 Amit Sinha1
Amit Sinha1 Zhiru Li1
Zhiru Li1 Catherine B. Poole1
Catherine B. Poole1 Richard D. Morgan1
Richard D. Morgan1 Laurence Ettwiller1Nathália F. Lima2
Laurence Ettwiller1Nathália F. Lima2 Marcelo U. Ferreira2,3Fanny F. Fombad4
Marcelo U. Ferreira2,3Fanny F. Fombad4 Samuel Wanji4
Samuel Wanji4 Clotilde K. S. Carlow1*
Clotilde K. S. Carlow1*- 1Genome Biology Division, New England Biolabs, Ipswich, MA, United States
- 2Department of Parasitology, Institute of Biomedical Sciences, University of Saão Paulo, Saão Paulo, Brazil
- 3Global Health and Tropical Medicine, Institute of Hygiene and Tropical Medicine, NOVA University of Lisbon, Lisbon, Portugal
- 4Department of Microbiology and Parasitology, University of Buea, Buea, Cameroon
The filarial parasites Mansonella ozzardi and Mansonella perstans, causative agents of mansonellosis, infect hundreds of millions of people worldwide, yet remain among the most understudied of the human filarial pathogens. M. ozzardi is highly prevalent in Latin American countries and Caribbean Islands, while M. perstans is predominantly found in sub-Saharan Africa as well as in a few areas in South America. In addition to the differences in their geographical distribution, the two parasites are transmitted by different insect vectors, as well as exhibit differences in their responses to commonly used anthelminthic drugs. The lack of genome information has hindered investigations into the biology and evolution of Mansonella parasites and understanding the molecular basis of the clinical differences between species. In the current study, high quality genomes of two independent clinical isolates of M. perstans from Cameroon and two M. ozzardi isolates one from Brazil and one from Venezuela are reported. The genomes are approximately 76 Mb in size, encode about 10,000 genes each, and are largely complete based on BUSCO scores of about 90%, similar to other completed filarial genomes. These sequences represent the first genomes from Mansonella parasites and enabled a comparative genomic analysis of the similarities and differences between Mansonella and other filarial parasites. Horizontal DNA transfers (HDT) from mitochondria (nuMTs) as well as transfers from genomes of endosymbiotic Wolbachia bacteria (nuWTs) to the host nuclear genome were identified and analyzed. Sequence comparisons and phylogenetic analysis of known targets of anti-filarial drugs diethylcarbamazine (DEC), ivermectin and mebendazole revealed that all known target genes were present in both species, except for the DEC target encoded by gon-2 gene, which is fragmented in genome assemblies from both M. ozzardi isolates. These new reference genome sequences will provide a valuable resource for further studies on biology, symbiosis, evolution and drug discovery.
1 Introduction
A large part of the world’s population is infected with one or more filarial nematode parasites resulting in several distinct diseases often referred to as a group under the general term filariasis. The filarial parasites Mansonella ozzardi and Mansonella perstans are the two main causes of mansonellosis in humans. Infections are transmitted by infected biting midges (mostly of the genus Culicoides) or blackflies (genus Simulium) that release infective stage larvae during a blood meal. Adult male and female worms have been observed rarely in humans since they are presumed to live in the serous body cavities and/or subcutaneous tissues. Viviparous females release unsheathed microfilaria which circulate in the blood, available for ingestion by an appropriate vector during feeding. Unlike some of the other human filarial diseases, Mansonella infections do not present with a distinct pathology or specific clinical picture. Infected individuals are often asymptomatic or may have a variety of clinical features including subcutaneous swellings, aches, pain, fever, headache, pruritis, corneal lesions and eosinophilia (1–5). Other manifestations include immunosuppression (6) with important ramifications such as increased susceptibility to other infections and reduced efficacy of vaccine programs (7).
Even though mansonellosis ranks first in prevalence among the human filariases, affecting hundreds of millions of people in Africa and Central and South America, it remains one of the least studied and most neglected of all filarial diseases (1, 3, 4, 7–11). Indeed, much of the somewhat limited information available has been obtained serendipitously during studies on other filarial parasites or malaria, in which individuals were co-infected with Mansonella parasites. M. perstans is considered the most common filariasis in Central Africa with infection prevalence often very high (up to 96%) in endemic areas, even among children (1, 7, 8). It is also found in northern regions of the Amazon rainforest and the Caribbean coast in South America (7, 9), with molecular evidence supporting introduction of M. perstans to the American continent from Africa because of the slave trade (12). M. ozzardi infections have been reported from many countries in South America, Central America, and some Caribbean Islands (5, 7, 12–14). Co-infections of M. perstans and M. ozzardi, together with other filarial parasites, have also been reported in South America (15, 16), making their treatment challenging as the two species respond differently to the commonly used anti-filarial drugs such as ivermectin, diethylcarbamazine (DEC) or mebendazole (7, 17). The recent development of highly specific, simple molecular tests which can distinguish M. perstans and M. ozzardi, and do not cross-react with other filarial parasites (18), will facilitate an effective treatment regimen. The antibiotic doxycycline, targeting the endosymbiotic Wolbachia bacteria of M. perstans and M. ozzardi, has been shown to clear the microfilarial stage from the blood (19–21) highlighting the importance of Wolbachia in these species. The Wolbachia genomes, wMpe from M. perstans, and wMoz from M. ozzardi have recently been published (22), providing a valuable resource for the discovery of new antibiotics and studies on the symbiotic relationship and evolution.
Here we provide the first de novo assemblies of the nuclear genomes from M. perstans and M. ozzardi using combined PacBio long-read and Illumina paired-end sequencing data. We used customized metagenomic assembly and binning pipelines, and extensive quality checks to ensure accurate assemblies from a mixture of DNA from the Mansonella parasites, their human host, and the associated microbiome. These methods were applied to generate genomes from two clinical isolates of each species, providing a unique opportunity for intra-species and inter-species comparisons and exploring features of sequence evolution of these parasites. We analyzed the genomic regions containing horizontally transferred DNA from mitochondria (nuMTs), as well horizontally transferred DNA from their endosymbiont Wolbachia (nuWTs). Phylogenetic analysis of orthologs of genes that encode drug targets of common anti-filarial drugs such as DEC, ivermectin and mebendazole revealed intriguing differences in the presence and absence of these targets between M. perstans and M. ozzardi that might have clinical implications for the treatment of mansonellosis.
2 Methods
2.1 Ethics statement
All research involving human subjects was approved by the appropriate committee and performed in accordance with all relevant guidelines and regulations. Informed consent was obtained from all participants or their legal guardians. These samples have also been described and utilized in previous studies (18, 22).
Ethical clearance for M. perstans samples was obtained from the National Institutional Review board, Yaoundé (REF: N° 2015/09/639/CE/CNERSH/SP) and administrative clearance from the Delegation of Public Health, South-West region of Cameroon (Re: R11/MINSANTE/SWR/RDPH/PS/259/382). Approval for the study was granted by the “National Ethics Committee of Research for Human Health” in Cameroon. Special consideration was taken to minimize the health risks to which any participant in this study was exposed. The objectives of the study were explained to the consenting donor after which they signed an informed consent form. The participant’s documents were given a code to protect the privacy of the study subject. At the end of the study, the donor received a cure of mebendazole (100 mg twice a day for 30 days).
The study protocols for M. ozzardi were approved by the Institutional Review Board of the Institute of Biomedical Sciences, University of São Paulo, Brazil (1133/CEP, 2013) as part of a previous study (13). Written informed consent was obtained from all patients, or their parents or guardians if participants were minors younger than 18 years. Diagnosed infections were treated with a single dose of 0.2 mg/kg of ivermectin after blood sampling (13).
2.2 Illumina library construction and sequencing
The parasite materials, DNA extractions, Illumina library preparation and sequencing have been described in detail previously (18, 22). Briefly, two clinical isolates of M. perstans, named Mpe-Cam-1 and Mpe-Cam-2, were obtained as microfilariae in a blood draw from consenting individuals in Cameroon. One clinical isolate of M. ozzardi, named Moz-Brazil-1, was obtained from a consenting individual as part of a previous study in Brazil, while another isolate from Venezuela, named Moz-Venz-1, was generously donated by Izaskun Petralanda in 1989. The DNA samples from Mpe-Cam-1, Mpe-Cam-2, and Moz-Brazil-1 were treated with the NEBNext Microbiome DNA enrichment to reduce human DNA contamination before being used for library preparation. Illumina libraries for all 4 isolates were constructed using the NEBNext Ultra II FS DNA Library Prep Kit (New England Biolabs Inc., USA) as per standard protocol and sequenced using the Illumina MiSeq and NextSeq500 platforms (paired end, 150 bps). Bioinformatic analysis programs were run with their default parameters unless otherwise stated. The bioinformatic analysis pipeline and scripts are available from https://github.com/aWormGuy/Mansonella-Genomes-Sinha-et-al.-2023.
2.3 PacBio library sequencing and de novo assembly
A total of 250 ng of genomic DNA from M. perstans isolate Mpe-Cam-1 was used for sequencing on PacBio RSII platform as per manufacturer’s instructions. Consensus reads from raw PacBio reads were obtained using the protocol RS_PreAssembler.1 (parameters minLen=1000, minQual=0.80, genomeSize=100000000) on PacBio SMRT Portal (smrtanalysis_2.3.0.140936.p5.167094). Reads originating from human genomic DNA were removed by first by mapping the consensus reads to the human genome (grch38) using minimap2 v2.17-r941 (23) followed by discarding all mapped reads (MAPQ score > 20). The remaining reads were assembled using canu v2.2 (24) (parameters genomeSize=90m correctedErrorRate=0.045). The assembly was iteratively polished over 12 rounds using “Resequencing” protocol on PacBio SMRT Portal (v 6.0.0.47841) (parameters –minMatch 12 –bestn 10 –minPctSimilarity 70.0 –refineConcordantAlignments) till no new variants could be detected by the Resequencing pipeline. The assembly contiguity was further improved by using finisherSC software v2.1 (25), a scaffolder that uses raw PacBio reads while accounting for any genomic repeats in the assembly. The assembly was polished using the Illumina data for the same isolate via the polca software (26). Finally, gap-filling and heterozygosity removal was performed on this assembly using Illumina reads as an input to the Redundans pipeline v0.14a (27). The assembly quality was evaluated using quast (28) at each step.
2.4 Metagenomic assembly using Illumina reads
Illumina raw reads were processed using the BBTools package (https://jgi.doe.gov/data-and-tools/bbtools/). Duplicate raw reads and bad tiles were removed using the clumpify.sh and filterbytile.sh utilities. Adapter trimming, removal of phiX reads, and reads with 13-nt or longer stretches of the same nucleotide (poly-G, poly-C, poly-A, poly-T) was performed using bbduk.sh. Human host reads were removed by mapping against the human genome (grch38) using bbmap.sh utility. The quality metrics of the processed reads at each step were assessed using FastqC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads from different runs of the libraries prepared from the same genomic DNA sample were combined and used as an input for the assembly of the metagenome using metaSpades (29).
2.5 Metagenomic binning to identify Mansonella contigs
Binning of metagenomic contigs to identify sequences originating from Mansonella genomes was carried out based on the BlobTools software (30) and additional curation, as described previously (22). Briefly, for each of the isolates Mpe-Cam-2, Moz-Brazil-1 and Moz-Venz-1, the Illumina reads were mapped back to the metagenomic contigs produced by metaSpades using bowtie2 (31). For the PacBio assembly obtained from the Mpe-Cam-1 sample, the corresponding Illumina reads were mapped to this assembly using bowtie2. Sequence similarities of assembled contigs to the NCBI nt database and Uniport database was carried out using blastn and diamond in BlobTools package. The bins annotated as “Nematoda” and “Proteobacteria” by BlobTools were analyzed further to retrieve sequences originating from Mansonella. Contigs that were marked as Proteobacteria but co-localized with the Nematoda blobs in the blobplots, were included in the Mansonella bins as they represented potential examples of horizontal gene transfer from Wolbachia to Mansonella host. These HGT candidates were validated by inspection of blastn and blastx hits of these contigs to the NCBI nt and nr databases respectively.
Contigs originating from mitochondrial DNA were identified by analysis of nucmer alignments between assembled sequences and the previously reported mitochondrial genomes, GenBank accessions MT361687.1 and KX822021.1 for M. perstans (32) and M. ozzardi (33) respectively. The contigs with more than 98% sequence identity to published mitochondrial genomes were marked as mtDNA and removed from the nuclear assembly.
2.6 Assembly refinements and heterozygosity removal for Illumina assemblies
For each Illumina-only assembly, scaffolding, gap-filling and heterozygosity removal was performed using the corresponding Illumina reads in the Redundans pipeline v0.14a (27).
2.7 Genome annotations and comparative analysis
Identification of repetitive elements in the assembled genomes were performed using RepeatModeler version 2.0.3 (34) and classified against the Dfam database (35). First round of repeat annotations was performed on a fasta file combining all assembled genomes. The repeat sequence families identified from this run were used as a reference library in RepeatMasker 4.1.2-p1 (http://repeatmasker.org/) to mask repeats in each of the 4 genome assemblies independently. The outputs from RepeatMasker were processed with parseRM.pl utility (36) to generate summary statistics on repeats classified as DNA transposons and retroelements.
Gene predictions were carried out using the braker2 pipeline v2.2.6 (37), with a reference proteome comprised of all metazoan proteins in OrthoDB version 10.1 (38) and all filarial proteomes downloaded from WormBase ParaSite (39). To ensure that the same parameters and hmm models are applied to all assemblies, the 4 assemblies were combined into a single file, and braker2 pipeline was run on this composite dataset. The genes predicted from this run were then assigned to respective isolates by parsing the gff3 output from braker2. The completeness of the protein-coding content of genomes was assessed using the BUSCO pipeline v5.0 beta using the “nematoda_odb10” reference dataset (40). OrthoFinder v2.4 (41) was used to infer orthologs of protein coding genes across multiple nematode genomes. The set of proteomes included all 17 filarial species with publicly available genomes. The species Setaria digitata and Thelazia callipaeda from the same infraorder Spiruromorpha were included as the immediate outgroups, while Caenorhabditis elegans and Pristionchus pacificus were included as relatively more distant outgroups. The protein and transcript sequences were downloaded from WormBase parasite where available. Although the genome sequences for filarial nematodes Cruorifilaria tuberocauda, Dipetalonema caudispina, Dirofilaria immitis, Litomosoides brasiliensis and Madathamugadia hiepei (42) were available from NCBI (GenBank accessions GCA_013365365.1, GCA_013365325.1, GCA_013365355.1, GCA_013365375.1 and GCA_013365335.1 respectively), the corresponding gene annotations for these assemblies were not available in NCBI. Hence, repeat-modeling, repeat-masking and gene annotation using braker2 pipeline was performed on each of these species as described for M. perstans and M. ozzardi above, and have been made available at https://github.com/aWormGuy/Mansonella-Genomes-Sinha-et-al.-2023/tree/main/gene_predictions_on_Lefoulon2020_genomes.
The source databases, the corresponding accession numbers, filenames, citations and BUSCO scores for all the genomes used in comparative analysis are listed in Supplementary Material Table 1.
Whole-genome alignments for global sequence similarity and synteny between various repeat-masked genomes were performed using minimap2 v2.17-r941 (23) and visualized using the JupiterPlot tool (https://github.com/JustinChu/JupiterPlot). For global sequence comparisons across multiple filarial genomes, the average nucleotide identity (ANI) scores were calculated using the OrthoANIu tool (43).
Comparison and analysis of sequence differences, including structural difference, between different pairs of genomes, namely (A) Mpe-Cam-1 versus Mpe-Cam-2 (B) Moz-Brazil-1 versus Moz-Venz-1 (C) Mpe-Cam-1 versus Moz-Brazil-1 were performed using nucdiff version 2.0.3 (44). The localization of the sequence variants within gene bodies and exons was calculated through the intersectBed tool in the bedtools package v2.29.2 (45). The effect of sequence variants on the coding potential of affected genes was determined using snpEff version 5.1d (46).
2.8 Phylogenetic analysis
For genome-wide phylogenetic analysis, single copy orthologs across various nematodes were identified from the OrthoFinder analysis described above. The corresponding protein sequences were aligned using mafft v7.149b (47) and trimmed using Gblocks v0.91b (48). Phylogenetic analysis of the resulting supermatrix of 823,832 amino acids was carried out using iqTree v2.1.2 (49). The best fit substitution model was chosen automatically using ModelFinder implementation within iqTree (50). Bootstrap support values were calculated based on (i) the SH-like approximate likelihood ratio test (51), and (ii) ultrafast bootstrap (52) with 1000 replicates each (iqTree command-line options ‘-B 1000 -alrt 1000 -abayes -lbp 1000’).
For phylogenetic analysis of specific genes and gene families, their homologs and the corresponding protein sequences were obtained from the OrthoFinder output. The protein domains for all the sequences were annotated using NCBI CD search and the NCBI CDD database (53). The protein sequences which lacked the domains present in the C. elegans homologs, or were too short in length (less than 60% of the length of the C. elegans homolog) were inspected and excluded from the final analysis. The multiple sequence alignments and tree inference was carried out using mafft and iqtree as described above. The consensus trees were edited and annotated using the iTOL webserver (54) and Adobe Illustrator.
2.9 Identifying nuWTs and nuMTs
The assembled nuclear genomes were aligned to the respective Wolbachia wMpe or wMoz (22) genomes using nucmer v4.0.0beta2 (55). Aligned fragments (“MuM”s) that were separated by less than 50 bp, were combined into a single nuWT locus. For manual validation of HDT of Wolbachia sequences into filarial contigs, the Wolbachia regions were verified to be flanked by nematode-like sequences and protein coding genes from nematodes.
A similar strategy was employed for nuMT identification, with nucmer alignments against respective mitochondrial sequences, (32, 33), and blastx against nematode nuclear proteomes. The nuMT loci with more than 85% AT-content were removed to avoid spurious matches to low-complexity regions.
Comparison of nuMT or nuWT loci for conservation of their sequence and position across different genomes was based on their sequence comparisons using megablast coupled with the synteny analysis described above. The sequences for nuMT or nuWT loci were extracted for each genome and compared using megablast, and only hits with more than 90% identity over 90% of the sequences were analyzed further as candidates for conserved loci. In the next step, if these candidate loci were present on syntenic contigs, they were marked as conserved loci across strains or species. A blastx analysis of the nuWT or nuMT sequences against the respective mitochondrial or Wolbachia proteomes was performed to identify any intact protein coding genes.
3 Results
3.1 PacBio sequencing and de novo assembly generates a highly contiguous M. perstans genome
PacBio sequencing and de novo genome assembly of genomic DNA isolated from M. perstans isolate “Mpe-Cam-1”, together with BlobTools analysis for metagenomic binning resulted in a high-quality M. perstans genome. The genome assembly is comprised of 581 scaffolds with a total size of 76 Mb, with the largest scaffold greater than 1.2 Mb and a N50 size greater than 285 kb (Table 1). Based on these quality metrics, the Mpe-Cam-1 assembly ranks highly in comparison to all filarial genomes available so far (Supplementary Material Table 1). Repetitive elements account for 9.7% of the genome, of which about 27% of the repeat sequence length was comprised of low complexity and simple repeats. Long terminal repeat (LTR) retrotransposons comprised 17% of all the repeats, while 50% of the repeat content is comprised of unknown, novel repeat families (Supplementary Material Table 2). The genome has 9,477 protein coding genes, with a BUSCO score of 93.2% (Supplementary Figure 1) when evaluated against a set of 3,131 core proteins conserved across the nematode phylum, further indicating the high quality of this genome assembly.
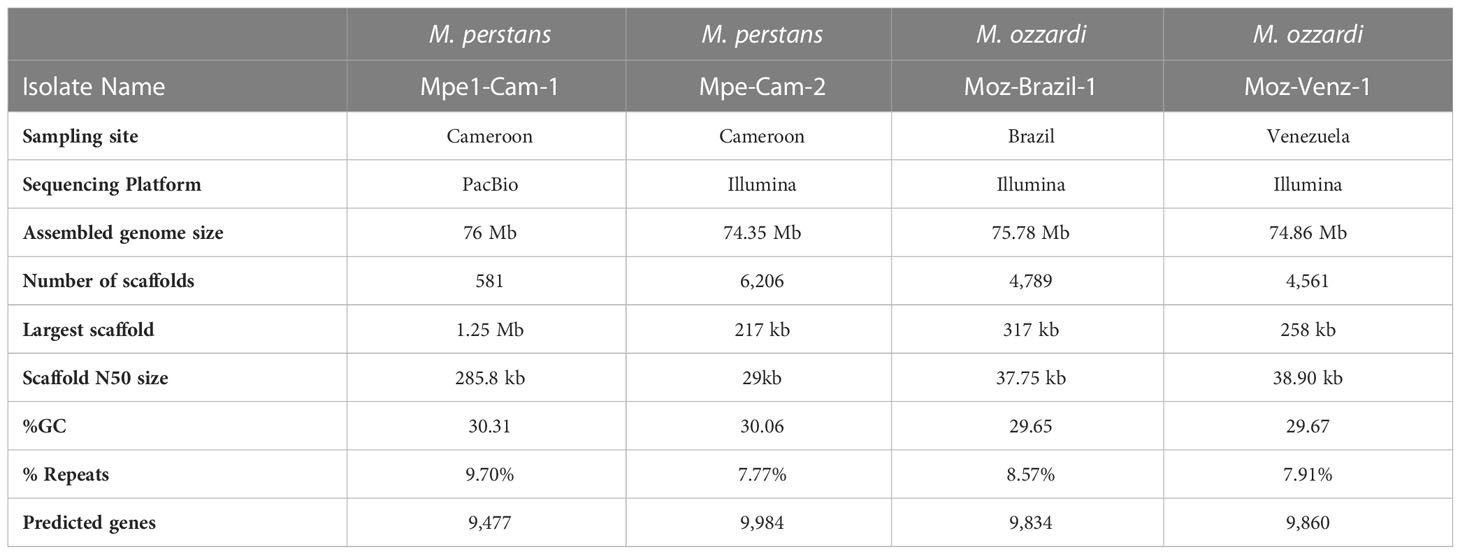
Table 1 Summary statistics of Mansonella genome assemblies.
Another clinical isolate from Cameroon, “Mpe-Cam-2” was sequenced and assembled de novo based on Illumina sequencing. Although the quality metrics of this Illumina short-reads assembly are lower than the PacBio long-read Mpe-Cam-1 assembly (Table 1), the repeat content, (Supplementary Material Table 2), the number of predicted genes and their BUSCO scores are similar to Mpe-Cam-1 (Supplementary Figure 1).
3.2 Genome sequencing and assembly of M. ozzardi from South America
For M. ozzardi, two genome assemblies from Latin American isolates, “Moz-Brazil-1” from Brazil and “Moz-Venz-1” from Venezuela, were generated based on Illumina sequencing reads. These assembled genomes are about 76 Mb (Table 1), similar in size to the M. perstans genomes described above. The number of predicted genes, their BUSCO scores (Supplementary Figure 1) and repeat content (Supplementary Material Table 2) are also similar to those present in M. perstans genomes.
3.3 Analysis of genome variation across M. perstans and M. ozzardi and among multiple clinical isolates
The availability of genomes from two Mansonella species and multiple isolates provided a unique opportunity for analysis of whole genome sequence variation both between and within the two species. The genome-wide average nucleotide identity (gANI) scores for isolates within species are 99.5% (Table 2) in comparison to the score of 89.69% between the two Mansonella species. Visualizations of whole genome alignments between isolates as Jupiter plots (Figure 1) reveal high levels of synteny. The isolates Mpe-Cam-1and Mpe-Cam-2, collected from the same geographical region within a few years apart are most similar to each other (Figure 1A). Relatively more genomic re-arrangements (represented as crisscrossing lines in Jupiter plots) can be seen between M. ozzardi isolates Moz-Brazil-1 and Moz-Venz-1 (Figure 1B), which have been collected from different geographical regions (Brazil, Venezuela) and at about 30 years apart. This indicates that the evolution of Mansonella genomes is dependent on geographical and temporal separation. Whole-genome alignments comparing M. perstans and M. ozzardi genomes also demonstrate high levels of synteny (Figure 1C), although lower than the within-species levels, consistent with the trend in their gANI scores.
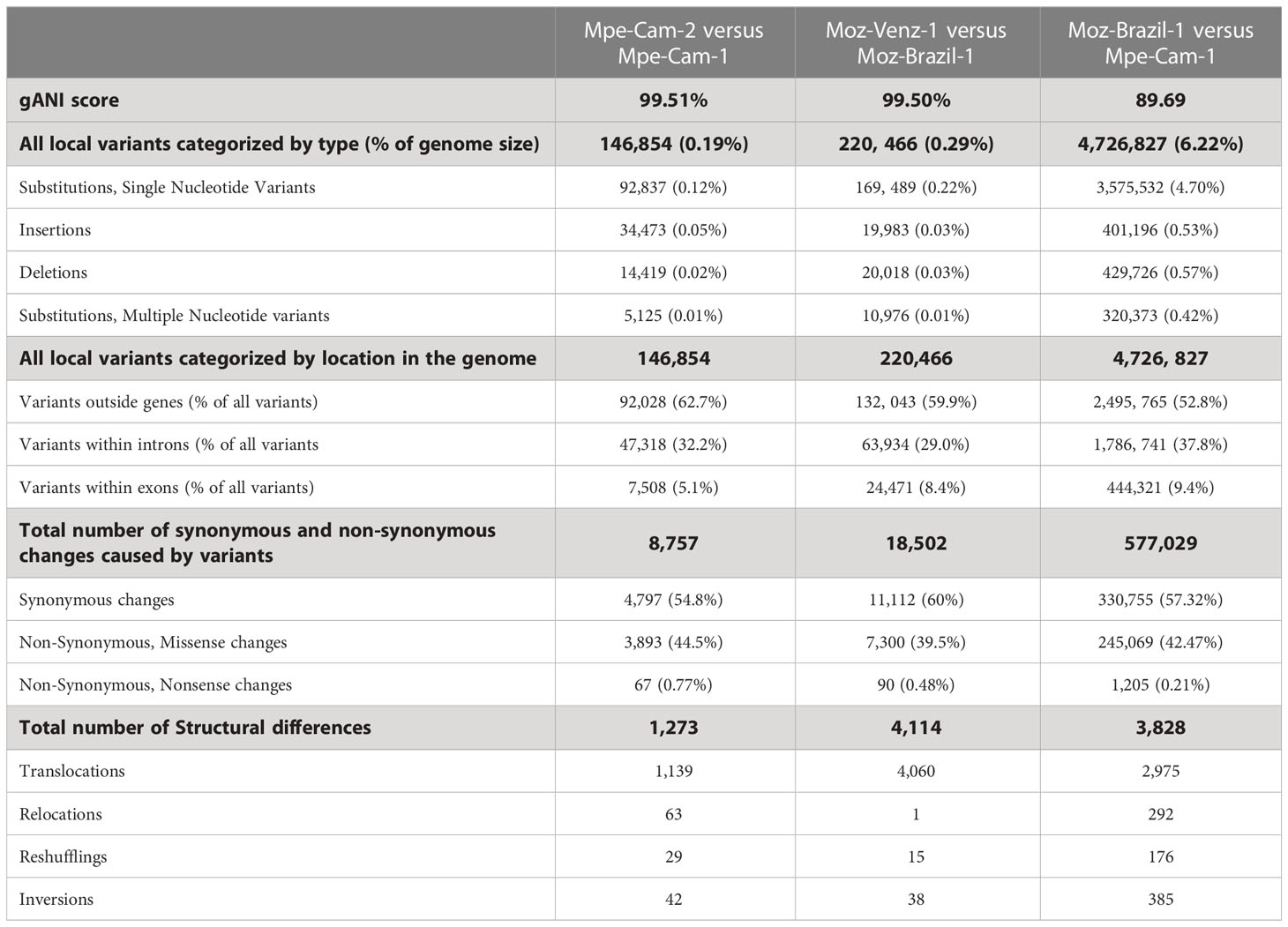
Table 2 Structural and local differences between Mansonella genomes.
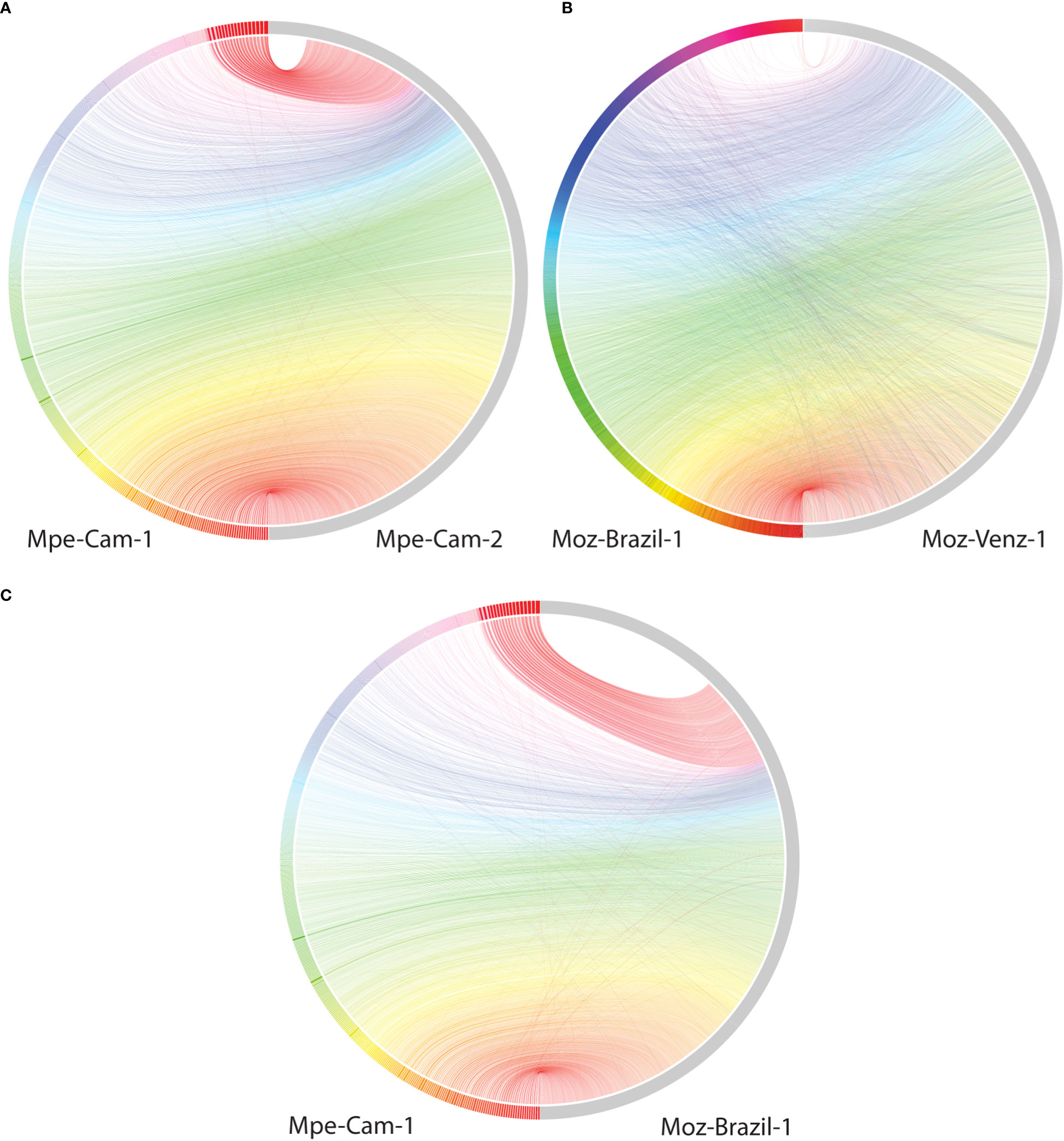
Figure 1 Comparisons of syntenic regions across the assembled Mansonella genomes. Whole genome alignments were performed using minimap with default parameters. Syntenic regions were visualized using Jupiter plots, comparing (A) The two isolates of M. perstans (B) The two isolates of M. ozzardi, and (C) the cross-species comparison between Mpe-Cam-1 and Moz-Brazil-1.
At the nucleotide level, the numbers of simple substitutions, insertions and deletions (Table 2) were 10 to 30-fold higher in across-species comparisons (6.22%), as compared to within-species (0.2% and 0.29%). In all the pairwise comparisons, most of the sequence variation (>52.8%) was localized to the intergenic portions of the genomes outside the protein-coding genes CDS regions. The proportion of variants within protein-coding genes ranged from about 5% to 10% (Table 2). Most of the nucleotide changes (54% to 60%) that occurred within coding regions, were mutationally silent as they resulted in synonymous amino-acid changes, 39 to 44% of coding variants did result in missense or non-synonymous changes in the predicted protein sequences and only a small fraction (0.21% to 0.77%) of the coding variants resulted in a nonsense mutation. Overall, the intra-species differences between the two M. ozzardi isolates from different geographical areas were higher than the corresponding intra-species differences of two M. perstans isolates which were both collected in Cameroon. A similar trend was observed for structural differences as well, as the number of relocations, reshufflings and inversions of contig fragments was observed to be much higher in cross-species comparisons (Table 2).
3.4 Orthology and phylogenomic analysis of filarial nematodes
An Orthofinder analysis of proteomes from all available filarial genomes (Supplementary Material Table 1), with S. digitata and T. callipaeda as immediate outgroups from the sub-order Spiruromorpha, and C. elegans and P. pacificus as distant outgroups identified 3,829 orthogroups conserved across all these nematodes (Figure 2). This analysis also highlighted 110 orthogroups specific to Mansonella genus, 320 orthogroups specific to Onchocerca genus, and 130 orthogroups specific to Brugia. Most of these proteins had only weak similarities to other nematode proteins (median sequence identity and coverage both around 33%), making them good candidates for genus-specific biomarkers (Supplementary Material Table 3).
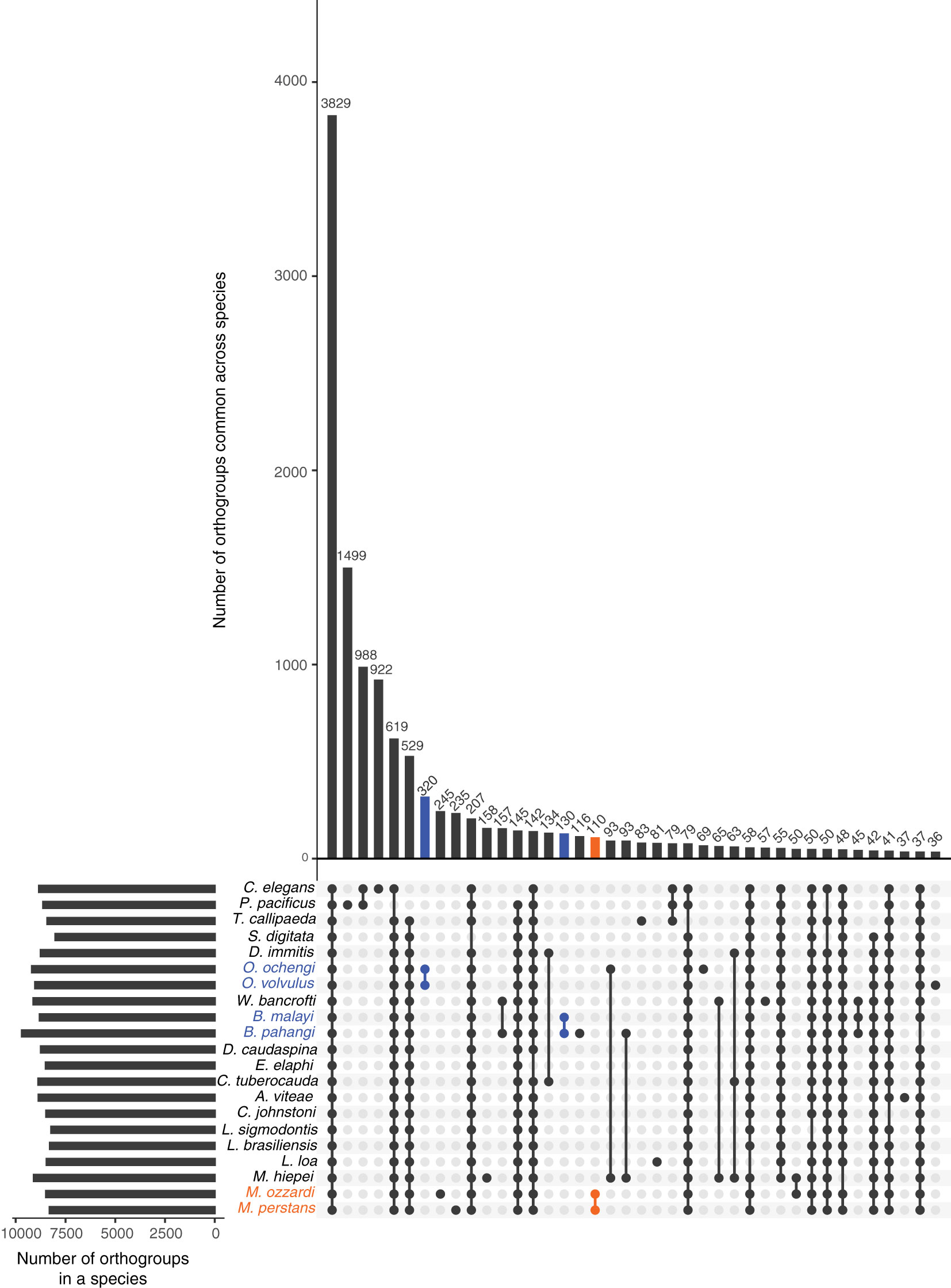
Figure 2 Orthology analysis across filarial parasites. Homologs conserved across various filarial species, as well as the Onchocercid nematodes S. digitata and T. callipaeda, and free-living model nematodes P. pacificus and C. elegans from Clade V were identified using OrthoFinder. The set of all 263,019 proteins from these nematode genomes were grouped into 16,564 orthogroups. The number of orthogroups identified in each species are represented by horizontal bars in the Upset plot, and the number of orthogroups common across different species are represented by vertical bars. The presence of an orthogroup in a species is indicated by a filled, colored dot while the absence is indicated by empty gray dots. Genus-specific orthogroups are shown as orange dots/bars for Mansonella, and blue dots/bars for Onchocerca and Brugia.
Among the orthogroups conserved across all nematode genomes analyzed here, 465 orthogroups comprised of single copy orthogroups (SCOs) and were used for analysis. The phylogenomic tree (Figure 3) generated using these 465 orthogroups recapitulated the phylogenetic relationships based on analysis of mitochondrial genomes (56) and the monophyletic feature of major clades ONC2, ONC3, ONC4 and ONC5 based on 7 genes (57).
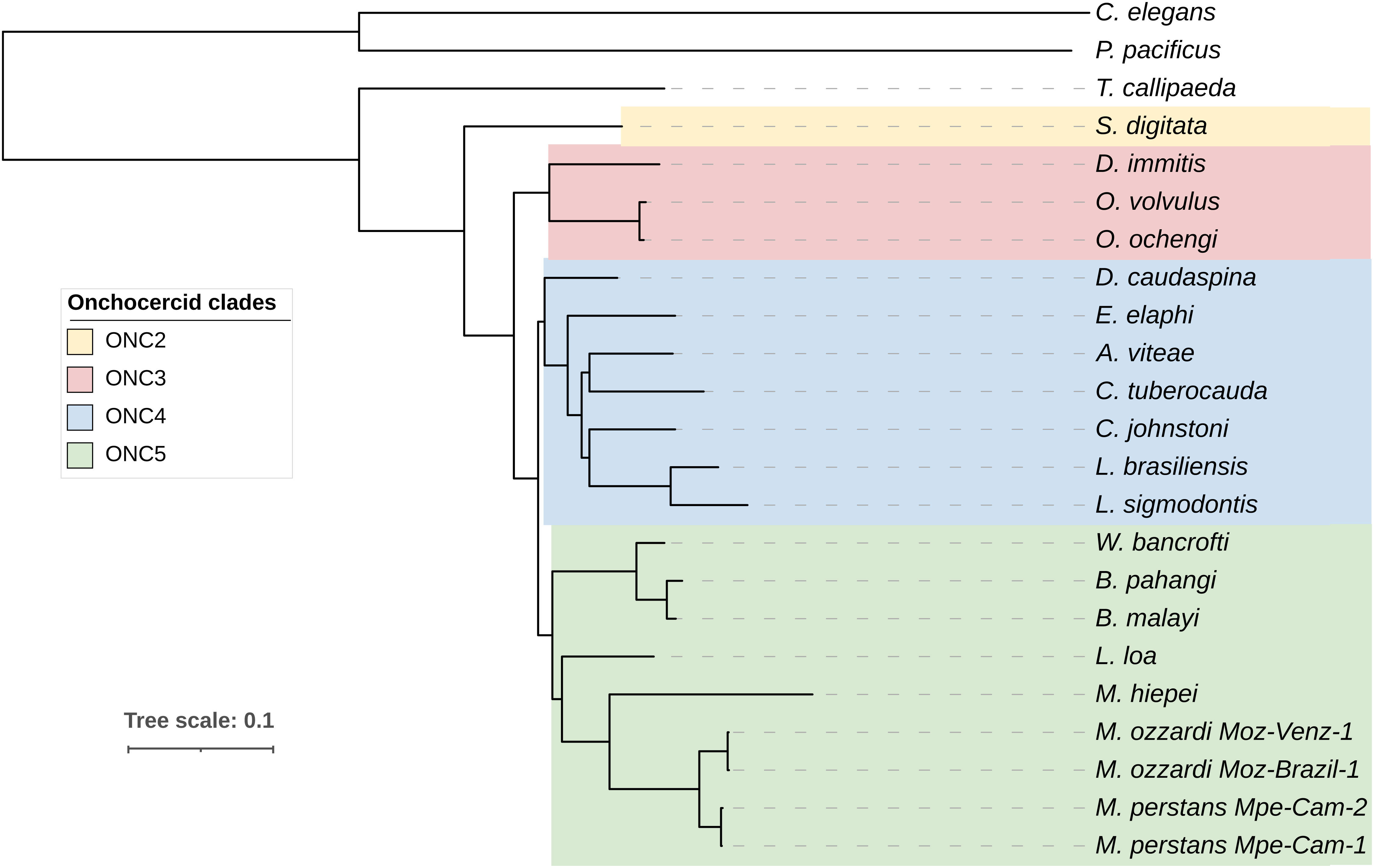
Figure 3 Phylogenomic analysis of filarial parasites. Phylogenomic tree was generated based on protein sequences of 465 single copy orthologs identified across all nematode genomes. Onchocercid specie S. digitata and T. callipaeda were used as immediate outgroups, while free-living nematodes C. elegans and P. pacificus were used as more distant outgroups. The previously described Onchocercid clades ONC2 to ONC5 are highlighted.
3.5 Analysis of nuMTs, horizontal transfers of mitochondrial DNA into nuclear genomes
Transfer of mitochondrial DNA fragments into the nuclear genome has been observed for multiple filarial nematodes (58) as well other eukaryotes (59). Since mitochondrial markers are often used as barcodes for species identification and phylogenetic analysis (33), it is important to distinguish between genuine mitochondrial sequences versus nuMTs (33, 60). In the newly sequenced Mansonella genomes, 39 to 94 nuMT loci with median sizes ranging around 250 bp were found across different isolates, with the largest loci ranging from 2.35 kb to 3.69 kb in size (Table 3). None of the nuMTs was found to carry an intact protein coding gene. The total number and the combined size of nuMTs in was observed to be higher in M. ozzardi as compared to M. perstans (Table 3). The locations and sequences of nuMTs were poorly conserved across M. perstans and M. ozzardi. For example, in a comparison of the 48 Mpe-Cam-1 nuMTs with the 82 Moz-Brazil-1 nuMTs, only 2 conserved loci were found (Supplementary Material Table 4). Even within the isolates of each species, approximately 35% and 60% of nuMT loci were conserved in M. perstans and M. ozzardi respectively (Supplementary Material Table 4), while the remainder were isolate-specific, indicating a fast turnover of these elements. The existence of multiple cox1 derived nuMT pseudogenes has been reported in M. ozzardi (33). Similarly, multiple fragments of the cox1 gene were also observed in both M. ozzardi isolates (Supplementary Figure 2). However, an exact match for these sequences could not be found in either of the Moz-Brazil-1 or Moz-Venz-1 assemblies, providing further evidence for poor conservation of such nuMTs across different isolates.
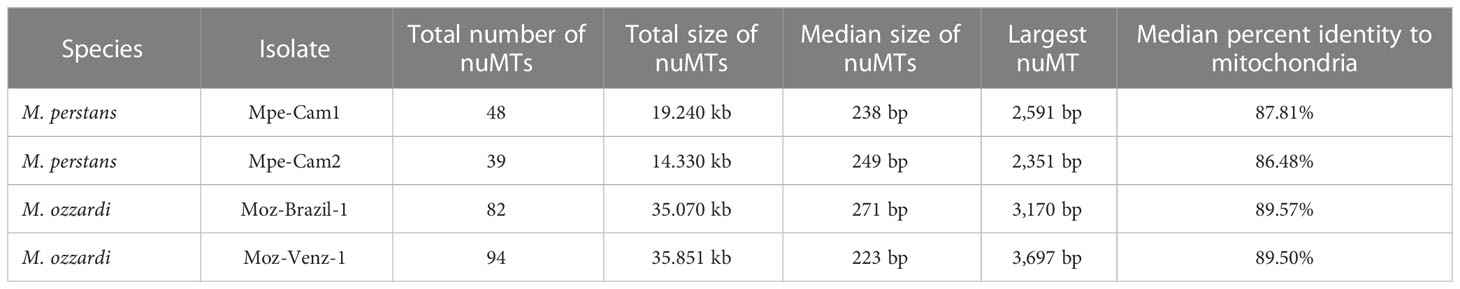
Table 3 Nuclear mitochondria Transfers (nuMTs) annotated across all genomes.
When the nuMT sequences from each isolate were mapped back to the mitochondrial genomes, they were found to cover almost the entire mitochondrial genome, without any preference for specific regions to be transferred into the nuclear genome (Supplementary Figures 2, 3).
3.6 Analysis of nuWTs, horizontal transfers of Wolbachia endosymbiont DNA into host nuclear genomes
Instances of transfer of DNA sequences from Wolbachia endosymbionts into the nuclear genomes of their hosts, termed “nuWTs” for nuclear-Wolbachia transfers, have been observed frequently in filarial nematodes (58, 61–63) as well as arthropod hosts (64, 65). The Mansonella isolates reported here harbor about 300 nuWT loci in each isolate, with median size about 180 bp, adding up to a total of 76 kb to 88 kb), with the largest nuWT (2.73 kb) observed in Mpe-Cam-1 (Table 4). None of these nuWTs were found to harbor any intact protein coding gene. Analysis of synteny and sequence identity of nuWTs across species identified only 34 conserved nuWTs between M. perstans and M. ozzardi (Supplementary Material Table 5). Interestingly, about 80% (260 from M. perstans and 280 from M. ozzardi) were found to be conserved in within-species comparisons of M. perstans and M. ozzardi respectively (Supplementary Material Table 5).
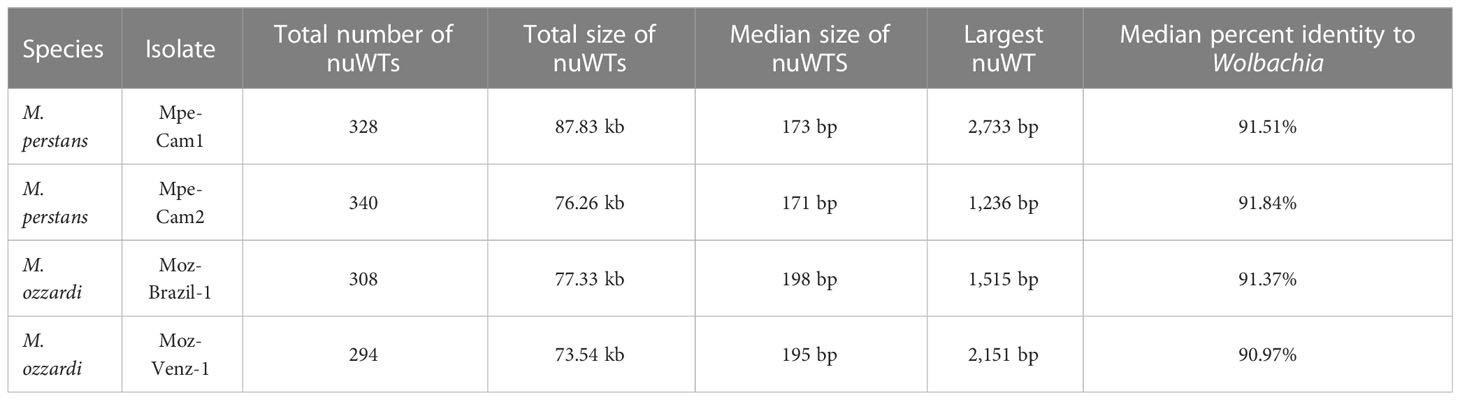
Table 4 Nuclear Wolbachia Transfers (nuWTs) annotated across all genomes.
3.7 Analysis and comparison of genes encoding drug targets for the filaricides diethylcarbamazine, ivermectin and albendazole
The main preventive intervention for filarial infections is based on mass drug administration (MDA) using diethylcarbamazine (DEC), ivermectin (IVM) and albendazole (ALB) (1). Although DEC has been successfully used to treat M. perstans infections, the drug has been found to be ineffective against M. ozzardi (17). To investigate potential reasons for differences in drug susceptibility between M. perstans and M. ozzardi, the genes encoding the drug targets for DEC, ivermectin and albendazole, were identified and analyzed. A search for the DEC target gon-2 (66), a gene that encodes a Transient Receptor Protein M (TRPM) channel subunit, identified two potential homologs in M. perstans but one in M. ozzardi. Phylogenetic analysis of the gon-2 homologs in Mansonella and other nematode genomes revealed two distinct, paralogous clades, denoted as Clade 1 and Clade 2, which represent filarial orthologs of Cel-gon-2 and Cel-gtl-2 respectively (Figure 4). The nematodes known to be susceptible to DEC, including C. elegans and filarial parasites B. malayi L. loa, and W. bancrofti, all have a gon-2 ortholog in Clade 1. Interestingly, M. perstans, which is treatable with DEC, also has a corresponding ortholog in Clade 1 while in M. ozzardi, which is refractory to DEC, the orthologous gene was found in two fragments on the ends of two separate contigs in each of the two isolates (Supplementary Figure 4). Artificially combining the two gene fragments could reconstruct the full gene except for a few missing bases between exons 19 and 20 (Supplementary Figures 5, 6). It is possible that the gene is intact in M. ozzardi but is split over two contigs due to incomplete genome assembly. To explore this further, the raw Illumina reads from each isolate were mapped to a synthetic contig constructed by joining the contigs carrying the gon-2 fragments in appropriate orientations with a 50 bp spacer marked by Ns, and all read-pairs spanning the potentially un-assembled gap were investigated (Supplementary Figures 7, 8). This analysis showed an abrupt drop in read coverage in the potential gap region, even though the contig terminals had sufficient coverage. Although a few read-pairs (less than 10 counts) were observed to span across the gap, the size of gap estimated via the insert size of the paired-end reads was around 2.5 kb, much larger than the insert size of the sequencing library. In contrast, the read-coverage across the corresponding region in M. perstans is uniform (Supplementary Figure 9). Together, these observations indicate that the synthetic contig is unlikely to represent the true genomic sequence, and therefore the gon-2 gene is fragmented in both M. ozzardi isolates. Both M. perstans and M. ozzardi do possess orthologs of Cel-gtl-2 in Clade 2, which share only 52% sequence identity with the C. elegans GON-2 protein in Clade 1 and are unlikely to share their functions. Similar analysis for the other known DEC target genes, trp-2, ced-11 and slo-1 identified the corresponding one-to-one orthologs for all three genes in all the filarial species including both isolates of M. perstans and M. ozzardi (Supplementary Figures 10-12). Taken together, this suggests that the absence the gon-2 ortholog in M. ozzardi is most likely responsible for the ineffectiveness of DEC against this parasite.
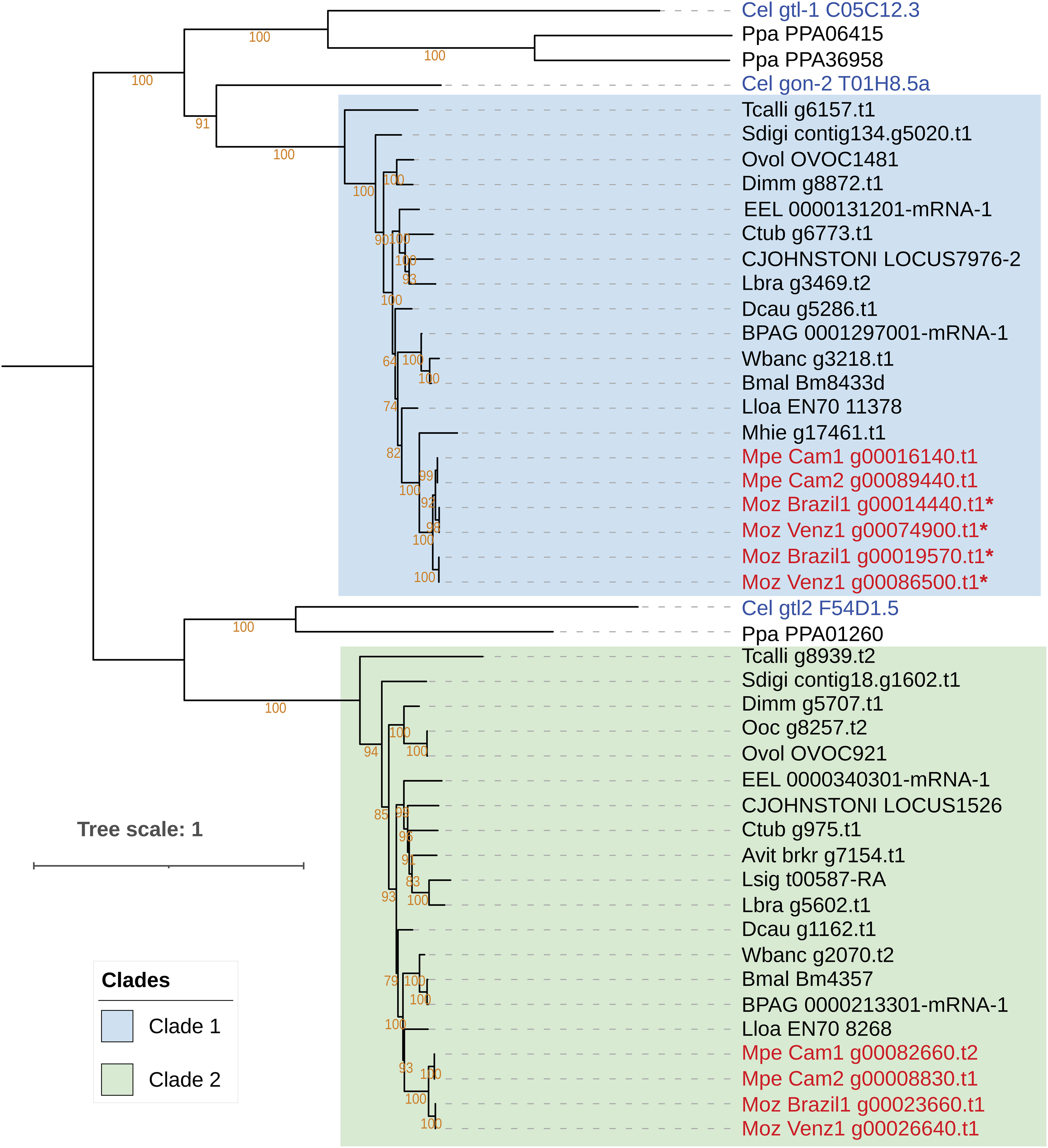
Figure 4 Phylogenetic analysis of genes encoding the targets of the anti-filarial drug Diethylcarbamazine (DEC). Phylogenetic analysis of the gon-2, gtl-1 and gtl-2 gene family was performed based on their protein sequences and the tree was rooted at the midpoint. The orthologs of the Cel-gon-2 gene are placed in Clade 1, highlighted in blue, and orthologs of Cel-gtl-2 orthologs are placed in Clade 2, highlighted green. The gtl-1 gene has no orthologs in filarial nematodes. Genes from M. perstans and M. ozzardi are in red, and C. elegans genes are in blue. Asterisks (*) next to gene names indicate fragmented gene structures.
The targets of ivermectin as identified through genetic studies in C. elegans include various subunits of the Glutamate‐gated Cl− channels (GluCl), encoded by genes glc-1, glc-2, glc-3, glc-4, avr-14 and avr-15 (67, 68). Ivermectin is effective in treating M. ozzardi infections (69), but is reported to be ineffective against M. perstans (70). Phylogenetic analysis of these genes across filarial parasites shows that the homologs of glc-1, glc-2, glc-3, avr-14 and avr-15 form a family of paralogous genes with complex patterns of homology across filarial parasites (Figure 5), while glc-4 has one-to-one orthologs across all genomes analyzed (Supplementary Figure 13). Orthologs for Cle-glc-1, Cel-glc-2 and Cel-avr-15 are not present in either of the Mansonella species (Figure 5). Both isolates of M. perstans and M. ozzardi have one ortholog most closely related to the Cel-glc-3, and one ortholog most closely related to to Cel-avr-14,
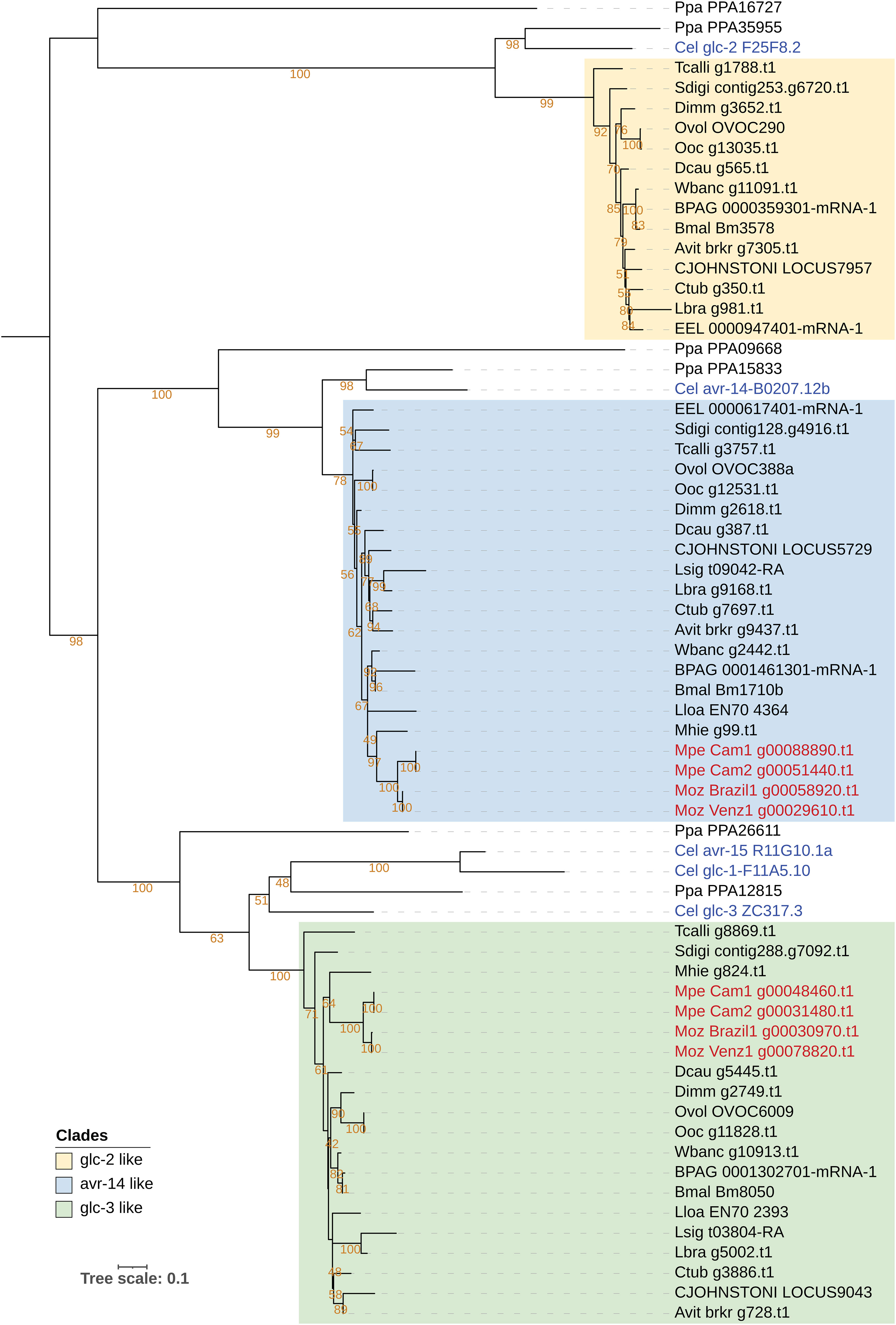
Figure 5 Phylogenetic analysis of genes encoding Glutamate Chloride channel subunits, targets of ivermectin. Phylogenetic analysis of the GluCl gene family was based on their protein sequences and the tree was rooted at the midpoint. The clade with Cel-glc-2 orthologs is highlighted in yellow, clade with Cel-avr-14 orthologs is highlighted in blue, and the clade with Cel-glc-3 orthologs is highlighted in green. M. perstans and M. ozzardi genes are in red, and C. elegans genes are in blue.
The anthelminthic drug mebendazole acts by binding to beta-tubulin protein and inhibiting microtubule formation (71). Mebendazole is commonly used in combination DEC or ivermectin for treating M. perstans infections (70) but its use for M. ozzardi has not been reported. The beta-tubulin gene family in C. elegans consists of 6 different members (ben-1, tbb-1, tbb-2, tbb-4, tbb-6, and mec-7) all of which have varying effects on drug sensitivity (72, 73). A phylogenetic analysis of the beta-tubulin gene family in Mansonella and other nematodes reveals a complex pattern of expansion and contraction of this gene family across filarial species, with no direct orthologs for ben-1 identifiable in any filarial nematodes (Supplementary Figure 14). The exact roles of different beta-tubulin homologs on drug susceptibility in Mansonella will require further study.
4 Discussion
To gain some insight into the neglected pathogens responsible for human mansonellosis, we report the first de novo genome assemblies from the filarial parasites M. perstans and M. ozzardi, providing reference genomes for each species. The genomes encode approximately 10,000 genes each and are high quality and largely complete based on universally conserved orthologs (BUSCO scores greater than 90%) relative to other filarial species. The availability and sequencing of two isolates for M. perstans and M. ozzardi also allowed higher confidence in calling presence or absence of genes in each species as it could be confirmed in two independent isolates. The Mansonella genomes average 76 Mb in length and are similar in size to the genomes described from other human and animal filarial parasites (56, 74–79). The sequencing of two clinical isolates of M. perstans and M. ozzardi also enabled a quantification of the genetic variability both between species as well in the same parasite from different regions. Most of the genetic differences observed between isolates and species are in the intergenic regions, and only a small fraction (10%) of variants affects coding exons. Relatively little is known about natural variation in the nuclear genomes of filarial parasites (80, 81). In the heartworm Dirofilaria immitis, a closely related filarial parasite of dogs and cats, sequencing of two isolates from Europe and the United States revealed very little genetic variation in their nuclear or mitochondrial genomes (75). Other studies of genetic variation within isolates of a filarial species have used mitochondrial cox1 gene (80) or the mitochondrial genomes (81). Our study presents a more comprehensive analysis of variation in nuclear genomes within isolates. It will be interesting to investigate how these patterns evolve in other human filarial species.
Transfer of mitochondrial DNA fragments into the nuclear genome has been observed for multiple filarial nematodes (58). These nuMT loci are annotated for all Mansonella isolates using their reported mitochondrial genomes (32, 33). Most filarial parasites possess the intracellular bacterial symbiont, Wolbachia, capable of transferring genetic material to the nuclear genome of its host. The genomes of Wolbachia endosymbionts wMpe and wMoz from M. perstans and M. ozzardi have also been reported (22), enabling an analysis of nuWTs, regions of DNA transfers from Wolbachia to its host nuclear genomes. While reported in other filarial species, the biological function and significance of such nuclear transfer events are not well understood. Moreover, the levels of conservation of such loci across multiple isolates of a species, or closely related species has not been previously studied. Our analysis combining synteny and sequence identity shows that the nuMT loci are poorly conserved in Mansonella even within the same species, and rarely harbor intact coding genes, suggesting that most of these transfers are not evolutionarily selected, and non-functional. For the nuWTs, relatively more loci were found to be conserved across the two species, but isolate specific acquisition of these nuWT fragments was also observed. However, none of these loci harbored any intact protein-coding genes, suggesting a lack of functional constraints on their evolution. This observation is consistent with the detection of mostly non-functional nuWTs in other filarial parasites (58) and suggests that these loci most likely arise randomly and degrade via genetic drift over shorter time scales, as seen in other organisms (59, 65, 82).
Despite their genetic relatedness, M. perstans and M. ozzardi differ in their geographical distribution, adaptation to insect vectors, and in their response to drug treatments. We have used these new genomes to explore the genetic basis for the reported differences in their response to commonly used anti-filarial drugs. Our analysis of the sequences and phylogeny of known gene products targeted by DEC revealed that M. ozzardi does not have an intact gon-2 gene, one of the targets of this drug. In contrast, M. perstans as well as other filarial parasites that are susceptible to DEC have a gon-2 ortholog. It can therefore be hypothesized that the absence of gon-2 ortholog in M. ozzardi has rendered it unsusceptible to DEC. Future research focusing on this gene sequence and its variation between different isolates of the two species can shed further light on its function.
All the genes known to be targets of ivermectin were found in both M. perstans and M. ozzardi. The observed differences in ivermectin susceptibility of the two parasites could not be readily explained by the subtle differences in sequences of these targets. There could be additional ivermectin targets that are not discovered yet but could play a role in the differential response of M. perstans and M. ozzardi. Although mebendazole is used for the treatment M. perstans, it was found to lack an ortholog of one of the targets encoded by the beta-tubulin gene tbb-4, while the corresponding ortholog is present in M. ozzardi. Given the complex patterns of homology for this gene family, and the absence of any reported effects of mebendazole on M. ozzardi, the significance of this observation remains to be determined. In addition to these observations, there could be yet other undiscovered filarial genes that have also have effects on drug susceptibility and could be responsible for differences between M. perstans and M. ozzardi. A functional validation of these hypotheses regarding drug and their target genes is currently not feasible, but future advances may be able to test these hypotheses. At least one of the parasites, M. perstans, can now be successfully cultured in vitro enabling functional investigations into its biology (83). Advances in genome editing techniques for non-model organisms including helminth parasites open further possibilities of genetic and functional studies, and control of parasites (84–89). The availability of the new Mansonella genomes reported here will be a critical genomic resource for such endeavors and for further studies on drug targets, as well as enabling insights into the biology and evolution of these parasites.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA917716, https://www.ncbi.nlm.nih.gov/, PRJNA917721, https://www.ncbi.nlm.nih.gov/, PRJNA917722, https://www.ncbi.nlm.nih.gov/, PRJNA917766.
Ethics statement
The studies involving human participants were reviewed and approved by National Institutional Review Board, Yaoundé (REF: N° 2015/09/639/CE/CNERSH/SP), Delegation of Public Health, South-West region of Cameroon (Re: R11/MINSANTE/SWR/RDPH/PS/259/382), National Ethics Committee of Research for Human Health, Cameroon, Institutional Review Board of the Institute of Biomedical Sciences, University of São Paulo, Brazil (1133/CEP, 2013). The patients/participants provided their written informed consent to participate in this study.
Author contributions
NL, MF, FF, SW collected the clinical samples. AS, ZL, CP, LE, RM performed the sequencing experiments and bioinformatics analysis. AS, ZL, CP, LE, RM, CC analysed the data. AS, ZL, CP, CC wrote the initial manuscript. AS, ZL, CP, LE, RM, MF, SW, CC and all authors contributed to the final manuscript.
Funding
Research funding was provided by New England Biolabs, Ipswich, MA 01938, USA to AS, ZL, LE, RM, CP, CC. FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) supported the field research in Brazil through a research grant to M.U.F (2013/12723-7) and a doctoral scholarship to NL (2013/26928-0).
Acknowledgments
The authors thank the DNA sequencing core at New England Biolabs. The authors are grateful to the late Don Comb for initiating and inspiring research into NTDs at NEB.
Conflict of interest
Authors AS, ZL, CP, LE, RM, and CC are employees at New England Biolabs Inc., Ipswich, MA 01938 USA, which manufactures biotechnology reagents including those used in Next Generation Sequencing.
The remining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2023.1139343/full#supplementary-material
References
1. Simonsen PE, Onapa AW, Asio SM. Mansonella perstans filariasis in Africa. Acta Tropica (2011) 120:S109–20. doi: 10.1016/j.actatropica.2010.01.014
2. Vianna LMM, Martins M, Cohen MJ, Cohen JM, Belfort R. Mansonella ozzardi corneal lesions in the Amazon: a cross-sectional study. BMJ Open (2012) 2:e001266. doi: 10.1136/bmjopen-2012-001266
3. Lima NF, Aybar CAV, Juri MJD, Ferreira MU. Mansonella ozzardi: a neglected new world filarial nematode. Pathog Global Health (2016) 110:97–107. doi: 10.1080/20477724.2016.1190544
4. Mediannikov O, Ranque S. Mansonellosis, the most neglected human filariasis. New Microbes New Infect (2018) 26:S19–22. doi: 10.1016/j.nmni.2018.08.016
5. Ferreira MU, Crainey JL, Luz SLB. Mansonella ozzardi. Trends Parasitol (2021) 37:90–1. doi: 10.1016/j.pt.2020.03.005
6. Ritter M, Ndongmo WPC, Njouendou AJ, Nghochuzie NN, Nchang LC, Tayong DB, et al. Mansonella perstans microfilaremic individuals are characterized by enhanced type 2 helper T and regulatory T and b cell subsets and dampened systemic innate and adaptive immune responses. PloS Negl Trop Dis (2018) 12:e0006184. doi: 10.1371/journal.pntd.0006184
7. Ta-Tang T-H, Crainey JL, Post RJ, Luz SL, Rubio JM. Mansonellosis: Current perspectives. Res Rep Trop Med (2018) 9:9–24. doi: 10.2147/RRTM.S125750
8. Downes B, Jacobsen K. A systematic review of the epidemiology of mansonelliasis. Afr J Infect Dis (2010) 4:7–14. doi: 10.4314/ajid.v4i1.55085
9. Bélard S, Gehringer C. High prevalence of Mansonella species and parasitic coinfections in Gabon calls for an end to the neglect of Mansonella research. J Infect Dis (2021) 223:187–8. doi: 10.1093/infdis/jiaa671
10. Sandri TL, Kreidenweiss A, Cavallo S, Weber D, Juhas S, Rodi M, et al. Molecular epidemiology of Mansonella species in Gabon. J Infect Dis (2021) 223:287–96. doi: 10.1093/infdis/jiaa670
11. Ta-Tang T-H, Luz SL, Crainey JL, Rubio JM. An overview of the management of mansonellosis. RRTM (2021) 12:93–105. doi: 10.2147/RRTM.S274684
12. Tavares da Silva LB, Crainey JL, Ribeiro da Silva TR, Suwa UF, Vicente ACP, Fernandes de Medeiros J, et al. Molecular verification of new world Mansonella perstans parasitemias. Emerging Infect Dis (2017) 23:545–7. doi: 10.3201/eid2303.161159
13. Lima NF, Gonçalves-Lopes RM, Kruize YCM, Yazdanbakhsh M, Ferreira MU. CD39 and immune regulation in a chronic helminth infection: The puzzling case of Mansonella ozzardi. PloS Negl Trop Dis (2018) 12:e0006327. doi: 10.1371/journal.pntd.0006327
14. Raccurt CP. Mansonella ozzardi and its vectors in the new world: An update with emphasis on the current situation in Haiti. J Helminthol. (2018) 92:655–61. doi: 10.1017/S0022149X17000955
15. Kozek WJ, Palma G, Henao A, García H, Hoyos M. Filariasis in Colombia: Prevalence and distribution of Mansonella ozzardi and Mansonella (=Dipetalonema) perstans infections in the comisaría del guainía. Am J Trop Med Hygiene (1983) 32:379–84. doi: 10.4269/ajtmh.1983.32.379
16. Calvopina M, Chiluisa-Guacho C, Toapanta A, Fonseca D, Villacres I. High prevalence of Mansonella ozzardi infection in the Amazon region, Ecuador. Emerging Infect Dis (2019) 25:2081–3. doi: 10.3201/eid2511.181964
17. Chadee DD, Tilluckdharry CC, Rawlins SC, Doon R, Nathan MB. Mass chemotherapy with diethylcarbamazine for the control of bancroftian filariasis: A twelve-year follow-up in northern Trinidad, including observations on Mansonella ozzardi. Am J Trop Med Hygiene (1995) 52:174–6. doi: 10.4269/ajtmh.1995.52.174
18. Poole CB, Sinha A, Ettwiller L, Apone L, McKay K, Panchapakesa V, et al. In silico identification of novel biomarkers and development of new rapid diagnostic tests for the filarial parasites Mansonella perstans and Mansonella ozzardi. Sci Rep (2019) 9:10275. doi: 10.1038/s41598-019-46550-9
19. Coulibaly YI, Dembele B, Diallo AA, Lipner EM, Doumbia SS, Coulibaly SY, et al. A randomized trial of doxycycline for Mansonella perstans infection. New Engl J Med (2009) 361:1448–58. doi: 10.1056/NEJMoa0900863
20. Debrah LB, Phillips RO, Pfarr K, Klarmann-Schulz U, Opoku VS, Nausch N, et al. The efficacy of doxycycline treatment on Mansonella perstans infection: An open-label, randomized trial in Ghana. Am J Trop Med Hygiene (2019) 101:84–92. doi: 10.4269/ajtmh.18-0491
21. Crainey JL, Costa CHA, de Oliveira Leles LF, Ribeiro da Silva TR, de Aquino Narzetti LH, Serra dos Santos YV, et al. Deep sequencing reveals occult mansonellosis coinfections in residents from the Brazilian Amazon village of são Gabriel da cachoeira. Clin Infect Dis (2020) 71:1990–3. doi: 10.1093/cid/ciaa082
22. Sinha A, Li Z, Poole CB, Ettwiller L, Lima NF, Ferreira MU, et al. Multiple origins of nematode-Wolbachia symbiosis in supergroup F and convergent loss of bacterioferritin in filarial Wolbachia. bioRxiv (2022). doi: 10.1101/2021.03.23.436630
23. Li H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics (2018) 34:3094–100. doi: 10.1093/bioinformatics/bty191
24. Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res (2017) 27:722–36. doi: 10.1101/gr.215087.116
25. Lam K-K, LaButti K, Khalak A, Tse D. FinisherSC: A repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics (2015) 31:3207–9. doi: 10.1093/bioinformatics/btv280
26. Zimin AV, Salzberg SL. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PloS Comput Biol (2020) 16:e1007981. doi: 10.1371/journal.pcbi.1007981
27. Pryszcz LP, Gabaldón T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res (2016) 44:e113–3. doi: 10.1093/nar/gkw294
28. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics (2013) 29:1072–5. doi: 10.1093/bioinformatics/btt086
29. Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res (2017) 27:824–34. doi: 10.1101/gr.213959.116
30. Laetsch DR, Blaxter ML. BlobTools: Interrogation of genome assemblies. F1000Research (2017) 6:1287. doi: 10.12688/f1000research.12232.1
31. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods (2012) 9:357–9. doi: 10.1038/nmeth.1923
32. Chung M, Aluvathingal J, Bromley RE, Nadendla S, Fombad FF, Kien CA, et al. Complete mitochondrial genome sequence of Mansonella perstans. Microbiol Resour Announc (2020) 9. doi: 10.1128/MRA.00490-20
33. Crainey JL, Marín MA, da Silva TRR, de Medeiros JF, Pessoa FAC, Santos YV, et al. Mansonella ozzardi mitogenome and pseudogene characterisation provides new perspectives on filarial parasite systematics and CO-1 barcoding. Sci Rep (2018) 8. doi: 10.1038/s41598-018-24382-3
34. Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS (2020) 117:9451–7. doi: 10.1073/pnas.1921046117
35. Storer J, Hubley R, Rosen J, Wheeler TJ, Smit AF. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mobile DNA (2021) 12:2. doi: 10.1186/s13100-020-00230-y
36. Kapusta A., Suh A., Feschotte C. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci (2017) 114:E1460–E1469. doi: 10.1073/pnas.1616702114
37. Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinf (2021) 3. doi: 10.1093/nargab/lqaa108
38. Kriventseva E. V., Kuznetsov D., Tegenfeldt F., Manni M., Dias R., Simão F. A., et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucl Acids Res (2019) 47:D807–11. doi: 10.1093/nar/gky1053
39. Howe KL, Bolt BJ, Shafie M, Kersey P, Berriman M. WormBase ParaSite – a comprehensive resource for helminth genomics. Mol Biochem Parasitol (2017) 215:2–10. doi: 10.1016/j.molbiopara.2016.11.005
40. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics (2015) 31:3210–2. doi: 10.1093/bioinformatics/btv351
41. Emms DM, Kelly S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol (2019) 20:238. doi: 10.1186/s13059-019-1832-y
42. Lefoulon E, Clark T, Guerrero R, Cañizales I, Cardenas-Callirgos JM, Junker K, et al. Diminutive, degraded but dissimilar: Wolbachia genomes from filarial nematodes do not conform to a single paradigm. Microbial Genomics (2020) 6:e000487. doi: 10.1099/mgen.0.000487
43. Yoon S-H, Ha S, Lim J, Kwon S, Chun J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek (2017) 110:1281–6. doi: 10.1007/s10482-017-0844-4
44. Khelik K, Lagesen K, Sandve GK, Rognes T, Nederbragt AJ. NucDiff: In-depth characterization and annotation of differences between two sets of DNA sequences. BMC Bioinf (2017) 18:338. doi: 10.1186/s12859-017-1748-z
45. Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics (2010) 26:841–2. doi: 10.1093/bioinformatics/btq033
46. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (2012) 6:80–92. doi: 10.4161/fly.19695
47. Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics (2018) 34:2490–2. doi: 10.1093/bioinformatics/bty121
48. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biol (2007) 56:564–77. doi: 10.1080/10635150701472164
49. Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol (2015) 32:268–74. doi: 10.1093/molbev/msu300
50. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat Methods (2017) 14:587–9. doi: 10.1038/nmeth.4285
51. Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biol (2010) 59:307–21. doi: 10.1093/sysbio/syq010
52. Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: Improving the ultrafast bootstrap approximation. Mol Biol Evol (2018) 35:518–22. doi: 10.1093/molbev/msx281
53. Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res (2020) 48:D265–8. doi: 10.1093/nar/gkz991
54. Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res (2021) 49:W293–6. doi: 10.1093/nar/gkab301
55. Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: A fast and versatile genome alignment system. PloS Comput Biol (2018) 14:e1005944. doi: 10.1371/journal.pcbi.1005944
56. McCann K, Grant W, Doyle SR. The genome sequence of the Australian filarial nematode, Cercopithifilaria johnstoni. Wellcome Open Res (2021) 6:259. doi: 10.12688/wellcomeopenres.17258.2
57. Lefoulon E, Bain O, Makepeace BL, d’Haese C, Uni S, Martin C, et al. Breakdown of coevolution between symbiotic bacteria Wolbachia and their filarial hosts. PeerJ (2016) 4:e1840. doi: 10.7717/peerj.1840
58. Grote A, Lustigman S, Ghedin E. Lessons from the genomes and transcriptomes of filarial nematodes. Mol Biochem Parasitol (2017) 215:23–9. doi: 10.1016/j.molbiopara.2017.01.004
59. Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PloS Genet (2010) 6:e1000834. doi: 10.1371/journal.pgen.1000834
60. Song H, Buhay JE, Whiting MF, Crandall KA. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc Natl Acad Sci (2008) 105:13486–91. doi: 10.1073/pnas.0803076105
61. McNulty SN, Foster JM, Mitreva M, Hotopp JCD, Martin J, Fischer K, et al. Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PloS One (2010) 5:e11029. doi: 10.1371/journal.pone.0011029
62. Cotton JA, Bennuru S, Grote A, Harsha B, Tracey A, Beech R, et al. The genome of Onchocerca volvulus, agent of river blindness. Nat Microbiol (2016) 2:16216. doi: 10.1038/nmicrobiol.2016.216
63. Hotopp JCD, Slatko BE, Foster JM. Targeted enrichment and sequencing of recent endosymbiont-host lateral gene transfers. Sci Rep (2017) 7:1–10. doi: 10.1038/s41598-017-00814-4
64. Klasson L, Kumar N, Bromley R, Sieber K, Flowers M, Ott SH, et al. Extensive duplication of the Wolbachia DNA in chromosome four of Drosophila ananassae. BMC Genomics (2014) 15:1097. doi: 10.1186/1471-2164-15-1097
65. Tvedte ES, Gasser M, Zhao X, Tallon LJ, Sadzewicz L, Bromley RE, et al. Accumulation of endosymbiont genomes in an insect autosome followed by endosymbiont replacement. Curr Biol (2022) 32:2786–2795.e5. doi: 10.1016/j.cub.2022.05.024
66. Verma S, Kashyap SS, Robertson AP, Martin RJ. Diethylcarbamazine activates TRP channels including TRP-2 in filaria, Brugia malayi. Commun Biol (2020) 3:1–11. doi: 10.1038/s42003-020-01128-4
67. Dent JA, Smith MM, Vassilatis DK, Avery L. The genetics of ivermectin resistance in Caenorhabditis elegans. Proc Natl Acad Sci U.S.A. (2000) 97:2674–9. doi: 10.1073/pnas.97.6.2674
68. Wolstenholme AJ, Rogers AT. Glutamate-gated chloride channels and the mode of action of the avermectin/milbemycin anthelmintics. Parasitology (2005) 131:S85–95. doi: 10.1017/S0031182005008218
69. de Basano SA, de Camargo JSAA, Fontes G, Pereira AR, Medeiros JF, de Laudisse MCO, et al. Phase III clinical trial to evaluate ivermectin in the reduction of Mansonella ozzardi infection in the Brazilian Amazon. Am J Trop Med Hygiene (2018) 98:786–90. doi: 10.4269/ajtmh.17-0698
70. Bregani ER, Rovellini A, Mbaïdoum N, Magnini MG. Comparison of different anthelminthic drug regimens against Mansonella perstans filariasis. Trans R Soc Trop Med Hygiene (2006) 100:458–63. doi: 10.1016/j.trstmh.2005.07.009
71. Lacey E. Mode of action of benzimidazoles. Parasitol Today (1990) 6:112–5. doi: 10.1016/0169-4758(90)90227-U
72. Driscoll M, Dean E, Reilly E, Bergholz E, Chalfie M. Genetic and molecular analysis of a Caenorhabditis elegans beta-tubulin that conveys benzimidazole sensitivity. J Cell Biol (1989) 109:2993–3003. doi: 10.1083/jcb.109.6.2993
73. Pallotto LM, Dilks CM, Park Y-J, Smit RB, Lu BT, Gopalakrishnan C, et al. Interactions of Caenorhabditis elegans β-tubulins with the microtubule inhibitor and anthelmintic drug albendazole. Genetics (2022) 221:iyac093. doi: 10.1093/genetics/iyac093
74. Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, et al. Draft genome of the filarial nematode parasite Brugia malayi. Science (2007) 317:1756–60. doi: 10.1126/science.1145406
75. Godel C, Kumar S, Koutsovoulos G, Ludin P, Nilsson D, Comandatore F, et al. The genome of the heartworm, Dirofilaria immitis, reveals drug and vaccine targets. FASEB J (2012) 26:4650–61. doi: 10.1096/fj.12-205096
76. Desjardins CA, Cerqueira GC, Goldberg JM, Hotopp JCD, Haas BJ, Zucker J, et al. Genomics of Loa loa, a Wolbachia-free filarial parasite of humans. Nat Genet (2013) 45(5):495–450. doi: 10.1038/ng.2585
77. Tallon LJ, Liu X, Bennuru S, Chibucos MC, Godinez A, Ott S, et al. Single molecule sequencing and genome assembly of a clinical specimen of Loa loa, the causative agent of loiasis. BMC Genomics (2014) 15:788. doi: 10.1186/1471-2164-15-788
78. Lau Y-L, Lee W-C, Xia J, Zhang G, Razali R, Anwar A, et al. Draft genome of Brugia pahangi: High similarity between B. pahangi and B. malayi. Parasites Vectors (2015) 8:451. doi: 10.1186/s13071-015-1064-2
79. Foster JM, Grote A, Mattick J, Tracey A, Tsai Y-C, Chung M, et al. Sex chromosome evolution in parasitic nematodes of humans. Nat Commun (2020) 11:1964. doi: 10.1038/s41467-020-15654-6
80. McNulty SN, Mitreva M, Weil GJ, Fischer PU. Inter and intra-specific diversity of parasites that cause lymphatic filariasis. Infection Genet Evol (2013) 14:137–46. doi: 10.1016/j.meegid.2012.11.002
81. Qing X, Kulkeaw K, Wongkamchai S, Tsui SK-W. Mitochondrial genome of Brugia malayi microfilariae isolated from a clinical sample. Front Ecol Evol (2021) 9:637805. doi: 10.3389/fevo.2021.637805
82. Bensasson D, Feldman MW, Petrov DA. Rates of DNA duplication and mitochondrial DNA insertion in the human genome. J Mol Evol (2003) 57:343–54. doi: 10.1007/s00239-003-2485-7
83. Njouendou AJ, Ritter M, Ndongmo WPC, Kien CA, Narcisse GTV, Fombad FF, et al. Successful long-term maintenance of Mansonella perstans in an in vitro culture system. Parasites Vectors (2017) 10:563. doi: 10.1186/s13071-017-2515-8
84. Lok JB, Shao H, Massey HC, Li X. Transgenesis in Strongyloides and related parasitic nematodes: Historical perspectives, current functional genomic applications and progress towards gene disruption and editing. Parasitology (2017) 144(3):327–42. doi: 10.1017/S0031182016000391
85. Zamanian M, Andersen EC. Prospects and challenges of CRISPR/Cas genome editing for the study and control of neglected vector-borne nematode diseases. FEBS J (2016) 283:3204–21. doi: 10.1111/febs.13781
86. Liu C, Grote A, Ghedin E, Unnasch TR. CRISPR-mediated transfection of Brugia malayi. PloS Negl Trop Dis (2020) 14:e0008627. doi: 10.1371/journal.pntd.0008627
87. Kwarteng A, Sylverken A, Asiedu E, Ahuno ST. Genome editing as control tool for filarial infections. Biomed. Pharmacother. (2021) 137:111292. doi: 10.1016/j.biopha.2021.111292
88. Sankaranarayanan G, Berriman M, Rinaldi G. An uneven race: genome editing for parasitic worms. Nat Rev Microbiol (2021) 19:621–1. doi: 10.1038/s41579-021-00625-5
Keywords: Mansonella, filaria, neglected tropical disease, genomics, evolution
Citation: Sinha A, Li Z, Poole CB, Morgan RD, Ettwiller L, Lima NF, Ferreira MU, Fombad FF, Wanji S and Carlow CKS (2023) Genomes of the human filarial parasites Mansonella perstans and Mansonella ozzardi. Front. Trop. Dis 4:1139343. doi: 10.3389/fitd.2023.1139343
Received: 06 January 2023; Accepted: 13 March 2023;
Published: 03 April 2023.
Edited by:
Catherine A Gordon, QIMR Berghofer Medical Research Institute, The University of Queensland, AustraliaReviewed by:
Stephen R Doyle, Wellcome Sanger Institute (WT), United KingdomAlexander Kwarteng, Kwame Nkrumah University of Science and Technology, Ghana
Copyright © 2023 Sinha, Li, Poole, Morgan, Ettwiller, Lima, Ferreira, Fombad, Wanji and Carlow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clotilde K. S. Carlow, Y2FybG93QG5lYi5jb20=