
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Trop. Dis., 12 July 2021
Sec. Antimicrobial Resistance
Volume 2 - 2021 | https://doi.org/10.3389/fitd.2021.691604
This article is part of the Research TopicAntimicrobial Resistance in Developing Countries: Impact, Interventions and ImplicationsView all 5 articles
 Rituparna De1,2*
Rituparna De1,2*Vibrio cholerae (VC) is the causative agent of the severe dehydrating diarrheal disease cholera. The primary treatment for cholera is oral rehydration therapy (ORT). However, in case of moderate to severe dehydration, antibiotics are administered to reduce morbidity. Due to the emergence of multidrug resistant (MDR) strains of VC routinely used antibiotics fail to be effective in cholera patients. Antimicrobial resistance (AMR) is encoded in the genome of bacteria and is usually acquired from other organisms cohabiting in the environment or in the gut with which it interacts in the gut or environmental niche. The antimicrobial resistance genes (ARGs) are usually borne on mobile genetic elements (MGEs) like plasmids, transposons, integrons and SXT constin. Horizontal gene transfer (HGT) helps in the exchange of ARGs among bacteria leading to dissemination of AMR. In VC the acquisition and loss of AMR to many antibiotics have been found to be a dynamic process. This review describes the different AMR determinants and mechanisms of resistance that have been discovered in VC. These ARGs borne usually on MGEs have been recovered from isolates associated with past and present epidemics worldwide. These are responsible for resistance of VC to common antibiotics and are periodically lost and gained contributing to its genetic evolution. These resistance markers can be routinely used for AMR surveillance in VC. The review also presents a precise perspective on the importance of the gut microbiome in the emergence of MDR VC and concludes that the gut microbiome is a potential source of molecular markers and networks which can be manipulated for the interception of AMR in the future.
Vibrio cholerae (VC) is the causative agent of the severe diarrheal illness, cholera. Among the 209 different serogroups of VC O1 and O139 have the potential to cause epidemic cholera. O1 has two biotypes, namely, the classical and the El Tor. The El Tor biotype is responsible for the ongoing seventh pandemic which started in the year 1961. Due to cholera 21000 to 143000 deaths occur annually (1, 2). Cholera is widespread among populations lacking access to clean water and sanitation (2). Morbidity in cholera patients can be improved with fluid replacement (2). Oral rehydration therapy using polymer-based or glucose-based rehydration solutions are complemented with antibiotic therapy to decrease morbidity and mortality (2). Antibiotics help in reduction of severity of symptoms, duration of illness and volume of stool (3). However, antibiotic resistance in bacteria leading to the emergence of multi-drug resistant (MDR) and extensively drug resistant (XDR) strains has challenged the effectiveness of antibiotic therapy including combination therapy (4). The latest weekly epidemiological report of cholera published by the World Health Organization has declared that the number of cholera cases have actually amplified in 2019 compared to that in 2018, stating the trend as retrogressive in cholera control (5). In this scenario, antibiotic therapy would serve as an essential component of treatment for cholera patients and therefore it evokes the requirement for alleviation of AMR in cholera for empirical drugs to continue to be effective while the search for newer drugs is ongoing. Therefore, controlling the spread of AMR in cholera is essential and for this step understanding the reservoirs, mechanisms and routes of AMR dissemination is the need of the hour.
Antimicrobial resistance (AMR), the phenomenon wherein, bacteria develop resistance to one or more antibiotics is a severe crisis worldwide and more so in countries like India which has a huge impoverished and uneducated population (6, 7). The AMR crisis in such countries has attained appalling heights due to their misuse and over-use. These are easily available over-the-counter without the requirement of a prescription of a registered physician. Due to the exorbitant use of antibiotics in agricultural, aquacultural, animal and poultry farms antibiotics have entered the food-chain leading to farm-to-fork transmission (8, 9). Moreover, failure of disposal of sewage and effluents contaminated with antibiotics flouting the correct methods of disposal have added to the nuisance and led to the accumulation of antibiotics in the environment (10). Antibiotics are also found naturally in the environment due to the presence of natural antibiotic producers (11). All these factors necessitated bacteria in different ecosystems to acquire genes that encode resistance to antibiotics and hence called antibiotic resistance genes (ARGs) for survival against antibiotic induced stress (10, 12). The threat of AMR is growing at an alarming rate and it is estimated that by 2050 there would occur 10 million deaths each year due to antibiotic-resistant pathogens (13).
Common bacterial pathogens have developed resistance to antibiotics including broad-spectrum ones belonging to all the major classes of antibiotics which have been empirically used for their treatment (12). The problem of AMR in VC is growing at a threatening pace (14). It is a common food-borne pathogen and the most common water-borne pathogen indicating its high chances of infecting the human population (14). Its resistance profile has been cautionary and calls for immediate action to revert the AMR crisis in VC (14, 15).
AMR in VC was not common till the early 1970s when the first reports on resistance in VC started emerging (16). Since the mid-20th century (1960’s) tetracycline and azithromycin have been the drugs of choice. These were superior to others like chloramphenicol and streptomycin resulting in pronounced improvement in morbidity of cholera victims, in both children and adults (17). Trimethoprim was effective against both classical and El Tor biotypes (18). Sulfamethoxazole was effective against El Tor and a combination of the two drugs proved synergistically effective against the classical and El Tor (18). VC gradually started developing resistance against these drugs (19). In December 1979, a strain of O1 VC was isolated from a cholera patient at the Matlab Hospital in Bangladesh which was found to be resistant to tetracycline, ampicillin, kanamycin, streptomycin, and trimethoprim-sulfamethoxazole (19). Strains collected 6 months prior to this also were found to be resistant to these drugs (19). Contemporary reports on resistance of VC against chloramphenicol, furazolidone, and sulphonamides also existed (20).
Thus, evidence of AMR in VC has been documented from the 1970s (16) and many of the AMR determinants were found to be carried on plasmids indicating the role that mobile genetic elements (MGEs) played towards introduction of AMR in VC (16, 19–21).
The epidemic causing O139 serogroup on its emergence in 1992 in India, was found to be resistant to major antibiotics like ampicillin, chloramphenicol and co-trimoxazole and also revealed significant difference in its antibiotic sensitivity pattern from that of O1 (22). Although the problem of AMR has been prevalent since the 1970s the crisis has aggravated over the years and is critical at present (15, 23). At present, 100% clinical VC isolates are resistant to nalidixic acid and sulfamethoxazole-trimethoprim (24). In clinical and environmental isolates ARGs which have never been detected in VC previously thereby conferring resistance to antibiotics which worked successfully in its treatment earlier have started appearing, like, the blaNDM-1 carbapenemase encoding gene isolated in environmental and clinical strains of VC (25, 26). Above and over that, novel MGEs have started being frequently isolated and the genetic analysis of these novel ARGs (25, 26) and MGEs (27, 28) and the frequency of their isolation indicate frequent genetic exchange of VC with other bacterial species (29).
Some common drugs which have been historically successfully used worldwide against VC are: ampicillin, azithromycin, chloramphenicol, ciprofloxacin, ceftriaxone, doxycycline, erythromycin, gentamycin, neomycin, nalidixic acid, norfloxacin, ofloxacin, streptomycin, cotrimoxazole, tetracycline, meropenam, spiromycin, aztreonem singly or in combination (30). Table 1 outlines the salient features of the major classes of antibiotics used against VC.
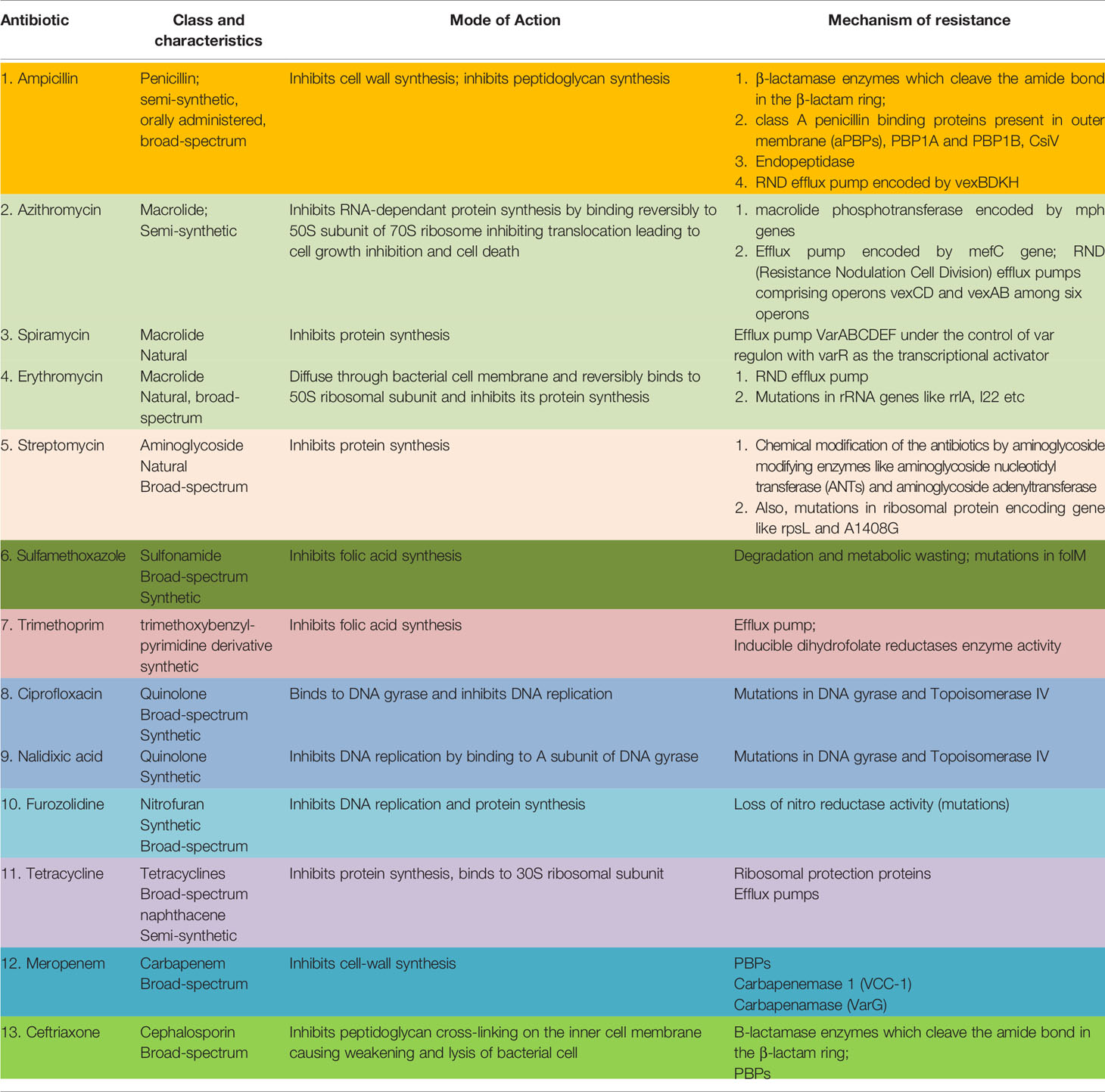
Table 1 Common antibiotics used against Vibrio cholerae, their mode of action and resistance mechanism
Due to the emergence of drug resistance to single (31) and multiple antibiotics (22) different patterns of sensitivity to these drugs have been reported from time to time revealing temporal variation. A classic example is that of Polymixin B resistance. The classical biotype has been sensitive to Polymixin B but the El Tor biotype was resistant (32, 33). This difference in the resistance pattern to Polymixin B served as an important phenotypic marker of demarcation between the two biotypes (32). Polymixin B and Polymixin E (colistin) have been considered to be the last resort drugs for the treatment of Gram-negative bacteria (34). Therefore, with the emergence of El Tor VC crept in the problem of Polymixin B resistance (32). Later, hybrid VC O1 strains like the Matlab I, II and III exhibited mixed sensitivity to polymixin B (35). Recently, O1 El Tor strains from different parts of India were found to be sensitive to Polymixin B (36). 260 clinical strains of VC O1 from 12 states in India tested for Polymixin B susceptibility using Kirby-Bauer disc diffusion method and Polymixin B plate susceptibility assay revealed 88.85% strains were sensitive (36). Another study conducted using 1200 VC O1 isolates collected from 1995-2019 revealed that sensitivity of VC O1 El Tor strains arose in 2005 (37). Additionally, on comparison of recent reports on the antibiotic sensitivity profile of VC strains (24) with that of strains isolated a decade ago (38) it was found that a stark difference in the overall antimicrobial resistance profile exists. These findings reveal that AMR in VC is a highly dynamic phenomenon and is prone to variation and evolution with time due to different selective pressure of antibiotics (39). Apart from mutations in genes involved in resistance mechanisms (40) the reason for temporal variation in the resistance pattern observed in the case of many antibiotics can be accredited to the role of the MGEs (23, 41). Most of the ARGs in VC are acquired from other organisms and are found on MGEs and are intermittently lost and gained (23). The microbiome plays a potential role in the dissemination of ARGs and in shaping the genome and hence the AMR profile of pathogens (24, 39). Most of the AMR determinants discovered in VC from time to time have been found to originate in taxonomically related and unrelated organisms (23, 28, 42). The spread of ARGs is facilitated in polymicrobial ecosystems like the gut and environment where the different inhabitants of the ecosystem are exposed to an interactive milieu conducive for genetic exchange of ARGs (24, 43). Genetic exchange occurs by horizontal gene transfer (HGT) and the major vehicles of HGT are the MGEs like plasmids, transposons, integrons and SXT constin.
AMR in VC is determined by traditional phenotypic methods like disk diffusion method on agar plates or by broth micro dilution methods (24). Alternatively, molecular methods like polymerase chain reaction (PCR) have been widely applied for rapid detection of AMR in VC (24). With the advent of next-generation sequencing (NGS), whole genome sequencing (WGS) is currently used in many molecular epidemiology laboratories (44). It is a non-targeted approach and provides information on the entire genomic composition of the isolates (23, 44). These methods are beneficial for epidemiological surveillance (44). The recent years have seen more refinement in metagenomic sequencing and analysis pipelines (45). This has yielded more precise information regarding abundance and taxonomic diversity of ARGs and has helped in identification of resistance determinants existing in extremely low concentrations below the detection level of the method used or detection of minority populations harboring resistance determinants (45). These developments taking place at a rapid pace have helped to uncover the enormous diversity of resistance genes and their sources (45). Metagenomic analysis of clinical and environmental samples has provided real-time information about the composition and genetic repertoire of different microbial communities (43). This has made resistome analysis conceivable and has generated immense information about the reservoirs of ARGs that facilitate dissemination into other vulnerable organisms like VC (43, 46). This information has made epidemiological tracking of AMR reservoirs easy and effective for containment measures (15). These analyses help us to foresee what kind of AMR trend may be encountered in the near future (24). This is significant for defining treatment modules for cholera patients. In addition, the innumerable molecular markers which are present in the microbiome and those components playing a significant role in the acquisition and expression of AMR can serve as potential agents for manipulation to be used to alleviate the AMR crisis in the future (47). These may include the plethora of bacteriocins and other antimicrobial secondary metabolites found in surplus in the gut microbiome and which have the potential to be developed into novel antimicrobial agents (43, 48). In addition, natural antagonism among bacterial communities can be used for the development of techniques and probiotics which will not only help in reducing diarrheal morbidity but also help to circumvent the problem posed by drug-resistant bacteria (47, 49). The review provides a precise insight into the different antibiotic resistance determinants found in MGEs and resistance mechanisms that have been reported in VC from time to time as a result of antimicrobial resistance surveillance and molecular typing. These resistance determinants have contributed to the evolution of its AMR (24). It also highlights the prominent role that gut and environmental microbiome have played in the evolution of the acquired AMR of VC and upholds the mechanisms at play within these communities which enable the transmission of these MGES and ARGs residing in them into VC leading to its evolution for its enhanced survival against the selective forces of nature (15, 46). The review concludes that by manipulation of the microbiome intervention strategies for interception of AMR can be attained (44, 47). Therefore, the microbiome is not only the source of AMR but also a potential tool for the containment of the AMR crisis (15, 23, 44, 47).
Resistance to antibiotics in VC arises by the following mechanisms: (i) Efflux proteins/ pumps (EPs) in the cell membrane. Many of the efflux pumps have additional functions apart from antibiotic and heavy metal efflux. These include virulence gene expression, adaptation and stress response (50). (ii) Point mutations or multiple mutations in genes which form the basis for antibiotic action (iii) Introduction of new antibiotic resistance gene cassettes. (iv) Many antibiotics themselves serve as inducers for activating SOS response in many bacteria and in turn undergo degradation (51). (v) The outer membrane and outer membrane proteins of VC help it to survive against high molecular weight antibiotics like vancomycin which fail to penetrate the outer membrane barrier (52). (vi) Enzymatic degradation of antibiotics. (vii) Target modification by enzymatic action.
Bacterial efflux systems have been classified into 5 families based on sequence similarity (53). These require an energy source for pumping the drug out of the bacterial cell and may be present in the plasma membrane or cytosol (53). These are MATE (multidrug and toxic compound extrusion), MFS (major facilitator superfamily), RND (resistance-nodulation-cell division), SMR (small multidrug resistance) and ABC (ATP-binding cassette) and have been reported in VC (53). RND efflux pumps have broad substrate specificity and have been shown to be involved in resistance against a host of antimicrobial agents like bile acids, antimicrobial peptides, and antibiotics in VC (54). There are 6 MATE family pumps in VC (55). These are NorM, VcrM, VcmA, VcmB, VcmD, VcmH and VcmN . Efflux activities of NorM, VcrM, VcmA, VcmB, VcmD, VcmH are Na+-dependent, but that of VcmN has been found to be Na+-independent (56). The putative multidrug efflux pump, EmrD-3, belonging to the MFS efflux pump was identified in the classical VC O395 (57) It was found to be involved in the efflux of ethidium (57). It was tested for activity against several antimicrobials and was found to enhance resistance against linezolid, rifampicin, tetraphenylphosphonium chloride, erythromycin, minocycline, trimethoprim, chloramphenicol, ethidium bromide and rhodamine (57).
One of the most important mechanisms of antimicrobial resistance in Gram-negative bacteria arises due to hydrolysis of β-lactam amide by β–lactamases (58). Different β–lactamases have been identified in VC encoding resistance to different β–lactam antibiotics like ampicillin, penicillin, carbenicillin, cephalosporin (30). β–lactamase SAR-1 was isolated from VC O1 isolates from Tanzania (59). The enzyme encoded resistance to carbenicillin and penicillin G (59). Carbapenemase and ESBL producing VC isolates have been reported worldwide (30). A recent isolate of VC from India was found to carry blaNDM-1 and blaDHA-1 genes (26). VC isolated from environmental water samples in New Delhi was found to carry blaNDM-1 gene (25).
Enzymatic modification of the drug target is another mechanism of antimicrobial resistance in VC. Polymixins are a class of detergent-like antimicrobial peptides which are the last line of defense in Gram-negative pathogens. The LPS of the outer membrane is the target of action for polymyxins. Polymyxin selectively binds to LPS. An electrostatic interaction occurs between the α,γ-diaminobutyric acid (Dab) residue of the positively charged polymyxin on one side and the phosphate groups of the negatively charged lipid A membrane on the other side. Consequently, divalent cations (Ca2+ and Mg2+) are displaced from the negatively charged phosphate groups of membrane lipids and LPS is therefore destabilized. This increases the permeability of the bacterial membrane, leading to leakage of the cytoplasmic content and ultimately causing cell death (60). Henderson et al. presented a Lipid A glycylation pathway in VC and mediated by the AlmEFG operon involved in elicitation of AMR due to enzymatic modification of the drug target (61).
Quinolone resistance due to point mutations in chromosome is an instance where chromosomal mutations lead to drug resistance (62). Quinolones are broad-spectrum antibiotics (62). These have been one of the most widely used antibiotics for clinical and veterinary practices and animal husbandry (62). They are naturally synthesized as secondary metabolites in many bacteria like the 2-alkyl-4(1H)-quinolones of P. aeruginosa (63, 64). They are also derived from plant alkaloids and are also chemically synthesized (62). The use and overuse of quinolones and its chemically modified synthetic derivatives fluoroquinolones have given rise to bacterial resistance against these drugs (65). Nalidixic acid, the first synthetic quinolone antibiotic (64) isolated as a by-product of chloroquine synthesis (66) and fluoroquinolones like norfloxacin, ciprofloxacin and ofloxacin are recommended drugs for the treatment of cholera (67–69). Till the late 1980s VC was sensitive to these antibiotics (70, 71). Nalidixic acid resistance in VC started in the 1990s (72) and today almost 100% isolates from any outbreak are found to be resistant to nalidixic acid (23, 65). In VC quinolone resistance occurs due to point mutations in the genes encoding DNA Gyrase (topoisomerase II) and Topoisomerase IV enzymes which play a vital role in DNA replication and repair mechanisms (62). These enzymes are the targets for the drug (62). Gyrase is composed of two subunits GyrA and GyrB (in Gram positive these are GrlA and GrlB respectively) (62). Topoisomerase IV is composed of two subunits ParC and ParE (62). Quinolones intercalate into DNA at both the cleaved bonds produced during replication and prevent ligation of the cut-ends thereby interfering with replication mechanism (62). The drug binds non-covalently at the enzyme-DNA interface at the cleavage-ligation active site. It uses serine at position 83 and acidic amino acid residues at position 87 as an anchoring point to form a water-metal ion bridge to reach the enzyme (62). The most commonly mutated amino acids are the serine and acidic amino acid residues (62). These mutations consequently disrupt the dug-enzyme binding leading to quinolone resistance (62). It has been found that the serine residue provides protection to bacterial gyrase against naturally occurring antibiotics. This is why it is highly conserved across the kingdom Bacteria (62). Sequence analysis of the enzyme subunits from quinolone resistant spontaneous mutants of E.coli KL16 showed that resistance occurred due to point mutations in the region between amino acid 67 and 106, especially in the vicinity of the amino acid 83 in GyrA protein (73, 74). The small region near the N-terminal region in the GyrA subunit in which the point mutations leading to quinolone resistance occurred and this region was close to the Tyr at position 122 which has been shown to be the site covalently bound to DNA (73). This region was termed as quinolone-resistance determining region (QRDR) (74). Later, Friedman et al. showed that even amino acid at 51 position was susceptible to mutation and Ala at this position was mutated to Val in 3 E.coli mutant strains which they studied, giving rise to quinolone resistance and providing cues for the expansion of the QRDR (75). Thus, these single nucleotide polymorphisms (SNPs) in chromosomal genes modify the structure of the target site of drugs leading to weaker drug-protein interactions (62) and leading to resistance against the drug.
In VC SNPs in the genes gyrAB and parCE in the quinolone-resistance determining (QRDR) regions have been reported frequently (24). These have been shown to interfere with the action of quinolones and fluoroquinolones rendering the strains resistant to these drugs (76). Kim et al. reported mutations in gyrA and parC genes in clinical isolates of VC from Bangladesh collected from 2002-2008. These mutations could be correlated to reduced susceptibility to ciprofloxacin. The mutations observed were in gyrA encoding Ser83Ile and in parC encoding Ser85Leu (76) Quilici et al. reported mutations in gyrA encoding Ser83Ile and in parC encoding Ser85Leu in VC O1 isolates from Nigeria (77). These mutations were responsible for reduced susceptibility to ciprofloxacin (77).
Usage of antibiotics is an ancient practice (78) and has existed before the discovery of antibiotics (penicillin) in 1928 by Alexander Fleming (79) and before the discovery of chemotherapy by Paul Ehrlich in 1909 (80). With these discoveries started the “antibiotic era”. The 1950s-1960’s when most of the antibiotics were discovered is called the “golden age of antibiotics” (79). However, for millennia antibiotics have been used to treat infections as evidenced from the practice of using molds, honey, soil and herbal extracts to cure infections in the ancient civilizations of the world (78, 80). These rituals were prominent factors which contributed to the accumulation of antibiotic resistance in human populations (80). The introduction of antibiotics in clinical practice was followed by the discovery of antibiotic resistance (AR). Discovery of sulfonamide resistance in the late 1930s and that of penicillinase in 1940 were the first instances of AMR discovery (79). Resistance against streptomycin was observed soon after its implementation in 1944 for the treatment of TB (Tuberculosis). Staphylococcus aureus was the first bacteria in which penicillin resistance was reported in 1947 four years after the mass-production of the drug started (79, 81). In the 1950s-1960s the accidental discovery of R factors (episomes) with functional similarity to F factors bearing multiple resistance genes in multi-drug resistant Shigella dysenteriae led the discovery for the first evidence of transmissible genetic element which could be disseminated by bacterial conjugation (82). AMR is governed by genetic determinants (79). Chakrabarty et al. in 1990 (83) and Webb and Davies in 1993 (84) reported about the presence of chromosomal DNA of fermentative organisms used for fermentative production of antibiotics in the preparations of antibiotics (84). They demonstrated the subsequent uptake of small amounts of these nucleic acids with intake of antibiotics (84). They proposed that under the selection pressure of the antibiotic, uptake of some of these resistance genes by some members of the microbial population of the host would occur (84). Subsequent inter- and intraspecific genetic transfers would ensue leading to acquisition of resistance in other microbes.
AMR has existed in the pre-antibiotic era (80). It is a natural phenomenon and has evolved alongside the producers of antibiotics and carriers of ARGs in nature (11, 85). These conclusions have been based on phylogenetic analysis of antimicrobial resistance genes and enzymes (80). Phylogeny of serine and metallo-β-lactamases helped to establish that these have existed in nature for over 2 billion years (80). The bla(OXY) gene of Klebsiella oxytoca was found to be evolving along with the host for almost 100 million years (85). Actinomycetes which are more ancestral to most bacteria including pathogenic Gram-positive, Gram-negative and Chlamydia sp. are believed to be the origin of antimicrobial resistance genes in pathogenic bacteria (86). This has been demonstrated by Woo et al. on the basis of phylogenetic analysis of 16s rRNA gene and 15 housekeeping genes from 90 bacterial genomes which established that Actinomycetes are more ancestral than pathogenic bacteria (86). The same study also showed that tetracycline resistance gene from Bifidobacterium longum is more ancestral to that of Actinomycetes (86). Molecular analysis of ARGs encoding β-lactamases isolated from Alaskan soil revealed that evolutionary forces lead to the evolution of ARGs in the environment (87). Apart from the environment, ARGs have been found even in the human gut and oral microbiome recovered from the medieval ages (88). From these relics putative antibiotic-resistance genes including beta-lactamases, penicillin-binding proteins, resistance to fosfomycin, chloramphenicol, aminoglycosides, macrolides, sulfa, quinolones, tetracycline and vancomycin, and multi-drug transporters were identified bearing evidence of human microbiome serving as a reservoir of AMR in the pre-antibiotic era (89). The environment has been the origin of AMR which later found its way into the clinic (88). The environment acts as a hub of origin and evolution of new mechanisms of AMR and new ARGs (90). In the environment like freshwater habitats allochthonous species introduced through effluents, sewage, agricultural run-off and autochthonous species which harbor intrinsic AMR mechanisms can easily engage in genetic exchange (90). This facilitates HGT often between taxonomically distant bacteria (11) and finally these ARGs enter into pathogenic and clinically relevant bacteria (90).
The evolutionary history of ARGs have been classified into macro- and micro-evolutionary periods (11). During the former and which took place in the pre-antibiotic era, and hence also called the “pre-antibiotic period”, the ARGs underwent evolutionary changes due to mutations and duplications and limited contribution from HGT (11). During the latter also called the “antibiotic period” due to the large-scale production and subsequent introduction of antibiotics to environmental ecosystems due to anthropogenic activities selective pressure shaped the selection and enrichment of ARGs in these ecosystems (11). These were disseminated from the environmental reservoirs into taxonomically diverse commensals and pathogens and HGT mediated by MGEs played a pivotal role in the process (11, 90).
In VC the majority of ARGs and gene cassettes which contribute to AMR are acquired by lateral gene transfer (LGT). These reside in the MGEs, usually have their source in the environment (91, 92) and often have been found to originate in bacterial and bacteriophage genomes (93). By HGT mechanisms like natural transformation by direct DNA uptake, by bacteriophage mediated transduction or by conjugation these ARGs become a part of the VC genome by transmission through MGEs like plasmids, integrons, transposons and integrative conjugative elements (ICE) (23).
HGT is the most decisive mechanism of AMR dissemination via MGEs. It helps in intra- and inter-species exchange of virulence genes and ARGs overriding taxonomic barriers and helping bacterial strains to assimilate genetic information from distantly related organisms and enrich their genetic diversity (94). The dissemination of ARGs is favored in biofilms (95) and in polymicrobial communities like the microbiome. It is stimulated in response to certain environmental signals and the presence of antibiotics (96–98). The three major mechanisms of HGT are conjugation, transformation and transduction and the vehicles facilitating HGT in VC are plasmids, integrons, SXT and transposons. MGEs carrying ARGs have been found to be associated with enhanced bacterial fitness which may explain the underlying impetus to the acquisition of ARGs (99).
In bacteria, conjugation, a unidirectional, multi-step process, occurs between mating pairs and requires cell-to-cell contact between these. By this process plasmids, both conjugative and non-conjugative, transposons and ICEs are transferred (100). It involves the transfer of DNA from the donor, which contains the F factor and hence designated F+ to the recipient which is F- through the conjugation tube/bridge also called the pilus formed between the two bacterial cells. In some bacterial mutants like Ti plasmid Vir mutants of Agrobacterium tumefaciens DNA transfer has been found to occur without the formation of pilus (101, 102). The major component of the pilus is the protein pilin (103). At the distal end of the pilus is the adhesin molecule which helps to make cell-to-cell contact (103). The type-IV secretion system (T4SS) is used by Gram-negative bacteria for DNA delivery and pilus biogenesis (101, 103). On formation of the mating pair one strand of the DNA of the F plasmid is cleaved and the single-stranded DNA is transported to the recipient (101, 103). Thereafter, in both the donor and the host DNA replication occurs by the rolling-circle model of DNA replication (104). The transport of the DNA requires the coupling protein (T4CP), VirD4 which binds to the relaxosome (the protein complex involved in the initiation of transfer of conjugative and mobilizable elements) and mediates the transfer of the nucleoprotein complex to the membrane channel (103). The mobilization of the DNA requires MOB genes, the tra genes apart from the transfer of the DNA has a host of other functions like biosynthesis of pilin, nicking and unwinding of the DNA before initiation of transfer, MPF genes are required for the mating pair formation (103, 105). Conjugative transposons however, are transferred via formation of a non-replicative circular intermediate which forms after the excision of the element from the chromosome (106). ARGs and genes determining resistance to heavy metals carried by the MGEs are transferred into the recipient by means of conjugative transfer (107). In this manner AMR has been found to be disseminated from environmental strains and commensals into pathogenic strains rendering the pathogens more virulent (46, 108). This process has contributed to the emergence of virulent MDR strains which have caused cholera epidemics worldwide (108, 109). The SXT element which was for the first time reported in O139 strains (110) responsible for the cholera epidemic in 1992 which started in Vellore (111) was later found to have actually originated in environmental strains (108). It was shown that by conjugative transfer the element harboring sulII, strAB and dfr18 and circulating before 1992 had spread to O139 and O1 strains (108). The SXT is transferable to a number of Gram-negative organisms, helps in transfer of certain mobilizable plasmids and also chromosomal DNA in an Hfr-like fashion (106).
Natural transformation (NT) is a mechanism of direct DNA uptake by bacteria involving the employment of a DNA uptake system and without the requirement of any extrachromosomal element (112, 113). The process involves internalization of foreign genetic material and its chromosomal integration by homologous recombination (112). To undergo transformation a bacterial cell must acquire competence (113). Competence is induced by a number of factors like environmental signals, quorum sensing, growth phase (97). The main objective of NT is generation of genetic diversity for genomic evolution or chromosomal repair and curing of their genome of their infectious parasitic MGEs as suggested by many authors (112–114). According to other authors, natural transformation, is a serendipitous occurrence accompanying bacterial adhesion and twitching motility which increase under stressful conditions, thereby facilitating DNA uptake in the process (115). Carvalho et al., has recently, successfully proposed through a computational model the underlying reason behind the occurrence of natural transformation by unifying all the above hypothesis (114). According to the authors, although costly for bacterial cells, MGEs encode functions which may also be beneficial to bacteria under stressful conditions like the presence of ARGs (114). In stochastic environments, an intermediate transformation rate maximizes bacterial fitness by allowing the reversible integration of MGEs carrying resistance genes, although the MGEs are costly for host cell replication (114). Based on this dual function (MGE acquisition and removal), transformation would be a key mechanism for stabilizing the bacterial genome in the long term, thereby explaining its striking conservation (114). Hence, natural transformation is a mechanism which enhances bacterial fitness for survival and leads to the acquisition of ARGs (115). Moreover factors inducing stress like antibiotics also promote DNA uptake thereby, enhancing the chances of acquisition of ARGs (115). In VC, it has been recently demonstrated that lysogenic strains use a prophage-dependent adaptive strategy wherein, they induce neighbor predation and acquisition of DNA by natural transformation in chitin microcosms (116). This conclusion was deduced by conducting an experiment wherein, the authors inactivated dprA (a gene that is essential for NT but is not required for transduction as phage-mediated transfer usually occurs by transduction) in VC O1 El Tor strains (116). It was concluded that the HGT observed is due to the transfer of DNA from the nonlysogen to the lysogen (116). Bacteria of diverse taxonomy and phylogenetically distant species share conserved uptake and processing proteins and may differ in inducing factors and regulatory mechanisms (112, 113). In VC the process has been well studied. In VC NT occurs in chitinous surfaces and later it was shown that it is induced by chitin, biopolymer of β- 1,4–linked N-acetylglucosamine (GlcNAc) (117). Competence requires a type IV pilus assembly complex, a putative DNA binding protein, and three convergent regulatory cascades, which are activated by chitin, increasing cell density, and nutrient limitation, a decline in growth rate, or stress (117). Chitin induces the expression of a 41-gene regulon involved in chitin colonization, digestion, transport, and assimilation, including genes predicted to encode a type IV pilus assembly complex (117). Meibom et al., demonstrated that chitin induces competence in VC (117). The authors grew VC O1 El Tor strain A1552 in a liquid minimal medium containing 2.5 mM (GlcNAc)6 and genomic DNA from the VC O1 El Tor strain VCXB21 which harbors a chromosomal gene for kanamycin resistance was added to the culture of A1552 (117). After 18 h of growth, the culture was plated onto antibiotic-free and kanamycin-containing LB agar which yielded a transformation frequency [kanamycin-resistant (Knr) colony-forming units (CFU)/ total CFU] of 2.7 x 10-5 (117). In the absence of donor DNA or when deoxyribonuclease (DNase) and donor DNA were added simultaneously, no Knr colonies were detected (118). The induction of competence was also tested using other carbohydrates including the chitin monomer GlcNAc, which does not upregulate the chitin regulon (117). Accordingly, only chitin induced the competence phenotype (117). When glucose was combined with (GlcNAc)6, competence was inhibited, which suggested catabolite repression of the competence phenotype (117). Chitin-induced NT with genomic DNA from the prototroph strain VCXB21 also restored prototrophy to two amino acid auxotrophic mutants that had deletions in either the proC or hisD gene and consequently were impaired of the ability to synthesize proline or histidine (117). The deleted version of the hisD gene was replaced by the wild-type copy from the donor DNA (117). Thus, the authors through their classical experiments demonstrated that the growth of VC O1 with a soluble chitin oligosaccharide induced transformation competence and the ability to acquire different genes (117). Chitin through TfoXVc induces the expression of a competence pseudopilus and other genes required for the degradation and uptake of chitin (117). Increasing cell density, along with effectors of increased RpoS abundance, enhance the expression of hapR, required for the positive regulation of competence (117). Both HapR and TfoXVc are required for the expression of the VC1917 gene that encodes a protein with a signal peptide and a motif homologous to the DNA-binding helix-hairpin-helix domain (117). The VC1917 was found to be required for competence (117). HapR and TfoX regulate the expression of another competence regulator, QstR (118). The molecular killing machinery T6SS (type VI secretion system) in VC also participates in NT in VC and is regulated by the same transcriptional regulators TfoX and QstR (119). It is used for killing neighboring non-immune cells resulting in the release of DNA and facilitating HGT (120). DNA released by an individual bacteria would be taken up by other members in a biofilm, thereby, indicating the role of biofilms in enhancing frequency of transformation and dissemination of genes like ARGs by NT (117). Quorum-sensing associated autoinducers control the degradation of exogenous DNA towards the uptake of intact DNA strands in competent-induced cells of VC (121, 122). comEA, which encodes a putative periplasmic DNA-binding protein and comEC, which encodes a DNA internalization-related competence protein are QS-regulated genes which are involved in DNA uptake and are strictly dependent on positive regulation by HapR (122). NT occurs in high cell density when dns gene is downregulated by higher HapR expression (122). Lloyd et al., has recently shown that NT can be induced in Classical VC which are naturally incapable of transformation due to a mutation in the master regulator hapR gene (118). The authors showed using a plasmid they constructed which expresses both TfoX and QstR which rendered the O395 Classical strain carrying a hapR plasmid capable of NT (118). Dalia et al., found the existence of a HGT transferred element, IdeA (ICE-encoded DNase) and globally distributed in VC which inhibits natural transformation even if all the genes required for NT are present in the genome (123). This element was discovered in the 2010 outbreak clinical isolates of VC O1 El Tor and was found to be present in the 97.7 kb ICE similar to VchInd5, a commonly occurring ICE (123). It was ubiquitously present in the Haitian strains (123). IdeA inhibits NT in cis via DNA endonuclease activity that is localized to the periplasm (123). The authors generated isogenic mutants of IdeA and observed that the mutants showed improved efficiency for NT (123). However, frequency of isolation of VC strains harboring this element has undergone a reduction in the recent years (123). This may consequently lead to higher chances of NT in VC and greater dissemination of ARGs.
Transduction is bacteriophage-mediated transfer of genes and gene cassettes from one bacteria to another. Three principal methods of transduction exist namely, generalized, specialized and lateral transduction (124). Among the HGT mechanisms, transduction is considered to be the major route of gene acquisition for their rapid adaptation to environmental challenges (124). The general mechanism of transduction encompasses the excision of host bacterial DNA, packaging them into transducing particles, infection of other cells and release of these particles into the recipient. The DNA either recombines into the recipient chromosome or replicates independently as a plasmid (124). In VC a number of pathogenicity islands like TCP, VPI-2, VSP-1 and VSP-2 exist which have been acquired by phage transduction (125). Phages have been found to be integrated in chromosome (126, 127) and plasmids in VC (128). Many phages have been found to confer the ability of conjugative transfer to the plasmids (128, 129). Phages are of common occurrence in VC and two types of bacteriophages have been identified (129). The lytic phages and lysogenic phages (129). An intermediate type has been reported too, the pseudotemperate lifestyle, wherein, the phage does not integrate with the host chromosome but remains as linear or circular plasmids (130). However, recent evidence suggests that upon infection the temperate phages can either be induced into a lytic life-cycle or a lysogenic one (131). Recently, it was shown by Silpe and Bassler that the QS AI determines which life-cycle would be activated in VC (131). VP882 phage encoded protein gp56 (VqmAPhage) which bears homology with the VC encoded VqmAVc receptor of AI 3,5-dimethylpyrazin-2-ol (DPO) can serve as a receptor for the VC QS AI DPO (131). It binds DPO and controls QS regulon in VC while propagating itself in the process as it switches on the lytic cycle with the mediation of another phage encoded antirepressor protein gp55 also known as Qtip (quorum-triggered inactivator of cI protein) (131). This inactivates the phage encoded repressor cI and inhibits its binding to q promoter DNA (131). However, if the temperate phage chooses to enter lysogeny then its repressor, (cI in Λ phage) inhibits transcription of its genes required for its lytic cycle and the prophage becomes quiescent and concomitantly its DNA is replicated with the host chromosome during cell cycle (130). The genetic evolution of VC is mostly associated with the lysogenic phages (132).
The existence of generalized transducing phages of VC were reported in 1982 and included the VcA-1, Vc-A2, Vc-A3, all temperate phages capable of lysogeny in both the biotypes and all were serologically related to the common kappa-type phage (129). They had the capability of randomly inserting into different sites in the VC chromosome (129). Later, the transducing phage CP-T1 was isolated from an apparently lysogenic El Tor strain (129). It had the ability to transduce widely separated markers and to propagate in both the biotypes (129). An association of lysogenic phages of VC and its virulence was shown by many researchers (129). In epidemic associated El Tor strains isolated between 1930 and 1960 it was found that those strains which released prophages were associated with disease severity while those which did not release prophage caused mild or asymptomatic cholera (129). Later, the chromosomally integrated CTXφ temperate phage was isolated and was shown to encode the major virulence factor of VC, cholera toxin (CTX) using toxin co-regulated pilus (TCP) as its receptor (126, 127). It could be transferred from pathogenic to benign cells contributing to transfer of virulence (126, 127). The CTX element under certain conditions is self-transmissible and replicates independently like a plasmid forming extracellular particles (CTXφ ) containing single-stranded DNA consisting entirely of CTX element (127). These findings point to the significant role that phage-mediated transduction plays in the genetic evolution of VC (126, 127, 132). Although phages have been associated with the propagation of AMR, there are rare reports of phage genome being linked with ARGs (133, 134). One such rare report has been presented by Moon et al., from a recent analysis involving the investigation of the presence of ARGs from viral contigs recovered from urban surface water viral metagenome data (134). They identified sequences of diverse ARGs, including polymyxin resistance genes, multidrug efflux proteins, and β-lactamases (134). On employment of a lenient threshold of e value of ≤ 1 × e-5 and query coverage of ≥ 60% in the Resfams database, novel β-lactamases blaHRV-1 and blaHRVM-1 were found (134). These genes had unique sequences and formed distinct clades of class A and subclass B3 β-lactamases, respectively (134). Minimum inhibitory concentration (MIC) analyses for E. coli strains harboring blaHRV-1 and blaHRVM-1 and catalytic kinetics of purified HRV-1 and HRVM-1 showed reduced susceptibility to penicillin, narrow- and extended-spectrum cephalosporins, and carbapenems (134). These genes were also found in bacterial metagenomes, indicating that they were harbored by actively infecting phages (134). The study showed that viruses in the environment carry novel functional ARGs, in small quantities suggesting that environmental bacteriophages could be reservoirs of diverse, unknown ARGs that could be disseminated via virus-host interactions (134). Enault et al., employed bioinformatics to detect ARGs in phage genomes (134). They found that ARG abundances in 1181 phage genomes were vastly overestimated using exploratory thresholds due to low similarities and matches to protein unrelated to AMR (134). They found experimentally that 4 of these ARGs predicted using exploratory thresholds failed to confer antibiotic resistance in E. coli (134). The authors on the basis of their findings reasserted the traditional view that phage genomes rarely encode ARGs (134). A recent study by Verma et al., was conducted to extensively characterize the genetic diversity and AMR traits of 443 isolates of VC collected from diarrheal patients in India from 2008 – 2015 and identified MDR, XDR strains, and acquired resistance traits based on the identification of ARGs physically linked to MGEs like plasmids, ICEs and transposons (22). Although prophages were present in these strains, none of the ARGs were physically linked to the phage genome (22). In this study the authors carried out WGS of 4 isolates encompassing MDR O1, O139, non-O1/non-O139 strains (22) and found that 5% of the strains harbored different MGEs like pathogenicity islands (PIs), metabolic islands, prophages, plasmids, and transposons that have been acquired by HGT from closely or distantly related bacterial species (22). Integration of most of these MGEs in VC chromosomes is reversible, and can be excised and propagated to other VC cells showing the potential of MGEs in the propagation of AMR (22). The MGEs linked to VC AMR and which have been derived as a result of extensive genetic analysis of VC strains isolated from the environment or associated with cholera have been discussed below. Table 2 outlines the key features of these elements helping to differentiate among them. Table 3 presents the various MGEs reported in VC, their resistance markers and the ARGs that they have been found to carry.
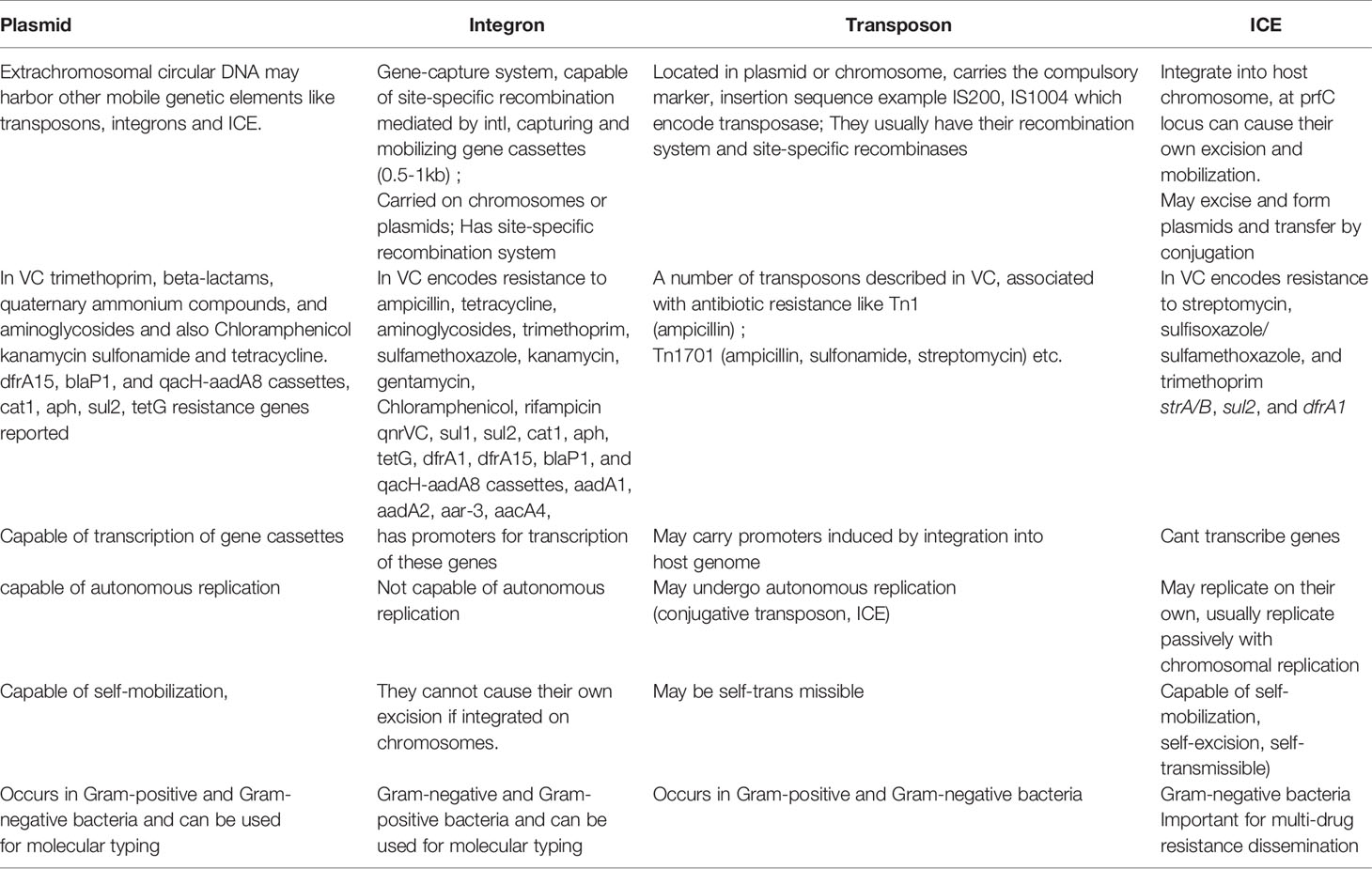
Table 2 Features of MGEs contributing to transmission of antibiotic resistance in V.cholerae.
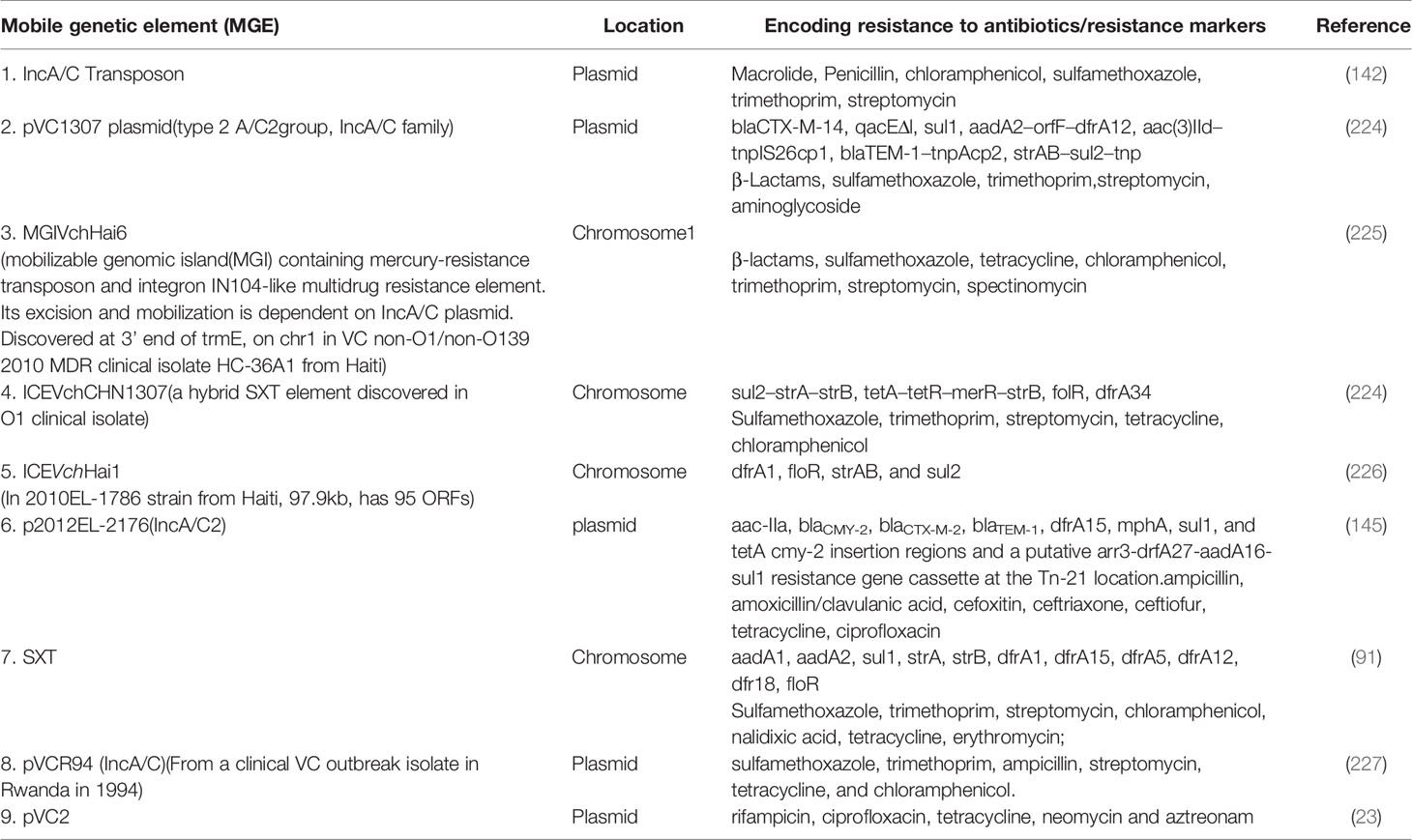
Table 3 Recent discoveries in V. cholerae mobilome associated with MDR.
Plasmids are extrachromosomal circular DNA capable of autonomous replication and may be self-mobilizable or non-mobilizable (135). The former is called conjugative plasmid while the latter is non-conjugative plasmid (135). The non-conjugative plasmids may be transferred with the help of self-conjugative systems/transfer systems of a co-resident conjugal plasmid or may use a process called conduction/conjugation to move from donor to recipient cells (136). In VC a conjugal system was discovered by Bhaskaran in 1958 and the fertility factor of VC corresponding to the F-factor of E.coli was called sex factor P (129). Plasmids are often associated with antibiotic and heavy metal resistance gene cassettes and may harbor other MGEs like transposons, integrons and ICE (29, 42, 107). Plasmids, thus encode various functions like antibiotic resistance (107), heavy metal resistance (107), toxin production (137), adhesin secretion (137) and cellular and metabolic functions like synthesis of membrane proteins and peptidoglycan (138). Many plasmids have been identified in VC in environmental reservoirs and also in clinical isolates of different serogroups including O1 and O139. Some examples are pVC, pSDH-1, pSDH-2 (138, 139). From a recent VC non-O1/non-O139 clinical strain, HC-1A2, collected from cholera outbreak in Haiti, Ceccarelli et al., characterized two novel circular plasmids and completely sequenced these (138). They are the pSDH-1(4985bp) and pSDH-2 (5580bp) (138). The plasmids were characterized using single-cell genomics (138). These were found to be self-mobilizable and in pSDH-2 a toxin- antitoxin (TA) system was identified (138). It was further concluded that this plasmid uses the ColE1 model of plasmid replication (138). Both the plasmids were found to be widespread among environmental non-O1/non-O139 isolates (138). On detailed analysis different genes borne on these were found to have similarity with homologues from V. parahemolyticus, Moraxella maccacae and Vibrio tasmaniensis (138). On the basis of these observations it was concluded that plasmids are propagated by conjugal transfer and contribute to clonal divergence as they harbor genes from diverse phylogenetic origin (138). In VC plasmids have been found to enhance virulence and pathogenicity (137) induce drug resistance and also help in mobilization and transfer of non-conjugative genomic islands harboring ARGs (29) thereby explaining the role of plasmids in genomic evolution by conferring the ability of enhanced survival and persistence.
Some recently described plasmids, playing a role in antibiotic resistance acquisition and dissemination in VC have been described below and which have been isolated from different geographical locations in clinical and environmental isolates. The p3iANG conjugative plasmid was reported by Ceccarelli et al. as a result of one such investigation (42). The plasmid was found carrying a set of 3 class1 integrons, harboring dfrA15, blaP1, and qacH-aadA8 cassettes, which code for resistance to trimethoprim, ß-lactams, quaternary ammonium compounds, and aminoglycosides and also chloramphenicol (cat1), kanamycin (aph), sulfonamide (sul2) and tetracycline (tetG) genes (42). The plasmid was detected as a result of a study aimed at resistance profiling and understanding its correlation with MGEs in the VC O1 clinical and VC O1 and non-O1 environmental isolates from different provinces of Angola and collected between 1991 and 1996 (42). The clinical O1 isolates were found to contain this large conjugative plasmid with a set of three class 1 integrons harboring the above mentioned ARGs clustered in a 19kb region (42). This plasmid was found to be present ubiquitously in isolates from all provinces from north to south and the integrons carried resistance cassettes not found in Africa before (42). dfrA15 and blaP1cassettes were identical to the cassettes observed by the same investigator previously in Thailand (42). These findings highlight the significant role plasmids play in the dissemination of ARGs among isolates of VC across different geographical locations (42). Toxigenic VC O1 MDR clinical isolates of 2008 from an outbreak in South Africa were found to harbor a ~140kb plasmid carrying blaTEM gene encoding TEM-63 β-lactamase which had been associated with nosocomial infections caused by K. pneumoniae in South Africa till then (140). This report revealed the role plasmids play in transfer of ARGs between bacteria of diverse taxonomy and their importance in the evolution of VC.
Another family of plasmid, the IncA/C conjugative plasmids have been often detected in VC and have been shown to play a pivotal role in MDR dissemination (142). It has been found to encode resistance to macrolide, penicillin, chloramphenicol, sulfamethoxazole, trimethoprim, streptomycin (142).
Wang et al., identified mega-plasmids from MDR VC O139 strains in China (141). One plasmid belonged to the IncA/C family and 10 ARGs were found in the MDR regions, including a blaTEM-20 gene and these conferred resistance to 7 antibiotics (141). This kind of plasmid was positive in 71.2% toxigenic O139 strains (141). Wang et al., identified an O1 El Tor strain isolated from a patient in 1998 which showed was intermediate susceptibility or resistance to 13 antibiotics and could potentially produce extended-spectrum β-lactamase (ESBL) (143). Three genetic elements were identified namely, a hybrid SXT element (ICEVchCHN1307), a new IncA/C plasmid (pVC1307) and a chromosomal integron (143). Twenty ARGs were located on them, including blaTEM-1, blaCTX-M-14 and phenotypically silenced tetRA genes (143). An IncA/C plasmid was isolated from two high minimum inhibitory concentration (MIC) azithromycin-resistant VC strains of the two serogroups, O1 and O139, isolated in China (31). In the 172 predicted open reading frames (ORFs), 16 genes were related to AMR, of which 5 were associated with macrolide resistance which were distributed in two clusters, mphR-mrx-mph(K) and mel-mph2, flanked by insertion sequence elements and involving two kinds of resistance mechanisms (31). Deletion of the complete region of the two clusters deceased the azithromycin MIC from ≥64 µg/mL to ≤0.5 µg/mL (31). In addition to 11 ARGS, 5 ARGs encoding macrolide resistance with other functions were accumulated repeatedly through transposition on one plasmid (31). This genotype could not be simply explained by antibiotic stress applied on the host from the environment or treatment (31). These phosphorylases and transmembrane transporters might be involved in the transport and metabolism of other non-antibiotic substances, enabling this kind of plasmid to propagate better in the host (31). This study highlights the importance of plasmid-mediated HGT for optimum adaptation of VC (31).
Walsh et al. detected NDM-1 gene encoding a carbapenemase in environmental VC strains from India. Walsh et al. was the first to report about the presence of NDM-1 gene in VC (25). The study encompassed the detection of NDM-1 gene for the evaluation of its prevalence in drinking water and seepage water samples from India (25). Two VC strains were isolated in the study from waste seepage water samples and both were found to carry transmissible NDM-1 genes by PCR and DNA probing and the existence of IncA/C plasmid in these strains was confirmed by Inc typing on blaNDM-1 positive plasmids (25). One of the strains was shown to carry chromosomal NDM-1 gene as well (25). Later, Mandal et al., reported about a plasmid-borne blaNDM-1 gene in a clinical strain isolated from the fecal sample of a cholera affected 2-year old patient in India (26). The strain was resistant to ampicillin, ceftriaxone, cotrimoxazole, and furoxone and sensitive to ciprofloxacin and tetracycline (26). The phenotypic tests for ESBL detection and PCR for detection of the ESBL genes were both negative (26). Therefore, the authors tested for the AmpC type of β-lactamase production (26). The strain, an AmpC β-lactamase producer, was found positive for the blaDHA gene by multiplex PCR (26). The blaDHA gene on sequencing was found to be 99% identical to Klebsiella pneumoniae β-lactamase blaDHA-1 gene (GenBank accession no. AY635140.1) (26). Carbapenems are considered the treatment of choice for AmpC-producing organisms (26). Therefore, the strain was tested for the production of carbapenemase (26). It was found to be a carbapenemase producer by a positive modified Hodge test and by the EDTA disk synergy test (26). The multiplex PCR for the detection of carbapenemase genes followed by sequencing confirmed it to be blaNDM-1 gene with 100% identity with E.coli strain HK-01 plasmid pNDM-HK (GenBank accession no. HQ451074.1) (26). The presence of a plasmid bearing the blaNDM-1 gene was confirmed by the isolation of the plasmid using the alkaline lysis method followed by subjecting it to multiplex PCR to detect the carbapenemase genes (26). It confirmed the presence of the blaNDM-1 gene (26).
A recent investigation on the antimicrobial susceptibility of the 2010 outbreak strains from cholera patients in Haiti was carried out (145). A report of the AMR traits of the typical outbreak strain (2010EL-1786) revealed resistance to streptomycin, sulfisoxazole, trimethoprim/sulfamethoxazole, and nalidixic acid, and decreased susceptibility to ciprofloxacin and chloramphenicol (145). Resistance was caused by mutations in the QRDR regions of the gyrA and parC genes and presence of ICEVchHai1 containing the dfrA1, floR, strAB, and sul2 resistance genes (145). Another isolate, 2012EL-2176, showed the typical resistance phenotype of the outbreak strain but additional resistance to ampicillin, amoxicillin/clavulanic acid, cefoxitin, ceftriaxone, ceftiofur and the tetracycline MIC was intermediate (145). PCR and WGS analysis with ResFinder helped to identify the original outbreak resistance determinants and additional determinants (aac(3)-IIa, blaCMY-2, blaCTX-M-2, blaTEM-1, dfrA15, mphA, sul1, and tetA) (145). Plasmid transfer by electroporation into E. coli (DH10B) confirmed that the ARGs were plasmid encoded (145). PCR-based replicon testing identified an IncA/C2 plasmid, and PCR and WGS confirmed that the plasmid encoded a unique set of resistance determinants (aac(3)-IIa, blaCMY-2, blaCTX-M-2, blaTEM-1, dfrA15, mphA, sul1, and tetA) and a second copy of the resistance genes floR, strAB, and sul2 identical to the ones found in ICEVchHai1 (145). Antimicrobial susceptibility testing of the transformant demonstrated transfer of the resistance profile and additional resistance to chloramphenicol, tetracycline, and decreased susceptibility to azithromycin (145). The plasmid was mobilizable by conjugation (conjugation efficiency = 1.3–1.4 × 10−2) when E. coli J53 was used as the recipient (145).
Hammerl et al. reported about the presence of VCC-1 carbapenemase gene blaVCC-1 borne on pVCC plasmid in four strains of non-toxigenic carbapenem resistant nonO1/non-O139 environmental strains of VC isolated from coastal waters in Germany, using WGS (144). The blaVCC-1-coding sequences and flanking nucleotide sequences were 100% identical among the strains (144). This study showed that environmental VC is a reservoir of carbapenem resistance and the authors have proposed that from the water these resistant strains may enter into mussels, fish and shrimps and may eventually enter the food chain (144).
A recent investigation by Verma et al. led to the identification of two large plasmids pVC1 and pVC2 in XDR VC strains from India (23). pVC2 was found to carry ARGs bla, ant(3′), and aac(3′) and was experimentally confirmed to confer resistance to rifampicin, ciprofloxacin, tetracycline, neomycin and aztreonam (23). Both the plasmids had a much lower GC content of ~ 40% compared to the host genome and both encoded mobility-associated genes like tra genes (23). Another large plasmid (∼94 kb) encoding β-lactamases, chloramphenicol acetyltransferase, aminoglycoside 3′-phosphotransferase, aminoglycoside N(3′) acetyltransferase, and bleomycin resistance protein were found in another strain (23).
The description of these plasmids and their genetic analysis proved that plasmids have played a substantial role in introducing different ARGs into the VC genome (25) helping in clonal divergence (138), enhanced adaptation (31) and dissemination of MDR traits across different bacterial species (138) and in different geographical locations (42).
Integrons are gene-capture systems, carried on chromosomes or plasmids and capable of site-specific recombination, capturing and mobilizing gene cassettes (0.5-1kb) (146–148). They have promoters for transcription of these genes (147, 148). They cannot cause their own excision if integrated on chromosomes (147, 148). The clinical integrons are borne on plasmids (146, 148). Superintegrons are located on chromosomes (146–148). In VC integrons are located in chromosome 2 (149, 150) and also plasmids like p3iANG have been found to carry integrons (28, 42). These MGEs have been found to encode resistance to ampicillin, tetracycline, aminoglycosides, trimethoprim, sulfamethoxazole, kanamycin, gentamycin, chloramphenicol, rifampicin in VC (42, 93). Resistance genes qnrVC, qnrVC1, qnrVC2, sul1, sul2, cat1, aph, tetG, dfrA1, dfrA15, blaP1 and qacH-aadA8 cassettes, aadA1, aadA2, aar-3, aacA4 have been frequently observed associated with integrons in VC (42, 93, 151). Integrons are more frequently found in Gram-negative bacteria than in Gram-positive bacteria (147). Five classes of integrons have been reported in bacteria till date (148, 152). In VC classes 1, 2 and 4 have been found (93). Shi et al. conducted an investigation for the detection of class 1, 2 and 4 integrons in 133 clinical strains of O1, O139 and non-O1 non-O139 VC isolates from Kolkata by PCR and sequencing methods (93). These strains were isolated from 1992 to 2000 (93). Class 1 integron harboring genes aadA1, aadA2, blaP1, aar-3, aacA4, and dfrA1 and dfrA15 were detected in 7 strains of O1, 1 strain of O139 and 6 strains of non-O1 non-O139 serogroups (93). None of the clinical isolates were found to carry class 2 integron (93). All the strains possessed class 4 integrons (93). This study revealed that class 4 integrons are present in all clinical strains of VC irrespective of serogroup while class 1 integron does not occur in all clinical strains (93). Class2 integrons harboring antibiotic resistance genes have been reported to be less frequently isolated in VC, however they are located in the chromosome of environmental and clinical isolates of non-O1/non-O139 serogroups like the strains RC121 (O27) and B0320 (O39) (153).
Several recent studies have reported about the detection of integrons in current isolates of VC from different regions of the world and experimentally deciphered their value in the genomic evolution and AMR of VC (27, 154–156). Aberkane et al. identified a non-O1/non-O139 VC isolate from cloacal swab sample of a yellow-legged gull in southern France producing both VIM-1 and VIM-4 carbapenemases (154). The blaVIM genes were found to be part of a class 1 integron structure located in an IncA/C plasmid (154). A PcS (strong) promoter variant, divergent to the integrase gene, was identified in the class 1 integron, with a functional P2 promoter located downstream of the PcS in the attI1 site as a consequence of insertion of three G residues (154). The PcS-P2 association is believed to confer high-level gene cassette expression (154). Wu et al., detected PER-1 extended-spectrum β-lactamase (ESBL) in a clinical non-O1/non-O139 VC strain from China (154). ISCR1-mediated bla(PER-1) was embedded in a complex In4 family class 1 integron belonging to the lineage of Tn1696 on a conjugative IncA/C plasmid (155). A free 8.98-kb circular molecule present with the ISCR1-bla(PER-1)-truncated 3'-conserved sequence (CS) structure was detected in this isolate (155). VC non-O1/non-O139 isolates recovered in Germany, from organs of domestic ducks with serious disease symptoms were investigated for phenotypic traits and by WGS (156). WGS data revealed only a distant phylogenetic relationship between the isolates determined using a CSI Phylogeny (version 1.4)-based single nucleotide polymorphism (SNP) tree (156). Three isolates showed susceptibility to a number of tested antimicrobials, and one strain possessed ARGs encoded on an integron (156). Accordingly, isolates were susceptible to most of the tested antimicrobial agents and one strain (CH415) displayed resistance to trimethoprim and sulfamethoxazole (156). A high MIC value against nalidixic acid was observed in the same strain (156). All isolates displayed resistance to colistin (156). In isolate 17-VB00405, only a catB9 gene was present (156). The nalidixic acid resistance of strain CH415 may be due to a mutation in the gyrA gene leading to an amino acid substitution (aspartic acid to glycine) in position 87 (156). The AMR genes sul1, aadA1 and dfrA1 of strain CH415 were physically linked to a class 1 integron integrase gene intl1 (156). Recently, Morita et al., detected integrons in the genomic islands of non-O1/non-O139 non-toxigenic VC isolated from cases of diarrhea-like illness in Kolkata, India, collected from 2007 to 2014 (28). Out of 25 strains analyzed 13 were antibiotic-resistant and 6 strains were resistant to more than 4 antibiotics (28). WGS was used to identify the ARGs (28). Accordingly, dfrA, floR,sulI, tet, mer operon, blaCARB-94 were detected (28). Four strains possessed Class 1 integrons in their genomes and 3 of these 4 integrons were found to be located in their genomic islands which were determined as novel types (28). IntI1 was detected in 4 strains and 1 strain possessed intSXT (28). the Class 1 integrons of 2 strains included tni module genes and hence, these Class 1 integrons were considered as members of a In16 family (28). Class 1 integron of the third isolate contained IS6100, indicating that this might be related to the In4 family and this isolate also contained IS26‐mphA‐mrx‐mphR‐IS6100 unit, an MGE (28). As the Class 1 integron of one of the strains contained the gene cassette, dfrA15‐qacEΔ1‐ sul1, the integron was regarded as the member of In192 (28). One of the strains possessed an SXT element harboring a mer operon encoding the mercury resistance gene (28). The authors detected GIs in 4 strains with Class 1 integron and in 3 of the strains it was located at the 3’ end of trmE in the VC0003 region of the VC genome (28). The arrangement of the GIs resembled that of AGI1 more than 99% which is a member of the SGI1 (Salmonella genomic island 1) family or contained regions from PGI1 in addition to the region from AGI1 (28). One of the strains possessed another GI which was 100% identical to GIVchHai8 of the VC strain HE‐45 from Haiti and the third strain also carried regions from this GI (28). The MDR region of the GIs harbored several ARGs which included a dfrA1, sul1, and mer operon; dfrA1, sul1, and mphA operon; and dfrA15, sul1, and mer operon (28).
Integrons play a significant role in the dissemination and emergence of MDR strains due to insertion or deletion of large portions of mobile gene cassettes (MGCs) or single MGCs (28, 149) and therefore, chromosomal integrons can be successfully employed for sensitive phylogenetic analysis of pandemic strains (149). Labbate et al. used PCR-based analysis of integron arrays to differentiate between pandemic and non-pandemic isolates (149). Differentiation was based on the MGC size class represented by the size of the PCR product (149). Seventh pandemic prototype O1 El Tor strain N16961 which contains 179 MGCs was used as the reference (149). The investigators arrived at the conclusion that pandemic VC strains arose from a common progenitor as the MGC composition was found to be relatively conserved among them (149). It helped to resolve the evolution of O139 from a subgroup of O1 El Tor. Overall, this method proved to possess greater sensitivity than single-gene based phylogeny and multi locus sequence analysis (149).
These are self-transmissible, integrative and conjugative elements, integrated into a single site (5’ end of the prfC locus) in the host chromosome and belong to the SXT/R391 family (106). They are not capable of autonomous replication (106, 157). They are transposon-like elements and are categorized under ICE (Integrative Conjugative Elements) and may carry integrons (157). These are hybrid structures bearing transposon associated ARGs and also ARGs borne on plasmids and phages (157). It is a 99,483bp long insert in VC, has 87 putative ORFs and encodes resistance to sulfamethoxazole, trimethoprim, streptomycin, chloramphenicol, nalidixic acid, tetracycline, erythromycin and have been found to carry ARGs like aadA1, aadA2, sul1, strA, strB, dfrA1, dfrA15, dfrA5, dfrA12, dfr18, floR, mphA (106, 158, 224). The ARGs are carried in a composite transposon-like element that interrupts the SXT encoded rumAB operon (159). The conjugative transfer of SXT utilizes a conjugation system related to the F plasmid (159). Two of its loci setC and setD encode regulators that activate the transcription of genes involved in SXT excision and transfer (159). Another regulatory gene setR whose product resembles the lambdoid phage CI repressors are involved in regulation of SXT gene expression (106). The setR encoded repressor represses the expression of SetCD, whose overexpression would otherwise be deleterious for the host (96). For optimal transfer of SXT, integration host factor (IHR) is an essential component required by VC for being a host or a recipient (160). IHF is a heterodimeric protein whose subunits are encoded by the himA and himD genes, and it helps regulate expression of F conjugation genes and stimulates the TraI-mediated cleavage of its origin of transfer (oriT) (160). IHF is required for both integration and excision of λ (160).
The SXT element was discovered in the O139 VC which is the only non-O1 serogroup that can cause epidemic cholera (96). It was isolated from MO10, an O139 strain, in 1993 (110). Later, the SXT element was found in O1 El Tor strains (161). However, the SXT element of the O1 El Tor designated as SXT ET did not carry the same resistance genes as SXT of VC O139 strain MO10 (159). The trimethoprim resistance gene was present 70kb away from the other resistance genes in SXT ET (159). The SXTMO10 acquired its ARGs and some adjacent sequences by a transposition event which introduced a 17.2-kbp region containing all five resistance genes, floR, dfr18, sulII, strA, strB into rumB, the second gene of the rumAB operon (159). The 17.2-kbp sequence is flanked both by an 8-bp direct repeat (corresponding to amino acids [aa] 76 to 78 of rumB) and by 16-bp imperfect inverted repeats, structures often found at the boundaries of transposons (159). The SXT element has been isolated from VC strains associated with epidemics around the world like Bangladesh, Mozambique, Kenya (159, 162, 163). Recent WGS analysis of environmental isolates of toxigenic O1 VC from Lake Victoria in Tanzania showed the existence of an SXT element with deletions in vital ARGs (164). Initial analysis using MyDbFinder1.2. revealed the absence of common genes like sul2, dfrA1, dfrA18, floR, strA, and strB in some of these strains (164). Subsequently, a detailed analysis was performed which revealed that fragments of strA, strB, Sul2, floR genes were present in the SXT and about 1100 bp gaps between nucleotide position 98500bp–102450 bp occurred in these strains (164). SXT elements are wide-spread in VC and other gram-negative bacteria (159) and newer insertion of gene cassettes in these elements are reported quiet frequently (158). SXT elements play a significant role in phylogenetic profiling helping in tracing the origin, transmission and dissemination of SXT-borne ARGs (158).
A number of variants of the VC O139 SXT element has been described by a number of authors (161, 165, 166). SXT elements have been later found in other Vibrio species as well (165). A variant of the VC SXT element was reported in a V. fluvialis strain H-08942 isolated from a 6-month aged infant with cholera-like diarrhea in India (165). The element was found to be shorter than that of the VC element and conferred resistance to the antibiotics typically displayed by SXT elements (165). Accordingly, genes strA, floR, sulII and dfr18 for resistance against streptomycin, chloramphenicol, sulfamethoxazole and trimethoprim were detected using PCR and the integrase gene and the attP attachment site for SXT were also found to be present in the strain (165). The integrase enzyme is required for excision of the SXT element from the chromosome and its site-specific integration into the host chromosome (106). The SXT encodes its own integrase gene which is related to integrases of the Λ family of phages (106). It is located at the 5’ end of the SXT element (106). The integrase Int is a recombinase which helps in the formation of the non-replicative, extrachromosomal circular form of SXT, which is an intermediate in the form in the transfer of SXT (106). The integrase gene of the V. fluvialis strain detected by Ahmed et al. in the study described above was 1242 bp in length and 413 amino acid residues and was found to be a variant of the SXT element of O139 VC (165). The attP site was shorter comprising 641bp as opposed to the 785bp long attP site of SXT of O139 VC (165). ICEs derived from different organisms on comparison have been found to have a standard size ranging from 79,733 bp to 108,623 bp and contain syntenous sets of 52 conserved core genes comprising their genetic backbone and each ICE also has genes specific for individual elements (167). There are variable regions of 676 to 29, 210 bp too, mainly concentrated in 5 hotspots which are intergenic regions of insertions/acquisitions of new DNA in the genetic backbone of the SXT element (167). These observations were derived by comparison of the SXT of VC O139 with that of R391 strain of Providencia rettgeri using the programs MAUVE and LAGAN which enable visualization of core and variable regions on a global scale (166, 167). The hotspots are HS1-HS5 (167). The first one occurs between sO43 and traL, the second one lies between trA and sO54, and the third one between sO73 and traF (166). Toma et al. identified a new SXT designated SXT LAOS that contains 2 novel open reading frames (ORFs) in the third hot spot (between sO73 and traF) (166). It is different from SXTET which contains a class 9 integron in hot spot sO73-traF that harbors dfrA1 as a gene cassette (166). In SXTMO10, the gene encoding trimethoprim resistance, dfr18 is encoded in the ≈17.2-kbp composite transposon-like element which interrupts the SXT-encoded rumAB operon (166). SXTLAOS does not encode dfr18 nor dfrA1 (166). On sequence analysis the authors found that the region between sO43 and traL showed 97% identity to the corresponding region of P. rettgeri R391 which encodes 2 hypothetical proteins (ORF 37 and ORF 38) (166). The region between traA and sO54 showed 97% identity to the corresponding region of SXTMO10 and that the region between sO26 and sO27 in SXTLAOS is also different from SXTMO10 (166). Wang et al. reported about the presence of two distinct types of ICEs in China among the 11 distinct SXT elements that they sequenced (224). These elements differed from one another on the basis of sequences of conserved genes (224). In addition, there was a deletion of 17.8kb region from s026 to s040 in all of the 11 SXT elements and all the 11 SXT elements were found to have insertions at H3 and H4 (224). Rashed et al., conducted molecular characterization and antibiotic susceptibility screening of 97 VC O1 strains from Bangladesh collected during 2010 and 2014 (168). Except two strains, all others were found to harbor the intSXT gene, the gene for SXT related integrase and 93% of the strains were MDR (168). 100% of the strains showed resistance to streptomycin (S) and sulfamethoxazole-trimethoprim (SXT) in 2010, 2011, and 2012 (168). However, S and SXT resistance fell to 96% the following 2 years, 2013 and 2014 (168). The SXT-related integrase (intSXT) was detected in all isolates resistant to S and SXT, suggesting that the SXT/R391 ICE has mediated resistance to S and SXT (168).
Recently, Sarkar et al. reported about two variants of SXT elements in strains from cholera patients in Kolkata, collected from 2008-2015 (27). The two elements were designated as ICE TET and ICE GEN and were the major determinants of AMR in these strains (27). ICE TET carried tetA, sulII, strAB, dfrA1 while ICE GEN carried floR, sulII, strAB, dfrA1 (27). ICEGEN resembled the ICEVchInd5 with 99% identity at 100% query coverage while the ICETET had only 99% identity at 70% query coverage (27). ICEGEN was found to be 96.7 kb while ICE TET was 91.5kb (27). The findings clearly depict that recombination in SXT is a frequent event and HGT of SXT is a common mechanism of dissemination of AMR in VC. Beaber et al., had shown that although the G+C content of the SXT region was 47.1% similar to that of the VC genome (47.6%), there were certain regions within the SXT element with different G+C composition like the ARGs embedded in the transposon-like structure which has a G+C content of 51.9% (106). Beaber et al., has demonstrated how environmental factors and antibiotics induce the activation of SOS response which in turn alleviates the expression of SetR, the repressor of activators of genes used for SXT transfer (96). Therefore, antibiotics via SOS response increases the expression of genes required for SXT transfer and hence increases the frequency of transfer (96). This phenomenon was demonstrated by growing VC cells in the presence of ciprofloxacin and also mitomycin C, a known DNA damaging agent. The transfer frequency of SXT element increased via induction of the SOS response (96). The authors proposed a regulatory pathway to demonstrate the SOS induction mediated augmentation of SXT transfer in VC (96). Accordingly, the co-protease activity of RecA protein becomes activated during the SOS response (96). Activated RecA facilitates the autocleavage of SetR and alleviates the repression of s086, setD and setC expression (96). Increased levels of SetC and SetD lead to the autoactivation of setD and setC and also of the tra and int loci (96). The studies described have provided with a clear perspective on the structural aspects of the SXT ICE and the mechanism it utilizes to excise and transfer from one host to another thereby contributing to the dissemination of MDR traits which are constantly undergoing rearrangement (27, 166) in response to the driving force for dissemination, namely, antibiotics and other environmental signals (96, 106, 157).
These are mobile genetic elements that integrate into non-homologous target sites in plasmids or chromosomes (169–171). Some transposons show target site selectivity while others show little selectivity wherein they avoid certain sites or regions (169). Target site selectivity ensures safe propagation of these elements and optimum host-element relationship (169, 170). Transposons were originally discovered as MGEs which confer antibiotic resistance (170). They exist either as part of a plasmid or on a chromosome (171). They can jump from one plasmid to another, from plasmid to a chromosome or from one part of a chromosome to another part or from one chromosome to another (171). They contain inverted repeat (IR) sequences and a recognizable IS (insertion sequence) element flanking an ARG (170). The genes for drug resistance and other functions like heavy metal resistance and those enhancing metabolic plasticity (172) appear between the IR sequences (170). The IR along with these genes is called a transposon (Tn) which is therefore, longer than an IS element as a Tn contains the protein-coding genes (170). In many instances IR sequences have been found to be a pair of IS elements (170). Transposons and IS elements are grouped together under Transposable elements (170). Other genetic sequences associated with transposons are transposase gene (tnpA), resolution site (res) and resolvase gene (tnpR) (173) which are involved in target site selection. tnpA encodes the transposase, which is usually the element-encoded recombinase which promotes transposition (169). There are two mechanisms of recombination called transposition and retrotransposition respectively (169). During transposition the ends of the mobile element is cleaved from the donor site and these cleaved ends are joined to the target site by DNA strand transfer reactions (169). Prokaryotic TEs use this pathway, usually (174). In the other pathway, DNA, RNA and reverse transcriptase participate in the recombination (169, 174). Elements which employ this pathway are called PolyA+ or non-LTR retrotransposons (169). During retrotransposition DNA cleavage takes place at the target site of element insertion, exposing a 3’-OH which provides a primer for reverse transcription, uses an element RNA as the template and finally the DNA form of the element is inserted at this site (169). A number of transposons have been described in VC, associated with antibiotic resistance like Tn1 (ampicillin) (175). Transposons have been classified into retrotransposons (class I) and DNA transposons (class II) (176). Bacterial transposons are class II transposons (176).
Transposons have been traditionally used for DNA fingerprinting of VC due to the IS elements that are characteristic traits of these MGEs (177). Bik et al., characterized a novel IS element IS1004 in Vibrio sp (177). It is a 628bp element containing an ORF whose product showed a large degree of sequence identity with the Tpase encoded by IS200 (177). It was successfully used to differentiate between O1 and non-O1 strains, and within the O1 serotype it was possible to distinguish between the classical and El Tor biotypes (177). IS elements are usually involved in no other function other than their mobility, contain sequences which participate in recombination and define the boundary of the element (178). They usually do not contain any other genetic sequences other than that encoding the transposase (Tpase) which processes these recombinationally active ends (178). These terminal IR sequences are a short stretch of 10bp-40bp (178). ISs are classified according to (i) the length and sequence of IR, (ii) the length and sequence of direct target DNA repeats (DR), (iii) the organization of their open reading frames (ORFs), (iv) target sites, (v) Tpases (174).
Apart from being used for molecular typing and as genetic manipulation tools (169) transposons help to disseminate ARGs (175, 179). Goldstein et al., found the existence of a transposon Tn1527 in the VC El Tor strain BM2508 which was isolated from the feces of a child suffering from severe diarrhea (179). It was 14kb and closely related to Tn7 (179). This transposon was found to confer resistance to trimethoprim and O/129 (179). Resistance to the former was due to a dihydrofolate reductase type I and resistance to streptomycin-spectinomycin was due to a 3'',9-aminoglycoside-aminocyclitol adenylyltransferase (179). The ARGs were not transferable to E.coli and were found to reside on the chromosome (179). The ARGs were transposed to multiple sites of plasmids belonging to incompatibility groups 6-C and P and introduced into BM2508 and subsequently transferred to other Gram-negative bacteria (179).
Recently, in a V. cholerae non-O1/non-O139 clinical isolate HC-36A1 from Haiti, isolated in 2010, a mobilizable genomic island MGIVchHai6 was discovered in chromosome 1 by Carraro et al. (142). MGIVchHai6 contains a mercury resistance transposon and an integron In104-like multidrug resistance element (142). It was found to confer resistance to β-lactams, SXT, tetracycline, chloramphenicol and streptomycin/spectinomycin (142). It was shown that MGIVchHai6 excises from the chromosome in an AcaCD-dependent manner and is mobilized by ACPs (142). ICEs described in the preceding section 4c carry resistance determinants against an array of antibiotics (106). They are conjugative transposons and may replicate autonomously by rolling circle model of replication and require the mediation of relaxase and specific helicase (180).
The analysis of the resistance determinants in VC show that most of these have been acquired from other organisms from time to time like the blaTEM encoding TEM-63 β-lactamase borne by a plasmid and which had been associated with nosocomial infections due to K. pneumoniae in South Africa and later acquired by VC O1 (140). Fonsesca et al. isolated Class 1 integron borne qnr-like genes (qnrVC1 and qnrVC2) in clinical O1 VC strains from Brazil (151). qnr genes have been found mainly in the family Enterobacteriaceae, and borne on plasmids (151). Vibrionaceae is considered to be the source of these genes (151). However, TMQR (transferable mechanisms of quinolone resistance) had not been earlier reported in VC (181) till 1998 when the first TMQR, QnrA was reported (181). These findings indicate that the transferable qnr genes have been introduced into VC from another organism with the help of MGEs and therefore, qnr genes borne on MGEs have started being isolated (151, 181). These findings revealed the importance of the MGEs in dissemination of resistance determinants overriding taxonomic boundaries and introducing novel resistance determinants and mechanisms of resistance in VC (151, 181). The dissemination of ARGs occur in response to antibiotics and other environmental signals and may also be induced by predatory mechanisms of VC (96, 119). VC has been found to employ its T6SS machinery for neighbor predation and in the process, the DNA that is released from the destroyed non-immune neighboring cells induces NT in VC (119). Many of the genes encoding antibiotic resistance help to enhance fitness and survival of VC enabling it to tide over the deleterious antibiotics (31, 114). These genetic exchanges have been facilitated by the ecological interactions of these organisms in different ecosystems like the gut and the aquatic ecosystem which is the natural habitat of VC (14, 15, 39, 144).
Revolutionary discoveries on the structure and function of the gut and environmental microbiome where these interactions occur have been facilitated by NGS techniques like metagenomic analysis (39, 44, 182). These have provided an insight into the ecological role of the organisms that constitute the microbiota and engage in the social and adaptive interactions (15, 43). It is now evident that organisms do not exist in isolation but reside in a milieu in which they have to closely interact with other organisms (15, 43, 182, 183). These interactions often shape their core and pan-genome and contribute to their genomic evolution (46, 184). Recent evidence provided by extensive genetic analysis of VC strains from diarrheal patients has shown the presence of a wide diversity of AMR determinants including novel ARGs and novel MGEs in the VC genome (26, 27). The MGEs on which the ARGs reside have the capacity to excise and transfer to other cells by the various HGT mechanisms already discussed in the review (23). Therefore, this is how VC acquires ARGs from other organisms resulting in development of AMR and consequently, of variable pattern of its AMR (23). The microbiota has been found to exert significant influence on the generation of genomic diversity of VC w.r.t. its virulence and MDR traits (46, 185, 186). Therefore, it plays a crucial role in determining virulence, pathogenicity and its response to antibiotics (14, 185). Genetic exchange in the microbiota does not occur randomly (187). It is facilitated only between certain groups of bacteria engaging in social interaction (187). These groups comprise what is known as exchange communities (182, 183, 187). HGT does not occur by random connivance but due to need (96, 115). This has been demonstrated by Jain et al., who analyzed 20,000 ORFs (Open Reading Frames) in 8 free-living prokaryotic genomes and concluded that HGT occurs between communities which share similar factors and is influenced by internal and environmental factors (187) which include genome size, G/C composition, carbon utilization, oxygen tolerance, synergism and competitive inhibition and selective forces in the environment (39, 187, 188). These are the same factors, therefore, which also facilitate HGT of ARGs (115, 187). Therefore, VC is also engaged in ecological relationship with certain members of the microbiota which influence its genetic evolution (189). Recent studies have clearly demonstrated this (189). Recent studies have shown that cholera is accompanied by a dysbiosis of the gut microbiome (189). The severe dehydration in cholera due to profuse efflux of water and electrolytes into the gut lumen results due to the effect of the enterotoxin CT (cholera toxin) (190). Before the onset of cholera, the microbiota bears a specific signature revealed from comparison of gut microbiome of cholera patients in the recovery phase with that of healthy adults and children (189). This signature has been found to consist of Bacteroides, Prevotella, Ruminococcus/Blautia, and Faecalibacterium (e.g., B. vulgatus, P. copri, R. obeum, and Faecalibacterium prausnitzii) and the relative abundance of these bacteria was found to increase with a shift from diarrheal to healthy adult state (191). This is disrupted by the entry of VC (191). These microbes can contribute significantly to healing of cholera and assumed to be involved in repair of the wounded gut (191). These members of the healthy gut is resurrected when the infection subsides (189). When VC enters the gut, it is found to eliminate the inherent flora by ejection of rice watery stool and comprises 50% of the bacterial burden in adult cholera stool in the diarrheal phase (189) and declines within hours of administration of ORS, where in Streptococcus sp. or Fusobacterium sp. or Campylobacter sp. become dominant (191). The organisms with which VC co-exists during diarrhea, may exert influence on VC w.r.t various aspects like niche competition, colonization resistance mechanisms, such as antimicrobial peptide production, nutrient competition, and intestinal barrier maintenance while VC senses those signals and modulates the expression of relevant genes to circumvent those stresses during infection, leading to successful colonization on the surface of small intestinal epithelial cells by employment of T6SS, QS, reactive oxygen species (ROS)/pH stress and bioactive metabolites (192). It is also in this milieu that optimal exchange of ARGs can occur (46) as exchange of ARGs is stress induced (96, 115, 119). This is possible, because the microbiome houses diverse ARGs of diverse taxonomic origin (43). It also has a high abundance of ARGs as revealed by several novel studies (45).
Advanced software are available for metadata analysis and these have helped in enhanced characterization of the microbiome, enabling us to get a clear picture of the taxonomic composition and also the transcriptome and proteome (44, 45). Its benefit has been revealed in the successful detection and estimation of higher abundance and diversity of resistance determinants in the gut than previously thought of (45). Lanza et al., developed ResCap, a targeted sequence capture platform based on SeqCapEZ (NimbleGene) technology, which includes probes for 8667 canonical resistance genes (7963 ARGs and 704 genes conferring resistance to metals or biocides), and 2517 relaxase genes (plasmid markers) and 78,600 genes homologous to the previous identified targets (47,806 for antibiotics and 30,794 for biocides or metals) (45). Its outcome was compared with that of metagenomic shotgun sequencing method in 17 fecal samples comprising 9 humans and 8 swine) (45). ResCap was proved to have enhanced capacity of identifying higher gene abundance and diversity and also helped in the identification of novel genes involved in antimicrobial and biocide resistance (45). With the aid of advanced platforms and by virtue of recent investigations it has been concluded that gut microbiome harbors a rich diversity of ARGs (45). Mu et al. used shotgun metagenomic sequencing of a human fecal sample from a nosocomial outbreak and a neural network algorithm based on tetranucleotide frequency profiling and reconstructed the genome of Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae in the patient fecal sample, thereby confirming the causative agent of the nosocomial outbreak and also a vancomycin-resistant Enterococcus faecium as a persistent colonizer of the patient (193). The study showed that the gut harbors ARGs against β-lactams, glycopeptides, macrolides, aminoglycosides, and tetracyclines (193). Another report based on whole-genome shotgun metagenomics performed using the Illumina MiSeq platform and analyzed with ABRicate showed that diarrheal fecal samples contain a pool of resistance determinants against the major classes of antibiotics and that Eschericia sp. was found to be the origin of most of these ARGs even in the gut metagenome obtained from cholera patients (43).
A recent study showed that the fecal gene pool contained ARGs many of which were found to be absent in the VC strains isolated from the same fecal sample (24). Molecular typing methods PCR and Sanger sequencing were aptly employed to study ARG diversity in diarrheal fecal samples (24). ARGs against 6 classes of antibiotics (macrolides, tetracyclines, aminoglycosides, sulphamethoxazole, trimethoprim, amphenicol) which are indispensable for diarrheal treatment were detected (24). This study showed that the gut microbiome serves as a reservoir of ARGs (24). The same study also reported the existence of markers of integrons and transposons which are essential for HGT (24). These conditions may facilitate the HGT of ARGs from the gut microbiome gene pool into VC genome (46). Several studies have demonstrated that the gut microbiota facilitates the transmission of ARGs into bacterial genome by HGT (194). Liu et al. identified 13,514 HGT genes in 308 human microbes and constructed an HGT event network which suggested that the human microbes could be divided into specific communities which only partly overlap their distribution in human body (194). The study showed that the human microbiome may facilitate frequent HGT among bacteria in the human body (194). Therefore, the role of the microbiome in the acquisition of AMR and consequently in its evolution becomes evident from these studies (24, 43, 194). Among the organisms with which VC was found to co-exist in the gut microbiome during the diarrheal phase (191) many commensals like F. prausnitzii and Bifidobacterium sp. have been found to be resident members of the commensal gut flora (43, 195). These commensals have often been found to be carriers of ARGs which reside in MGEs (195, 196). The microbiota having abundant resistance determinants (43, 45) facilitate transformation of VC in the gut in a contact-dependent manner or by direct uptake of exogenous DNA (197, 198). Evidence in favor of this hypothesis has already been provided by a recent study showing the significant role that the microbiome plays in the transfer of AMR to VC (46). The study involved characterization of AMR phenotypes and genotypes of five dominant commensal enteric bacteria isolated from the gut of adult healthy Indians and also characterization of the resistome of the fecal samples (46). The commensals included were, Faecalibacterium prausnitzii Indica, Megasphaera elsdenii Indica, Prevotella copri Indica, Collinsella aerofaciens Indica, Bifidobacterium longum Indica (46). In the resistome genes associated with tetracycline resistance were the most abundant in all the six metagenomes (46). β-lactamases were the second most abundant resistance genes across all the metagenomes (46). Genes that encoded resistance against macrolides, streptogramin, lincosamide, aminoglycosides, vancomycin, and fluoroquinolones were also detected in all the metagenomes (46). Bacteroides was the major genus contributing to the resistome, followed by Bifidobacterium, Escherichia, and Campylobacter (46). The enteric commensals were found to be MDR too when tested for antibiotic susceptibility and also on WGS analysis (46). More than 25 AMR genes that can encode resistance against β-lactam, aminoglycoside, macrolides, tetracycline, and fluoroquinolones were identified (46). The genes encoding antibiotic resistance were found to be physically linked with MGEs, namely, IS elements, complex transposons, bacteriophages, and GIs and could disseminate vertically to the progeny and laterally to the distantly related microbial species (46). VC was experimentally shown to take up these resistance genes by natural transformation from the pool of genomic DNA of enteric bacteria (46). The study successfully demonstrated that ARGs present in the chromosome of commensal gut bacteria was a source of resistance in VC thereby, clearly proving the role of the gut microbiome in shaping the acquired resistome of VC (46).
The VC genome comprises regions composed of genetic material acquired from exogenous sources (125). Analysis of these acquired genetic fragments have helped in the accumulation of evidence on the different HGT events and MGEs which have contributed towards this mosaic structure (137, 138). MGEs are the major vehicles of evolution, are propagators of ARGs and influence survival of the bacteria in its ecological niche in the environment and hosts (107). These assimilate genetic information from a multitude of organisms overriding taxonomic boundaries eventually leading to the evolution of the host cell acquiring it (29, 138). ARGs are exchanged within exchange communities and are induced by a host of internal and external factors (187). In VC genetic exchange with other organisms occur in the environment and the gut (14, 199). It also occurs among VC strains (14). Garg et al. found that non-O1/non-O139 and O1 VC influence the AMR profile of O139 strains circulating at the same time and spread rapidly giving rising to MDR strains (22). There are several reports which have revealed that ARGs located on MGEs have been introduced into VC by HGT (200). MGEs like plasmids and integrons carrying ARGs have been found to occur across different members of γ-Proteobacteria like the plasmid bearing the blaTEM gene encoding TEM-63 β-lactamase which was initially found in K. pneumoniae in South Africa and later isolated from VC in South Africa (140). Homologues of many ARGs which have been found to originate in Enterobacteriaceae have also been detected in VC (142, 201). The cat (chloramphenicol acetyltransferase) orthologs are distributed across γ-Proteobacteria (200). cat1 gene is found in Enterobacteriaceae and catB9 gene in Vibrionaceae (200). However, Ceccarelli et al., showed the presence of cat1 gene in VC (42). These findings have proved that transmission of ARGs across different taxonomic levels like bacterial families, genera and species occur and which are brought about by HGT and the same mechanisms affect the AMR evolution of VC (142). HGT occurs when presence of signals like stress (115), scarcity of nutrients (114), necessity for chromosomal repair (114), presence of antibiotics (96) stimulate the need for acquisition of genes that will help in adaptation and survival (115, 192). Exchange of genes, occurs within exchange communities within the microbiome (187) which provides the optimum milieu for genetic exchange by providing the optimum ambience and parameters required to induce HGT like niche competition, stress and adaptation (192). However, there are also several barriers to HGT and therefore, often, introduction of exogenous DNA into VC is a challenging affair (129). There are several decisive factors influencing the success of HGT mechanisms (202) . For conjugation to occur, the presence of F-factor is a prerequisite for mating pair formation (203). The frequency of transfer of a gene depends on its distance from the origin (203). Strains deficient in F-plasmid are incompetent to engage in conjugal transfer (203). Plasmid transfer efficiency depends on a number of biotic and abiotic parameters like temperature, nutrient concentration, pH, moisture, population densities, cell signaling, cell physiology, type of plasmid, donor, or recipient, and growth on surfaces versus in well-mixed liquids (202). Since conjugation relies on cell-cell contact and biofilms facilitate this physical contact therefore previously, it was considered that biofilms were hotspots of conjugation, however, this paradigm has been ruled out due to observations disfavoring this hypothesis (202). Accordingly, it was found that conjugation occurred with high frequency in certain well-defined narrow zones, in the initial stages of contact between distinct colonies of donors and recipients (202). Cellular segregation of individuals with distinct genotypes occur while only closest neighboring cells come in physical contact (202). Apart from these requirements environmental and cellular signals are required to induce transfer competence in donors of conjugative plasmids (202). Moreover, if non-conjugative elements are involved in conjugation, these have to be associated with the regulatory machinery of a conjugative plasmid for its mobilization and transfer thereby highlighting the stringent circumstances essential for the success of HGT (29). Regulatory mechanisms in response to environmental and physiological signals such as QS, the SOS response, extracytoplasmic stress and gene silencing by histone-like nucleoid structuring protein (H-NS) also affect conjugative transfer of some plasmids (202). Apart from these parameters, due to divergent evolution the machinery for conjugative transfer is often not compatible between different bacteria thereby serving as a barrier to conjugation (135).
NT too, have several prerequisites for DNA uptake to occur like the availability of chitin, inhibition of DNAses and restriction modification systems, availability of non-degraded intact DNA strands, activation of DNA transfer genes, activation of QS, presence of autoinducers, high cell density in biofilms with higher expression of the master regulator HapR which also represses the dns gene (122, 204, 205). However, MGEs like IncC plasmid, a common conjugative plasmid of Vibrionaceae and Enterobacteriaceae and a common propagator of MDR in VC has been found to have the capacity to overcome the inhibition by restriction-modification systems, including the CRISPR-Cas system of VC which accords adaptive immunity with the help of a conserved operon containing five genes (vcrx089-vcrx093) that confer a novel host defense evasion (hde) phenotype (205). vcrx089-vcrx090 promote resistance against type I restriction-modification while vcrx091-vcxr093 promote CRISPR-Cas evasion by repairing double-strand DNA breaks via recombination between short sequence repeats (205). vcrx091, vcrx092 and vcrx093 encode a single-strand binding protein, and a single-strand annealing recombinase and double-strand exonuclease related to Redβ and λExo of bacteriophage λ, respectively (205). Homologous genes of the ICE R391 also confer CRISPR-Cas evasion ability (205). Therefore, the conserved hde operon helps to expand the host range of large families of MGEs helping in dissemination of MDR (205). These findings reveal adaptations of MGEs occasionally found in VC which help these to override the barriers posed on HGT (205). The IncC plasmid is a common spreader of MDR in VC (29). Its conjugative transfer is activated by AcaCD, the master activator which also controls the excision and transfer of chromosomally integrated GIs like MGIVchHai6 (29). The latter is a MDR GI integrated at the 3′ end of trmE (mnmE or thdF) in chromosome 1 of non-O1/non-O139 VC clinical isolates from the cholera outbreak of 2010, in Haiti (29). In the presence of an IncC plasmid expressing AcaCD, MGIVchHai6 excises from the chromosome and transfers at high frequency (29). AcaCD drives the expression of genes, including xis and mobIM, involved in excision and mobilization and which are borne by the GI (29). A 49-bp fragment upstream of mobIM serves as the minimal origin of transfer (oriT) of MGIVchHai6 (29). IncC plasmid-encoded factors, including the relaxase TraI, were found to be required for GI transfer (29). In silico exploration of γ-Proteobacteria genomes led to the identification of 47 novel related and potentially AcaCD-responsive GIs in 13 different genera (29). These GIs integrate at trmE, yicC, or dusA and carry a diverse cargo of genes involved in phage resistance (29). However, drug-resistance genes were not found in these GIs suggesting their ancient origin and that MDR has been acquired recently (29). Thus, VC has to encounter innumerable adversities in the polymicrobial ecological niche it occupies in the gut and environment which necessitates the acquisition of these ARGs as an outcome of social adaptive behavior (206). These mechanisms have enabled the bacteria to successfully thrive against the forces of extinction for years and evolve its pathogenicity and acquire resistance from time to time necessitated by requirement for adaptation to the changing environmental scenario and continue its struggle against extinction due to natural antagonism and human intervention with antibiotics.
AMR in pathogens has emerged as one of the most critical problems in the domain of health-care (13). The indiscriminate use of antibiotics for clinical, veterinary, poultry, animal husbandry and agricultural usage, consumption of antibiotics in sub-lethal doses, disposal of industrial run-off from antibiotic manufacturing units and usage of non-pharmaceutical antibiotics have led to the severe AMR crisis challenging the antibiotic treatment regime today (8). The World Health Organization (WHO) recommends the use of azithromycin, erythromycin, tetracycline for the treatment of cholera (207). Recent studies showed VC strains to be susceptible to these drugs in a cholera endemic region (24), however, the gut microbiota was found to harbor genetic determinants for resistance against macrolides, aminoglycosides and tetracyclines in the fecal samples from which the VC strains were isolated (24). Given, the existence of MGEs like IncA/C plasmids which can overcome the barriers of HGT, it is not unlikely that most of these ARGs would enter the VC genome (205). Other studies discussed in the review also have shown the abundance of ARGs against different classes of antibiotics in the gut (193). At present isolates of VC from outbreaks in different geographical regions of the world are MDR (27, 28, 42, 142). Acquisition of drug-resistance is being triggered due to increased environmental pollution by antibiotics and other bacterial stress inducing factors (15). Environmental strains of VC belonging to serotypes other than O1 and O139 from water samples, animals and birds have also been found to harbor a wide range of ARGs located on MGEs including those encoding carbapenemases and other ß-lactamases (25, 154, 155). These findings show that environmental strains are serving as a reservoir of ß-lactamases in VC (13, 155). Some of these ARGs like NDM-1 are new for VC AMR (25). Bacterial species belonging to the family Enterobacteriaceae have been found to be challenged with the development of critical ß-lactamase resistance (208). Genetic analysis of VC AMR has shown that many of these ARGs have originated from Enterobacteriaceae (26) indicating that genetic exchange between members of Enterobacteriaceae and VC occurs in the different ecosystems (140) and the influence of Enterobacteriaceae on the genetic evolution of VC (140). NDM-1 in VC was first reported by Walsh et al. who detected the gene in seepage water samples from India and found it to be borne on the IncA/C plasmid (25). These have also found their way into clinical strains of VC (26). This is of tremendous concern for the health care domain (25). ARGs from environmental sources are a terrible nuisance (144). These enter into the food-chain from environmental strains (14, 144) as VC is associated with phytoplanktons, zooplanktons and also copepods in its natural habitat (14, 209). Copepods serve as food for human populations (14). Clinical strains may also contaminate environmental strains (25). The discovery of MDR environmental isolates of VC without MGEs are also of major concern (210, 211). Chromosomal inheritance of genes acquired by HGT and integrated with the chromosome are vertically transferred leading to inherited AMR traits and highlights the eventual disastrous consequences of HGT lying in chromosomal inheritance of laterally acquired traits (212). As already mentioned, need for adaptation leads to acquisition of genes of metabolic importance by HGT (11, 39). These integrate with the chromosome and are vertically inherited by later generations (212). Therefore, acquisition of AMR by HGT (210, 211) leads to persistence of AMR against common antibiotics in future generations (212). Tables 3–5 enumerate the different resistance markers associated with VC antibiotic resistance (AR) and which are useful for rapid screening and molecular detection of AMR. MGEs are major agents of VC evolution (125) and their contribution in the emergence of MDR VC has been acknowledged in the review (23, 138). In many instances it was found that the MGEs were novel indicating newer sources of introduction of AMR at play in VC indicating not only the evolution of AMR in VC but also of MGEs (27, 28, 166).
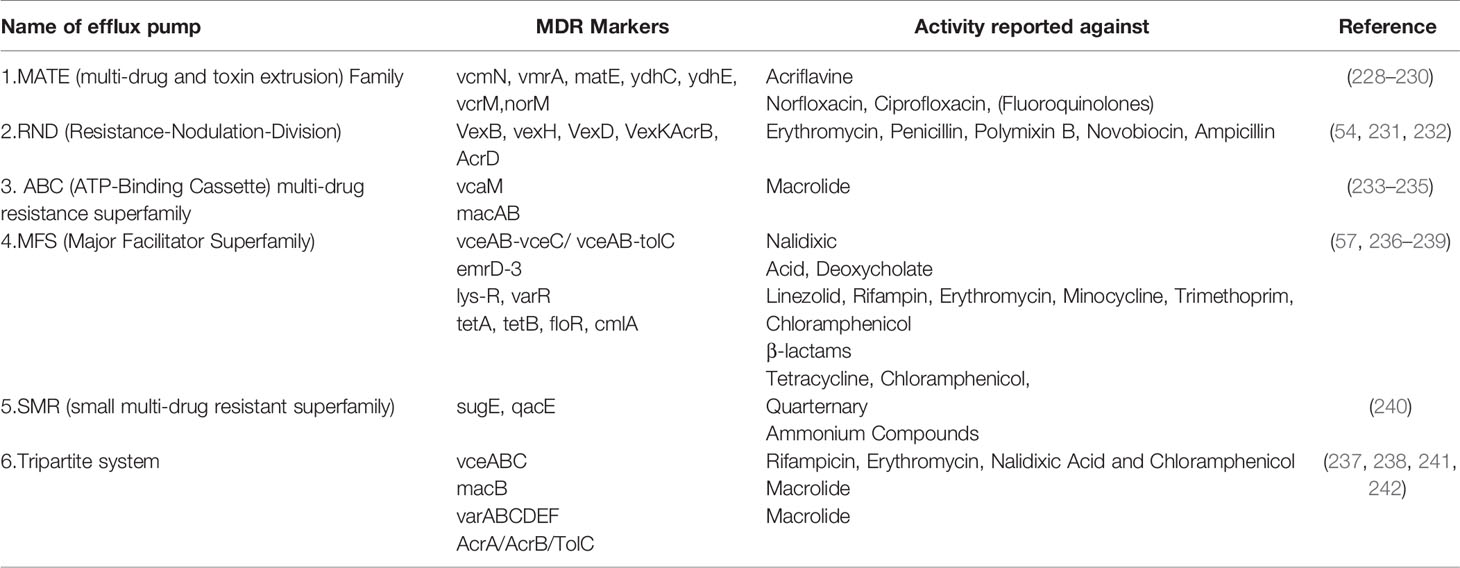
Table 4 Multidrug efflux pumps in V. cholerae and their genetic markers.

Table 5 Resistance genetic markers.
The review attempted to address the different resistance determinants that have been identified in VC and explore the mechanism of their acquisition and acknowledge the significance of MGEs and reservoirs of ARGs in the microbiome that have shaped the evolution of AMR in VC. It was evident that numerous environmental factors drive the occurrence of HGT and although, ARG transfer may not be the primary goal of ARG dissemination always, acquisition of resistance to antibiotics occurs as a side-effect of other adaptive strategies like adhesion and twitching motility induced by stress (115) although the selection pressure of antibiotics and other stress inducing parameters in the aquatic environment and the gut are crucial drivers for the spread and acquisition of resistance genes in VC (15, 23). AMR has emerged as one of the health-care priorities globally and efforts aiming to alleviate the crisis are being rigorously leveraged (13, 215). In the wake of these efforts the demand for new antimicrobials and alternative treatment with probiotics have been evoked (48, 215, 216). De et al., carried out WGS on Illumina MiSeq system and assembly of raw reads using metaSPAdes v3.9.1 to study the gut resistome of diarrheal patients and reported about the presence of 491 resistance determinants (43). Biosynthetic gene clusters (BGCs) associated with secondary metabolites were also recovered from the resistome and annotated with the help of antiSMASH algorithm (43). The authors found that bacteriocins and nonribosomal peptide synthetase (NRPS) were highly abundant in the diarrheal resistome (43), not only indicating antagonism in the gut (43) but also revealing the gut to be a source of novel antimicrobials (4, 217). Swift et al., investigated the potential of anaerobic gut fungi to synthesize natural products that could regulate the gut microbiome (216). They generated a catalog of natural products of anaerobic gut fungi and identified 146 genes that encode biosynthetic enzymes for diverse types of natural products like bacteriocins, NRPS and polyketide synthases (216). Experimentally the authors confirmed that 26% of total core biosynthetic genes in all the strains were transcribed (216). 30% of total biosynthetic gene products were detected via proteomics when grown on cellobiose (216). Liquid chromatography-tandem mass spectrometry (LC-MS/MS) helped to detect 72 likely natural products from A. robustus alone (216). A compound produced by all the strains of anaerobic fungi was putatively identified as the polyketide-related styrylpyrone baumin (216). The authors also detected three groups of natural products which were unique to anaerobic fungi (216). Bacteriocins are antimicrobial peptides and have been considered as promising alternative novel antimicrobials (218) and their potential of serving as probiotics has already been demonstrated (217). Bacteriocins have also been found to be able to inhibit growth of antibiotic-resistant pathogens without affecting the commensal flora and reestablishing resistance to the growth of the resistant pathogen, thereby, helping to inhibit AMR (219). Kim et al., showed that a four-strained consortium of commensal bacteria that contained Blautia producta BPSCSK was able to reverse antibiotic-induced susceptibility to vancomycin-resistant Enterococcus faecium (VRE) infection (219). The authors showed that BPSCSK reduced growth of VRE by secreting a lantibiotic similar to the nisin-A produced by Lactococcus lactis (219). Growth of VRE was inhibited by BPSCSK and L. lactis in vitro (219). BPSCSK colonized the colon and reduced VRE density in vivo (219). Compared to nisin-A, the BPSCSK lantibiotic had reduced activity against intestinal commensal bacteria (219). Several new techniques of reverting AMR have been demonstrated (220, 221) . One such technique is based on DNA sequence of the pathogen (221). Guanines in DNA or RNA assemble to form G-quadruplex (GQ) structures which have shown the potential of serving as drug targeting sites for pathogenic bacteria and viruses (221). Shankar et al., performed a genome-wide screening in VC using a bioinformatic approach (221). The authors observed ∼85 G-quadruplex forming motifs (VC-PGQs) in chromosome I and ∼45 putative G-quadruplexes (PGQs) in chromosome II of VC (221). Ten such motifs (VC-PGQs) were selected on the basis of conservation throughout the genus and functional analysis was performed revealing their location in the essential genes encoding bacterial proteins like methyl-accepting chemotaxis protein, orotate phosphoribosyl transferase protein, amidase proteins, etc. (221). The predicted VC-PGQs were validated using different biophysical techniques like Nuclear Magnetic Resonance (NMR) spectroscopy, Circular Dichroism (CD) spectroscopy, and electrophoretic mobility shift assay (EMSA) (221). These demonstrated the formation of highly stable GQ structures in VC (221). The interaction of these VC-PGQs with specific known GQ ligand, TMPyP4, was analyzed using ITC and molecular dynamics studies which displayed the stabilization of the VC-PGQs by the GQ ligands (221). The authors demonstrated the potential of this technique to serve as therapeutic strategy against VC by inhibiting the PGQ harboring gene expression, thereby inhibiting bacterial growth and virulence and has the potential to be used to treat the MDR problem of VC (221).
Apart from the currently available strategies being used for containing the AMR crisis, newer methods targeting the interruption of HGT mechanisms within the microbiome could be focused as HGT is the major route of AMR acquisition. Considerable amount of research is already being undertaken and steady progress has already been achieved in this direction (47). Few recent work include use of sex pilus-specific (SPS) phage to reduce the carriage of AMR plasmids (220), targeted killing of pathogenic bacteria (222), use of pathogen-microbiota antagonism and exploiting the barriers of HGT (47), plasmid curing and anti-plasmid methods (223).
Colom et al. using lytic bacteriophages which use plasmid-associated sex pili as attachment points demonstrated that SPS phage can kill drug-resistant E. coli and select for AMR plasmid loss in vitro (220). The authors demonstrated that SPS phage can inhibit the spread of resistant Salmonella enteritidis infection in chickens and shift the bacterial population towards antibiotic sensitivity (220). The authors used a model system comprising the Flac plasmid in a laboratory E. coli strain, and the SPS RNA phage MS2 30 to study plasmid loss in vitro (220). They also transferred the plasmid to a Salmonella enterica ser. Enteritidis strain to study phage activity and plasmid loss in vivo in a chicken model of infection (220).
Lopez-Igual et al., has exploited the targeted killing of pathogenic bacteria without affecting the beneficial members of the host microbiota to cure infections, rectify antimicrobial-related dysbiosis and inhibit the development of AMR by engineering toxins that are split by inteins and delivering them to a mixed population of bacteria (222). The authors reported that the toxin-intein antimicrobial was activated only in bacteria which harbored certain specific transcription factors and used the technology to specifically target and kill antibiotic-resistant VC in mixed populations with the outcome that 100% antibiotic-resistant VC receiving the plasmid were killed (222). Escape mutants were found to be extremely rare (10−6–10−8) and the authors showed that conjugation and specific killing of targeted bacteria could be achieved in the microbiota of zebrafish and crustacean larvae, the natural hosts for Vibrio sp. (222).
Baumgartner et al. has exploited the gut microcosm to quantify the effect of three human gut microbiome communities on growth and resistance evolution of a focal strain of E. coli and found that the resident microbial communities not only showed antagonistic effect by suppressing the growth and colonization by focal E. coli but also prevented it from evolving AMR upon exposure to a ß-lactam antibiotic (47). It was observed that with samples from all three human donors, the focal E. coli strain used in the study only evolved AMR in the absence of the resident microbial community, although resistance genes, including a highly effective resistance plasmid, in resident microbial communities were found (47). Physical constraints on plasmid transfer was responsible for inhibition of acquisition of ARGs by the focal E. coli strain and the authors observed that some chromosomal resistance mutations were only beneficial in the absence of the resident microbiota (47). The study revealed that depending on in situ gene transfer dynamics, interactions with resident microbiota can inhibit AMR evolution of individual species (47).
Most of these novel strategies have been developed utilizing the potential of the microbiome and aim to strike at the root of the problem by targeting HGT (47). Although, these are still in their infancy and confined to the experimental stage, being trialed and tested, appear to be promising. More focus should be cast on such strategies which if soon become available in the clinic would help to alleviate the AMR crisis in VC.
The author confirms being the sole contributor of this work and has approved it for publication.
This work was supported by the Department of Health Research, India, grant number R12015/01/2018-HR.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author thanks Dr. G. Balakrish Nair for his encouragement towards this work and Dr. Shanta Dutta, Director, NICED, for her invaluable support and edits.
1. Ali M, Nelson AR, Lopez AL, Sack DA. Updated Global Burden of Cholera in Endemic Countries. PLoS Negl Trop Dis (2015) 9(6):e0003832. doi: 10.1371/journal.pntd.0003832
2. World Health Organization database. (2021). Available at: https://www.who.int/news-room/fact-sheets/detail/cholera (Accessed May 22 2021).
3. Hsueh BY, Waters CM. Combating Cholera. F1000Res (2019) 8:589. doi: 10.12688/f1000research.18093.1
4. Rashed SM, Mannan SB, Johura FT, Islam MT, Sadique A, Watanabe H, et al. Genetic Characteristics of Drug-Resistant Vibrio Cholerae O1 Causing Endemic Cholera in Dhaka, 2006-2011. J Med Microbiol (2012) 61:1736–45. doi: 10.1099/jmm.0.049635-0
5. World Health Organization database. Weekly Epidemiological Record, No 37, 95 (2020). Available at: http://www.who.int/wer (Accessed 22 May 2021).
6. Veeraraghavan B, Walia K. Antimicrobial Susceptibility Profile & Resistance Mechanisms of Global Antimicrobial Resistance Surveillance System (GLASS) Priority Pathogens From India. Indian J Med Res (2019) 149:87–96. doi: 10.4103/ijmr.IJMR_214_18
7. Iskandar K, Molinier L, Hallit S, Sartelli M, Hardcastle TC, Haque M, et al. Surveillance of Antimicrobial Resistance in Low- and Middle-Income Countries: A Scattered Picture. Antimicrob Resist Infect Control (2021) 10:63. doi: 10.1186/s13756-021-00931-w
8. Aarestrup FM, Wegener HC, Collignon P. Resistance in Bacteria of the Food Chain: Epidemiology and Control Strategies. Expert Rev Anti Infect Ther (2008) 6:733–50. doi: 10.1586/14787210.6.5.733
9. Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, et al. Global Trends in Antimicrobial Use in Food Animals. Proc Natl Acad Sci USA (2015) 112:5649–54. doi: 10.1073/pnas.1503141112
10. Karkman A, Do TT, Walsh F, Virta MPJ. Antibiotic-Resistance Genes in Waste Water. Trends Microbiol (2018) 26(3):220–8. doi: 10.1016/j.tim.2017.09.005
11. Aminov RI. The Role of Antibiotics and Antibiotic Resistance in Nature. Environ Microbiol (2009) 11:2970–88. doi: 10.1111/j.1462-2920.2009.01972.x
12. Shin H, Kim Y, Han D, Hur HG. Emergence of High Level Carbapenem and Extensively Drug Resistant Escherichia Coli ST746 Producing NDM-5 in Influent of Wastewater Treatment Plant, Seoul, South Korea. Front Microbiol (2021) 12:645411. doi: 10.3389/fmicb.2021.645411
13. World Health Organization. No Time to Wait: Securing the Future From Drug-Resistant Infections (2019). Available at: https://www.who.int/antimicrobial-resistance/interagency-coordination-group/final-report/en/ (Accessed 20 May 2021).
14. Chen D, Li X, Ni L, Xu D, Xu Y, Ding Y, et al. First Experimental Evidence for the Presence of Potentially Toxic Vibrio Cholerae in Snails, and Virulence, Cross-Resistance and Genetic Diversity of the Bacterium in 36 Species of Aquatic Food Animals. Antibiotics (Basel) (2021) 10:412. doi: 10.3390/antibiotics10040412
15. Hendriksen RS, Lukjancenko O, Munk P, Hjelmsø MH, Verani JR, Ng’eno E, et al. Pathogen Surveillance in the Informal Settlement, Kibera, Kenya, Using a Metagenomics Approach. PLoS One (2019) 14:e0222531. doi: 10.1371/journal.pone.0222531
16. Bogdanova MI, Mishan’kin BN, Lebedeva SA. [Episomal Resistance of the Cholera Vibrio to Penicillin]. Antibiotiki (1971) 16:630–2.
17. Lindenbaum J, Greenough WB, Islam MR. Antibiotic Therapy of Cholera. Bull World Heal Organ (1967) 36:871–83.
18. Northrup RS, Doyle MA, Feeley JC. In Vitro Susceptibility of El Tor and Classical Vibrio Cholerae Strains to Trimethoprim and Sulfamethoxazole. Antimicrob Agents Chemother (1972) 1:310–4. doi: 10.1128/AAC.1.4.310
19. Glass RI, Huq I, Alim AR, Yunus M. Emergence of Multiply Antibiotic-Resistant Vibrio Cholerae in Bangladesh. J Infect Dis (1980) 142:939–42. doi: 10.1093/infdis/142.6.939
20. O’Grady F, Lewis MJ, Pearson NJ. Global Surveillance of Antibiotic Sensitivity of Vibrio Cholerae. Bull World Health Organ (1976) 54:181–5.
21. Poraziková T, Michalus M, Krcméry V. R Plasmids in Vibrionaceae - Beta-Lactamases in Vibrio Cholerae (NAG-Heiberg II) and A. Hydrophyla. Zentralblatt fur Bakteriol Parasitenkunde Infekt und Hyg Erste Abteilung Orig R A Medizinische Mikrobiol und Parasitol (1978) 242:481–6.
22. Garg P, Chakraborty S, Basu I, Datta S, Rajendran K, Bhattacharya T, et al. Expanding Multiple Antibiotic Resistance Among Clinical Strains of Vibrio Cholerae Isolated From 1992-7 in Calcutta, India. Epidemiol Infect (2000) 124:393–9. doi: 10.1017/s0950268899003957
23. Verma J, Bag S, Saha B, Kumar P, Ghosh TS, Dayal, et al. Genomic Plasticity Associated With Antimicrobial Resistance in Vibrio Cholerae. Proc Natl Acad Sci USA (2019) 116:6226–31. doi: 10.1073/pnas.1900141116
24. De R, Mukhopadhyay AK, Dutta S. Molecular Analysis of Selected Resistance Determinants in Diarrheal Fecal Samples Collected From Kolkata, India Reveals an Abundance of Resistance Genes and the Potential Role of the Microbiota in Its Dissemination. Front Public Heal (2020) 11:61. doi: 10.3389/fpubh.2020.00061
25. Walsh TR, Weeks J, Livermore DM, Toleman MA. Dissemination of NDM-1 Positive Bacteria in the New Delhi Environment and Its Implications for Human Health: An Environmental Point Prevalence Study. Lancet Infect Dis (2011) 11:355–62. doi: 10.1016/S1473-3099(11)70059-7
26. Mandal J, Sangeetha V, Ganesan V, Parveen M, Preethi V, Harish BN, et al. Third-Generation Cephalosporin-Resistant Vibrio Cholerae, India. Emerg Infect Dis (2012) 18:1326–8. doi: 10.3201/eid1808.111686
27. Sarkar A, Morita D, Ghosh A, Chowdhury G, Mukhopadhyay AK, Okamoto K, et al. Altered Integrative and Conjugative Elements (Ices) in Recent Vibrio Cholerae O1 Isolated From Cholera Cases, Kolkata, India. Front Microbiol (2019) 10:2072. doi: 10.3389/fmicb.2019.02072
28. Morita D, Takahashi E, Morita M, Ohnishi M, Mizuno T, Miyoshi SI, et al. Genomic Characterization of Antibiotic Resistance-Encoding Genes in Clinical Isolates of Vibrio Cholerae Non-O1/Non-O139 Strains From Kolkata, India: Generation of Novel Types of Genomic Islands Containing Plural Antibiotic Resistance Genes. Microbiol Immunol (2020) 64:435–44. doi: 10.1111/1348-0421.12790
29. Rivard N, Colwell RR, Burrus V. Antibiotic Resistance in Vibrio Cholerae: Mechanistic Insights From IncC Plasmid-Mediated Dissemination of a Novel Family of Genomic Islands Inserted at Trme. mSphere (2020) 5:e00748–20. doi: 10.1128/mSphere.00748-20
30. Leibovici-Weissman Y, Neuberger A, Bitterman R, Sinclair D, Salam MA, Paul M. Antimicrobial Drugs for Treating Cholera. Cochrane Database Syst Rev (2014) 2014:CD008625. doi: 10.1002/14651858.CD008625.pub2
31. Wang R, Liu H, Zhao X, Li J. and Wan K. IncA/C Plasmids Conferring High Azithromycin Resistance in Vibrio Cholerae. Int J Antimicrob Agents (2018) 51:140–4. doi: 10.1016/j.ijantimicag.2017.09.009
33. Mandal TK. Electron Microscopic Study of the Polymyxin Treated Vibrio Cholerae Cells. Z Naturforsch C J Biosci (1990) 45:902–10. doi: 10.1515/znc-1990-7-824
34. Zavascki AP, Goldani LZ, Li J, Nation RL. Polymyxin B for the Treatment of Multidrug-Resistant Pathogens: A Critical Review. J Antimicrob Chemother (2007) 60:1206–15. doi: 10.1093/jac/dkm357
35. Nair GB, Faruque SM, Bhuiyan NA, Kamruzzaman M, Siddique AK, Sack DA. New Variants of Vibrio Cholerae O1 Biotype El Tor With Attributes of the Classical Biotype From Hospitalized Patients With Acute Diarrhea in Bangladesh. J Clin Microbiol (2002) 40:3296–9. doi: 10.1128/JCM.40.9.3296-3299.2002
36. Samanta P, Saha RN, Chowdhury G, Naha A, Sarkar S, Dutta S, et al. Dissemination of Newly Emerged Polymyxin B Sensitive Vibrio Cholerae O1 Containing Haitian-Like Genetic Traits in Different Parts of India. J Med Microbiol (2018) 67:1326–33. doi: 10.1099/jmm.0.000783
37. Pa BB, Behera DR, Nayak SR, Nayak AK, Biswal B, Pati S. Dissemination of Polymyxin B Sensitivity in El Tor Vibrio Cholerae O1 Strains in Odisha, India. Jpn J Infect Dis (2021) 74:169–71. doi: 10.7883/yoken.JJID.2020.592
38. Bhattacharya K, Kanungo S, Sur D, Lal Sarkar B, Manna B, Lopez AL, et al. Tetracycline-Resistant Vibrio Cholerae O1, Kolkata, India. Emerg Infect Dis (2011) 17:568–9. doi: 10.3201/eid1703.101176
39. Aminov RI. Horizontal Gene Exchange in Environmental Microbiota. Front Microbiol (2011) 2:158. doi: 10.3389/fmicb.2011.00158
40. Samanta P, Mandal RS, Saha RN, Shaw S, Ghosh P, Dutta S, et al. A Point Mutation in Carr Is Involved in the Emergence of Polymyxin B-Sensitive Vibrio Cholerae O1 El Tor Biotype by Influencing Gene Transcription. Infect Immun (2020) 88:e00080–20. doi: 10.1128/IAI.00080-20
41. Das B, Martínez E, Midonet C, Barre FX. Integrative Mobile Elements Exploiting Xer Recombination. Trends Microbiol (2013) 21:23–30. doi: 10.1016/j.tim.2012.10.003
42. Ceccarelli D, Salvia AM, Sami J, Cappuccinelli P, Colombo MM. New Cluster of Plasmid-Located Class 1 Integrons in Vibrio Cholerae O1 and a dfrA15 Cassette-Containing Integron in Vibrio Parahaemolyticus Isolated in Angola. Antimicrob Agents Chemother (2006) 50:2493–9. doi: 10.1128/AAC.01310-05
43. De R, Mukhopadhyay AK, Dutta S. Metagenomic Analysis of Gut Microbiome and Resistome of Diarrheal Fecal Samples From Kolkata, India, Reveals the Core and Variable Microbiota Including Signatures of Microbial Dark Matter. Gut Pathog (2020) 12:32. doi: 10.1186/s13099-020-00371-8
44. De R. Metagenomics: Aid to Combat Antimicrobial Resistance in Diarrhea. Gut Pathog (2019) 11:47. doi: 10.1186/s13099-019-0331-8
45. Lanza VF, Baquero F, Martínez JL, Ramos-Ruíz R, González-Zorn B, Andremont A, et al. In-Depth Resistome Analysis by Targeted Metagenomics. Microbiome (2018) 6:11. doi: 10.1186/s40168-017-0387-y
46. Bag S, Ghosh TS, Banerjee S, Mehta O, Verma J, Dayal M, et al. Molecular Insights Into Antimicrobial Resistance Traits of Commensal Human Gut Microbiota. Microb Ecol (2019) 77:546–57. doi: 10.1007/s00248-018-1228-7
47. Baumgartner M, Bayer F, Pfrunder-Cardozo KR, Buckling A. And Hall, a. R. Resident Microbial Communities Inhibit Growth and Antibiotic-Resistance Evolution of Escherichia Coli in Human Gut Microbiome Samples. PLoS Biol (2020) 18:e3000465. doi: 10.1371/journal.pbio.3000465
48. Garcia-Gutierrez E, Mayer MJ, Cotter PD, Narbad A. Gut Microbiota as a Source of Novel Antimicrobials. Gut Microbes (2019) 10:1–21. doi: 10.1080/19490976.2018.1455790
49. Mao N, Cubillos-Ruiz A, Cameron DE, Collins JJ. Probiotic Strains Detect and Suppress Cholera in Mice. Sci Transl Med (2018) 10:eaao2586. doi: 10.1126/scitranslmed.aao2586
50. Bina XR, Howard MF, Taylor-Mulneix DL, Ante VM, Kunkle DE, Bina JE. The Vibrio Cholerae RND Efflux Systems Impact Virulence Factor Production and Adaptive Responses Via Periplasmic Sensor Proteins. PLoS Pathog (2018) 14:e1006804. doi: 10.1371/journal.ppat.1006804
51. Baharoglu Z, Krin E, Mazel D. Rpos Plays a Central Role in the SOS Induction by Sub-Lethal Aminoglycoside Concentrations in Vibrio Cholerae. PLoS Genet (2013) 9:e1003421. doi: 10.1371/journal.pgen.1003421
52. Dörr T, Delgado F, Umans BD, Gerding MA, Davis BM, Waldor MK. A Transposon Screen Identifies Genetic Determinants of Vibrio Cholerae Resistance to High-Molecular-Weight Antibiotics. Antimicrob Agents Chemother (2016) 60:4757–63. doi: 10.1128/AAC.00576-16
53. Kitaoka M, Miyata ST, Unterweger D, Pukatzki S. Antibiotic Resistance Mechanisms of Vibrio Cholerae. J Med Microbiol (2011) 60:397–407. doi: 10.1099/jmm.0.023051-0
54. Bina XR, Provenzano D, Nguyen N, Bina JE. Vibrio Cholerae RND Family Efflux Systems Are Required for Antimicrobial Resistance, Optimal Virulence Factor Production, and Colonization of the Infant Mouse Small Intestine. Infect Immun (2008) 76:3595–605. doi: 10.1128/IAI.01620-07
55. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, et al. DNA Sequence of Both Chromosomes of the Cholera Pathogen Vibrio Cholerae. Nature (2000) 406:477–83. doi: 10.1038/35020000
56. Begum A, Rahman MM, Ogawa W, Mizushima T, Kuroda T, Tsuchiya T. Gene Cloning and Characterization of Four MATE Family Multidrug Efflux Pumps From Vibrio Cholerae Non-O1. Microbiol Immunol (2005) 49:949–57. doi: 10.1111/j.1348-0421.2005.tb03690.x
57. Smith KP, Kumar S, Varela MF. Identification, Cloning, and Functional Characterization of EmrD-3, a Putative Multidrug Efflux Pump of the Major Facilitator Superfamily From Vibrio Cholerae O395. Arch Microbiol (2009) 191:903–11. doi: 10.1007/s00203-009-0521-8
58. Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, Takebayashi Y, et al. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J Mol Biol (2019) 431:3472–500. doi: 10.1016/j.jmb.2019.04.002
59. Reid AJ, Amyes SG. Plasmid Penicillin Resistance in Vibrio Cholerae: Identification of New Beta-Lactamase SAR-1. Antimicrob Agents Chemother (1986) 30:245–7. doi: 10.1128/aac.30.2.245
60. Poirel L, Jayol A, Nordmann P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin Microbiol Rev (2017) 30:557 LP – 596. doi: 10.1128/CMR.00064-16
61. Henderson JC, Fage CD, Cannon JR, Brodbelt JS, Keatinge-Clay AT. And Trent, M. SAntimicrobial Peptide Resistance of Vibrio Cholerae Results From an LPS Modification Pathway Related to Non Ribosomal Peptide Synthetases. ACS Chem Biol (2014) 9:2382–92. doi: 10.1021/cb500438x
62. Aldred KJ, Kerns RJ, Osheroff N. Mechanism of Quinolone Action and Resistance. Biochemistry (2014) 53:1565–74. doi: 10.1021/bi5000564
63. Pesci EC, Milbank JB, Pearson JP, McKnight S, Kende AS, Greenberg EP, et al. Quinolone Signaling in the Cell-to-Cell Communication System of Pseudomonas Aeruginosa. Proc Natl Acad Sci USA (1999) 96:11229–34. doi: 10.1073/pnas.96.20.11229
64. Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Cámara M. Quinolones: From Antibiotics to Autoinducers. FEMS Microbiol Rev (2011) 35:247–74. doi: 10.1111/j.1574-6976.2010.00247.x
65. Rijal N, Acharya J, Adhikari S, Upadhaya BP, Shakya G, Kansakar P, et al. Changing Epidemiology and Antimicrobial Resistance in Vibrio Cholerae: AMR Surveillance Findings (2006-2016) From Nepal. BMC Infect Dis (2019) 19:801. doi: 10.1186/s12879-019-4432-2
66. Lesher GY, Froelich EJ, Gruett MD, Bailey JH, Brundage RP. 1,8-NAPHTHYRIDINE Derivatives. A New Class of Chemotherapeutic Agents. J Med Pharm Chem (1962) 91:1063–5. doi: 10.1021/jm01240a021
67. Shungu DL, Weinberg E, Gadebusch HH. In Vitro Antibacterial Activity of Norfloxacin (MK-0366, AM-715) and Other Agents Against Gastrointestinal Tract Pathogens. Antimicrob Agents Chemother (1983) 23:86–90. doi: 10.1128/aac.23.1.86
68. Morris JGJ, Tenney JH, Drusano GL. In Vitro Susceptibility of Pathogenic Vibrio Species to Norfloxacin and Six Other Antimicrobial Agents. Antimicrob Agents Chemother (1985) 28:442–5. doi: 10.1128/aac.28.3.442
69. Grüneberg RN, Felmingham D, O’Hare MD, Robbins MJ, Perry K, Wall RA, et al. The Comparative In-Vitro Activity of Ofloxacin. J Antimicrob Chemother (1988) 22 Suppl C:9–19. doi: 10.1093/jac/22.supplement_c.9
70. Ichinose Y, Ehara M, Watanabe S, Shimodori S, Waiyaki PG, Kibue AM, et al. The Characterization of Vibrio Cholerae Isolated in Kenya in 1983. J Trop Med Hyg (1986) 89:269–76.
71. Nakasone N, Iwanaga M, Eeckels R. Characterization of Vibrio Cholerae 01 Recently Isolated in Bangladesh. Trans R Soc Trop Med Hyg (1987) 81:876–8. doi: 10.1016/0035-9203(87)90059-9
72. Saraswathi K, Deodhar LP. A Study of V. Cholerae Strains Isolated in Bombay. J Postgrad Med (1990) 36:128–30.
73. Yoshida H, Kojima T, Yamagishi J, Nakamura S. Quinolone-Resistant Mutations of the Gyra Gene of Escherichia Coli. Mol Gen Genet MGG (1988) 211:1–7. doi: 10.1007/BF00338386
74. Yoshida H, Bogaki M, Nakamura M, Nakamura S. Quinolone Resistance-Determining Region in the DNA Gyrase Gyra Gene of Escherichia Coli. Antimicrob Agents Chemother (1990) 34:1271–2. doi: 10.1128/aac.34.6.1271
75. Friedman SM, Lu T, Drlica K. Mutation in the DNA Gyrase A Gene of Escherichia Coli That Expands the Quinolone Resistance-Determining Region. Antimicrob Agents Chemother (2001) 45:2378–80. doi: 10.1128/AAC.45.8.2378-2380.2001
76. Kim HB, Wang M, Ahmed S, Park CH, LaRocque RC, Faruque ASG, et al. Transferable Quinolone Resistance in Vibrio Cholerae. Antimicrob Agents Chemother (2010) 54:799–803. doi: 10.1128/AAC.01045-09
77. Quilici ML, Massenet D, Gake B, Bwalki B, Olson DM. Vibrio Cholerae O1 Variant With Reduced Susceptibility to Ciprofloxacin, Western Africa. Emerg Infect Dis (2010) 16:1804–5. doi: 10.3201/eid1611.100568
78. Gould K. Antibiotics: From Prehistory to the Present Day. J Antimicrob Chemother (2016) 71:572–5. doi: 10.1093/jac/dkv484
79. Davies J, Davies D. Origins and Evolution of Antibiotic Resistance. Microbiol Mol Biol Rev (2010) 74:417–33. doi: 10.1128/MMBR.00016-10
80. Aminov RI. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front Microbiol (2010) 1:134. doi: 10.3389/fmicb.2010.00134
81. Chambers HF, Deleo FR. Waves of Resistance: Staphylococcus Aureus in the Antibiotic Era. Nat Rev Microbiol (2009) 7:134. doi: 10.1038/nrmicro2200
82. Davies J. Vicious Circles: Looking Back on Resistance Plasmids. Genetics (1995) 139:1465–8. doi: 10.1093/genetics/139.4.1465
83. Chakrabarty AN, Dastidar SG, Ganguli M, Chattopadhyay D. ‘DNA’ as Contaminants in Antibiotics and Its Capacity to Transform Bacteria to Drug Resistance. Indian J Exp Biol (1990) 28:58–62.
84. Webb V, Davies J. Antibiotic Preparations Contain DNA: A Source of Drug Resistance Genes? Antimicrob Agents Chemother (1993) 37:2379–84. doi: 10.1128/AAC.37.11.2379
85. Fevre C, Jbel M, Passet V, Weill FX, Grimont PA, Brisse S. Six Groups of the OXY beta-Lactamase Evolved Over Millions of Years in Klebsiella Oxytoca. Antimicrob Agent Chemother (2005) 49:3453–62. doi: 10.1128/AAC.49.8.3453-3462.2005
86. Woo PCY, Lau SKP, Huang Y, Yuen K. Genomic Evidence for Antibiotic Resistance Genes of Actinomycetes as Origins of Antibiotic Resistance Genes in Pathogenic Bacteria Simply Because Actinomycetes Are More Ancestral Than Pathogenic Bacteria. Med Hypotheses (2006) 67:1297–304. doi: 10.1016/j.mehy.2005.12.053
87. Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J. Functional Metagenomics Reveals Diverse β-Lactamases in a Remote Alaskan Soil. ISME J (2009) 3:243–51. doi: 10.1038/ismej.2008.86
88. Perry J, Waglechner N, Wright G. The Prehistory of Antibiotic Resistance. Cold Spring Harb Perspect Med (2016) 6:a025197. doi: 10.1101/cshperspect.a025197
89. Santiago-Rodriguez TM, Fornaciari G, Luciani S, Dowd SE, Toranzos GA, Marota I, et al. Gut Microbiome of an 11th Century A.D. Pre-Columbian Andean Mummy. PLoS One (2015) 10:e0138135. doi: 10.1371/journal.pone.0138135
90. Lupo A, Coyne S, Berendonk TU. Origin and Evolution of Antibiotic Resistance: The Common Mechanisms of Emergence and Spread in Water Bodies. Front Microbiol (2012) 3:18. doi: 10.3389/fmicb.2012.00018
91. Thungapathra M, Amita, Sinha KK, Chaudhuri SR, Garg P, Ramamurthy T, et al. Occurrence Ofantibiotic Resistance Gene Cassettesaac(6’) -Ib, dfrA5, dfrA12, and ereA2 in Class Iintegrons in Non-O1, Non-O139 Vibrio Cholerae Strains in India. Antimicrob Agents Chemother (2002) 46:2948–55. doi: 10.1128/AAC.46.9.2948-2955.2002
92. Mala W, Faksri K, Samerpitak K, Yordpratum U, Kaewkes W, Tattawasart U, et al. Antimicrobial Resistance and Genetic Diversity of the SXT Element in Vibrio Cholerae From Clinical and Environmental Water Samples in North-Eastern Thailand. Infect Genet Evol (2017) 52:89–95. doi: 10.1016/j.meegid.2017.04.013
93. Shi L, Fujihara K, Sato T, Ito H, Garg P, Chakrabarty R, et al. Distribution and Characterization Ofintegronsin Various Serogroups of Vibrio Cholerae Strains Isolated From Diarrhoeal Patients Between 1992 and 2000 in Kolkata, India. J Med Microbiol (2006) 55:575–83. doi: 10.1099/jmm.0.46339-0
94. Ochman H, Lawrence JG, Groisman EA. Lateral Gene Transfer and the Nature of Bacterial Innovation. Nature (2000) 18:299–304. doi: 10.1038/35012500
95. Abe K, Nomura N, Suzuki S. Biofilms: Hot Spots of Horizontal Gene Transfer (HGT) in Aquatic Environments, With a Focus on a New HGT Mechanism. FEMS Microbiol Ecology (2020) 96:fiaa031. doi: 10.1093/femsec/fiaa031
96. Beaber J, Hochhut B, Waldor M. SOS Response Promotes Horizontal Dissemination of Antibiotic Resistance Genes. Nature (2003) 427:72–4. doi: 10.1038/nature02241
97. Liao J, Chen Y, Huang H. Effects of CO2 on the Transformation of Antibiotic Resistance Genes Via Increasing Cell Membrane Channels. Environ Pollut (2019) 254:113045. doi: 10.1016/j.envpol.2019.113045
98. Wu HY, Shi DY, Yang D, Yin J, Yang ZW, Li JW, et al. Putative Environmental Levels of Levofloxacin Facilitate the Dissemination of Antibiotic-Resistant Escherichia Coli Via Plasmid-Mediated Transformability. Ecotoxicol Environ Saf (2020) 195:110461. doi: 10.1016/j.ecoenv.2020.110461
99. Lee H, Shin J, Chung YJ, Park M, Kang KJ, Baek JY, et al. Co-Introduction of Plasmids Harbouring the Carbapenemase Genes, blaNDM-1 and blaOXA-232, Increases Fitness and Virulence of Bacterial Host. J BioMed Sci (2020) 27:8. doi: 10.1186/s12929-019-0603-0
100. Weiss E, Scher C, Haas R, Fischer W. Excision and Transfer of an Integrating and Conjugative Element in a Bacterial Species With High Recombination Efficiency. Sci Rep (2019) 9:8915. doi: 10.1038/s41598-019-45429-z
101. Llosa M, Gomis-Rüth FX, Coll M, de la Cruz Fd F. Bacterial Conjugation: A Two-Step Mechanism for DNA Transport. Mol Microbiol (2002) 45:1–8. doi: 10.1046/j.1365-2958.2002.03014.x
102. Sagulenko E, Sagulenko V, Chen J, Christie PJ. Role of Agrobacterium Virb11 ATPase in T-Pilus Assembly and Substrate Selection. J Bacteriol (2001) 183:5813–25. doi: 10.1128/JB.183.20.5813-5825.2001
103. Cabezón E, Ripoll-Rozada J, Peña A, de la Cruz F, Arechaga I. Towards an Integrated Model of Bacterial Conjugation. FEMS Microbiol Rev(2015) 39:81–95. doi: 10.1111/1574-6976.12085
104. Wawrzyniak P, Płucienniczak G, Bartosik D. The Different Faces of Rolling-Circle Replication and Its Multifunctional Initiator Proteins. Front Microbiol (2017) 8:2353. doi: 10.3389/fmicb.2017.02353
105. Zatyka M, Thomas CM. Control of Genes for Conjugative Transfer of Plasmids and Other Mobile Elements. FEMS Microbiol Rev (1998) 21:291–319. doi: 10.1111/j.1574-6976.1998.tb00355.x
106. Beaber JW, Hochhut B, Waldor MK. Genomic and Functional Analyses of SXT, an Integrating Antibiotic Resistance Gene Transfer Element Derived From Vibrio Cholerae. J Bacteriol (2002) 184:4259–69. doi: 10.1128/JB.184.15.4259-4269.2002
107. Siddiqui MT, Mondal AH, Gogry FA, Husain FM, Alsalme A, Haq QMR. Plasmid-Mediated Ampicillin, Quinolone, and Heavy Metal Co-Resistance Among ESBL-Producing Isolates From the Yamuna River. New Delhi, India. Antibiotics (Basel) (2020) 9:826. doi: 10.3390/antibiotics9110826
108. Mohapatra SS, Mantri CK, Mohapatra H, Colwell RR, Singh DV. Analysis of Clonally Related Environmental Vibrio Cholerae O1 El Tor Isolated Before 1992 From Varanasi, India Reveals Origin of SXT-ICEs Belonging to O139 and O1 Serogroups. Environ Microbiol Rep (2010) 2:50–7. doi: 10.1111/j.1758-2229.2009.00051.x
109. Li Z, Pang B, Wang D, Li J, Xu J, Fang Y, et al. Expanding Dynamics of the Virulence-Related Gene Variations in the Toxigenic Vibrio Cholerae Serogroup O1. BMC Genomics (2019) 20:360. doi: 10.1186/s12864-019-5725-y
110. Waldor MK, Tschape H, Mekalanos JJ. A New Type of Conjugative Transposon Encodes Resistance to Sulfamethoxazole, Trimethoprim, and Streptomycin in Vibrio Cholerae O139. J Bacteriol (1996) 178:4157–65. doi: 10.1128/jb.178.14.4157-4165.1996
111. John TJ, Jesudason MV. The Spread of Vibrio Cholerae O139 in India. J Infect Dis (1995) 171:759–60. doi: 10.1093/infdis/171.3.759
112. Johnston C, Martin B, Fichant G, Polard P, Claverys JP. Bacterial Transformation: Distribution, Shared Mechanisms and Divergent Control. Nat Rev Microbiol (2014) 12:181–96. doi: 10.1038/nrmicro3199
113. Attaiech L, Charpentier X. Silently Transformable: The Many Ways Bacteria Conceal Their Built-in Capacity of Genetic Exchange. Curr Genet (2017) 63:451–5. doi: 10.1007/s00294-016-0663-6
114. Carvalho G, Fouchet D, Danesh G, Godeux AS, Laaberki MH, Pontier D, et al. Bacterial Transformation Buffers Environmental Fluctuations Through the Reversible Integration of Mobile Genetic Elements. mBio (2020) 11:e02443–19. doi: 10.1128/mBio.02443-19
115. Bakkali M. Could DNA Uptake be a Side Effect of Bacterial Adhesion and Twitching Motility? Arch Microbiol (2013) 195:279–89. doi: 10.1007/s00203-013-0870-1
116. Molina-Quiroz RC, Dalia TN, Camilli A, Dalia AB, Silva-Valenzuela CA. Prophage-Dependent Neighbor Predation Fosters Horizontal Gene Transfer by Natural Transformation. mSphere (2020) 5:e00975–20. doi: 10.1128/mSphere.00975-20
117. Meibom KL, Blokesch M, Dolganov NA, Wu CY, Schoolnik GK. Chitin Induces Natural Competence in Vibrio Cholerae. Science (2005) 310:1824–7. doi: 10.1126/science.1120096
118. Lloyd CJ, Mejia-Santana A, Dalia TN, Dalia AB, Klose KE. Natural Transformation in a Classical-Biotype Vibrio Cholerae Strain. Appl Environ Microbiol (2021) 87:e00060–21. doi: 10.1128/AEM.00060-21
119. Borgeaud S, Metzger LC, Scrignari T, Blokesch M. The Type VI Secretion System of Vibrio Cholerae Fosters Horizontal Gene Transfer. Science (2015) 347:63–7. doi: 10.1126/science.1260064
120. Jaskólska M, Stutzmann S, Stoudmann C, Blokesch M. QstR-Dependent Regulation of Natural Competence and Type VI Secretion in Vibrio Cholerae. Nucleic Acids Res (2018) 46:10619–34. doi: 10.1093/nar/gky717
121. Scrudato L, Blokesch M. The Regulatory Network of Natural Competence and Transformation of Vibrio Cholerae. PLoS Genet (2012) 8:e1002778. doi: 10.1371/journal.pgen.1002778
122. Blokesch M. A Quorum Sensing-Mediated Switch Contributes to Natural Transformation of Vibrio Cholerae. Mob Genet Elements (2012) 2:224–7. doi: 10.4161/mge.22284
123. Dalia AB, Seed KD, Calderwood SB, Camilli A. A Globally Distributed Mobile Genetic Element Inhibits Natural Transformation of Vibrio Cholerae. Proc Natl Acad Sci USA (2015) 112:10485–90. doi: 10.1073/pnas.1509097112
124. Chiang YN, Penadés JR, Chen J. Genetic Transduction by Phages and Chromosomal Islands: The New and Noncanonical. PLoS Pathog (2019) 15:e1007878. doi: 10.1371/journal.ppat.1007878
125. Faruque SM, Mekalanos JJ. Phage-Bacterial Interactions in the Evolution of Toxigenic Vibrio Cholerae. Virulence (2012) 3(7):556–65. doi: 10.4161/viru.22351
126. Williams N. Phage Transfer: A New Player Turns Up in Cholera Infection. Science (1996) 272:1869–70. doi: 10.1126/science.272.5270.1869
127. Waldor MK, Mekalanos JJ. Lysogenic Conversion by a Filamentous Phage Encoding Cholera Toxin. Science (1996) 272:9110–1914. doi: 10.1126/science.272.5270.1910
128. Johnson SR, Romig WR. Vibrio Cholerae Conjugative Plasmid pSJ15 Contains Transposable Prophage Dvca1. J Bacteriol (1981) 146:632–6388. doi: 10.1128/JB.146.2.632-638.1981
129. Guidolin A, Manning PA. Genetics of Vibrio Cholerae and Its Bacteriophages. Microbiol Rev (1987) 51(2):285–98. doi: 10.1128/mr.51.2.285-298.1987
130. Fortier LC, Sekulovic O. Importance of Prophages to Evolution and Virulence of Bacterial Pathogens. Virulence (2013) 4:354–65. doi: 10.4161/viru.24498
131. Silpe JE, Bassler BL. A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell (2019) 176:268–280.e13. doi: 10.1016/j.cell.2018.10.059
132. Kim EJ, Yu HJ, Lee JH, Kim JO, Han SH, Yun CH, et al. Replication of Vibrio Cholerae Classical CTX Phage. Proc Natl Acad Sci USA (2017) 114:2343–8. doi: 10.1073/pnas
133. Enault F, Briet A, Bouteille L, Roux S, Sullivan MB, Petit MA. Phages Rarely Encode Antibiotic Resistance Genes: A Cautionary Tale for Virome Analyses. ISME J (2017) 11:237–47. doi: 10.1038/ismej.2016.90
134. Moon K, Jeon JH, Kang I, Park KS, Lee K, Cha CJ, et al. Freshwater Viral Metagenome Reveals Novel and Functional Phage-Borne Antibiotic Resistance Genes. Microbiome (2020) 8:75. doi: 10.1186/s40168-020-00863-4
135. Smillie C, Garcillán-Barcia MP, Francia MV, Rocha EPC, de la Cruz F. Mobility of Plasmids. Microbiol Mol Biol Rev (2010) 74:434–52. doi: 10.1128/MMBR.00020-10
136. Clark AJ, Warren GJ. Conjugal Transmission of Plasmids. Annu Rev Genet (1979) 13:99–125. doi: 10.1146/annurev.ge.13.120179.000531
137. Hamood AN, Sublett RD, Parker CD. Plasmid-Mediated Changes in Virulence of Vibrio Cholerae. Infect Immun (1986) 52:476–83. doi: 10.1128/IAI.52.2.476-483.1986
138. Ceccarelli D, Garriss G, Choi SY, Hasan NA, Stepanauskas R, Pop M, et al. Characterization of Two Cryptic Plasmids Isolated in Haiti From Clinical Vibrio Cholerae Non-O1/Non-O139. Front Microbiol (2017) 8:2283. doi: 10.3389/fmicb.2017.02283
139. Zhang R, Wang Y, Leung PC, Gu J-D. pVC, a Small Cryptic Plasmid From the Environmental Isolate of Vibrio Cholerae MP-1. J Microbiol (2007) 45:193–8.
140. Ismail H, Smith AM, Sooka A, Keddy KH. Genetic Characterization of Multidrug-Resistant, Extended-Spectrum- β-Lactamase-Producing Vibrio Cholerae O1 Outbreak Strains, Mpumalanga, South Africa, 2008. J Clin Microbiol (2011) 49:2976–9. doi: 10.1128/JCM.00293-11
141. Wang R, Yu D, Zhu L, Li J, Yue J, Kan B. IncA/C Plasmids Harboured in Serious Multidrug-Resistant Vibrio Cholerae Serogroup O139 Strains in China. Int J Antimicrob Agents (2015) 45:249–54. doi: 10.1016/j.ijantimicag.2014.10.021
142. Carraro N, Rivard N, Ceccarelli D, Colwell RR, Burrus V. IncA/C Conjugative Plasmids Mobilize a New Family of Multidrug Resistance Islands in Clinical Vibrio Cholerae Non-O1/Non-O139. Isolates From Haiti. mBio (2016) 7:e00509–16. doi: 10.1128/mBio.00509-16
143. Wang R, Li J, Kan B. Sequences of a Co-Existing SXT Element, a Chromosomal Integron (CI) and an IncA/C Plasmid and Their Roles in Multidrug Resistance in a Vibrio Cholerae O1 El Tor Strain. Int J Antimicrob Agents (2016) 48:305–9. doi: 10.1016/j.ijantimicag.2016.05.020
144. Hammerl JA, Jäckel C, Bortolaia V, Schwartz K, Bier N, Hendriksen RS, et al. Carbapenemase VCC-1-Producing Vibrio Cholerae in Coastal Waters of Germany. Emerg Infect Dis (2017) 23:1735–7. doi: 10.3201/eid2310.161625
145. Folster JP, Katz L, McCullough A, Parsons MB, Knipe K, Sammons SA, et al. Multidrug-Resistant IncA/C Plasmid in Vibrio Cholerae From Haiti. Emerg Infect Dis (2014) 20:1951–3. doi: 10.3201/eid2011.140889
146. Hall RM, Collis CM. Mobile Gene Cassettes and Integrons: Capture and Spread of Genes by Site-Specific Recombination. Mol Microbiol (1995) 15:593–600. doi: 10.1111/j.1365-2958.1995.tb02368.x
147. Hall RM. Mobile Gene Cassettes and Integrons: Moving Antibiotic Resistance Genes in Gram-Negative Bacteria. Ciba Found Symp (1997) 207:192–5. doi: 10.1002/9780470515358.ch12
148. Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile Genetic Elements Associated With Antimicrobial Resistance. Clin Microbiol Rev (2018) 31:e00088–17. doi: 10.1128/CMR.00088-17
149. Labbate M, Boucher Y, Joss MJ, Michael CA, Gillings MR, Stokes HW. Use of Chromosomal Integron Arrays as a Phylogenetic Typing System for Vibrio Cholerae Pandemic Strains. Microbiology (2007) 153:1488–98. doi: 10.1099/mic.0.2006/001065-0
150. Gillings MR. Integrons: Past, Present, and Future. Microbiol Mol Biol Rev (2014) 78:257–77. doi: 10.1128/MMBR.00056-13
151. Fonseca EL, Dos Santos Freitas F, Vieira VV, Vicente ACP. New Qnr Gene Cassettes Associated With Superintegron Repeats in Vibrio Cholerae O1. Emerg Infect Dis (2008) 14:1129–31. doi: 10.3201/eid1407.080132
152. Clark CA, Purins L, Kaewrakon P, Focareta T, Manning PA. The Vibrio Cholerae O1 Chromosomal Integron. Microbiology (2000) 146:2605–12. doi: 10.1099/00221287-146-10-2605
153. Ahmed AM, Kawaguchi F, Shimamoto T. Class 2 Integrons in Vibrio Cholerae. J Med Microbiol (2006) 55:643–4. doi: 10.1099/jmm.0.46378-0
154. Aberkane S, Compain F, Barraud O, Ouédraogo AS, Bouzinbi N, Vittecoq M, et al. Non-O1/non-O139 Vibrio Cholerae Avian Isolate From France Cocarrying the Bla(VIM-1) and Bla(VIM-4) Genes. Antimicrob Agents Chemother (2015) 59:6594–6. doi: 10.1128/AAC.00400-15
155. Wu J, Xie L, Zhang F, Ni Y, Sun J. Molecular Characterization of ISCR1-Mediated blaPER-1 in a Non-O1, Non-O139 Vibrio Cholerae Strain From China. Antimicrob Agents Chemother (2015) 59:4293–5. doi: 10.1128/AAC.00166-15
156. Hirsch N, Kappe E, Gangl A, Schwartz K, Mayer-Scholl A, Hammerl JA, et al. Phenotypic and Genotypic Properties of Vibrio Cholerae Non-O1, Non-O139 Isolates Recovered From Domestic Ducks in Germany. Microorganisms (2020) 8:1104. doi: 10.3390/microorganisms8081104
157. Beaber JW, Burrus V, Hochhut B, Waldor MK. Comparison of SXT and R391, Two Conjugative Integrating Elements: Definition of a Genetic Backbone for the Mobilization of Resistance Determinants. Cell Mol Life Sci (2002) 59:2065–70. doi: 10.1007/s000180200006
158. Pang B, Du P, Zhou Z, Diao B, Cui Z, Zhou H, et al. The Transmission and Antibiotic Resistance Variation in a Multiple Drug Resistance Clade of Vibrio Cholerae Circulating in Multiple Countries in Asia. PLoS One (2016) 11:e0149742. doi: 10.1371/journal.pone.0149742
159. Hochhut B, Lotfi Y, Mazel D, Faruque SM, Woodgate R, Waldor MK. Molecular Analysis of Antibiotic Resistance Gene Clusters in Vibrio Cholerae O139 and O1 SXT Constins. Antimicrob Agents Chemother (2001) 45:2991–3000. doi: 10.1128/AAC.45.11.2991-3000.2001
160. McLeod SM, Burrus V, Waldor MK. Requirement forIntegration Host Factor in Conjugative DNA Transfer. Journ Bact (2006) 188:5704–11. doi: 10.1128/JB.00564-06
161. Amita, Chowdhury SR, Thungapathra M, Ramamurthy T, Nair GB, Ghosh A. Class I Integrons and SXT Elements in El Tor Strains Isolated Before and After 1992 Vibrio Cholerae O139 Outbreak, Calcutta, India. Emerg Infect Dis (2003) 9:500–2. doi: 10.3201/eid0904.020317
162. Dalsgaard A, Forslund A, Sandvang D, Arntzen L, Keddy K. Vibrio Cholerae O1 Outbreak in Mozambique and South Africa in 1998 Are Multiple-Drug Resistant, Contain the SXT Element and the aadA2 Gene Located on Class 1 Integrons. J Antimicrob Chemother (2001) 48:827–38. doi: 10.1093/jac/48.6.827
163. Kiiru JN, Saidi SM, Goddeeris BM, Wamae NC, Butaye P, Kariuki SM. Molecular Characterisation of Vibrio Cholerae O1 Strains Carrying an SXT/R391-like Element From Cholera Outbreaks in Kenya: 1994-2007. BMC Microbiol (2009) 29:275. doi: 10.1186/1471-2180-9-275
164. Hounmanou YMG, Leekitcharoenphon P, Hendriksen RS, Dougnon TV, Mdegela RH, Olsen JE, et al. Surveillance and Genomics of Toxigenic Vibrio Cholerae O1 From Fish, Phytoplankton and Water in Lake Victoria, Tanzania. Front Microbiol (2019) 10:901. doi: 10.3389/fmicb.2019.00901
165. Ahmed AM, Shinoda S, Shimamoto T. A Variant Type of Vibrio Cholerae SXT Element in a Multidrug-Resistant Strain of Vibrio Fluvialis. FEMS Microbiol Lett (2005) 15:241–7. doi: 10.1016/j.femsle.2004.11.012
166. Toma C, Nakasone N, Song T, Iwanaga M. Vibrio Cholerae SXT Element, Laos. Emerg Infect Dis (2005) 11:346–7. doi: 10.3201/eid1102.040794
167. Wozniak RA, Fouts DE, Spagnoletti M, Colombo MM, Ceccarelli D, Garriss G. Comparative ICE Genomics: Insights Into the Evolution of the SXT/R391 Family of Ices. PLoS Genet (2009) 5:e1000786. doi: 10.1371/journal.pgen.1000786
168. Rashed SM, Hasan NA, Alam M, Sadique A, Sultana M, Hoq MM, et al. Vibrio Cholerae O1 With Reduced Susceptibility to Ciprofloxacin and Azithromycin Isolated From a Rural Coastal Area of Bangladesh. Front Microbiol (2017) 8:252. doi: 10.3389/fmicb.2017.00252
169. Craig NL. Target Site Selection in Transposition. Annu Rev Biochem (1997) 66:437–74. doi: 10.1146/annurev.biochem.66.1.437
170. Griffiths AJF, Miller JH, Suzuki DT, et al. An Introduction to Genetic Analysis. In: Freeman WH, editor. Prokaryotic Transposons, 7th edition. New York (2000). Available at: https://www.ncbi.nlm.nih.gov/books/NBK21818/.
171. Magagnin CM, Campos JC, da Rocha DA, Sampaio SCF, Rozáles FP, Barth AL, et al. Dissemination of Bla(OXA-370) Is Mediated by IncX Plasmids and the Tn6435 Transposon. Eur J Clin Microbiol Infect Dis (2018) 37:2165–9. doi: 10.1007/s10096-018-3356-x
172. Blackwell G, Iqbal Z, Thomson N. Evolution and Spread of Bacterial Transposons. Access Microbiol (2019) 1. doi: 10.1099/acmi.ac2019.po0568
173. El Salabi A, Walsh TR, Chouchani C. Extended Spectrum β-Lactamases, Carbapenemases and Mobile Genetic Elements Responsible for Antibiotics Resistance in Gram-Negative. Crit Rev Microbiol (2013) 39:113–22. doi: 10.3109/1040841X.2012.691870
174. Siguier P, Gourbeyre E, Chandler M. Bacterial Insertion Sequences: Their Genomic Impact and Diversity. FEMS Microbiol Rev (2014) 38:865–91. doi: 10.1111/1574-6976.12067
175. Johnson SR, Romig WR. Vibrio Cholerae Hybrid Sex Factor That Contains Ampicillin Transposon Tn1. J Bacteriol (1979) 137:531–6. doi: 10.1128/JB.137.1.531-536.1979
176. Babakhani S, Oloomi M. Transposons: The Agents of Antibiotic Resistance in Bacteria. J Basic Microbiol (2018) 58:905–17. doi: 10.1002/jobm.201800204
177. Bik EM, Gouw RD, Mooi FR. DNA Fingerprinting of Vibrio Cholerae Strains With a Novel Insertion Sequence Element: A Tool to Identify Epidemic Strains. J Clin Microbiol (1996) 34:1453–61. doi: 10.1128/JCM.34.6.1453-1461.1996
178. Makałowski W, Gotea V, Pande A, Makałowska I. Transposable Elements: Classification, Identification, and Their Use As a Tool for Comparative Genomics. In: Anisimova M, editor. Evolutionary Genomics: Statistical and Computational Methods. New York, NY: Springer New York (2019). p. 177–207. doi: 10.1007/978-1-4939-9074-0_6
179. Goldstein F, Gerbaud G, Courvalin P. Transposable Resistance to Trimethoprim and 0/129 in Vibrio Cholerae. J Antimicrob Chemother (1986) 17:559–69. doi: 10.1093/jac/17.5.559
180. Lee CA, Babic A, Grossman AD. Autonomous Plasmid-Like Replication of a Conjugative Transposon. Mol Microbiol (2010) 75:268–79. doi: 10.1111/j.1365-2958.2009.06985.x
181. Ruiz J. Transferable Mechanisms of Quinolone Resistance From 1998 Onward. Clin Microbiol Rev (2019) 32:e00007–19. doi: 10.1128/CMR.00007-19
182. Yaffe E, Relman DA. Tracking Microbial Evolution in the Human Gut Using Hi-C Reveals Extensive Horizontal Gene Transfer, Persistence and Adaptation. Nat Microbiol (2020) 5:343–53. doi: 10.1038/s41564-019-0625-0
183. Lerner A, Matthias T, Aminov R. Potential Effects of Horizontal Gene Exchange in the Human Gut. Front Immunol (2017) 8:1630. doi: 10.3389/fimmu.2017.01630
184. Levade I, Terrat Y, Leducq JB, Weil AA, Mayo-Smith LM, Chowdhury F, et al. Vibrio Cholerae Genomic Diversity Within and Between Patients. Microb Genom (2017) 3:e000142. doi: 10.1099/mgen
185. Midani FS, Weil AA, Chowdhury F, Begum YA, Khan AI, Debela MD, et al. Human Gut Microbiota Predicts Susceptibility to Vibrio Cholerae Infection. Jour Infect Dis (2018) 218:645–53. doi: 10.1093/infdis/jiy192
186. Alavi S, Mitchell JD, Cho JY, Liu R, Macbeth JC, Hsiao A. Interpersonal Gut Microbiome Variation Drives Susceptibility and Resistance to Cholera Infection. Cell (2020) 181:1533–46.e13. doi: 10.1016/j.cell.2020.05.036
187. Jain R, Rivera MC, Moore JE, Lake JA. Horizontal Gene Transfer Accelerates Genome Innovation and Evolution. Mol Biol Evol (2003) 20:1598–602. doi: 10.1093/molbev/msg154
188. van Elsas JD, Bailey MJ. The Ecology of Transfer of Mobile Genetic Elements. FEMS Microbiol Ecol (2002) 42:187–97. doi: 10.1111/j.1574-6941.2002.tb01008.x
189. Hay AJ, Zhu J. Microbiota Talks Cholera Out of the Gut. Cell Host Microbe (2014) 16:549–50. doi: 10.1016/j.chom.2014.10.011
190. Kaper JB, Morris JGJ, Levine MM. Cholera. Clin Microbiol Rev (1995) 8:48–86. doi: 10.1128/CMR.8.1.48-86.1995
191. Hsiao A, Ahmed AMS, Subramanian S, Griffin NW, Drewry LL, Petri, et al. Members of the Human Gut Microbiota Involved in Recovery From Vibrio Cholerae Infection. Nature (2014) 515:423–6. doi: 10.1038/nature13738
192. Qin Z, Yang X, Chen G, Park C, Liu Z. Crosstalks Between Gut Microbiota and Vibrio Cholerae. Front Cell Infect Microbiol (2020) 10:582554. doi: 10.3389/fcimb.2020.582554
193. Mu A, Kwong JC, Isles NS, Gonçalves da Silva A, Schultz MB, Ballard SA, et al. Reconstruction of the Genomes of Drug-Resistant Pathogens for Outbreak Investigation Through Metagenomic Sequencing. mSphere (2019) 4:e00529–18. doi: 10.1128/mSphere.00529-18
194. Liu L, Chen X, Skogerbø G, Zhang P, Chen R, He S, et al. The Human Microbiome: A Hot Spot of Microbial Horizontal Gene Transfer. Genomics (2012) 100:265–70. doi: 10.1016/j.ygeno.2012.07.012
195. Bag S, Ghosh TS, Das B. Complete Genome Sequence of Faecalibacterium Prausnitzii Isolated From the Gut of a Healthy Indian Adult. Genome Announc (2017) 5:e01286–17. doi: 10.1128/genomeA.01286-17
196. Duranti S, Lugli GA, Mancabelli L, Turroni F, Milani C, Mangifesta M, et al. Prevalence of Antibiotic Resistance Genes Among Human Gut-Derived Bifidobacteria. Appl Environ Microbiol (2017) 83:e02894–16. doi: 10.1128/AEM.02894-16
197. Bernardy EE, Turnsek MA, Wilson SK, Tarr CL, Hammer BK. Diversity of Clinical and Environmental Isolates of Vibrio Cholerae in Natural Transformation and Contact-Dependent Bacterial Killing Indicative of Type VI Secretion System Activity. Appl Environ Microbiol (2016) 82:2833–42. doi: 10.1128/AEM.00351-16
198. Veening J-W, Blokesch M. Interbacterial Predation as a Strategy for DNA Acquisition in Naturally Competent Bacteria. Nat Rev Microbiol (2017) 15:621–9. doi: 10.1038/nrmicro.2017.66
199. Campos LC, Zahner V, Avelar KES, Alves RM, Pereira DSG, Vital BJM, et al. Genetic Diversity and Antibiotic Resistance of Clinical and Environmental Vibrio Cholerae Suggests That Many Serogroups Are Reservoirs of Resistance. Epidemiol Infect (2004) 132:985–92. doi: 10.1017/s0950268804002705
200. Schwarz S, Kehrenberg C, Doublet B, Cloeckaert A. Molecular Basis of Bacterial Resistance to Chloramphenicol and Florfenicol. FEMS Microbiol Rev (2004) 28:519–42. doi: 10.1016/j.femsre.2004.04.001
201. Oyelade AA, Adelowo OO, Fagade OE. Bla(NDM-1)-Producing Vibrio Parahaemolyticus and V. Vulnificus Isolated From Recreational Beaches in Lagos, Nigeria. Environ Sci Pollut Res Int (2018) 25:33538–47. doi: 10.1007/s11356-018-3306-2
202. Stalder T, Top E. Plasmid Transfer in Biofilms: A Perspective on Limitations and Opportunities. NPJ Biofilms Microbiomes (2016) 2:16022. doi: 10.1038/npjbiofilms.2016.22
203. Griffiths AJF, Miller JH, Suzuki DT, et al. An Introduction to Genetic Analysis. In: Freeman WH, editor. Bacterial Conjugation, 7th edition. New York (2000).
204. Bellanger X, Payot S, Leblond-Bourget N, Guédon G. Conjugative and Mobilizable Genomic Islands in Bacteria: Evolution and Diversity. FEMS Microbiol Rev (2014) 38:720–60. doi: 10.1111/1574-6976.12058
205. Roy D, Huguet KT, Grenier F, Burrus V. Incc Conjugative Plasmids and SXT/R391 Elements Repair Double-Strand Breaks Caused by CRISPR-Cas During Conjugation. Nucleic Acids Res (2020) 18:48(16):8815–8827. doi: 10.1093/nar/gkaa518
206. Koraimann G, Wagner MA. Social Behavior and Decision Making in Bacterial Conjugation. Front Cell Infect Microbiol (2014) 4:54. doi: 10.3389/fcimb.2014.00054
207. Global Task Force on Cholera Control. Prevention and Control of Cholera Outbreaks: WHO Policy and Recommendations. (2021). Available at: https://www.who.int/cholera/technical/prevention/control/en/). (Accessed May 21 2021).
208. Islam MA, Islam M, Hasan R, Hossain MI, Nabi A, Rahman M, et al. Environmental Spread of New Delhi Metallo-β-Lactamase-1-Producing Multidrug-Resistant Bacteria in Dhaka, Bangladesh. Appl Environ Microbiol (2017) 83(15):e00793–17. doi: 10.1128/AEM.00793-17
209. Islam MS, Zaman MH, Islam MS, Ahmed N, Clemens JD. Environmental Reservoirs of Vibrio Cholerae. Vaccine (2020) 38(Suppl 1):A52–62. doi: 10.1016/j.vaccine.2019.06.033
210. Baron S, Larvor E, Chevalier S, Jouy E, Kempf I, Granier SA, et al. Antimicrobial Susceptibility Among Urban Wastewater and Wild Shellfish Isolates of Non-O1/Non-O139 Vibrio Cholerae From La Rance Estuary (Brittany, France). Front Microbiol (2017) 8:1637. doi: 10.3389/fmicb.2017.01637
211. Lepuschitz S, Baron S, Larvor E, Granier SA, Pretzer C, Mach RL, et al. Phenotypic and Genotypic Antimicrobial Resistance Traits of Vibrio Cholerae Non-O1/Non-O139 Isolated From a Large Austrian Lake Frequently Associated With Cases of Human Infection. Front Microbiol (2019) 10:2600. doi: 10.3389/fmicb.2019.02600
212. Florez-Cuadrado D, Moreno MA, Ugarte-Ruíz M, Domínguez L. Antimicrobial Resistance in the Food Chain in the European Union. Adv Food Nutr Res (2018) 86:115–36. doi: 10.1016/bs.afnr.2018.04.004
213. Mangat CS, Boyd D, Janecko N, Martz S-L, Desruisseau A, Carpenter M, et al. Characterization of VCC-1, a Novel Ambler Class A Carbapenemase From Vibrio Cholerae Isolated From Imported Retail Shrimp Sold in Canada. Antimicrob Agents Chemother (2016) 60:1819–25. doi: 10.1128/AAC.02812-15
214. Petroni A, Corso A, Melano R, Cacace ML, Bru AM, Rossi A, et al. Plasmidic Extended-Spectrum Beta-Lactamases in Vibrio Cholerae O1 El Tor Isolates in Argentina. Antimicrob Agents Chemother (2002) 46:1462–8. doi: 10.1128/aac.46.5.1462-1468.2002
215. World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics (2017). Available at: https://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/ (Accessed 20 May 2021).
216. Swift CL, Louie KB, Bowen BP, Olson HM, Purvine SO, Salamov A, et al. Anaerobic Gut Fungi Are an Untapped Reservoir of Natural Products. Proc Natl Acad Sci USA (2021) 118:e2019855118. doi: 10.1073/pnas.2019855118
217. Umu ÖC, Bäuerl C, Oostindjer M, Pope PB, Hernández PE, Pérez-Martínez G, et al. The Potential of Class II Bacteriocins to Modify Gut Microbiota to Improve Host Health. PLoS One (2016) 11:e0164036. doi: 10.1371/journal.pone.0164036
218. Cotter PD, Ross RP, Hill C. Bacteriocins - A Viable Alternative to Antibiotics? Nat Rev Microbiol (2013) 11:95–105. doi: 10.1038/nrmicro2937
219. Kim SG, Becattini S, Moody TU, Shliaha PV, Littmann ER, Seok R, et al. Microbiota-Derived Lantibiotic Restores Resistance Against Vancomycin-Resistant Enterococcus. Nature (2019) 572:665–9. doi: 10.1038/s41586-019-1501-z
220. Colom J, Batista D, Baig A, Tang Y, Liu S, Yuan F, et al. Sex Pilus Specific Bacteriophage to Drive Bacterial Population Towards Antibiotic Sensitivity. Scien Rep (2019) 9:12616. doi: 10.1038/s41598-019-48483-9
221. Shankar U, Jain N, Majee P, Kodgire P, Sharma TK, Kumar A. Exploring Computational and Biophysical Tools to Study the Presence of G-Quadruplex Structures: A Promising Therapeutic Solution for Drug-Resistant Vibrio Cholerae. Front Gen (2020) 11:935. doi: 10.3389/fgene.2020.00935
222. López-Igual R, Bernal-Bayard J, Rodríguez-Patón A, Ghigo J, Mazel D. Engineered Toxin–Intein Antimicrobials can Selectively Target and Kill Antibiotic-Resistant Bacteria in Mixed Populations. Nat Biotechnol (2019) 37:755–60. doi: 10.1038/s41587-019-0105-3pasteur-02558499
223. Buckner MMC, Ciusa ML, Piddock LJV. Strategies to Combat Antimicrobial Resistance: Anti-Plasmid and Plasmid Curing. FEMS Microbiol Rev (2018) 42:781–804. doi: 10.1093/femsre/fuy031
224. Wang R, Yu D, Yue J, Kan B. Variations in SXT Elements in Epidemic Vibrio Cholerae O1 El Tor Strains in China. Sci Rep (2016) 6:22733. doi: 10.1038/srep22733
225. Pugliese N, Maimone F, Scrascia M, Materu SF, Pazzani C. SXT-Related Integrating Conjugative Element and IncC Plasmids in Vibrio Cholerae O1 Strains in Eastern Africa. J Antimicrob Chemother (2009) 63:438–42. doi: 10.1093/jac/dkn542
226. Sjölund-Karlsson M, Reimer A, Folster JP, Walker M, Dahourou GA, Batra DG, et al. Drug-Resistance Mechanisms in Vibrio Cholerae O1 Outbreak Strain, Haiti, 2010. Emerg Infect Dis (2011) 17:2151–4. doi: 10.3201/eid1711.110720
227. Carraro N, Sauvé M, Matteau D, Lauzon G, Rodrigue S, Burrus V. Development of Pvcr94ΔX From Vibrio Cholerae, a Prototype for Studying Multidrug Resistant IncA/C Conjugative Plasmids. Front Microbiol (2014) 5:44. doi: 10.3389/fmicb.2014.00044
228. Kusakizako T, Tanaka Y, Hipolito CJ, Kuroda T, Ishitani R, Suga H, et al. LCP Crystallization and X-ray Diffraction Analysis of VcmN, a MATE Transporter From Vibrio Cholerae. Acta Crystallogr. Sect F Struct Biol Commun (2016) 72:552–7. doi: 10.1107/S2053230X16008931
229. Huda MN, Chen J, Morita Y, Kuroda T, Mizushima T, Tsuchiya T. Gene Cloning and Characterization of VcrM, a Na+-Coupled Multidrug Efflux Pump, From Vibrio Cholerae Non-O1. Microbiol Immunol (2003) 47:419–27. doi: 10.1111/j.1348-0421.2003.tb03379.x
230. Singh AK, Haldar R, Mandal D, Kundu M. Analysis of the Topology of Vibrio Cholerae NorM and Identification of Amino Acid Residues Involved in Norfloxacin Resistance. Antimicrob Agents Chemother (2006) 50:3717–23. doi: 10.1128/AAC.00460-06
231. Taylor DL, Bina XR, Bina JE. Vibrio Cholerae VexH Encodes a Multiple Drug Efflux Pump That Contributes to the Production of Cholera Toxin and the Toxin Co-Regulated Pilus. PLoS One (2012) 7:e38208. doi: 10.1371/journal.pone.0038208
232. Elkins CA, Nikaido H. Substrate Specificity of the RND-type Multidrug Efflux Pumps AcrB and AcrD of Escherichia Coli Is Determined Predominantly by Two Large Periplasmic Loops. J Bacteriol (2002) 184:6490–8. doi: 10.1128/jb.184.23.6490-6499.2002
233. Lu WJ, Lin HJ, Janganan TK, Li CY, Chin WC, Bavro VN, et al. ATP-Binding Cassette Transporter VcaM From Vibrio Cholerae Is Dependent on the Outer Membrane Factor Family for Its Function. Int J Mol Sci (2018) 19:1000. doi: 10.3390/ijms19041000
234. Huda N, Lee E-W, Chen J, Morita Y, Kuroda T, Mizushima T, et al. Molecular Cloning and Characterization of an ABC Multidrug Efflux Pump, VcaM, in Non-O1 Vibrio Cholerae. Antimicrob Agents Chemother (2003) 47:2413–7. doi: 10.1128/aac.47.8.2413-2417.2003
235. Lu S, Zgurskaya HI. Role of ATP Binding and Hydrolysis in Assembly of MacAB-TolC Macrolide Transporter. Mol Microbiol (2012) 86:1132–43. doi: 10.1111/mmi.12046
236. Vediyappan G, Borisova T, Fralick JA. Isolation and Characterization of VceC Gain-of-Function Mutants That Can Function With the AcrAB Multiple-Drug-Resistant Efflux Pump of Escherichia Coli. J Bacteriol (2006) 188:3757–62. doi: 10.1128/JB.00038-06
237. Colmer JA, Fralick JA, Hamood AN. Isolation and Characterization of a Putative Multidrug Resistance Pump From Vibrio Cholerae. Mol Microbiol (1998) 27:63–72. doi: 10.1046/j.1365-2958.1998.00657.x
238. Lin H-TV, Massam-Wu T, Lin C-P, Wang Y-JA, Shen Y-C, Lu W-J, et al. The Vibrio Cholerae Var Regulon Encodes a Metallo-β-Lactamase and an Antibiotic Efflux Pump, Which Are Regulated by VarR, a LysR-type Transcription Factor. PLoS One (2017) 12:e0184255. doi: 10.1371/journal.pone.0184255
239. Coyne S, Courvalin P, Périchon B. Efflux-Mediated Antibiotic Resistance in Acinetobacter Spp. Antimicrob Agents Chemother (2011) 55(3):947–53. doi: 10.1128/AAC.01388-10
240. Bay DC, Turner RJ. Diversity and Evolution of the Small Multidrug Resistance Protein Family. BMC Evol Biol (2009) 9:140. doi: 10.1186/1471-2148-9-140
241. Borges-Walmsley MI, Du D, McKeegan KS, Sharples GJ, Walmsley AR. Vcer Regulates the vceCAB Drug Efflux Pump Operon of Vibrio Cholerae by Alternating Between Mutually Exclusive Conformations That Bind Either Drugs or Promoter DNA. J Mol Biol (2005) 349:387–400. doi: 10.1016/j.jmb.2005.03.045
242. Greene NP, Kaplan E, Crow A, Koronakis V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front Microbiol (2018) 9:950. doi: 10.3389/fmicb.2018.00950
243. Nonaka L, Maruyama F, Suzuki S, Masuda M. Novel Macrolide-Resistance Genes, Mef(C) and Mph(G), Carried by Plasmids From Vibrio and Photobacterium Isolated From Sediment and Seawater of a Coastal Aquaculture Site. Lett Appl Microbiol (2015) 61:1–6. doi: 10.1111/lam.12414
244. Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. Characterization of the Vibrio Cholerae vexAB and vexCD Efflux Systems. Arch Microbiol (2006) 186:171–81. doi: 10.1007/s00203-006-0133-5
245. Rahman MM, Matsuo T, Ogawa W, Koterasawa M, Kuroda T, Tsuchiya T. Molecular Cloning and Characterization of All RND-Type Efflux Transporters in Vibrio Cholerae Non-O1. Microbiol Immunol (2007) 51:1061–70. doi: 10.1111/j.1348-0421.2007.tb04001.x
246. Sun J, Zhou M, Wu and Q, Ni Y. Characterization of Two Novel Gene Cassettes, dfrA27 and aadA16, in a Non-O1, Non-O139 Vibrio Cholerae Isolate From China. Clin Microbiol Infect (2010) 16:1125–9. doi: 10.1111/j.1469-0691.2009.03060.x
247. Po KHL, Wong MHY, Chen S. Identification and Characterisation of a Novel Plasmid-Mediated Quinolone Resistance Gene, qnrVC7, in Vibrio Cholerae of Seafood Origin. Int J Antimicrob Agents 45:667–8. doi: 10.1016/j.ijantimicag.2015.02.002
248. Zhou Y, Yu L, Li J, Zhang L, Tong Y, Kan B. Accumulation of Mutations in DNA Gyrase and Topoisomerase IV Genes Contributes to Fluoroquinolone Resistance in Vibrio Cholerae O139 Strains. Int. J Antimicrob Agents (2013) 42:72–5. doi: 10.1016/j.ijantimicag.2013.03.004
249. Okuda J, Hayashi N, Wakahara Y, Gotoh N. Reduced Expression of the vca0421 Gene of Vibrio Cholerae O1 Results in Innate Resistance to Ciprofloxacin. Antimicrob Agents Chemother (2010) 54:4917–9. doi: 10.1128/AAC.01652-09
250. Kumar P, Thomas S. Presence of qnrVC3 Gene Cassette in SXT and Class 1 Integrons of Vibrio Cholerae. Int J Antimicrob Agents (2011) 37:280–1. doi: 10.1016/j.ijantimicag.2010.12.006
251. Nfsa NADPH-dependent Nitroreductase NfsA [Escherichia Coli Str. K-12 Substr. MG1655]. Available at: https://www.ncbi.nlm.nih.gov/gene/945483 (Accessed June7 2021).
252. Kim YH, Jun LJ, Park S-H, Yoon SH, Chung JK, Kim JC, et al. Prevalence of Tet(B) and Tet(M) Genes Among Tetracycline-Resistant Vibrio Spp. In the Aquatic Environments of Korea. Dis Aquat Organ (2007) 75:209–16. doi: 10.3354/dao075209
Keywords: Vibrio cholerae, resistance, antimicrobial, microbiome, SXT, plasmid, integron, transposon
Citation: De R (2021) Mobile Genetic Elements of Vibrio cholerae and the Evolution of Its Antimicrobial Resistance. Front. Trop. Dis 2:691604. doi: 10.3389/fitd.2021.691604
Received: 06 April 2021; Accepted: 03 June 2021;
Published: 12 July 2021.
Edited by:
Alex Owusu-Ofori, Kwame Nkrumah University of Science and Technology, GhanaCopyright © 2021 De. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rituparna De, cml0dXBhcm5hMjZAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.