 Nicholas Robert Bates
Nicholas Robert Bates- 1Bermuda Institute of Ocean Sciences, St. George’s, Bermuda
- 2Department of Ocean and Earth Science, National Oceanography Centre, University of Southampton, Southampton, United Kingdom
The US GEOTRACES Eastern Pacific Zonal Transect in 2013 that sampled in the South Pacific Ocean has provided an opportunity to investigate the biogeochemical cycling of trace elements and isotopes (TEIs) and seawater carbon dioxide (CO2)–carbonate chemistry. Across the Peru to Tahiti section, the entire water column was sampled for total alkalinity (TA) and dissolved inorganic carbon (DIC), in addition to core hydrographic and chemical measurements conducted as part of the GEOTRACES cruise. From the nutrient-rich, low-oxygen coastal upwelling region adjacent to Peru to the oligotrophic central Pacific, very large horizontal gradients in marine carbonate chemistry were observed. Near the coast of Peru, upwelling of CO2-rich waters from the oxygen-deficient zone (ODZ) impinged at the surface with very high partial pressures of CO2 (pCO2; >800–1,200 μatm), and low pH (7.55–7.8). These waters were also undersaturated with respect to aragonite, a common calcium carbonate (CaCO3) mineral. These chemical conditions are not conducive to pelagic and shelf calcification, with shelf calcareous sediments vulnerable to CaCO3 dissolution, and to the future impacts of ocean acidification. A comparison to earlier data collected from 1991 to 1994 suggests that surface seawater DIC and pCO2 have increased by as much as 3 and 20%, respectively, while pH and saturation state for aragonite (Ωaragonite) have decreased by as much as 0.063 and 0.54, respectively. In intermediate waters (∼200–500 m), dissolved oxygen has decreased (loss of up to -43 μmoles kg-1) and nitrate increased (gain of up to 5 μmoles kg-1) over the past 20 years and this likely reflects the westward expansion of the ODZ across the central Eastern South Pacific Ocean. Over the same period, DIC and pCO2 increased by as much as +45 μmoles kg-1 and +145 μatm, respectively, while pH and Ωaragonite decreased by -0.091 and -0.45, respectively. Such rapid change in pH and CO2–carbonate chemistry over the past 20 years reflects substantial changes in the ODZ and water-column remineralization of organic matter with no direct influence from uptake of anthropogenic CO2. Estimates of anthropogenic carbon (i.e., CANT) determined using the TrOCA method showed no significant changes between 1993 and 2014 in these water masses. These findings have implications for changing the thermodynamics and solubility of intermediate water TEIs, but also for the marine ecosystem of the upper waters, especially for the vertically migrating community present in the eastern South Pacific Ocean.
Introduction
The primary goal of the international GEOTRACES program has been “to understand the sources, sinks, and internal cycling of trace elements and isotopes (TEIs) based on their distributions across the global ocean” (GEOTRACES, 2006). As part of the effort to understand the sources and sinks of TEIs, it is helpful to place TEI data into the context of entire water column hydrographic properties (i.e., temperature, salinity, nutrients) and their preformed distributions. Such information elucidates the processes that influence TEI in the ocean, and the physical and biogeochemical processes that influence their distributions.
As part of the context for deciphering the physical and biogeochemical dynamics of TEIs, it is also important to understand the controls imparted by changes in seawater carbonate chemistry, and pH in particular. Seawater pH has a strong influence on the speciation of trace elements (TEs; e.g., Turner et al., 1981; Byrne et al., 1988; Millero et al., 1995, 2009; Zeebe and Wolf-Gladrow, 2001; Byrne, 2002; Millero, 2013), thermodynamics and kinetic activity of metal TEs (e.g., Millero, 2001a,b), and potentially the influence of TEs complexed to organic ligands, complexation in seawater, and growth of marine organisms. Those trace elements that form strong complexes such as Copper (Cu2+) and rare earth elements are those that are the most strongly influenced by pH. For example, a decrease in pH due to natural water-column changes or ocean acidification (OA) induced change will increase free ionic Cu by ∼30% (with a change in pH from 8.1 to 7.4; Millero et al., 2009), a change that is significant considering that free Cu is potentially toxic to marine organisms (e.g., Sunda and Ferguson, 1983). A shift in pH also influences the solubility of many TEs, and, for example, Fe(III) solubility increases by 40% with a decrease in pH from 8.1 to 7.4 (Millero et al., 2009). Chemical reaction rates such as those that influence Fe(II) are also influenced by pH conditions. In addition, the thermodynamic influence of pH on trace element complexed organic matter is not well characterized or understood due to a lack of knowledge about the composition and reactivity of organic ligands (GEOTRACES, 2006).
The increase in atmospheric carbon dioxide (CO2) due to human activities and its uptake by the global ocean (Sabine et al., 2004; Khatiwala et al., 2013) has altered ocean chemistry and the marine CO2–carbonate system over the past few decades (e.g., Bates and Peters, 2007; González-Dávila et al., 2007; Dore et al., 2009; Olafsson et al., 2009, 2010; González-Dávila et al., 2010; Bates et al., 2012, 2014). The observed decline in ocean pH and saturation states of calcium carbonate (CaCO3) minerals (i.e., Ω) as a result of OA and future changes in ocean chemistry (e.g., Caldeira and Wickett, 2003; Orr et al., 2005) has potential direct and indirect consequences for marine biogeochemical cycles, and ocean organisms, and taxa, especially given that many trace elements are biochemically important for marine phytoplankton (Morel et al., 2003; Shi et al., 2010).
The US GEOTRACES Eastern Pacific Zonal Transect (i.e., GEOTRACES cruise GP16) conducted in 2013 has not only provided new information about the physical and biochemical cycling of TEIs in a region with one of the largest, permanently stratified oxygen-deficient zones (ODZs) on the planet and venting of hydrothermal fluids from the East Pacific Rise (e.g., Resing et al., 2015; Jenkins et al., 2018; Moffett and German, 2018, studies therein; Schlitzer et al., 2018), but also context for understanding such TEI variability relative to pH and seawater carbonate chemistry. Across the Peru to Tahiti section, there are very large horizontal gradients in hydrography and biogeochemistry, including the region of nutrient-rich coastal upwelling region adjacent to the coastline of Peru, the presence of the thermocline ODZ on the eastern Pacific Ocean, and westward across the oligotrophic central Pacific Ocean. Across this part of the eastern South Pacific Ocean, there are also very large horizontal gradients in upper-ocean (e.g., Feely et al., 1995, 2006; Takahashi et al., 2009; Quay et al., 2011) and vertical changes in seawater carbonate chemistry and pH, in particular. For example, pH variability ranges from 7.9 to 8.2 in surface waters (depending on pCO2 values of upwelled water), while pH decreases significantly in waters of the ODZ in this part of the Eastern Pacific Ocean.
The primary objectives of this paper are to report the observations of seawater carbonate chemistry across the ODZ of the Eastern South Pacific Ocean, and in particular, document the high pCO2 and low pH values found in the anoxic zone. Although the region has relatively sparse data, the GEOTRACES data collected in 2013 are compared to data collected a couple of decade earlier. This comparison is used to determine if the marine carbon cycle has changed and the ODZ expanded recently, and, if so, to assess the rates of changes of seawater carbonate chemistry such as DIC, TA, pCO2, pH, and the saturation states for calcium carbonate (CaCO3) minerals such as aragonite and calcite. The TrOCA method updated by Touratier et al. (2007) is used to calculate anthropogenic CO2 (CANT) contributions to the water column, if changes have occurred between 1994 and 2013, and if anthropogenic CO2 has contributed to substantial changes in pCO2 and low pH, for example, over the past 20 years.
Sampling, Methods, and Analytical Protocols
Cruise Information
The US GEOTRACES cruise GP16 was conducted in the Eastern Pacific Ocean from the R/V Thomas G. Thompson during October to December 2013 (Peters et al., 2018; data repository given therein). In the paper by Peters et al. (2018), details on the sampling and analysis of hydrography (e.g., potential temperature, salinity, inorganic nutrients, dissolved oxygen, and TEIs) with analytical methods and uncertainties given therein. The metadata and data for cruise GP16 are publically disseminated by the Biological and Chemical Oceanography Data Management Office1, while the entire GEOTRACES program data have been reported elsewhere (Schlitzer et al., 2018). As discussed later, the fractional contributions of different water masses in the region through the water column have been determined (Peters et al., 2018) and partially summarized in Table 1.
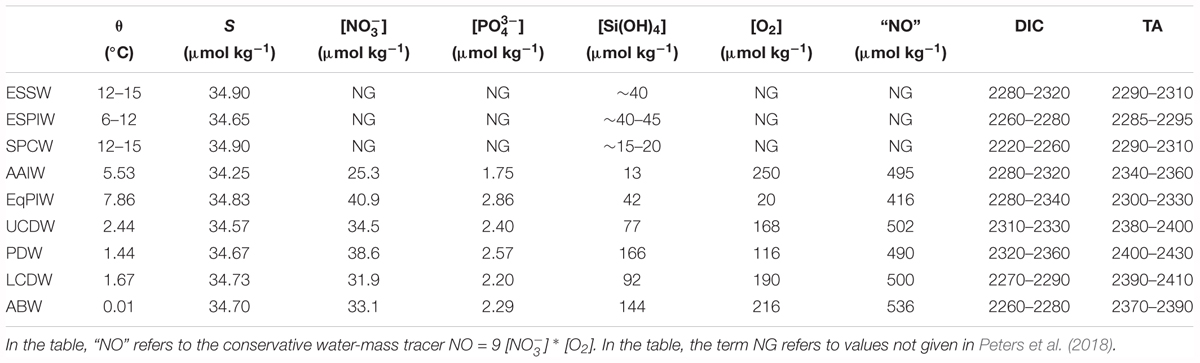
TABLE 1. End-member definitions for water types included in the upper ocean, intermediate (I) and deep (D) OMPAs reported by Peters et al. (2018), with mean DIC and TA ranges given.
The US GEOTRACES cruise GP16 cruise began in the nutrient-rich coastal upwelling region adjacent to the coastline of Peru, transited across the ODZ approximately at a latitude of ∼11–13°S, and finished in oligotrophic of the South Pacific subtropical gyre at ∼150°W and approximately 5° north of Tahiti (Figure 1). The deviation southward of the cruise track reached ∼14°S.
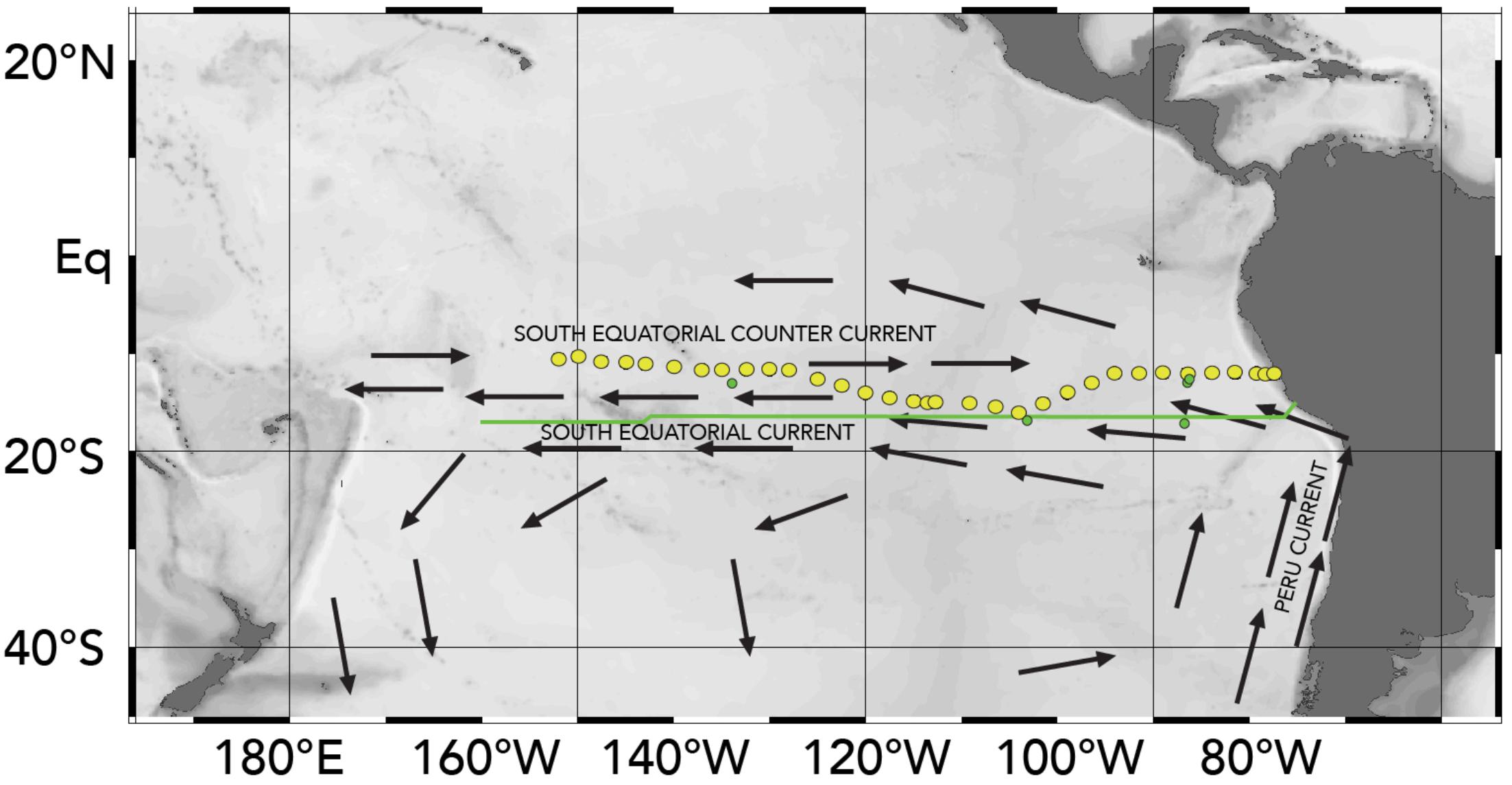
FIGURE 1. A map of the GEOTRACES GP16 cruise track (filled yellow dots) conducted in the Eastern Pacific Ocean from the R/V Thomas G. Thompson during October to December 2013. The green line denotes the cruise track from 1994 (Millero et al., 1994; cruise details), and cross-over stations (green circles; Feely et al., 1995; Takahashi et al., 1995). The approximate positions of the Peru Current and its transition to the South Equatorial Current are shown. Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
Considerations of Seawater Carbonate Chemistry
In this paper, the description of seawater carbonate chemistry follows long-established knowledge (e.g., Zeebe and Wolf-Gladrow, 2001; Dickson et al., 2007; Millero, 2013). Of the measurable parameters of the seawater carbonate system, the chemical description of DIC is as follows (e.g., Zeebe and Wolf-Gladrow, 2001; Dickson et al., 2007):
In this equation, the term [] represents the summed concentration of dissolved H2CO3 and CO2. The chemical description of alkalinity of seawater (TA) is defined as (e.g., Zeebe and Wolf-Gladrow, 2001; Dickson et al., 2007):
In Equation (2), the summed concentrations of [] + 2[] + [] represent the major chemical components of total alkalinity (TA) and the proton/charge balance of seawater. Here, other chemical species that contribute to alkalinity are typically minor constituents with negligible impact on TA (e.g., Zeebe and Wolf-Gladrow, 2001; Dickson et al., 2007; Millero, 2013). Given long-established guidelines (Dickson et al., 2007; and references therein); DIC and TA are both expressed as μmoles kg-1. In this paper, pCO2 is used rather than fugacity of CO2 (i.e., fCO2; note that this parameter was also computed in data files), and this term refers to the partial pressure of CO2 in equilibrium with water with units of μatm. pH in seawater is dimensionless and expressed on the total seawater scale (Dickson et al., 2007).
In well-established chemical understanding of the seawater carbonate system, the chemical equations governing the formation and dissolution of calcium carbonate (CaCO3) minerals are as follows (e.g., Zeebe and Wolf-Gladrow, 2001; Dickson et al., 2007; Millero, 2013):
In typical seawater, the rates of CaCO3 production and dissolution vary in general terms depending on CaCO3 saturation state (Ω). The saturation state for CaCO3 minerals such as aragonite and calcite is chemically defined by the following equations and relationships between calcium and carbonate ion concentrations, thus:
In Equations (4) and (5), the solubility product for CaCO3, , and , varies primarily as a function of temperature, salinity, and pressure conditions. In general terms, although aragonite and calcite dissolution is favored in seawater when Ωaragonite and Ωcalcite values that are lower than one (i.e., Ω < 1), and aragonite and calcite formation generally occurs at Ωaragonite and Ωcalcite values that are greater than one (i.e., Ω > 1), dissolution and production of CaCO3 can occur above 1 and below 1, respectively.
Sampling Protocols for Seawater Carbonate Chemistry
Seawater samples for carbonate chemistry were collected for the entire water column at all of the GEOTRACES CTD-hydrocast stations. Samples for both TA and DIC were withdrawn from the Niskin samplers (after dissolved oxygen and salinity) using well-established gas sampling methods (Bates et al., 1996a; Dickson et al., 2007), and collected into the clean 500 ml size Pyrex glass reagent bottles. For each sample, a headspace of <1% of the bottle volume was left to allow for water expansion and contraction during storage and shipping. All of the TA and DIC samples were poisoned with ∼100 μl volume of saturated HgCl2 solution in order to prevent biological alteration, bottles were then sealed with Apiezon silicon vacuum grease and ground-glass stoppers, and stored in the dark. TA and DIC data here are not corrected for the volume of HgCl2 added. Samples were returned to the marine biogeochemistry laboratory at the Bermuda Institute of Ocean Sciences (BIOS) for analysis. Similar sampling protocols were established at BIOS for sampling at the BATS (Bates et al., 1996a,b; Bates, 2001). Samples were typically analyzed within 3 months of collection in the laboratory at BIOS.
Analytical Determination of Seawater DIC and TA
Seawater DIC contents of each sample were measured using a well-established gas extraction/coulometric method [see Bates et al. (1996a,b) for details]. At BIOS, two nearly identical automated instruments (SOMMA; Single-Operator Multi-Metabolic Analyzer; VINDTA 3C, Marianda Com) were used to deliver a known volume of sample seawater, and with weak phosphoric acid, convert all carbonate species to dissolved CO2. UIC CO2 coulometer detectors were then used to measure the amount of CO2 evolved from the seawater sample in order to determine DIC content (Bates et al., 2012). Replicate sample analyses of >5,000 seawater samples analyzed at BIOS from 1992 to present indicate the analytical precision of DIC at better than 0.025% (∼0.5 μmoles kg-1), with more than 50 replicates for this cruise giving similar results. In addition, seawater certified reference materials (CRMs; prepared by A.G. Dickson, Scripps Institution of Oceanography) were routinely analyzed to provide an estimate of analytical accuracy of DIC at 0.05% (∼1 μmoles kg-1). Such precision and accuracy are typical for more than 10,000 DIC samples analyzed in the laboratory at BIOS over the past 20 years. This is due in part to analyses being primarily devoted to time-series analysis (e.g., Bates et al., 2014), rigorous methodologies, and a very stable analytical environment in the laboratory.
For, each sample, TA was measured by potentiometric titration with a weak solution of HCl [see Bates et al. (1996a,b, 2012) for details]. Replicate sample analyses of >5,000 seawater samples analyzed at BIOS indicated an analytical precision for TA of 0.04% (∼1 μmoles kg-1), while concurrent analyses of CRM samples typically gave results within 0.15% (∼1–2 μmoles kg-1) of certified TA values reported by A.G. Dickson (Scripps Institution of Oceanography2). The use of CRM samples for both DIC and TA analyses is a critically important approach for assessing seawater chemistry over time and accurate calculation of pCO2 and pH for seawater samples (e.g., Bates et al., 2012, 2014). CRM batch numbers 134 and 140 were used for this suite of DIC and TA sample analyses.
Seawater CO2–Carbonate Chemistry Computations and Propagation of Uncertainty
The full suite of seawater carbonic acid system parameters (i.e., [CO2], [H2CO3], [], [], [H+]) can be determined from the measurement of two parameters (i.e., DIC, TA, pCO2, and pH), along with temperature and salinity (e.g., Zeebe and Wolf-Gladrow, 2001; Millero, 2013). In this paper, the seawater carbonic acid system parameters of [], [], pCO2, fCO2, pH, Ωaragonite, and Ωcalcite values were determined from DIC, TA, temperature, and salinity data using the dissociation constants of Mehrbach et al., 1973 (as refit by Dickson and Millero, 1987), and borate dissociation constant of Dickson (1990), and the software program of Robbins et al. (2010). In this paper, pCO2 was used rather than fCO2, with small differences of ∼1–3 μatm between both parameters, while SOCAT data are expressed as fCO2 (e.g., Bakker et al., 2016). pH is expressed on the seawater scale. Nitrate data were not used in the computation which introduced very minor uncertainty for the calculation of pCO2, pH, Ωaragonite, and Ωcalcite values.
The error estimates for calculation of pCO2, pH, Ωaragonite, and Ωcalcite values were undertaken using standard procedures for propagation of uncertainty. Given the analytical uncertainty of the two seawater carbonate chemistry parameters (i.e., DIC and TA; ±1 μmoles kg-1) from replicate measurements of CRMs, the error is estimated at 3 μatm, 0.003, 0.021, and 0.021, for pCO2, pH, and Ωaragonite and Ωaragonite for surface waters, respectively. The calculation error is larger for high pCO2 (>1,000 μatm)/low pH (<7.7) waters at up to 15 μatm, 0.004, 0.023, and 0.026, for pCO2, pH, and Ωaragonite and Ωaragonite, respectively. Please note that pH, Ωaragonite, and Ωaragonite are dimensionless and thus do not have units. The use of other sets of dissociation constants for calculation of the complete seawater carbonic acid system would introduce additional range of calculation error due to uncertainties in the dissociation constants used, but prescriptions of the calculations are included in the prior paragraph.
Estimates of the Anthropogenic Carbon
Estimates of anthropogenic carbon (i.e., CANT) were determined using the TrOCA (Tracer combining Oxygen, inorganic Carbon, and TA) methods first proposed by Touratier and Goyet (2004a,b) and updated by Touratier et al. (2007). This approach allows comparison with recent estimates determined for the western South Pacific Ocean by Wagener et al. (2018). In the Touratier et al.’s (2007) method, changes in the quasi-conservative tracer, TrOCA in individual water masses accounts for changes in carbonate chemistry due to biological influences (e.g., remineralization of organic matter and dissolution of CaCO3). Differences between current and pre-industrial TrOCA values provide an estimate of anthropogenic carbon (i.e., CANT) contribution to the water mass. Here, the formulation proposed by Touratier et al. [2007; their equation (11)] is used:
where Φ is the potential temperature. The uncertainty of the estimate is ∼6 μmoles kg-1 (Touratier et al., 2007) with the limitations of the method discussed in this paper and other subsequent papers (e.g., Wagener et al., 2018).
CANT estimates determined using the TrOCA method for the mixed layer cannot be considered or verified due to euphotic zone biological activity and air–sea gas exchange (Touratier et al., 2007). As with the recent analysis of CANT by Wagener et al. (2018) in the western South Pacific, CANT estimates for the Peru–Tahiti section are given below ∼100 m depth.
Comparison With Other Marine Carbonate Chemistry Datasets
In the section “Discussion,” the GEOTRACES data collected herein are compared with historical data collected in the region. As with other analyses of historical data (e.g., GLODAP-v2; Key et al., 2015; Olsen et al., 2016), deep water is assumed to remain unaltered over the past couple of decades. In the deep eastern Pacific Ocean (<3,000 m deep), Peters et al. (2018) identified the presence of varying amounts of Pacific Deep Water (PDW), Lower Circumpolar Deep Water (LCDP), and Antarctic Bottom Water (ABW). In these deep-water masses, no systematic differences of greater that 0.01 and 0.003 for potential temperature and salinity, and ≤2 μmoles kg-1 for DIC and TA between recent and historical data were determined. Thus, there does not appear to be any systematic analytical bias or offsets between historical and recent data for deep water or waters shallower than 3,000 m. As such, the historical data were not corrected for potential offsets given the absence of evidence for such systematic biases.
Results
Water Mass Identification
In a recent paper, Peters et al. (2018) identified nine different water masses below the seasonal thermocline (up to ∼200 m) using Optimum Multiparameter Analysis (OMPA; Tomczak, 1981). This method provides the fractional composition of a water mass for each sample using definitions of hydrographic properties for individual water mass end members (Table 1). The OMPA identification of water mass using hydrographic properties of potential temperature, salinity, inorganic nutrients (i.e., nitrate, phosphate, silicate), and dissolved oxygen (i.e., Peters et al., 2018) provides useful context for understanding seawater carbonate chemistry across the section from 77°W to 155°W (Figure 2A).
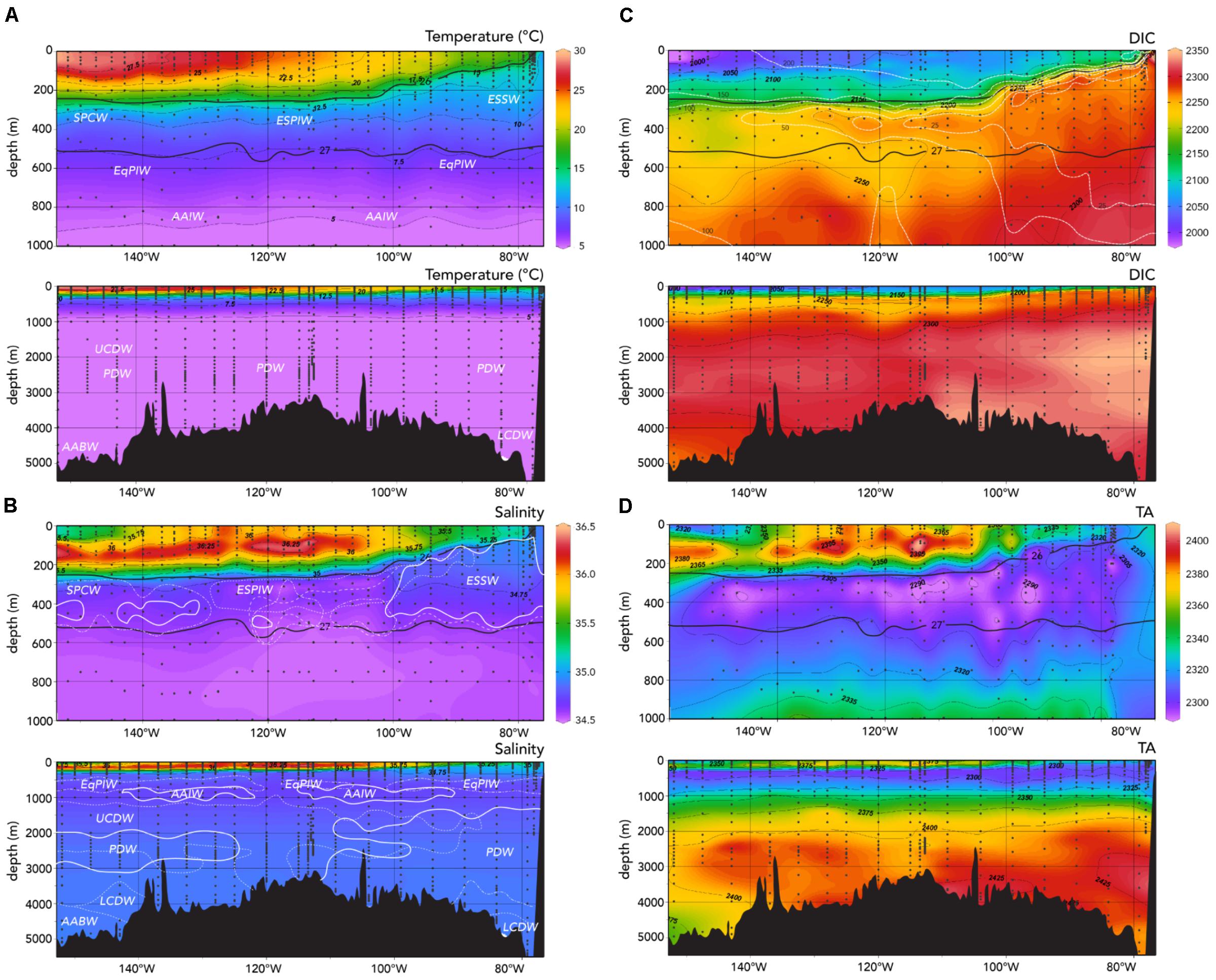
FIGURE 2. Observed sections of (A) potential temperature (°C); (B) salinity; (C) dissolved inorganic carbon, DIC (μmoles kg-1); and (D) total alkalinity, TA (μmoles kg-1) along the GP16 section. The depth ranges of 0–1,000 and 0–5,000 m are shown for each physical or biogeochemical property. In panels (A)–(D), isopycnal surfaces that define the thermocline layer of σ0 = 26.0–27.0 are contoured for ease of reference (Peters et al., 2018). In panel B, the approximate core depths of water masses identified by Peters et al. (2018) are shown (white and dashed lines). In panel C, approximate contours for dissolved oxygen are also shown to highlight the extent of the ODZ (white dashed lines). Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
In the thermocline, between the approximate sigma theta density horizons of 26.0–27.0 kg m-3, Equatorial Subsurface Water (ESSW) dominates the upper ocean and in particular, it constitutes the low oxygen waters of the eastern tropical South Pacific (∼100–500 m deep; contributions of greater than 60% to the water mass). Moving westward, the water mass merges with Eastern South Pacific Intermediate Water (ESPIW; ∼220–500 m deep) which tends to contribute up to 30% of the water present. In the westernmost part of the section (∼130–155°W) is dominated by South Pacific Central Water (SPCW; ∼220–500 m deep; Figure 2B); in thermocline waters that are typically less dense than 27.0 kg m-3, and with a fractional contribution up to 60% (Peters et al., 2018). The end-member properties are given in Table 1.
Nutrient-rich intermediate waters below the thermocline (between the approximate sigma theta density horizon of 27.0 and 27.8 kg m-3) can be characterized by the contributions of: (1) Equatorial Pacific Intermediate Waters (EqPIW; ∼600–800 m deep); and (2) Antarctic Intermediate Water (AAIW; ∼800–1000 m deep). Other deeper waters included PDW, Upper Circumpolar Deep Water (UCDW), LCDW, and ABW (Figure 2A; see Peters et al., 2018).
Horizontal Gradients in Upper Ocean Variability of Carbonate Chemistry
Surface waters across the Peru–Tahiti section can be conveniently subdivided into three zones, from east to west: (1) upwelling zone near the Peru Coast; (2) Peru current (∼80–90°W); and (3) central eastern South Pacific waters (∼90–150°W).
Surface Waters in the Upwelling Zone Near Peru (∼76–80°W)
The US GEOTRACES Peru–Tahiti section fortunately sampled the upwelling zone close to the Peru coast. In the zone of upwelling, water mass properties of the mixed layer and upper 100 m were as expected for an ODZ influenced region with cooler (∼15°C; Figure 2A) and fresher (∼35) waters (Figure 2B) that were low in dissolved oxygen (<25 μmoles kg-1; Figure 2C) and high in inorganic nutrients (i.e., nitrate >20 μ moles kg-1).
In this upwelling region, there is very little prior data about marine carbonate chemistry with the exception of earlier transects in 1994 (Millero et al., 1994) and 2009 (Uchida et al., 2011). In the fresher upwelled waters, TA was low (<2,310 μmoles kg-1; Figure 2D), while DIC was high at >2,200 μmoles kg-1 (Figure 2C) relative to surface waters elsewhere.
In the upper 100 m, very high pCO2 values of >1,200 μatm were observed with surface values as much as >800 μatm (Figure 3A). Such high pCO2 values would be expected with the very low TA:DIC ratios (∼1.055:1.000) of the upwelled waters compared to surface waters further west in the central Eastern South Pacific with higher TA:DIC ratios (∼1.15:1.000; Figure 4). The large longitudinal change in surface water carbonate chemistry from the upwelling zone to the central Eastern South Pacific is also shown in Figure 4 (data shown in the figure under the gray region) and Figure 5. Surface waters in the upwelling zone have much higher DIC (by as much as 200 μmoles kg-1) compared to central Eastern South Pacific waters, without much change in alkalinity reflecting the long-term gain of CO2 in deep and intermediate waters through remineralization of organic matter back to CO2. In the plots of Figure 5, the relatively cool (15–20°C) and fresh (<35.25), high DIC (>2,100 μmoles kg-1) and pCO2 (>600 μatm), and low pH (<7.95) waters were observed in the uppermost 5 m in the upwelling region (∼76–80°W).
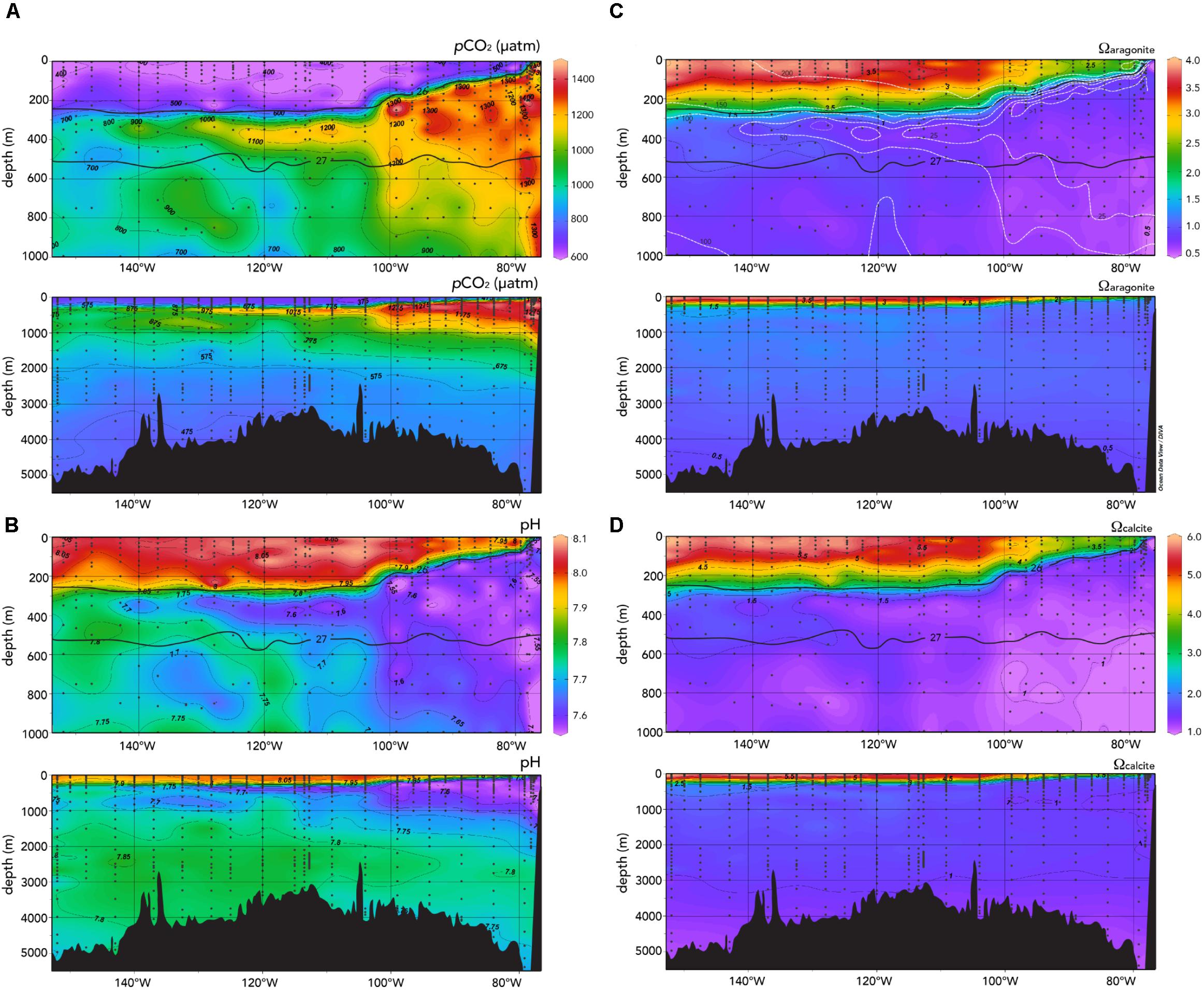
FIGURE 3. Observed sections of (A) pCO2 (μatm); (B) pH; (C) Ωaragonite; and (D) Ωcalcite along the GP16 section. The depth ranges of 0–1,000 and 0–5,000 m are shown for each physical or biogeochemical property. In panels (A)–(D), isopycnal surfaces that define the thermocline layer of σ0 = 26.0–27.0 are contoured for ease of reference (Peters et al., 2018). In panel C, approximate contours for dissolved oxygen are also shown to highlight the extent of the ODZ (white dashed lines). In panels C and D, values less than 1 denote potential for CaCO3 dissolution. Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
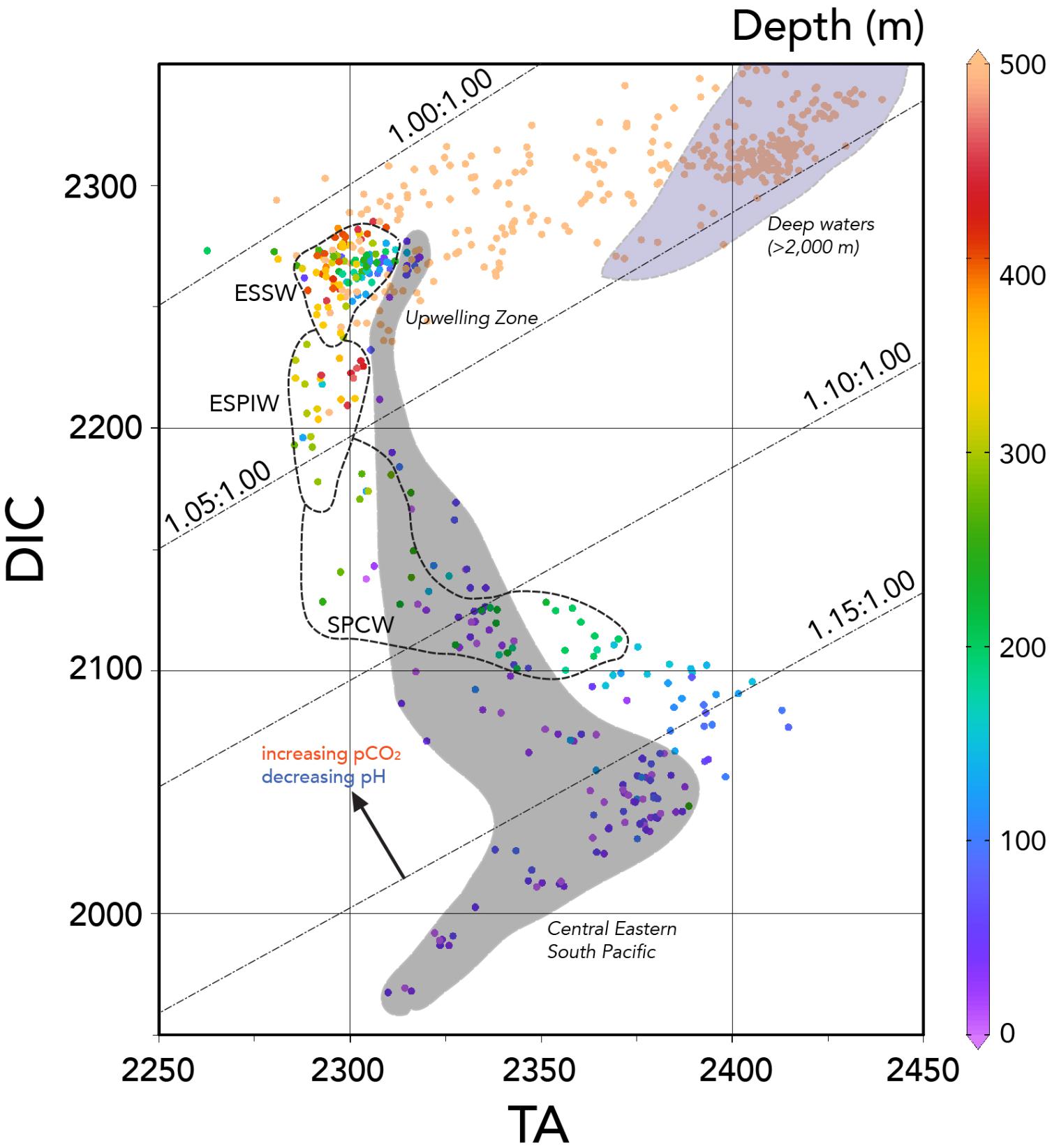
FIGURE 4. Scatter plot of TA (μmoles kg-1) against DIC (μmoles kg-1) for the entire water column of the Peru–Tahiti section. The depth ranges of 0–500 m are color-contoured in the plot. Surface waters (i.e., 0 to ∼100 m deep) along Peru–Tahiti section typically fall within the light gray area shown in the figure, while the cluster of deep water (>2,000 m) samples are highlighted in the pale blue area. The approximate TA:DIC properties of three intermediate waters (∼200–500 m deep) are also shown (i.e., ESSW, ESPIW, and SPCW). In the plot, the hatched lines show the approximate isolines for the TA:DIC ratios of 1.00:1.00, 1.05:1.00, 1.10:1.00, and 1.15:1.00, respectively. As shown by the arrow (diagonal to the isolines of TA:DIC), a decrease in TA:DIC ratio of water masses typically results in an increase in pCO2 and decrease in pH (and Ωaragonite and Ωcalcite, not shown). Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
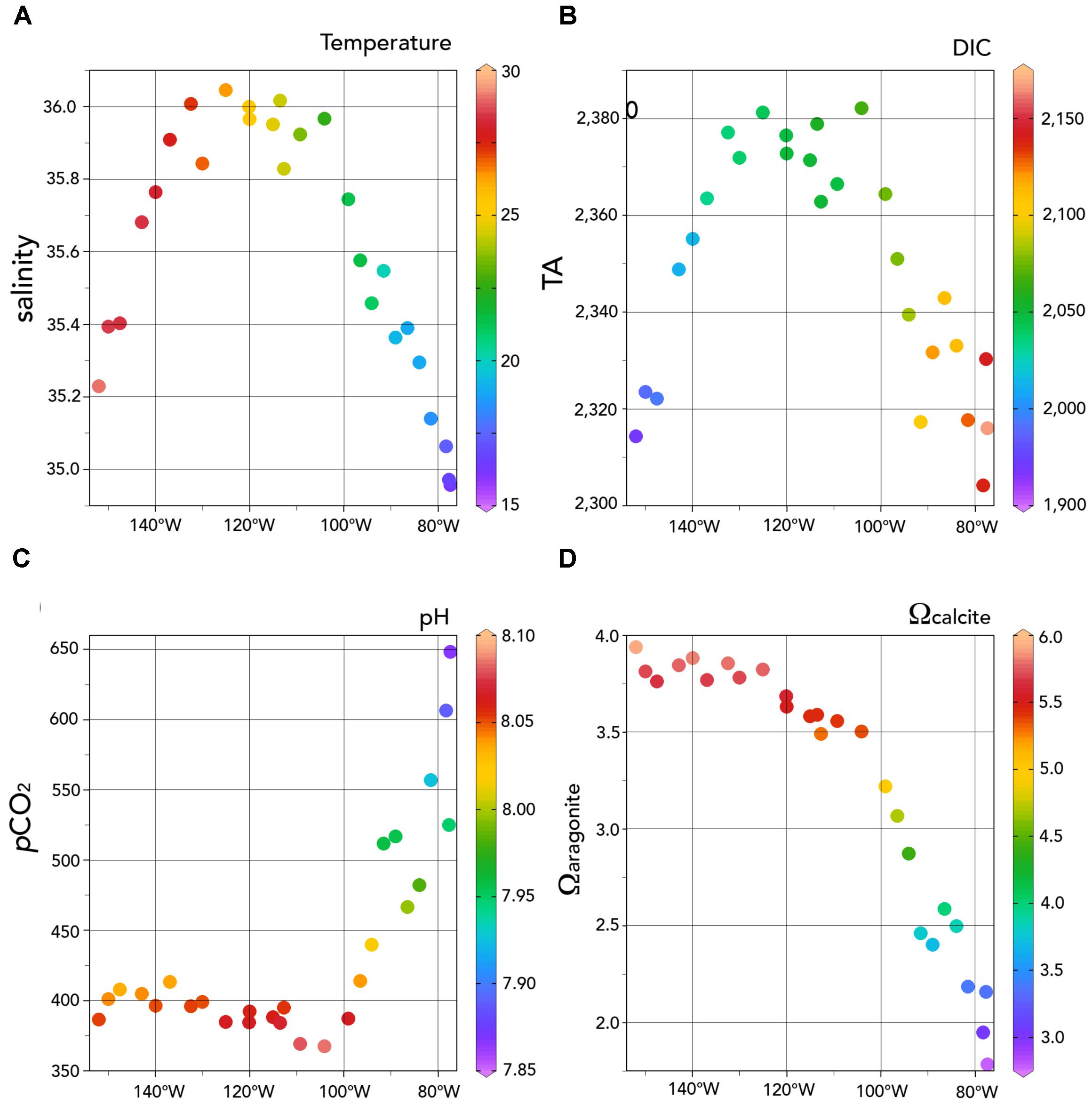
FIGURE 5. Surface variations of carbonate chemistry for the upper 5 m of the water-column for the 2013 Peru-Tahiti section. (A) salinity and temperature (°C); (B) TA and DIC (μmoles kg-1); (C) pCO2 (μatm) and pH; and (D) Ωcalcite and Ωaragonite. Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
Previously, surface pCO2 measurements have not been collected in the upwelling zone close to the Peru coast (i.e., within 2° of latitude of the coast). Limited surface data compiled as part of the Surface Ocean Carbon Atlas effort (e.g., SOCAT, Bakker et al., 2014, 2016) reveal the highest pCO2 values a little farther offshore at ∼600 μatm (Figure 6). The US GEOTRACES Peru–Tahiti section observations of the upwelling zone indicate that surface waters have much higher pCO2 (>600 μatm) than the atmosphere (∼395 μatm; mean atmospheric pCO2 values from Easter Island from the NOAA flask sampling network3) with the driving force of air–sea CO2 gas exchange strongly directed toward a loss of CO2 from the ocean.
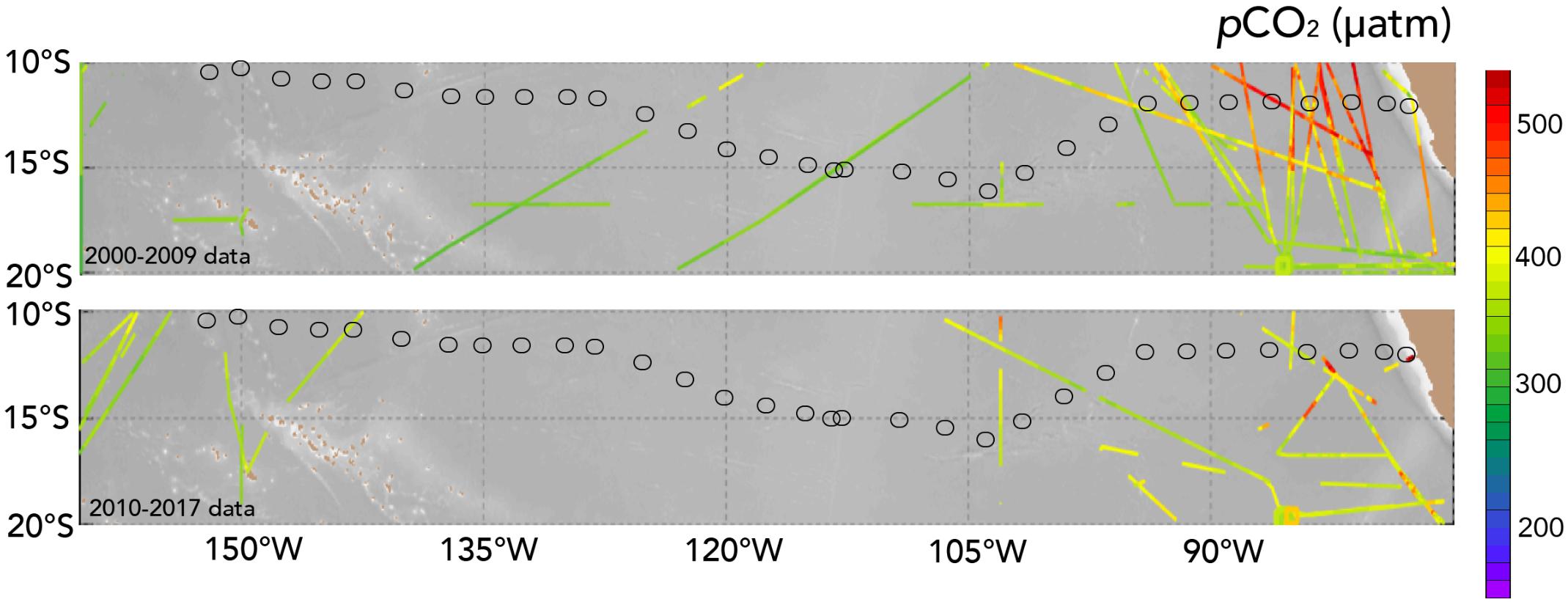
FIGURE 6. Surface scatter plot of pCO2 taken from the SOCAT database (Bakker et al., 2014, 2016; version 6) with data from 2000–2009 and 2010–2017. The cruise stations of the Peru–Tahiti section are shown in both panels.
The upwelled water also exhibited pH values lower than 7.55 in the upper 100 m, and less than 7.80 in surface waters (Figures 3B, 5). Furthermore, the CaCO3 saturation states of the upwelled water were close to 1 and 2.5 for Ωaragonite and Ωcalcite, respectively (Figures 3C,D). Ωaragonite values lower than 1 were observed at 50 m deep indicating conditions favorable for aragonite dissolution. The implication of this finding is that the upwelling is highly likely to bring waters that are potentially corrosive for aragonite (but not calcite) onto the shelf of the Peru coast. As with other coastal zones influenced by upwelling (e.g., Feely et al., 2008), this part of the Peru coast and its marine ecosystem (particularly benthic or pelagic calcifying taxa) will be vulnerable to CaCO3 dissolution and any augmentation of this driver by OA.
The US GEOTRACES Peru–Tahiti section data revealed high DIC and pCO2, and low pH waters in the upwelling zone of the eastern South Pacific Ocean. Such ranges in ocean carbonate chemistry are not shown in the interpolated and extrapolated global ocean mean climatologies of DIC, TA, pCO2, pH, and Ω (e.g., Takahashi et al., 2014), and undersampling of the region.
Peru Current Region Surface Waters (∼80–90°W)
Away from the upwelling zone near the Peru coast, the Peru–Tahiti section sampled across waters associated with the northward flow of the Peru Current (from ∼80 to 90°W). In this zone, salinity was about 35.2 and temperatures ranged from 18 to 20°C (Figures 2A,B, 5A). The TA values (∼2,330 μmoles kg-1) were higher and DIC (∼2,100–2,125 μmoles kg-1) concentrations lower than the upwelling zone, but both generally lower than the central eastern South Pacific west of 90°W (Figures 2C,D).
Surface and mixed layer pCO2 values ranged from about 400 to 450 μatm (Figure 3A). Such pCO2 values are similar to previous north–south observations collected in the SOCAT data (Figure 5, ∼450–550 μatm; Bakker et al., 2014, 2016). The pH of these northward flowing waters ranged from 7.95 to 8.00 (Figure 3B), while the Ωaragonite and Ωcalcite values ranged from 2.0 to 2.5 and 3.5 to 4.0, respectively (Figures 3C,D).
Central Eastern South Pacific Surface Waters (∼90–150°W)
In the central Eastern South Pacific, salinity ranged from 35.5 to 36.0 (highest between 100 and 140°W; Figure 5B), while west of the Peru Current, surface temperatures increased from ∼20 to >28°C (Figures 2A,B, 5A). TA distributions followed a similar pattern to salinity across the section from 90°W (ranging from 2,330 to 2,390 μmoles kg-1; Figure 5D), but DIC decreased from ∼2,100 to 2,000 μmoles kg-1 westward toward 150°W (Figures 2C,D, 5C).
Interestingly, across the section from 90 to 150°W, surface pCO2 remained fairly constant in the range of ∼390–420 μatm (Figures 3C, 5E), while pH ranged from ∼8.00 to 8.10 (Figure 5F). In Figure 4, surface waters in the central Eastern South Pacific generally lie along a TA:DIC ratio line of ∼1.16:1.00. This underlying chemical state primarily pre-conditions surface waters to have similar pCO2, pH, Ωaragonite, and Ωcalcite values with secondary modulation of these properties by temperature variations across the region. As shown in Figure 6, there is little pCO2 data across the region except for transects around 120°W, with this sparse data showing similar ranges to observations collected from the Peru–Tahiti section.
The lack of east to west variability of surface water pCO2 in the central Eastern South Pacific reflects a competition between a couple of dominant processes: (1) pCO2 increasing due to increasing temperature and (2) decreasing due to loss of DIC from east to west. For example, warming acts to increase pCO2 by ∼4.2% per °C due to thermodynamics of the carbonate system (Zeebe and Wolf-Gladrow, 2001). Thus, a warming of ∼8°C will increase by ∼160 μatm. However, the westward loss of DIC of ∼100 μmoles kg-1 acts oppositely to decrease pCO2 by ∼170 μatm. The range of surface pCO2 values was similar to atmospheric pCO2 with a small driving force for gas exchange. This finding suggests that CO2 sink-sources status of the region is close to neutral. Across this zone, pH is also fairly constant (∼8.05; Figures 3B, 5F) as are Ωaragonite (>3.5) and Ωcalcite (>5.5), with strong oversaturation with respect to both CaCO3 minerals (Figures 3C,D).
Three intermediate waters have been identified as present across the Peru–Tahiti section, including ESSW, ESPIW, and SPCW (Figure 2A; Peters et al., 2018). As the source for upwelled waters, the ESSW waters found near the Peru coast represent the core region of the ODZ (Figure 2C). The carbonate chemistry in the core of the ODZ (<50 m to nearly 600 m) has relatively low TA values relative to other water masses in the Eastern South Pacific (<2,310 μmoles kg-1; Figure 2D), and high DIC values at >2,200 μmoles kg-1 (Figure 2C). The TA and DIC properties of the ESSW in the core of the ODZ were very similar to upwelled water (Figure 4). The only difference between these two water masses is that the TA value of surface waters is approximately 10–20 μmoles kg-1 higher. This can be explained by the difference in nitrate concentrations between the ESSW (∼20–35 μmoles kg-1) and surface waters (∼10–15 μmoles kg-1). Since nitrate contributes to alkalinity, the TA + nitrate values from the ESSW and surface waters were very similar.
The OMPA analyses of Peters et al. (2018) indicated a transition westward from ESSW waters to ESPIW water, and father west merging into SPCW. Across the Peru–Tahiti section, DIC decreases by ∼100–150 μmoles kg-1 from the waters of the ESSW to the SPCW, while TA does not change substantively (Figure 4). In concert with these latitudinal water mass changes, and changes in DIC and TA, pCO2 values decrease westward along the section (Figure 3A), while pH, Ωaragonite, and Ωcalcite all increase (Figures 3B–D).
Deep Water Variability of Carbonate Chemistry
The deeper water masses of the Eastern South Pacific have also been identified through the OMPA analyses of Peters et al. (2018). As one would expect, the deep water DIC and TA increase with depth (Figures 2C,D, 7A,B) reflecting the gain over time of CO2 from remineralization of sinking organic matter, dissolution of CaCO3 minerals. The increase of TA to DIC is approximately in the ratio of two to one (i.e., ∼100 μmoles kg-1 TA increase compared to ∼50 μmoles kg-1 DIC increase in deep waters compared to intermediate waters). While there is source water variability in the carbonate chemistry of the different deep–water masses, dissolution of CaCO3 at depth seems to dominate relative to the biological pump of carbon. The highest pCO2 (>1,500 μatm; Figure 7C) and lowest pH values (Figure 7D) were observed in intermediate waters (∼200–1,000 m deep) across the section. The profiles of Ωcalcite and Ωaragonite shown in Figures 7C,D show highly variable depth ranges for lysocline depth (i.e., Ω = 1). In the ODZ region, the lysocline depths are shallow for calcite (∼100 m) and aragonite (∼100 m). They are much deeper in the central Pacific at >4,000 and ∼1,000 m for calcite and aragonite, respectively.
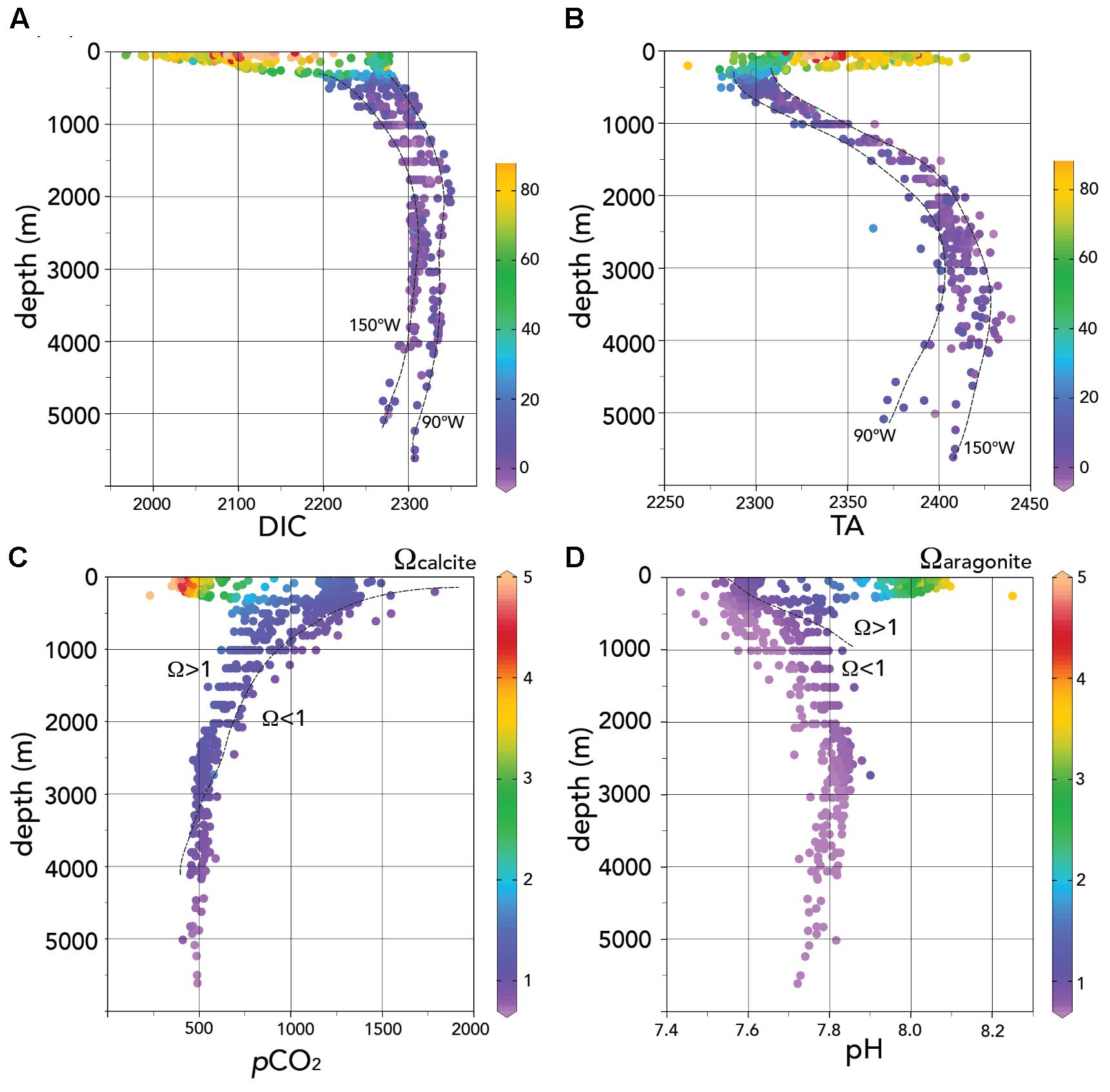
FIGURE 7. Water column profiles of seawater carbonate chemistry data for the entire 2013 Peru–Tahiti section. (A) DIC (μmoles kg-1) shown against depth (m). The color range indicates the CANT values (μmoles kg-1) determined using the TrOCA approach (Touratier et al., 2007). (B) TA (μmoles kg-1) shown against depth (m). The color range indicates the CANT values (μmoles kg-1) determined using the TrOCA approach (Touratier et al., 2007). (C) pCO2 (μatm) shown against depth (m). The color range indicates the Ωcalcite values. (D) pH shown against depth (m). The color range indicates the Ωaragonite values. In panels A and B, the easternmost and westernmost profiles are shown by the dashed lines. In panels C and D, Ω values of approximately 1 are shown by the dashed lines (with Ω > 1 and Ω < 1 shown on the plots). Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
In the deep waters below 2,000 m, there is little change of pCO2, pH, Ωaragonite, and Ωcalcite relative to the shallower intermediate waters (Figures 3A–D, 7C,D).
Is Different Seawater Carbonate Chemistry Observable in the Deep Hydrothermal Plume Originating at the East Pacific Rise?
As reported previously, a large trace element and isotope enriched hydrothermal plume (i.e., Eastern Boundary Plume, EBP; Peters et al., 2018), originating from the East Pacific Rise (located at ∼113°W), has been shown to extend 4,000 km westward across the Pacific (Resing et al., 2015; Lupton and Jenkins, 2017; Jenkins et al., 2018; Figures 2C,D). Hydrothermal plume tracers such as 3He, radiocarbon, and other noble gases were primary found at depths of 2,500±200 m, and associated with altered TEI chemistry and scavenging in the plume waters (Fitzsimmons et al., 2017; Buck et al., 2018; Moffett and German, 2018; Pavia et al., 2018). Co-located at depth with the EBP, the DIC and TA, and other carbonate parameters were not markedly different to shallower and deeper waters of the PDW. Thus, the EBP was not observable with marine carbonate parameters on this section.
Discussion
Changes in Carbonate Chemistry Across the Section
The Eastern South Pacific Ocean has been significantly undersampled compared to other regions of the global ocean with regard to marine biogeochemical properties and carbonate chemistry. Over the last 30 years, only two other cruises collecting high-quality carbonate data have been conducted (Millero et al., 1994; Uchida et al., 2011) at similar latitude to the US GEOTRACES Peru–Tahiti section. The CLIVAR P6 cruise sampled in the South Pacific at 30°S but considerably south of the GEOTRACES section (∼10–15°S). Other data include north–south section collected between 1991 and 1994 (e.g., Feely et al., 1994; Takahashi et al., 1995; Rubin et al., 1998). The sampling in 1991, 1994, and 2013 also occurred during neutral El Ninõ-Southern Oscillation (ENSO) phases, and thus do not represent periods undergoing climatic episodes of either El Niño or La Niña phases.
As demonstrated by Santer et al. (2011), it can take a decade or more for significant changes in the ocean and atmosphere to become evident upon data analysis. Keller et al. (2014) have demonstrated pCO2 and pH observations needed to be made for at least 12 years and ∼10–30 years for DIC in the ocean, while long-term open-ocean time-series have demonstrated decadal trends with data collected for more than 20 years (e.g., Bates et al., 2014). Here, the Peru to Tahiti section data seawater carbonate chemistry data are compared to the Millero et al.’s (1994) data and crossover data to establish if marine biogeochemical changes can be determined over the past 20 years. The sampling interval between the Uchida et al. (2011) data collection are, however, too short to establish more than direction and possible magnitude of change in the region of the US GEOTRACES Peru–Tahiti section data.
In other ocean basins, the repeat hydrography and carbonate chemistry sampling has been very useful for demonstrating inventories of anthropogenic CO2 using tracers such as C∗ (Gruber et al., 1996; Sabine et al., 2004), TrOCA (Touratier et al., 2007), and other tracers (e.g., Khatiwala et al., 2013). In the Pacific Ocean, changes in these tracers over time using data from sections very close to each other have been used to examine rates of anthropogenic CO2 uptake (e.g., Azouzi et al., 2009; Murata et al., 2010; Kouketsu et al., 2013; Carter et al., 2017; Wagener et al., 2018). The Millero et al. (1994) sampling across the Eastern South Pacific in 1994 was conducted farther south by about 4° of latitude (Figure 1), while the mean difference in station longitude was about 0.2°, compared to the 2013 cruise track. The sampling intervals were also different with higher vertical resolution of the upper water column undertaken during the US GEOTRACES Peru–Tahiti section.
Here, in the first approach, dissolved oxygen, nitrate, and carbonate chemistry data are compared between both sections (1994 and 2013) by binning data into three depth zones of interest (i.e., 0–100; 100–200; and 200–500 m), and grids of 10° longitude. This provides sufficient data to compare surface (0–100 m, the section “Horizontal Gradients in Upper Ocean Variability of Carbonate Chemistry”) and intermediate waters (200–500 m; the section “Deep Water Variability of Carbonate Chemistry”) across the Peru–Tahiti section (Figure 8).
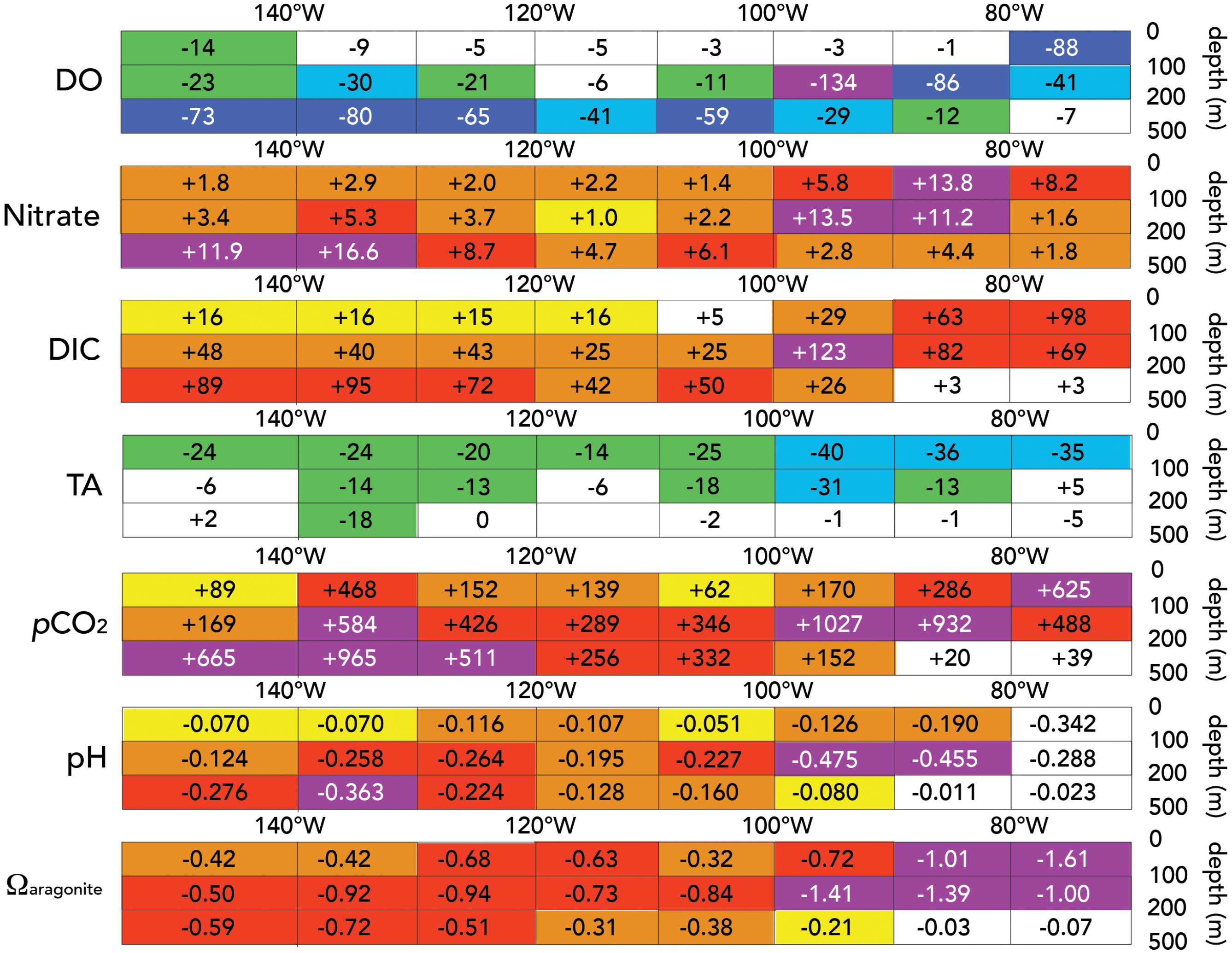
FIGURE 8. Changes in biogeochemical properties and carbonate chemistry over the past 20 years (1994–2013) in the Eastern South Pacific Ocean. The data mean is shown in each of three depth horizons, i.e., (0–100; 100–200; and 200–500 m), and in approximately 10° longitude grids except for the upwelling zone and the larger 140–152°W grid. The units shown for mean change are: (1) dissolved oxygen (μmoles kg-1 20 years-1); (2) Nitrate (μmoles kg-1 20 years-1); (3) DIC (μmoles kg-1 20 years-1); (4) TA (μmoles kg-1 20 years-1); (5) pCO2 (μatm 20 years-1). Please note that pH and Ωaragonite parameters are dimensionless and thus do not have units.
In Figure 8, a comparison of Millero et al. (1994) and GEOTRACES data (this data) are shown for dissolved oxygen, nitrate, DIC, TA, pCO2, pH, and Ωaragonite for the three depth zones of interest. There were small differences in dissolved oxygen content of surface waters (0–100 m) with a greater mean decrease of ∼14 μmoles kg-1 in the westernmost part of the section (Figure 8). However, in surface of the upwelling zone, DO contents were substantially lower by an average ∼88 μmoles kg-1. In transition waters (below 100 m) and the intermediate waters from ESSW to SPCW, very significant differences in dissolved oxygen of ∼40–134 μmoles kg-1 were observed (Figure 8). Nitrate contents also showed significant differences from ∼2.8 to 16.6 μmoles kg-1 higher in 2013 (Figure 8). The largest DO nitrate differences appeared to occur in the core of the ESSW and the SPCW at the westernmost part of the Peru–Tahiti section.
Seawater DIC was also higher in 2013 across the Peru to Tahiti section. In contrast, TA had much smaller changes (Figure 8) with the mean change over the section close to zero. In the surface waters, higher values of ∼16 μmoles kg-1 west of 100°W in 2013 compared to 1994. Such findings were expected given that similar changes would be expected from uptake of anthropogenic CO2 by the ocean (Figure 8; Bates et al., 2014). However, in the intermediate waters, DIC was higher in 2013 compared to 1994 by ∼40–120 μmoles kg-1 across the Peru–Tahiti section (Figure 8C), about three to eight times that expected from uptake of anthropogenic CO2 into the ocean from the atmosphere. The mean pCO2 in intermediate waters appeared higher by ∼200–1,000 μatm (Figure 8E), particularly west of 80°W and in the region of the SPCW (∼120–155°W; Peters et al., 2018). The pH of intermediate waters was also lower in 2013 by 0.10 to as much as 0.45 (Figure 8F), while Ωaragonite was lower by 0.2 to 1.4 (Figure 8).
Such changes in dissolved oxygen, nitrate, and carbonate chemistry appear difficult to reconcile with previous estimate of DO increases in the ODZ (e.g., Stramma et al., 2008). Direct comparison of the 1994 and 2013 sections may not be applicable here due to the latitudinal difference of at least 4° of latitude between the two sections. It seems likely that the 1994 section did not sample the core of the ODZ, whereas the 2013 US GEOTRACES section did.
The second approach is to compare stations from the 2013 US GEOTRACES section with proximal crossover stations sampled between 1991 and 1994 (i.e., Feely et al., 1994; Millero et al., 1994; Takahashi et al., 1995; Rubin et al., 1998; as part of the World Ocean Circulation Experiment, WOCE, datasets). From east to west, these crossover locations approximate the locations of the three intermediate waters, ESSW, ESPIW, and SPCW. A comparison to earlier data collected from 1991 to 1994 suggests that surface DIC and pCO2 have increased by as much as 3 and 20%, respectively, while pH and saturation state for aragonite (Ωaragonite) have decreased by as much as 0.063 and 0.54, respectively (Table 2).
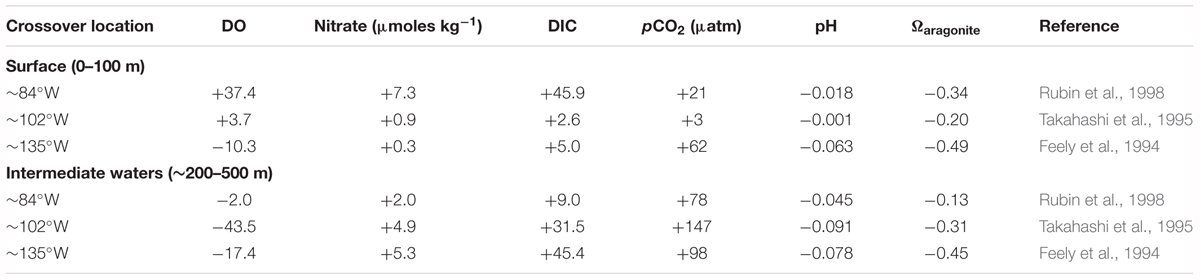
TABLE 2. Carbonate chemistry changes from 1991/1994 to 2013 at select crossover locations in the South Pacific Ocean.
In intermediate waters (∼200–500 m), dissolved oxygen has decreased (loss of up to -43 μmoles kg-1) and nitrate increased (gain of up to 5 μmoles kg-1) over the past 20 years (Table 2). This coincides with previous reports of increased denitrification, increased anoxia, and expansion of Pacific Ocean and other ODZs (e.g., Stramma et al., 2008; Keeling et al., 2010; Stramma et al., 2010; Bopp et al., 2013; Horak et al., 2016). The comparison of GEOTRACES data here with older data appears to confirm a westward expansion of the ODZ across the central Eastern South Pacific Ocean. Such changes have substantial implications for the studies of TEIs, their variability, and scavenging, in the Eastern Pacific Ocean (e.g., Black et al., 2018; Buck et al., 2018; Lam et al., 2018; Moffett and German, 2018). Such changes in the DIC content of intermediate waters (∼200–500 m) suggest the expansion of high-DIC waters westward in conjunction with the expansion of the ODZ from off the Peru coast. Over the same period, DIC and pCO2 increased by as much as +45 μmoles kg-1 and +145 μatm, respectively (Table 2), while pH and Ωaragonite decreased by -0.091 and -0.45, respectively (Table 2). These changes are much greater than the open-ocean surface water pH changes (e.g., 0.017 decade-1) over the past 30 years associated with uptake of anthropogenic CO2 and OA (Bates et al., 2014). The changes in the seawater carbonate chemistry of intermediate waters (∼200–500 m) in the eastern Pacific Ocean suggest the expansion of high-DIC and high CO2, and low pH and low Ωaragonite waters westward in conjunction with the expansion of the ODZ from off the Peru coast.
The Potential Contribution of Anthropogenic CO2 to the Observed Changes in the Eastern South Pacific Ocean
A third approach is to use the TrOCA method (Touratier et al., 2007) to determine the contribution of anthropogenic CO2 to carbonate chemistry as undertaken for the South and tropical Pacific Ocean (e.g., Murata et al., 2007; Azouzi et al., 2009; Kouketsu et al., 2013; Wagener et al., 2018). The TrCOA values of CANT for the 2013 US GEOTRACES Peru–Tahiti are shown in Figure 9. The highest values of CANT range from ∼40 to 70 μmoles kg-1 in the upper 100 to 200 m which is similar to TrOCA-derived CANT values reported by Wagener et al. (2018) in the western South Pacific Ocean. The TrOCA values of CANT values rapidly decrease with depth in the ∼300–600 m zone (i.e., in σ𝜃 range of 26–27; Figure 9A). Below ∼600 m (and especially in deep waters) and in water masses with a density higher than 27.0, CANT values are low (<7 to -3 μmoles kg-1; Figures 9A,B and Table 3). Such CANT values are within the error of the TrOCA method (e.g., Touratier et al., 2007; Wagener et al., 2018) and indicate negligible contributions of anthropogenic CO2 to water mass carbonate chemistry.
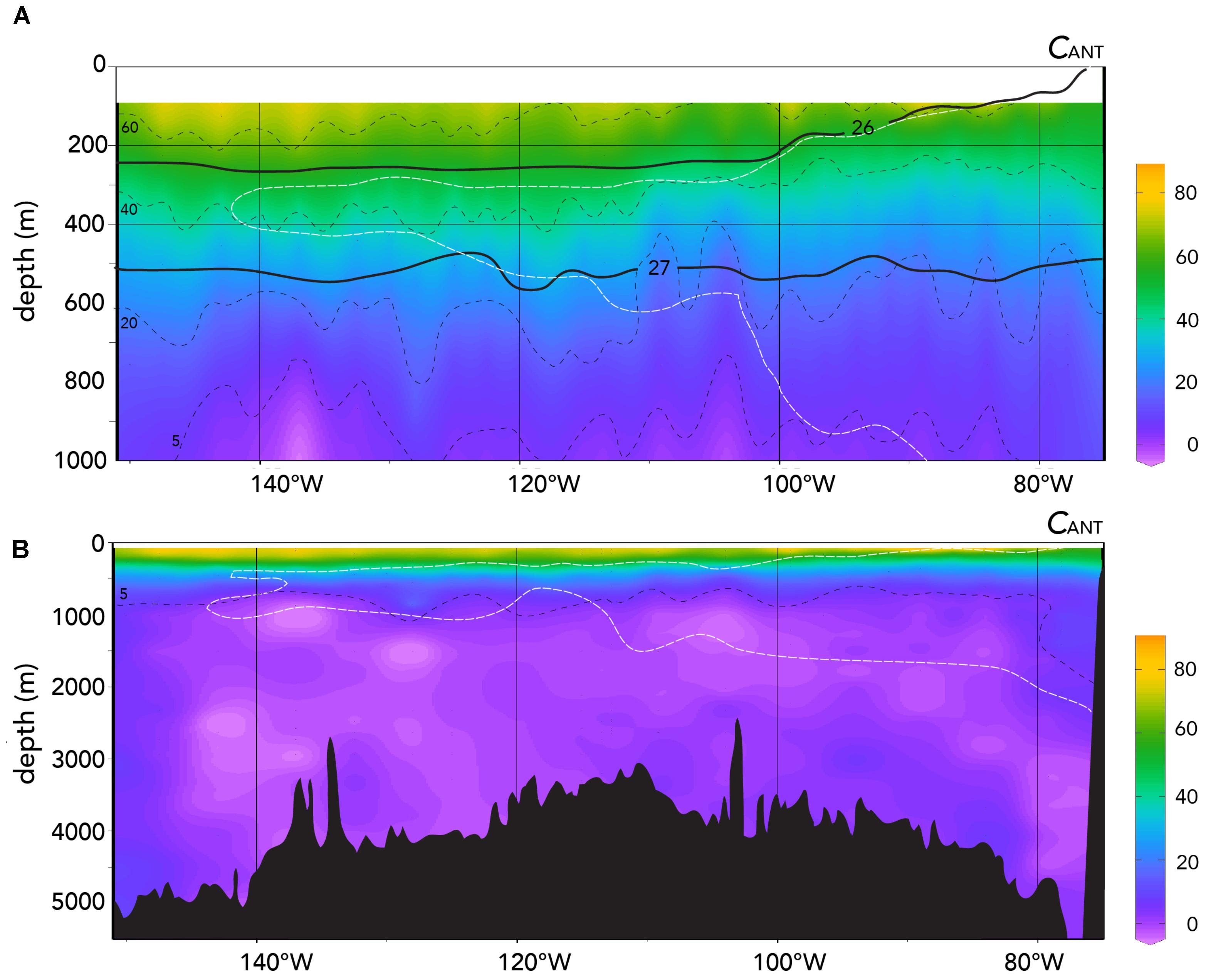
FIGURE 9. Water column CANT values (μmoles kg-1) determined using the TrOCA approach (Touratier et al., 2007) for the entire 2013 Peru–Tahiti section. The black dashed lines refer to approximately contours of CANT μmoles kg-1. The white dashed lines denote the approximate extent of the ODZ. (A) CANT (μmoles kg-1) shown against depth (0–1,000 m). (B) CANT (μmoles kg-1) shown against depth (0 to >5,000 m). Ocean Data View (R. Schlitzer; Ocean Data View; http://odv.awi.de, 2016) was used to generate the base plots and modified with Adobe Illustrator.
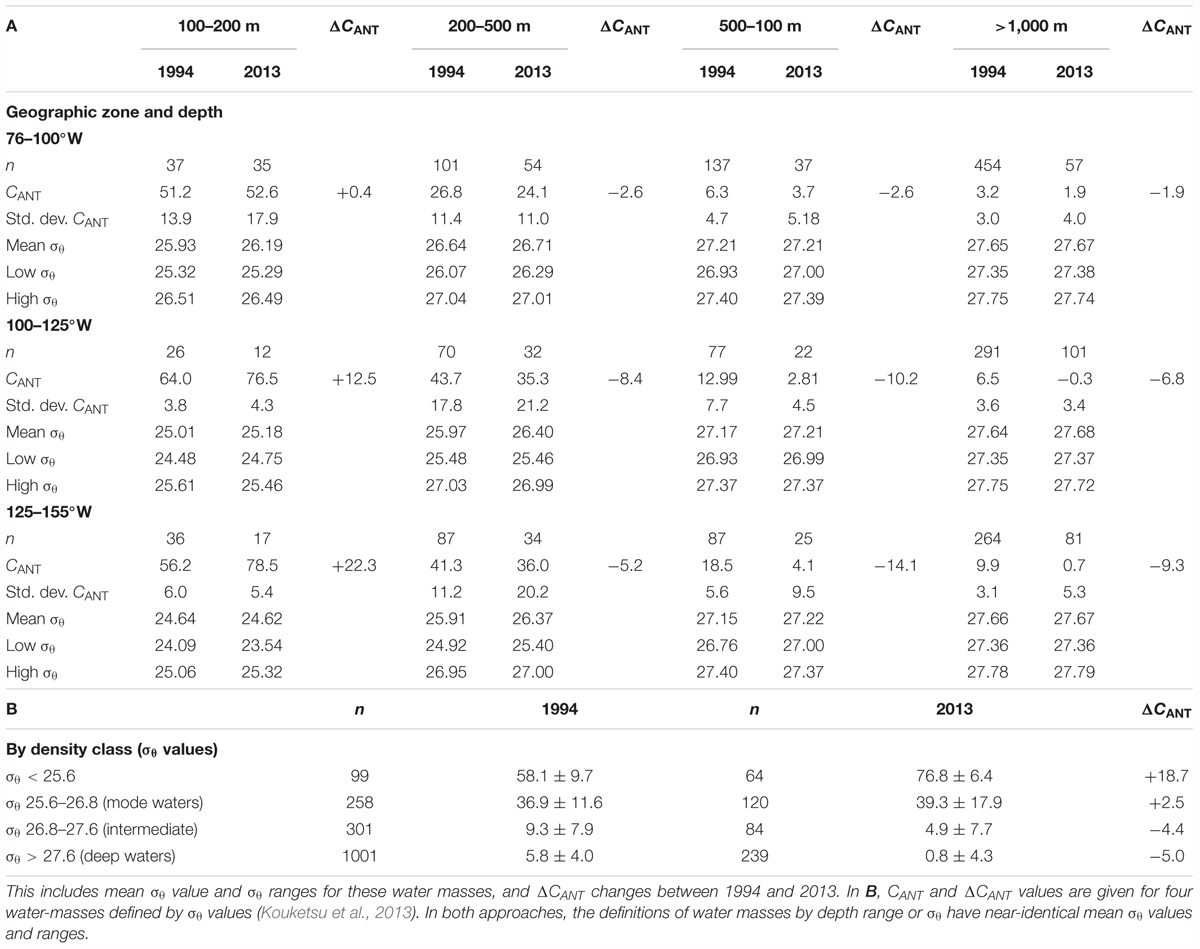
TABLE 3. Mean and standard deviation estimates of CANT values for our depth zones (i.e., 100–200; 200–500; 500–1,000; and >1,000 m depth) for 1994 and 2013 data (A).
A comparison of TrOCA values for CANT for 1994 (Millero et al., 1994) and 2013 US GEOTRACES Peru–Tahiti sections in the eastern South Pacific Ocean is given in Table 3. The values for CANT and its change of the 1994–2013 period (i.e., ΔCANT) were determined for four depth zones (i.e., 100–200; 200–500; 500–1000; >1,000 m) for three geographic regions (i.e., 76–100; 100–125; and 125–125°W), and for density classes previously used and defined as a range of σ𝜃 values by Kouketsu et al. (2013). The classification of CANT and ΔCANT by depth zone given in Table 3 for 1994 and 2013 data also coincides with near-identical density classes and ranges. Although the 1994 and 2013 Peru–Tahiti sections did not overlap geographically (Figure 1), both datasets for the depth ranges chosen, and σ𝜃 definitions had nearly identical mean and range σ𝜃 values (Table 3). The similarity of water mass density provides confidence that near-identical water masses are compared here between 1994 and 2013.
Surface Water (100–200 m) Attribution of Carbonate Chemistry Changes
In the region of upwelling and the Peru current (i.e., 76–100°W), the ΔCANT change between 1994 and 2013 in the 100–200 m depth zone is close to zero (+0.4 μmoles kg-1; Table 3). The near zero change in ΔCANT compares to observed DIC changes of 69–123 μmoles kg-1 over the 1994–2014 period (Figure 8) indicating that anthropogenic CO2 uptake has not influenced DIC and other carbonate chemistry changes, but likely reflects changes in the upwelling and expansion of the ODZ in the region (given the decreases in oxygen and increases in inorganic nutrients; Figure 8).
To the west of 100°W, CANT values are higher in the 75–80 μmoles kg-1 range (Table 3A) with the mean ΔCANT increasing from +12.5 to +22.5 μmoles kg-1 in the 125–155°W zone for the 1994–2013 period. By density class, waters with σ𝜃 values of <25.6 (that approximate the 100–200 m depth) show an increase of ΔCANT of 18.7 μmoles kg-1 (Table 3B). Such increases in anthropogenic CO2 contents of the uppermost waters (below the mixed layer) are about 50% of the observed DIC changes of ∼25–48 μmoles kg-1 (Figure 8). In the 125–155°W, the ΔCANT increase is about 1.1 μmoles kg-1 year-1 increase between 1994 and 2013, and very similar to the upper ocean estimate of 1.1–1.2 μmoles kg-1 year-1 increase due to anthropogenic CO2 determined for the western South Pacific Ocean by Azouzi et al. (2009) and Wagener et al. (2018). Such changes in carbonate chemistry due to the uptake of anthropogenic CO2 are equivalent to decreases of pH and Ωaragonite of -0.0018 year-1 and -0.01 year-1 in the South Pacific Ocean. Such recent changes of pH and Ωaragonite are lower by 20–50% than observed in the central Pacific by Carter et al. (2017) and the western South Pacific Ocean by Wagener et al. (2018), but within the typical range observed at the global ocean time-series sites over the past 30 years (Bates et al., 2014).
Waters examined in the region with σ𝜃 values of <25.6, ranged in thickness from <100 to 200 m at most across the Peru–Tahiti section. The ΔCANT changes observed between 1994 and 2013 are equivalent to a change of approximately +0.1 to +0.2 mol m-2 year-1 due to anthropogenic CO2 uptake. This rate is significantly lower than the rates determined by Kouketsu et al. (2013; +0.4 to +0.6 mol m-2 year-1) and Murata et al. (2010; +0.5 to +1.0 mol m-2 year-1) in other regions of the South Pacific Ocean.
Upper Ocean (200–500 m) Attribution of Carbonate Chemistry Changes
In waters with σ𝜃 values of 25.6–26.8 range (Table 3B), or waters at depths of 200–500 m (Table 3A), the ΔCANT changes observed between 1994 and 2013 are slightly positive (+2.46 μmoles kg-1 for σ𝜃 of 25.6–26.8) to negative (-2.6 to -8.4 μmoles kg-1, 200–500 m). Similar observations are reported by Kouketsu et al. (2013) for mode and intermediate waters of the South Pacific Ocean. In the 125–150°W, the reduction of anthropogenic CO2 in these water masses partly mitigates the substantial increases in DIC of 72–95 μmoles kg-1 (Figure 8) by 10–20%. The substantial changes in carbonate chemistry in waters with σ𝜃 values of 25.6–26.8 or found at 200–500 m depth appear primarily related to the shoaling of the high DIC and pCO2, and low pH and Ωaragonite waters of the ODZ.
The caveat to the above finding of changes in the chemistry of the ODZ is that there remains disagreement about the expansion of global ODZs and timing relative to past contractions of ODZs (e.g., Deutsch et al., 2014; Tems et al., 2014; Yang et al., 2017). If such rapid change in pH and CO2–carbonate chemistry has occurred over the past 20 years in the central Eastern South Pacific (and especially in waters of the ODZ), then there are significant implications for changing the thermodynamics and solubility of intermediate water TEIs in the south Pacific Ocean (e.g., Moffett and German, 2018).
Intermediate and Deep Ocean (>500 m) Attribution of Carbonate Chemistry Changes
In deeper waters, classified by depth range or σ𝜃 the ΔCANT changes are all negative from -1.9 to -14.1 μmoles kg-1 (almost -0.7 μmoles kg-1 year-1; Table 3). The contribution of anthropogenic CO2 to the water column below 500 m also decreases from east to west. Such findings of negative ΔCANT values over the past 20 years suggest that intermediate and deep water may be more influenced by mixing and entrainment of older waters through PDW circulation with negligible anthropogenic CO2 contributions to carbonate chemistry.
Conclusion
The carbonate chemistry observations of the 2013 US GEOTRACES Peru–Tahiti section show a substantial change from 1994 to 2013 in the chemical environment (e.g., DIC, pCO2, pH), of intermediate waters across the Eastern South Pacific associated with the apparent expansion of the ODZ. In the shallow waters masses (∼200–800 m), such as the ESSW in the eastern section of the South Pacific, pCO2 has increased over the past 20 years by as much as 20–40 μatm, with decreases in pH of 0.011–0.023, and Ωaragonite of 0.03. In waters with the greatest contribution of SCPW, ESPIW and to a lesser extent ESSW water masses, the westward expansion of the anoxic zone has substantial changed the chemical environment of waters resident between 200 and 600 m deep. In such waters, pCO2 has increased over the past 20 years by as much as 600 μatm, with pH decreasing by up to 0.36, and Ωaragonite by 0.72. Given such chemical changes in the 200–600 m deep zone of the South Pacific, greater remineralization of sinking organic carbon (and perhaps minor contributions from dissolution of CaCO3) would be expected. Particle production, aggregation, and disaggregation dominate carbon cycling and, interception and transformation of the sinking particle flux in this region (Lam et al., 2018). As such, and in light of the chemical changes in the relatively shallow thermocline, substantial alteration of the rain-ratio of organic carbon and CaCO3 has likely occurred over the past two decades. It appears that the contribution of anthropogenic CO2 to waters deeper than 200 m or σ𝜃 values greater than 25.6 has actually declined of the past 20 years. As such, uptake of anthropogenic CO2 has not contributed to the substantial changes in carbonate chemistry observed in this region of the eastern South Pacific Ocean. The exception to this finding is that uptake of anthropogenic CO2 has contributed ∼50% to carbonate chemistry changes in the uppermost 100–200 m (or waters with σ𝜃 <25.6), and similar to the previous findings in the Pacific Ocean (e.g., Azouzi et al., 2009; Murata et al., 2010; Kouketsu et al., 2013; Carter et al., 2017; Wagener et al., 2018).
An additional conclusion is that, as Millero et al. (2009) and others have suggested, such large decreases in pH and Ωaragonite observed in the ODZ of this region will have influenced the TEI solubilities and kinetics in intermediate waters across the Peru–Tahiti section. It is also expected that the upper ocean and intermediate water marine ecosystem will have been influenced by these pH and Ωaragonite changes, and in particularly, the vertically migrating zooplankton community in the eastern South Pacific Ocean (e.g., Siebel, 2011; Wishner et al., 2013; Maas et al., 2016).
Author Contributions
NB designed the study, analyzed and interpreted the results, produced the figures and tables, and wrote the manuscript.
Funding
The following award from the National Science Foundation (NSF OCE 1233706) to NB provided funding for this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The following people are thanked for their contribution to sampling and analyses of samples as part of this project. Rebecca Garley is thanked for sample analyses at the BIOS. All the crew and scientific participants of the GEOTRACES cruise are thanked for their contributions. NSF OCE 1233706 (NRB) provided funding for this work. The contributors to GLODAP-V2 are thanked for use of data, and in particular, Frank Millero, Taro Takahashi, and Dick Feely and their teams for use of historical inorganic carbon data collected in the South Eastern Pacific Ocean. The contributors to SOCAT are thanked for the use of data in this region. The Surface Ocean CO2 Atlas (SOCAT) is an international effort, endorsed by the International Ocean Carbon Coordination Project (IOCCP), the Surface Ocean Lower Atmosphere Study (SOLAS), and the Integrated Marine Biosphere Research (IMBeR) program, to deliver a uniformly quality-controlled surface ocean CO2 database. The many researchers and funding agencies responsible for the collection of data and quality control are thanked for their contributions to SOCAT.
Footnotes
- ^ https://www.bco-dmo.org/project/499723
- ^ http://www.dickson.ucsd.edu
- ^ https://www.esrl.noaa.gov/gmd/ccgg/flask.php
References
Azouzi, L., Goyet, C., Gonçalves Ito, R., and Touretier, F. (2009). Anthropogenic carbon distribution in the eastern Pacific Ocean. Biogeosciences 6, 149–156.
Bakker, D. C. E., Pfeil, B., Landa, C. S., Metzl, N., O’Brien, K. M., Olsen, A., et al. (2016). A multi–decade record of high quality fCO2 data in version 3 of the Surface Ocean CO2 Atlas (SOCAT). Earth Syst. Sci. Data 8, 383–413. doi: 10.5194/essd-8-383-2016
Bakker, D. C. E., Pfeil, B., Smith, K., Hankin, S., Olsen, A., Alin, S. R., et al. (2014). An update to the surface ocean carbon atlas (SOCAT version 2). Earth Syst.Sci. Data 6, 69–90. doi: 10.5194/essd-6-69-2014
Bates, N. R. (2001). Interannual variability of oceanic CO2 and biogeochemical properties in the Western North Atlantic subtropical gyre. Deep Sea Res. 48, 507–501. doi: 10.1016/S0967-0645(00)00151-X
Bates, N. R., Astor, Y. M., Church, M. J., Currie, K., Dore, J. E., Gonzalez–Davila, M., et al. (2014). Changing ocean chemistry: a time–series view of ocean uptake of anthropogenic CO2 and ocean acidification. Oceanography 27, 121–141. doi: 10.5670/oceanog.2014.16
Bates, N. R., Best, M. H., Neely, K., Garley, R., Dickson, A. G., and Johnson, R. J. (2012). Indicators of anthropogenic carbon dioxide uptake and ocean acidification in the North Atlantic Ocean. Biogeosciences 9, 2509–2522. doi: 10.5194/bg-9-2509-2012
Bates, N. R., Michaels, A. F., and Knap, A. H. (1996a). Alkalinity changes in the Sargasso Sea: geochemical evidence of calcification? Mar. Chem. 51, 347–358. doi: 10.1016/0304-4203(95)00068-2
Bates, N. R., Michaels, A. F., and Knap, A. H. (1996b). Seasonal and interannual variability of oceanic carbon dioxide species at the US JGOFS Bermuda Atlantic Time-series Study (BATS) site. Deep Sea Res. 43, 347–383. doi: 10.1016/0967-0645(95)00093-3
Bates, N. R., and Peters, A. J. (2007). The contribution of atmospheric acid deposition to ocean acidification in the subtropical North Atlantic Ocean. Mar. Chem. 107, 547–558. doi: 10.1016/j.marchem.2007.08.002
Black, E. E., Buesseler, K. O., Pike, S. M., and Lam, P. J. (2018). 234Th as a tracer of particulate export and remineralization in the southeastern tropical Pacific. Mar. Chem. 201, 35–50. doi: 10.1016/j.marchem.2017.06.009
Bopp, L., Resplandy, L., Orr, J., Doney, S., Dunne, J., Gehlen, M., et al. (2013). Multiple stressors of ocean ecosystems in the 21st century: projections with CMIP5 models. Biogeosciences 10, 6225–6245. doi: 10.5194/bg-10-6225-2013
Buck, K., Sedwick, P., Sohst, B., and Carlson, C. (2018). Organic complexation of iron in the eastern tropical South Pacific: results from US GEOTRACES Eastern Pacific Zonal Transect (GEOTRACES cruise GP16). Mar. Chem. 201, 229–241. doi: 10.1016/j.marchem.2017.11.007
Byrne, R. H. (2002). Inorganic speciation of dissolved elements in seawater: the influence of pH on concentration ratios. Geochem. Trans. 3, 11–16. doi: 10.1186/1467-4866-3-11
Byrne, R. H., Kump, L. R., and Cantrell, K. J. (1988). The influence of temperature and pH on trace metal speciation in seawater. Mar. Chem. 25, 163–181. doi: 10.1016/0304-4203(88)90062-X
Caldeira, K., and Wickett, M. F. (2003). Anthropogenic carbon and ocean pH. Nature 425:365. doi: 10.1038/425365a
Carter, B. R., Feely, R. A., Mecking, S., Cross, J. N., MacDonald, A. M., Siedlecki, S. A., et al. (2017). Two decades of Pacific anthropogenic carbon storage and ocean acidification along Global Ocean Ship-based Hydrographic Investigations Program sections P16 and P02. Glob. Biogeochem. Cycles 31, 306–327. doi: 10.1002/2016GB005485
Deutsch, C., Berelson, W., Thunell, R., Weber, T., Tems, C., McManus, J., et al. (2014). Centennial changes in North Pacific anoxia linked to tropical trade winds. Science 345, 665–668. doi: 10.1126/science.1252332
Dickson, A. G. (1990). Thermodynamics of the dissociation of boric acid in synthetic seawater from 273.15 to 318.15 K. Deep Sea Res. 37, 755–766. doi: 10.1016/0198-0149(90)90004-F
Dickson, A. G., and Millero, F. J. (1987). A Comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media. Deep Sea Res. 34(Part A), 733–731. doi: 10.1016/0198-0149(87)90021-5
Dickson, A. G., Sabine, C. L., and Christian, J. R. (2007). Guide to Best Practices for Ocean CO2 Measurements. Sidney: North Pacific Marine Science Organization.
Dore, J. E., Lukas, R., Sadler, D. W., Church, M. J., and Karl, D. M. (2009). Physical and biogeochemical modulation of ocean acidification in the central North Pacific. Proc. Natl. Acad. Sci. U.S.A. 106, 235–212. doi: 10.1073/pnas.0906044106
Feely, R., Bullister, J., and Roberts, M. (1994). Hydrographic, Chemical and Carbon Data Obtained During the R/V Discoverer cruise in the Pacific Ocean during WOCE Section P18 (EXPOCODE 31DSCG94_1,2,3). Washington, DC: US Department of Energy.
Feely, R. A., Sabine, C. L., Hernandez-Ayon, J. M., Ianson, D., and Hales, B. (2008). Evidence for upwelling of corrosive “acidified” water onto the continental shelf. Science 320, 490–491. doi: 10.1126/science.1155676
Feely, R. A., Takahashi, T., Wanninkhof, R., McPhaden, M. J., Cosca, C. E., Sutherland, S. E., et al. (2006). Decadal variability of the air-sea CO2 fluxes in the equatorial Pacific Ocean. J. Geophys. Res. Oceans 111:C08S90. doi: 10.1029/2005JC003129
Feely, R. A., Wanninkhof, R., Cosca, C. E., Murphy, P. P., Lamb, M. F., and Steckley, M. D. (1995). CO2 distributions in the equatorial Pacific during the 1991-1992 ENSO event. Deep Sea Res. 42, 365–386. doi: 10.1016/0967-0645(95)00027-N
Fitzsimmons, J. N., John, S. G., Marsay, C. M., Hoffman, C. L., Nicholas, S. L., Toner, B. M., et al. (2017). Iron persistence in a distal hydrothermal plume supported by dissolved-particulate exchange. Nat. Geosci. 10, 195–201. doi: 10.1038/ngeo2900
GEOTRACES (2006). GEOTRACES: an international study of marine biogeochemical cycles of trace elements and their isotopes: a Science Plan. Sci. Committee Ocean Res. 67, 85–131.
González-Dávila, M., Santana-Casiano, J. M., and González-Dávila, E. F. (2007). Interannual variability of the upper ocean carbon cycle in the northeast Atlantic Ocean. Geophys. Res. Lett. 34:L07608. doi: 10.1029/2006GL028145
González-Dávila, M., Santana-Casiano, J. M., Rueda, M. J., and Llinas, O. (2010). The water column distribution of carbonate system variables at the ESTOC site from 1995 to 2004. Biogeosciences 7, 67–63. doi: 10.5194/bg-7-3067-2010
Gruber, N., Sarmiento, J. L., and Stocker, T. F. (1996). An improved method for detecting anthropogenic CO2 in the oceans. Glob. Biogeochem. Cycles 10, 809–837. doi: 10.1029/96GB01608
Horak, R. E. A., Reuf, W., Ward, B. B., and Devol, A. H. (2016). Expansion of denitrification and anoxia in the easter tropical North Pacific from 1972 to 2012. Geophys. Res. Lett. 43, 5252–5260. doi: 10.1002/2016GLO68871
Jenkins, W. J., Lott, D. E., German, C. R., Cahill, K. L., Goudreau, J., and Longworth, B. (2018). The deep distributions of helium isotopes, radiocarbon, and noble gases along the U.S. GEOTRACES East Pacific Zonal Transect (GP16). Mar. Chem. 201, 167–182. doi: 10.1016/j.marchem.2017.03.009
Keeling, R., Körtzinger, A., and Gruber, N. (2010). Ocean deoxygenation in a warming world. Ann. Rev. Mar. Sci. 2, 199–229. doi: 10.1146/annurev.marine.010908.163855
Keller, K. M., Joos, F., and Raible, C. C. (2014). Time of emergence of trends in ocean biogeochemistry. Biogeosciences 11, 3647–3659. doi: 10.5194/bg-11-3647-2014
Key, R. M., Olsen, A., van Heuven, S., Lauvset, S. K., Velo, A., Lin, X., et al. (2015). Global Ocean Data Analysis Project, Version 2 (GLODAPv2), ORNL/CDIAC-162, ND-P093. Oak Ridge, TN: Oak Ridge National Laboratory. doi: 10.3334/CDIAC/OTG.NDP093_GLODAPv2
Khatiwala, S., Tanhua, T., Mikaloff Fletcher, S., Gerber, M., Doney, S. C., Graven, H. D., et al. (2013). Global ocean storage of anthropogenic carbon. Biogeosciences 10, 2169–2191. doi: 10.5194/bg-10-2169-2013
Kouketsu, S., Murata, A., and Doi, T. (2013). Decadal changes in dissolved inorganic carbon in the Pacific Ocean. Glob. Biogeochem. Cycles 27, 65–76.
Lam, P. J., Lee, M.-J., Heller, M. I., Mehic, S., Xiang, Y., and Bates, N. R. (2018). Size-fractionated distributions of suspended particle concentration and major phase composition from the U.S. GEOTRACES Eastern Pacific Zonal Transect (GP16). Mar. Chem. 201, 90–107. doi: 10.1016/j.marchem.2017.08.013
Lupton, J. E., and Jenkins, W. J. (2017). Evolution of the south Pacific helium plume over the past three decades. Geochem. Geophys. Geosyst. 18, 1810–1823. doi: 10.1002/2017GC006848
Maas, A. E., Lawson, G. L., and Wang, A. L. (2016). The metabolic response of thecosome pteropods from the North Atlantic and North Pacific oceans to high CO2 and low O2. Biogeosciences 13, 6191–6210. doi: 10.5194/bg-13-6191-2016
Mehrbach, C., Culberson, C. H., Hawley, J. E., and Pytkowicz, R. M. (1973). Measurement of the apparent dissociation constants of carbonic acid in seawater at atmospheric pressure. Limnol. Oceanogr. 18, 897–907. doi: 10.4319/lo.1973.18.6.0897
Millero, F., Winn, C., and Goyet, C. (1994). Hydrographic, Chemical and Carbon Data Obtained During the R/V Melville cruise in the Pacific Ocean during WOCE Section P21EW (EXPOCODE 318MWESTW_4_5). Oak Ridge, TN: Oak Ridge National Laboratory. doi: 10.3334/CDIAC/otg.318MWESTW_4_5
Millero, F. J. (2001b). Speciation of metals in natural water. Geochem. Trans. 2, 56–64. doi: 10.1186/1467-4866-2-57
Millero, F. J., Woosley, R., DiTrolio, B., and Waters, J. (2009). Effect of ocean acidification on the speciation of metals in seawater. Oceanography 22, 72–85. doi: 10.5670/oceanog.2009.98
Millero, F. J., Yao, W., and Aicher, J. (1995). The speciation of Fe(II) and Fe(III) in natural waters. Mar. Chem. 50, 21–39. doi: 10.1016/0304-4203(95)00024-L
Moffett, J. W., and German, C. R. (2018). The U.S. GEOTRACES eastern tropical Pacific transect. Mar. Chem. 201, 1–5. doi: 10.1016/j.marchem.2017.12.001
Morel, F. M. M., Milligan, A. J., and Saito, M. A. (2003). Marine bioinorganic chemistry: The role of trace metals in the oceanic cycles of major nutrients. Treatise Geochem. 6, 113–143. doi: 10.1016/B0-08-043751-6/06108-9
Murata, A., Kumamoto, Y., Sasaki, K., Watanabe, S., and Fukasawa, M. (2010). Decadal increases in anthropogenic CO2 along 20°S in the South Indian Ocean. J. Geophys. Res. 115:C12. doi: 10.1029/2010JC006250
Olafsson, J., Olafsdottir, S. R., Benoit-Cattin, A., Danielsen, M., Arnarson, T. S., and Takahashi, T. (2009). Rate of Iceland Sea acidification from time series measurements. Biogeosciences 6, 661–662. doi: 10.5194/bg-6-2661-2009
Olafsson, J., Olafsdottir, S. R., Benoit-Cattin, A., and Takahashi, T. (2010). The Irminger Sea and the Iceland Sea time series measurements of sea water carbon and nutrient chemistry 1983–2006. Earth Syst. Sci. Data 2, 99–104. doi: 10.5194/essd-2-99-2010
Olsen, A., Key, R. M., van Heuven, S., Lauvset, S. K., Velo, A., Lin, X., et al. (2016). The Global Ocean Data Analysis Project version 2 (GLODAPv2) - an internally consistent data product for the world ocean. Earth Syst. Sci. Data 8, 297–323. doi: 10.5194/essd-8-297-2016
Orr, J. C., Fabry, V. J., Aumont, O., Bopp, L., Doney, S. C., Feely, R. A., et al. (2005). Anthropogenic ocean acidification over the twenty-first century and its impacts on calcifying organisms. Nature 437, 681–686. doi: 10.1038/nature04095
Pavia, F., Anderson, R., Vivancos, S., Fleisher, M., Lam, P., Lu, Y., et al. (2018). Intense hydrothermal scavenging of 230Th and 231Pa in the deep Southeast Pacific. Mar. Chem. 201, 212–228. doi: 10.1016/j.marchem.2017.08.003
Peters, B. D., Casciotti, K. I., Jenkins, W. J., Swift, J. H., German, C. R., James, W., et al. (2018). Water mass analysis of the 2013 US GEOTRACES Eastern Pacific Zonal Transect (GP16). Mar. Chem. 201, 6–19. doi: 10.1016/j.marchem.2017.09.007
Quay, P. D., Sutsman, J., Feely, R. A., and Juranek, L. W. (2011). Net community production rates across the subtropical and equatorial Pacific Ocean estimated from air-sea δ13C disequilibrium. Glob. Biogeochem. Cycles 23:GB2006. doi: 10.1029/2008GB003193
Resing, J. A., Sedwick, P. N., German, C. R., Jenkins, W. J., Moffett, J. W., Sohst, B., et al. (2015). Basin-scale transport of hydrothermal dissolved metals across the South Pacific Ocean. Nature 523, 200–203. doi: 10.1038/nature14577
Robbins, L. L., Hansen, M. E., Kleypas, J. A., and Meylan, S. C. (2010). CO2calc: a User-Friendly Seawater Carbon Calculator for Windows, Max OS X, and iOS (iPhone): U.S. Geological Survey Open-File Report, 2010–1280. Available at: http:pubs.usgs.gov/of/2010/1280/
Rubin, S., Goddard, J. G., Chipman, D. W., Takahashi, T., Sutherland, S. C., Reid, J. L., et al. (1998). Carbon Dioxide, Hydrographic, and Chemical Data Obtained in the South Pacific Ocean (WOCE Sections P16A/P17A, P17E/P19S, and P19C, R/V Knorr, October 1992-April 1993. ORNL/CDIAC-109, NDP-065. Oak Ridge, TN: Oak Ridge National Laboratory. doi: 10.3334/CDIAC/otg.ndp065
Sabine, C. L., Feely, R. A., Gruber, N., Key, R. M., Lee, K., Bullister, J. L., et al. (2004). The oceanic sink for anthropogenic CO2. Science 305, 367–371. doi: 10.1126/science.1097403
Santer, B. D., Mears, C., Doutriaux, C., Caldwell, P., Gleckler, P. J., Wigley, T. M. L., et al. (2011). Separating signal and noise in atmospheric temperature changes: the importance of timescales. J. Geophys. Res. Atmos. 116:D22105. doi: 10.1029/2011JD016263
Schlitzer, R., Anderson, R. F., Masferrer Dodas, E., Lohan, M., Geibert, W., Tagliabue, A., et al. (2018). The GEOTRACES Intermediate data product 2017. Chem. Geol. 493, 210–223. doi: 10.1016/j.chemgeo.2018.05.040
Shi, D. L., Xu, Y., Hopkinson, B. M., and Morel, F. M. M. (2010). Effect of ocean acidification on iron availability to marine phytoplankton. Science 327, 676–679. doi: 10.1126/science.1183517
Siebel, B. A. (2011). Critical oxygen levels and metabolic suppression in oceanic oxygen minimum zones. J. Exp. Biol. 214, 326–336. doi: 10.1242/jeb.049171
Stramma, L., Johnson, G. C., Firing, E., and Schmidtko, S. (2010). Eastern Pacific oxygen minimum zones: supply paths and multidecadal changes. J. Geophys. Res. 115:C09011. doi: 10.1029/2009JC005976
Stramma, L., Johnson, G. C., Sprintall, J., and Mohrholz, V. (2008). Expanding oxygen-minimum zones in the tropical oceans. Science 320, 655–658. doi: 10.1126/science.1153847
Sunda, W. G., and Ferguson, R. L. (1983). “Sensitivity of natural bacterial communities to additions of copper and to cupric ion activity: a bioassay of copper complexation in seawater,” in Trace Metals in Seawater, eds C. S. Wong, E. Boyle, K. W. Bruland, J. D. Burton, and E. D. Goldberg (New York, NY: Plenum), 871–891.
Takahashi, T., Goddard, J. G., Rubin, S., Chipman, D. W., Sutherland, S. C., and Goyet, C. (1995). Carbon Dioxide, Hydrographic, and Chemical Data Obtained in the Central South Pacific Ocean (WOCE Sections P17S and P16S) During the TUNES-2 Expedition of the R/V Thomas Washington, July-August, 1991. Oak Ridge, TN: Oak Ridge National Laboratory. doi: 10.3334/CDIAC/otg.ndp054
Takahashi, T., Sutherland, S. C., Chipman, D. W., Goddard, J. G., Ho, C., Newberger, T., et al. (2014). Climatological distributions of pH, pCO2, total CO2, alkalinity, and CaCO3 Saturation in the global surface ocean, and temporal changes at selected locations. Mar. Chem. 164, 95–124. doi: 10.1016/j.jmarchem.2014.06.004
Takahashi, T., Sutherland, S. C., Wanninkhof, R., Sweeney, C., Feely, R. A., Chipman, D. W., et al. (2009). Climatological mean and decadal change in surface ocean pCO2, and net sea–air CO2 flux over the global oceans. Deep Sea Res. 56, 554–577. doi: 10.1016/j.dsr2.2008.12.009
Tems, C., Berelson, W., and Prokopenko, M. (2014). δ15N in laminated marine sediments provide a proxy for mixing between the California Undercurrent and the California Current: a proof of concept. Geophys. Res. Lett. 42, 419–427. doi: 10.1002/2014GL061993
Tomczak, M. (1981). A multi-parameter extension of temperature/salinity diagram techniques for the analysis of non-isopycnal mixing. Progr. Oceanogr. 10, 147–171. doi: 10.1016/0079-6611(81)90010-0
Touratier, F., Azouzi, L., and Goyet, C. (2007). CFC-11, Δ14C and 3He tracers as a means to assess anthropogenic CO2 concentrations in the ocean. Tellus 59B, 318–325. doi: 10.1111/j.1600-0889.2006.00247.x
Touratier, F., and Goyet, C. (2004a). Definition, properties, and Atlantic distribution of the new tracer TrOCA. J. Mar. Syst. 46, 169–179. doi: 10.1016/j.jmarsys.2003.11.016
Touratier, F., and Goyet, C. (2004b). Applying the new TrOCA approach to assess the distribution of anthropogenic CO2 in the Atlantic Ocean. J. Mar. Syst. 46, 181–197. doi: 10.1016/j.jmarsys.2003.11.020
Turner, D. R., Whitfield, M., and Dickson, A. G. (1981). The equilibrium speciation of dissolved components of freshwater and seawater at 25°C and 1 atm pressure. Geochim. Cosmochim. Acta 44, 855–881. doi: 10.1016/0016-7037(81)90115-0
Uchida, H., Murata, A., and Doi, T. (2011). Carbon Dioxide, Hydrographic, and Chemical Data Obtained During the R/V Mirai Repeat Hydrography Cruise in the Pacific Ocean: CLIVAR CO2 Section P21_2009 (10 April - 3 July, 2009). Oak Ridge, TN: Oak Ridge National Laboratory.
Wagener, T., Metzl, N., Caffin, M., Fin, J., Helias Nunige, S., Lo Monaco, C., et al. (2018). Carbonate system distribution, anthropogenic carbon, and acidification in the western tropical South Pacific (OUTPACE 2015 section). Biogeosciences 15, 5221–5236. doi: 10.5194/bg-15-5221-2018
Wishner, K. F., Outram, D. M., Seibel, B. A., Daly, K. L., and Williams, R. L. (2013). Zooplankton in the eastern tropical north Pacific: Boundary effects of oxygen minimum zone expansion. Deep Sea Res. 79, 122–140. doi: 10.1016/j.dsr.2013.05.012
Yang, S., Gruber, N., Long, M. C., and Vogt, M. (2017). ENSO-driven variability of denitrification and suboxia in the Eastern Tropical Pacific Ocean. Glob. Biogeochem. Cycles 31, 1470–1487. doi: 10.1002/2016GB005596
Keywords: seawater carbonate chemistry, marine biogeochemistry, GEOTRACES, Pacific Ocean, oxygen deficient zone, anthropogenic carbon
Citation: Bates NR (2018) Seawater Carbonate Chemistry Distributions Across the Eastern South Pacific Ocean Sampled as Part of the GEOTRACES Project and Changes in Marine Carbonate Chemistry Over the Past 20 Years. Front. Mar. Sci. 5:398. doi: 10.3389/fmars.2018.00398
Received: 21 August 2018; Accepted: 09 October 2018;
Published: 07 November 2018.
Edited by:
Javier Arístegui, Universidad de Las Palmas de Gran Canaria, SpainReviewed by:
Wei-dong Zhai, Shandong University, ChinaNicolas Metzl, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2018 Bates. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas Robert Bates, nick.bates@bios.edu