
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Trop. Dis. , 05 February 2024
Sec. Neglected Tropical Diseases
Volume 5 - 2024 | https://doi.org/10.3389/fitd.2024.1276210
 Aristide Toussaint Nguélé1
Aristide Toussaint Nguélé1 Matteo Mozzicafreddo1,2
Matteo Mozzicafreddo1,2 Hongliang Chen1,3
Hongliang Chen1,3 Angela Piersanti1Salum Seif Salum4Said M. Ali5
Angela Piersanti1Salum Seif Salum4Said M. Ali5 Junjie Zhang6
Junjie Zhang6 Cristina Miceli1*
Cristina Miceli1*Large intestine-dwelling helminths affect microbiome composition. In sub-Saharan Africa, where helminth infections are endemic, the use of chemotherapeutic drugs is the primary strategy for controlling soil-transmitted helminthiases (STHs). However, the emergence of anthelmintic resistance necessitates the urgent exploration of alternative and complementary treatments to achieve the World Health Organization’s goal of eliminating STHs. One promising avenue involves the manipulation of gut microbiota in at-risk populations. This study aimed to enhance the understanding of the interplay between Trichuris trichiura and the gut microbiome. In this study, we used the Mini-FLOTAC technique for parasitological analyses and a shotgun metagenomic sequencing approach to investigate the effect of T. trichiura on the gut microbiome by comparing infected and non-infected women of reproductive age (WRA) from Pemba. Structural and functional analyses of the gut microbiome revealed that T. trichiura infection shaped the host gut microbiome in WRA. Some taxa vary according to infection status. Prevotella genus was more abundant in healthy participants, whereas species such as Weissella cibaria, Leuconostoc citreum (new emergent probiotics), and Leuconostoc lactis (starter) decreased in infected individuals, suggesting the use of potential probiotic treatments to mitigate dysbiosis induced by STHs. Furthermore, the overall number of common fungi, irrespective of species, was significantly higher in the mycobiome of Trichuris infected participants. Functional analysis revealed significant differences in metabolic pathways (p < 0.05), with cholesterol metabolism and pathogenic infections being more abundant in the infected samples than in the non-infected samples. In conclusion, this study sheds light on the intricate interactions between helminth infections and the gut microbiome in the WRA, particularly in STH-endemic regions. The identified associations between specific gut microbial changes and T. trichiura infection may pave the way for innovative complementary treatments to effectively combat STHs.
Soil-transmitted helminth (STH) infections are among the most common infections worldwide, with an estimated 1.5 billion infected people, or 24% of the world’s population. The main species that infect humans are roundworms (Ascaris lumbricoides), whipworms (T. trichiura), and hookworms (Necator americanus and Ancylostoma duodenale). According to the World Health Organization (WHO), heavy infections can cause a variety of symptoms, including intestinal manifestations (diarrhoea and abdominal pain), malnutrition, general malaise and weakness, impaired growth, and physical development. School-going children are most affected by STH infections, resulting in poor school performance and impaired cognitive function, among other detrimental effects (1). Recent estimates indicate that > 880 million children require treatment for these parasites. The population at risk in the African region according to the World Health Organization is estimated to be 350 million (www.who.int/health-topics/helminthiasis). Polyparasitism involving STHs and Schistosoma blood flukes is common in low- and middle-income countries. These helminths affect the gut environment and may cause changes in the gut microbiome composition (2, 3). Gastrointestinal pathogens can alter the host gut microbiome and affect bacterial diversity and abundance, brain function, digestive health, immune function, and development (4).
Studies on the human intestinal microbiota have been neglected for several years (5), although they are at the interface between ingested food and the gut epithelium. They are also in contact with the first pool of immune cells and the second pool of neural cells in the body. The gut microbiota is now gaining recognition as an organ that plays a major role in health and disease (6). However, it remains true that many structural analyses are limited on gut bacteria ignoring other gut microbial members for which important roles during health and disease have become increasingly more appreciated (7). A pilot study revealed that changes in gut microbial composition and structure occur in T. trichiura infected individuals compared to uninfected individuals (8). Regarding the relationship between Trichuris sp and the gut microbiota, studies reveal that the hatching of some Trichuris eggs is favoured in presence of specific bacteria such as Escherichia coli and Lactobacillus reuteri (9). A recent study by Rosa et al. (2021) showed significant positive associations for seven taxa, including Escherichia, which has been shown to induce whipworm egg hatching and Bacteroides, which has previously been identified as a major component of the whipworm internal microbiome (10). Another study revealed a relationship between success- and failure-associated enterotypes. Using a survival analysis, this investigation confirmed that patients presenting an enterotype rich in Eubacterium coprostanoligenes and Ruminococcus torques before the treatment are more likely to be more efficiently cured from the T. trichiura infestation by using the albendazole and ivermectin-based treatment than those presenting an enterotype rich in Prevotella, Roseburia and Coprococcus, or rich in Faecalibacterium and Escherichia/Shigella (11).
With the emergence of resistance to deworming drugs (12), it is necessary to better understand the relationship between the gut microbiota and helminths to optimise the fight against STH/Trichuris infections. This study was conducted in an endemic area of helminth infections. Results of a survey published in 2021 revealed that the prevalence of STH was evaluated at 80% (95% CI 78.1–81.5) and most of the STH cases were due to T. trichiura (13). In Unguja and Pemba, the two main islands that form the Zanzibar Archipelago, STH infections were recognised as a major public health problem in the early 1990s. In 1994, the Ministry of Health and Social Welfare of Zanzibar, in collaboration with the WHO, established an action plan for controlling STHs and urinary schistosomiasis (14). Chemotherapy consisting of albendazole has yielded interesting results by reducing the intensity of STH infections and morbidity. However, the burden of STH infection remains a public health challenge in Unguja and even more so in Pemba. Hence, it is imperative to explore complementary strategies for their use in conjunction with the administration of chemical drugs to combat helminth infections. One promising avenue involves manipulation of the gut microbiota of at-risk populations. This study aimed to enhance our understanding of the interplay between T. trichiura and the gut microbiome. This study aimed to investigate the effects of T. trichiura on the gut microbiome of women of reproductive age (WRA), where soil-transmitted helminths (STHs) are highly prevalent. This study employed shotgun metagenomic sequencing technology to compare the gut microbiomes of infected and uninfected individuals.
The project was approved by the Zanzibar Health Research Institute (ZAHRI; protocol number: ZAMREC/001/SEPT/018)) and written informed consent was obtained from all participants enrolled in the study.
This pilot cross-sectional study was conducted as secondary analysis of previously collected samples. Participants were categorised into two groups each consisting of 5 individuals classified as either positive or negative for T. trichiura infection (All samples were coded as shown in Table 1). As previously reported (8), women aged from 23 to 45 years were enrolled from sanitary centres in Pemba Island (Tanzania) where helminth infection is endemic and interviewed in Swahili with the help of nurses and personnel of the Public Health Laboratory Ivo de Carneri (PHL-IdC). Each participant completed a questionnaire (reported in 8), signed the informed consent, and provided faecal samples. The inclusion criteria for all the individuals were as follows: very similar origin of food and diet, mainly consisting of banana fruit, cassava, rice, cassava leaves as vegetable and dagaa fish, no HIV infection, no diarrhea, no fever, no diabetes, no malaria, and no antibiotics or anthelmintic treatment in the previous 3 months.
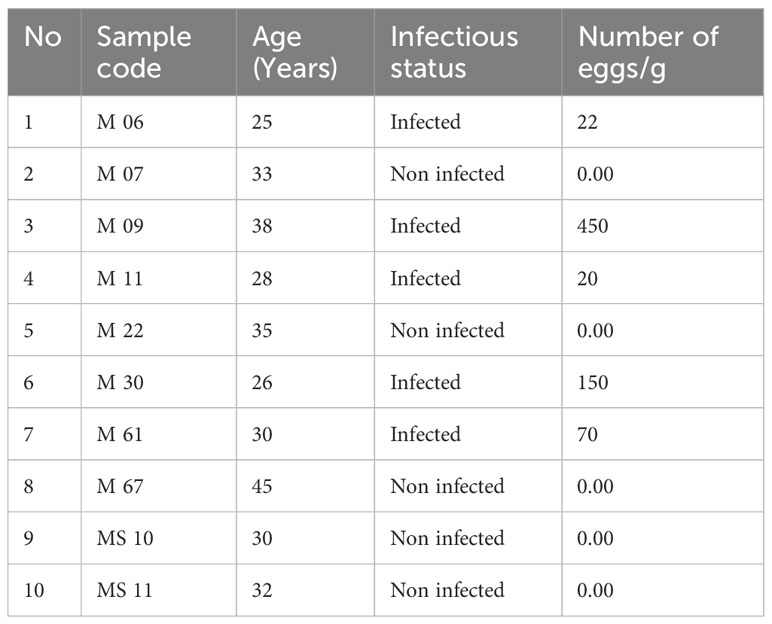
Table 1 Characteristics of the selected samples in relation to Trichuris infection status and detected number of eggs/g.
Parasitological analyses aimed at detecting helminth infections, employed flotation techniques, and microscopic observations were independently conducted by two experts (8). The samples included in this study were randomly chosen based on the criteria of absence of co-infection with helminth species, non-pregnant, and having not used any antiparasitic treatments including herbal medications.
Genomic DNA was extracted from frozen faecal samples using the NORGEN BIOTEK Stool DNA Isolation Kit, according to the manufacturer’s instructions. DNA quality and quantity were determined using a NanoDrop spectrophotometer and direct gel electrophoresis, respectively. DNA integrity was confirmed by analysing 300 ng of DNA by electrophoresis and visualisation under ultraviolet (UV) light. The extracted DNA was stored at -20°C. The purified DNA was sent to the SynBiotec laboratory (spin-off of the University of Camerino) for shotgun sequencing, which was carried out using the Illumina Miseq platform 2×150PE. Prior to library construction, the DNA was quantified using a Qubit 4.0 Fluorometer. A DNA library was prepared according to the Illumina DNA Prep Guide for Illumina paired-end-indexed sequencing. The extracted DNA (150–500 ng) were used as the library input. Bead-Linked Transposomes (BLT) were used to fragment and tag DNA with adapter sequences. The adapter-tagged DNA was washed with BLT before polymerase chain reaction (PCR) amplification, using a limited-cycle PCR program comprising five cycles. The amplified library was subjected to double-sided bead purification. To confirm the size distribution, the resulting libraries were validated using an Agilent Bioanalyzer 2100 (High-Sensitivity DNA kit). The library concentrations were quantified using a Quibit 4.0 Fluorometer. The indexed DNA libraries were normalised to 4 nM and combined into equal volumes. The samples were then sequenced using an Illumina Miseq, 2×150bp V2 paired end run. On average, each sample yields 1,282,162.5 reads with an average read length of 146.825 bp.
Bioinformatics and statistical analyses were performed using the OmicsBox software (version 3.0.30). The step of cleaning from contaminants such as the human DNA was performed. All read data were cleaned of human and phiX DNA using the Bowtie2 tool. Next, kraken2 classifier, a taxonomic classification system that allows high accuracy and fast classification (database version 2019.06), was used for taxonomic profiling. Paired-end reads were selected as the type of input data, and a confidence filter was enabled and set at 0.05. A Stacked bar chart was used to provide a view for inter-sample comparison separated at the main taxonomic level. The average operational taxonomic units (OTUs) were ordered by abundance from high to low. Only the 500 largest OTUs are shown for each sample; the remaining were grouped into an extra group called others. OTUs groups were also studied by analysing Krona pie charts using the same software. A rarefaction curve was generated to determine whether the sequencing coverage was sufficiently deep to obtain an accurate estimate of the total number of OTUs present in a specific sample. A Diversity Curve was generated and used to evaluate the benefits of microbial diversity, including additional samples, in the dataset. Principal Coordinate Analysis (PCoA plot), a two-dimensional plot reporting the Bray-Curtis distances between samples, was generated to perceive the distance between samples according to Trichuris infection status. Differential Abundance Analysis of Taxa was performed by dividing samples into two groups and comparing features using the edgeR module of OmicsBox. Non-infected samples were assigned to the reference group and compared with infected samples set as the contrast group. A false discovery rate (FDR) <0.05 was considered significant. For each sample, a genome assembly was generated using the assembly pipelines of meta-SPAdes, based on the de Bruijn graph included in OmicsBox, and setting the K-mer sizes as automated. In the following step, gene prediction was performed for each assembly using the application FragGeneScan, also available in OmicsBox, which is capable of predicting intact and incomplete ORFs in short sequencing reads. For functional investigation, Clusters of Orthologous Genes (COG) and protein family (Pfam) databases were used for metabolic pathway analysis and gene annotation. The contrast-infected group was compared with the reference uninfected group to determine the differences in terms of over- or under-represented metabolic pathways. Statistical significance was set at P < 0.05. T test analysis and plotting were performed using GraphPad Prism (version 9.5.1).
Principal coordinate analysis (PCoA) mapping was performed to better understand the relationships between the participants’ gut microbiota and to identify similarities and differences. PCoA facilitated the visualisation of clusters based on the infection status, as highlighted in Figure 1. In addition, PCoA allowed the identification of subclusters emerging in response to parasitic loads. Notably, subjects M06 and M11, with similar loads (22 and 20 eggs/g, respectively), showed a close relationship, implying greater similarities in their gut microbiota composition. In addition, subjects M61 and M30, who carried slightly elevated parasitic loads (70 and 150 eggs/g, respectively), formed another subcluster. Intriguingly, subject M09, bearing the heaviest load (450 eggs/g), appeared to be isolated, although closer to the group of non-infected subjects. This suggests a different impact of the worm on the gut microbiota depending on the infection load. Samples from non-infected individuals showed more cohesive clustering than those from infected individuals. This suggests that the gut microbiota of the non-infected samples shared greater similarity.
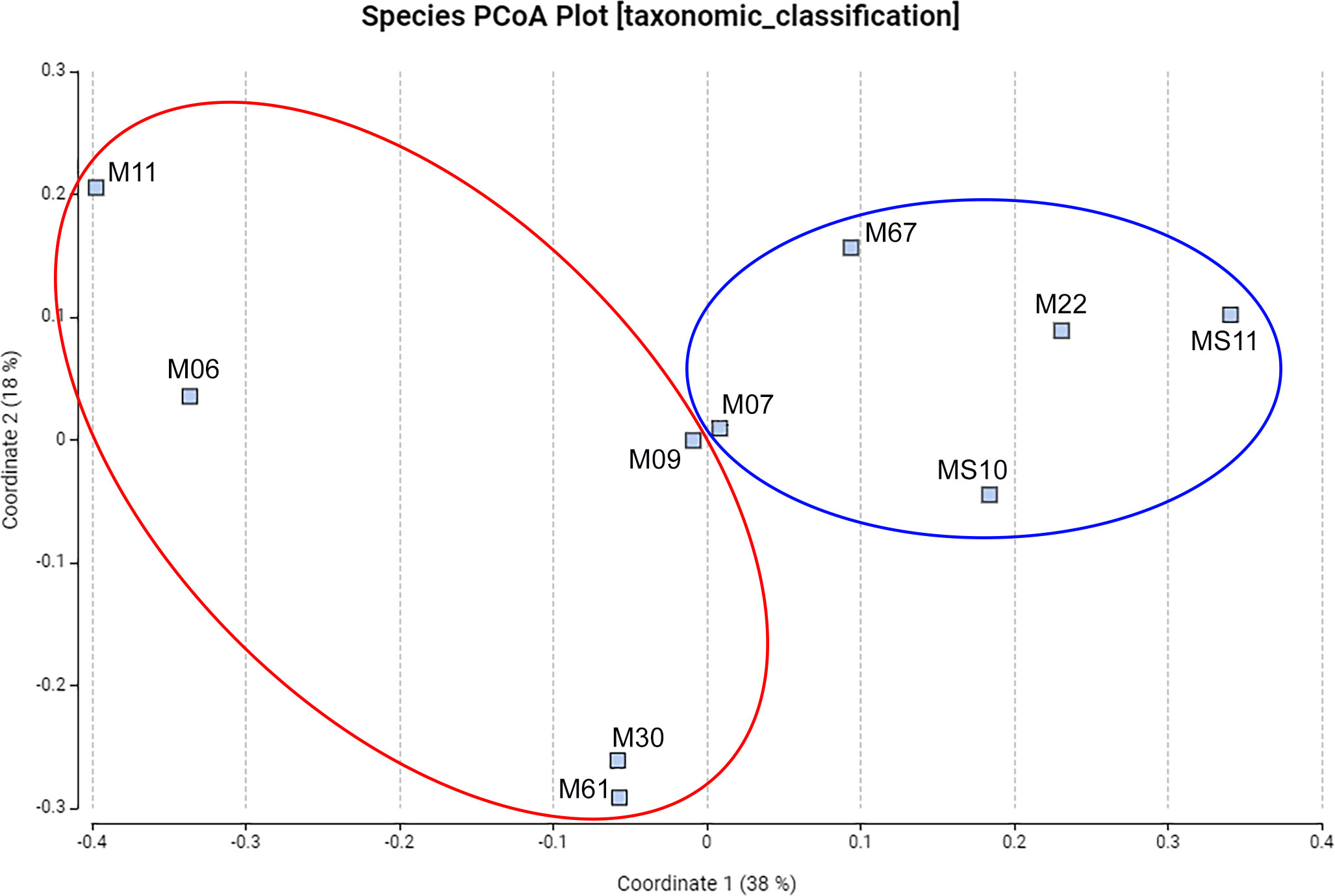
Figure 1 Class PCoA plot showing different clusters between non-infected and infected samples. Non-infected samples M07, M67, M22, MS10, MS11 are indicated by blue color circle and infected samples M06, M09, M11, M30, M61 by red color circle.
Shotgun metagenomic sequencing of ten selected samples yielded a total number of 12,652,028 reads. On average, each sample contained 1,265,203 reads, with an overall average read length of 146.87. Distribution of the top 10 hits revealed that bacteria accounted for more than 90 percent of each sample, followed by eukaryotes, archaea, and viruses. The microbiome composition, visualised up to the species level in each sample using Krona charts, remained unclassified in approximately 50% of the reads. This indicates that a considerable number of sequenced genes remained unannotated. This phenomenon is well known and can be attributed to different reasons, such as limitations in the databases with respect to the high microbial biodiversity of the gut, specifically for eukaryotic microbes that may not have matches in the existing databases. This can also be attributed to the software’s limitations in accommodating (annotating) genes and taxa with a limited presence, which cannot be easily interpreted by sequencing with short read lengths.
In addition, an intriguing observation emerged: the percentage of classified reads was relatively higher in non-infected samples than in infected samples (see Supplementary Material, Table l: Percentage of classification). The rarefaction curve provided evidence that the richness was adequate in the sequenced samples (Supplementary Material, Figure 1). The diversity curve used to assess and compare the diversity across populations confirmed that the number of samples was sufficient to obtain the expected diversity (Supplementary Material, Figure 2).
Taxonomic analysis revealed that, at the phylum level (Figure 2), the community was dominated by Firmicutes (also named Bacillota), Bacteroidetes, Proteobacteria, and Actinobacteria. Notably, the Firmicutes/Bacteroidetes ratio (as shown in Figure 3A), a widely used indicator for assessing the state of intestinal microbiota health, was significantly low in subjects without T. trichiura infection. In infected samples, a higher relative abundance of Firmicutes was observed than in noninfected samples, whereas noninfected samples showed a higher relative abundance of the phylum Bacteroidetes, except for sample M09. Proteobacteria, known to include more pathogenic species, displayed a higher relative abundance in Trichuris infected samples (Figure 3B).
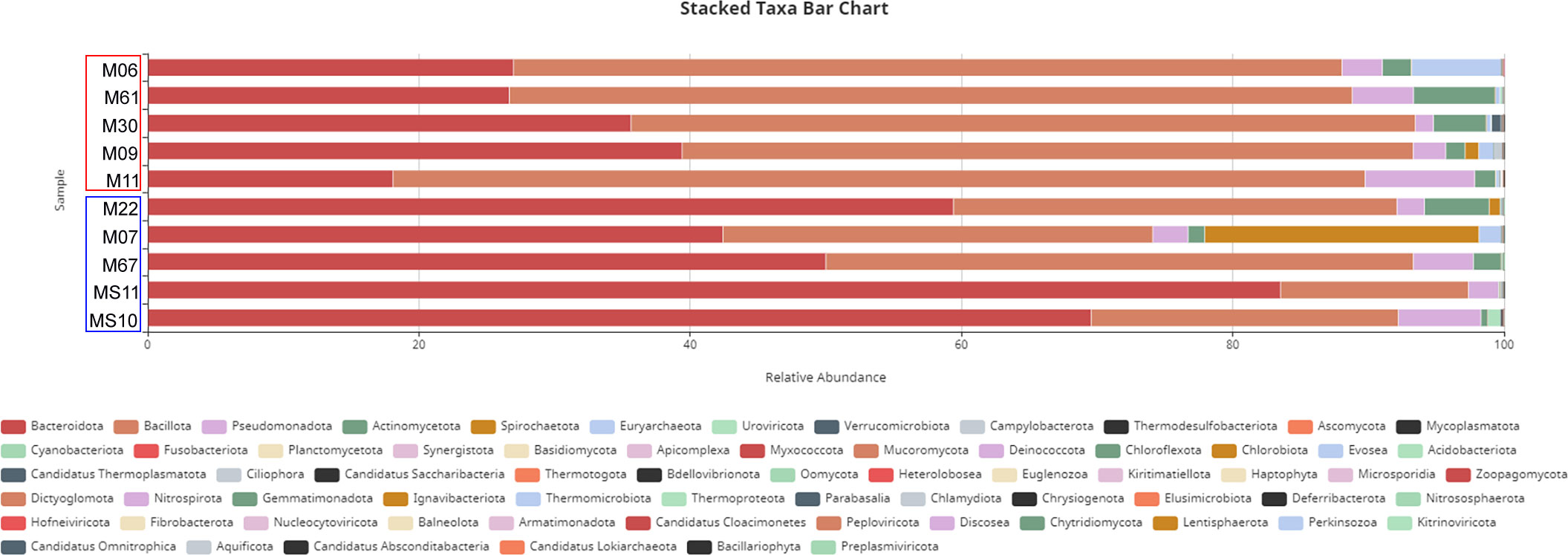
Figure 2 Taxa bar chart analysed at the phylum level. Non-infected samples M07, M67, M22, MS10, MS11 are indicated by blue color rectangle and infected samples M06, M09, M11, M30, M61 by red color rectangle.
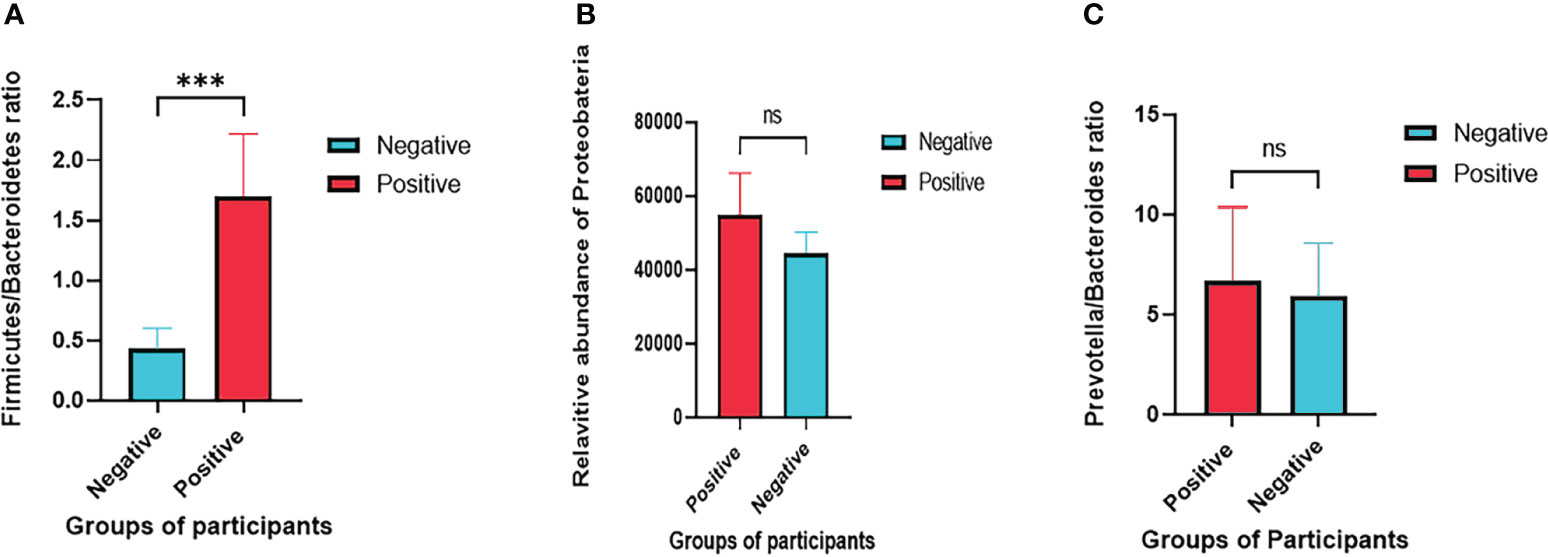
Figure 3 Analysis of markers of the healthy condition of the gut microbiota. (A) Firmicutes/Bacteroidetes ratio in Trichuris-negative vs Trichuris-positive samples (unpaired t. test, P = 0.0009); (B) relatively higher abundance of Proteobacteria in Trichuris-positive samples (P = 0.1099); (C) the Prevotella/Bacteroides ratio is higher in infected samples (P = 0.6979). *** stands for a P value ≤ 0.001 and ns stands for P value ≥ 0.05.
Analysis of the community composition at the genus level unveiled Prevotella as the most abundant genus, followed by Faecalibacterium, Bacteroides and Roseburia (Figure 4). The ratio of Prevotella/Bacteroides (Figure 3C), both members of the phylum Bacteroidetes, was only slightly higher in Trichuris infected samples. The abundance of these two bacterial genera is driven by distinct dietary preferences and contributes to the classification of different enterotypes. Furthermore, this analysis also revealed that some bacteria responsible of short chain fatty acids (SCFAs) production, including Prevotella, Prevotella 9, and Ruminococcus showed a lower relative abundance in infected samples in comparison to healthy samples (as illustrated in Figure 4 for samples MS10, MS11, M07, M22, and M67). In contrast, Faecalibacterium was significantly more prevalent in infected samples than in non-infected samples.
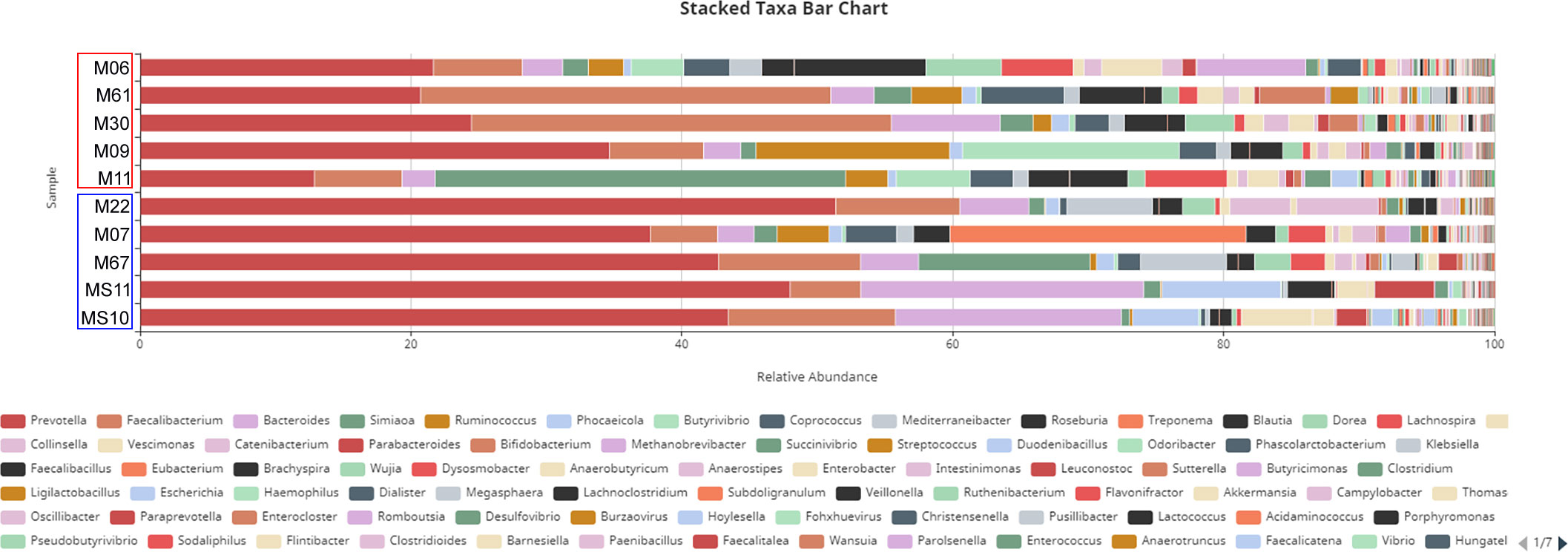
Figure 4 Taxa bar chart analysed at the genus level. Infected and non-infected samples are indicated by red and blue columns, respectively.
Species level analysis revealed that Prevotella copri, Simiaoa sunii and Faecalibacterium prausnitzii were the predominant species (Figure 5). These were followed by other species, including Ruminococcus torques, Ruminococcus SP.JE7A12, Roseburia intestinalis, Lachnospira eligens, and Treponema succinifaciens.
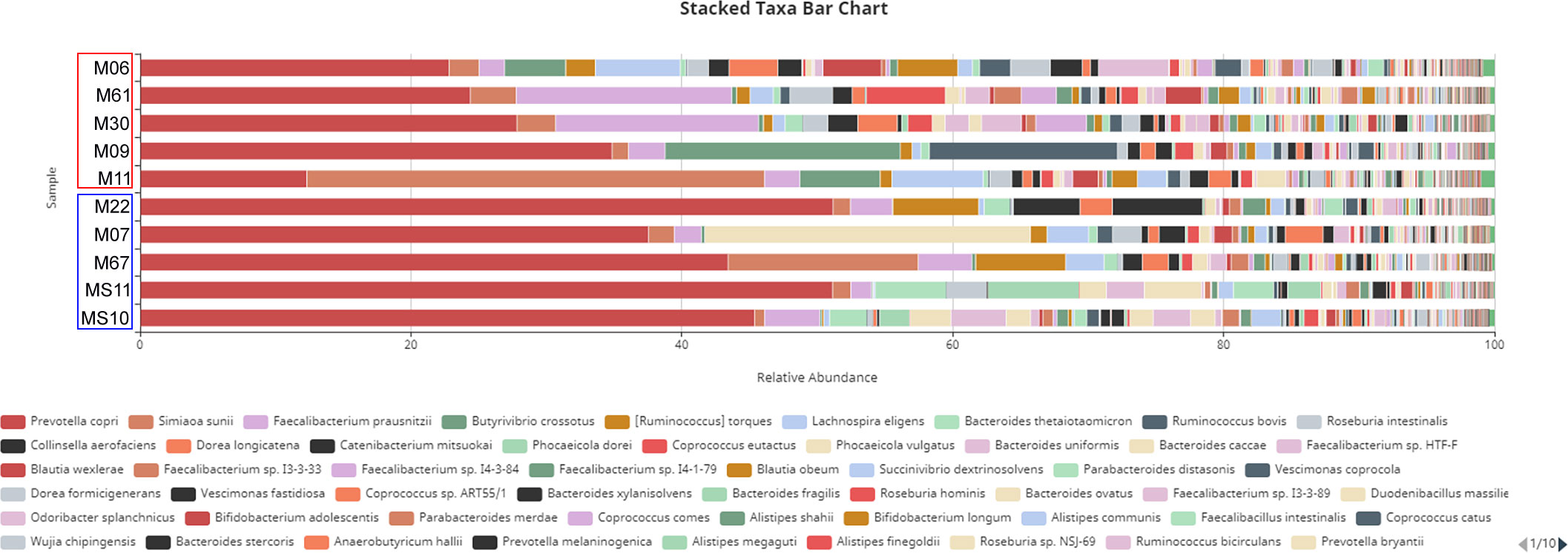
Figure 5 Taxa bar chart at the species level. Infected and non-infected samples are indicated by red and blue columns, respectively.
The differential taxonomic abundance analyses conducted at the species level yielded distinct taxonomic compositions based on the Trichuris infection status in the WRA. Species such as Weissella cibaria, Weissella paramesenteroides and Leuconostoc citreum (emerging probiotics) and Leuconostoc lactis (used as a starter in fermentation) were underrepresented in infected samples compared with healthy samples. Species of Bacteroides genus, such as B. stercoris and B. fragilis were also underrepresented in the same group. Conversely, other species displayed contrasting patterns, such as Butyrivibrio crossotus, Bifidobacterium bifidum, Ligalactobacillus salivaris, and Methanobrevibacter woesei that were more abundant in T. trichiura infected subjects when compared to healthy individuals. (Figure 6). Potentially pathogenic species, such as Treponema succinifaciens and Streptococcus gallolyticus, the latter being the main causative agents of septicaemia and infective endocarditis in elderly and immunocompromised individuals, and strongly associated with colorectal cancer (15), were notably present in infected samples.
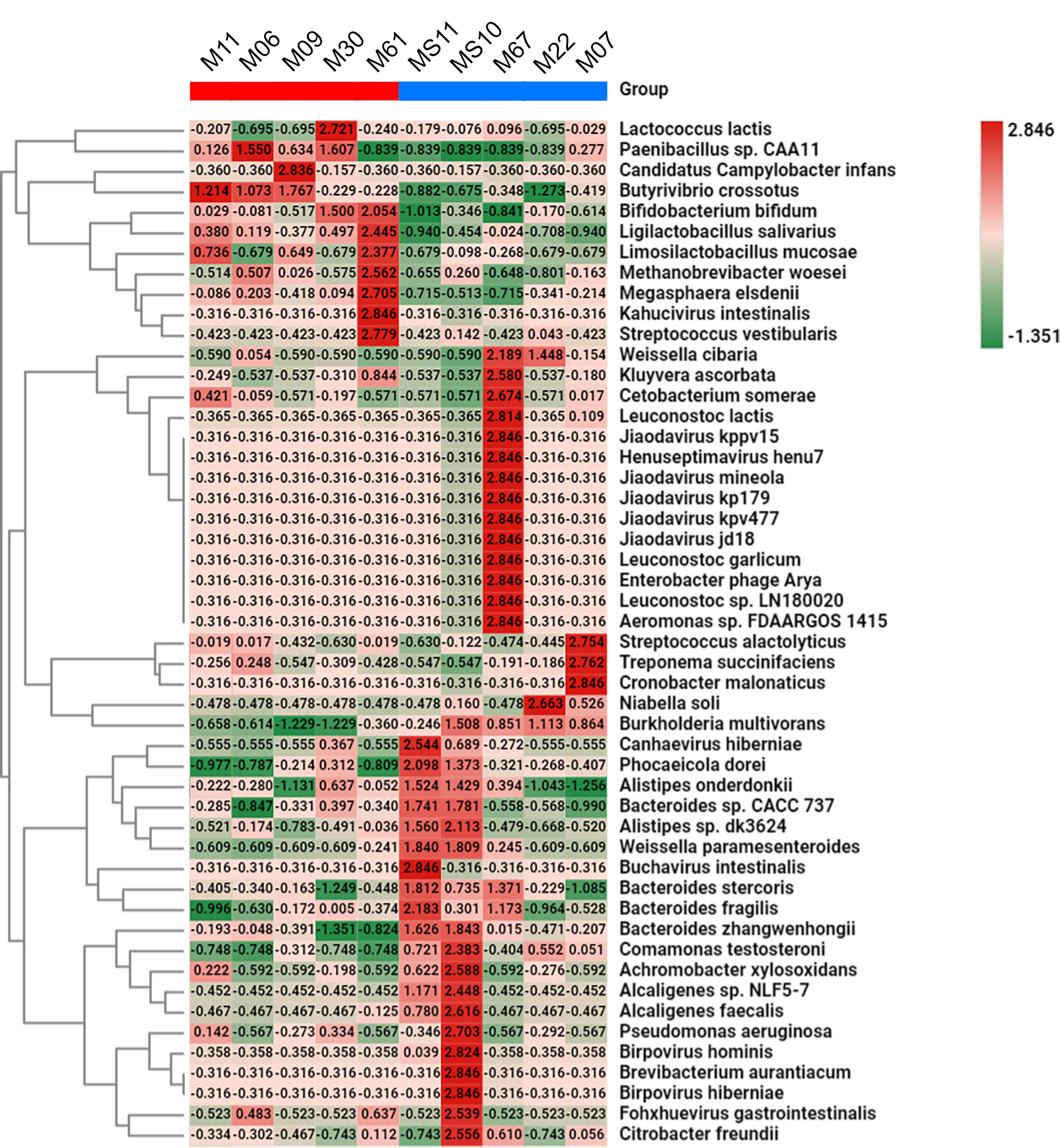
Figure 6 Heat map of relative abundances of species. The bar at the top indicates the infected (in red) and non-infected (in blue) samples. Numbers indicate the differential abundances between the two groups.
The differential abundance analysis of metabolic pathways (Table 2) using COG and Pfam databases showed significant over-representation and no under-represented pathways in Trichuris infected compared to non-infected women (P < 0.05). Additionally, eight COG metabolic pathways were significantly overrepresented. These pathways include Human T-cell leukaemia virus 1 infection, cholesterol metabolism, and pathogenic E. coli infection. This finding indicates that Trichuris infection not only influences the composition of the gut microbiome, as highlighted by structural analysis, but also has a discernible impact on its functional pathways. The effect of Trichuris infection on the function of the gut microbiome was further confirmed by differential analysis of protein families. This analysis revealed several protein families that were either over-represented or under-represented based on the Trichuris infection status of the participants (Table 3). Notably, most of these proteins remain functionally uncharacterized, except for the endothelial cell-specific chemotaxis regulator ATP synthase subunit C, Restriction endonuclease, and putative tight adherence pilin protein family, which are overrepresented in infected WRA. In contrast, certain protein families were less abundant in infected samples. They are nodavirus V-methyltransferase, four C-terminal TMM regions of protein-O-mannosyltransferase, and the GldH Lipoprotein families (as detailed in Table 3).
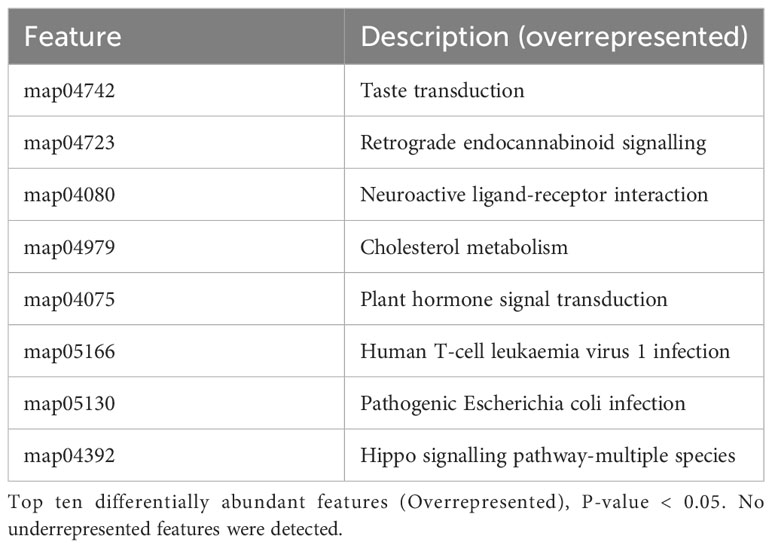
Table 2 Differential abundance analysis of metabolic pathways (COG).
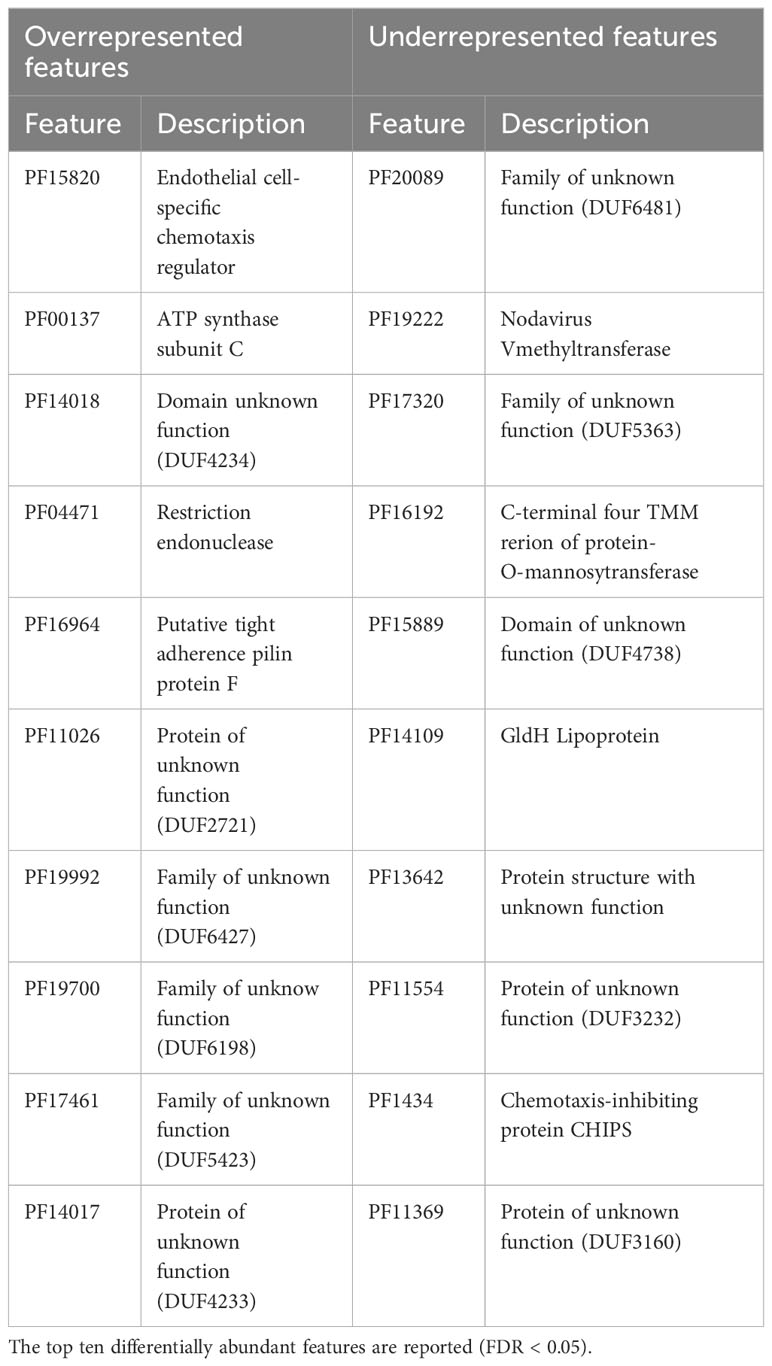
Table 3 Differential abundance analysis of pfam families.
To further investigate the microbiome composition and influence of Trichuris infection, we conducted an analysis of the mycobiome, with specific attention directed toward common gut fungi, including Candida spp, Aspergillus spp., Saccharomyces spp, and Malassezia (Figure 7B). Candida albicans was the dominant species (Figure 7B). The cumulative fungal content, irrespective of species, displayed a higher abundance in the mycobiome of Trichuris non-infected individuals (see Figure 7A). Although we did not observe beneficial Saccharomyces boulardii, known for its positive effects on human health, our findings showed distinct trends. Candida albicans, Candida dublinensis and Aspergillus fumigatus were more abundant in T. trichiura infected samples than in non-infected samples (Figure 7B). Conversely, Saccharomyces cerevisiae, Saccharomyces paradoxus were more abundant in healthy individuals (Figure 7B). No significant differences were observed between groups.
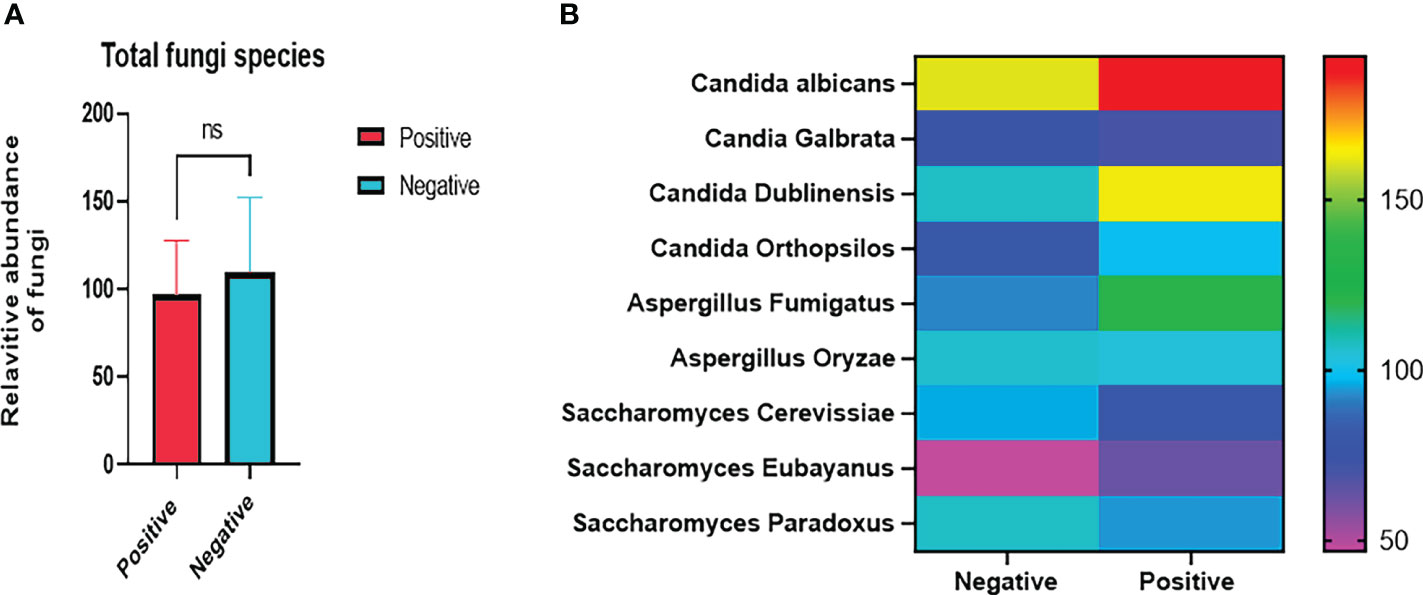
Figure 7 Selective mycobiome analyses. (A) Relative increase of the total fungal species in negative samples (unpaired t test P = 0.4875); (B) Heatmap showing variation of selected common gut fungal species in positive and negative samples; positive and negative refer to T. trichiura-infected and non-infected samples, respectively. ns stands for P value ≥ 0.05.
This study highlights the distinct nature of gut microbiota composition according to T. trichiura infection status. Principal coordinate analysis revealed the subdivision of the samples into two distinct clusters: infected and non-infected. In particular, samples within the latter cluster had a tighter grouping than the Trichuris infected samples, implying a higher degree of similarity in their gut microbiota composition. This is in agreement with a previous study, in which significant differences in both alpha and beta diversities were found between helminth-positive and-negative groups (8). Another study demonstrated microbial shifts associated with helminthiases in endemic regions of Thailand, manifested in both faecal and saliva microbiota (16). Another study also explored the interplay between Trichuris and gut microbiota, showing interconnections (17). Furthermore, the exploration of potential interactions between Trichuris and the gut microbiota revealed that embryonated eggs incubated with gut explants containing caecal bacteria or bacterial cultures of E. coli and/or Staphylococcus aureus, provided the microbial cues necessary for successful worm larval hatching at the onset of infection. Moreover, investigations on antibiotic treatment prior to T. muris infection showed a significant reduction in the gut microbiota, with a consequent decrease in hatching rates. This provides evidence of a symbiotic relationship between Trichuris and the gut microbiota, which is crucial for initiating infection (18).
The Firmicutes/Bacteroidetes ratio observed in this study is higher in the microbiota of infected participants in contrast to a lower ratio in individual not affected by the parasite. This ratio has attracted attention as a marker for assessing the health status of gut microbiota and has been extensively investigated in relation to obesity and gut inflammation. Alterations in the dominant phyla Firmicutes and Bacteroidetes depletion have been described in obese animals (19). A calorie-restricted diet for 1 year has shown an increase in Bacteroidetes abundance and the restoration of the Firmicutes/Bacteroidetes ratio (20). For patients with irritable bowel syndrome (IBS), an increased Firmicutes/Bacteroidetes ratio has been reported (21). The divergence in the Firmicutes/Bacteroidetes ratio between Trichuris-infected and non-infected individuals holds potential significance. This finding suggests that modulation of the gut microbiota of infected individuals through dietary interventions can potentially reduce this ratio, promoting the development of a resilient and healthy gut microbiota.
Comparative analysis at the genus level revealed that some SCFA-producing bacteria, Prevotella, Prevotella 9, and Ruminococcus, were relatively decreased in infected individuals and were overrepreseted in healthy subjects. Moreover, the Prevotella/Bacteroides ratio was higher in healthy samples and lower in Trichuris infected samples, which is in agreement with previous research on the same population (8) and another study on the Bantu population in Central Africa (22), where Prevotella was found to be the most representative genus in the gut microbiome. Prevotella and Bacteroides are members of the phylum Bacteroidetes and are known to be driven by different types of diets. A diet rich in carbohydrates is associated with an elevated representation of Prevotella, whereas a diet rich in protein and animal fat is associated with an elevated proportion of Bacteroides (23). Schneeberger et al. (2022) reported that knowing the pretreatment enterotype or gut microbiota composition can be predictive of anthelminthic treatment outcomes (11). In that study, a Prevotella rich enterotype was associated with a lower efficiency of the albendazole-ivermectin treatment. In a context where helminth infection is endemic and the effect of treatments is limited owing to drug efficacy issues (24), the Prevotella/Bacteroides ratio could be better investigated with the objective of modulating the gut microbiota to facilitate drug efficacy. Thus, it is possible to investigate the effects on drug treatment by different Prevotella species. In this study, Prevotella copri appeared to be the dominant species in both infected and non-infected samples.
Differential abundance analysis also revealed that some species known as emergent probiotics, such as Weissella cibaria, Leuconostoc citreum and Leuconostoc lactis, were significantly underrepresented in infected samples and overrepresented in healthy samples. Bacteria belonging to the genus Weissella may be important in controlling foodborne diseases through the production of bacteriocins and hydrogen peroxide. This genus has a great potential for use in the food industry (25).
Functional analysis of the metagenomic data showed significant differences in metabolic pathways (P-value < 0.05), such as cholesterol metabolism, pathogenic E. coli infection, and human T-cell leukaemia virus 1 infection, which were overrepresented in the infected samples. An association between gut microbiota and cholesterol metabolism has been reported in the literature. However, there is little evidence on how and which bacteria are involved in this metabolism. Some researchers have reported coprostanol-forming bacteria such as Eubacterium coprostanoligenes, Bacteroides dorei, Lactobacillus sp., and Bifidobacterium spp (26–29). According to Lawson et al. (17), helminths require bacteria to hatch eggs. The absence of microbiota (germ-free mice) prevents Trichuris hatching and infection, whereas the presence of highly diverse microbiota enhances host susceptibility to infection. Further investigation is required to understand how bacterial metabolism affects Trichuris infections. Differential analysis of the protein families revealed significant differences. This confirms that Trichuris infection not only modulates the composition, but also the function of the gut microbiome.
Mycobiome analysis revealed that Candida albicans was the dominant species in the population. This agrees with a previous study in which Candida spp were the dominant fungal species in the human gut (30). However, Hoffman et al. (31) found Saccharomyces to be the most prevalent genus, followed by Candida and Cladosporium. On the other hand, the discrepancy regarding the abundances of Candida and Saccharomyces might be simply explained by the fact that fungi are poor gut colonizers, but transient in the gut that rapidly changes according to the diet (31). In a non-human primate study, Barelli et al. (32) found that in red colobus monkeys and yellow baboons, Trichuris infection was associated to different gut fungal compositions. These findings suggest that greater attention should be given to gut fungi.
To the best of our knowledge, this is the first human gut microbiome analysis, including functional investigations, in the Zanzibar population, an area with a high prevalence of STH infection. The differences in the Firmicutes/Bacteroidetes and the Prevotella/Bacteroides ratios suggest that the modulation of the gut microbiota with diet and/or probiotics could be used as complementary approach to fight against helminths infections in Pemba. This approach may also produce a higher efficacy for deworming drug administration.
Although shotgun metagenomic sequencing generates a large amount of data, we must acknowledge that this approach has limitations, such as the missed characterisation of approximately 50% of genomic sequences that remain unannotated, and that they may include viruses and protozoan parasites, for which the contribution to the gut microbiome is less studied and less present in databases.
The purpose of this study was to assess the influence of Trichuris infection on the composition and function of the microbiome in women of reproductive age using a shotgun metagenomic sequencing approach. Our results confirmed that T. trichiura infection significantly shapes the gut microbiome structure and function in WRA. Notably, we identified markers of the gut microbiome health that were changed in infected participants. These indicators can be used as targets for the modulation of gut microbiota in this population. Some taxa known for beneficial characteristics and less abundant in infected participants require further attention in order to see if they can be used as probiotics in prevention or therapeutic support against helminth infection. Therefore, a well-elaborated and sustainable diet with the aim of modulating the gut microbiome and favouring the development of beneficial species can be used as a preventive and complementary approach against T. trichiura infection in Pemba and potentially in other areas where this infection is endemic. Since this was a pilot cross-sectional study with a relatively small sample size, a larger cohort investigation with the same population is needed to better understand the results found so far.
The data presented in the study are deposited in the ENA METAGENOME repository, accession number: PRJEB71403.
The studies involving humans were approved by Zanzibar Health Research Institute (ZAHRI with protocol number: ZAMREC/001/SEPT/018). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
AN: Conceptualization, Investigation, Methodology, Writing – original draft. MM: Data curation, Formal Analysis, Methodology, Writing – original draft. HC: Investigation, Resources, Writing – review & editing. AP: Formal Analysis, Investigation, Methodology, Writing – review & editing. SS: Resources, Supervision, Writing – review & editing. SA: Project administration, Resources, Supervision, Writing – review & editing. JZ: Investigation, Methodology, Writing – review & editing. CM: Conceptualization, Funding acquisition, Methodology, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding sources were provided by the University of Camerino, Grant BVI000082-FAR, to CM, by the School of Advanced Studies, University of Camerino PhD Grant to ATN and HC, and by the Erasmus plus Project IT02-KA107-077890 for covering the mobility of researchers.
We thank the technical staff of Public Health Laboratory Ivo de Carneri and of the School of Health and Medical Sciences (State University of Zanzibar) for their great help in identifying and interviewing participants and in helminth detection, and the Ivo de Carneri Foundation for their support. We would like to thank Editage (www.editage.com) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2024.1276210/full#supplementary-material
1. Pasaribu AP, Alam A, Sembiring K, Pasaribu S, Setiabud D. Prevalence and risk factors of soil transmitted helminthiasis among school children living in an agricultural area of North Sumatera, Indonesia. BMC Public Health (2019) 19:1066. doi: 10.1186/s12889-019-7397-6
2. Gordon CA, Krause L, McManus DP, Morrison M, Weerakoon KG, Connor MC, et al. Helminths, polyparasitism, and the gut microbiome in the Philippines. Int J Parasitol (2020) 50(3):217–25. doi: 10.1016/j.ijpara.2019.12.008
3. Kupritz J, Angelova A, Thomas B, Nutman TB, Gazzinelli-Guimaraes PH. Helminth-induced human gastrointestinal dysbiosis: a systematic review and meta-analysis reveals insights into altered taxon diversity and microbial gradient collapse. mBio (2021) 12(6):e02890–21. doi: 10.1128/mBio.02890-21
4. Stracke K, Adisakwattana P, Phuanukoonnon S, Yoonuan T, Poodeepiyasawat A, Dekumyoy P, et al. Field evaluation of the gut microbiome composition of pre-school and school-aged children in Tha Song Yang, Thailand, following oral MDA for STH infections. PloS Negl Trop Dis (2021) 15(7):e0009597. doi: 10.1371/journal.pntd.0009597
5. Szajewska H. Gut microbiota: no longer the forgotten organ. Ann Nutr Metab (2021) 77(suppl 3):1–2. doi: 10.1159/000519223
6. Malard F, Dore J, Gaugler B, Mohty M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol (2021) 14:547–54. doi: 10.1038/s41385-020-00365-4
7. Jaswal K, Todd OA, Behnsen J. Neglected gut microbiome: interactions of the non-bacterial gut microbiota with enteric pathogens. Gut Microbes (2023) 15(1):2226916. doi: 10.1080/19490976.2023.2226916
8. Chen H, Mozzicafreddo M, Pierella E, Carletti V, Piersanti A, Ali SM, et al. Dissection of the gut microbiota in mothers and children with chronic Trichuris trichiura infection in Pemba Island. Tanzania Parasites Vectors (2021) 14(62). doi: 10.1186/s13071-021-04580-1
9. Vejzagić N, Adelfio R, Keiser J, Kringel H, Thamsborg SM, Kapel CMO. Bacteria-induced egg hatching differs for Trichuris muris and Trichuris suis. Parasites Vectors (2015) 8(371). doi: 10.1186/s13071-015-0986-z
10. Rosa BA, Snowden C, Martin J, Fischer K, Kupritz J, Beshah E, et al. Whipworm-associated intestinal microbiome members consistent across both human and mouse hosts. Front Cell Infect Microbiol (2021) 11:637570. doi: 10.3389/fcimb.2021.637570
11. Schneeberger PHH, Gueuning M, Welsche S, Hürlimann E, Dommann J, Häberli C, et al. Different gut microbial communities correlate with efficacy of albendazole-ivermectin against soil-transmitted helminthiases. Nat Commun (2022) 13(1):1063. doi: 10.1038/s41467-022-28658-1
12. Geerts S, Gryseels B. Anthelmintic resistance in human helminths: a review. Trop Med Int Health (2001) 6(11):915–21. doi: 10.1046/j.1365-3156.2001.00774.x
13. Ame S, Kabole F, Nanai AM, Mwinzi P, Mupfasoni D, Ali SM, et al. Impact of preventive chemotherapy on transmission of soiltransmitted helminth infections in Pemba Island, United Republic of Tanzania, 1994–2021. PloS Negl Trop Dis (2022) 16(6):e0010477. doi: 10.1371/journal.pntd.0010477
14. Knopp S, Mohammed KA, Rollinson D, Stothard JR, Khamis I S, Utzinger J, et al. Changing patterns of soil-transmitted helminthiases in zanzibar in the context of national helminth control programs. Am J Trop Med Hyg (2009) 81(6):1071–8. doi: 10.4269/ajtmh.2009.09-0377
15. Pasquereau-Kotula E, Martins M, Aymeric L, Dramsi S. Significance of Streptococcus gallolyticus subsp. Gallolyticus Association with Colorectal Cancer. Front Microbiol (2018) 9:614. doi: 10.3389/fmicb.2018.00614
16. Gobert GN, Atkinson LE, Lokko A, Yoonuan T, Phuphisut O, Poodeepiyasawat A, et al. Clinical helminth infections alter host gut and saliva microbiota. PloS Negl Trop Dis (2022) 16(6):e0010491. doi: 10.1371/journal.pntd.0010491
17. Lawson MAE, Roberts IS, Grencis RK. The interplay between Trichuris and the microbiota. Parasitology (2021) 148:1–8. doi: 10.1017/S0031182021000834
18. Hayes KS, Bancroft AJ, Goldrick M, Portsmouth C, Roberts IS, Grencis RK. Exploitation of the intestinal microflora by the parasitic nematode trichuris muris. Science (2010) 328(5984):1391–4. doi: 10.1126/science.1187703
19. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology. Human gut microbes associated with obesity. Nature (2006) 444(7122):1022–3. doi: 10.1038/nature4441022a
20. Nicole de Wit N, Derrien M, Bosch-Vermeulen H, Oosterink E, Keshtkar S, Duval C, et al. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am J Physiol Liver Physiol (2012) 303:G589–99. doi: 10.1152/ajpgi.00488.2011
21. Duan R, Zhu S, Wang B, Duan L. Alterations of gut microbiota in patients with irritable bowel syndrome based on 16S rRNA-targeted sequencing: A systematic review. Clin Trans Gastroenterol (2019) 10. doi: 10.14309/ctg.0000000000000012
22. Gomez A, Petrzelkova KJ, Burns MB, White BA, Leigh SR, Blekhman R, et al. Gut microbiome of coexisting baAka pygmies and bantu reflects gradients of traditional subsistence patterns. Cell Rep (2016) 14:2142–53. doi: 10.1016/j.celrep.2016.02.013
23. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (2011) 334(6052):105–8. doi: 10.1126/science.1208344
24. Truscott JE, Turner HC, Anderson RM. What impact will the achievement of the current World Health Organization targets for anthelmintic treatment coverage in children have on the intensity of soil transmitted helminth infections? Parasites Vectors (2015) 8:551. doi: 10.1186/s13071-015-1135-4
25. Teixeira CG, da Silva RR, Fusieger A, Martins E, de Freitas R, de Carvalho AF. The Weissella genus in the food industry: A review. Res Soc Dev (2021) 10(5):e8310514557. doi: 10.33448/rsd-v10i5.14557
26. Freier TA, Beitz DC, Li L, Hartman PA. Characterization of Eubacterium coprostanoligenes sp. nov., a Cholesterol-Reducing Anaerobe. Int J Systematic Bacteriol (1994) 44:137–42. doi: 10.1099/00207713-44-1-137
27. Gerard P, Lepercq P, Leclerc M, Gavini F, Raibaud P, Juste C. Bactéroïdes sp. Strain D8, the first cholesterol-reducing bacterium isolated from human feces. Appl Environ Microbiol (2007) 73(18):5742–9. doi: 10.1128/AEM.02806-06
28. Lye HS, Rusul G, Liong MT. Removal of cholesterol by lactobacilli via incorporation and conversion to coprostanol. J Dairy Sci (2010) 93:1383–92. doi: 10.3168/jds.2009-2574
29. Kenny DJ, Plichta DR, Shungin D, Koppel N, Hall AB, Fu B, et al. Cholesterol metabolism by uncultured human gut bacteria influences host cholesterol level. Cell Host Microbe (2020) 28:245–57. doi: 10.1016/j.chom.2020.05.013
30. Pauline D. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of feces. ISME J (2008) 2:1183–93. doi: 10.1038/ismej.2008.76
31. Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PloS One (2013) 8(6):e66019. doi: 10.1371/journal.pone.0066019
Keywords: helminths, women of reproductive age, Trichuris trichiura, gut microbiome, mycobiome, fungi
Citation: Nguélé AT, Mozzicafreddo M, Chen H, Piersanti A, Salum SS, Ali SM, Zhang J and Miceli C (2024) Trichuris trichiura infection is associated with changes in gut microbiome composition and function among women of reproductive age from Pemba, Tanzania. Front. Trop. Dis 5:1276210. doi: 10.3389/fitd.2024.1276210
Received: 30 August 2023; Accepted: 05 January 2024;
Published: 05 February 2024.
Edited by:
Manuela Berto Pucca, Sao Paulo State Universty, BrazilReviewed by:
Collins Okoyo, Kenya Medical Research Institute (KEMRI), KenyaCopyright © 2024 Nguélé, Mozzicafreddo, Chen, Piersanti, Salum, Ali, Zhang and Miceli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Miceli, Y3Jpc3RpbmEubWljZWxpQHVuaWNhbS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.