsl.2005.01.004
Liu, D., Dong, H., Bishop, M. E., Zhang, J., Wang, H., Xie, S., et al. (2012). Microbial reduction of structural iron in interstratified illite-smectite minerals by a sulfate-reducing bacterium. Geobiology 10, 150–162. doi: 10.1111/j.1472-4669.2011.00307.x
Manceau, A., Lanson, M., and Geoffroy, N. (2007). Natural speciation of Ni, Zn, Ba, and Az in ferromanganese coatings on quartz using X-ray fluorescence, absorption, and diffraction. Geochim. Cosmochim. Acta 71, 95–128. doi: 10.1016/j.gca.2006.08.036
McKinley, J. P., Zachara, J. M., Smith, S. C., and Liu, C. (2007). Cation exchange reactions controlling desorption of 90Sr2+ from coarse-grained contaminated sediments at the Hanford site, Washington. Geochim. Cosmochim. Acta 71, 305–325. doi: 10.1016/j.gca.2006.09.027
Michalopoulos, P., and Aller, R. C. (1995). Rapid clay mineral formation in amazon delta sediments: reverse weathering and oceanic elemental cycles. Science 270, 614–617. doi: 10.1126/science.270.5236.614
Moore, D. M., and Reynolds, R. C. (1997). X-Ray Diffraction and the Identification and Analysis of Clay Minerals. Oxford: Oxford University Press, 378.
Nozaki, Y., and Alibo, D. S. (2003). Importance of vertical geochemical processes in controlling the oceanic profiles of dissolved rare earth elements in the northeastern Indian Ocean. Earth Planet. Sci. Lett. 205, 155–172. doi: 10.1016/S0012-821X(02)01027-0
Oelkers, E. H., Gislason, S. R., Eiriksdottir, E. S., Jones, M., Pearce, C. R., and Jeandel, C. (2011). The role of riverine particulate material on the global cycles of the elements. Appl. Geochem. 26, S365–S369. doi: 10.1016/j.apgeochem.2011.03.062
Oelkers, E. H., Schott, J., Gauthier, J.-M., and Herrero-Roncal, T. (2008). An experimental study of the dissolution mechanism and rates of muscovite. Geochim. Cosmochim. Acta 72, 4948–4961. doi: 10.1016/j.gca.2008.01.040
Osborne, A. H., Hathorne, E. C., Schijf, J., Plancherel, Y., Böning, P., and Frank, M. (2017). The potential of sedimentary foraminiferal rare earth element patterns to trace water masses in the past. Geochem. Geophys. Geosyst. 18, 1550–1568. doi: 10.1002/2016GC006782
Palmer, M. R., and Elderfield, H. (1985). Variations in the Nd isotopic composition of foraminifera from Atlantic Ocean sediments. Earth Planet. Sci. Lett. 73, 299–305. doi: 10.1016/0012-821x(85)90078-0
Pearce, C. R., Jones, M. T., Oelkers, E. H., Pradoux, C., and Jeandel, C. (2013). The effect of particulate dissolution on the neodymium (Nd) isotope and rare earth element (REE) composition of seawater. Earth Planet. Sci. Lett. 36, 138–147. doi: 10.1016/j.epsl.2013.03.023
Piper, D. F. (1974). Rare-earth elements in ferromanganese nodules and other marine phases. Geochim. Cosmochim. Acta 38, 1007–1022. doi: 10.1016/0016-7037(74)90002-7
Poulton, S. W., and Canfield, D. E. (2005). Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221. doi: 10.1016/j.chemgeo.2004.09.003
Poulton, S. W., and Raiswell, R. (2005). Chemical and physical characteristics of iron oxides in riverine and glacial meltwater sediments. Chem. Geol. 218, 203–221. doi: 10.1016/j.chemgeo.2005.01.007
Rahman, S., Aller, R. C., and Cochran, J. K. (2017). The Missing silica sink: revisiting the marine sedimentary Si cycle using cosmogenic 32Si. Glob. Biogeochem. Cycles 31, 1559–1578. doi: 10.1002/2017GB005746
Rao, V. P. (1987). Mineralogy of polymetallic nodules and associated sediments from the Central Indian Ocean Basin. Mar. Geol. 74, 151–157. doi: 10.1016/0025-3227(87)90011-9
Ren, J. B., Yao, H. Q., Zhu, K. C., He, G. W., Deng, X. G., Wang, H. F., et al. (2015). Enrichment mechanism of rare earth elements and yttrium in deep-sea mud of Clarion-Clipperton Region. Earth Sci. Front. 22, 200–211. doi: 10.13745/j.esf.2015.04.021
Rickli, J., Gutjahr, M., Vance, D., Fisher-Gödde, M., Hillenbrand, C.-D., and Kuhn, G. (2014). Neodymium and hafnium boundary contributions to seawater along the West Antarctic continental margin. Earth Planet. Sci. Lett. 394, 99–110. doi: 10.1016/j.epsl.2014.03.008
Roberts, N. L., and Piotrowski, A. M. (2015). Radiogenic Nd isotope labelling of the northern NE Atlantic during MIS 2. Earth Planet. Sci. Lett. 423, 125–133. doi: 10.1016/j.epsl.2015.05.011
Rousseau, T. C. C., Sonke, J. E., Chmeleff, J., van Beek, P., Souhaut, M., Boaventura, G., et al. (2015). Rapid neodymium release to marine waters from lithogenic sediments in the Amazon estuary. Nat. Commun. 6:7592. doi: 10.1038/ncomms8592
Rutberg, R. L., Hemming, S. R., and Goldstein, S. L. (2000). Reduced North Atlantic deep water flux to the glacial Southern Ocean inferred from neodymium isotope ratios. Nature 405, 935–938. doi: 10.1038/35016049
Sa, R., Sun, X., He, G., Xu, L., Pan, Q., Liao, J., et al. (2018). Enrichment of rare earth elements in siliceous sediments under slow deposition: a case study of the central North Pacific. Ore Geol. Rev. 94, 12–23. doi: 10.1016/j.oregeorev.2018.01.019
Schacht, U., Wallman, J., and Kutterolf, S. (2010). The influence of volcanic ash alteration on the REE composition of marine pore waters. J. Geochem. Explor. 106, 176–187. doi: 10.1016/j.gexplo.2010.02.006
Schott, J., Pokrovsky, O. S., and Oelkers, E. H. (2009). The link between mineral dissolution/precipitation kinetics and solution chemistry. Rev. Mineral. Geochem. 70, 207–258. doi: 10.2138/rmg.2009.70.6
Schott, J., Pokrovsky, O. S., Spalla, O., Devreux, F., Gloter, A., and Mielczarksy, J. A. (2012). Formation, growth and transformation of leached layers during silicate minerals dissolution: the example of wollastonite. Geochim. Cosmochim. Acta 98, 259–281. doi: 10.1016/j.gca.2012.09.030
Schwertmann, U., and Pfab, G. (1994). Structural vanadium in synthetic goethite. Geochim. Cosmochim. Acta 58, 4349–4352. doi: 10.1016/0016-7037(94)90338-7
Sholkovitz, E. R., Elderfield, H., Szymczak, R., and Casey, K. (1999). Island weathering: river sources of rare earth elements to the Western Pacific Ocean. Mar. Chem. 68, 39–57. doi: 10.1016/S0304-4203(99)00064-X
Sholkovitz, E. R., Landing, W. M., and Lewis, B. L. (1994). Ocean particle chemistry: the fractionation of rare earth elements between suspended particles and seawater. Geochim. Cosmochim. Acta 58, 1567–1579. doi: 10.1016/0016-7037(94)90559-2
Singh, S. P., Singh, S. K., Goswami, V., Bhushan, R., and Rai, V. K. (2012). Spatial distribution of dissolved neodymium and εNd in the Bay of Bengal: role of particulate matter and mixing of water masses. Geochim. Cosmochim. Acta 94, 38–56. doi: 10.1016/j.gca.2012.07.017
Skinner, L. C., Sadekav, A., Brandon, M., Greaves, M., Plancherel, Y., de la Fuente, M., et al. (2019). Rare Earth Elements in early-diagenetic foraminifer ‘coatings’: Pore-water controls and potential palaeoceanographic applications. Geochim. Cosmochim. Acta 245, 118–132. doi: 10.1016/j.gca.2018.10.027
Stewart, J. A., Gutjahr, M., James, R. H., Anand, P., and Wilson, P. A. (2016). Influence of the Amazon River on Nd isotope composition of deep water in the western equatorial Atlantic during the Oligocene-Miocene transition. Earth Planet. Sci. Lett. 454, 132–141. doi: 10.1016/j.epsl.2016.08.037
Stichel, T., Hartman, A. E., Duggan, B., Goldstein, S. L., Scher, H., and Pahnke, K. (2015). Separating biogeochemical cycling of neodymium from water mass mixing in the Eastern North Atlantic. Earth Planet. Sci. Lett. 412, 245–260. doi: 10.1016/j.epsl.2014.12.008
Stoffyn-Egli, P., and Mackenzie, F. T. (1984). Mass balance of dissolved lithium in the oceans. Geochim.Cosmochim. Acta 48, 859–872. doi: 10.1016/0016-7037(84)90107-8
Tachikawa, K., Athias, V., and Jeandel, C. (2003). Neodymium budget in the modern ocean and paleo-oceanographic implications. J. Geophys. Res. 108, 3254–3267. doi: 10.1029/1999JC000285
Tachikawa, K., Roy-Barman, M., Michard, A., Thouron, D., Yeghicheyan, D., and Jeandel, C. (2004). Neodymium isotopes in the Mediterranean Sea: comparison between seawater and sediment signals. Geochim. Cosmochim. Acta 68, 3095–3106. doi: 10.1016/j.gca.2004.01.024
Tachikawa, K., Toyofuku, T., Basile-Doelsch, I., and Delhaye, T. (2013). Microscale neodymium distribution in sedimentary planktonic foraminiferal tests and associated mineral phases. Geochim. Cosmochim. Acta 100, 11–23. doi: 10.1016/j.gca.2012.10.010
Takahashi, Y., Hayasaka, Y., Morita, K., Kashiwabara, T., Nakada, R., Marcus, M. A., et al. (2015). Transfer of rare earth elements (REE) from manganese oxides to phosphates during early diagenesis in pelagic sediments inferred from REE patterns, X-ray absorption spectroscopy, and chemical leaching method. Geochem. J. 49, 653–674. doi: 10.2343/geochemj.2.0393
Taylor, R. M., and McKenzie, R. M. (1966). The association of trace elements with manganese minerals in Australian soils. Aust. J. Soil Res. 2, 235–248.
Taylor, S. R., and McLennan, S. M. (1985). The Continental Crust: Its Composition and Evolution. An Examination of the Geochemical Record Preserved in Sedimentary Rocks. Oxford: Blackwell Scientific Publications.
Toyoda, K., and Tokonami, M. (1990). Diffusion of rare earth elements in fish teeth from deep-sea sediments. Nature 345, 607–609. doi: 10.1038/345607a0
Trotter, J. A., Barnes, C. R., and McCraken, A. D. (2016). Rare earth elements in conodont apatite: seawater or pore-water signatures? Palaeogeogr. Palaeoclimatol. Palaeoecol. 462, 92–100. doi: 10.1016/j.palaeo.2016.09.007
Turner, D. R., Whitfield, M., and Dickson, A. G. (1981). The equilibrium speciation of dissolved components in freshwater and seawater at 25°C and 1 atm pressure. Geochim. Cosmochim. Acta 45, 855–881. doi: 10.1016/0016-7037(81)90115-0
van de Flierdt, T., Griffiths, A. M., Lambelet, M., Little, S. H., Stichel, T., and Wilson, D. J. (2016). Neodymium in the oceans: a global database, a regional comparison and implications for palaeoceanographic research. Philos. Trans. R. Soc. 374:20150293. doi: 10.1098/rsta.2015.0293
van der Zee, C., Roberts, D. R., Rancourt, D. G., and Slomp, C. P. (2003). Nanogoethite is the dominant reactive oxyhydroxide phase in lake and marine sediments. Geology 31, 993–996. doi: 10.1130/G19924.1
Van Olphen, H., and Fripiat, J. J. (1979). Data Handbook for Clay Minerals and other Non Metallic Materials. Oxford: Pergamon Press, 346.
Vance, D., and Burton, K. (1999). Neodymium isotopes in planktonic foraminifera: a record of the response of continental weathering and ocean circulation rates to climate change. Earth Planet. Sci. Lett. 173, 365–379. doi: 10.1016/S0012-821X(99)00244-7
Vance, D., Scrivner, A. E., Beney, P., Staubwasser, M., Henderson, G. M., and Slowey, N. C. (2004). The use of foraminifera as a record of past neodymium isotope composition of seawater. Paleoceanography 19:A2009. doi: 10.1029/2003PA000957
VanLaningham, S., Duncan, R. A., Pisias, N. G., and Graham, D. W. (2008). Tracking fluvial response to climate change in the Pacific Northwest: a combined provenance approach using Ar and Nd isotopic systems on fine-grained sediments. Quat. Sci. Rev. 27, 497–517. doi: 10.1016/j.quascirev.2007.10.018
Viers, J., Roddaz, M., Filizola, N., Guyot, J.-L., Sondag, F., Brunet, P., et al. (2008). Seasonal and provenance controls on Nd-Sr isotopic compositions of Amazon rivers suspended sediments and implications for Nd and Sr fluxes exported to the Atlantic Ocean. Earth Planet. Sci. Lett. 274, 511–523. doi: 10.1016/j.epsl.2008.08.011
Vigier, N., Decarreau, A., Millot, R., Carignan, J., Petit, S., and France-Lanord, C. (2008). Quantifying Li isotope fractionation during smectite formation and implications for the Li Cycle. Geochim. Cosmochim. Acta 3, 780–792. doi: 10.1016/j.gca.2007.11.011
von Blanckenburg, F. (1999). Tracing past ocean circulation? Science 286, 1862–1863. doi: 10.1126/science.286.5446.1862b
Vorhies, J. S., and Gaines, R. R. (2009). Microbial dissolution of clay minerals as a source of iron and silica in marine sediments. Nat. Geosci. 2, 221–225. doi: 10.1038/ngeo441
Wilson, D. J., Piotrowski, A. M., Galy, A., and Clegg, J. A. (2013). Reactivity of neodymium carriers in deep sea sediments: implications for boundary exchange and paleoceanography. Geochim. Cosmochim. Acta 109, 197–221. doi: 10.1016/j.gca.2013.01.042
Wilson, D. J., Piotrowski, A. M., Galy, A., and McCave, I. N. (2012). A boundary exchange influence on deglacial neodymium isotope records from the deep western Indian Ocean. Earth Planet. Sci. Lett. 34, 35–47. doi: 10.1016/j.epsl.201206.009
Wood, S. A. (1990). The aqueous geochemistry of the rare-earth elements and yttrium: 1. review of available low-temperature data for inorganic complexes and the inorganic REE speciation of natural waters. Chem. Geol. 82, 159–186. doi: 10.1016/0009-2541(90)90080-q
Yang, J., and Haley, B. (2016). The profile of the rare earth elements in the Canada Basin, Arctic Ocean. Geochem. Geophys. Geosyst. 17, 3241–3253. doi: 10.1002/2016gc006412
Zhang, J., Dong, H., Liu, D., Fischer, T. B., Wang, S., and Huang, L. (2012). Microbial reduction of Fe(III) in illite–smectite minerals by methanogen Methanosarcina mazei. Chem. Geol. 292, 35–44. doi: 10.1016/j.chemgeo.2011.11.003
Zhang, J., and Nozaki, Y. (1996). Rare earth elements and yttrium in seawater: ICP-MS determinations in the East Caroline, Coral Sea, and South Fiji basins of the wester South Pacific Ocean. Geochim. Cosmochim. Acta 60, 4631–4644. doi: 10.1016/s0016-7037(96)00276-1
Zhang, L., Algeo, T. J., Cao, L., Zhao, L., Chen, Z.-C., and Li, Z. (2016). Diagenetic uptake of rare earth elements by conodont apatite. Palaeogeogr. Palaeoclimatol. Palaoecol. 458, 176–197. doi: 10.1016/j/palaeo.2015.10.049
Zhang, Y., Lacan, F., and Jeandel, C. (2008). Dissolved rare earth elements trace lithogenic inputs over the Kerguelen Plateau (Southern Ocean). Deep Sea Res. I 55, 638–652. doi: 10.1016/j.dsr2.2007.12.028
Keywords: rare earth elements, diagenesis, clay minerals, neodymium isotopes, isotope geochemistry, benthic source, sediment phases, paleoclimate
Citation: Abbott AN, Löhr S and Trethewy M (2019) Are Clay Minerals the Primary Control on the Oceanic Rare Earth Element Budget? Front. Mar. Sci. 6:504. doi: 10.3389/fmars.2019.00504
Received: 30 November 2018; Accepted: 29 July 2019;
Published: 20 August 2019.
Edited by:
Catherine Jeandel, UMR5566 Laboratoire d’Etudes en Géophysique et Océanographie Spatiales (LEGOS), FranceReviewed by:
Will Homoky, University of Oxford, United KingdomCopyright © 2019 Abbott, Löhr and Trethewy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: April N. Abbott, YXByaWwuYWJib3R0QG1xLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.