 Cavin Mgawe1,2,3
Cavin Mgawe1,2,3 Clement Shilluli2
Clement Shilluli2 Steven Nyanjom1Eddy Odari4
Steven Nyanjom1Eddy Odari4 Jacqueline C. Linnes5
Jacqueline C. Linnes5 Bernard N. Kanoi2
Bernard N. Kanoi2 Jesse Gitaka2*†
Jesse Gitaka2*† Lucy Ochola4*†
Lucy Ochola4*†- 1Department of Biochemistry, Jomo Kenyatta University of Agriculture and Technology (JKUAT), Juja, Kenya
- 2Centre for Malaria Elimination, Institute of Tropical Medicine, Mount Kenya University, Thika, Kenya
- 3Department of Tropical and Infectious Diseases, Institute of Primate Research, Nairobi, Kenya
- 4Department of Medical Microbiology, Jomo Kenyatta University of Agriculture and Technology (JKUAT), Juja, Kenya
- 5Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN, United States
Introduction: Malaria remains a significant health concern, particularly in regions with widespread prevalence. As the transmission rates decrease, there is a rise in low-density infections with the causative parasite, P. falciparum, that often escape detection through standard point-of-care diagnostic tools. In-low transmission areas, even few undetected cases can trigger outbreaks, necessitating rapid and sensitive diagnostics. Loop-mediated isothermal Amplification (LAMP) stands out as a nucleic acid technique that can easily utilizes un-processed samples such of saliva, urine, and lysed whole blood templates for a sensitive detection. However, most nucleic acid tests detect genes with few copies per parasite making it difficult to detect low-density parasitaemia.
Methods: We selected Pfr364 multi-copy repeats of the P. falciparum genome as a target for amplification due to their higher copy number, ideal for rapid amplification, addressing amplification drawbacks of limited parasites DNA. We used a sequence clustering approach to design a novel set of LAMP primers, capable of binding to multiple sites. Subsequently, we developed a hydroxynaphthol blue (HNB) colorimetric LAMP assay, using genomic DNA obtained from the 3D7 strain cultivated in vitro. This assay’s performance was validated using archived clinical samples of both whole blood and matched saliva, ensuring accuracy through comparative analysis against gold standard, nested PCR, targeting the 18S RNA gene.
Results: The HNB-LAMP assay achieved rapid amplification within 15 minutes and exhibited high sensitivity with a limit of detection of 1 parasite. Further, the LAMP assay was robust in whole blood lysed with Triton X-100 and heat-treated saliva clinical samples. Against nested PCR, the assay showed sensitivity of 100% for whole blood and 40% for saliva samples. Moreover, co-analysis with the nested PCR showed a perfect agreement between the two techniques. (K = 0.99 for whole blood, and 0.66 for saliva).
Conclusion: Our study presents a method for detecting P. falciparum using LAMP, which results in increased sensitivity, shorter assay times, and a simpler workflow than nucleic acid tests relying on conventional DNA extraction and additional equipment for result interpretation. These findings hold great promise for improved malaria diagnosis, especially in settings where low-density parasitaemia is prevalent and rapid and accurate malaria detection is crucial.
Introduction
Malaria is a global public health challenge, causing significant morbidity and mortality worldwide (WHO, 2022). Although the implementation of multiple interventions have led to a decline in malaria incidence and burden, the detection of sub-microscopic and asymptomatic infections remains a challenge. These infections are characterized by low parasites loads that fall below the detection limit of conventional microscopy of 100 parasites/µL, and malaria antigen-based rapid diagnostic tests (mRDTs) with a similar detection limit as microscopy. According to the World Health Organization (WHO), diagnostic tools developed for this purpose must be capable to detect parasite loads of <10 parasites/µL. Moreover, the emergence and spread of HRP2/HRP3 gene deletions among the deadliest malaria causing Plasmodium species, P. falciparum, (Koita et al., 2012; Akinyi et al., 2013; Abdallah et al., 2015; Amoah et al., 2016; Berhane et al., 2017; Kozycki et al., 2017) can significantly decrease the sensitivity of antigen-based mRDTs. Therefore, accurate diagnosis of low-density infections requires alternative and more sensitive diagnostic techniques, such as nucleic acid-based PCR. However, the high cost of equipment and long turnaround time of PCR limits its up take at the point-of-care.
Loop-mediated isothermal amplification (LAMP) technology, developed in 2000 by Notomi et al. (2000), has demonstrated that it can be simpler to implement than PCR for field and point-of-care tests. LAMP utilizes strand displacement via the Bst DNA polymerase and six (6) set of primers that recognizes up to eight (8) distinct sequences on target DNA sequence. This tool has been extensively utilized for the detection of tropical infectious diseases, including Leishmania spp. (Adams et al., 2010), Trypanosoma (Thekisoe et al., 2007), P. falciparum (Poon et al., 2006) and various bacterial strains (Techathuvanan et al, 2010; Nurul Najian et al, 2016; Martzy et al., 2017), among other pathogens. The original LAMP technique, employed four (4) primers that are specific to six regions on the target DNA. In the years since its invention, several improvements to LAMP primers design have been made, these include the addition of loop primers (Nagamine et al., 2002), STEM primers (Gandelman et al., 2011), and SWAM primers (Martineau et al., 2016). Although additional primers have improved response time, and sensitivity, they add complexity to the primer design and optimization process, and may act as amplifiable structures that can lead to false positive results (Wang et al., 2015; Meagher et al., 2018; Odiwuor et al., 2022).
Recent data suggest that LAMP sensitivity can be enhanced by targeting multi-copy genes or tandem repeats (Hofmann et al., 2015; Kolluri et al., 2021; Nolasco et al., 2021) and high copy number genes for detection and amplification. Our study aimed to enhance sensitivity in a shorter amplification time by utilizing multiple primer sites across the P. falciparum genome using core four LAMP primers, and further simplified visual detection of amplified products through a closed HNB colorimetric system to eliminate the need for further downstream analysis aimed at interpreting the results. We hypothesized that such an approach would improve response time and sensitivity without the need for additional LAMP primers. We selected Pfr364 tandem repeat sequences (Demas et al., 2011) which provides multiple binding sites for LAMP primers across the 41 Pfr364 repeats. These repeats are located within sub-telomeric region 2 of the P. falciparum chromosomes and have been previously utilized in PCR assays (Demas et al., 2011; Waggoner et al., 2015; Amaral et al., 2019) and LAMP assays (Nolasco et al., 2021). However, the repeats are highly diverse in region with averagely 50 polymorphic sites along the 1500 nucleotides length of each Pfr364 repeats, which could result in primers with primer-template mismatches which can significantly extend amplification time (Wang, 2016) therefore we further employed a sequence clustering approach to maximize repeat sequence similarity for designing novel highly effective LAMP primers.
Materials and methods
Ethics statement
Malaria samples, which include whole blood and saliva were obtained from the specimen biobank of the Foundation of Innovative New Diagnostics (FIND) as a kind donation. For specificity analysis, clinical isolates of P. falciparum, P. ovale, and P. malariae were acquired from the biobank at the Center for Malaria Elimination, Mount Kenya University. This study was approved by Institute of Primate Research Ethics Review Board (ISERC/08/2022).
Malaria in vitro cultures
The P. falciparum strain 3D7 (obtained through the Malaria Research Reference Resource Center (MR4) as part of the BEI Resources Repository, NIAID, NIH) was cultured in human O+ erythrocytes using standard methods described in Trager, 1976. Cultures were maintained in complete RPMI 1640 (supplemented with 37.5mM HEPES, 7mM D-glucose, 6 mM NaOH, 25µg/ml of gentamicin sulphate, 2mM L-glutamine, and 10% human serum at pH 7.2) gassed with a mixture of 95% nitrogen, 5% carbon dioxide, and 5% oxygen and incubated at 37°C. Parasite DNA was extracted using Magazhorb mini-prep (Promega, Madison WI, USA) following the guideline provided by the manufacturer. Extracted DNA from these 3D7 cultures served as standard to evaluate LAMP primers optimization, specificity, and lower limit of detection (LLoD).
Retrieval of Pfr364 repeats and design of LAMP Primers
The repeat sequences of Pfr364 were retrieved from PlasmoDB, utilizing repeat coordinates as described by Demas et al., 2011. Multiple sequence alignment was performed using R/msa package (Bodenhofer et al., 2015) and phylogenetic analysis using R/ape package (Paradis et al., 2004) to cluster repeats sequences into related groups. Subsequently, the sequences within each group were aligned using MUSCLE (Edgar, 2004) to obtain a more robust consensus alignment for the design of LAMP primers. The Primer Explorer V5 tool (Eiken Chemical Co. LTD, Tokyo, Japan; http://primerexplorer.jp/e) was utilized to design three sets of LAMP primers from the consensus sequences generated from three clusters, following guidelines of Tomita et al. (2008).
In silico validation of primers specificity
To ensure primer robustness, the primers were evaluated for the formation of secondary structures such as 3’ hairpins and homodimers. To further assess the potential for cross-reactivity, we conducted in silico analysis by aligning primer sequences against the sequences of other plasmodium species using BLAST algorithm (Altschul et al., 1990). The LAMP primer set was aligned with sequences from P. falciparum strains representing various malaria-endemic regions of the worldwide. This primer set designed is robust, having multiple binding sites to strains from Togo, Guinea, Senegal, Mali, Congo, Kenya, and Cambodia. (Supplementary Figure 3). To assess potential cross-reactivity with other Plasmodium strains causing malaria, P. falciparum, P. ovale, and P. malariae from a biobank of Centre of malaria elimination, Mount Kenya University as well as P. vivax samples from FIND biobank, were used. However, this primer set has not been evaluated against P. knowlesi.
Evaluation of the performance of different primer sets
Three LAMP primer sets designed were evaluated in a reaction mixture with a total volume of 25 µL. The reaction mixture contained 2.5 µL of 10× isothermal amplification buffer which was composed of (20 mmol/L Tris-HCl pH 8.8, 10 mmol/L KCl, 2 mmol/L MgSO4, 10 mmol/L (NH4)2SO4 and 0.1% Triton X-100). In addition, the mixture contained 1.6 µM of FIP and BIP primers, 0.2 µM of F3 and B3 primers; 8 U of Bst 2.0 Warmstart, 1.4 mM of dNTPs, 6mM of MgSO4, and 0.4 M Betaine. Template DNA from 3D7 cultures and non-template control (NTC- distilled water) were added in a volume of 2.0 µL. The reaction mixture was incubated at 65°C for 60 minutes in Loopamp LA-320c real time turbidimeter (Eiken Co, Japan). The primer set that gave faster response among the three primer sets was selected for subsequent analysis. All the LAMP reagents were obtained from New England Biolabs (MA, USA).
Further analysis of amplification time to achieve a high-faster response time
The Loopamp LA-320c turbidimeter (Eiken Chemical Co., Tokyo, Japan) is specifically designed for the detection of magnesium pyrophosphate, a byproduct of LAMP amplification. In order to be detected by the turbidimeter, the accumulation of magnesium pyrophosphate must reach a specific threshold. Hence, to assess the amplification rate without relying on the production of the byproduct, we periodically checked for the presence of LAMP amplicons by conducting incubation at specific time intervals. These incubations were performed in a dry heat block incubator LF-160 (Eiken Chemical Co., Tokyo, Japan) set at 65 °C, and the amplified products were visualized through 1.5% gel electrophoresis.
Optimizing reaction temperature
After determining the optimal amplification time for the assay, we proceeded to test different temperatures by incubating the reaction at 65°C, 63°C, and 60°C using the Loopamp LA-320c real-time turbidimeter (Eiken Chemical Co., Tokyo, Japan). The resulting turbidity was measured to evaluate how these temperatures would affect amplification.
Experimental determination of specificity and cross-reactivity
Primer specificity was evaluated against bio-banked samples of P. falciparum, P. ovale, P. malariae, and P. vivax. DNA was extracted using Promega Magazhorb mini kits. The extracted parasite DNA was first validated by nested PCR targeting the 18S RNA gene using species-specific primers (Snounou et al., 1993). LAMP amplification followed with amplification products being visualized on 1.5% gel electrophoresis.
Lower limit of detection
The lower limit of detection of the assay was determined by using DNA obtained from 3D7 cultures quantified using Qubit 3 fluorimeter Invitrogen (CA, USA) then serially diluted 10-fold. The dilutions were then amplified in 5 replicates each. The amplified products were visualized by 1.5% gel electrophoresis. The lower limit of detection was determined as the lowest dilution at which the replicates gel bands were detected with 95% confidence. Probit regression analysis was then performed in R 4.3.1 (CRAN) using ratio of successful reactions to total number of reactions performed for each dilution.
Establishing hydroxynaphthol blue colorimetric assay for end point detection of the LAMP assay
After determining LLoD, we proceeded to establish colorimetric LAMP assay using hydroxy naphthol blue dye (HNB) from Sigma-Aldrich (Wis, USA) which is a suitable endpoint indicator for Loop-mediated isothermal amplification (LAMP) (Goto et al., 2009; Mohon et al. 2014). The serially diluted 3D7 DNA used for determining LLoD were subsequently subjected to HNB-LAMP assay using in a total reaction mixture of 12.5 µL containing 1.25 µL of 10× isothermal amplification buffer which was composed of 10 mmol/L Tris-HCl pH 8.8, 5 mmol/L KCl, 1 mmol/L MgSO4, 5 mmol/L (NH4)2SO4 and 0.1% Triton X-100. In addition, the mixture contained 1.6 µM of FIP and BIP primers, 0.2 µM of F3 and B3 primers; 8 U of Bst 2.0 Warmstart, 1.4 mM of 10 mM of dNTPs, 6 mM of MgSO4, and 120 µM of 2 M HNB dye. 1 µL template of 3D7 DNA was added and incubated at 65°C for 15 minutes. The HNB color intensity of the serial dilutions was measured using imageJ. For each dilution, the mean value of both background signal and the negative control were subtracted mean values of background signal and the negative control from the mean value of color intensity. These resulting values were then plotted on a bar graph for visualization.
Samples for assay validation
A total of 84 samples were sourced for validating the assay, comprising 42 whole blood and 42 matching saliva samples. These samples were obtained from Foundation of Innovative New Diagnostics (FIND) (www.finddx.org) Bio-bank and comprise of samples from Nigerian and Peruvian 2017 cohorts. As summarized in the Table 1.
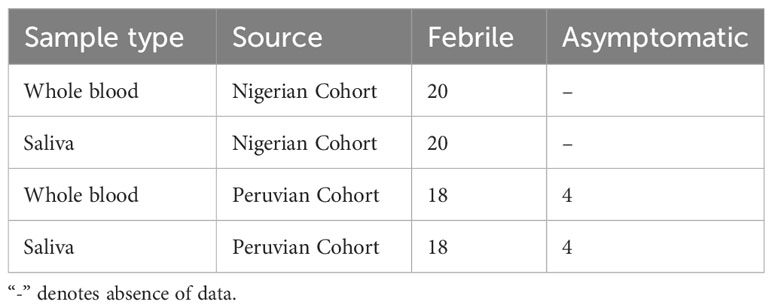
Table 1 Summary of the archived malaria sample.
DNA extraction from whole blood and saliva
The DNA from clinical whole blood and saliva was extracted using Magazhorb mini kits. The extraction was performed on 100 µL of whole blood and 200 µL of saliva samples. DNA was eluted in 50 µL of nuclease-free water.
Closed-tube HNB-LAMP assay on DNA extracted from whole blood and saliva
The LAMP assay reaction was carried out on the extracted DNA using HNB dye as indicator for amplification. The reaction was carried out in a total reaction mixture of 12.5 µL containing 1.25 µL of 10× isothermal amplification buffer which was composed of (10 mmol/L Tris-HCl pH 8.8, 5 mmol/L KCl, 1 mmol/L MgSO4, 5 mmol/L (NH4)2SO4 and 0.05% Triton X-100). In addition, the mixture contained 1.6 µM of FIP and BIP primers, 0.2 µM of F3 and B3 primers; 4 U of Bst 2.0 Warmstart, 1.4 mM of 10 mM of dNTPs, 6 mM of MgSO4, and 120 µM of 2 mM HNB dye. 2 µL template of DNA from whole blood was added to the reaction mixture. The tubes were then sealed and incubated at 65°C for 15 minutes.
Validation of DNA through nested polymerase chain reaction
Subsequently, DNA extracted from both whole blood and saliva were validated through nested PCR, targeting the 18S RNA gene as described by Snounou et al. (1993). In brief, the first round of amplification was carried out in 25 µL reaction volume containing 1 µL of the template, 0.2 µM of the RPLU5 and RPLU6 primers, 12.5 µL of the onetaq 2X master mix with standard buffer (New England Biolabs, MA, USA) The template was denatured at 94°C for 3 minutes, followed by 35 cycles of amplification (94°C for 30 seconds, 54°C for 30 seconds, 68°C for 1 minutes 15 seconds) and a final extension at 68°C for 5 minutes. 0.5 µL from the product of the first round was used for the second round of amplification, containing 0.2 µM of rFAL1 and rFAL2 primers at denaturation of 94°C for 1 minutes followed by 35 cycles of 94°C for 30 seconds, 55°C for 30 seconds, 72°C for 20 seconds and final extension of 5 minutes at 72°C. Amplification was then visualize on 1.5% agarose gel electrophoresis.
LAMP amplification of Triton x-100 treated whole blood and crude saliva
For these analyses, a total of nine samples of whole blood were selected. These samples had been confirmed as positive for P. falciparum through both nested PCR and LAMP assay. 40 µL of these samples were mixed with 1% Triton X-100 in PBS. The mixture was then incubated in heating block at 95°C for 5 minutes, followed by a centrifugation performed at 16000 xg for 4 minutes. The resulting supernatant which contains the DNA was collected and used as template for LAMP reaction, a modification of Sowmya et al. (2012); Hayashida et al. (2015), and Dixit et al. (2018). Subsequently, 4 µL of the supernatant was added to the HNB-LAMP reaction and incubated at 65°C for 15 minutes. Similarly, 40 µL of the saliva samples were heated at 95°C for 5 minutes to break the complexity and thickness. After the heating step, 4 µL of the resulting solution was added to the HNB-LAMP assay as template and incubated at 65°C for 15 minutes.
Serial dilution of whole blood and saliva samples for further evaluation of sensitivity
To further determine the lowest concentration at which the assay could reliably detect the presence of P. falciparum DNA in clinical samples, 10-fold serial dilutions of lysed whole blood supernatant was prepared. For each dilution point, 4 µL of the supernatant was added to the LAMP reaction as the template. Likewise, for the saliva sample, a similar approach was followed. Initially, the saliva samples were heated at 95°C for 5 minutes to ensure proper sample preparation. Serial dilutions of the heated saliva were prepared, and each dilution was subjected to HNB-LAMP amplification.
Statistical analysis
The data obtained from the experiments were entered, stored, and formatted in Microsoft Excel (Microsoft Corp.). The Circa software was used to generate primer binding map, ImageJ was used to quantify color intensity of HNB-LAMP for serial dilutions, Kappa agreement measurement was used to assess agreement between nPCR and LAMP in whole blood and saliva samples. Probit analysis was used to determine LLoD at 95% confidence interval. Data analyses and visualization were performed using R 4.3.1 (CRAN).
Results
Primer design and synthesis
Sequences of three clusters with a higher number of Pfr364 repeats were selected, and their consensus sequences were utilized to generate LAMP primer sequences using primer Explorer V5 (Supplementary Figure 1). in silico validation to ascertain their potential for off-target binding, formation, and stability of 3’ hairpin structure for F3, FIP, BIP, and B3 was carried out. Top scored primers set (Table 2 and Supplementary Table 1), were synthesized and then optimized to select the set giving best amplification result for further analysis.

Table 2 LAMP Primers generated by primer explorer V5 Primer set1.
Performance of the designed LAMP primers
To assess primer performance, progress of LAMP amplification was monitored in real-time using a LA-320c turbidimeter (Eiken Chemical Co., Tokyo, Japan). The differential value of turbidity was recorded to assess efficiency of amplification process as shown in two independent experiments performed (Figure 1A). The obtained amplification times from each repetition were recorded and average time calculated by summing individual times and dividing by the number of runs (Table 3). The amplified products were visualized in 1.5% agarose gel (Figure 1B). Among the three evaluated primer sets, Primer Set 1 showed the highest efficiency, yielding results within approximately 18 minutes. Therefore, it was chosen for subsequent analysis.
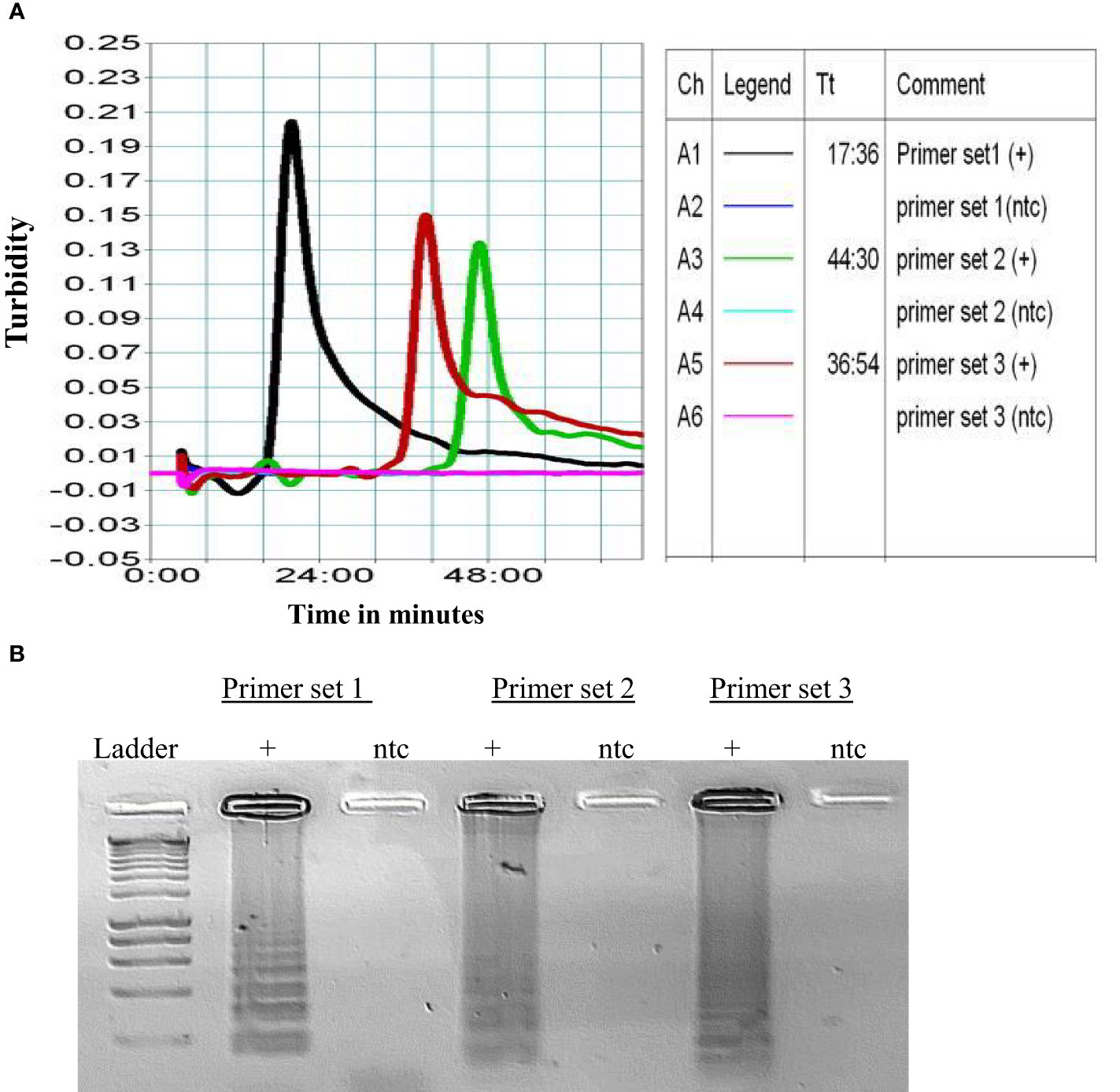
Figure 1 (A) Graph for the amplification of three primer sets. Ch- is reaction channel, A1-A6 are the channels of the reaction block. (B) The amplified products were further visualized in 1.5% agarose gel. This experiment was repeated and average Tt (Threshold time) calculated as shown in Table 3.
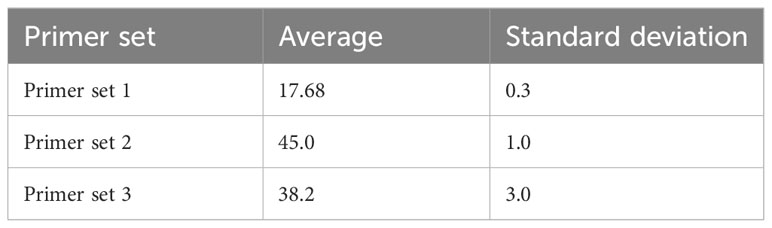
Table 3 Reproducibility of Tt values obtained from the real-time turbidimeter.
Optimizing reaction temperature
Following the initial incubation at 65°C, the temperatures were subsequently adjusted to 63°C, and 60°C; to evaluate how these adjustments would affect the reaction rate. We observed that incubation at 65°C resulted in over ten-fold increased turbidity compared to 63°C, while incubation at 60°C did not generate any turbidity within the first 20 minutes of incubation. (Supplementary Figure 3).
Primer set 1 demonstrated faster response rate for LAMP amplification
Additional incubations conducted at specific time intervals of 10, 13, and 15 minutes in dry heat block incubator LF-160 (Eiken Chemical Co., Tokyo, Japan) to evaluate generation of LAMP amplicons, revealed that incubation at 15 minutes yielded an adequate amount of LAMP amplifications (Figure 2A).
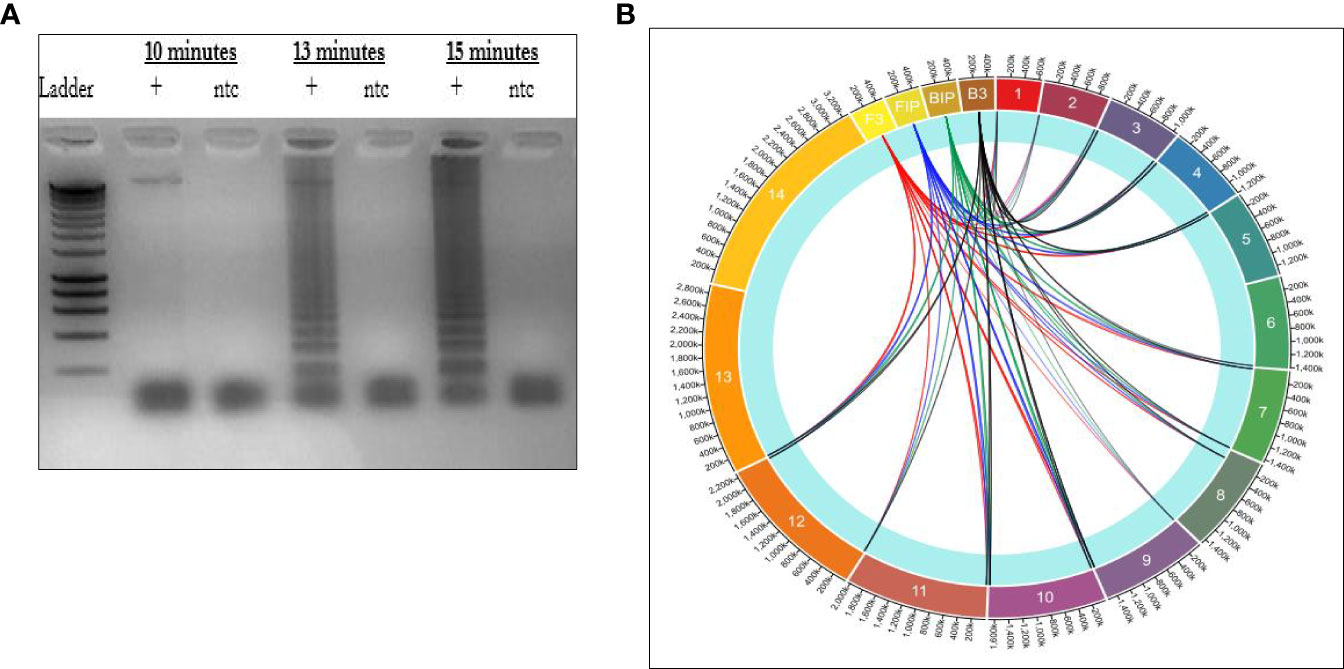
Figure 2 (A) Gel image illustrating rapid amplification rate for the LAMP primers. (B) Circa plot of distribution of LAMP primers across P. falciparum chromosomes. The block layers (1-14) indicates the P. falciparum chromosomes. The lines in red, blue, green, and black shows how each primer connects to each chromosome.
Multiple primer binding sites across P. falciparum enhance faster response time for amplification
The sequences of primer set 1 were subjected to BLAST against PlasmoDB and NCBI databases to generate binding sites across all the chromosomes. The data generated was then used to map the distribution of primers across all the 3D7 chromosomes using circa software as illustrated in Figure 2B.
Specificity of the LAMP primers
The nested PCR successfully amplified all four Plasmodium strains, whereas the LAMP assay specifically amplified the positive control 3D7 and P. falciparum (Supplementary Figures 4A, B).
Determination of Limit of detection and reproducibility
Next, we determined lower limit of detection using 10-fold serial dilutions starting with 5.0x10^6 copies of genomic DNA of 3D7, and each dilution was amplified in five replicates. The resulting amplified products were visualized using 1.5% gel electrophoresis. Successful amplification was achieved across various dilution levels. However, in the dilution with approximately 5x10^-1 copies of genomic DNA, stochastic amplification was observed among the replicates. Replicates amplified and the probability of detection was recorded (Supplementary Table 2) Based on this result, we conducted a probit regression analysis using R 4.3.1 (CRAN) and defined the lower limit of detection as the concentration at which the replicates consistently detected the target with 95% confidence (Figure 3A). We estimated the lower limit of detection (LLoD) to be approximately 19.51 fg/µL, which correspond to approximately 1 parasite based on calculation outlined by Dolezel et al. (2003). Next, we examined the reliability of the HNB colorimetric read-out for LAMP across these dilutions (Figure 3B) and quantified mean color intensity (Figure 3C).
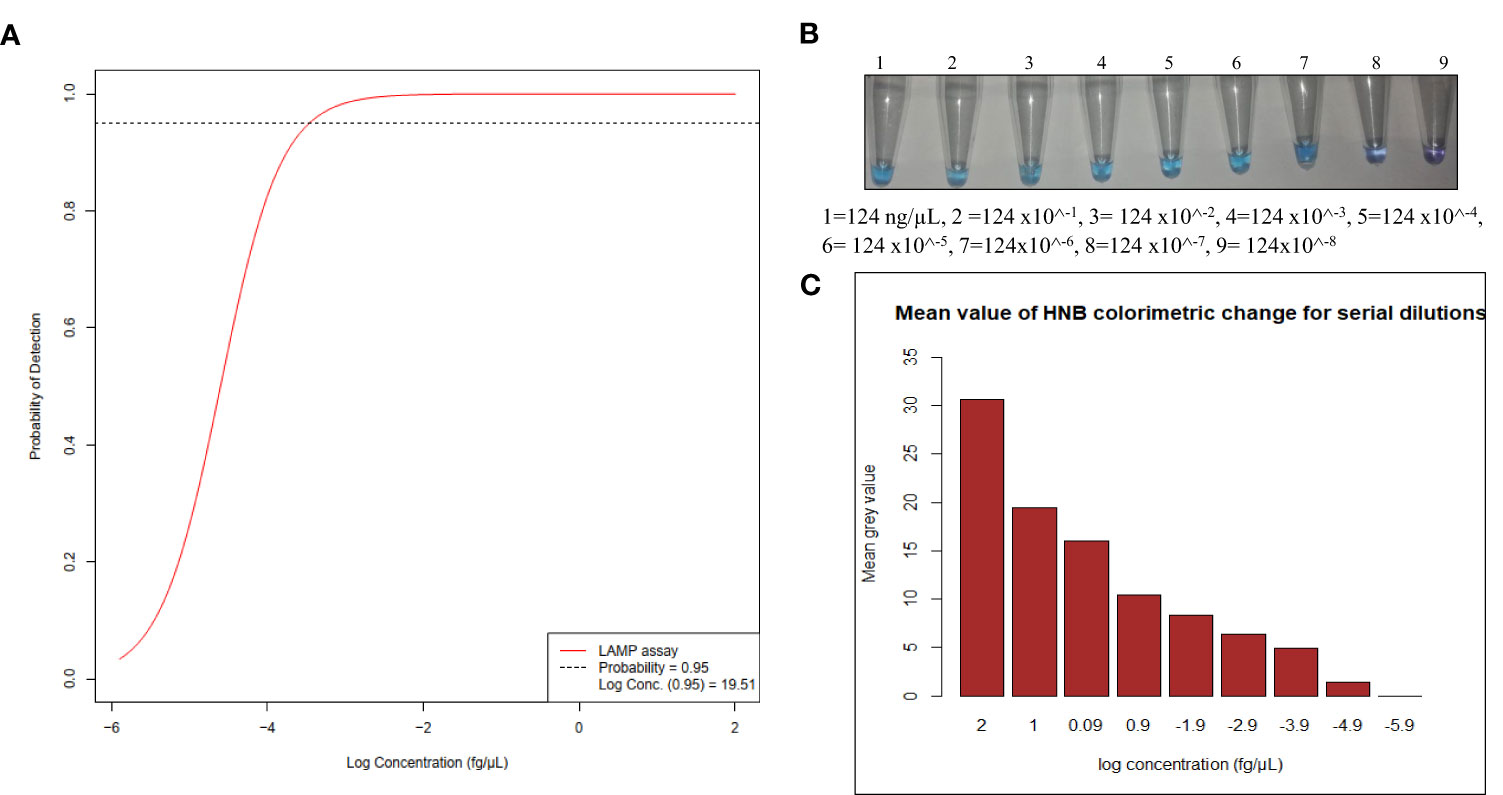
Figure 3 (A) Probit regression analysis resulting in estimated LLoD of 19.51 fg/µL, (B) HNB Colorimetric LAMP assay tested on serial dilutions (C) Bar graph of quantified values of HNB colorimetric on serial dilutions.
The assay effectively amplifies DNA from clinical isolates in whole blood and saliva
To assess the performance of the HNB-LAMP assay on clinical isolates of P. falciparum, we analyzed a total of 84 clinical samples from malaria-endemic regions of Peru and Nigeria. This sample set included 4 asymptomatic individuals and 38 febrile patients (Table 1). These clinical samples, initially diagnosed for P. falciparum by microscopy (Supplementary Table 6), consisted of both whole blood and matching saliva samples. For all 84 samples, we initially conducted an analysis of extracted DNA from whole blood and saliva using our newly developed LAMP assay. Among the 42 whole blood samples that tested positive by microscopy, two of them tested negative when tested by the LAMP assay. These same DNA samples underwent further analysis using nested PCR, and the same two samples that were negative by the LAMP assay were also not detected by PCR. (Supplementary Figures 5 and 6).
It’s important to note that, for both the nested PCR method and LAMP assay methods, whole blood samples consistently yielded more positive results compared to the saliva samples. Specifically, the positivity rates were 95.2% for whole blood versus 21.4% for saliva in nested PCR, and 95.2% for whole blood versus 35.7% for saliva in the LAMP assay. A chi-square test of association for the two tests revealed statistically significant associations both overall and in saliva samples (Supplementary Table 3) at an alpha level of 0.05. In both assays, whole blood consistently showed a higher proportion of positive results (Figure 4A), even at low parasitemia counts (median=1,925 parasites/µL) for both LAMP assay and PCR. Saliva samples of the matching whole blood samples with low parasitemia, yielded more negative results for both assays. However, when the parasite counts increased (median = 10,262 parasites/µL) positivity for saliva samples significantly increased, for the LAMP assay (Figures 4B, C).
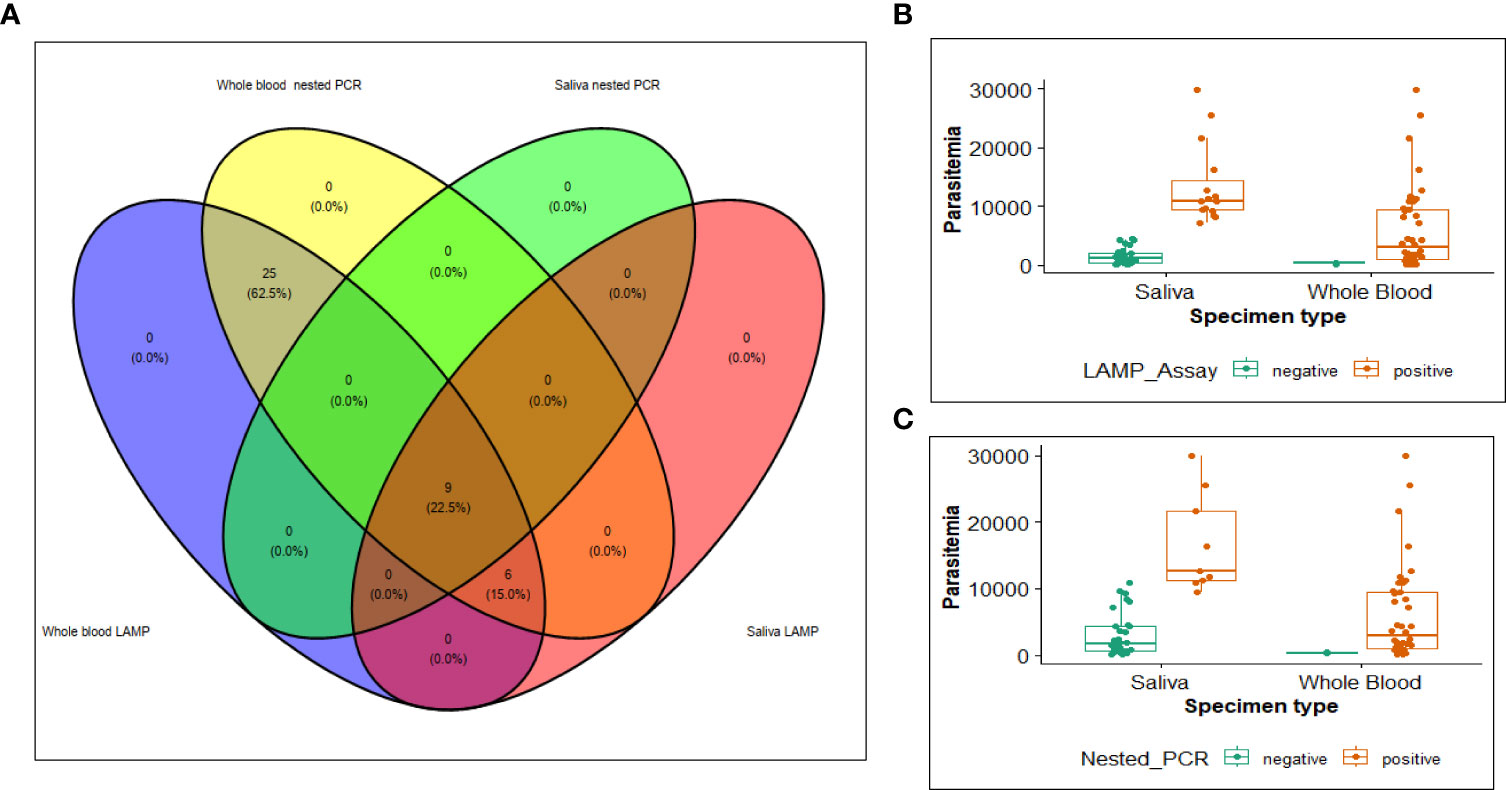
Figure 4 Venn diagram (A) illustrating samples detected as positive by nested PCR and LAMP method. Detection of parasites in relation to parasite densities for (B) LAMP assay (C) Nested PCR.
HNB-LAMP assay had sensitivity similar to nested PCR for the extracted DNA
The sensitivity observed for detecting P. falciparum from whole blood sample indicates that the HNB-LAMP assay can detect P. falciparum in 100% of DNA extracted from whole blood and 40% of DNA extracted from the saliva samples. HNB-LAMP assay successfully detected P. falciparum in all 40 samples that had tested positive using nested PCR method. Additionally, it correctly identified two samples as negative which were also negative in the nested PCR. In the case of saliva samples, LAMP assay detected P. falciparum in six additional samples compared to the nested PCR. To assess the level of agreement between the two tests, we calculated a kappa agreement measurement. The results indicated that the LAMP assay and PCR had a near-perfect agreement for the overall sample set and a perfect agreement for whole blood samples (Supplementary Table 3).
HNB-LAMP assay efficiently detected P. falciparum from the supernatant of whole blood and crude saliva
To further demonstrate the utility of the assay, we analyzed supernatants from 9 whole blood samples that tested positive in both nested PCR and LAMP assay. 4µL of the supernatant was added as template to the LAMP assay. Similarly, 4 µL of the corresponding heat-treated saliva samples were added as template to the LAMP assay. This assay successfully amplified DNA from supernatant of whole blood as well as from crude saliva (Figures 5A, B).
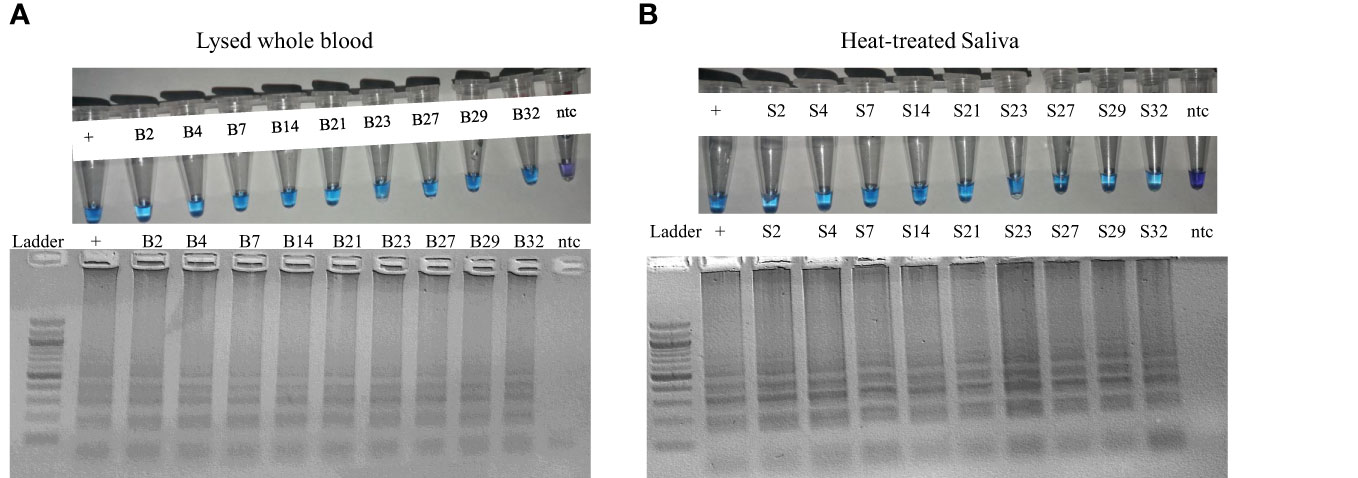
Figure 5 LAMP assay result obtained from (A) lysed whole blood, (B) heat-treated saliva of the archived clinical samples.
Finally, we conducted an additional sensitivity evaluation using serial dilutions supernatant obtained from lysed whole blood and heat-treated saliva samples. To achieve this, we carried out 10-fold serial dilutions of supernatant of whole blood sample initially containing 11,799 parasites/µL, along with matched saliva sample. We specifically chose this sample because its parasite count matched the calculated median range for the positivity of saliva saliva samples in the LAMP assay method. This assay succesfully amplified diluted supernatant at a 10,000 fold diltuion, or 1.2 parasites/uL. For the heat-treated saliva samples, LAMP assay detected amplification up to 100 fold dilutions (118.0 parasites/uL). (Supplementary Figures 5A, B).
Discussion
Efforts to combat malaria have seen remarkable progress over the years, with significant advancements in diagnostic techniques. Despite these advancements, malaria diagnosis still faces several limitations to detect low-level infections or asymptomatic carriers that serve as reservoirs for ongoing transmission, hindering malaria control efforts. Increased highly reliable and field deployable tools capable of identifying these reservoirs would be crucial for effective disease surveillance and targeted interventions. One avenue to explore involves the use of easily obtainable samples such as saliva, as source for detecting P. falciparum (Mharakurwa et al., 2006; Nwakanma et al., 2009; Lloyd et al., 2018).
In this study, we developed a set of LAMP primers targeting a total of 156 binding sites across the 3D7 genome, with a specific focus on improving sensitivity and assay turnaround time. We first establish LAMP amplification process as illustrated in Figure 1. Following this, we determine the optimal incubation temperature (Supplementary Figure 3), based on this result, we determined the optimal incubation temperature for this LAMP assay as 65 °C. To better understand the amplification rate, we observed the production of LAMP amplicons within 15-minute timeframe via colorimetric changes. It’s worth noting that during this period, the Loopamp LA-320c turbidimeter (Eiken Chemical Co., Tokyo, Japan) did not indicate accumulation of magnesium pyrophosphate. Our observation of the gel images Figure 2B revealed that at the 10-minutes mark, no LAMP products were detectable; at the 13-minutes mark, LAMP amplicons began to accumulate; and these amplicons became more pronounced by the 15-minute mark. Moreover, colorimetric read out of serial dilutions at 15 minutes confirmed that HNB dye serves as a reliable colorimetric indicator for detecting low concentrations of P. falciparum DNA. These insights provided an understanding of the reaction rate, and established a baseline for determining the optimal incubation time.
Through further optimization, we achieved an impressive sensitivity level of detecting as little as 1 parasite within a 15 minutes incubation period and extra 15 minutes for template preparation. This LAMP assay successfully detected P. falciparum from archived samples, both from asymptomatic and symptomatic individuals, in two countries (Peru and Nigeria) within malaria-endemic regions of the world. When compared to the gold standard nested PCR assay, LAMP had a strong agreement with 40% sensitivity for saliva, and 100% sensitivity for whole blood with a corresponding specificity of 100% in both sample matrices. This phenomenon mirrors a trend reported by Singh et al. (2013). However, we also observed that in both nested PCR and LAMP, the sensitivity of detecting P. falciparum from saliva was low when the parasite density was low. Similarly, the two tests exhibited higher sensitivity for saliva samples from individuals with a high parasite density. As for whole blood samples, we observed a consistently high sensitivity across various parasite densities.
Previous study had indicated that different genes within P. falciparum can yield amplification with varying assay sensitivity and limit of detection (Zhang et al., 2017). For instance, the LAMP assays focusing on the apicoplast genome with 15 copies in P. falciparum demonstrated a limit of detection of 2 parasites/µL for the 3D7 strain, with overall sensitivity of 60% on archived DNA samples (Oriero et al., 2015). The initial LAMP assay based on Pfr364 repeats (Nolasco et al., 2021) showed a limit of detection of 2.7 parasites/µL and an amplification time of 1 hour. Another LAMP targeting the amplification of 18S RNA pan-genes detected 5 parasites/µL (Han et al., 2007). Meanwhile, mitochondrial genes (Polley et al., 2010) demonstrated limit of detection of 5 parasites/µL. In contrast, the LAMP primer sets developed in our study achieved a lower limit of detection (1 parasite/μL) within a significantly shorter time frame of just 15 minutes, representing a significant time advantage. Furthermore, use of the closed hydroxynaphthol blue (HNB) colorimetric detection system eliminated the need for additional gel electrophoresis, simplifying the process, reducing the risk of cross-contamination, and enhancing potential for point-of-care deployment.
The LAMP method is known for its robustness and has proven resilient against PCR inhibitors such as hemoglobin (Thekisoe et al., 2009; Njiru et al., 2008). This robustness has prompted numerous studies to explore the possibility of bypassing DNA extraction steps (Linnes et al., 2014, Hayashida et al., 2015; Xu G et al., 2016; Dixit et al., 2018). Dried blood spot (DBS) samples, which eliminate the need for cold-chain storage, offer excellent feasibility at point-of-care. Over the years, numerous studies have demonstrated that accurate and sensitive detection of Plasmodium parasites from DBS is achievable. For instance, Zainabadi et al., 2017, achieved a sensitivity of 1 parasite/µL for every 50 µL of blood spotted on Whatman paper using PCR. Subsequently, Hashimoto et al. (2019) demonstrated that Whatman FTA cards can provide high-quality template DNA, allowing for LoD of 1.6 parasite/µL. Moreover, Hailemeskel et al., 2021 showed enhanced sensitivity in detecting asymptomatic Plasmodium reservoirs through nested PCR. Similarly, LAMP-SNP assay (Khammanee et al., 2021) achieved 100% sensitivity in detecting malaria parasites in cases low parasitemia using DBS samples.
In our study, we were able to successfully detect 1.2 parasites/µL DNA in the supernatant of whole blood lysed with Triton x-100, and 118 parasites/µL of heat-treated saliva samples. These results demonstrate that the quantity of parasite DNA obtained from lysed whole blood is approximately a hundred-fold higher than the expected amount in saliva. These findings suggest that the assay has the potential to offer high sensitivity for the detection of P. falciparum from dried blood spot (DBS), making it a suitable option for sample storage in point-of-care settings. However, we recommend that future studies should focus on evaluating the use of dried blood spots (DBS) and detection of submicroscopic infections.
Conclusion
The sequence clustering approach to multisite LAMP binding and simplified single-tube HNB LAMP workflow allowed for rapid, robust, and sensitive amplification with visual detection. This streamlined approach demonstrated the possibility of processing and analyzing a sample within 30 minutes, and could greatly facilitate its use in point-of-care settings. Our findings highlight the exquisite sensitivity of a single parasite for closed HNB-LAMP detection of P. falciparum, shortened assay time, and simplified workflow. These findings hold great promise for enhanced malaria diagnosis, particularly in resource-limited settings, where rapid and accurate detection is essential for effective for disease management and surveillance.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institute of Primate Research Ethics Review Board (ISERC/08/2022). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from gifted from another research group. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
CM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. CS: Investigation, Methodology, Validation, Writing – review & editing, Data curation. SN: Methodology, Supervision, Writing – review & editing. EO: Methodology, Supervision, Writing – review & editing. JL: Methodology, Writing – review & editing. BK: Formal Analysis, Methodology, Writing – review & editing. JG: Methodology, Resources, Writing – review & editing, Funding acquisition. LO: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was financially supported by the Early Career Research Grant of Royal Society of Tropical medicine and Hygiene (RSTMH) in partnership with the National Institute for Health Research (NIHR) as part of 2021 Small Grants Program.
Acknowledgments
We express our gratitude to the Foundation of Innovative new Diagnostics (FIND) for generously providing the clinical samples utilized in this study. Additionally, we extend our appreciation to Royal Society of Tropical Medicine and Hygiene (RSTMH) for providing the grant that supported this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) LO, JG, BNK declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmala.2023.1303980/full#supplementary-material
Abbreviations
PCR, Polymerase chain reaction; nPCR, nested Polymerase chain reaction; LAMP, Loop-mediated isothermal amplification; HNB, hydroxynaphthol blue dye; DNA, Deoxyribonucleic acid; LLoD, Lower limit of detection; 3D7, Laboratory strain of P. falciparum; MR4, Malaria Research Reference Resource Center.
References
Abdallah J. F., Okoth S., Fontecha G. A., Torres R., Banegas E. I., Matute M., et al. (2015). Prevalence of pfhrp2 and pfhrp3 gene deletions in Puerto Lempira, Honduras. Malaria J. 14, 19. doi: 10.1186/s12936-014-0537-7
Adams E. R., Schoone G. J., Ageed A. F., Safi S. E., Schallig H. D. (2010). Development of a reverse transcriptase loop-mediated isothermal amplification (LAMP) assay for the sensitive detection of Leishmania parasites in clinical samples. Am. J. Trop. Med. Hyg. 82 (4). doi: 10.4269/ajtmh.2010.09-0369
Akinyi S., Hayden T., Gamboa D., Torres K., Bendezu J., Abdallah J. F., et al. (2013). Multiple genetic origins of histidine-rich protein 2 gene deletion in Plasmodium falciparum parasites from Peru. Sci. Rep. 3, 1–8. doi: 10.1038/srep02797
Altschul F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3). doi: 10.1016/S0022-2836(05)80360-2
Amaral L. C., Robortella D. R., Guimarães L. F., Limongi J. E., Fontes C. J. F., Pereira D. B., et al. (2019). Ribosomal and non-ribosomal PCR targets for the detection of low-density and mixed malaria infections. Malaria J. 18 (1). doi: 10.1186/s12936-019-2781-3
Amoah L. E., Abankwa J., Oppong A. (2016). Plasmodium falciparum histidine rich protein-2 diversity and the implications for PfHRP 2: based malaria rapid diagnostic tests in Ghana. Malaria J. 15, 1–8. doi: 10.1186/s12936-016-1159-z
Berhane A., Russom M., Bahta I., Hagos F., Ghirmai M., Uqubay S. (2017). Rapid diagnostic tests failing to detect Plasmodium falciparum infections in Eritrea: an investigation of reported false negative RDT results. Malaria J. 16, 105. doi: 10.1186/s12936-017-1752-9
Bodenhofer U., Bonatesta E., -Kainrath C. H., Hochreiter S. (2015). msa: an R package for multiple sequence alignment. Bioinformatics. 31 (24). doi: 10.1093/bioinformatics/btv494
Demas A., Oberstaller J., DeBarry J., Lucchi N. W., Srinivasamoorthy G., Sumari D., et al. (2011). Applied genomics: data mining reveals species-specific malaria diagnostic targets more sensitive than 18S rRNA. J. Clin. Microbiol. 53 (11). doi: 10.1128/JCM.02603-10
Dixit K. K., Verma S., Singh O. P., Singh D., Singh A. P., Gupta R., et al. (2018). Validation of SYBR green I based closed tube loop mediated isothermal amplification (LAMP) assay and simplified direct-blood-lysis (DBL)-LAMP assay for diagnosis of visceral leishmaniasis (VL). PloS Negl. Trop. Dis. 129 (11). doi: 10.1371/journal.pntd.0006922
Dolezel J., Bartos J., Voglmayr H., Greilhuber J. (2003). Nuclear DNA content and genome size of trout and human. Cytometry A 51 (2). doi: 10.1002/cyto.a.10013
Edgar R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5, 113. doi: 10.1186/1471-2105-5-113
Gandelman O., Jackson R., Kiddle G., Tisi L. (2011). Loop-mediated amplification accelerated by stem primers. Int. J. Mol. Sci. 12 (12). doi: 10.3390/ijms12129108
Goto M., Honda E., Ogura A., Nomoto A., Hanaki K. (2009). Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 46 (3). doi: 10.2144/000113072
Hailemeskel E., Tebeje S. K., Behaksra S. W., Shumie G., Shitaye G., Keffale M., et al. (2021). The epidemiology and detectability of asymptomatic plasmodium vivax and plasmodium falciparum infections in low, moderate and high transmission settings in Ethiopia. Malar J. 20 (1), 59. doi: 10.1186/s12936-021-03587-4
Han E. T., Watanabe R., Sattabongkot J., Khuntirat B., Sirichaisinthop J., Iriko H., et al. (2007). Detection of four plasmodium species by genus- and species-specific loop-mediated isothermal amplification for clinical diagnosis. J. Clin. Microbiol. 45 (8). doi: 10.1128/JCM.02117-06
Hashimoto M., Bando M., Kido J. I., Yokota K., Mita T., Kajimoto K., et al. (2019). Nucleic acid purification from dried blood spot on FTA elute card provides template for polymerase chain reaction for highly sensitive plasmodium detection. Parasitol. Int. 73, 101941. doi: 10.1016/j.parint.2019.101941
Hayashida K., Kajino K., Hachaambwa L., Namangala B., Sugimoto C. (2015). Direct blood dry LAMP: A rapid, stable, and easy diagnostic tool for human african trypanosomiasis. PloS Negl. Trop. Dis. 9 (3). doi: 10.1371/journal.pntd.0003578
Hofmann N., Mwingira F., Shekalaghe, Robinson L. J., Mueller I., Felger I. (2015). UltraSensitive detection of plasmodium falciparum by amplification of multi-copy subtelomeric targets. PloS Med. 12 (3). doi: 10.1371/journal.pmed.1001788
Khammanee T., Sawangjaroen N., Buncherd H., Tun A. W., Thanapongpichat S. (2021). A LAMP-SNP assay detecting C580Y mutation in pfkelch13 gene from clinically dried blood spot samples. Korean J. Parasitol. 59 (1). doi: 10.3347/kjp.2021.59.1.15
Koita O. A., Doumbo O. K., Ouattara A., Tall L. K., Konaré A., Diakité M., et al. (2012). False-negative rapid diagnostic tests for malaria and deletion of the histidine-rich repeat region of the hrp2 gene. Am. J. Trop. Med. Hyg 86 (2). doi: 10.4269/ajtmh.2012.10-0665
Kolluri N., Kamath S., Lally P., Zanna M., Galagan J., Gitaka J., et al. (2021). Development and clinical validation of iso-IMRS: A novel diagnostic assay for p. falciparum malaria. Anal. Chem. 93 (4), 2097–2105. doi: 10.1021/acs.analchem.0c03847
Kozycki C. T., Molise N., Rulisa S., Mwikarago E. I., Musabyimana J. P., Habimana J. P., et al. (2017). False-negative malaria rapid diagnostic tests in Rwanda: impact of Plasmodium falciparum isolates lacking hrp2 and declining malaria transmission. Malaria J. 16 (123). doi: 10.1186/s12936-017-1768-1
Linnes J. C., Fan A., Rodriguez N. M., Lemieux B., Kong H., Klapperich C. M. (2014). Paper-based molecular diagnostic for chlamydia trachomatis. RSC 4 (80), 42245–42251. doi: 10.1039/C4RA07911F
Lloyd Y. M., Esemu L. F., Antallan J., Thomas B., Yunga S. T., Obase B., et al. (2018). PCR-based detection of Plasmodium falciparum in saliva using mitochondrial cox3 and varATS primers. Trop. Med. Health. 46 (22). doi: 10.1186/s41182-018-0100-2
Martineau R. L., Murray S. A., Ci S., Gao W., Chao S. H., Meldrum D. R. (2016). Improved performance of loop-mediated isothermal amplification assays via swarm priming. Anal. Chem. 89 (1), 625–632. doi: 10.1021/acs.analchem.6b02578
Martzy R., Kolm C., Brunner K., Mach R. L., Krska R., Šinkovec H., et al. (2017). A loop-mediated isothermal amplification (LAMP) assay for the rapid detection of Enterococcus spp. in water. Water Res. 122, 62–69. doi: 10.1016/j.watres.2017.05.023
Meagher R. J., Priye A., Light Y. K., Huang C., Wang E. (2018). Impact of primer dimers and self-amplifying hairpins on reverse transcription loop-mediated isothermal amplification detection of viral RNA. Analyst. 143 (8). doi: 10.1039/c7an01897e
Mharakurwa S., Simoloka C., Thuma P. E., Shiff C. J., Sullivan D. J. (2006). PCR detection of Plasmodium falciparum in human urine and saliva samples. Malaria J. 5, 103. doi: 10.1186/1475-2875-5-103
Mohon A. N., Elahi R., Khan W. A., Haque R., Sullivan D. J. Jr, Alam M. S. (2014). A new visually improved and sensitive loop mediated isothermal amplification (LAMP) for diagnosis of symptomatic falciparum malaria. Acta Trop. 134. doi: 10.1016/j.actatropica.2014.02.016
Nagamine K., Watanabe K., Ohtsuka K., Hase T., Notomi T. (2002). Loop-mediated isothermal amplification reaction using a nondenatured template. Clinic. Chem. 47, 9. doi: 10.1093/clinchem/47.9.1742
Njiru Z. K., Mikosza A. S., Armstrong T., Enyaru J. C., Ndung'u J. M., Thompson A. R. (2008). Loop-mediated isothermal amplification (LAMP) method for rapid detection of Trypanosoma brucei rhodesiense. PloS Negl. Trop. Dis. 2 (2). doi: 10.1371/journal.pntd.0000147
Nolasco O., Montoya J., Rosales Rosas A. L., Barientos S., Rosanas-Urgell A., Gamboa D., et al. (2021). Multicopy targets for Plasmodium vivax and Plasmodium falciparum detection by colorimetric LAMP. Malaria J. 20 (1). doi: 10.1186/s12936-021-03753-8
Notomi T., Okayama H., Masubuchi H., Yonekawa T., Watanabe K., Amino N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28 (13). doi: 10.1093/nar/28.12.e63
Nurul Najian A. B., Engku Nur Syafirah E. A., Ismail N., Mohamed M., Yean C. Y. (2015). Development of multiplex loop mediated isothermal amplification (m-LAMP) label-based gold nanoparticles lateral flow dipstick biosensor for detection of pathogenic Leptospira. Anal. Chim. Acta. 903. doi: 10.1016/j.aca.2015.11.015
Nwakanma D. C., Gomez-Escobar N., Walther M., Crozier S., Dubovsky F., Malkin E., et al. (2009). Quantitative detection of plasmodium falciparum DNA in saliva, blood, and urine. J. Infect. Dis. 5 (103). doi: 10.1086/598856
Odiwuor N., Xiong J., Ogolla F., Hong W., Li X., Khan F. M., et al. (2022). A point-of-care SARS-CoV-2 test based on reverse transcription loop-mediated isothermal amplification without RNA extraction with diagnostic performance same as RT-PCR. Anal. Chim. Acta. 1200. doi: 10.1016/j.aca.2022.339590
Oriero C. E., van Geertruyden J.-P., Jacobs J., D'Alessandro U., Nwakanma. D. (2015). Validation of an apicoplast genome target for the detection of Plasmodium species using polymerase chain reaction and loop mediated isothermal amplification. Clin. Microbiol. Infection. 21 (7). doi: 10.1016/j.cmi.2015.02.025
Paradis E., Claude J., Strimmer K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 20 (2). doi: 10.1093/bioinformatics/btg412
Polley S. D., Mori Y., Watson J., Perkins M. D., González I. J., Notomi T., et al. (2010). Mitochondrial DNA targets increase sensitivity of malaria detection using loop-mediated isothermal amplification. J. Clin. Microbiol. 48 (8). doi: 10.1128/JCM.00355-10
Poon L. L., Wong B. W., Ma E. H., Chan K. H., Chow L. M., Abeyewickreme W., et al. (2006). Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin. Chem. 52 (2). doi: 10.1373/clinchem.2005.057901
Singh R., Savargaonkar D., Bhatt R., Valecha N. (2013). Rapid detection of Plasmodium vivax in saliva and blood using loop mediated isothermal amplification (LAMP) assay. J. Infect. 67 (3). doi: 10.1016/j.jinf.2013.04.016
Snounou G., Viriyakosol S., Zhu X. P., Jarra W., Pinheiro L., do Rosario V. E., et al. (1993). High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol. Biochem. Parasitol. 61 (2). doi: 10.1016/0166-6851(93)90077
Sowmya N., Thakur M. S., Manonmani H. K. (2012). Rapid and simple DNA extraction method for the detection of enterotoxigenic Staphylococcus aureus directly from food samples: comparison of PCR and LAMP methods. J. Appl. Microbiol. doi: 10.1111/j.1365-2672.2012.05315
Techathuvanan C., Draughon F. A., D'Souza D. H. (2010). Loop-mediated isothermal amplification (LAMP) for the rapid and sensitive detection of Salmonella Typhimurium from pork. J. Food Sci. 75 (5). doi: 10.1111/j.1750-3841.2010.01554.x
Thekisoe O. M., Kuboki N., Nambota A., Fujisaki K., Sugimoto C., Igarashi I., et al. (2007). Species-specific loop-mediated isothermal amplification (LAMP) for diagnosis of trypanosomosis. Acta Tropica. 102, 3. doi: 10.1016/j.actatropica.2007.05.004
Thekisoe O. M., Bazie R. S. B., coronel-servian A. M., sugimoto c., kawazu s.-i., et al. (2009). Stability of loop-mediated isothermal amplification (LAMP) reagents and its amplification efficiency on crude trypanosome DNA templates. J. Veterinary Med. Sci. 71 (4). doi: 10.1292/jvms.71.471
Tomita N., Mori Y., Kanda H., Notomi T. (2008). Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 3 (5), 877–882. doi: 10.1038/nprot.2008.57
Waggoner J. J., Abeynayake J., Balassiano I., Lefterova M., Sahoo M. K., Liu Y., et al. (2015). Multiplex nucleic acid amplification test for diagnosis of dengue fever, malaria, and leptospirosis. J. Clin. Microbiol. 52 (2). doi: 10.1128/JCM.00341-14
Wang D.-G., Brewster J. D., Paul M., Tomasula P. M. (2015). Two methods for increased specificity and sensitivity in loop-mediated isothermal amplification. Molecules. 20 (4). doi: 10.3390/molecules20046048
Wang D. (2016). Effect of internal primer–template mismatches on loop-mediated isothermal amplification. Biotechnol. Biotechnolog. Equip. 30, 2. doi: 10.1080/13102818.2015.1125765
WHO. (2022). Fact sheet: world malaria day 2022 (Geneva, Switzerland: WHO). Available at: http://www.who.int/malaria/media/world-malaria-day-2022/en/.
Xu G., Nolder D., Reboud J., Oguike M. C., van Schalkwyk D. A., Sutherland C. J., et al. (2016). Paper-origami-based multiplexed malaria diagnostics from whole blood. Angew Chem. Int. Ed Engl. 55 (49). doi: 10.1002/anie.201606060
Zainabadi K., Adams M., Han Z. Y., Win H. L., Han T., Ouattara A., et al. (2017). A novel method for extracting nucleic acids from dried blood spots for ultrasensitive detection of low-density Plasmodium falciparum and Plasmodium vivax infections. Malar J. 16 (377). doi: 10.1186/s12936-017-2025-3
Keywords: loop mediated isothermal amplification (LAMP), saliva, multi-copy repeat, P. falciparum, HNB-colorimetric LAMP, LAMP performance in whole blood and saliva
Citation: Mgawe C, Shilluli C, Nyanjom S, Odari E, Linnes JC, Kanoi BN, Gitaka J and Ochola L (2023) Application of multiple binding sites for LAMP primers across P. falciparum genome improves detection of the parasite from whole blood samples. Front. Malar. 1:1303980. doi: 10.3389/fmala.2023.1303980
Received: 28 September 2023; Accepted: 08 November 2023;
Published: 04 December 2023.
Edited by:
Andre Lin Ouedraogo, Bill and Melinda Gates Foundation, United StatesReviewed by:
Inke Nadia D. Lubis, University of North Sumatra, IndonesiaIsaac Kweku Quaye, Regent University College of Science and Technology, Ghana
Copyright © 2023 Mgawe, Shilluli, Nyanjom, Odari, Linnes, Kanoi, Gitaka and Ochola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesse Gitaka, amdpdGFrYUBta3UuYWMua2U=; Lucy Ochola, bGFvY2hvbGFAZ21haWwuY29t
†These authors share senior authorship