
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 22 September 2016
Sec. Marine Biogeochemistry
Volume 3 - 2016 | https://doi.org/10.3389/fmars.2016.00172
This article is part of the Research TopicOrganic ligands - A key control on trace metal biogeochemistry in the oceanView all 22 articles
 Imelda B. Velasquez1
Imelda B. Velasquez1 Enitan Ibisanmi1
Enitan Ibisanmi1 Elizabeth W. Maas2†
Elizabeth W. Maas2† Philip W. Boyd3,4
Philip W. Boyd3,4 Scott Nodder2
Scott Nodder2 Sylvia G. Sander1,4*
Sylvia G. Sander1,4*The microbial degradation of marine particles is an important process in the remineralization of nutrients including iron. As part of the GEOTRACES process study (FeCycle II), we conducted incubation experiments with marine particles obtained from 30 to 100 m depth at two stations during austral spring in the subtropical waters east of the North Island of New Zealand. The particles were collected using in-situ pumps, and comprised mainly of suspended and slow sinking populations along with associated attached heterotrophic bacteria. In treatments with live bacteria, increasing concentrations of Fe binding ligands were observed with an average stability constant of logKFeL, Fe3+ = 21.11 ± 0.37 for station 1 and 20.89 ± 0.25 for station 2. The ligand release rates varied between 2.54 and 11.8 pmol L−1 d−1 (calculated for ambient seawater particle concentration) and were similar to those found in two Southern Ocean subsurface studies from ~110 m depths in subpolar and polar waters. Dissolved iron (DFe) was released at a rate between 0.33 and 2.09 pmol Fe L−1 d−1 with a column integrated (30–100 m) flux of 107 and 58 nmol Fe m−2 day−1 at station 1 and 2, respectively. Given a mixed layer DFe inventory of ~48 μmol m−2 and ~4 μmol m−2 at the time of sampling for station 1 and 2, this will therefore result in a DFe residence time of 1.2 and 0.18 years, assuming particle remineralization was the only source of iron in the mixed layer. The DFe release rates calculated were comparable to those found in the previously mentioned study of Southern Ocean water masses. Fe-binding ligand producing bacteria (CAS positive) abundance was found to increase throughout the duration of the experiment of 7–8 days. For the first time ferrioxamine type siderophores, including the well-known ferrioxamine B and G, have been quantified using chemical assays and LC-ESI-MS. Our subtropical study corroborates prior reports from the Southern Ocean of particle remineralization being an important source of DFe and ligands, and adds unprecedented detail by revealing that siderophores are probably an important component of the ligands released into subsurface waters during particle remineralisation.
Iron is an essential metal required for many microbial enzymatic activities (Geider and La Roche, 1994). It plays a central role in controlling primary production over much of the world's ocean. Hence, many studies have focused on better understanding the biogeochemical cycle of iron in the ocean (Rue and Bruland, 1995, 1997; Behrenfield et al., 1996; Boyd et al., 2000, 2005; Morel and Price, 2003; Moore et al., 2013). Previous studies revealed that over 99.9% of dissolved iron in the ocean is bound by iron-complexing ligands (Gledhill and van den Berg, 1994; Van den Berg, 1995; Tian et al., 2006; Sander et al., 2015). These iron-binding ligands are separated into operational classes based on trends in conditional stability constants—L1 being the strongest Fe-binding ligand and L2,L3 etc. subsequently weaker ones (Rue and Bruland, 1995; Sander et al., 2011).
Many studies have investigated the production and sources of Fe-binding ligand classes (Gledhill and Buck, 2012). Siderophores and possibly other strong iron binding ligands with conditional stability constants similar to L1 class ligands have been shown to be produced, and released, by heterotrophic bacteria and cyanobacteria in response to iron limitation (Reid et al., 1993; Wilhelm and Trick, 1994; Martinez et al., 2001; Gledhill et al., 2004; Mawji et al., 2008a) or in response to episodic Fe supply to take up Fe opportunistically (Adly et al., 2015). Ligands, although probably not siderophores, were also released or produced during micro and meso-zooplankton grazing on phytoplankton during an in-situ mesoscale iron enrichment experiments conducted in the western subarctic Pacific (Moore and Braucher, 2008) and our present FeCycle II study (Boyd et al., 2012). Other potential sources for iron binding ligand classes (L1 and weaker ligands) include virus lysis (Poorvin et al., 2011), exopolysaccharides (Hassler et al., 2011), and humic substances (Laglera and van den Berg, 2009; Abualhaija and van den Berg, 2014; Mahmood et al., 2015).
Particulate iron (PFe) is often the dominant form of iron in the ocean (Frew et al., 2006; Lenes et al., 2009; Ellwood et al., 2014, 2015). Hunter and Boyd (2007) pinpointed the importance of understanding the particle dynamics of iron in an effort to better understand the origins of iron-complexing ligands. They proposed the so-called “ligand soup hypothesis,” which suggested that almost any particulate organic matter, after suitable mixing and timescale, will most likely generate iron-complexing ligands. Some potential sources of the “ligand soup” include the breakdown of sinking particles in biological material and residual terrestrial organic matter transported from the continental shelves (Hunter and Boyd, 2007). Aspects of the “ligand soup” hypothesis were confirmed in a series of experiments in Southern Ocean sub- and polar-waters in which incubation of suspended and slow-settling particles resulted in the release of both DFe and L2 iron-binding ligands (Boyd et al., 2010).
The vertical distributions of iron-complexing ligands also provide insights as to the provenance of the ligand classes. Early papers already eluded to the possible linkage of the ligand distribution and microorganisms, due to a strong correlation between microbial abundance and ligand concentrations (Rue and Bruland, 1995; Boye et al., 2001). Ibisanmi et al. (2011) reported two ligand classes—termed sum of all ligand class (ΣL) and a strong ligand class (L1). L1 was restricted to the upper ocean (i.e., < 200 m depth), whereas ΣL was observed throughout the water column down to 1000 m. These depth-dependent trends, showing the presence of different ligand classes in the upper stratum vs. throughout the water column were observed by researchers in the central Pacific Ocean although different terminologies were used to describe some of these observed ligand classes (Rue and Bruland, 1995, 1997; Buck et al., 2007; Bundy et al., 2014). As part of the GEOTRACES program, the entire ocean basins have been sampled and analyzed systematically for Fe-binding ligands. Results from some of these studies such as the one conducted in the North Atlantic Ocean (Buck et al., 2015) reported up to three ligand classes throughout the water column. Improved titration methodology and data evaluation is another reason for better ligand class classification and resolution (Wells et al., 2013; Pižeta et al., 2015).
The evidence of joint iron and ligand release during a remineralization experiments was first published by Boyd et al. (2010), but no other parameters such as siderophore production, POC, nutrients or the presence of culturable heterotrophic and CAS active bacteria were measured in that pilot study. Therefore, we present the findings of a new framework of a remineralization experiment of marine particles performed during the FeCycle II GEOTRACES process study that targeted the spring bloom in subtropical waters east of the North Island of New Zealand. Special focus was drawn on the inter-relationship between POC, PFe, DFe stocks, and the inventory and chemical characterization of iron-complexing ligands during particle remineralization. The findings will help improve our conceptual understanding of the biogeochemical cycling of iron.
This study took place in high iron mesotrophic sub-tropical waters located off the east coast of the North Island of New Zealand as part of FeCycle II quasi-Lagrangian biogeochemical experiment (see Boyd et al., 2012; Weller et al., 2013 for further details). Two stations were visited during austral spring (September, 2008) on board the New Zealand research vessel RV Tangaroa to determine productivity and carbon export in New Zealand waters. The sampling location for FeCycle II was in open ocean waters (>2000 m bottom depth), ~200 km east of New Zealand. Station 1 at 39.21°S; 181.21°E (Figure 1) was sampled on the 20th September 2008, julian day 264 and corresponded to phase I of the study of an algal bloom. Station 2 at 39.40°S; 181.42°E (Figure 1) was sampled on the 27th September 2008, julian day 272, corresponding to phase II of FeCycle II (Boyd et al., 2012; Weller et al., 2013; LeCleir et al., 2014). Mixed layer depths for the entire cruise are given in Boyd et al. (2012) and Weller et al. (2013). For station 1, the mixed layer was ~78 m and for station 2 the mixed layer was < 65 m.
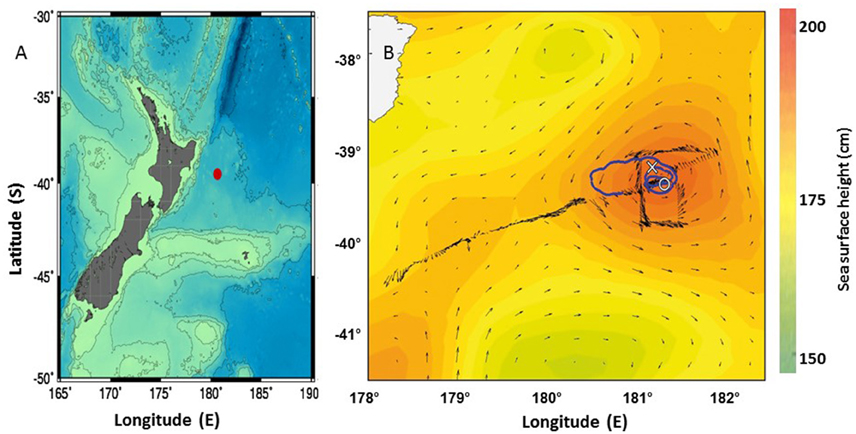
Figure 1. Maps for the larger area around New Zealand with the FeCycle II- spring bloom voyage sited marked as a red dot (A) and the closer area showing the track of the RV Tangaroa (blue solid line) during the FeCycle II- spring bloom voyage (14 September 2008 to 7 October 2008) starting from the center of an eddy. Stations 1 and 2 are marked (X) and (0) respectively (B).
All seawater samples were collected using a General Oceanics model 1018 autonomous intelligent rosette deployed on a Kevlar hydroline (Turner et al., 2004). All equipment used throughout this work had been rigorously acid cleaned following the protocols recommended by (Sander et al., 2009) and experimental samples were stored in acid cleaned polyethylene bottles (Nalgene, Rochester, NY). Particles for the remineralization experiment as well as for PFe and POC analysis were taken by trace metal clean in-situ McLane pumps.
Samples collected at both sites were taken from 30 to 100 m to be able to calculate column integrated fluxes. Particles were collected by in-situ filtration. At station 1, 192 L (30 m depth) and 286 L (100 m) of seawater were filtered, whereas 195 L (30 m) and 196 L (100 m) of seawater were filtered at station 2. Seawater samples from each depth were filtered through a 5.0 μm acid washed polycarbonate filter with a diameter of 14 cm. This pore size was chosen in order to retain most particles and their associated attached bacteria while excluding free-living bacteria (~1 μm). This pore size also allowed for better simplification of experimental outcomes and to ascribe any increases in DFe and L for ratio comparison particle attached bacteria. Using information from the Frew et al. (2006) study in subantarctic waters off New Zealand, we assumed that ~70% of all particles in the mixed layer are >5 μm. The particles were re-suspended into 250 mL of 0.2 μm unfiltered seawater collected from depths and station corresponding to pump stations. Ten milliliters aliquots of concentrated particle suspension were then further diluted with unfiltered seawater to produce 18 incubation solutions of 250 mL. This resulted in concentration factors of 29 for station 1 at 30 m, 43 for station 1 at 100 m, and 30 for both depths at station 2.
All experiments were run in triplicate for each timepoint. Half of the incubation solutions (i.e., 9) were sterilized using a domestic microwave adjusted to full power for 6 min prior to incubation (Boyd et al., 2010). Microwave sterilized samples from day 0 were filtered after cooling down to room temperature. Filter sterilized samples without particles (i.e., ambient seawater from 30 or 100m) were only analyzed at day 0, but not incubated because of previous results from the SAZ-Sense study had already shown that samples without particle enrichment did not produce significant amounts of ligands or DFe on a timescale of 8 days (Boyd et al., 2010). The first sets of experiments performed at station 1 were incubated close to ambient temperature of 14°C in the dark for 8 days using three samples per treatment (i.e., sterilized and unsterilized) per time point 0 (time-zero, immediately after particle resuspension), 4 and 8 days. However, due to logistical constraints the second experiment at station 2 was incubated for 7 days with time points at 0, 4, and 7 days.
All samples were packed in black bags before initiating the incubation experiments to avoid exposure to light and subsequent photochemical breakdown of ligands (Barbeau et al., 2001) and kept at 14°C, the ambient temperature at 30 and 100 m depth. At each time-point 3 samples of sterilized and unsterilized samples were filtered using a 0.2 μm acid washed polycarbonate filter membrane for DFe and iron-binding ligand analyses.
The remainder of the concentrated particle samples (i.e., 70 ml) from each experiment were re-suspended in 10 L of unfiltered seawater from the station and depth where the particles were taken using the in-situ pumps. The re-suspended samples were then incubated in the dark for 8 or 7 days at 14°C for molecular characterization of Fe-binding ligands (Velasquez et al., 2011).
Five additional surface samples (which were not incubated, see Supplementary Information Table S1) were taken throughout the cruise and pre-concentrated in a 300 mL column filled with XAD-16 resin. These were extracted and analyzed parallel to the incubation experiment samples using chemical assays and LC-ESI-MS. The samples were taken for a separate study and results were not discussed in this paper. However, the results were used to verify absence/presence of ferrioxamine B and G and other masses with fragmentation pattern similar to known siderophores before the particle incubation experiments were done.
Particulate organic carbon (POC) measurements were done on parts of the same filter collected for the bioremineralization experiment as well as on full filters taken at 30 and 100 m using the McLane pumps. POC were measured at the National Institute of Water and Atmospheric Research (NIWA) laboratory in New Zealand. POC analyses were conducted on a Thermo-Finnigan Delta Plus Mass spectrometer using procedure MAM, 01–1090. Dissolved nutrients (N, P, Si) were analyzed on board ship by automated analysis (after Frew et al., 2001).
Bacteria abundances were determined by flow cytometry using a FACS Calibur instrument (Becton Dickinson) with CellQuest Vers. 3.3 software following the methods described in Lebaron et al. (1998). Samples were run on the high flow setting (60 μL min−1) with a minimum of 10,000 counts per sample. The bacterial population was identified using a FL1 (green light, 530/30 BP filter, log scale) vs. SSC (Cell size, linear scale) scatter plot.
Samples for the determination of culturable bacteria were serially diluted in sterile phosphate buffer solution (PBS) and were spread plated in duplicate on Marine Agar (MA; Difco Laboratories) and Chrome azurol S (CAS) Agar plates. CAS plates were prepared as described by Schwyn and Neilands (1987). Both Marine Agar and CAS Agar plates were incubated at 15°C and were counted after 4 weeks for colony forming units.
Iron-complexing ligands were determined by competing ligand equilibration-adsorptive cathodic stripping voltammetry (CLE-AdCSV) with 2-(2-Thiazolylazo)-p-cresol (TAC) as the competing ligand (Croot and Johansson, 2000). The voltammetric equipment consisted of a Metrohm VA 663 stand (Herisau, Switzerland) an IME 663 interface and a μAutolab type III potentiostat (both Metrohm Autolab, Utrecht, The Netherlands). The working electrode was a hanging mercury drop electrode (HMDE) with a mercury drop size of 2 (0.4 mm2 ± 10%). The reference electrode was Ag∣AgCl∣3 M KCl, and the counter electrode was a glassy carbon rod. All samples were contained in Teflon cells (Metrohm) during AdCSV analysis and stirred with the inbuilt all-PTFE Teflon stirring tip (1500 rpm) of the VA 663. The voltammetry system was computer controlled using the GPES software (Version 4.9, 2001–2009).
All sample manipulations were performed in a Class 100 laminar airflow bench at room temperature. Milli-Q ultrapure water (MQ, 18 MΩ) was used for the preparation of chemicals and rinsing throughout this study. Buffer solution of N-(2-hydroxyethyl)-piperazine-N′-2-propanesulfonic acid (EPPS, Sigma-Aldrich, St Louis, MO) was prepared in ammonium hydroxide (NH4OH) to give a pH of 8.1. NH4OH used for pH adjustments of the buffer was purified by passive vapor phase equilibration. A 10 mL Fe (III) stock solution was prepared from a standard solution of 1 g L−1 FeCl3·6H2O (Sigma), and acidified to ≤ pH 2 with quartz distilled HCL (q-HCl). A working iron standard solution (0.4 μM) was prepared daily from the stock 10 μM solution and acidified to ≤ pH 2 with q-HCl. 5.5 μM stock solution of TAC (Aldrich) was prepared in quartz distilled methanol (q-MeOH) every 2–4 weeks. All chemicals were kept in the fridge.
For the Fe-ligand titration, subsamples (12 mL) of seawater were pipetted into a series of 14 Teflon bottles and buffered at pH 8.1 with 50 μL of EPPS buffer (1.0 M). Iron was added to twelve of the bottles, yielding concentrations from 0 to 6 nM. After 1h equilibration, TAC was added to a final concentration of 3.5 μM, and the sample was left to equilibrate for at least 8 h. The samples were measured by DP-AdCSV from the lowest to the highest concentration in the usual manner with parameters as described by Ibisanmi et al. (2011) without rinsing in between aliquots. Cell cups were rinsed with Milli-Q water between titrations to avoid carry-over. Specific Teflon bottles were always used for the same iron concentration added and they were only rinsed with either Milli-Q or the sample itself in between titrations to keep them in equilibrium. Two to three scans were usually obtained for each sample. The DFe in freshly thawed ligand samples were determined by UV digesting it for 6 h using a Photoreactor Model APQ 40 (Photochemical Reactors) with a 400-W medium-pressure Hg lamp. Samples that were not freshly thawed were acidified to pH 2 prior to UV digestion. The pH of the samples was brought back to pH 8.0 with isothermally distilled NH4OH prior to analysis using the AdCSV protocols.
Ligand classifications were made according to the methods described by Ibisanmi et al. (2011). Briefly, L1 is operationally defined as a strong ligand having logKFeL1, Fe3+≥22 and L1 > DFe, whereas ΣL is operationally defined as the sum of all iron-complexing ligands detectable under the chosen analytical window. Ligand parameters were calculated using ProMCC software (Omanović et al., 2015).
Siderophore-like compounds were pre-concentrated by passing the filtered sample through an in-house prepared solid phase extraction (SPE) column containing 30 g pre-conditioned Amberlite XAD-16 resin (Sigma; Velasquez et al., 2011). The columns were loaded with organic compounds from the 0.2 μm filtered 10 L sample at the end of the incubation period. After loading the columns were washed with 500 mL MQ to remove salt. The organic compounds were subsequently eluted with three bed volumes of 95% methanol (Merck, HPLC grade). The eluents were collected in q-HCl cleaned Teflon bottles and kept refrigerated for further processing.
Chemical assays were done to test for the presence of siderophore moieties in the methanolic extracts of the XAD-16 column. The chrome azurol S (CAS) test for general iron binding capability (Schwyn and Neilands, 1987), Arnow and Rioux tests for catecholates (Arnow, 1937; Rioux et al., 1983) and Csaky test for hydroxamate type compounds (Gillam et al., 1981) were applied.
The methanolic XAD-16 extracts were dried using a SAVANT Speed Vac Plus SC210A and ~1.0 mg of sample was re-dissolved in 1.0 mL 5% acetonitrile (HPLC grade, Merck). The sample was passed through a 1.0 g SPE column (VARIAN Bond Elut C18-SPE). The sample was then eluted off sequentially with three bed-volumes of solutions with increasing methanol: water ratio (i.e., 10, 20, 30, and 70%). The different eluents were collected separately. The various methanol fractions were transferred to acid-cleaned Eppendorf tubes, and then evaporated until the volume of the sample remainder was reduce to < 10 μL, using the speed vac. The preconcentrated samples were diluted with 50 μL MQ and centrifuged for 3 min. The supernatant was injected into the HPLC.
The HPLC and mass spectrometry analyses were performed on an analytical HPLC system (DIONEX Ultimate 3000 System) attached to a Quadrupole Time-of-Flight mass spectrometer QTOF-MS; micrOTOF-Q, Bruker Daltonics). Chromatographic separations were carried out using a reversed phase column (250 × 4.6 mm, GraceSmart RP 18 5 μ, Grace Davison Discovery Sciences). Twenty microliters of sample was injected onto the column by means of an auto sampler. The HPLC separation was done in 52 min using a linear gradient of 6–90% acetonitrile:water with 0.1% formic acid. Two wavelengths (220 and 435 nm) were monitored to detect total hydrolysis products and ferric hydroxamate complexes, respectively.
The QTOF-MS used in this study utilized electrospray ionization (ESI) as its ion source and operated under positive mode. The scan range was set from 50 to 1000 m/z. The capillary voltage was 4500 V and the end plate offset was −500 V. The collision cell radio frequency (RF) was 90.0 voltage per pole (Vpp), the nebulizer pressure was set at 3.0 bar and the dry heater temperature was at 200°C. Throughout the analysis the dry gas flow rate was set at 7.0 L min−1.
For the determination of the fragmentation pattern of the molecule, the instrument was optimized and set to MSMS Auto mode. Since the concentration of the siderophore-like compounds present in the sample were expected to be very low, the MSMS (second mass spectra obtained after fragmentation of the highest peak) absolute threshold was set to an intensity of 500. To ensure that all masses of interest underwent MSMS analysis the machine was set to include chromatographic peaks with peak widths ≥12 s. The setting takes into account fast rising chromatographic peaks which can include siderophore-like ions. A summation factor of 3 was used to increase the intensity of MSMS spectra. The data for both single MS and MSMS analyses were obtained and processed using Brucker Software.
The mass spectrometer was calibrated using sodium formate calibrant. However, to ensure an accuracy of < 10 ppm, desferrioxamine B (DFB) was also injected directly to the mass spectrometer for calibration. In this calibration, an accuracy of < 2 ppm was generally obtained for the analysis. DFB was routinely used as a standard in the HPLC-MSMS analysis because it is commonly used as model siderophore. Other standards like mixed ferrioxamine, enterobactin, and aerobactin were interchangeably used as additional standards for the HPLC-MSMS analysis.
The natural Fe-siderophore complexes were identified using the natural Fe-isotopic ratio, which is a ratio of 56Fe/54Fe of 100/6.35 (De Hoffman and Stroobant, 1991). he criteria we looked for in details were: (1) ions that are bound to Fe should exhibit the Fe-isotopic pattern; (2) the 54Fe isotope is confirmed if natural abundance is within the range of 1–7% and the mass accuracy is within < 10 ppm (parts per million) and; (3) no molecular ion that is bound to Fe is present at the relevant retention time were observed in the blank. However, we are also reporting precursor ions that show the fragmentation pattern of ferrioxamines even in the absence of the Fe-complex.
DFe in microwave-sterilized particles treatments from both stations were not significantly different to the “no-particle” control treatment at the start of the experiment (Figure 2 and Table 1). DFe in microwaved-sterilized samples collected from 100 m at station 2 were accidentally not analyzed. L1 was detected in some of the sterilized samples but in low concentrations. Significant increases over 7 or 8 days in DFe and ΣL for the “live” particle samples were observed, while no L1−ligands were detectable in any of the treatments from station 1 and 2 on day 8 or 7, respectively (Table 1). The total DFe and ΣL released or produced at both stations 1 and 2 were generally higher at 30 m depth as compared to the 100 m depth samples (Table 2). While DFe was released throughout all four incubations, ΣL release or production turned into removal or consumption after day 4 at station 2 for 100 m, which was the only sample taken from below the mixed layer.
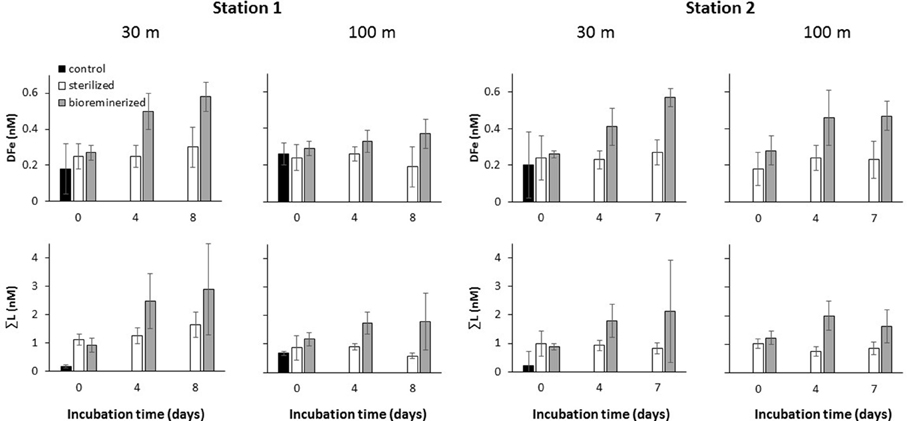
Figure 2. Release of DFe and production of ΣL. from re-suspended marine particles >5.0 μm during dark incubation at ambient seawater temperature of specified depths (i.e., 30 and 100 m) at station 1 and 2. Control filtered seawater and microwave sterilized particle suspensions were treated in the same manner.
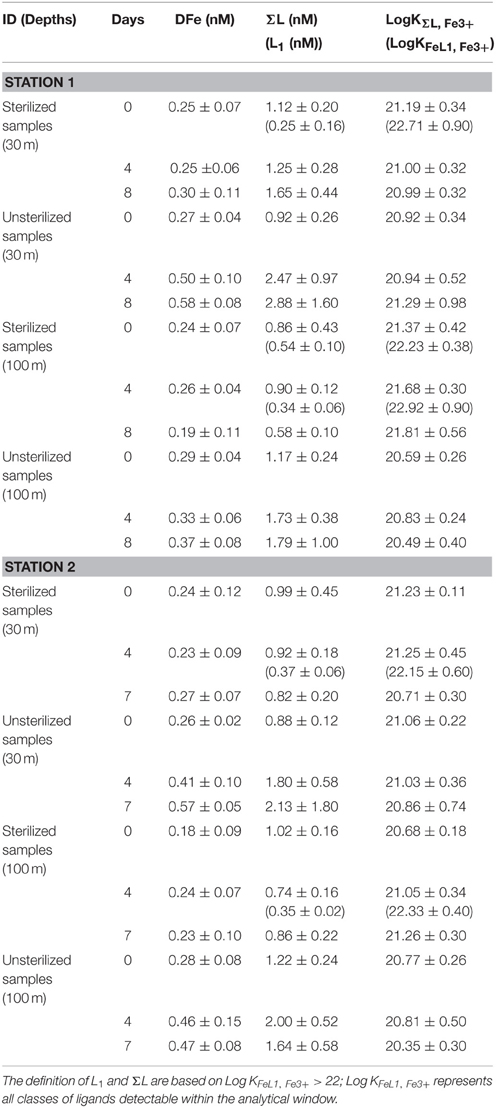
Table 1. Iron speciation data showing the concentration of dissolved iron (DFe), sum of ligand (ΣL) and L1, in brackets, and their conditional stability constants with respect to Fe3+ (LogKFeL1, Fe3+ and Log KΣL, Fe3+) observed during the remineralization experiments at 30 and 100 m of station 1 and 2.
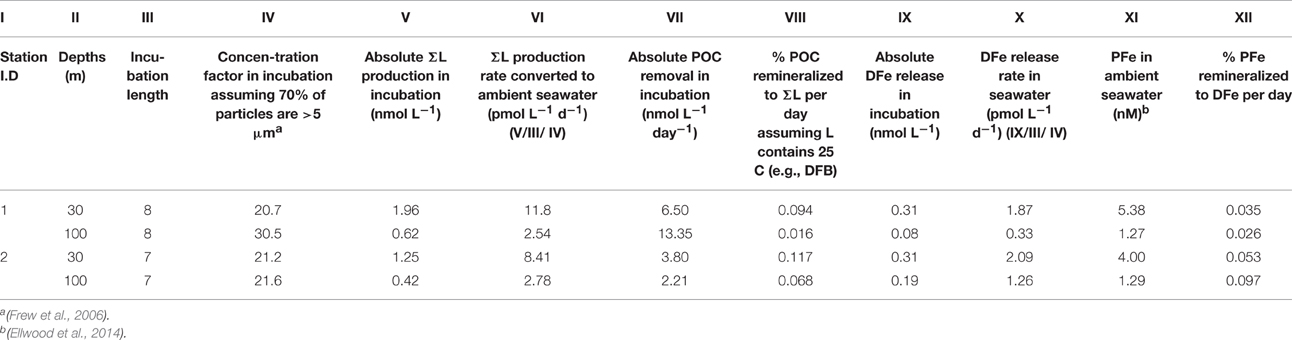
Table 2. ΣL production and DFe release rates from particle mineralization experiments.
The time-series of bacterial abundances in the samples with incubated particles is shown in Figure 3. For station 1, heterotrophic bacterial abundance was not significantly different on day 4 and 8. However, unfortunately no abundance data are available for day 0. At station 2, the 30 and 100 m samples both exhibited similar trends of peaking on day 4 and declining to close to initial abundances by day 7. Bacterial abundance did not change significantly over the duration of the treatment with sterilized particles. Bacterial abundance in the sterilized particles may represented dead or ghost cells counted by the flow cytometer. In the bacterial agar plates, the responsive bacteria in the community showed the same trend for all stations and depths, i.e., an increase in culturable heterotopic bacteria, as determined on marine agar for the duration of the experiment. Likewise, the CAS active bacteria also exhibited significant increases in abundances between start and end of the experiment (see Figure 4).
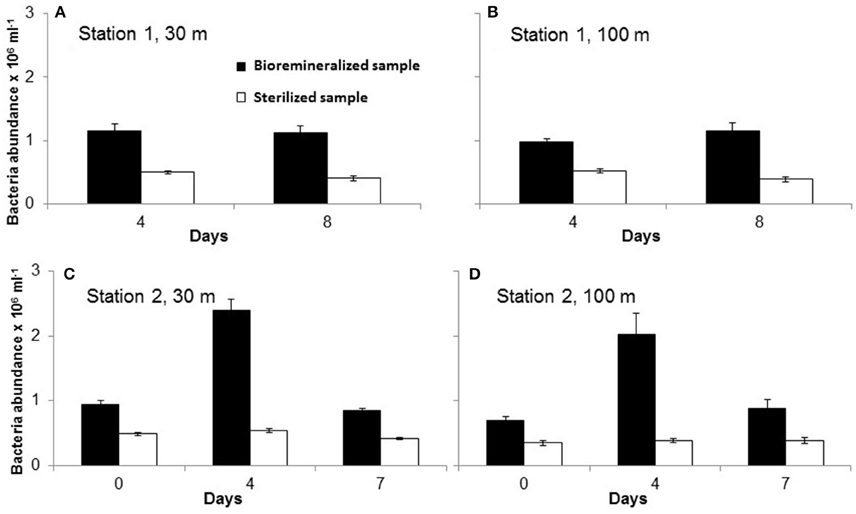
Figure 3. Abundance of bacteria counted in samples at different during the particle reminera lization experiment in unsterilized (filled black bars) and sterilized (unfilled bars) samples from 30 m (A,C) and 100 m (B,D) of station 1 (A,B) and Station 2 (C,D). Note that for station 1 day-0 samples were not measured due to instrument issues.
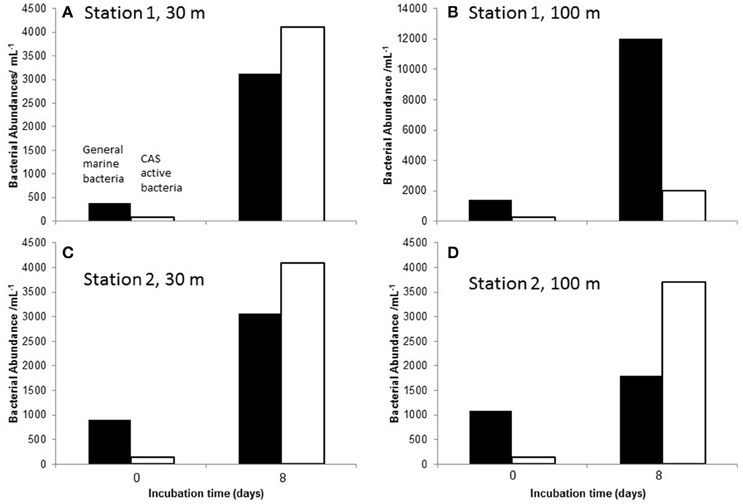
Figure 4. The number of heterotrophic marine bacteria (MA, black bars) and CAS active bacteria (unfilled bar) at the start (day 0) and the end of incubation period [day 8, or day 7 station 1 (A,B) and station 2 (C,D) respectively]. Note different scale in (B).
Nutrient analyses were not done in replicates due to sample volume limitation. Since observed changes in nutrients throughout the experiments were quite small and no errors or outliers were determined, we are not showing data here. Nitrate in station 1 incubation experiment was 3 times higher at 30 m than at 100 m, but did not change with time. Ammonia clearly increased in the “live” samples at station 1 for both depths. Nutrients analyzed from samples of station 2 are even more variable throughout and no clear trends were observed. The POC concentration measured in the beginning of the experiment from sections of the (5 μm) in-situ filter were converted to ambient concentrations using the concentration factors for each station and depth. Ambient seawater POC samples were taken from the same filters as used by the in-situ pump at the same day and depth where particles were taken for the remineralization experiment. POC concentrations were 5.1 and 5.6 μmol L−1 at 30 m and 2.5 and 2.2 μmol L−1 at 100 m for stations 1 and 2 respectively. No significant changes of the above mentioned parameters were observed in the sterilized particle treatments.
Results of different chemical assays run on extracts from samples incubated with particles and from large volume ambient seawater were CAS assay positive, confirming the presence of Fe-binding ligands in incubations from both depths at each station (see Table 3 and Supplementary Material Table S1). Similarly, hydroxamate moieties were found, using the Csaky assay, in all treatments. At station 1, no catecholate type compounds were detected by the Rioux assay from the treatments at the both depths. However, concentrations around the detection limit of the method were found in treatments of both depths from station 2. The detection limit of Rioux assay was 0.16 μM in the methanolic extract, which translates to a concentration in the treatment of 1.4 nM. The results are summarized in Table 3.
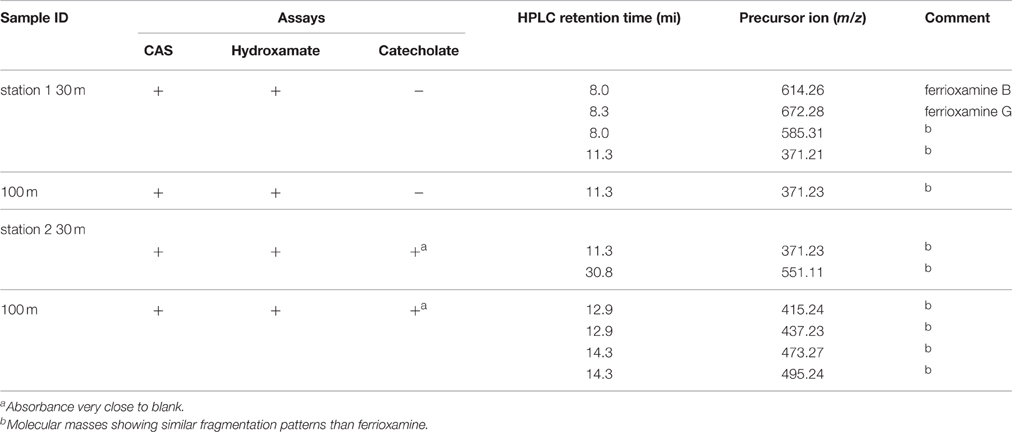
Table 3. Summary of results from the characterization of siderophore type compounds extracted from incubation experiments of marine particles at the end of the treatment.
Since the total amounts of dissolved organic material extracted from the particle remineralization experiments were very small, the intensities of the tandem MSMS analyses were relatively low but above the detection limit (45 nM based on DFB analyses with intensity of 102). From the 20% methanol extract, three siderophore-type compounds with m/z 585.31, 614.26 and 672.28 eluted off the C18-reverse phase column between 8 and 9 min (station 1, 30 m) while m/z 371.21 was observed at 11.3 min (station 1, 100 m). The natural Fe-isotopic pattern was observed in m/z 614.26 and 672.28 ([M+H+Fe-3H]+). The tandem MSMS analysis of m/z 614.26 yielded low intensity fragments (see Supplementary Material Figures SI4–SI6). However, the mass differences were similar to those commonly observed in hydroxamate siderophores. The simulated molecular formula for m/z 614.26 was within 10 ppm accuracy compared to the known linear ferrioxamine B (DFB; Figure 5A). Likewise, MSMS of m/z 672.28 yielded very low intensity fragments but the simulated molecular formula has mass accuracy within 10 ppm and is comparable to the known linear hydroxamate siderophore, ferrioxamine G (DFG; Figure 5B). For quality assurance, ferrioxamine B and G in a mixed ferrioxamine standard were analyzed and found to co-elute at ~8.0 min (Figure 5C). The ion peaks m/z 585.31 and m/z 371.21 ([M+H]+) did not exhibit the Fe-isotopic pattern but the MSMS fragments were equal to those of known a hydroxamate-type siderophores. However, these peaks may also be from non-siderophore compounds. The MSMS analysis of the m/z 371.21 precursor ion yielded very low intensity fragments and it was very difficult to evaluate fragment ions (see Supplementary Material Figure SI7). However, high intensity Na adducts of the compound were observed ([M+Na] m/z 393; [M+2Na] m/z 415; [M+3Na] m/z 437; [M+4Na] m/z 459). The MS spectra of 30% SPE methanol fraction in 100 m sample of station 1 yielded m/z 371.23 and its Na adducts. The mass differences found in the MSMS of [M+3Na] m/z 437 is also indicative of a hydroxamate-type siderophore (Feistner et al., 1993; Mawji et al., 2008b).
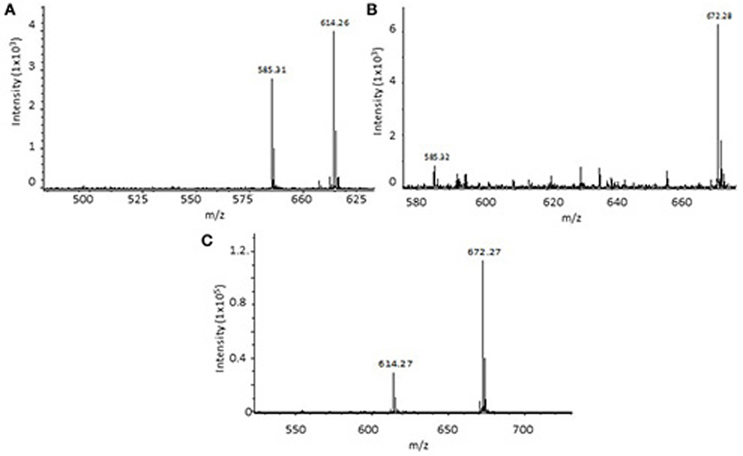
Figure 5. Mass spectra of particle bioremineralization sample station 1, 30 m at the end of the incubation, eluting at retention time (RT) 8.0 and 8.3 min, respectively. (A) mass resembling ferrioxamine B and (B) ferrioxamine G. Mass spectra of [M+H+Fe-3H]+ ferrioxamine B (m/z 614) and G (m/z 672) from a mixed ferrioxamine standard (RT 8.2 min) (C).
For the second incubation experiment (at station 2), the MS of the sample from day 7 of the 30 m treatment, likewise showed presence of m/z 371.21 (Table 3). An additional peak m/z 551.11 [M+H]+ was observed together with its [M+Na]+ m/z 573.09 at a retention time of 30.8 min. Mass differences of fragment ions indicated the presence of hydroxamate moieties, typical for ferrioxamine fragmentation (Feistner et al., 1993; Mawji et al., 2008b). The sample taken from day 7 of the 100 m treatment of station 2 yielded sodiated m/z 371.21 ([M+2Na] m/z 415.24; [M+3Na] m/z 437.23) and two ferri-siderophore-type ions ([M+H+Fe-3H]+) m/z 473.27 and m/z 495.24 (Table 3). The MSMS fragments of these two precursor ions suggests that the parent molecule may possibly be a ferrioxamine hydroxamate-type siderophore (Feistner et al., 1993; Mawji et al., 2008b). Further MS and MSMS spectra are given in the Supplementary Material.
In this study, we demonstrate that samples with marine particles in the presence of live particle-attached culturable bacteria produce or release iron binding ligands and DFe, whereas treatments with sterilized particles did not. In a previous remineralization experiment during a Southern Ocean study (subpolar and polar waters, SAZ-Sense study), it was also shown that in filter-sterilized samples (< 0.2 μm) neither the concentration of iron binding ligand, nor that of DFe increased significantly (Boyd et al., 2010). Unsurprisingly, it can be concluded that marine particles incubated in the dark were remineralized by attached bacteria (as the free-living bacteria were removed from the sea water, See Materials and Methods) and as a result Fe-binding ligands were produced at pM rates per day.
Table 2 shows calculations performed to estimate ligand production and DFe release rates in the concentrated particle suspension and converted to ambient seawater concentrations. The daily ligand production rate in seawater ranges from 2.54 pmol L−1 d−1 (100 m depth) to 11.8 pmol L−1 d−1 (30 m) for station 1, and rates for station 2 were slightly lower (see Table 2). Consideration of the estimated molecular size of Fe-binding ligands, in the order of 500–700 Da (e.g., DFB with 25 carbon atoms), means the ligand production rate can be converted into a DOC concentration and linked to a consumption of POC (see Table 2). For station 1, less POC was converted to L (0.094 and 0.016% for 30 and 100 m) than for station 2 where 0.117 and 0.068% of POC was converted to L per day.
The remineralization of PFe and the resulting release of DFe have also been calculated (Table 2). Since no PFe analysis was done directly in the incubations, the PFe concentrations from ambient seawater at the same depth one day after or before our sampling were used and extrapolated to our remineralization experiment. Errors associated with this extrapolation may be in the order of ±10%, but orders of magnitude of the ratio of PFe being remineralized to DFe per day can still be ascertained of being between 0.026 and 0.097%.
In the present study, ~2 pM L−1 d−1 of ligands were released or produced at a depth of 100 m, and around 10 pM L−1 d−1 at 30 m (see Table 2) in the subtropical waters East of New Zealand. In contrasting water masses (polar HNLC Southern Ocean, and a complex transition zone between subantarctic and subtropical waters), release rates were comparable in subsurface waters from ~110 m depth, i.e., 16.2 and 7.2 pM L−1 d−1 (Boyd et al., 2010). These values were all calculated for ambient seawater particle concentrations and display a surprising constancy across contrasting water masses.
Similarly, the DFe release rates were very comparable in all three water masses (subtropical, subantarctic, polar) ranging from 0.3 pmol L−1day−1 at 100 m in station 1 of the present study up to 3.6 pmol L−1day−1 at 111 m depth (subantarctic, Boyd et al., 2010). Interestingly PFe concentrations in the present study were one order of magnitude higher than in the water masses used in the Boyd et al. (2010) study. This discrepancy raises the question of which PFe pool will most likely be available for bacteria to be remineralized. In Boyd et al. (2010), it was postulated that detrital PFe, which contributes mostly to PFe export flux in the Southern Ocean waters (i.e., PFZ study site; Frew et al., 2006; Sarthou et al., 2008) would be the source of DFe during remineralization over a greater depth horizon, while algal PFe would probably be restricted mainly in the stratum within the mixed layer. This however cannot explain the release rates observed here as all but station 2, 100 m were within the mixed layer.
The column integrated DFe release calculated from values in Table 2 is 107 nmol m−2 day−1 at station 1 and 58.1 nmol m−2 day−1 at station 2 (for 30–100 m). These values compare well with results from the previous remineralization experiment (Boyd et al., 2010), i.e., 42 nmol m−2 day−1 and 108 nmol m−2 day−1 at the polar and subantarctic sites, respectively.
Given a mixed layer DFe inventory of ~48 μmol m−2 and ~4 μmol m−2 at the time of sampling for station 1 and 2 (Boyd et al., 2010), it would result in a DFe residence time of 1.2 and 0.18 years assuming that particle remineralization was the only source of iron to the mixed layer. These calculated residence times compares well with results from similar particle remineralization experiments in the PFZ and NSAZ of 0.4 and 0.2 year, respectively (Boyd et al., 2010) and those from a model simulation of the upper 103 m of a low-iron high-nutrient region, which were 1.4 year (Moore and Braucher, 2008). Our calculations are rough estimates for a body of water in the mixed layer right where we sampled. The lateral or vertical advection of DFe and any other biological process from biota >5 μm were not taken into account. The approximately one order of magnitude variability within 8 days in the same water mass demonstrates the dynamic of the process. Although it could be said that the residence times of iron binding ligands may be in the same order of magnitude we refrain from giving number here as the ligand concentration changed significantly throughout the bloom development and unfortunately we did not sample for a full ligand depth profile on day 254. Taking the ligand depth distribution from day 263 would make the residence time calculations meaningless.
The cell counts of both culturable bacteria types [i.e., heterotrophic bacterial community (MA) and Fe-binding ligand producing bacteria (CAS)] increased throughout all “live” particle treatment remineralization experiments (see Figure 4). The CAS assay is often used by microbiologist to test for siderophore production, however in general it detects any strong Fe-binding compound regardless of the function of the molecule. Thus, the increase in CAS active bacterial numbers may not necessarily prove that siderophore-producing bacteria are present, but the increase in cell abundances at the end of the incubation provides evidence that Fe-binding ligand-producing bacteria are increasing in numbers in the suspension of particles (at greater than ambient concentrations) and these ligands may be siderophores. The failure to detect strong L1-type ligands in any of the treatments may mean that the concentration of siderophores produced is too low to be detected at the given DFe concentration or that the siderophores produced have stability constants < logKFeL1, Fe3+ = 22 (Maldonado et al., 2005).
Although the bacterial community in the remineralization experiment was not characterized directly, such data was produced from ambient seawater and in-situ sediment trap incubations throughout the FeCycleII-Springbloom cruise. Results showed a very dynamic response of the community in this water mass throughout the bloom (LeCleir et al., 2014). They showed that the microbial community in the water column and particles collected in sediment traps (at 5 and 100 m depth) were very similar in their bacterial composition, and only bacterial abundances varied between the two different bloom phases and with depth. LeCleir et al. (2014) proposed the presence of a “core” microbial community in this locale. They also found that heterotrophic bacteria of the Roseobacter clade and some other bacteria rapidly colonized the particles from 5 to 100 m in high abundance. Roseobacter have been shown to produce siderophores (Poorvin et al., 2011) which may be a source of the ligands produced during our particle remineralization experiments.
The chemical characterization of Fe-binding ligands produced in the “live” particle treatments was done semi-quantitively in methanolic extracts of the XAD-16. The CAS test is widely used due to its comprehensive, sensitive nature and can specifically detect Fe3+ binding ligands. In our study, CAS test gave positive results in all incubations at the end of the experiment. The negative results for the catecholate specific Arnow test may indicate either the absence of any cathecol type of siderophore, or it could also mean that the catechol-functionality is sterically hindered. Samples from station 1 at 30 and 100 m, which were collected at the end (8 days) of the particle remineralization experiment were positive to Csaky (hydroxamate) and negative to both the catecholate sensitive Arnow and Rioux tests (Table 3). This suggests that the Fe-binding ligands produced during both incubations are likely hydroxamate-type siderophores. In particle remineralization incubations at station 2, two hydroxamate and a catechol type siderophores or a mixed type of siderophore may have been produced (Table 3).
The detected hydroxamate siderophores in the remineralization experiments only ranged from 9 to 24% of the [ΣL]. It may be an underestimate because the calculations were based on the concentration of hydroxamate compounds detected in the pre-concentrated XAD-16 methanol extracts and the extraction efficiency is < 100%. It is not known what makes up the other 91–76% of Fe-binding ligands detected in our experiment, but exopolysaccharides, humic substances, porphyrins and others have also been shown to bind Fe at stability constants very similar to that detected in our experiments (Hutchins et al., 1999; Laglera and van den Berg, 2009; Hassler et al., 2011; Abualhaija and van den Berg, 2014). We did not test for the presence of any of these compounds in this study.
Molecular ions m/z 614.26 and m/z 672.28 were detected at the end of station 1's 30 m particle remineralization experiment. The mass spectrometry analysis suggests that these correspond to the well-known linear hydroxamate ferri-ferrioxamine B (DFB) [M+H+Fe-3H]+ and ferri-ferrioxamine G (DFG) [M+H+Fe-3H]+. The compounds were present at low intensity, however the Fe-isotopic pattern was very clear and the retention times fall within the elution time of the ferrioxamine B and G standards (Figures 5A–C). Feistner et al. (1993) reported that biosynthetic capabilities of an organism to produce siderophores can be flexible and considered the open chained ferrioxamines as biosynthetic intermediates of cyclic ferrioxamines (Feistner et al., 1993; Mawji et al., 2008b). But however, until to date, the presence of ferrioxamine B and G associated with the remineralization of marine particles had not been reported.
The ion m/z 585.31 did not exhibit the Fe-isotopic pattern, but, it was previously shown by (Feistner and Hsieh, 1995) that m/z 585 can be observed as a result of protonation at the N-terminal of the diaminopentane group, then followed by a charge-driven fragmentation in the N-terminal of the hydroxamate bond. The ion peak m/z 371.21 was found in station 1 at 30 and 100 m, and in station 2 at 30 m. Although it did not exhibit the Fe-isotopic pattern, the mass difference of its MSMS fragments were similar to those also obtained for the ferrioxamines (Feistner et al., 1993; Mawji et al., 2008b). In station 2 at the 100 m treatment, only the 2 and 3 Na adducts (m/z 415.24 and 437.23) of m/z 371.21 were obtained at the end of the experiment (7 days). The ion peak m/z 551.11 [M+H]+ found in station 2 did not show the iron isotopic pattern (i.e., was not bound to Fe) in the 30 m sample, but the mass difference observed for the MSMS fragments were identical to those shown by Feistner et al. (1993) for ferrioxamines. Fe-bound ligands were observed for 100 m sample at station 2 which again showed fragmentation pattern of the hydroxamate ferrioxamine type siderophores. It is possible that the siderophores that we detected were photoproducts of siderophore molecules that were present in low levels. The Fe(III)-siderophore complex may have been truncated or undergone ligand oxidation due to photolysis leading to a lower binding strength (Barbeau et al., 2001). Molecular ions m/z 371, 551, 437, 415, 43, and 495 were also found in samples collected from ambient water which were not incubated (see Supplementary Material). No trace of m/z 585, 614, and 672 were observed in any extracts from the ambient samples.
It is clear that the siderophores found for both depths in each experiment are hydroxamates, but the known siderophores ferrioxamine B and G were only found in the incubation experiments of surface samples (30 m) from station 1. A shift in the dominant groups in the microbial community (LeCleir et al., 2014) will likely have an effect on the ligands present in the water and in the particulates. However, the information available from our study is insufficient to clearly link the relationship between the microbes and the siderophore-type compounds that were identified.
In this study, two marine particle remineralization experiments were performed to investigate the production of ligands and the release of DFe in seawater at two depths at 30 and 100 m. Particulate samples were collected from the subtropical waters east of the North Island of New Zealand as part of the FeCycle II GEOTRACES process study. Significant ligand production was observed in treatments containing live particle attached heterotrophic bacteria. Hence, it can be concluded that some of the iron-complexing ligands measured in the ocean originated from the remineralization of particles.
Two main classes of ligands, namely strong (L1) and sum of all ligands (ΣL), have been identified in our study. Open chained ferrioxamine B and G, as well as other unknown hydroxamate siderophores have been identified in samples from both particle remineralization experiments. Two other unidentified siderophore-types with their Na adducts were observed. The MSMS spectra of these compounds suggest that they are possibly hydroxamate types similar to ferrioxamines. The presence of siderophore-producing bacteria and results from other chemical assays suggest the mass spectrometric observations of siderophore production in the experimental treatments. The electrochemical analysis of L failed to detect L1 from the samples, although siderophores were detected by MS. This finding questions whether siderophores present in the ocean are truly L1 or their concentration and contribution into the ligand pool may be too small compared to other weaker organic ligands produced during the remineralization and throughout in the open ocean.
IBV and SGS ran the experiment on board ship, PWB helped design the experiment. EWM measured bacterial data and helped their interpretation. EI performed voltammetric analysis, SN took particle samples and ran POC analysis. SGS, IBV, and EI wrote the manuscript which was commented by PWB and EWM.
EI and IBV received a University of Otago PhD scholarship. SGS and PWB were supported by Marsden grant UOO1117.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Special thanks to the officers and crew members of RV Tangaroa during the FeCycle II study for making this work possible. We thank Michael Ellwood (PFe), Karen Thompson (nutrients and flow cytometry), and the trace metal clean sampling team onboard. We acknowledge financial support from the University of Otago, the National Institute of Water and Atmospheric Research (NIWA), and the Hanse-Wissenschaftskolleg Institute for Advanced Study, Delmenhorst, Germany.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00172
Abualhaija, M. M., and van den Berg, C. M. G. (2014). Chemical speciation of iron in seawater using catalytic cathodic stripping voltammetry with ligand competition against salicylaldoxime. Mar. Chem. 164, 60–74. doi: 10.1016/j.marchem.2014.06.005
Adly, C. L., Tremblay, J.-E., Powell, R. T., Armstrong, E., Peers, G., and Price, N. M. (2015). Response of heterotrophic bacteria in a mesoscale iron enrichment in the northeast subarctic Pacific Ocean. Limnol. Oceanogr. 60, 136–148. doi: 10.1002/lno.10013
Arnow, L. E. (1937). Colorimetric determination of the components of 3,4-dihydroxyphenylalanine-tyrosine mixtures. J. Biol. Chem. 118, 531–537.
Barbeau, K., Rue, E. L., Bruland, K. W., and Butler, A. (2001). Photochemical cycling of iron in the surface ocean mediated by microbial iron (III) -binding ligands. Nature 413, 409–413. doi: 10.1038/35096545
Behrenfield, M. J., Bale, A. J., Kolber, Z. S., Aiken, J., and Falkowski, P. G. (1996). Confirmation of iron limitation of phytoplankton photosynthesis in the equatorial Pacific Ocean. Nature 383, 508–511. doi: 10.1038/383508a0
Boyd, P., Watson, A., Law, C., Abraham, E., Trull, T., Murdoch, R., et al. (2000). A mesoscale phytoplankton bloom in the polar Southern Ocean stimulated by iron fertilization. Nature 407, 695–702. doi: 10.1038/35037500
Boyd, P. W., Ibisanmi, E., Sander, S. G., Hunter, K. A., and Jackson, G. A. (2010). Remineralization of upper ocean particles: implications for iron biogeochemistry. Limnol. Oceanogr. 55, 1271–1288. doi: 10.4319/lo.2010.55.3.1271
Boyd, P. W., Law, C. S., Hutchins, D. A., Abraham, E. R., Croot, P. L., Ellwood, M., et al. (2005). FeCycle: attempting an iron biogeochemical budget from a mesoscale SF6 tracer experiment in unperturbed low iron waters. Global Biogeochem. Cycles 19:GB4S20. doi: 10.1029/2005GB002494
Boyd, P. W., Strzepek, R., Chiswell, S., Chang, H., DeBruyn, J. M., Ellwood, M., et al. (2012). Microbial control of diatom bloom dynamics in the open ocean. Geophys. Res. Lett. 39:L18601. doi: 10.1029/2012GL053448
Boye, M., van den Berg, C., de Jong, J., Leach, H., Croot, P. L., and de Baar, H. J. W. (2001). Organic complexation of iron in the Southern Ocean. Deep Sea Res. 48, 1477–1497. doi: 10.1016/S0967-0637(00)00099-6
Buck, K. N., Lohan, M. C., Berger, C. J. M., and Bruland, K. W. (2007). Dissolved iron speciation in two distinct river plumes and an estuary: implications for riverine iron supply. Limnol. Oceanogr. 52, 843–855. doi: 10.4319/lo.2007.52.2.0843
Buck, K. N., Sohst, B., and Sedwick, P. N. (2015). The organic complexation of dissolved iron along the U.S. GEOTRACES (GA03) North Atlantic Section. Deep Sea Res. Part II 116, 152–165. doi: 10.1016/j.dsr2.2014.11.016
Bundy, R. M., Biller, D. V., Buck, K. N., Bruland, K. W., and Barbeau, K. A. (2014). Distinct pools of dissolved iron-binding ligands in the surface and benthic boundary layer of the California Current. Limnol. Oceanogr. 59, 769–787. doi: 10.4319/lo.2014.59.3.0769
Croot, P. L., and Johansson, M. (2000). Determination of Iron Speciation by Cathodic Stripping Voltammetry in Seawater Using the Competing Ligand 2-(2-Thiazolylazo)-p-cresol (TAC). Electroanalysis 12, 565–576. doi: 10.1002/(SICI)1521-4109(200005)12:8<565::AID-ELAN565>3.0.CO;2-L
De Hoffman, K., and Stroobant, V. (1991). Fast atom bombardment Tandem Mass spectrometric analysis of hydroxamate siderophores. Biol. Mass Spectrom. 20, 142–152. doi: 10.1002/bms.1200200308
Ellwood, M. J., Hutchins, D. A., Lohan, M. C., Milne, A., Nasemann, P., Nodder, S. D., et al. (2015). Iron stable isotopes track pelagic iron cycling during a subtropical phytoplankton bloom. Proc. Natl. Acad. Sci. U.S.A. 112, E15–E20. doi: 10.1073/pnas.1421576112
Ellwood, M. J., Nodder, S. D., King, A. L., Hutchins, D. A., Wilhelm, S. W., and Boyd, P. W. (2014). Pelagic iron cycling during the subtropical spring bloom, east of New Zealand. Mar. Chem. 160, 18–33. doi: 10.1016/j.marchem.2014.01.004
Feistner, G. J., and Hsieh, L. L. (1995). On the collision-activated fragmentation of proferrioxamines: evidence for a succinimide-mediated mechanism. J. Am. Soc. Mass Spectrom. 6, 836–846. doi: 10.1016/1044-0305(95)00324-7
Feistner, G. J., Stahi, D. C., and Gabrik, A. H. (1993). Proferrioxamine siderophores of Erwinia Amylovora. A capillary liquid chromatographic/Electrospray tandem mass spectrometric study. J. Mass Spectrom. 28 163–175.
Frew, R., Bowie, A., Croot, P., and Pickmere, S. (2001). Nutrient and tracemetal geochemistry of an in situ iron-induced Southern Ocean bloom. Deep Sea Res. Part II: Top. Stud. Oceanogr. 48, 2467–2481. doi: 10.1016/S0967-0645(01)00004-2
Frew, R. D., Hutchins, D. A., Nodder, S., Sanudo-Wilhelmy, S., Tovar-Sanchez, A., Leblanc, K., et al. (2006). Particulate iron dynamics during FeCycle in subantarctic waters southeast of New Zealand. Global Biogeochem. Cycles 20:GB1S93. doi: 10.1029/2005GB002558
Geider, R. J., and La Roche, J. (1994). The role of iron in phytoplankton photosynthesis, and the potential for iron-limitation of primary productivity in the sea. Photosyn. Res. 39, 275–301. doi: 10.1007/BF00014588
Gillam, A. H., Lewis, A. G., and Andersen, R. J. (1981). Quantitative determination of hydroxamic acids. Anal. Chem. 53, 841–844. doi: 10.1021/ac00229a023
Gledhill, A., McCormack, P., Ussher, S., Achterberg, E., Mantoura, R. F. C., and Worsfold, P. J. (2004). Production of siderophore type chelates by mixed bacterioplancton populations in nutrient enriched seawater incubations. Mar. Chem. 88, 75–83. doi: 10.1016/j.marchem.2004.03.003
Gledhill, M., and Buck, K. N. (2012). The organic complexation of iron in the marine environment: a review. Front. Microbiol. 3:69. doi: 10.3389/fmicb.2012.00069
Gledhill, M., and van den Berg, C. M. G. (1994). Determination of complexation of iron(III) with natural organic complexing ligands in seawater using cathodic stripping voltammetry. Mar. Chem. 47, 41–54. doi: 10.1016/0304-4203(94)90012-4
Hassler, C. S., Schoemann, V., Nichols, C. M., Butler, E. C. V., and Boyd, P. W. (2011). Saccharides enhance iron bioavailability to Southern Ocean phytoplankton. Proc. Natl. Acad. Sci. U.S.A. 108, 1076–1081. doi: 10.1073/pnas.1010963108
Hunter, K. A., and Boyd, P. W. (2007). Iron-binding ligands and their role in the ocean biogeochemistry of iron. Environ. Chem. 4, 221–232. doi: 10.1071/EN07012
Hutchins, D. A., Witter, A. E., Butler, A., and Luther, G. W. (1999). Competition among marine phytoplankton for different chelated iron species. Nature (London) 400, 858–861.
Ibisanmi, E., Sander, S. G., Boyd, P. W., Bowie, A. R., and Hunter, K. A. (2011). Vertical distributions of iron-(III) complexing ligands in the Southern Ocean. Deep Sea Res. II: Top. Stud. Oceanogr. 58, 2113–2125. doi: 10.1016/j.dsr2.2011.05.028
Laglera, L. M., and van den Berg, C. M. G. (2009). Evidence for geochemical control of iron by humic substances in seawater. Limnol. Oceanogr. 54, 610–619. doi: 10.4319/lo.2009.54.2.0610
Lebaron, P., Ghiglione, J.-F., Fajon, C., Batailler, N., and Normand, P. (1998). Phenotypic and genetic diversity within a colony morphotype. FEMS Microbiol. Lett. 160, 137–143. doi: 10.1111/j.1574-6968.1998.tb12903.x
LeCleir, G. R., DeBruyn, J. M., Maas, E. W., Boyd, P. W., and Wilhelm, S. W. (2014). Temporal changes in particle-associated microbial communities after interception by nonlethal sediment traps. FEMS Microbiol. Ecol. 87, 153–163. doi: 10.1111/1574-6941.12213
Lenes, J. M., Walsh, J. J., Prospero, J. M., and Byrne, R. H. (2009). Response to “Aerosol iron deposition to the surface ocean - Modes of iron supply and biological responses” by PW Boyd, DS Mackie, and KA Hunter. Mar. Chem. 116, 56–57. doi: 10.1016/j.marchem.2009.07.006
Mahmood, A., Abualhaija, M. M., van den Berg, C. M. G., and Sander, S. G. (2015). Organic speciation of dissolved iron in estuarine and coastal waters at multiple analytical windows. Mar. Chem. 177(Pt 5), 706–719. doi: 10.1016/j.marchem.2015.11.001
Maldonado, M. T., Strzepek, R. F., Sander, S., and Boyd, P. W. (2005). Acquisition of iron bound to strong organic complexes-with different Fe binding groups and photochemical reactivities by plankton communities in Fe-limited subantarctic waters. Global Biogeochem. Cycles 19:GB4S23. doi: 10.1029/2005GB002481
Martinez, J. S., Haygood, M. G., and Butler, A. (2001). Identification of a natural desferrioxamine siderophore produced by a marine bacterium. Limnol. Oceanogr. 46(Suppl. 7), 420–424. doi: 10.4319/lo.2001.46.2.0420
Mawji, E., Gledhill, M., Milton, J. A., Tarran, G. A., Ussher, S., Thompson, A., et al. (2008a). Hydroxamate Siderophores: Occurrence and Importance in the Atlantic Ocean. Environ. Sci. Technol. 42, 8675–8680. doi: 10.1021/es801884r
Mawji, E., Gledhill, M., Worsfold, P. J., and Achterberg, E. P. (2008b). Collision-induced dissociation of three groups of hydroxamate siderophores: ferrioxamines, ferrichromes and coprogens/fusigens. Rapid Commun. Mass Spectrom. 22, 2195–2202. doi: 10.1002/rcm.3604
Moore, C. M., Mills, M. M., Arrigo, K. R., Berman-Frank, I., Bopp, L., Boyd, P. W., et al. (2013). Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 6, 701–710. doi: 10.1038/ngeo1765
Moore, J. K., and Braucher, O. (2008). Sedimentary and mineral dust sources of dissolved iron to the world ocean. Biogeosciences 5, 631–656. doi: 10.5194/bg-5-631-2008
Morel, F. M. M., and Price, I. G. (2003). The biogeochemical cycles of trace metals in the oceans. Science 300, 944–947. doi: 10.1126/science.1083545
Omanović, D., Garnier, C., and Pižeta, I. (2015). ProMCC: an all-in-one tool for trace metal complexation studies. Mar. Chem. 173, 25–39. doi: 10.1016/j.marchem.2014.10.011
Pižeta, I., Sander, S. G., Hudson, R. J. M., Omanović, D., Baars, O., Barbeau, K. A., et al. (2015). Interpretation of complexometric titration data: an intercomparison of methods for estimating models of trace metal complexation by natural organic ligands. Mar. Chem. 173, 3–24. doi: 10.1016/j.marchem.2015.03.006
Poorvin, L., Sander, S. G., Velasquez, I., Ibisanmi, E., LeCleir, G. R., and Wilhelm, S. W. (2011). A comparison of Fe bioavailability and binding of a catecholate siderophore with virus-mediated lysates from the marine bacterium Vibrio alginolyticus PWH3a. J. Exp. Mar. Biol. Ecol. 399, 43–47. doi: 10.1016/j.jembe.2011.01.016
Reid, R. T., Live, D. H., Faulkner, D. J., and Butler, A. (1993). A siderophore from a marine bacterium with an exeptional ferric iron affinity constant. Nature (London) 366, 455–458. doi: 10.1038/366455a0
Rioux, C., Jordan, D. C., and Rattray, J. B. M. (1983). Colorimetric determination of catechol siderophores in microbial cultures. Anal. Biochem. 133, 163–169. doi: 10.1016/0003-2697(83)90238-5
Rue, E. L., and Bruland, K. W. (1995). Complexation of iron (III) by natural organic-ligands in the Central North Pacific as determined by a new competitive ligand equilibration adsorptive cathodic stripping voltammetric method. Mar. Chem. 50, 117–138. doi: 10.1016/0304-4203(95)00031-L
Rue, E. L., and Bruland, K. W. (1997). The role of organic complexation on ambient iron chemistry in the Equatorial Pacific Ocean and the response of a mesoscale iron addition experiment. Limnol. Oceanogr. 42, 901–910. doi: 10.4319/lo.1997.42.5.0901
Sander, S. G., Hunter, K. A., Harms, H., and Wells, M. (2011). Numerical approach to speciation and estimation of parameters used in modeling trace metal bioavailability. Environ. Sci. Technol 45, 6388–6395. doi: 10.1021/es200113v
Sander, S. G., Tian, F., Ibisanmi, E. B., Currie, K. I., Hunter, K. A., and Frew, R. D. (2015). Spatial and seasonal variations of iron speciation in surface waters of the Subantarctic front and the Otago Continental Shelf. Mar. Chem. 173, 114–124. doi: 10.1016/j.marchem.2014.09.001
Sander, S., Hunter, K., and Frew, R. (2009). “Sampling and measurement of trace metals in seawater,” in Practical Guidelines for the Analysis of Seawater, ed O. Wurl (Boca Raton, FL: CRC Press), 305–328.
Sarthou, G., Vincent, D., Christaki, U., Obernoster, I., Timmermans, K. R., and Brussard, C. P. D. (2008). The fate of biogenic iron during a phytoplankton bloom induced by natural fertilisation: impact of copepod grazing. Deep Sea Res. II 55, 734–752. doi: 10.1016/j.dsr2.2007.12.033
Schwyn, B., and Neilands, J. B. (1987). Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160, 47–56. doi: 10.1016/0003-2697(87)90612-9
Tian, F., Frew, R., Strzepek, R., and Ellwood, M. (2006). “Iron organic complexation in the oligotrophic subtropical waters in the Tasman Sea,” in Advances in Geoscience, eds W.-H. Ip and Y.-T. Chen (Singapore: World Scientific Publishing Company), 75–89.
Turner, S. M., Harvey, M. J., Law, C. S., Nightingale, P. D., and Liss, P. S. (2004). Iron-induced changes in oceanic sulfur biogeochemistry. Geophys. Res. Lett. 31:L14307. doi: 10.1029/2004GL020296
Van den Berg, C. M. G. (1995). Evidence for organic complexation of iron in seawater. Mar. Chem. 50, 139–157. doi: 10.1016/0304-4203(95)00032-M
Velasquez, I., Nunn, B. L., Ibisanmi, E., Goodlett, D. R., Hunter, K. A., and Sander, S. G. (2011). Detection of hydroxamate siderophores in coastal and Sub-Antarctic waters off the South Eastern Coast of New Zealand. Mar. Chem. 126, 97–107. doi: 10.1016/j.marchem.2011.04.003
Weller, D. I., Law, C. S., Marriner, A., Nodder, S. D., Chang, F. H., Stephens, J. A., et al. (2013). Temporal variation of dissolved methane in a subtropical mesoscale eddy during a phytoplankton bloom in the southwest Pacific Ocean. Prog. Oceanogr. 116, 193–206. doi: 10.1016/j.pocean.2013.07.008
Wells, M., Buck, K. N., and Sander, S. G. (2013). New approach to analysis of voltammetric ligand titration data improves understanding of metal speciation in natural waters. Limnol. Oceanogr. Methods 11, 450–465. doi: 10.4319/lom.2013.11.450
Keywords: siderophores, voltammetry, dfb, particle remineralization, iron, particulate matter
Citation: Velasquez IB, Ibisanmi E, Maas EW, Boyd PW, Nodder S and Sander SG (2016) Ferrioxamine Siderophores Detected amongst Iron Binding Ligands Produced during the Remineralization of Marine Particles. Front. Mar. Sci. 3:172. doi: 10.3389/fmars.2016.00172
Received: 07 May 2016; Accepted: 30 August 2016;
Published: 22 September 2016.
Edited by:
Claire Mahaffey, University of Liverpool, UKReviewed by:
Martha Gledhill, GEOMAR Helmholtz-Zentrum für Ozeanforschung, GermanyCopyright © 2016 Velasquez, Ibisanmi, Maas, Boyd, Nodder and Sander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvia G. Sander, c3lsdmlhLnNhbmRlckBvdGFnby5hYy5ueg==
†Present Address: Elizabeth W. Maas, Ministry for Primary Industries, Napier, New Zealand
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.