 Till Röthig†
Till Röthig† Rúben M. Costa†
Rúben M. Costa† Fabia Simona
Fabia Simona Sebastian Baumgarten
Sebastian Baumgarten Ana F. Torres
Ana F. Torres Anand Radhakrishnan
Anand Radhakrishnan Manuel Aranda
Manuel Aranda Christian R. Voolstra*
Christian R. Voolstra*- Division of Biological and Environmental Science and Engineering (BESE), Red Sea Research Center, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
Coral reefs are in decline. The basic functional unit of coral reefs is the coral metaorganism or holobiont consisting of the cnidarian host animal, symbiotic algae of the genus Symbiodinium, and a specific consortium of bacteria (among others), but research is slow due to the difficulty of working with corals. Aiptasia has proven to be a tractable model system to elucidate the intricacies of cnidarian-dinoflagellate symbioses, but characterization of the associated bacterial microbiome is required to provide a complete and integrated understanding of holobiont function. In this work, we characterize and analyze the microbiome of aposymbiotic and symbiotic Aiptasia and show that bacterial associates are distinct in both conditions. We further show that key microbial associates can be cultured without their cnidarian host. Our results suggest that bacteria play an important role in the symbiosis of Aiptasia with Symbiodinium, a finding that underlines the power of the Aiptasia model system where cnidarian hosts can be analyzed in aposymbiotic and symbiotic states. The characterization of the native microbiome and the ability to retrieve culturable isolates contributes to the resources available for the Aiptasia model system. This provides an opportunity to comparatively analyze cnidarian metaorganisms as collective functional holobionts and as separated member species. We hope that this will accelerate research into understanding the intricacies of coral biology, which is urgently needed to develop strategies to mitigate the effects of environmental change.
Introduction
Coral reefs are biodiversity hotspots of enormous ecological and economic importance. In these ecosystems, corals are the foundation species that build the calcium carbonate skeletons that give rise to the massive three-dimensional reef structures providing a habitat for millions of species (Reaka-Kudla et al., 1996) and economic activity worth around US$ 5.7 billion each year for Australia's Great Barrier Reef alone (Hoegh-Guldberg, 2015). However, reef ecosystems are under threat due to a combination of local (e.g., overfishing, pollution) and global (e.g., ocean warming and acidification) factors (Hughes et al., 2003). While unusually high sea surface temperatures cause coral bleaching (i.e., the disruption of the coral-algal symbiosis resulting in algal expulsion and tissue whitening), pollution may cause coral disease and facilitate bleaching susceptibility from high nutrient loads or other toxic substances (Negri et al., 2011; Vega Thurber et al., 2014). In the Caribbean, 80% of coral cover has been lost over the last decades (Gardner et al., 2003). Despite a reasonably good understanding of the environmental conditions that are harmful to corals, we are still missing knowledge on the cellular and molecular basis of coral bleaching and disease, and the contributions of microbes to stress resilience (Mouchka et al., 2010; Bourne et al., 2016), information that is critical to conceive strategies for mitigating future reef loss.
The basic functional unit of stony corals is the coral holobiont, consisting of the cnidarian-animal host, its intracellular dinoflagellate algae of the genus Symbiodinium, and a specific consortium of associated microbes, including bacteria, archaea, fungi, and viruses (among other organisms) (Rohwer et al., 2002). While the dependency on a functional symbiosis between the animal host and its photosynthetic algae has long been acknowledged (Trench, 1993), the importance of bacterial microbes has only recently been elucidated in more detail (Rosenberg et al., 2007; Raina et al., 2009; Ritchie, 2011; Jessen et al., 2013; Rädecker et al., 2015; Röthig et al., 2016; Ziegler et al., 2016). Sparked by the development of new genomic tools (e.g., next-generation sequencing), recent years have brought a changing understanding in life sciences (Mcfall-Ngai et al., 2013). The common notion is that all animals and plants are metaorganisms that critically depend on living together with a highly diverse and specific group of microbes that provide functions related to metabolism, immunity, and environmental adaptation, among others (Mcfall-Ngai et al., 2013). These metaorganisms or holobionts cannot be understood in isolation, but must be studied as a consortium of organisms, i.e., as hosts and associated microbes. Consequently, interactions and communication mechanisms among holobiont members presumably play a major role in maintaining host health and microbiome stability.
One of the reasons why progress is slow on gaining a better insight into the molecular mechanisms governing holobiont function is due to the difficulties of working with corals. For instance, corals are difficult to grow in culture, have long generation times, and are difficult to be kept without their associated algal symbionts, prohibiting the study of a non-symbiotic “control” or “reference” state (Voolstra, 2013). To this end, the sea anemone Aiptasia has emerged as a tractable laboratory model to study coral symbiosis (Weis et al., 2008). A key aspect is Aiptasia's ease of culturing and flexibility in its symbioses (e.g., Aiptasia can host the same algal symbionts as corals), allowing the comparative analysis of symbiotic and non-symbiotic states side-by-side in a laboratory context (Voolstra, 2013). In this regard, the recent assembly and analysis of the Aiptasia genome provides a foundation for its role as a model for coral biology (Baumgarten et al., 2015), but characterization of the associated bacterial microbial community is missing.
In order to further contribute to the establishment of Aiptasia as a model system for coral symbiosis and to contribute to the characterization of the entire Aiptasia holobiont, we set out to analyze the bacterial community associated with Aiptasia. To do this, we compared bacterial communities from Aiptasia strain CC7 that are aposymbiotic and symbiotic with the Symbiodinium strain SSB01 (species S. minutum) (Xiang et al., 2013b; Baumgarten et al., 2015) to investigate how microbial assemblages may change with symbiotic state. Last, we report on the generation of culturable isolates from bacterial taxa of the microbial community providing the opportunity to study host-microbe interactions in detail.
Materials and Methods
Animal Rearing
Aposymbiotic and symbiotic Aiptasia of the clonal strain CC7 were generated and reared as described previously (Baumgarten et al., 2015). Briefly, aposymbiotic animals were obtained through repetitive cold-shock by addition of 4°C cold autoclaved freshly collected seawater (AFSW) from the Red Sea and subsequent incubation at 4°C for 4 h. Anemones were then treated for 1–2 days with 50 μM of the photosynthesis inhibitor diuron (Sigma-Aldrich, St. Louis, MO, USA) at 25°C in AFSW. Aposymbiotic Aiptasia were raised in 1 liter AFSW-tanks at 25°C in the dark for more than 1 year, fed Artemia twice weekly, and supplied with AFSW the day after feeding. Symbiotic Aiptasia were generated by infecting aposymbiotic animals with the clade B Symbiodinium strain SSB01 (Xiang et al., 2013a) at a final concentration of 104 algal cells mL−1. Following infection, symbiotic animals were transferred to a 12 h light: 12 h dark incubator (20–40 μmol photons m−2 s−1 of photosynthetically active radiation) at 25°C and fed Artemia twice weekly. Two weeks prior to the start of the experiment, aposymbiotic and symbiotic Aiptasia were cultured in 6 multiwell cell culture plates (3–5 organisms per well in 6 mL AFSW), kept on a 12 h light: 12 h dark cycle at 25°C, and repeatedly tested for Symbiodinium re-infection by fluorescent microscopy (Leica DMI3000 B). Additionally, aposymbiotic Aiptasia were regularly tested for the presence of Symbiodinium via PCRs with Symbiodinium-specific primers. Five days prior to experiments food supply was ceased to avoid Artemia contamination.
Bacterial Microbiome - DNA Isolation and 16S rRNA Gene Sequencing
For bacterial DNA isolation from anemones, five aposymbiotic and five symbiotic Aiptasia polyps of ~0.8 cm length were collected from the respective multiwell plates with a Pasteur pipette and transferred into 1.5 mL microtubes, washed thrice with AFSW, and remaining water was carefully removed. All 10 microtubes holding the polyps were transferred to −20°C. Aiptasia samples were crushed while thawing using a 10 μL pipette tip, and subsequently 400 μL AP1 buffer (DNeasy Plant Mini Kit, Qiagen) were added. DNA extraction was performed according to the manufacturer's instructions. For bacterial DNA isolation from water, 300 mL water were collected from each AFSW-container in which symbiotic and aposymbiotic anemones were reared. The collected water was firstly filtered through a 40 μm cell strainer (BD, Franklin Lakes, NJ, USA) to remove debris, and then through a 0.22 μm Durapore PVDF filter (Millipore, Billerica, MA, USA). Filters were frozen at −20°C, thawed, cut in strips using a sterile razorblade, and transferred into 2 mL microtubes. 400 μL AP1 buffer were added (DNeasy Plant Mini Kit, Qiagen, Hilden, Germany) and the microtubes were incubated on a rotating wheel for 20 min. Further procedure followed the manufacturer's instructions (DNeasy Plant Mini Kit, Qiagen). DNA concentrations of samples were quantified on a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). To generate 16S rRNA gene amplicons for sequencing, we targeted the variable regions 5 and 6 of the 16S rRNA gene using the primer pair 784F [5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-AGGATTAGATACCCTGGTA 3′] and 1061R [5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-CRRCACGAGCTGACGAC 3′] (Andersson et al., 2008) with Illumina (San Diego, CA, USA) adaptor overhangs (underlined above). For each sample, PCRs were performed in triplicate using the Qiagen Multiplex PCR kit, between 10 and 80 ng template DNA, a primer concentration of 0.5 μM, and a final reaction volume of 25 μL. PCRs were performed as follows: One cycle at 95°C for 15 min, 27 cycles each at 95°C for 30 s, 55°C for 90 s, and 72°C for 30 s, followed by a final extension step at 72°C for 10 min. Triplicate PCRs for each sample were pooled and cleaned with the Agencourt AMPure XP magnetic bead system (Beckman Coulter, Brea, CA, USA), and subsequently underwent an indexing PCR to add Nextera XT barcoded sequencing adapters (Illumina) according to the manufacturer's instructions. Indexed PCR products were cleaned using the Invitrogen SequalPrep normalization plate kit (Thermo Fisher Scientific, Carlsbad, CA, USA) following the manufacturer's instructions and eluted at normalized concentrations (~4 nM) in 20 μl elution buffer and pooled in equimolar ratios. Pooled samples were quality checked on the BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA) for presence of primer dimers. The library was sequenced at 8 pM with 10% phiX on the Illumina MiSeq, 2*300 bp paired-end version 3 chemistry according to the manufacturer's specifications.
Bacterial Microbiome—Analysis
The sequence data set comprised 2.48 million sequence reads. Reads were demultiplexed and adapters and barcodes were removed in MiSeq Reporter (v. 2.4.60.8). Data were imported into mothur version 1.36.1 (Schloss et al., 2009) and 1,239,574 contigs were assembled using the “make.contigs” command. Contigs were quality trimmed, i.e., sequences with ambiguous nucleotides, sequences with excessively long homopolymers (>5), and sequences of insufficient length were removed. Additionally 432,543 singletons were removed. Remaining sequences were aligned against SILVA release 119 (Pruesse et al., 2007), preclustered (2 bp difference) (Huse et al., 2010), and chimeric sequences were removed using UCHIME (Edgar et al., 2011). Sequences were classified against the Greengenes database (release gg_13_8_99) with a minimum bootstrap of 60 (Mcdonald et al., 2012), and unwanted sequences (i.e., unknown, eukaryota, archaea, mitochondria, and chloroplasts) were removed. From the remaining 575,354 sequences alpha diversity indices for bacterial communities were calculated in mothur, and the composition of samples was compared on the family level by creating stack column plots in R (R Core Team, 2014). For taxon-based analysis, samples were subsampled to 11,000 sequences and clustered into Operational Taxonomic Units (OTUs) using a 97% similarity cutoff. Rarefaction curves, non-metric multidimensional scaling (nMDS), and analysis of molecular variance (AMOVA) (Excoffier et al., 1992) were conducted as implemented in the software mothur. Differences between alpha diversity indices of samples were assessed after testing for normality and homoscedasticity (Shapiro-Wilk and Levene's test performed in R) using one-way ANOVAs (STATISTICA 10, StatSoft Inc.). nMDS results were plotted in SigmaPlot 11 (Systat Software, Point Richmond, CA, USA). The commands make.shared, classify.OTU, and get.OTUrep were used to create a list of all OTUs and their distribution across samples. Based on these data, we obtained a putative “core microbiome” (i.e., all OTUs present in 100% of all Aiptasia polyps), an aposymbiotic microbiome or “apobiome” (i.e., all OTUs present in 100% of all aposymbiotic polyps), and a symbiotic microbiome or “symbiome” (i.e., all OTUs present in 100% of all symbiotic polyps). Of note, the respective OTUs may be members of multiple “biomes” and can be present in the water samples. To identify previous occurrences of identical or highly similar bacteria, the representative sequence of each OTU occurring in at least one “biome” was BLASTed against NCBI's GenBank nr and the three best matches were considered (e-value cutoff e−20). Putative functions encoded in the microbial communities of anemones were based on phylogenetic inference and assessed using METAGENassist for automated taxonomic-to-phenotypic mapping (Arndt et al., 2012). We created input files in mothur using the make.shared and classify.OTU commands. During data processing, OTUs present in anemones were assigned, mapped, and condensed into 236 functional taxa in METAGENassist. Data were further filtered based on interquartile range (Hackstadt and Hess, 2009), and the remaining 225 functional taxa were normalized across samples by sum and over taxa by Pareto scaling. We analyzed the dataset for “metabolism by phenotype” using the Spearman distance measure to cluster the 15 most differentially abundant metabolic processes.
Generation of Bacterial Cultivates
Reared and starved aposymbiotic and symbiotic anemones (see above) were collected in 1.5 mL microtubes with 500 μL of sterile seawater, crushed using a pestle, and subsequently spread out on either M1 (MO) Agar (10 g Starch, 4 g yeast extract, 2 g peptone, 18 g agar, 1 L sterile seawater) or Marine (MA) Agar (55.1 g Difco™ Marine Agar 2216 in 1 L sterile seawater) plates and incubated at 28°C for up to 24 h. To determine the identity of cultured isolates, bacterial colonies were picked from the agar plates into 96 well plates using sterile 10 μL pipette tips. Each well contained 10 μl PCR mix (5 μl Qiagen Multiplex PCR kit, 1 μM of 27F and 1492R primers, adjusted to the final volume with dH2O). The PCR conditions were set as follows: 95°C for 15 min, followed by 35 cycles of each: 30 s at 95°C, 90 s at 55°C, and 90 s at 72°C. A final extension step was set at 72°C for 10 min. PCR reactions were cleaned using Illustra ExoStar 1-Step (GE Healthcare, Little Chalfont, UK) according to manufacturer's instructions. Sanger sequencing for 16S rRNA gene products was performed by the Bioscience Core Lab (BCL) at KAUST using the primer 1492R to yield a 16S rRNA gene partial sequence that aligns with the MiSeq amplicon (see above). Sequencing analysis was conducted using CodonCode Aligner (v.3.7.1.1). Briefly, *.ab1 files were imported and sequence ends were clipped using default quality parameters. To obtain matches between cultured isolates and OTUs, a BLAST database (Altschul et al., 1990) was created from all OTU sequences, and only hits with 100% similarity were considered.
Results
Bacterial Community of Aiptasia and Rearing Water
We produced 12 16S rRNA gene libraries containing a total of 1,239,574 sequences from 5 aposymbiotic and 5 symbiotic Aiptasia animals and 2 water samples (from both rearing conditions, i.e., 1 aposymbiotic and 1 symbiotic). After quality trimming and removal of singletons and unwanted sequences, 575,354 sequences with an average length of 292 bp were available for subsequent analyses. Classification of sequences on the family level revealed noticeable differences between the microbial community associated with aposymbiotic and symbiotic anemones (Figure 1). On average, aposymbiotic Aiptasia were overall dominated by Alteromonadaceae (between 29% and 52%, mean 47%), Rhodobacteraceae (between 6% and 15%, mean 11%), and Oceanospirillaceae (between 1% and 22%, mean 12%). In contrast, microbial communities from symbiotic anemones showed an increased amount of Pseudomonadaceae (between 17% and 24%, mean 20%) and Dermabacteraceae (between 10% and 15%, mean 12%), but contained noticeably less Alteromonadaceae (between 16% and 23%, mean 19%). By comparison, water samples were markedly different from all Aiptasia samples and also different from each other. On average, water samples were more diverse, i.e., more bacterial families with a more even abundance were present (e.g., Alteromonadaceae, Rhodobacteraceae, and unclassified families of the order Flavobacteriales and the class Gammaproteobacteria made up >50% of sequences).
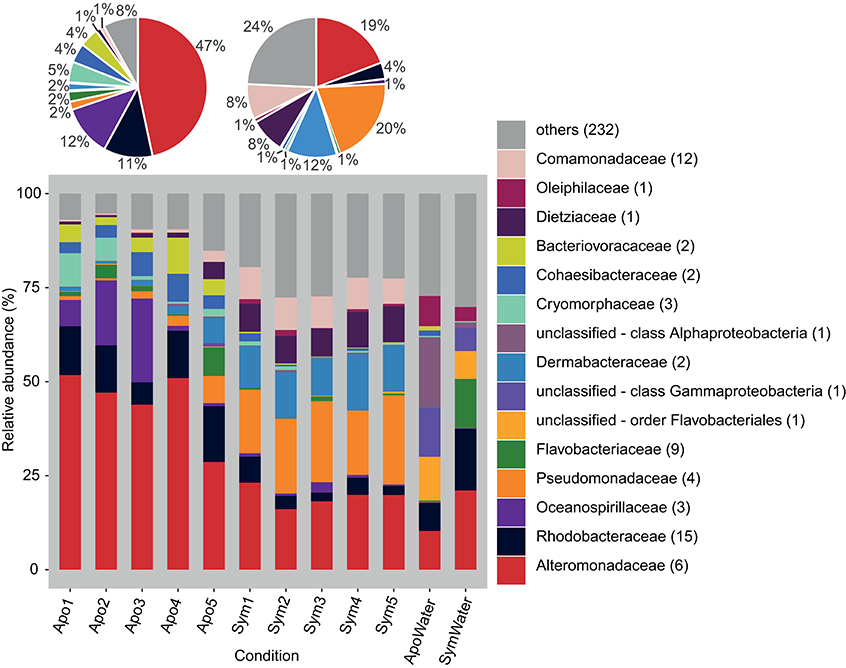
Figure 1. Bacterial community composition on the phylogenetic level of family (Greengenes database, bootstrap ≥60). Each color represents one of the 15 most abundant families across all samples. Less abundant families are grouped under “others.” Pie charts display average bacterial community composition of aposymbiotic (left) and symbiotic (right) Aiptasia. Sequences unclassified on the family level are denoted at the next higher classified taxonomic level. Numbers in parenthesis demark the number of different taxa within the respective families. Apo, aposymbiotic Aiptasia; Sym, symbiotic Aiptasia; WaterApo, water from rearing of aposymbiotic Aiptasia; WaterSym, water from rearing of symbiotic Aiptasia.
To assess differences between bacterial communities of aposymbiotic and symbiotic Aiptasia in more detail, we clustered sequences into operational taxonomic units (OTUs) at a 97% similarity cutoff after subsampling to 11,000 reads and calculated alpha diversity indices (Table 1, Supplementary File S1). We identified a total of 486 OTUs, 379 associated with Aiptasia (251 OTUs were exclusively found in Aiptasia) and 235 found in water (of these 107 exclusively in water) (Supplementary File S2). Average Chao1 estimator of species richness was significantly higher for aposymbiotic samples than for symbiotic samples (average 166 vs. 131, respectively) (t-test <0.05). Simpson's evenness and the inverse Simpson index, however, were significantly higher (t-test <0.05) in symbiotic samples (average 0.095 and 11.8, respectively) than in aposymbiotic samples (average of 0.053 and 8.0, respectively). Water samples showed a higher Chao1 (average 257) and inverse Simpson index (average 14.7), but a similar evenness (average 0.059) in comparison to Aiptasia samples. Differences in bacterial communities from aposymbiotic and symbiotic Aiptasia and water samples were visualized in a non-metric multidimensional scaling (nMDS) plot based on the Yue & Clayton theta similarity coefficient (Supplementary File S3). As expected, we found a clear separation between the water samples and all Aiptasia samples (PAMOVA = 0.014) demonstrating the presence of a specific and selected microbiome associated with Aiptasia. To focus on differences between apo- and symbiotic Aiptasia, we excluded water samples from subsequent analyses.
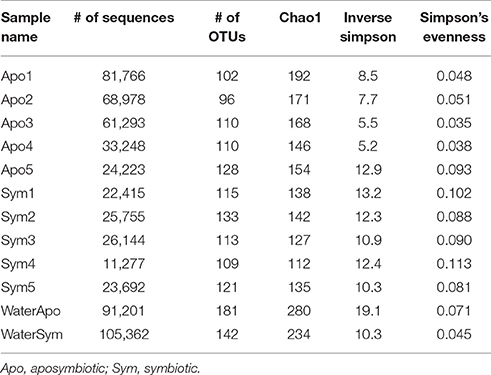
Table 1. Summary statistics of 16S rRNA gene bacterial community sequencing of Aiptasia.
Distinct Bacterial Communities of Aposymbiotic and Symbiotic Aiptasia
Bacterial communities associated with aposymbiotic and symbiotic Aiptasia were significantly different in an OTU framework (PAMOVA = 0.008). To further identify OTUs associated with different symbiotic states, we determined the “core microbiome” (i.e., all OTUs present in 100% of all Aiptasia samples), the aposymbiotic microbiome or “apobiome” (i.e., all OTUs present in 100% of aposymbiotic Aiptasia), and the symbiotic microbiome or “symbiome” (i.e., all OTUs present in 100% of symbiotic Aiptasia) (Figure 2).
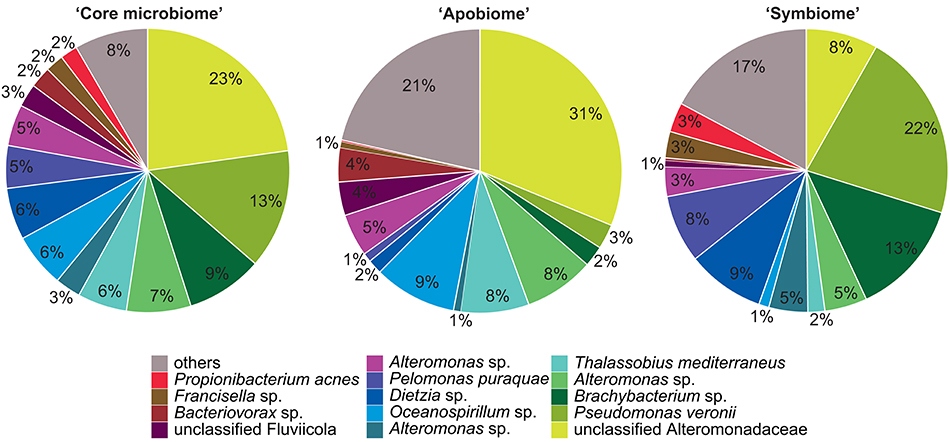
Figure 2. Aiptasia “core microbiome”, “apobiome” (aposymbiotic microbiome), and “symbiome” (symbiotic microbiome). Bacterial members were determined by assessing presence of OTUs over samples. Only OTUs present in all anemones, all aposymbiotic anemones, and all symbiotic anemones were included in the “core microbiome”, “apobiome” (aposymbiotic microbiome), and “symbiome” (symbiotic microbiome), respectively. Each color represents a distinct OTU of the 14 most abundant taxa; 49 rare OTUs have been summarized in gray in the category “others.”
We identified 24 OTUs in the core microbiome (Table 2, Supplementary File S2), which included the 10 most abundant OTUs, comprising >60% of all OTU sequence counts. We next looked for patterns of differential abundance among core microbiome members in aposymbiotic and symbiotic Aiptasia, since their relative abundance may indicate functional differences (Figure 2, Table 2). Interestingly, only three OTUs showed a comparatively modest fold-change between 1.2- and 1.7-fold (OTU004, OTU010, OTU024), while the remaining 21 OTUs, i.e., the vast majority of all core microbiome taxa, showed marked differences in abundance (between 2.4- to 18-fold) between aposymbiotic and symbiotic anemones. For the “apobiome”, we identified 50 distinct OTUs, including 11 OTUs that were exclusively found in aposymbiotic animals (Supplementary File S2). The 50 bacterial taxa of the “apobiome” represented abundant and rare members of the microbiome (mean abundance of 1–3318 sequence counts in aposymbiotic conditions). Similarly, the “symbiome” consisted of 37 OTUs, including only 1 OTU that was exclusively found in symbiotic anemones (Supplementary File S2). The average abundance of OTUs from the “symbiome” ranged between 6 and 2173 sequence counts in symbiotic Aiptasia.
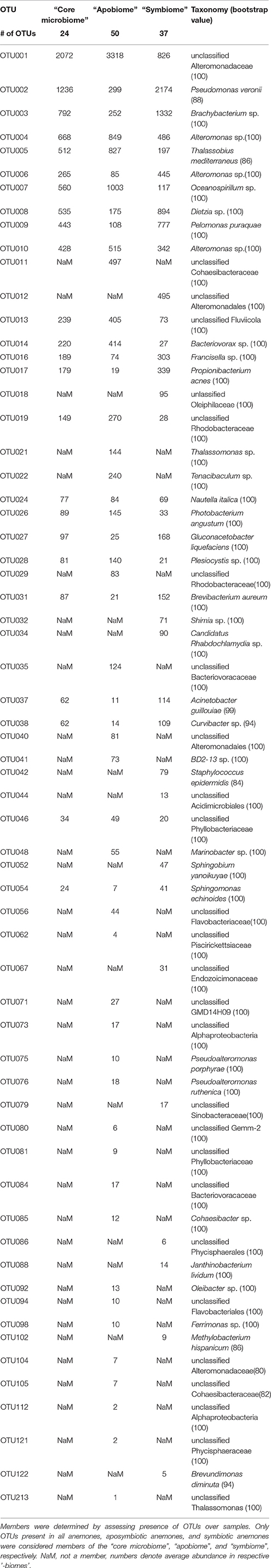
Table 2. Aiptasia “core microbiome”, “apobiome” (aposymbiotic microbiome), and “symbiome” (symbiotic microbiome).
Taxonomy-Based Functional Profiling of Bacterial Communities in Aiptasia
To assess putative functional changes underlying the different bacterial communities in aposymbiotic and symbiotic Aiptasia, we used METAGENassist (Figure 3, Supplementary File S4). Symbiotic Aiptasia clustered together tightly indicating homogeneity in enrichment and depletion of functions. By comparison, aposymbiotic samples seemed more diverse and did not cluster together. In particular, one of the samples (Apo5, Figure 3) exhibited higher similarity to the symbiotic samples as indicated by the clustering of this sample with symbiotic Aiptasia. In general, we found processes to be enriched in aposymbiotic and depleted in symbiotic samples (e.g., “Sulfate reducer”, “Sulfide oxidizer”, “Selenate reducer”, “Denitrifying”) or vice versa enriched in symbiotic and depleted in aposymbiotic samples (e.g., “Sulfur oxidizer”, “Chlorophenol degrading”, “Degrades aromatic hydrocarbons”, “Sulfur metabolizing”, “Naphthalene degrading”), besides some processes that were more inconsistent (e.g., “Xylan degrader”, “Atrazine metabolism”, “Iron oxidizer”) (Figure 3, Supplementary File S4).
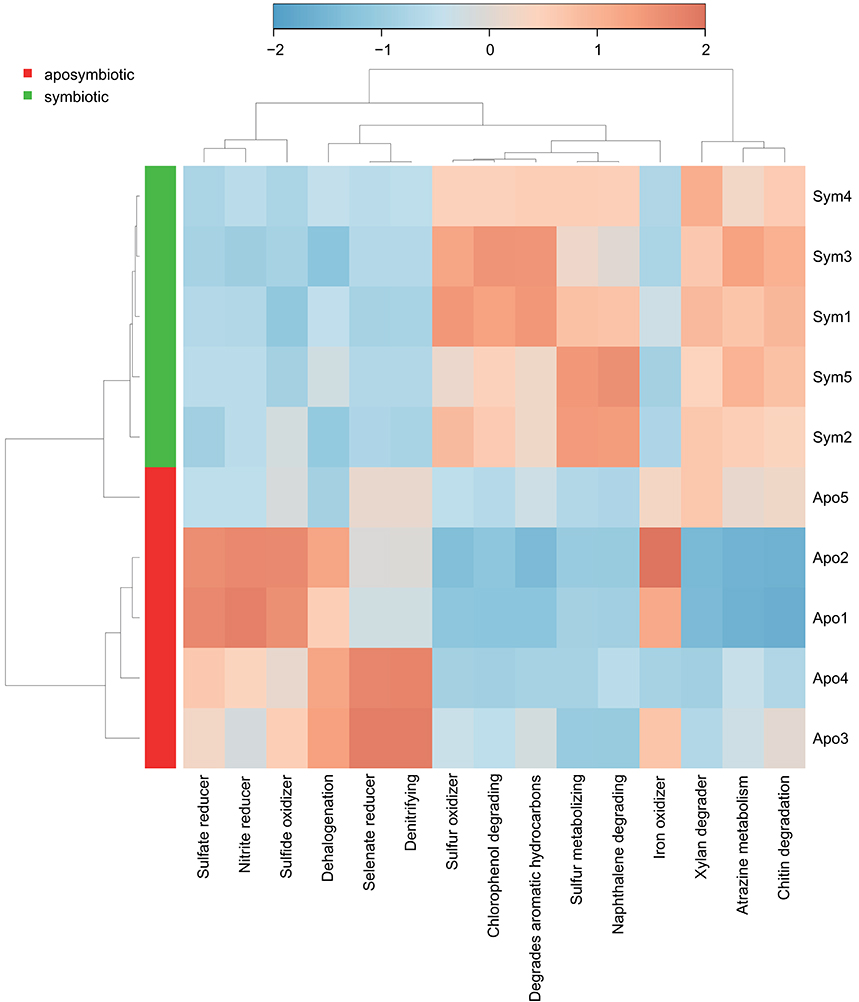
Figure 3. Taxonomy-based functional profiling of bacterial communities. Heatmap displaying putative functional differences based on the bacterial community composition of aposymbiotic and symbiotic Aiptasia. Changes are displayed on a relative scale with enrichment in red and depletion in blue. Sym, symbiotic Aiptasia; Apo, aposymbiotic Aiptasia.
Cultured Isolates of Aiptasia-Associated Bacteria
In order to enable functional studies on bacteria-host interactions in Aiptasia, it is of great benefit to have culturable isolates of bacterial associates, as previously demonstrated for Hydra (Fraune et al., 2015). To obtain cultured isolates, we used lysates of aposymbiotic and symbiotic animals and compared the isolated bacteria to the native microbial community. We retrieved approximately 700 bacterial colonies. Subsequent 16S marker gene sequencing and comparison to the native microbiome revealed about 200 distinct cultivates with a similarity of ≥97% (data not shown). Importantly, 14 cultivates displayed a similarity of 100% to the 16S rRNA gene amplicon, which were further considered (Table 3). These 14 OTUs included 3 of the 10 most abundant bacteria (i.e., OTU001, OTU004, and OTU006) and were members of the most abundant family Alteromonadaceae in aposymbiotic and symbiotic anemones (Table 3, Supplementary File S2). Importantly, we could culture the most abundant member (OTU001) from the core microbiome and identified it to the genus Glaciecola, which was possible based on the longer Sanger sequence (~900 bp) in comparison to the MiSeq amplicon. The 14 OTUs contained 6 OTUs (25%) of the core microbiome, 9 OTUs (18%) of the apobiome, and 7 OTUs (19%) of the symbiome. The use of two different growth media retrieved different cultures. For instance, a bacterial cultivate representing OTU001 was obtained from bacterial colonies grown on Marine Agar, but not M1 Agar. Further, while Marine Agar retrieved a higher taxonomic diversity, M1 Agar showed an increased selectivity for the genera Alteromonas and Pseudoalteromonas.
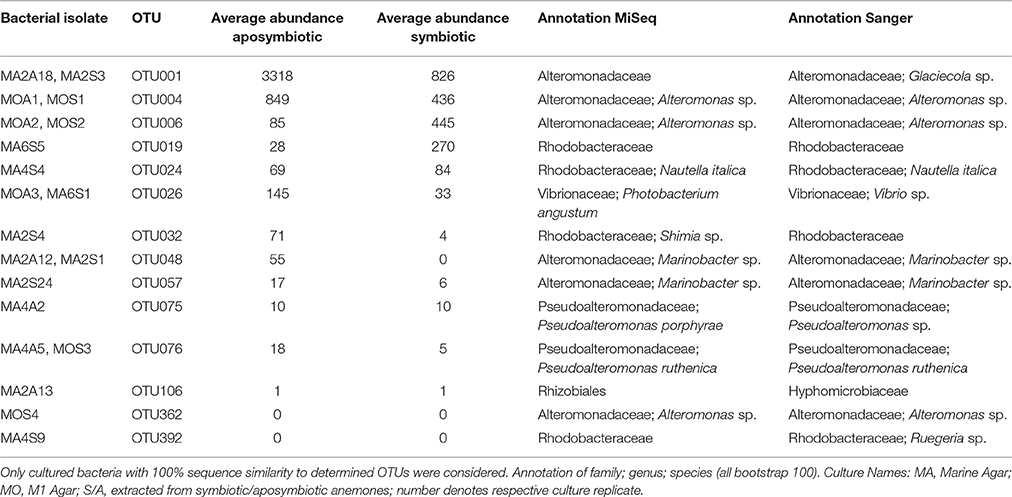
Table 3. Overview of cultured isolates of Aiptasia-associated bacteria.
Discussion
The Microbiome of Aiptasia
Despite the importance of bacteria to animal and plant function (Mcfall-Ngai et al., 2013), the microbiome of model systems has only begun to be studied in earnest over the past few years. While studies in Hydra magnipapillata show that co-operation between host-selected microbes exist (Fraune et al., 2015), the microbiome of Nematostella vectensis has only been characterized very recently and functional studies are not yet available (Har et al., 2015). Here we describe the microbiome of Aiptasia associated with aposymbiotic and symbiotic states. We find that the bacterial microbiome, irrespective of the symbiotic state with Symbiodinium, is comprised of a fairly consistent number of OTUs (between 96 to 133 OTUs). In Hydra a similar number of OTUs (~100) has been found in 15 week old polyps (Franzenburg et al., 2013). In corals, numbers of associated OTUs vary more pronouncedly between species and prevailing environmental conditions, but are also on the order of tens to hundreds of OTUs (Bayer et al., 2013; Jessen et al., 2013; Roder et al., 2014, 2015; Neave et al., 2016; Röthig et al., 2016; Ziegler et al., 2016).
Although the core microbiome was comparably small, the 24 bacterial taxa made up the majority of sequence counts. The ubiquity and high abundance of these OTUs suggest functional importance to the animal host, regardless of the symbiotic state. Yet, the majority of core microbiome taxa considerably differed in their abundance in aposymbiotic and symbiotic anemones. At present, it is unknown why these bacteria display differential abundance, but these data suggest a link between the bacterial community and the cnidarian-algal symbiosis, strongly arguing to integrate bacterial communities in research of the cnidarian-algal symbiosis.
Interestingly, Pseudomonas veronii was identified as a highly abundant member of the core microbiome (Table 3). P. veronii was strongly enriched in a recent study on fungid corals exposed to hypersalinity in the Red Sea (Röthig et al., 2016). The repeated association with different symbiotic cnidarians might point toward the functional importance of this bacterial taxon and makes it an interesting candidate for cultivation and functional studies. We also identified Nautella italica, a bacterial pathogen able to colonize and invade different algae (Fernandes et al., 2011; Gardiner et al., 2015), which at least hypothetically shows how host-associated bacteria can affect the animal host-algal symbiosis. Many of the remaining core microbiome members were found previously associated with corals, sponges, echinoids, algae, and sediments (Supplementary File S2).
In the future, improved resolution of taxonomical classification could be obtained by compiling databases harboring sequences specifically associated with cnidarians, as done for members of the human intestinal microbiota (Ritari et al., 2015). Recent efforts to establish cnidarian-specific databases include the Coral Microbiome Portal (CMP) at https://vamps.mbl.edu/portals/coral_microbe/coral.php and reefgenomics.org (Liew et al., 2016) at http://reefgenomics.org that, besides serving as a data repository for genomics data associated with reef organisms, also anticipates to hold microbial data such as those arising from the ReFuGe 2020 consortium (Voolstra et al., 2015).
Functional Differences Associated with the Microbial Community
Interestingly, 4 out of the 15 most pronounced differences in metabolic processes were involved in sulfur cycling (i.e., “Sulfate reducer”, “Sulfide oxidizer”, “Sulfur oxidizer”, “Sulfur metabolizing”). Sulfur utilization is enhanced by the presence of Symbiodinium in juvenile corals (Yuyama et al., 2016). Similarly, we argue that symbiotic Aiptasia have higher levels of dimethylsulfoniopropionate (DMSP), which accordingly provides a source of sulfur for the bacterial community (Supplementary File S5). In support, aposymbiotic Aiptasia seem unable to produce DMSP as it was only found in symbiotic animals (Van Alstyne et al., 2009). Taken together, DMSP is an important substrate of bacterial sulfur cycling (Raina et al., 2010), and its increased synthesis in symbiotic Aiptasia likely explains the enrichment of sulfur cycling bacteria, as shown previously for coral holobionts (Frade et al., 2016). Besides differential abundance of functions related to sulfur cycling, we identified differences in nitrogen cycling (Supplementary File S5). Nitrogen is a limiting nutrient in the coral holobiont and algal symbiont densities are controlled, in part, by nitrogen availability (Falkowski et al., 1993; Rädecker et al., 2015). The bacterial processes “nitrite reduction” and “denitrification” were increased in aposymbiotic Aiptasia, indicating either increased nitrogen availability and/or increased recycling. Given that Symbiodinium is the major sink for nitrogen compounds released by the host in symbiotic coral holobionts (Pernice et al., 2012), nitrogen may no longer be a limiting factor in aposymbiotic animals. Hence, excess nitrogen availability may stimulate growth of denitrifying bacteria, allowing for the efficient removal of these nitrogen compounds from the holobiont. Future studies using metagenomics and metatranscriptomics to study aposymbiotic and symbiotic states have the potential to provide further insight and a more direct assessment of the functional attributes of the microbiome (see e.g., Daniels et al., 2015).
Cultured Isolates of Aiptasia-Associated Bacteria—Toward Functional Microbiome Studies
Even though functional studies of corals exist (Lema et al., 2015; Pollock et al., 2015), a laboratory model is needed in order to conduct more elaborate studies, such as experimental replacement of native bacteria in order to assess functional contribution of a specific bacterial species. For this type of experiment, it is essential to obtain bacterial cultivates that represent key microbial symbionts. In this study, we could culture a range of abundant and rare Aiptasia-associated bacteria, including isolates that were specific to the aposymbiotic or symbiotic condition. The cultured isolates here present a starting point for functional studies, especially with regard to the notion that abundant and rare bacteria in cnidarians are functionally important (Bosch, 2013; Golberg et al., 2013; Fraune et al., 2015; Glasl et al., 2016). Of note, this is an ongoing effort, and we anticipate that further application of different culture media and conditions will enable a much more complete cultivation of Aiptasia-associated bacteria. These efforts will be complemented by whole genome sequencing of key bacterial associates, as conducted by Har et al. (2015), in order to gain further understanding of the putative functions encoded and provided by the bacterial microbiome. In addition, an important accompanying step to culturing and characterization of bacterial isolates is the generation of axenic Aiptasia that may then be used for infection studies with bacterial cultivates in order to unequivocally assign function (Fraune et al., 2015), with the ultimate aim of identifying bacteria that affect holobiont traits of significance to environmental change, such as those that confer increased thermotolerance (Moran and Yun, 2015).
Conclusions
The unprecedented decline of coral reef cover in the last decades and in particular in recent years has heightened the need to better understand the mechanistic and molecular underpinnings of coral holobiont function. The growing popularity of the Aiptasia coral model promises to yield new insights and allows for the design of novel experiments, such as the comparison of aposymbiotic and symbiotic states. Our data show that aposymbiotic and symbiotic Aiptasia harbor distinct bacterial microbiomes with strong implications for the coral holobiont, namely that bacteria putatively play an important role in the coral-algal symbiosis and that the entire holobiont adjusts to the symbiotic condition. This is further corroborated by taxonomy-based functional profiling indicating that the bacterial microbiome of symbiotic Aiptasia is highly structured, less variant, and enriched for functions of putative relevance to the algal symbiosis. We hope that cultivation of members of the bacterial community of Aiptasia provides a foundation to conduct functional studies with the aim of better understanding the contributions of bacteria to holobiont function and identifying the members that are critical for environmental resilience of Aiptasia, and by extension of stony corals.
Author Contributions
TR, RC, FS, and CRV designed and conceived the experiments. TR, RC, SB, and FS generated the data. TR, RC, and CRV analyzed and interpreted the data. AT, MA, AR, and CRV contributed reagents/materials/analysis tools. TR, RC, and CRV wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by baseline funds to CRV by King Abdullah University of Science and Technology (KAUST) and by the Center Competitive Funding (CCF) Program FCC/1/1973-18-01.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Mohammad Bin Sarhan for preliminary work on cultural isolates and Craig T. Michell for MiSeq library generation.
Data Accessibility
Sequence data determined in this study have been deposited on NCBI under BioProject accession no. PRJNA325476 (http://www.ncbi.nlm.nih.gov/bioproject/325476).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00234/full#supplementary-material
Supplementary Data Sheet 1. Rarefaction curves illustrating OTU richness as a function of sequencing depth for subsampled dataset (n = 11,000 sequences per sample).
Supplementary Data Sheet 2. OTU abundance counts over samples with annotation, reference OTU sequence, and affiliation to “core microbiome”, “apobiome” (aposymbiotic microbiome), and “symbiome” (symbiotic microbiome). For OTUs constituting a member of any “biome,” the closest BLASTn match for the reference OTU sequence including source environment and available literature is denoted.
Supplementary Data Sheet 3. Non-metric multidimensional scaling (nMDS) plot of bacterial communities of aposymbiotic and symbiotic Aiptasia and water samples. Clustering of samples based on Yue & Clayton theta similarity coefficient of microbial community abundances (R2 = 0.95, lowest stress = 0.108).
Supplementary Data Sheet 4. Taxonomy-based functional profiling of bacterial communities on average bacterial community composition of aposymbiotic and symbiotic Aiptasia. Heatmap displaying putative functional differences based on the bacterial community composition of aposymbiotic and symbiotic Aiptasia. Changes are displayed on a relative scale with enrichment in red and depletion in blue. Sym, symbiotic Aiptasia, Apo, aposymbiotic Aiptasia.
Supplementary Data Sheet 5. Conceptual model of cnidarian holobiont functioning and differences between aposymbiotic and symbiotic states (model extended from Rohwer et al., 2002). Functions proposed in the original conceptual holobiont model are in black, putative functions related to the presence of Symbiodinium in green, functions enriched in aposymbiotic Aiptasia in red, and functions present in the aposymbiotic and symbiotic state in gray.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836. doi: 10.1371/journal.pone.0002836
Arndt, D., Xia, J., Liu, Y., Zhou, Y., Guo, A. C., Cruz, J. A., et al. (2012). METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. 40, 88–95. doi: 10.1093/nar/gks497
Baumgarten, S., Simakov, O., Esherick, L. Y., Liew, Y. J., Lehnert, E. M., Michell, C. T., et al. (2015). The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc. Natl. Acad. Sci. U.S.A. 112, 11893–11898. doi: 10.1073/pnas.1513318112
Bayer, T., Neave, M. J., Alsheikh-Hussain, A., Aranda, M., Yum, L. K., Mincer, T., et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated endozoicomonas bacteria. Appl. Environ. Microbiol. 79, 4759–4762. doi: 10.1128/AEM.00695-13
Bosch, T. C. G. (2013). Cnidarian-microbe interactions and the origin of innate immunity in metazoans. Annu. Rev. Microbiol. 67, 499–518. doi: 10.1146/annurev-micro-092412-155626
Bourne, D. G., Morrow, K. M., and Webster, N. S. (2016). Coral holobionts: insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Daniels, C., Baumgarten, S., Yum, L. K., Michell, C. T., Bayer, T., Arif, C., et al. (2015). Metatranscriptome analysis of the reef-buidling coral Orbicella faveolata indicates holobiont response to coral disease. Front. Mar. Sci. 2:62. doi: 10.3389/fmars.2015.00062
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491.
Falkowski, P. G., Dubinsky, Z., Muscatine, L., and Mccloskey, L. (1993). Population control in symbiotic corals. Bioscience 43, 606–611.
Fernandes, N., Case, R. J., Longford, S. R., Seyedsayamdost, M. R., Steinberg, P. D., Kjelleberg, S., et al. (2011). Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. PLoS ONE 6:e27387. doi: 10.1371/journal.pone.0027387
Frade, P. R., Schwaninger, V., Glasl, B., Sintes, E., Hill, R. W., Simó, R., et al. (2016). Dimethylsulfoniopropionate in corals and its interrelations with bacterial assemblages in coral surface mucus. Environ. Chem. 13, 252–265. doi: 10.1071/EN15023
Franzenburg, S., Fraune, S., Altrock, P. M., Künzel, S., Baines, J. F., Traulsen, A., et al. (2013). Bacterial colonization of hydra hatchlings follows a robust temporal pattern. ISME J. 7, 781–790. doi: 10.1038/ismej.2012.156
Fraune, S., Anton-Erxleben, F., Augustin, R., Franzenburg, S., Knop, M., Schröder, K., et al. (2015). Bacteria-bacteria interactions within the microbiota of the ancestral metazoan Hydra contribute to fungal resistance. ISME J. 9, 1543–1556. doi: 10.1038/ismej.2014.239
Gardiner, M., Thomas, T., and Egan, S. (2015). A glutathione peroxidase (GpoA) plays a role in the pathogenicity of Nautella italica strain R11 towards the red alga Delisea pulchra. FEMS Microbiol. Ecol. 91:fiv021. doi: 10.1093/femsec/fiv021
Gardner, T. A., Côté, I. M., Gill, J. A., Grant, A., and Watkinson, A. R. (2003). Long-term region-wide declines in caribbean corals. Science 301, 958–960. doi: 10.1126/science.1086050
Glasl, B., Herndl, G. J., and Frade, P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292. doi: 10.1038/ismej.2016.9
Golberg, K., Pavlov, V., Marks, R. S., and Kushmaro, A. (2013). Coral-associated bacteria, quorum sensing disrupters, and the regulation of biofouling. Biofouling 29, 669–682. doi: 10.1080/08927014.2013.796939
Hackstadt, A. J., and Hess, A. M. (2009). Filtering for increased power for microarray data analysis. BMC Bioinformatics 10:11. doi: 10.1186/1471-2105-10-11
Har, J. Y., Helbig, T., Lim, J. H., Fernando, S. C., Reitzel, A. M., Penn, K., et al. (2015). Microbial diversity and activity in the Nematostella vectensis holobiont: insights from 16S rRNA gene sequencing, isolate genomes, and a pilot-scale survey of gene expression. Front. Microbiol. 6:818. doi: 10.3389/fmicb.2015.00818
Hoegh-Guldberg, O. (2015). Reviving the Ocean Economy: The Case for Action–2015. Gland: WWF International.
Hughes, T. P., Baird, A. H., Bellwood, D. R., Card, M., Connolly, S. R., Folke, C., et al. (2003). Climate change, human impacts, and the resilience of coral reefs. Science 301, 929–933. doi: 10.1126/science.1085046
Huse, S. M., Welch, D. M., Morrison, H. G., and Sogin, M. L. (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
Jessen, C., Villa Lizcano, J. F., Bayer, T., Roder, C., Aranda, M., Wild, C., et al. (2013). In-situ effects of eutrophication and overfishing on physiology and bacterial diversity of the red sea coral Acropora hemprichii. PLoS ONE 8:e62091. doi: 10.1371/annotation/be4a3168-5284-4083-b5ed-5cd0f4630823
Lema, K. A., Clode, P. L., Kilburn, M. R., Thornton, R., Willis, B. L., and Bourne, D. G. (2015). Imaging the uptake of nitrogen-fixing bacteria into larvae of the coral Acropora millepora. ISME J. 10, 1804–1808. doi: 10.1038/ismej.2015.229
Liew, Y. J., Aranda, M., and Voolstra, C. R. (2016). Reefgenomics.org - a repository for reefgenomics data. Database 1–4, 10.1093/database/baw152
Mcdonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., Desantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Mcfall-Ngai, M., Hadfield, M. G., Bosch, T. C. G., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
Moran, N. A., and Yun, Y. (2015). Experimental replacement of an obligate insect symbiont. Proc. Natl. Acad. Sci. U.S.A. 112, 2093–2096. doi: 10.1073/pnas.1420037112
Mouchka, M. E., Hewson, I., and Harvell, C. D. (2010). Coral-associated bacterial assemblages: current knowledge and the potential for climate-driven impacts. Integr. Comp. Biol. 50, 662–674. doi: 10.1093/icb/icq061
Neave, M. J., Rachmawati, R., Xun, L., Michell, C. T., Bourne, D. G., Apprill, A., et al. (2016). Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J. [Epub ahead of print]. doi: 10.1038/ismej.2016.95
Negri, A. P., Flores, F., Röthig, T., and Uthicke, S. (2011). Herbicides increase the vulnerability of corals to rising sea surface temperature. Limnol. Oceanogr. 56, 471–485. doi: 10.4319/lo.2011.56.2.0471
Pernice, M., Meibom, A., Van Den Heuvel, A., Kopp, C., Domart-Coulon, I., Hoegh-Guldberg, O., et al. (2012). A single-cell view of ammonium assimilation in coral-dinoflagellate symbiosis. ISME J. 6, 1314–1324. doi: 10.1038/ismej.2011.196
Pollock, F. J., Krediet, C. J., Garren, M., Stocker, R., Winn, K., Wilson, B., et al. (2015). Visualization of coral host–pathogen interactions using a stable GFP-labeled Vibrio coralliilyticus strain. Coral Reefs 34, 655–662. doi: 10.1007/s00338-015-1273-3
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Rädecker, N., Pogoreutz, C., Voolstra, C. R., Wiedenmann, J., and Wild, C. (2015). Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol. 23, 490–497. doi: 10.1016/j.tim.2015.03.008
Raina, J.-B., Dinsdale, E. A., Willis, B. L., and Bourne, D. G. (2010). Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol. 18, 101–108. doi: 10.1016/j.tim.2009.12.002
Raina, J.-B., Tapiolas, D., Willis, B. L., and Bourne, D. G. (2009). Coral-Associated bacteria and their role in the biogeochemical cycling of sulfur. Appl. Environ. Microbiol. 75, 3492–3501. doi: 10.1128/AEM.02567-08
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Reaka-Kudla, M. L., Wilson, D. E., and Wilson, E. O. (1996). Biodiversity II: Understanding and Protecting our Biological Resources. Washington, DC: Joseph Henry Press.
Ritari, J., Salojärvi, J., Lahti, L., and De Vos, W. M. (2015). Improved taxonomic assignment of human intestinal 16S rRNA sequences by a dedicated reference database. BMC Genomics 16:1056. doi: 10.1186/s12864-015-2265-y
Ritchie, K. (2011). “Bacterial symbionts of corals and symbiodinium,” in Beneficial Microorganisms in Multicellular Life Forms, eds E. Rosenberg and U. Gophna (Berlin; Heidelberg: Springer-Verlag), 139–150.
Roder, C., Arif, C., Bayer, T., Aranda, M., Daniels, C., Shibl, A., et al. (2014). Bacterial profiling of white plague disease in a comparative coral species framework. ISME J. 8, 31–39. doi: 10.1038/ismej.2013.127
Roder, C., Bayer, T., Aranda, M., Kruse, M., and Voolstra, C. R. (2015). Microbiome structure of the fungid coral Ctenactis echinata aligns with environmental differences. Mol. Ecol. 24, 3501–3511. doi: 10.1111/mec.13251
Rohwer, F., Seguritan, V., Azam, F., and Knowlton, N. (2002). Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10. doi: 10.3354/meps243001
Rosenberg, E., Koren, O., Reshef, L., Efrony, R., and Zilber-Rosenberg, I. (2007). The role of microorganisms in coral health, disease and evolution. Nat. Rev. Microbiol. 5, 355–362. doi: 10.1038/nrmicro1635
Röthig, T., Ochsenkühn, M. A., Roik, A., Van Der Merwe, R., and Voolstra, C. R. (2016). Long-term salinity tolerance is accompanied by major restructuring of the coral bacterial microbiome. Mol. Ecol. 25, 1308–1323. doi: 10.1111/mec.13567
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Van Alstyne, K. L., Dominique, V. J., and Muller-Parker, G. (2009). Is dimethylsulfoniopropionate (DMSP) produced by the symbionts or the host in an anemone–zooxanthella symbiosis? Coral Reefs 28, 167–176. doi: 10.1007/s00338-008-0443-y
Vega Thurber, R. L., Burkepile, D. E., Fuchs, C., Shantz, A. A., Mcminds, R., and Zaneveld, J. R. (2014). Chronic nutrient enrichment increases prevalence and severity of coral disease and bleaching. Glob. Change Biol. 20, 544–554. doi: 10.1111/gcb.12450
Voolstra, C. R. (2013). A journey into the wild of the cnidarian model system Aiptasia and its symbionts. Mol. Ecol. 22, 4366–4368. doi: 10.1111/mec.12464
Voolstra, C. R., Miller, D. J., Ragan, M. A., Hoffmann, A., Hoegh-Guldberg, O., Bourne, D., et al. (2015). The ReFuGe 2020 consortium - Using 'omics' approaches to explore the adaptability and resilience of coral holobionts to environmental change. Front. Mar. Sci. 2:68. doi: 10.3389/fmars.2015.00068
Weis, V. M., Davy, S. K., Hoegh-Guldberg, O., Rodriguez-Lanetty, M., and Pringle, J. R. (2008). Cell biology in model systems as the key to understanding corals. Trends Ecol. Evol. 23, 369–376. doi: 10.1016/j.tree.2008.03.004
Xiang, T., Hambleton, E. A., Denofrio, J. C., Pringle, J. R., and Grossman, A. R. (2013a). Isolation of clonal axenic strains of the symbiotic dinoflagellate Symbiodinium and their growth and host specificity1. J. Phycol. 49, 447–458. doi: 10.1111/jpy.12055
Xiang, T., Hambleton, E. A., Denofrio, J. C., Pringle, J. R., and Grossman, A. R. (2013b). Isolation of clonal axenic strains of the symbiotic dinoflagellate Symbiodinium and their growth and host specificity(1). J. Phycol. 49, 447–458. doi: 10.1111/jpy.12055
Yuyama, I., Higuchi, T., and Takei, Y. (2016). Sulfur utilization of corals is enhanced by endosymbiotic algae. Biol. Open. 5, 1299–1304. doi: 10.1242/bio.020164
Keywords: coral reef, cnidarian-dinoflagellate symbiosis, microbial community profiling, 16S rRNA gene, functional profiling
Citation: Röthig T, Costa RM, Simona F, Baumgarten S, Torres AF, Radhakrishnan A, Aranda M and Voolstra CR (2016) Distinct Bacterial Communities Associated with the Coral Model Aiptasia in Aposymbiotic and Symbiotic States with Symbiodinium. Front. Mar. Sci. 3:234. doi: 10.3389/fmars.2016.00234
Received: 18 June 2016; Accepted: 01 November 2016;
Published: 18 November 2016.
Edited by:
Thomas Carl Bosch, University of Kiel, GermanyReviewed by:
Simon K. Davy, Victoria University of Wellington, New ZealandMathieu Pernice, University of Technology, Australia
Copyright © 2016 Röthig, Costa, Simona, Baumgarten, Torres, Radhakrishnan, Aranda and Voolstra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian R. Voolstra, Y2hyaXN0aWFuLnZvb2xzdHJhQGthdXN0LmVkdS5zYQ==
†These authors have contributed equally to this work.