
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Trop. Dis., 13 July 2023
Sec. Major Tropical Diseases
Volume 4 - 2023 | https://doi.org/10.3389/fitd.2023.1094286
This article is part of the Research TopicThe Transition of Novel Genetic Control Strategies into Reality for Vector-Borne Tropical Disease Control and Prevention; Research Advances and next StepsView all 12 articles
 Sana Tamim1
Sana Tamim1 Julius Nwobegahay2,3Armelle Gaelle Fepa Kwesseu4
Julius Nwobegahay2,3Armelle Gaelle Fepa Kwesseu4 Ida Marlene Guiateu Tamo4
Ida Marlene Guiateu Tamo4 Marceline Djuidje Ngounoue4,5*
Marceline Djuidje Ngounoue4,5*Introduction: In humans, RNA viruses are responsible for a wide range of acute, chronic, emerging and re-emerging infections. Human Immunodeficiency virus (HIV) and hepatitis C virus (HCV) rank as some of the most important public health challenges affecting Africa.
Methods: We performed enzyme-linked immune-sorbent assays to confirm positive specimens, and the genomic characterization on two cohorts of people living with HIV in Douala and Yaoundé for the periods 2005-2006 and 2015-2016. These groups were tested for co-infection with HCV using the enzyme-linked immunosorbent assays. Viral RNA was extracted from positive patients’ plasma samples by QIAGEN method, and specific primers were used to amplify the genes of interest on HIV and HCV genomes. The amplification products were subsequently cloned and sequenced. The nucleotide sequences were aligned, genotyped and phylogenetically analyzed.
Results: The HIV isolate identified in this study belongs to HIV-1 group M Subtype A1. The HCV subtypes characterized in this study are 1h and 4t corresponding to the dominant strains that circulate in Cameroon. Phylogenetic analysis of the HCV NS5B gene showed that the study viruses cluster with Gabonese, Canadian, and previously sequenced viruses from Cameroon.
Conclusion and perspectives: These results shed light on the genetic diversity of HIV and HCV in Cameroon. Virulent HCV infections are common in Cameroon, and therefore there is a great need for further analysis of the viral evolutionary and spatio-temporal patterns.
The acquired immune deficiency syndrome (AIDS) pandemic has long captured the attention of the medical world, but despite various strategies it still causes significant morbidity and mortality. There were more than 38.4 million people globally living with human immunodeficiency virus (HIV) in 2021; 1.5 million became newly infected with HIV and 650,000 people died from AIDS-related illnesses in the same year. Furthermore, since the start of the HIV epidemic, 84.2 million people have become infected with HIV, and 40.1 million have died from AIDS-related illnesses. In Western and Central Africa, 5.0 million people were living with HIV in 2021 (1). Unlike HIV/AIDS, viral hepatitis has been understudied in tropical Africa, but, fortunately, disease management has been given greater significance following the endorsement of a new Global Health Sector Strategy on HIV in the 2016–2021 sustainable development goals (SDGs) (2). SDG Target 3.3 aims to end the epidemics of AIDS, tuberculosis, malaria, and neglected tropical diseases, and combat hepatitis, water-borne diseases, and other communicable diseases.
Viral hepatitis infection can cause life-threatening liver disease. Six types of viruses are known to be responsible for hepatitis: A, B, C, Delta, E, and G. Hepatitis B virus (HBV) and hepatitis C virus (HCV) are the most virulent species, with infections among the top 10 causes of death worldwide and therefore can be considered an international concern. HBV and HCV are responsible for most liver cirrhosis and cancers (3). There is currently no vaccine available against HCV. Antiviral medicines can cure approximately 90% of people with HCV, thereby reducing the risk of death from liver cirrhosis and cancer (3–5). The introduction of direct acting antivirals (DAAs), such as daclatasvir (NS5A inhibitor), symeprevir (NS3 inhibitor), and sofosbuvir (NS5B inhibitor), against HCV is revolutionizing HCV treatment and improving the prognosis of HCV infections and also coinfections with other viruses such as HIV. However, the HCV treatment protocol and the duration and the success of treatment with DAAs depend on the stage of the infection (acute or chronic), and more importantly on the HCV genotypes/subtypes (6). Hence, there is a need to always perform HCV genotyping before the initiation of the treatment. The most recent national survey in Cameroon indicates that HCV infections are endemic, with a prevalence of 1.03% in the general population. However, HCV infection prevalence is higher in older populations: 3% between those aged 45–55 years and 7% in people aged above 55 years (7, 8). Additionally, studies show that the HCV epidemic in Cameroon is characterized by significant genetic diversity (9–11).
HCV is a positive-strand RNA virus with a genome of approximately 9.6 kb. It has a single open-reading frame (ORF) that is flanked by 5′ and 3′ untranslated regions (UTRs). The ORF encodes a polyprotein precursor of about 3,010 amino acids that is cleaved by cellular and viral proteases into structural proteins (core, E1 and E2, P7) and non-structural proteins (NS2–5B) (12, 13). HCV isolates are classified into seven major genotypes, each containing several subtypes that differ by about 20% at the amino-acid level (14). Studies conducted in Cameroon have described the dominance of HCV genotypes 1, 2, and 4 (9–11). However, limited data are available for a full-length complete genome analysis of Cameroonian HCV isolates. Coinfection with other viruses is an important hallmark of HCV infection that can contribute to differences in pathogenesis mechanisms and treatment outcomes. For instance, it is possible that the presence of other viruses such as HIV and HBV may exacerbate the pathogenic features of an HCV infection and thereby the clinical and/or therapeutic outcomes. As a matter of fact, it has been observed in a case report study that DAA treatment in patients with an HCV/HBV coinfection may lead to a life-threatening reactivation of an HBV infection (15).
In this study, we performed genomic surveillances and phylogenetic analyses of circulating HIV and HCV mono-infected and coinfected isolates from Cameroonian patients in 2005–2006 and 2015–2016. The evolutionary patterns detected in this analysis are valuable to the monitoring of disease progression and can inform the development of (diagnosis) tools and HCV therapeutics vaccines.
Based on previous reports on HIV and HCV seroprevalence in Cameroon (7, 8), and also a systematic review with meta-analysis (16), we collected representative mono-infected/coinfected samples for the molecular analysis. The positivity of the specimens was confirmed by enzyme-linked immunosorbent assays (ELISAs). From 2005 to 2006, 36 blood specimens from patients who had tested positive for HIV (22 from volunteer blood donors and 14 from newly diagnosed people living with HIV/AIDS not yet on HIV treatment) were collected in Laquintinie Hospital, Douala. From 2015 to 2016, blood samples were collected from 149 HIV-positive patients from Yaoundé Military Hospital and Bastos Clinic, aged 18–64 years. Samples from both cohorts were confirmed as HIV positive by a sandwich ELISA (GENSCREEN PLUS HIV, BioRad, France): GENSCREEN PLUS HIV Ag-Ab is an enzyme immunoassay for the detection of HIV infections; it is based on the principle of the sandwich technique, and thus on the detection of the HIV-1 antigen and the various antibodies associated with HIV-1 and/or HIV-2 virus in human plasma. The solid phase is coated with the following monoclonal antibodies against p24 HIV-1 antigen and purified antigens: gp160 recombinant protein with an artificial functional consensus polypeptide, which is composed of variable sequences of the virus inserted into well-preserved sequences of HIV-1 group O strains and a peptide mimicking the immunodominant epitope of the HIV-2 envelope protein. The GENSCREEN PLUS HIV Ag-Ab conjugates are based on the use of biotinylated polyclonal antibodies to HIV antigens, Avidin, and HIV antigens peroxidase conjugate (gp41) and gp36 mimicking the immunodominant epitopes of the HIV-1 and HIV-2 envelope glycoproteins, and the same functional consensus polypeptide of HIV-1 used for the solid phase. To determine whether these samples were HCV positive they were screened for capsid antigens and antibodies associated with HCV infections using the ORTHO HCV 3.0 ELISA Test System with Enhanced SAVe (Ortho-Clinical Diagnostics Kit, USA). This is a semi-quantitative ELISA for the detection of antigens and antibodies of HCV in human plasma. It uses micro-wells coated with recombinant HCV-encoded antigens as the solid phase. Antigens and antibodies which become bound to the solid phase can be detected by complementary antibodies or antigens, labeled with an enzyme capable of acting on a chromogenic substrate. When an enzyme substrate is applied, the presence of antigens or antibodies can be detected by the development of the colored end-product, the absorbance of which can be read in the spectrophotometer. The assay is a three-stage test carried out in a micro-well coated with a combination of recombinant HCV antigens (c22–3, c200, and NS5).
To perform genotyping and phylogenetic analyses on HIV and HCV genes, viral RNA was isolated from HIV/HCV coinfected or mono-infected patients’ specimens in the above-mentioned reference hospitals located in Douala and Yaoundé, Cameroon. Because HIV and HCV are both RNA viruses, viral RNA was extracted from human plasma and served as basic genetic material for molecular studies. These molecular studies involved RNA isolation, cDNA synthesis, RT-PCR, nested PCR, semi-nested PCR, gene cloning, DNA sequencing, sequence data management, sequence data analysis, and phylogenetic analysis. To avoid co-purification of cellular DNA, a pre-treatment was necessary before the RNA extraction. For this, 200µL of human plasma samples were filtered through a 0.22µm filter (micrometer column) and viral particles were separated from host contaminants using centrifugation and filtration. To remove contaminated nucleic acids by digestion, the DNAse I enzyme was added for an hour at 37°C. Viral RNA was extracted using the QIAamp viral RNA QIAGEN Kit for viral RNA extraction according to the manufacturer’s instructions, in 50-µL concentrations, and used for both HIV and HCV genome amplification. To preserve the extracted viral RNA, specimens were immediately amplified, used for cDNA synthesis, or stored in absolute ethanol for subsequent analyses. The principal genes characterized were HIV Pol-RT and Env-c2v3, and also HCV Core, E2, and NS5B. Regions encompassing these genes were consecutively reverse transcribed and amplified by polymerase chain reaction (RT-PCR) using specific primers. Tables 1–3 display the primers used to amplify the different regions and the various positions on the viral genomes, as previously described (9–11). The viral RNA of HIV was reverse transcribed at 50°C for 50 min followed by 39 cycles of 94°C for 1 min, 54°C for 1 min, and an extension of 72°C for 1.5 min. The HCV RNA was amplified for 35 cycles of denaturation at 93°C for 30 s, annealing at 60°C for 30 s, and an extension at 72°C for 1 min. All PCR products were resolved in 1.5% agarose gel electrophoresis. PCR products were cloned into a TOPO pCR 2.1 vector using the TOPO pCR cloning technology (ThermoFisher). The resulting clones were used to transform chemically competent DH5-α cells, purified, and sequenced. Plasmid DNA obtained was used for DNA sequencing through the Applied Biosystems BigDye Terminator Sanger cycle sequencing as described by Iles et al., 2015 (11). The derived nucleotide sequences (n = 1 HIV sequences; n = 10 HCV sequences) were aligned using several different software programs: SeqMan implemented in DNASTAR in 2008, Genious version 12.2.3 in 2017, and Clustal W implemented in BioEdit. Consensus nucleotide sequences were compared with a database of reference sequences belonging to different genotypes/sub-genotypes available through NCBI BLASTN/homology search, and the genetic relatedness of the consensus sequences was inferred by phylogenetic analysis.
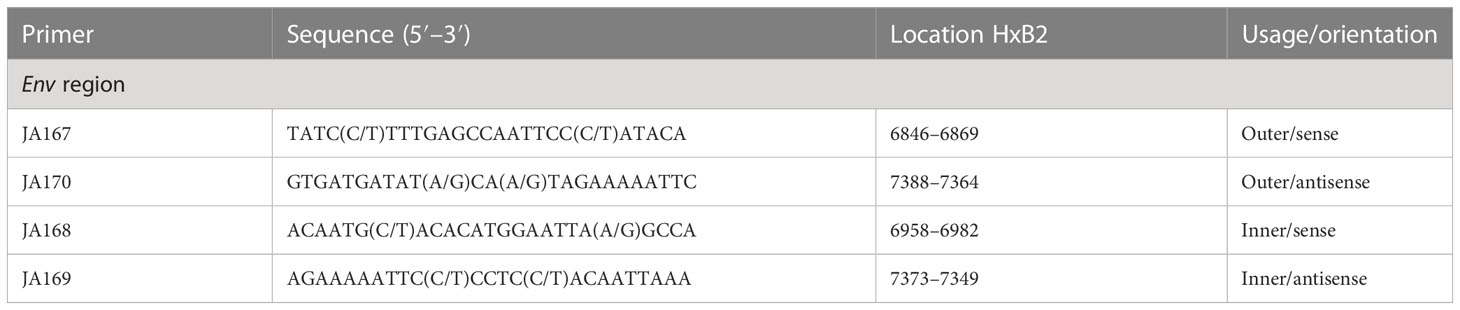
Table 1 Primers for cDNA synthesis and nested polymerase chain reaction (PCR) for the HIV-1 group M Env gene.
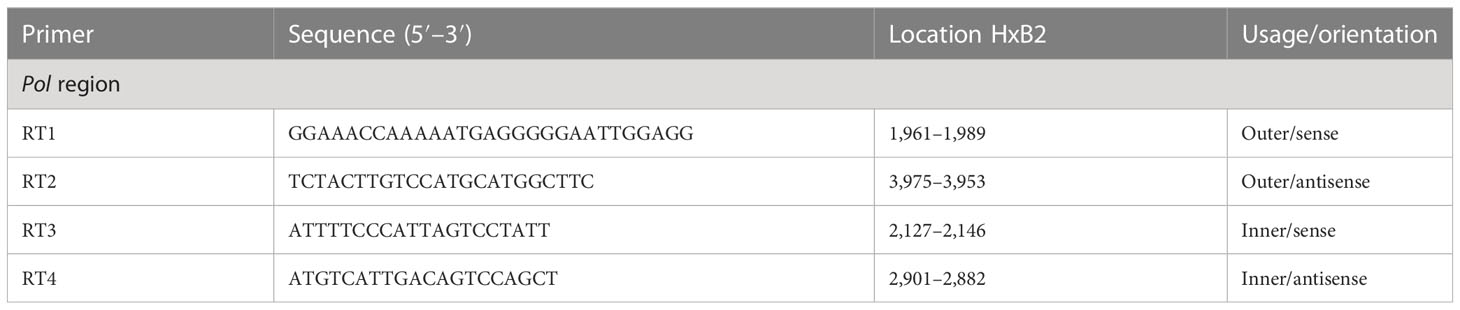
Table 2 Primers for the nested PCR to amplify the HIV Pol-RT gene.
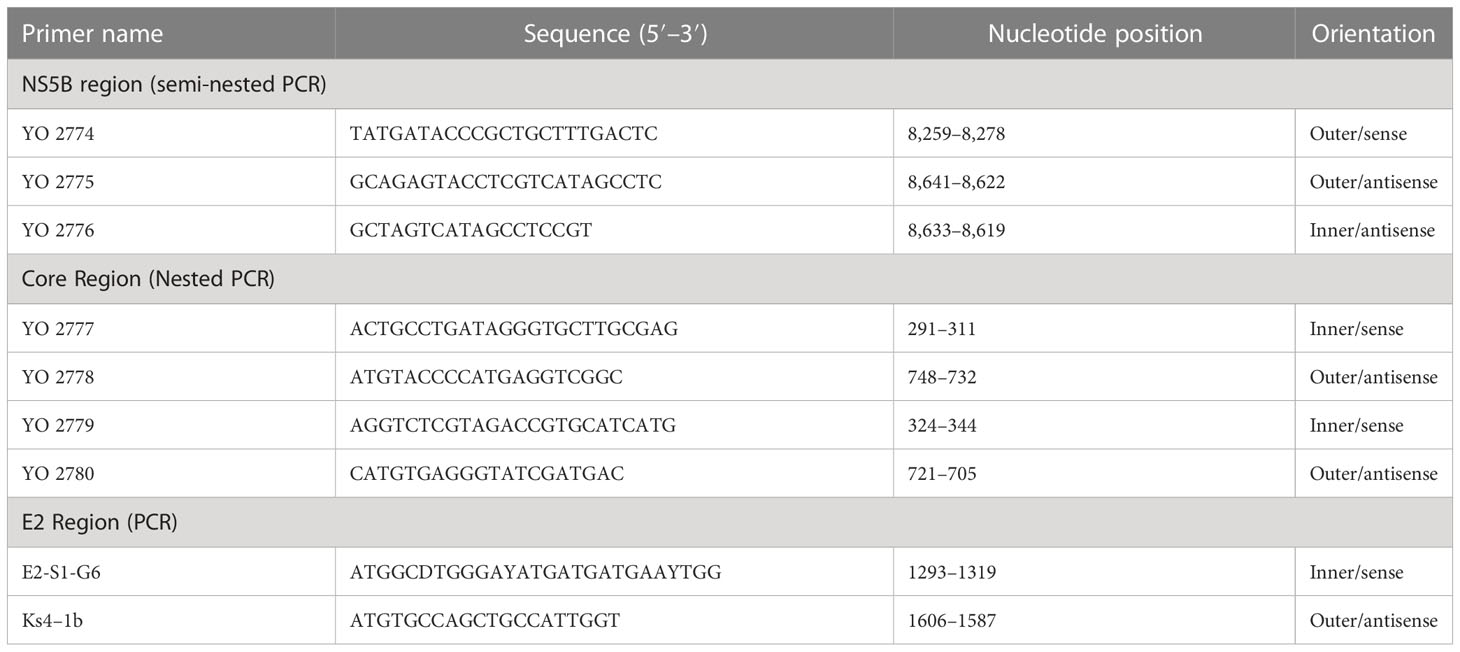
Table 3 Primers for the semi-nested PCR and nested PCR to amplified HCV genes.
In order to determine the evolutionary history of the study HCV and HIV sequences, reference sequences and background datasets were compiled using the NCBI Basic Local Alignment Search Tool (BLAST) database for HCV genes NS5B, E, and HIV pol gene. The first 20 sequences with the lowest e-value against each study sequence were retrieved and aligned with the reference sequences using the –add fragments option of MAFFT online (17). The final alignments were visually checked for artifacts produced during the alignment procedure using AliView (18). All alignments were analyzed for recombination with RDRP 4 software using default settings with the option of linear sequences (19) (https://rdp4.software.informer.com/4.1). Maximum likelihood trees of HCV genes NS5b and E2 were inferred using the web version of IQ-TREE software, with 1,000 ultrafast bootstrap (UFB) replicates generated per partition separately and then concatenated together (20, 21) (iqtree.cibiv.univie.ac.at). For the HIV pol gene, a maximum likelihood tree was built from the web version of IQ-TREE software, with 1,000 replicated of Shimodaira–Hasegawa approximate likelihood ratio test (SH-aLRT) as statistical support, since the length of the sequence was only 3% of the total length of the gene, which was too short for any bootstrap analysis (iqtree.cibiv.univie.ac.at).
Ethical considerations: The present study satisfied the national and international ethical standards: ethical clearance was obtained from the Cameroon National Research Ethics Committee for Human Health, before the study implementation; informed consent was obtained from study participants or through their legal representatives before enrollment and sample collection. The study was conducted in accordance with the CIOMS guidelines, and complied in accordance with the Declaration of Helsinki, 2013. Participants gave their authorization that samples could be transferred from Cameroon to the United States of America for future investigations in the field of genomics and cell biology.
A total of 185 HIV/HCV coinfected and mono-infected samples were analyzed from two study periods (2005–2006 and 2015–2016). We identified 73 samples that were positive for the capsid antigens and antibodies associated with HCV infections using ELISA. From these positive samples, 36 isolates were reverse transcribed and amplified by PCR, among which 17 HCV NS5B genes (47.22%), four HCV E genes (11.11%), and one HIV isolate (2.77%) were successfully sequenced. The alignments with the background dataset and the study sequences were inspected for recombination using RDRP4, with six different methods (RDP, GENECONV, MaxChi, Bootscan, Chimaera, and SiScan) that ran simultaneously against the sequences. The events detected by more than three of the above methods were considered positive. We determined that one sequence each in HCV NS5B and HIV background datasets had signals of recombination. We also identified nine study sequences for the HCV NS5B gene and two sequences for the HCV E gene that had significant genetic divergence. The recombinant and the divergent sequences were removed from the multi-sequence alignment and subsequent analyses.
The characterization of eight HCV NS5B genes showed that the study sequences from Cameroon belonged to genotypes 1 and 4 (Figure 1). Four HCV NS5B sequences belonged to genotype 1 subtype “h”. Sequences from samples labeled Cameroon 5 and Cameroon 6 clustered closely with each other and with the strains from Gabon and Canada (UFB=72), and sequences from samples labeled Cameroon 10 and 16 clustered together with each other and other previously sequenced HCV strains from Cameroon collected in 2016 (UFB=100). The study strains with identifiers Cameroon 1, 2, 12, and 14 belong to genotype 4 subtype “t” and clustered closely with each other and viruses from Canada, Gabon, and other local strains of HCV from Cameroon (UFB ranges from 82 to 100) that circulated during 2006–2014 (Figure 1). The HCV E gene sequences belong to genotype 2 (Figure 2) and cluster phylogenetically separately from each other. The isolate HCV_044E2 clusters with viruses from the United Kingdom (UFB=49), and HCV_036E2 was contiguous to a French isolate (UFB=26).
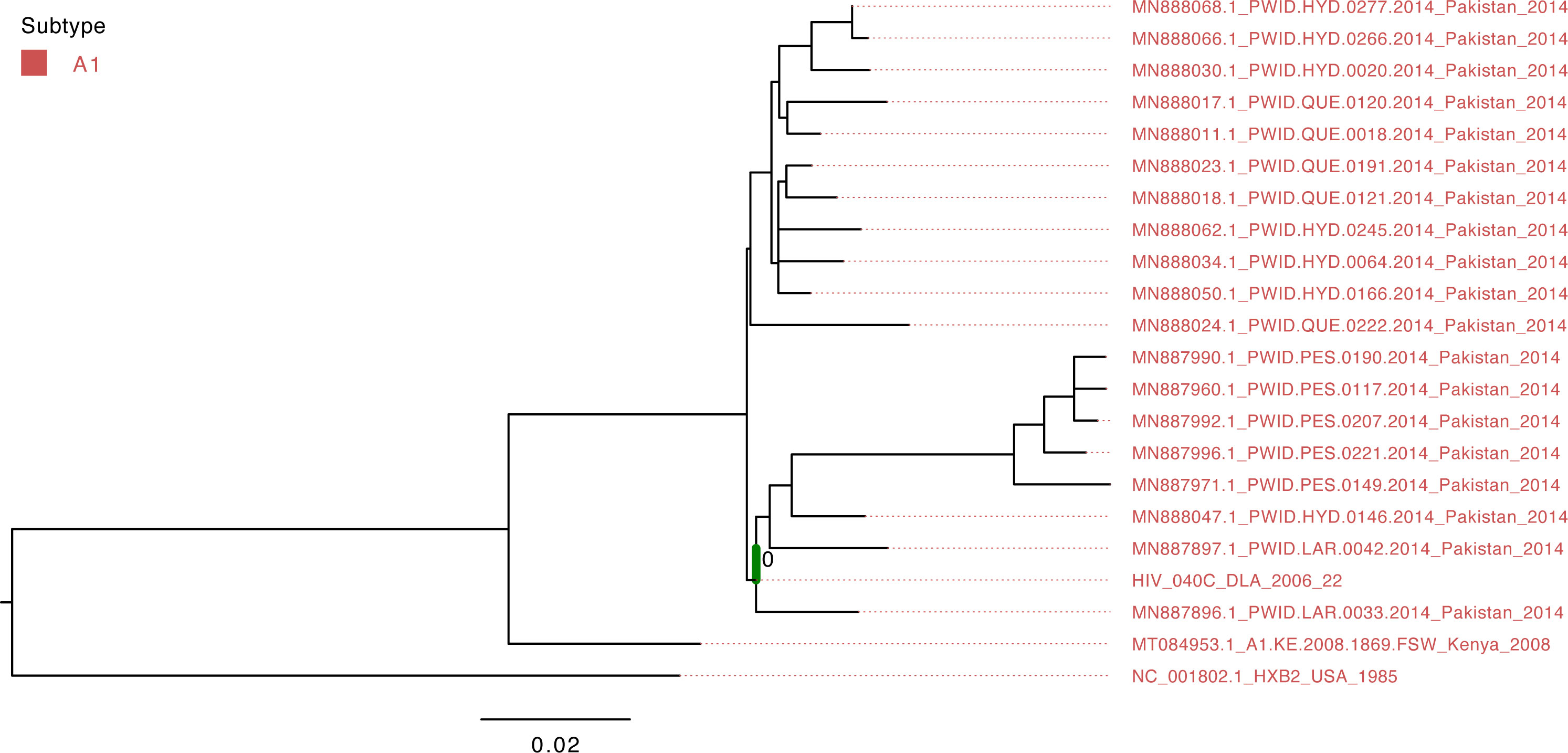
Figure 1 Maximum-likelihood tree of the HIV pol gene. The study sequence is represented by the bold green branch leading to the tip label. The tip colors represent HIV group M Subtype A1. The number at the internal node represents the SH-aLRT support of the ancestor node of the study sequence. The scale represents the nucleotide substitution per site.
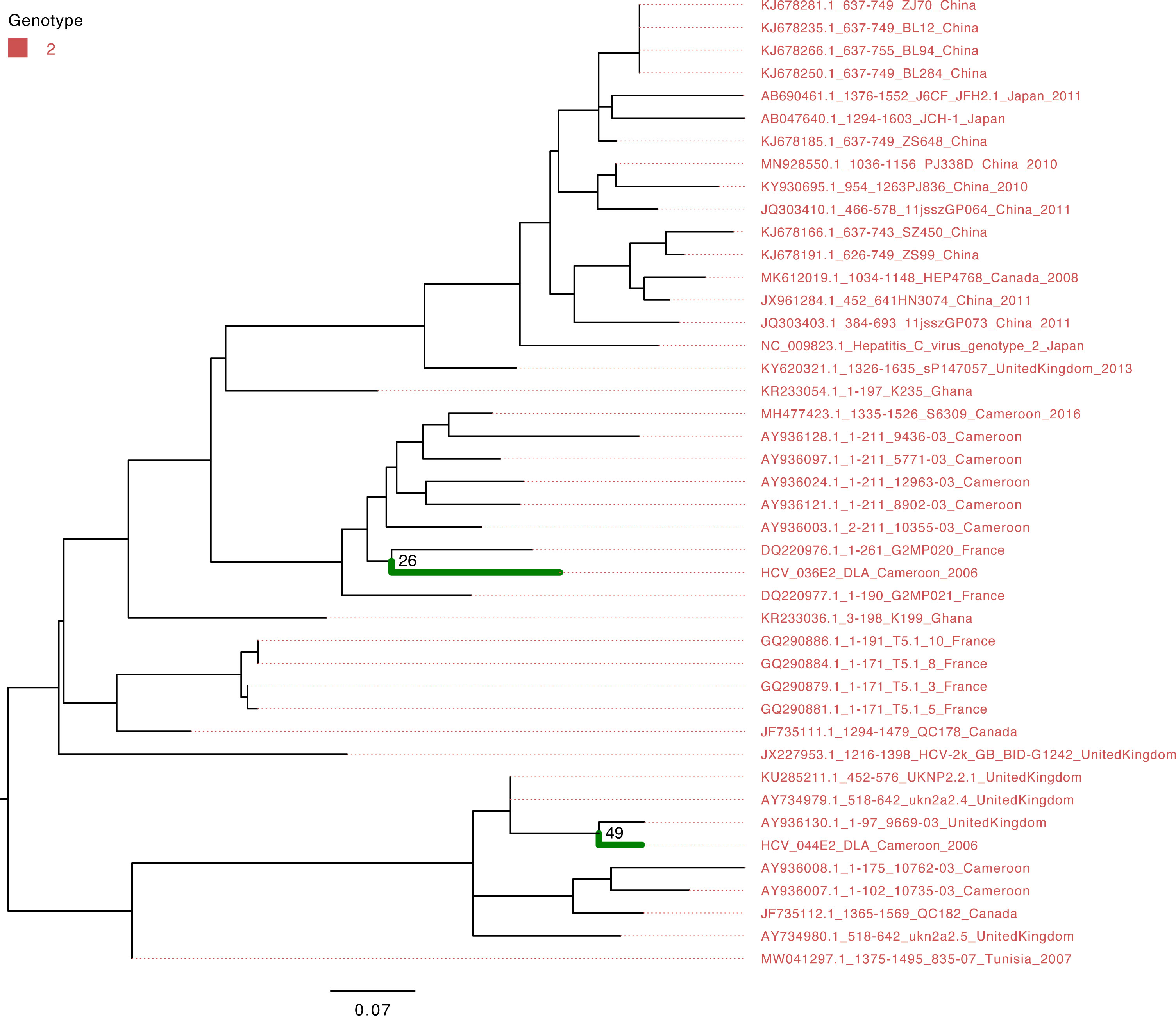
Figure 2 Maximum-likelihood tree of the HCV E gene. The study sequences are represented by the bold green branches leading to their tip labels. The tip colors represent HCV genotype 2. The numbers at the internal nodes represent the bootstrap support of the ancestor nodes of the study sequences. The scale represents the nucleotide substitution per site.
The isolate HIV_040C belonged to group M’s A1 subtype and clustered with viruses that circulated in Pakistan in 2014 (Figure 3); however, the support value is not significant (SH-aLRT=40.7) due to the short sequence length obtained from sequencing.
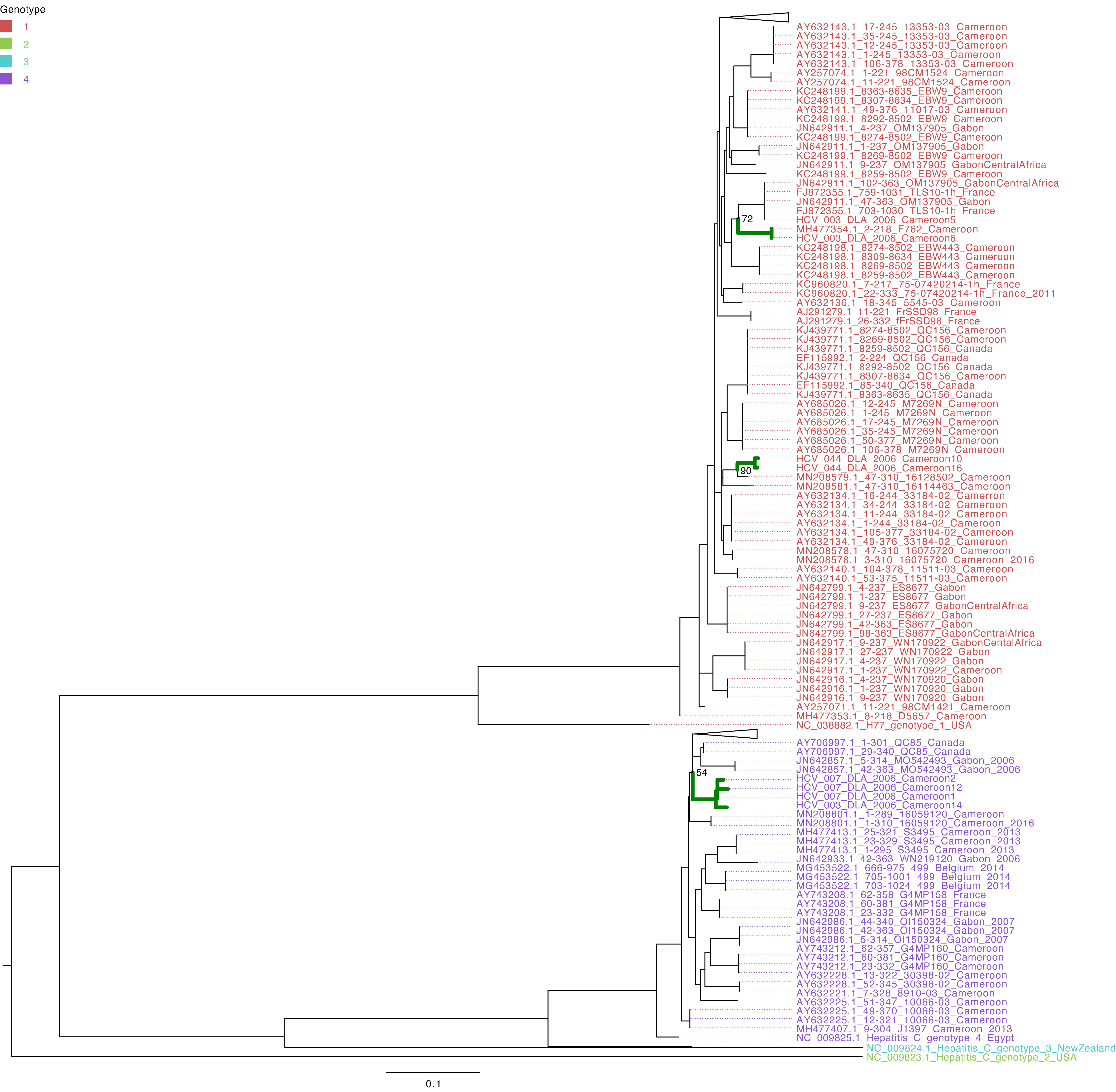
Figure 3 Maximum-likelihood tree of the HCV NS5B gene. The study sequences are represented by the bold green branches leading to their tip labels. The tip colors represent HCV genotypes 1, 2, 3, and 4. The numbers at the internal nodes represent the bootstrap support of the ancestor nodes of the study sequences. The scale represents the nucleotide substitution per site. Triangles in the phylogeny represent collapsed branches leading to sequences that are not relevant to the study dataset.
An age-stratified analysis shows that HCV-infected and HIV/HCV-coinfected individuals are, on average, older (35% patients above 55 years old) than HIV mono-infected (51% patients between 20 and 39 years old) (p-value <0.05).
The present findings corroborate and extend previous investigations on circulating HIV and HCV diversity in Africa. The results obtained from the present study show that the HIV isolate is from HIV-1 group M Subtype A1. Subtype A1 is among the most prevalent subtypes in Cameroon, after the circulating recombinant form CRF02_AG [Geography Search Interface (lanl.gov)].
The majority of studies with African isolates display the circulation of HCV genotypes 1, 4, and 5; thus, these strains most commonly occur in Africa (22–25). With the core and NS5B gene sequences, HCV genotypes 1 and 4 were identified in the present study, and our findings reveal that these strains belong to subtypes or sub-genotypes 1h and 4t, respectively. There are reports that fewer people with genotype 1 infections experience viral clearance with treatment than people with genotype 2 infections (6), whereas the same treatment protocol is recommended in people with genotypes 1 and 4. Genotypes 2 and 3 also circulate in Cameroon (9, 26), and the HCV E gene sequences in the present study were shown as belonging to genotype 2. This information was derived from analysis of the NS5B gene, as performed in other studies for genotyping and genetic diversity investigations (10, 11, 27). Furthermore, the HCV NS5B gene seems to be suitable for diagnostics, as amplification was efficient in more than 60% of the HIV/HCV coinfected isolates. In fact, the genetic heterogeneity in the HCV genome has been used to identify phylogenetically distinct genotypes HCV-1 to HCV-6 (28, 29) and many subtypes (30). A study conducted in the bordering country of Gabon also showed a high prevalence of HCV genotype 4, which might suggest some extent of transmission events between the countries. Since infection due to the mentioned genotypes is also known to be virulent (6), this might explain the great challenge of chronic hepatitis progression in the Central Africa sub-region. The phylogeny of HCV E2 presents long branches leading to E2 isolates, suggesting that additional data would be valuable for more accurate inferences of the evolutionary and transmission dynamics of HCV in Cameroon. Furthermore, we noticed that, regardless of whether the samples were collected in Douala or in Yaoundé within the various periods, there is no difference in terms of circulating HCV genotypes in Cameroon. The same genotypes 1 and 4 are identified as predominant even with NCBI BLAST. Because these strains are the most aggressive ones and are less likely thangenotypes 2 and 3 to respond to interferon treatment, WHO updated itsevidence-based recommendations for the use of DAAs, and this should beimplemented in these settings and the consequences of this evaluated.
Investigation also revealed that HIV/HCV-coinfected individuals are older on average than HIV mono-infected individuals, with an unusually high HCV prevalence (24.82%), and the majority are aged over 55 years. This corroborates the findings of a study in Cameroon from the late 1950s, which indicates that HCV is associated with age and access to medical care for patients aged over 50 years (7).
These results shed light on the genetic diversity of HIV and HCV in Cameroon. Virulent HCV infections are common in Cameroon, and therefore there is a need for further analysis of the viral strains driving these dynamics, as well as monitoring the disease progression in the sub-region. These findings are crucial for WHO policy and practice, as well as Cameroon’s Ministry of Health’s Strategic Plan. Comprehensive representation of the HCV and HIV diversity circulating in Africa can help inform the pre-clinical evaluation of vaccine candidates and therapeutics.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Cameroon National Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MN, JN, and ST significantly contributed to the methodology, results, and discussion implementation and writing. AK and IT contributed to the data collection. MN designed the study, supervised all steps of its implementation, and proposed the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Parts of the study were conducted at the J. Craig Venter Institute (Viral genomics Laboratory), U.S.A. with the grant offered by the National Institute of Allergy and Infectious Diseases (NIAID/NIH), and at Yale University through the Fulbright program that offered the opportunity for research scholarship.
The authors are grateful for the support of the J. Craig Venter Institute (Viral genomics Laboratory), USA, and the grant offered to us by National Institute of Allergy and Infectious Diseases (NIAID). We appreciate the help of the Laboratory of Hematology and Virology of the University of Yaoundé I, the staff of the Laquintinie Hospital, Douala, Cameroon, and all patients who participated in this study. The authors thank the head of Military Hospital in Cameroon for providing the participants, and CRESAR for the supervision of the seroprevalence study and transfer of the samples from Cameroon to the United States of America. The authors are grateful for the Fulbright program and Yale University, which offered the opportunity to transfer biological samples from Cameroon to the USA, under a duly executed material transfer agreement (MTA)/data sharing agreement (DSA) between the University of Yaoundé I and Yale University, as well as the CDC permit. We also would like to thank Dr. Nidia S. Trovao and Dr. David J. Spiro from the Fogarty International Center, National Institutes of Health, on their advice on genomic epidemiology and phylogenetics. The opinions expressed in this article are those of the authors and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government. Authors are also grateful of the support for research granted by the Cameroon Ministry of Higher Education to University Lecturers at the trimester basis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2023.1094286/full#supplementary-material
2. World Health Organization. Global health sector strategy on HIV: 2016-2021 (2016). Available at: https://www.who.int/publications/i/item/WHO-HIV-2016.05.
3. Keikha M, Eslami M, Yousefi B, Ali-Hassanzadeh M, Kamali A, Yousefi M, et al. HCV genotypes and their determinative role in hepatitis C treatment. Virus Dis (2020) 31(3):235–40. doi: 10.1007/s13337-020-00592-0
4. Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. American association for study of liver diseases. an update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American association for the study of liver diseases. Hepatology (2011) 54(4):1433–44. doi: 10.1002/hep.24641
5. Kumar A, Rajput MK, Paliwal D, Yadav A, Chhabra R, Singh S. Genotyping & diagnostic methods for hepatitis c virus: a need of low-resource countries. Indian J Med Res (2018) 147(5):445–55. doi: 10.4103/ijmr.IJMR_1850_16
6. Polish Group of Experts for HCV, Halota W, Flisiak R, Juszczyk J, Małkowski P, Pawłowska M, et al. Recommendations for the treatment of hepatitis C in 2017. Clin Exp Hepatol (2017) 3(2):47–55. doi: 10.5114/ceh.2017.67782
7. Njouom R, Siffert I, Texier G, Lachenal G, Tejiokem MC, Pépin J, et al. The burden of hepatitis C virus in Cameroon: spatial epidemiology and historical perspective. J Viral Hepat (2018) 25(8):959–68. doi: 10.1111/jvh.12894
8. Agbor VN, Tagny CT, Kenmegne JB, Awazi B, Ngansop C, Mbanya D, et al. Prevalence of anti-hepatitis C antibodies and its co-infection with HIV in rural Cameroon. BMC Res Notes (2018) 11:459. doi: 10.1186/s13104-018-3566-4
9. Ndjomou J, Pybus OG, Matz B. Phylogenetic analysis of hepatitis c virus isolates indicates a unique pattern of endemic infection in Cameroon. J Gen Virol (2003) 84:2333–41. doi: 10.1099/vir.0.19240-0
10. Njouom R, Pasquier C, Ayouba A, Tedjiokem MC, Vessiere A, Mfoupouendoun J, et al. Low risk of mother-to-child transmission of hepatitis C virus in Yaoundé, Cameroon: the ANRS 1262 study. Am J Trop Med Hyg (2005) 73:460–6. doi: 10.4269/ajtmh.2005.73.460
11. Iles JC, Njouom R, Foupouapouognigni Y, Bonsall D, Bowden R, Trebes A, et al. Characterization of hepatitis C virus recombination in Cameroon by use of nonspecific next-generation sequencing. J Clin Microbiol (2015) 53(10):3155–64. doi: 10.1128/JCM.00483-15
12. Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol (2000) 81(Pt 7):1631–48. doi: 10.1099/0022-1317-81-7-1631
13. Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol (2000) 242:55–84. doi: 10.1007/978-3-642-59605-6_4
14. Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology (2014) 59(1):318–27. doi: 10.1002/hep.26744
15. Ende AR, Kim NH, Yeh MM, Harper J, Landis CS. Fulminant hepatitis B reactivation leading to liver transplantation in a patient with chronic hepatitis C treated with simeprevir and sofosbuvir: a case report. J Med Case Rep (2015) 9:164. doi: 10.1186/s13256-015-0630-8
16. Bigna JJ, Amougou MA, Asangbeh SL, Kenne AM, Nansseu JR. Seroprevalence of hepatitis C virus infection in Cameroon: a systematic review and meta-analysis. BMJ Open (2017) 7:e015748. doi: 10.1136/bmjopen-2016-015748
17. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform (2019) 20(4):1160–6. doi: 10.1093/bib/bbx108
18. Larsson A. AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics (2014) 30(22):3276–8. doi: 10.1093/bioinformatics/btu531
19. Martin DP, Murrell B, Golden M, Khoosal A, Muhire BM. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol (2015) 1(1):vev003. doi: 10.1093/ve/vev003
20. Nei M, Xu P, Glazko G. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc Natl Acad Sci USA (2001) 98(5):2497–502. doi: 10.1073/pnas.051611498
21. Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. (2016) 44(W1):W232–5. doi: 10.1093/nar/gkw256
22. Karoney MJ, Siika AM. Hepatitis C virus (HCV) infection in Africa: a review. Pan Afr Med J (2013) 14:44. doi: 10.11604/pamj.2013.14.44.2199
23. Ali-Mahamat M, Njouom R. High rate of infection with hepatitis C virus genotype 4 in Chad, Central Africa. Indian J Med Microbiol (2015) 33:608–09. doi: 10.4103/0255-0857.167343
24. Njouom R, Frost E, Deslandes S, Mamadou-Yaya F, Labbe AC, Pouillot R, et al. Predominance of hepatitis C virus genotype 4 infection and rapid transmission between 1935 and 1965 in the Central African Republic. J Gen Virol (2009) 90:2452–56. doi: 10.1099/vir.0.011981-0
25. Prabdial-Sing N, Chirwa T, Thaver J, Smuts H, Vermeulen M, Suchard M, et al. Hepatitis C genotype distribution in patient and blood donor samples in South Africa for the period 2008–2012. J Viral Hepat (2016) 23:881–88. doi: 10.1111/jvh.12571
26. Ndjomou J, Kupfer B, Kochan B, Zekeng L, Kaptue L, Matz B. Hepatitis C virus infection and genotypes among human immunodeficiency virus high-risk groups in Cameroon. J Med Virol (2002) 66(2):179–86. doi: 10.1002/jmv.2128
27. Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatol (2005) 42(4):962–73. doi: 10.1002/hep.20819
28. Robertson DL, Hahn BH, Sharp PM. Recombination in AIDS viruses. J Mol Evol (1995) 40(3):249–59. doi: 10.1007/BF00163230
29. Li C, Lu L, Wu X, Wang C, Bennett P, Lu T, et al. Complete genomic sequences for hepatitis C virus subtypes 4b, 4c, 4d, 4g, 4k, 4l, 4m, 4n, 4o, 4p, 4q, 4r and 4t. J Gen Virol (2009) 90(Pt 8):1820–6. doi: 10.1099/vir.0.010330-0
Keywords: HIV, HCV, coinfection, genotypes, subtypes, phylogenetic analyses, Cameroon
Citation: Tamim S, Nwobegahay J, Fepa Kwesseu AG, Guiateu Tamo IM and Djuidje Ngounoue M (2023) Genomic analysis of circulating HIV and hepatitis C virus infections and coinfections in Cameroon: 2005–2006 and 2015–2016. Front. Trop. Dis 4:1094286. doi: 10.3389/fitd.2023.1094286
Received: 09 November 2022; Accepted: 12 June 2023;
Published: 13 July 2023.
Edited by:
Patricia Takako Endo, Universidade de Pernambuco, BrazilReviewed by:
Karina Salvatierra, Universidad Nacional de Misiones, ArgentinaCopyright © 2023 Tamim, Nwobegahay, Fepa Kwesseu, Guiateu Tamo and Djuidje Ngounoue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marceline Djuidje Ngounoue, bWFyY2VsbGluZS5kanVpZGplQGZhY3NjaWVuY2VzLXV5MS5jbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.