
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Lupus , 09 October 2023
Sec. Genetics and Genomics in Lupus
Volume 1 - 2023 | https://doi.org/10.3389/flupu.2023.1219277
This article is part of the Research Topic Advancements and Challenges in Epidemiology of Lupus View all 5 articles
 Kevin Yip1
Kevin Yip1 Meredith Akerman2Ruth Fernandez Ruiz1Nicole Leung3Huda Algasas1Yingzhi Qian4Jill P. Buyon3Jasmin Divers2Peter Izmirly3Michael Belmont3
Meredith Akerman2Ruth Fernandez Ruiz1Nicole Leung3Huda Algasas1Yingzhi Qian4Jill P. Buyon3Jasmin Divers2Peter Izmirly3Michael Belmont3 Ashira D. Blazer1*
Ashira D. Blazer1*
Background/purpose: African Americans (AA) with systemic lupus erythematosus (SLE) are at higher risk for both kidney disease and Osteonecrosis (ON). Two Apolipoprotein L1 (APOL1) risk variants (RV), G1 and G2, have been associated with chronic kidney disease (CKD), hypertension, and microvascular disease in AAs, which are independent risk factors for ON. Accordingly, we investigated the association between carriers of the APOL1 risk variants and the prevalence of ON in AA SLE patients.
Methods: A cohort of 121 adult participants of self-reported AA ancestry and meeting at least four of the American College of Rheumatology (ACR) revised criteria for SLE were recruited from a high volume urban SLE clinical site. PCR/sequencing was used to stratify participants by APOL1 genotype. Medical records, including clinical notes and imaging reports, were retrospectively reviewed for documentation of ON. Association between the number of APOL1 risk variants with time to first ON was tested.
Results: In our cohort, 18 individuals developed ON; across the APOL1 genotype groups, 2/37 0RV, 11/59 1RV, and 5/15 2RV participants were affected. The mean time to ON was 27 years, 22 years, and 18 years in 0RV, 1RV, and 2RV carriers, respectively. An adjusted Cox regression model showed that carrying the APOL1 risk variants associated with shorter ON free survival with hazard ratios (HR) of 3.1 (95% CI: 1.6–6.2) and 9.6 (95% CI 2.4–37.8) for 1RV and 2RV carriers, respectively. 2RV carriers more often exhibited multiple and bilateral joint sites affected by ON. Disease duration was longer in ON-affected participants at 20.5 years compared to 9.0 years in those unaffected (p < 0.001). In individuals who had received glucocorticoids, median cumulative prednisone equivalent dose was higher in ON-affected participants, though this did not reach statistical significance (18.7 g vs. 9.0 g; p-value = 0.3).
Conclusion: Our analysis suggests a higher risk of osteonecrosis among African American SLE patients who carry the APOL1 risk variants. In addition, disease duration increased the rate of ON. Given the high frequency of the APOL1 risk variants in African Americans, APOL1 high-risk genotype carriers may represent an ON-vulnerable subgroup within the AA population. Further work is necessary to uncover the mechanism of this association.
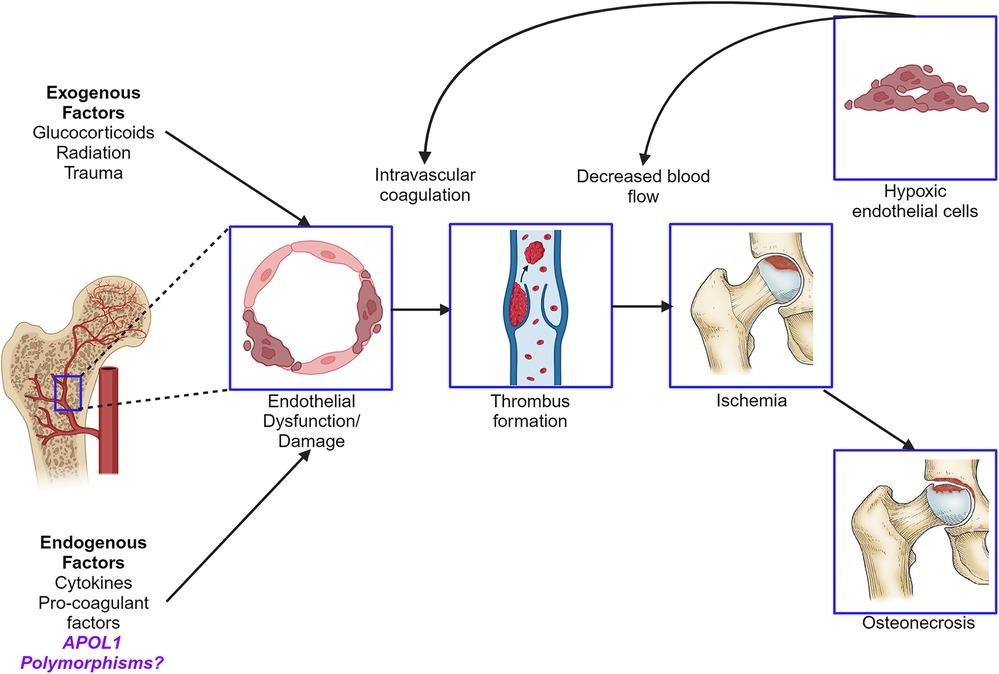
Graphical Abstract.
Osteonecrosis (ON), previously known as avascular necrosis, is a pathologic process of the bone involving reduced osseous and marrow perfusion and resultant atraumatic necrosis. Multiple conditions contributing to endothelial dysfunction, microangiitis, or marrow fat content appear to play a crucial role in ON (1). This risk is magnified in patients with systemic lupus erythematosus (SLE), where the estimated prevalence of symptomatic and asymptomatic ON reaches 9% and 29%, respectively (2). African American (AA) race, systemic steroid use, and antiphospholipid syndrome, along with comorbidities, such as hypertension (HTN), renal disease, and sickle cell anemia, are known risk factors for ON (2). Recent evidence has linked several polymorphisms to pathogenic mechanisms of ON, including mutations in nitric oxide synthase, prothrombin, and factor V Leiden, which are thought to influence the osseous microvasculature (3–5). While this growing genetic landscape offers clues about ON pathogenesis, the molecular mechanisms are not fully understood.
Two coding change variants in the Apolipoprotein L1 (APOL1) gene, G1 (S342G and I384M) and G2 (N388del; Y389del) have been identified in ancestrally African genomes (6). Approximately 13% of AA carry the APOL1 high-risk genotype (HRG), defined as two variant allele copies in any combination (G1/G1, G1/G2, or G2/G2) (6). The APOL1 HRG is most strongly associated with a higher incidence and severity of chronic kidney disease by multiple causes (7–10). Further, APOL1 HRG has been implicated in microvascular stroke and preeclampsia risk, suggesting a potential role in vascular dysfunction (11–13). We have previously identified several APOL1 risk traits in SLE, including lupus nephritis progression, hypertension, early cardiovascular disease, and organ damage accrual—all of which have been linked to SLE ON risk (14, 15). However, APOL1 as a candidate risk SNP for ON has not been studied.
APOL1 is expressed in vascular endothelial cells and arteriolar smooth muscle (16, 17). Multiple inflammatory stimuli increase APOL1 expression, leading to accumulation of the encoded protein and enhanced cytotoxicity in APOL1 HRG tissues (18, 19). In a recent endothelial cell culture model, treating HRG cells with interferon was shown to induce APOL1 expression and produce cytotoxicity by reducing mitochondrial energy production and disrupting autophagy, a critical cellular maintenance process (19). These data raise the possibility that APOL1 variants confer a genetic predisposition toward endothelial damage in the microvasculature. We hypothesize that APOL1 HRG may contribute to ON risk due to a cytotoxic effect on endothelial cells and its role in promoting known comorbid conditions associated with ON.
Considering the high proportion of AA carrying the APOL1 HRG and the known higher prevalence of SLE and renal disease in this population (20–22), we aimed to determine if APOL1 HRG independently associates with ON in a well-phenotyped African American SLE cohort. This study is the first to evaluate whether a common pathologic variant found exclusively in African genomes is associated with ON in this high risk population. These results could improve risk stratification among AA SLE patients, and offer new insights into ON pathogenesis. Moreover gene therapy for APOL1 HRG carriers is being developed (23); therefore identifying targetable disease phenotypes is an important step in furthering precision medicine for AA patients.
This was a nested retrospective study that enrolled participants from the ongoing, prospective NYU-wide Specimen and Matched Phenotype Linked Evaluation (SAMPLE) biorepository. This repository was initiated on February 14, 2014 with ongoing yearly renewal by the Institutional Review Boards of New York University Grossman School of Medicine (NYUGSoM) and Bellevue Hospital Center of the New York City Health and Hospitals Corporation. Written informed consent, which was available in English, Spanish, and Mandarin, was obtained from all participants in accordance with the Declaration of Helsinki principles. Participants were recruited from three high-volume SLE clinical sites. This nested sub-sample of eligible individuals were included, and retrospective chart reviews were completed. Inclusion criteria were individuals ≥18 years of age, of self-reported AA race, and meeting at least four of the American College of Rheumatology (ACR) revised criteria for SLE (24). Exclusion criteria were patients unwilling or unable to complete informed consent and patients with fewer than three clinical visits at least six months apart to ensure capture of longitudinal data. Self-reported ancestry was confirmed by ancestry informative markers as previously described (15), and participants without significant African admixture were excluded from analyses.
Genomic DNA was isolated from anti-coagulated whole blood collected in EDTA blood sample tubes using the Qiagen kit (Valencia, CA, USA) according to the manufacturer’s instructions. DNA was isolated from blood samples taken at clinical visits within 24–48 h of blood draw. DNA isolates were stored at −80°C. Batches of 10–15 DNA samples were evaluated and quantitated using a Nanodrop-1,000 spectrophotometer (Nanodrop Products, Wilmington, DE). One hundred nanograms (ng) of genomic DNA was used as a conventional polymerase chain reaction (PCR) template. A single 300-base-pair DNA segment containing the APOL1 polymorphisms, G1 (rs73885319 and rs60910145) and G2 (rs71785313), was amplified using AmpliTaq Gold 360 DNA Polymerase (Applied Biosystems, Foster City, CA). For quality control, DNA was elongated in both forward and reverse directions. Genotypes were analyzed using the GeneWiz online platform, as previously described (25).
We genotyped 121 study participants for APOL1; informative ancestry markers were completed, and a principal component analysis was run (26). Models were adjusted for African admixture using the
between principal components (PC) 1 and 2 of the AA samples and mean values for the African reference samples. This method excluded 10 participants who did not cluster with the African reference population. Of the 111 study participants remaining, longitudinal data capture was reviewed and supplemented with data from the electronic medical record where necessary. A study researcher (KY) blinded to the APOL1 status of the participants initially reviewed the medical charts, clinical notes, and imaging reports for documentation of evidence of ON, which was then validated by another co-researcher (AB) for accuracy. ON was defined by chart mention of a history of ON in physician notes. Mentions of Osteonecrosis in clinical notes were recorded and confirmed by imaging reports including xrays; and ultrasounds and MRIs where available. A screening workflow is presented in Figure 1.
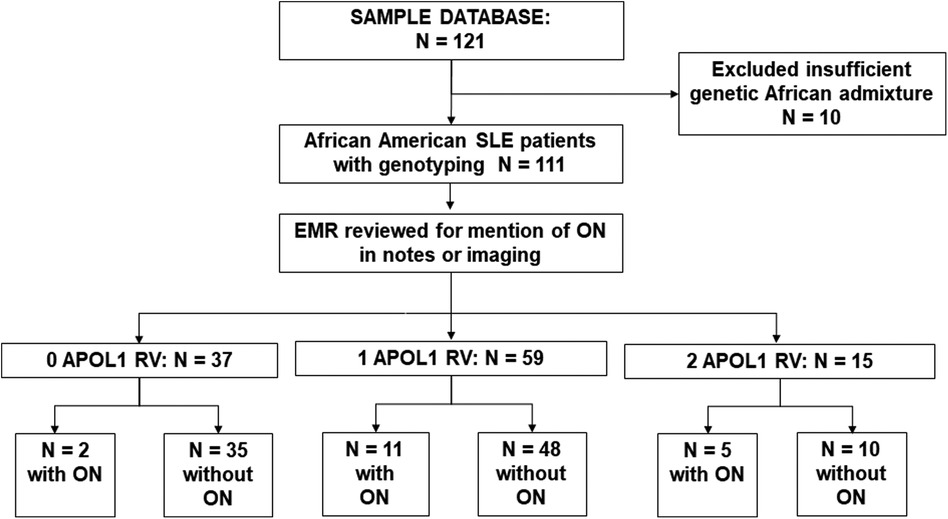
Figure 1. Screening workflow. We screened 123 Ancestrally African SLE patients; 10 with insufficient African admixture were excluded. Database and clinical charts were reviewed for the remaining 111 patients. Of 37 0RV carriers, 2 had osteonecrosis (ON), in 59 1RV carriers 11 had ON, and of 15 2RV carriers 5 had ON.
Longitudinal, prospective data captured as part of the NYU- division wide SLE (SAMPLE) repository was reviewed, including SLE ACR criteria, physical examination findings, medication history, and disease activity measured using the the SELENA-SLEDAI HYBRID index, which comprises all elements of the SELENA-SLEDAI instrument (27) but contains a modification of the definition of proteinuria (28) such that four points for proteinuria is always captured when the UPCR is >0.5. Information on smoking history, body mass index, diabetes, dyslipidemia, blood pressure, eGFR, and proteinuria, were collected through retrospective chart review. Values for vital signs and laboratory data were obtained at three visits at least six months apart and averaged. For all study participants, four independent reviewers (KY, NL, RF, AB) reviewed each SLE-related visit, including rheumatology outpatient visits, inpatient hospitalizations, infusion clinic visits, and nephrology outpatient visits, where applicable, from 2011 (first available electronic health records) to last study visit date. The prescribed corticosteroid dose was recorded and converted to prednisone equivalent and dose duration at each visit. Mentions of “pulse dose steroids” were assumed to indicate 1,000 mg of methylprednisolone for three days. Where available, doses were corroborated with pharmacy records. Once obtained, the prednisone equivalent dose was multiplied by the number of days prescribed. The resultant values were added to obtain the cumulative prednisone dose.
Descriptive statistics [mean and standard deviation or median (25th, 75th percentiles) for continuous variables; frequencies and percentages for categorical variables] were calculated separately by group (ON present vs. ON absent) for demographic information and comorbidities. The two groups were compared using the chi-square or Fisher's exact test, as deemed appropriate, for categorical variables and the two-sample t-test or Mann–Whitney test for continuous data.
To test the association between APOL1 genotype and ON, we treated the number of APOL1 risk variants (additive model) as the predictive variable. The primary outcomes were time from SLE disease diagnosis to first ON occurrence and the number of ON sites. The analysis of “time from SLE disease diagnosis to first ON occurrence” was accomplished b computing the Kaplan–Meier product limit curves, where the data were stratified by the number of APOL1 risk variants. In cases where the endpoint event had not yet occurred, data were censored at the time of death or at the date of the last study visit. Comparison across APOL1 genotypes were made using the log-rank test. Multiple imputations were performed using the Fully Conditional Specification approach as implemented in the MICE R package to create 10 imputed datasets (29, 30). Cox penalized regressions were fitted using each imputed dataset independently starting with an initial model that included the following variables: disease duration (D3_Disease_Duration), age (D1_Age), sex (D1_Sex), BMI (D2_BMI), eGFR (D2_eGFR), history of nephritis (D3_Nephritis), end stage kidney disease (D3_ESRD) average complement C3 level) (D3_C3), average complement C4 level (D3_C4), average SLEDAI score (D3_AVE_SLEDAI), APOL1 genotype (APOL1_Risk_allele), and cumulative prednisone dose (Total_pred_divided_by_visits) (31, 32). Variables consistently selected across the 10 imputed datasets were retained. A final model that only included variables that were consistently selected in the penalized cox regression across the 10 imputed datasets was fitted without the penalty term to provide unbiased parameter estimates. These variables were lupus nephritis, age, and APOL1 genotypes. We also included biological sex in the model due to the known sex differences in SLE. Ten sets of estimates, one for each imputed dataset, were obtained. These estimates were combined using Rubin's approach for combined results from multiple imputations (33–35). Model results are reported as hazard ratios with corresponding 95% confidence intervals. Unless otherwise specified, results were considered statistically significant at the 0.05 level of significance. Analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC), SPSS version 25, or R 3.6.3 platforms.
One hundred and eleven consented participants met the inclusion criteria. Demographic characteristics are shown in Table 1. Consistent with the reported higher incidence of SLE in women vs. men, 93% of study participants were female. The mean age was 43 ± 14 years, and mean BMI was 28.8 kg/m2. The mean age of SLE onset was 29 ± 12 years, and median disease duration was 10 (5, 18) years. Among those who had received glucocorticoids, the median cumulative prednisone equivalent dose was 9.2 g (3.7, 21.2). Fifty-six percent of study participants had a history of SLE nephritis, with 17% having received a course of cyclophosphamide therapy. The clinical characteristics of the cohort are described in Table 1. As shown in Figure 1, 37 participants carried 0RVs, 59 carried 1RV, and 15 carried 2RV. The allele frequencies of the ancestral allele, G0, and variant alleles, G1 and G2, were 0.6, 0.22, and 0.18, respectively.
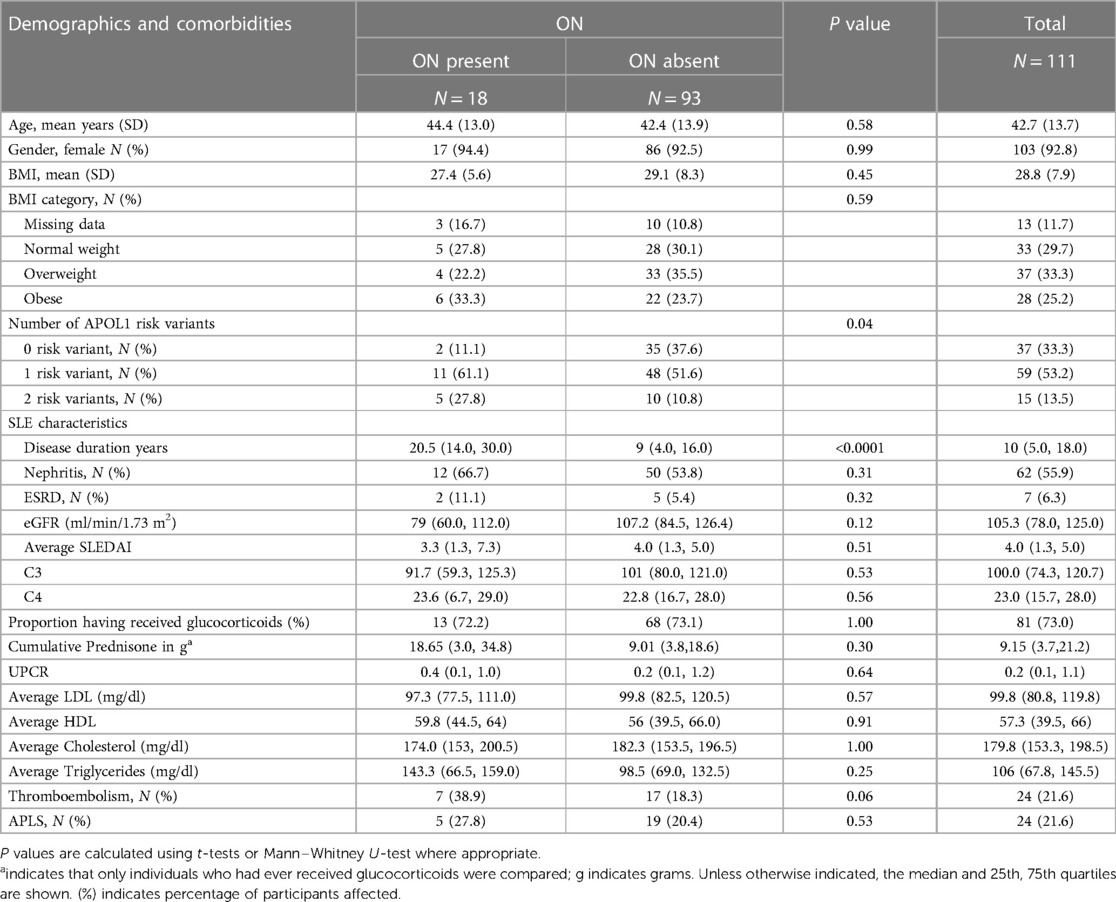
Table 1. Characteristics of the cohort by osteonecrosis (ON) status.
In our cohort 18 participants developed ON. Participants with ON had longer disease duration and carried more APOL1 RV copies with trends toward lower eGFR (Table 1). Across APOL1 genotype, 2 of the 37 0RV, 11 of the 59 1RV, and 5 of the 15 2RV participants developed ON (Figure 1). About 24% of the participants had no documented courses of glucocorticoids, both among those with and without ON. In participants who had received a glucocorticoid dose, the median cumulative prednisone equivalent dose was 18.6 g (2.0, 34.8) in those with ON and 9.0 g (3.8, 18.6) in those without ON. This trend did not meet statistical significance. The median disease duration in patients with vs. without ON was 9 (4, 16) and 20.5 (14, 30) years, respectively (Figure 2).
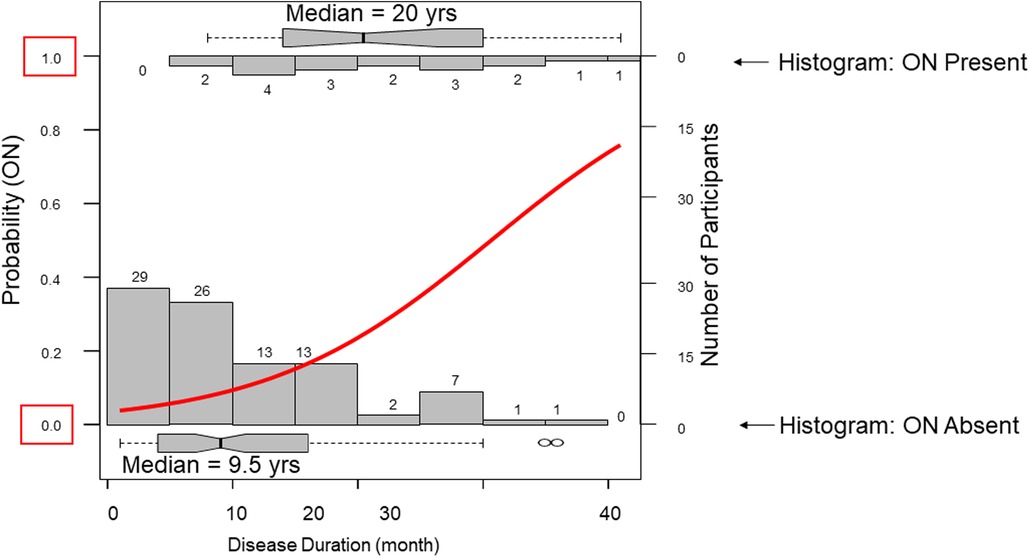
Figure 2. Disease duration associates with osteonecrosis. Two opposing histograms show disease duration frequencies in those with osteonecrosis (ON upper) and those without ON (lower). A regression curve is plotted in red. The median disease duration in those without ON was 9.5 years compared to 20 years in those wiht ON.
In time-to-event analysis, the proportion of individuals free from ON throughout SLE disease was assessed, and participants were censored at the age of the last follow-up visit or death. In an unadjusted ON-free survival model, 1RV and 2RV carriers developed ON earlier in SLE disease course, as presented by the Kaplan Meier Curves in Figure 3A. Across APOL1 genotype groups, the mean time to developing ON was 27 years in 0RV carriers, 22 years in 1RV carriers, and 18 years in 2 RV carriers (χ2 = 9.7; p = 0.005). The adjusted Cox regression model, 1RV and 2RV carriers exhibited shorter ON-free survival at hazard ratios of 3.1 (95% CI 1.6–6.2) and 9.6 (95% CI 2.4–37.8), respectively (Figure 3B).

Figure 3. (A) An unadjusted osteonecrosis (ON) free survival analysis is shown. The Y axis represents the proportion of individuals free of the outcome, and the X axis repesents disease duration over time. Individuals were censored at last follow-up or death. The number of individuals at risk at each time point is shown in the table below. (B) Adjusted Cox regression model for ON free survival, using an additive genetic model for APOL1 and adjusting for biological sex, age, and lupus nephritis status. Parameter estimates and hazard ratios are shown with 95% confidence intervals.
We lastly assessed the number of body sites and outcomes of ON in each of the APOL1 genotype groups. Figures 4A,B show that APOL1 variant carriers were more likely to have multiple and bilateral disease sites. Among the 18 individuals with ON, 40 joint sites were affected. The most reported sites were the femoral head, followed by the distal femur, proximal tibia, and ankles. There was one report of ON in the wrist. All 3 ON-affected joints in 0RV individuals occurred at a unilateral site. By contrast, 9 of 23 and 7 of 14 ON-affected joints in 1RV and 2RV individuals occurred at bilateral sites. Regarding ON frequency, 0RV carriers developed ON in 1–2 (mean = 1.5) joints compared to 1–3 (mean = 1.9) and 1–5 (mean = 2.8) joints in 1RV and 2RV carriers, respectively (χ2 = 19.0, p = 0.04, Figure 4B). The proportion of joints replaced was no different across the genotype groups.
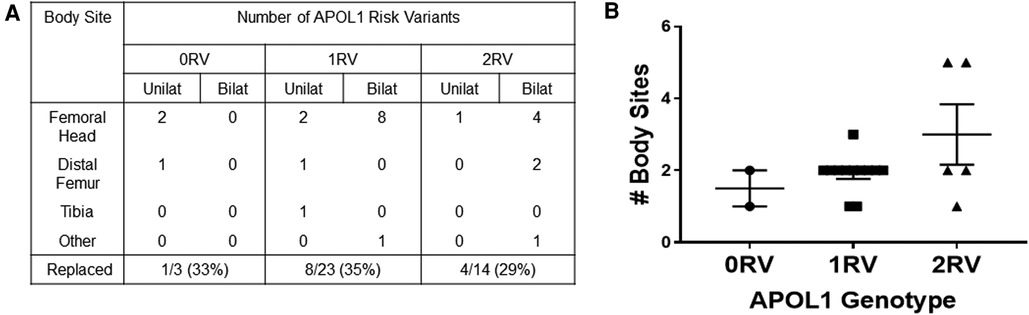
Figure 4. APOL1 risk variants associate with more osteonecrosis (ON) affected joint sites (A). In 18 individuals, 40 joints were affected by ON. The table shows the disease sites and laterality of joint sites by number of APOL1 risk variants (RV). The proportion of joints that were replaced is shown in the bottom row. (B) The number of body sites is shown on the y axis, and APOL1 genotype is shown on the X axis.
We have identified an association between APOL1 HRG and the development of ON in this well-phenotyped African Ancestry SLE cohort. APOL1 genotype associated with the presence of ON, time to first ON, and number of affected sites. This result was independent of disease duration, lupus nephritis status, and cumulative glucocorticoid dose. A higher proportion of 2RV carriers also developed ESRD relative to 0RV or 1RV carrying participants (2RV: 20% vs. 0RV: 8% and 1RV: 2%; p-value = 0.03). About 73% of individuals had documented corticosteroid doses; however the 1RV carriers had been prescribed a higher cumulative dose of glucocorticoids compred to 0RV or 1RV carrying participants. These participants also trended toward higher average SLEDAI scores than 0RV or 2RV participants. The APOL1 RV association best fit an additive model with an intermediate phenotype in the 1RV genotype group. Identifying this potential genetic risk for ON is a step forward in understanding ON pathobiology and identifying at-risk subpopulations within a vulnerable ancestral group.
Given that APOL1 HRG has been implicated in endothelial cell injury and is associated with predisposing comorbidities, an association with ON is biologically plausible (14, 19). APOL1 HRG has been shown to increase chronic kidney disease, hypertension, and microvascular disease risk (11, 12, 14, 15). ON pathogenesis involves an overlap of intravascular occlusion and extravascular compromise, leading to tissue ischemia and bone death (36). Immune complex deposition, intravascular lipid deposits, and thrombosis due to antiphospholipid syndrome all contribute to intravascular occlusion in SLE (37). Both endogenous interferons and hypoxia increase APOL1 expression (38). Therefore, it is plausible that interferon pathway activation and bony ischemia in SLE create a feedback loop with APOL1 HRG, whereby the accumulation of APOL1 variant protein induces endothelial damage. Endothelial cells highly express APOL1, and HRG-carrying cells exhibit an energetically senescent phenotype characterized by impaired autophagy (19). This critical cellular maintenance process allows cells to survive ischemic conditions (39). The APOL1 HRG endothelium may be more susceptible to ischemic injury characterized by ON.
Median cumulative corticosteroid dose in participants with ON was double that of those without ON, though this did not achieve statistical significance. It has long been shown that cumulative doses of corticosteroids, used to manage organ-threatening disease, increase ON risk (2). Various contributory mechanisms have been proposed. Systemic corticosteroids increase marrow fat content, which may lead to higher intraosseous pressure and lower circulation (40). Additional potential mechanisms include induction of a hypercoagulable state and inhibition of angiogenesis (41–43). High systemic corticosteroids are often used in lupus nephritis, the most common life-threatening SLE complication. Given that APOL1 HRG patients exhibit progressive proteinuric kidney disease despite standard of care LN treatment, these patients may be more likely to receive recurrent courses of glucocorticoids (44). Prospective cohorts better able to capture all inpatient and outpatient glucocorticoid dosing would help understand the relationship between HRG and clinical decision-making.
APOL1 HRG has been linked to systemic and aberrant intracellular lipid metabolism. APOL1 risk variants have been linked to dysfunctional reverse lipid transport and cholesterol accumulation in peripheral tissues and macrophages (45). Systemically, APOL1 HRG carriers exhibit higher levels of small HDL particles (46). Additional reports have found that the APOL1 HRG alters podocyte lipid metabolism; therefore, this role in ON development is a consideration (47, 48).
Strengths of our study include a detailed capture of clinical data, including cumulative corticosteroid dose and specific information regarding the frequency and laterality of ON. Our study is not without limitations. First, the small sample size may have limited the statistical power to identify other potential risk factors for ON, including hypertension, lupus nephritis, antiphospholipid syndrome, hemoglobin S and other β-globin chain abnormalities, radiation exposure, alcohol consumption and disease activity. Additionally, our study was not powered to understand whether or not the G1 and G2 alleles confer different phenotypes in SLE. Larger observational studies would be necessary to address these factors. Despite our attempt to control for the major known risk factors for ON, our data could still contain unassessed confounders. It is also possible that not all cases of ON were captured, given the study’s retrospective nature. Similarly, our retrospective review centered heavily on outpatient visits, pharmacy notes, and hospitalizations at one institution. Therefore historical glucocorticoid doses, or those given at outside emergency rooms and inpatient units, may have been missed resulting in an underestimation of the true cumulative glucocorticoid dose. Differences in parameters of steroid use, such as cumulative dose, maximum daily dose, and pulsed steroid dosing, may also pose a difference in the association between glucocorticoid use and ON. Despite these limitations, the biological plausibility and dose-dependent association between carrying these risk variants and ON is interesting and warrants additional study.
Our study found a higher risk of ON among African ancestry patients with SLE who carry APOL1 risk variants. This is a novel finding that should be studied in a multicenter, prospective study to address the limitations and confounders mentioned above. If confirmed, this association could provide important insights—particularly in light of forthcoming APOL1-targeted therapies. Further work is necessary to uncover the mechanism of association and help understand potential pathways of disease and treatment targets.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by New York University Grossman School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
AB and JB contributed to the conception and design of the study. AB, KY, RR, and NL made substantial contributions to the acquisition of data. AB, JD, MA, and YQ contributed to the analysis and interpretation of data. AB, KY, HA, PI, and MB drafted the article, and all authors were involved in revising it critically for important intellectual content. All authors approved the final version to be published. All authors contributed to the article and approved the submitted version.
Funding for this project was provided by the Rheumatology Research Foundation Scientist Development Award and, in part, by K23AI163359, both awarded to AB. This work was supported in part by the NYU CTSA grant 1UL1TR001445 from the National Center for the Advancement of Translational Science (NCATS), NIH, and The Colton Center at New York University School of Medicine. This work was supported in part by a Lupus Research Alliance Career Development Award to author AB.
The authors thank the SAMPLE biorepository study coordinator team for contributing to populating the SLE SAMPLE database and enrolling study participants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/flupu.2023.1219277/full#supplementary-material
1. Mankin HJ. Nontraumatic necrosis of bone (osteonecrosis). N Engl J Med. (1992) 326:1473–9. doi: 10.1056/NEJM199205283262206
2. Nevskaya T, Gamble MP, Pope JE. A meta-analysis of avascular necrosis in systemic lupus erythematosus: prevalence and risk factors. Clin Exp Rheumatol. (2017) 35:700–10.28240590
3. Kim HS, Bae SC, Kim TH, Kim SY. Endothelial nitric oxide synthase gene polymorphisms and the risk of osteonecrosis of the femoral head in systemic lupus erythematosus. Int Orthop. (2013) 37:2289–96. doi: 10.1007/s00264-013-1966-6
4. Chang JD, Hur M, Lee SS, Yoo JH, Lee KM. Genetic background of nontraumatic osteonecrosis of the femoral head in the Korean population. Clin Orthop Relat Res. (2008) 466:1041–6. doi: 10.1007/s11999-008-0147-1
5. Kim TH, Baek SH, Lim JO, Lee SH, Kim SY. Genetic variation in the coagulation factor V gene and risk of femoral head osteonecrosis. Mol Med Rep. (2015) 12:4434–40. doi: 10.3892/mmr.2015.4000
6. Limou S, Nelson GW, Kopp JB, Winkler CA. APOL1 kidney risk alleles: population genetics and disease associations. Adv Chronic Kidney Dis. (2014) 21:426–33. doi: 10.1053/j.ackd.2014.06.005
7. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (2011) 22:2129–37. doi: 10.1681/ASN.2011040388
8. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. (2010) 329:841–5. doi: 10.1126/science.1193032
9. Larsen CP, Beggs ML, Saeed M, Walker PD. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. J Am Soc Nephrol. (2013) 24:722–5. doi: 10.1681/ASN.2012121180
10. Lin CP, Adrianto I, Lessard CJ, Kelly JA, Kaufman KM, Guthridge JM, et al. Role of MYH9 and APOL1 in African and non-African populations with lupus nephritis. Genes Immun. (2012) 13:232–8. doi: 10.1038/gene.2011.82
11. Akinyemi R, Tiwari HK, Arnett DK, Ovbiagele B, Irvin MR, Wahab K, et al. APOL1, CDKN2A/CDKN2B, and HDAC9 polymorphisms and small vessel ischemic stroke. Acta Neurol Scand. (2018) 137:133–41. doi: 10.1111/ane.12847
12. Gutierrez OM, Irvin MR, Chaudhary NS, Cushman M, Zakai NA, David VA, et al. APOL1 nephropathy risk variants and incident cardiovascular disease events in community-dwelling black adults. Circ Genom Precis Med. (2018) 11:e002098. doi: 10.1161/CIRCGEN.117.002098
13. Miller AK, Azhibekov T, O'Toole JF, Sedor JR, Williams SM, Redline RW, et al. Association of preeclampsia with infant APOL1 genotype in African Americans. BMC Med Genet. (2020) 21:110. doi: 10.1186/s12881-020-01048-4
14. Blazer A, Dey ID, Nwaukoni J, Reynolds M, Ankrah F, Algasas H, et al. Apolipoprotein L1 risk genotypes in Ghanaian patients with systemic lupus erythematosus: a prospective cohort study. Lupus Sci Med. (2021) 8. doi: 10.1136/lupus-2020-000460
15. Blazer A, Wang B, Simpson D, Kirchhoff T, Heffron S, Clancy RM, et al. Apolipoprotein L1 risk variants associate with prevalent atherosclerotic disease in African American systemic lupus erythematosus patients. PLoS One. (2017) 12:e0182483. doi: 10.1371/journal.pone.0182483
16. Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. (2011) 22:2119–28. doi: 10.1681/ASN.2011010069
17. Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJ. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. (2002) 79:539–46. doi: 10.1006/geno.2002.6729
18. O'Toole JF, Bruggeman LA, Madhavan S, Sedor JR. The cell biology of APOL1. Semin Nephrol. (2017) 37:538–45. doi: 10.1016/j.semnephrol.2017.07.007
19. Blazer A, Qian Y, Schlegel MP, Algasas H, Buyon JP, Cadwell K, et al. APOL1 variant-expressing endothelial cells exhibit autophagic dysfunction and mitochondrial stress. Front Genet. (2022) 13. doi: 10.3389/fgene.2022.769936
20. Izmirly PM, Wan I, Sahl S, Buyon JP, Belmont HM, Salmon JE, et al. The incidence and prevalence of systemic lupus erythematosus in New York county (manhattan), New York: the Manhattan lupus surveillance program. Arthritis Rheumatol. (2017) 69:2006–17. doi: 10.1002/art.40192
21. Lewis MJ, Jawad AS. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology (Oxford. (2017) 56:i67–77. doi: 10.1093/rheumatology/kex200
22. Dummer PD, Limou S, Rosenberg AZ, Heymann J, Nelson G, Winkler CA, et al. APOL1 kidney disease risk variants: an evolving landscape. Semin Nephrol. (2015) 35:222–36. doi: 10.1016/j.semnephrol.2015.04.008
23. Egbuna O, Zimmerman B, Manos G, Fortier A, Chirieac MC, Dakin LA, et al. Inaxaplin for proteinuric kidney disease in persons with two APOL1 variants. N Engl J Med. (2023) 388:969–79. doi: 10.1056/NEJMoa2202396
24. Inês L, Silva C, Galindo M, López-Longo FJ, Terroso G, Romão VC, et al. Classification of systemic lupus erythematosus: systemic lupus international collaborating clinics versus American college of rheumatology criteria. A comparative study of 2,055 patients from a real-life, international systemic lupus erythematosus cohort. Arthritis Care Res (Hoboken). (2015) 67:1180–5. doi: 10.1002/acr.22539
25. Sesin CA, Yin X, Esmon CT, Buyon JP, Clancy RM. Shedding of endothelial protein C receptor contributes to vasculopathy and renal injury in lupus: in vivo and in vitro evidence. Kidney Int. (2005) 68:110–20. doi: 10.1111/j.1523-1755.2005.00385.x
26. Greenhagen RM, Crim BE, Shinabarger AB, Burns PR. Bilateral osteonecrosis of the navicular and medial cuneiform in a patient with systemic lupus erythematosus: a case report. Foot Ankle Spec. (2012) 5:180–4. doi: 10.1177/1938640012439605
27. Petri M, Kim MY, Kalunian KC, Grossman J, Hahn BH, Sammaritano LR, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med. (2005) 353:2550–8. doi: 10.1056/NEJMoa051135
28. Thanou A, James JA, Arriens C, Aberle T, Chakravarty E, Rawdon J, et al. Scoring systemic lupus erythematosus (SLE) disease activity with simple, rapid outcome measures. Lupus Sci Med. (2019) 6:e000365. doi: 10.1136/lupus-2019-000365
29. van Buuren S. Multiple imputation of discrete and continuous data by fully conditional specification. Stat Methods Med Res. (2007) 16:219–42. doi: 10.1177/0962280206074463
30. van Buuren S, Groothuis-Oudshoorn K. Mice: multivariate imputation by chained equations in R. J Stat Softw. (2011) 45:1–67. doi: 10.1097/00003086-199307000-00039
31. Tibshirani R. The lasso method for variable selection in the cox model. Stat Med. (1997) 16:385–95. doi: 10.1002/(SICI)1097-0258(19970228)16:4%3C385::AID-SIM380%3E3.0.CO;2-3
32. Fan J, Li R. Variable selection for cox’s proportional hazards model and frailty model. The Annals of Statistics. (2002) 30:74–99. doi: 10.1161/01.res.0000261924.76669.36
33. Rubin DB. Multiple imputation after 18+ years. J Am Stat Assoc. (1996) 91:473–89. doi: 10.1080/01621459.1996.10476908
34. Rubin DB. Handling nonresponse in sample surveys by multiple imputations. U.S. Department of commerce bureau of the ensus. Monograph. (1980) 1:75–112. doi: 10.1002/9780470316696
35. Mislevy RJ. Statistical analysis with missing data. Journal of Educational Statistics. (1991) 16:150–5. doi: 10.2307/1165119
36. Tsai H-L, Chang J-W, Lu J-H, Liu C-S. Epidemiology and risk factors for avascular necrosis in childhood systemic lupus erythematosus in a Taiwanese population. Sci Rep. (2020) 10:15563. doi: 10.1038/s41598-020-71923-w
37. Jones JP Jr. Fat embolism, intravascular coagulation, and osteonecrosis. Clin Orthop Relat Res. (1993) 292:294–308. doi: 10.1097/00003086-199307000-00039
38. Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, et al. Innate immunity pathways regulate the nephropathy gene apolipoprotein L1. Kidney Int. (2015) 87:332–42. doi: 10.1038/ki.2014.270
39. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circ Res. (2007) 100:914–22. doi: 10.1161/01.RES.0000261924.76669.36
40. Wang GJ, Cui Q, Balian G. The nicolas andry award. The pathogenesis and prevention of steroid-induced osteonecrosis. Clin Orthop Relat Res. (2000) 370:295–310. doi: 10.1097/00003086-200001000-00030
41. Fisher DE, Bickel WH, Holley KE. Histologic demonstration of fat emboli in aseptic necrosis associated with hypercortisonism. Mayo Clin Proc. (1969) 44:252–9.4888919
42. Boettcher WG, Bonfiglio M, Hamilton HH, Sheets RF, Smith K. Non-traumatic necrosis of the femoral head. I. relation of altered hemostasis to etiology. J Bone Joint Surg Am. (1970) 52:312–21. doi: 10.2106/00004623-197052020-00012
43. Smith DW. Is avascular necrosis of the femoral head the result of inhibition of angiogenesis? Med Hypotheses. (1997) 49:497–500. doi: 10.1016/S0306-9877(97)90067-0
44. Freedman BI, Langefeld CD, Andringa KK, Croker JA, Williams AH, Garner NE, et al. Nephritis-end-stage renal disease, end-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. (2014) 66:390–6. doi: 10.1002/art.38220
45. Ryu JH, Ge M, Merscher S, Rosenberg AZ, Desante M, Roshanravan H, et al. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS One. (2019) 14:e0211559. doi: 10.1371/journal.pone.0211559
46. Gutiérrez OM, Judd SE, Irvin MR, Zhi D, Limdi N, Palmer ND, et al. APOL1 Nephropathy risk variants are associated with altered high-density lipoprotein profiles in African Americans. Nephrol Dial Transplant. (2016) 31:602–8. doi: 10.1093/ndt/gfv229
47. Ge M, Molina J, Ducasa GM, Mallela SK, Varona Santos J, Mitrofanova A, et al. APOL1 risk variants affect podocyte lipid homeostasis and energy production in focal segmental glomerulosclerosis. Hum Mol Genet. (2021) 30:182–97. doi: 10.1093/hmg/ddab022
Keywords: systemic lupus erythematosus, osteonecrosis, APOL1, apolipoprotein 1, African Americans
Citation: Yip K, Akerman M, Fernandez Ruiz R, Leung N, Algasas H, Qian Y, Buyon JP, Divers J, Izmirly P, Belmont M and Blazer AD (2023) Osteonecrosis is associated with APOL1 variants in African Americans with systemic lupus erythematosus. Front. Lupus 1:1219277. doi: 10.3389/flupu.2023.1219277
Received: 8 May 2023; Accepted: 19 September 2023;
Published: 9 October 2023.
Edited by:
Beatrice Goilav, Children’s Hospital at Montefiore, United StatesReviewed by:
Teruhiko Yoshida, National Institute of Diabetes and Digestive and Kidney Diseases (NIH), United States© 2023 Yip, Akerman, Fernandez Ruiz, Leung, Algasas, Qian, Buyon, Divers, Izmirly, Belmont and Blazer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashira D. Blazer YmxhemVyYUBoc3MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.