 María Belén Fernández
María Belén Fernández Christoph Bleidorn3
Christoph Bleidorn3- 1Fundación para el Estudio de Especies Invasivas (FuEDEI), Hurlingham, Argentina
- 2Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina
- 3Animal Evolution and Biodiversity, Johann-Friedrich-Blumenbach Institute for Zoology and Anthropology, Georg-August-University Göttingen, Göttingen, Germany
Antagonistic interactions can affect population growth and dispersal of an invasive species. Wolbachia are intracellular endosymbiont bacteria that infect arthropod and nematode hosts and are able to manipulate reproduction, which in some cases leads to cocladogenesis. Moreover, the presence of the strictly maternally transferred Wolbachia in a population can indirectly induce selective sweeps on the hosts’ mitochondria. Ants have a Wolbachia infection rate of about 34%, which makes phylogenetic studies using mitochondrial markers vulnerable of being confounded by the effect of the endosymbiont. Nylanderia fulva is an invasive ant native to South America, considered a pest in the United States. Its distribution and biology are poorly known in its native range, and the taxonomic identity of this and its closely related species, Nylanderia pubens, has only recently been understood with the aid of molecular phylogenies. Aiming at estimating robust phylogenetic relationships of N. fulva in its native range, we investigated the presence and pattern of Wolbachia infection in populations of N. fulva from Argentina, part of its native range, to account for its possible effect on the host population structure. Using the ftsZ gene, 30 nests of N. fulva and four from sympatric Nylanderia species were screened for the presence of Wolbachia. We sequenced the MLST genes, the highly variable gene wsp, as well as glyQ, a novel target gene for which new primers were designed. Phylogeny of the ants was estimated using mtDNA (COI). We found supergroup A Wolbachia strains infecting 73% of N. fulva nests and two nests of Nylanderia sp. 1. Wolbachia phylogenetic tree inferred with MLST genes is partially congruent with the host phylogeny topology, with the exception of a lineage of strains shared by ants from different N. fulva clades. Furthermore, by comparing with Wolbachia sequences infecting other ants, we found that the strains infecting different N. fulva clades are not monophyletic. Our findings suggest there are three recent independent horizontally transmitted Wolbachia infections in N. fulva, and we found no evidence of influence of Wolbachia in the host mtDNA based phylogeny.
Introduction
In the past few years, there has been increasing evidence of different endosymbiotic bacteria capable of affecting the biology of a variety of arthropod species, and most prominent are their effects on host reproduction (1). Among these organisms, the most common arthropod endosymbiont is Wolbachia (1). The Wolbachia genus encompasses a large phylogenetic diversity, with deeply diverging supergroups infecting arthropod and nematode hosts (2), but due to this enormous diversity within the genus, there is still some controversy on whether and how species names can be applied (e.g. 3, 4).
These bacteria ensure their proliferation within host populations by vertical transmission. Their effect on the host biology varies among host species and can include parthenogenesis, male feminization up to male-killing (5). The effects of these bacteria on their hosts can be detrimental, which has been of importance in some cases where Wolbachia was adopted as an agent to control pest insects or diseases transmitted by mosquitoes (6, 7). From an evolutionary perspective, Wolbachia has been documented to experience co-cladogenetic events with its host (e.g. 8) or to transfer fragments of its genome to its host (6). The most common process to enhance the spread of the strictly maternally transmitted Wolbachia in the population is the induction of cytoplasmic incompatibility (CI) (3), which can lead to selective sweeps that subsequently reduce mitochondrial DNA polymorphism in the host population (9). Their effect becomes more evident when closely related infected and uninfected taxa are compared (10).
Ants have an overall Wolbachia infection rate of about 34% (11), and effects of these endosymbionts can be alterations in colony life cycles, nutrition, and in production of reproductive individuals (12, 13). In the case of the invasive ant Paratrechina longicornis, it has been documented that the presence of maternally transmitted Wolbachia in a population can indirectly induce selective sweeps on the hosts’ mitochondria (14). As a consequence, Wolbachia may cause an accelerated spread in an invasive ant species due to an indirect selection of mitochondrial allelic variants that favor its invasive capacity.
Nylanderia is an ecologically important ant genus with a nearly cosmopolitan distribution. This genus was recently validated as the result of a reassessment of morphological characters complemented by a molecular phylogeny, and belongs to the Prenolepis genus-group (15). Many Nylanderia species that moved outside their native range became invasive (16). Such is the case of the tawny crazy ant, Nylanderia fulva, considered invasive in southern U.S. and native to South America, where the exact limits of its distribution are not well understood (17). Recent interest in the Prenolepis genus-group has provided with some important revisional studies about species status, re-descriptions and the discovery of species complexes. Complementing traditional morphological studies with genetic markers has helped understand the cryptic species complex that comprises N. fulva and N. pubens (18).
Nylanderia fulva is a species of omnivorous ant that forms polygynous nests that contain up to hundreds of reproductive queens and thousands of workers, which contributes to its capacity to quickly expand through its invasive range (16). It has been reported to reproduce sexually both in its native and invasive populations, but its social organization is multicolonial in the former and supercolonial in the latter (19, 20). Contrary to what has been documented for many other invasive ants (e.g. Wasmannia auropunctata, Anoplolepis gracilipes, Paratrechina longicornis), N. fulva does not present parthenogenetic reproduction neither in native nor invasive populations (19).
Traditionally, information from both mitochondrial and nuclear DNA can help understand the evolutionary history of a group of organisms at different taxonomic levels. Intraspecific variability in mitochondrial DNA (mtDNA) can be attributed to various possible evolutionary scenarios. Particularly, in Wolbachia infected species, the possibility of a selective sweep or some form of reproductive alteration can be expected when exploring phylogenies based on mtDNA data. In a review of the usage of mtDNA as a marker to infer phylogenies, the authors found in about 90% of the studies a symbiont-driven effect on the host mtDNA, such as a reduction of diversity or paraphyly (21).
Wolbachia detection and strain characterization has changed over time. For the past 15 years, the multi-locus sequence typing (MLST) system of five housekeeping gene fragments (ftsZ, fbpA, gatB, hcpA and coxA) has been the most popular method (22), lately, with the increased accessibility of next-generation sequencing, it has shifted also to whole-genome approaches (23). Bleidorn and Gerth (23) evaluated the reliability of MLST data compared to whole-genome data, to find that MLST loci and the highly recombinant wsp (Wolbachia surface protein) gene do not perform well at differentiating between closely related Wolbachia strains and also do not match the phylogenetic relationships seen by analyzing whole-genome data. However, the simplicity of using a reduced set of genes already established is still the most cost and/or time effective way to answer some questions compared to genomic approaches.
With the aim to evaluate if N. fulva phylogenetic relationships in its native range are influenced by Wolbachia, we first investigated the prevalence of infection and diversity of Wolbachia strains infecting Nylanderia species from northern Argentina, southern limit of its native range. Second, we investigated the relationship between the hosts mitochondrial DNA and its associated Wolbachia strains. Finally, we designed primers for the best ranked gene suggested by Bleidorn and Gerth (23) and evaluated how this gene fragment performed compared to the MLST approach.
Methods
Nylanderia fulva Mitochondrial DNA
Ants were manually collected from 35 nests throughout northeastern Argentina. Identification was done up to morphospecies, since updated keys for Nylanderia species are not available for the studied region, resulting in: 30, 2, 2 and 1 samples of N. fulva, Nylanderia sp. 1, Nylanderia sp. 2 and P. longicornis, respectively (Table S1). Genomic DNA was extracted from a single worker ant per nest using Extract-N-Amp Tissue PCR kit (Sigma-Aldrich Inc., St. Louis, MO, USA). Ant cytochrome C oxidase subunit I (COI) gene fragments were amplified by PCR using Jerry and Pat primer pair (18) and sequencing was performed by Macrogen (Macrogen Inc., Seoul, South Korea).
Screening for Wolbachia Infection and Sequencing of MLST and wsp Genes
For detection of Wolbachia infection, genomic DNA was extracted from a single ant worker from each nest using GeneMATRIX Tissue & Bacterial DNA Purification Kit (EURx, Gdansk, Poland). To determine the infection status of the ants, 1-2 ants per nest were screened by PCR amplification of ftsZ gene. Some samples that did not amplify with ftsZ were cross-validated using coxA, which confirmed the same negative results in all cases (n=6). Positive samples were further amplified for the MLST genes described in Baldo et al. (22) and wsp gene, using primers and modified PCR protocols from Baldo et al. (22) (Table S2). Sequencing in both directions was provided by Microsynth (Microsynth Seqlab GmbH, Germany). Sequence typing was performed in the Wolbachia MLST database (https://pubmlst.org/bigsdb?db=pubmlst_wolbachia_seqdef).
For the sample N084, multiple peaks in the electropherogram profiles of genes coxA, ftsZ, hcpA, wsp and glyQ may indicate the presence of more than one Wolbachia variant. We used pGEM-T vector system (Promega) to clone these sample’s DNA fragments into a vector, and afterwards used primers previously described in this work to select colonies with the desired PCR insert. We obtained two variants for coxA, two for glyQ, one for ftsZ and one for wsp (cloning could not be achieved for hcpA). The glyQ variants are hereafter denoted N084a and N084b, and coxA variants N084c and N084d. Complete resolution of ftsZ, hcpA and wsp was not possible, so we present only one allele for each.
Development of a New Primer Set for Wolbachia glyQ Gene
Bleidorn and Gerth (23) evaluated the performance of Wolbachia MLST markers compared with 252 other single copy loci at strain differentiation, reflecting genetic diversity in the strains and as phylogenetic markers for Wolbachia. They suggested a rated list of these loci from which glycine-tRNA ligase subunit alpha (glyQ) is the best ranked. We designed a new set of primers for this locus using Primer Premier v. 6.25 software (Premier Biosoft International, San Francisco, CA, USA) and a group of eleven available Wolbachia sequences from the dataset of Bleidorn and Gerth (23): glyQF (forward) 5’-GCAATGGAATGGAAGTAACACAG-3’ and glyQR (reverse) 5’-YTCACACCAAGCACACCTCT-3’. We selected from the potential primer pairs according to their Tm, self- and cross-dimer formation, and potential to form secondary structure (hairpins). Sequences were obtained using the same PCR protocols as for other MLST loci (Table S1), and aligned as previously stated. Blast algorithm (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to corroborate that the sequenced fragments belonged to Wolbachia. Additional sequences were obtained from Wolbachia genomes deposited in Genbank (Table S3).
Phylogenetic Relationships
Chromatograms of the sequences were visually inspected in Chromas v2.6.6 (Technelysium Pty Ltd, Australia). Alignment was done in MEGA X (24) with the Muscle algorithm (25) using the default parameters for both Wolbachia and Nylanderia spp. data. DNA sequence diversity estimates were calculated in DnaSP v6 (26). Additional sequences from repositories were included as outgroups, for determination of supergroup identity and diversity of the Wolbachia samples (Table S3). Two gene sets were used to estimate Wolbachia trees: MLST loci (ftsZ, coxA, fbpA, hcpA and gatB) (2079 bp), and glyQ (333 bp). Wsp gene was not used to infer phylogenies due to its high recombination rate. Selection of the best-fitting evolutionary model, maximum likelihood (ML) analyses and bootstrapping (1000 replicates) were performed for all datasets with IQ-TREE v. 2 (27, 28). Finally, to evaluate the potential of the selected fragment of glyQ gene, we compared the phylogeny of Wolbachia based on MLST loci with one estimated with glyQ (Figure S1).
Results
After inspection of the presence of the Wolbachia ftsZ gene, we found infections in 41% of the 34 Nylanderia nests, and a high percentage of infected nests within N. fulva (68%) (Table 1). Distribution of the Wolbachia carrying nests spanned throughout northeastern Argentina (Figure 1). The sample of P. longicornis (PL150), resulted positive for Wolbachia, as well as Nylanderia sp. 1 with both nests infected. Nylanderia sp. 2 had no positive nests for Wolbachia.

Table 1 Prevalence of Wolbachia infection in Nylanderia spp. One to two workers per nest were tested for presence of Wolbachia. Clades are based on the ants’ phylogeny from this work (Figure 2).
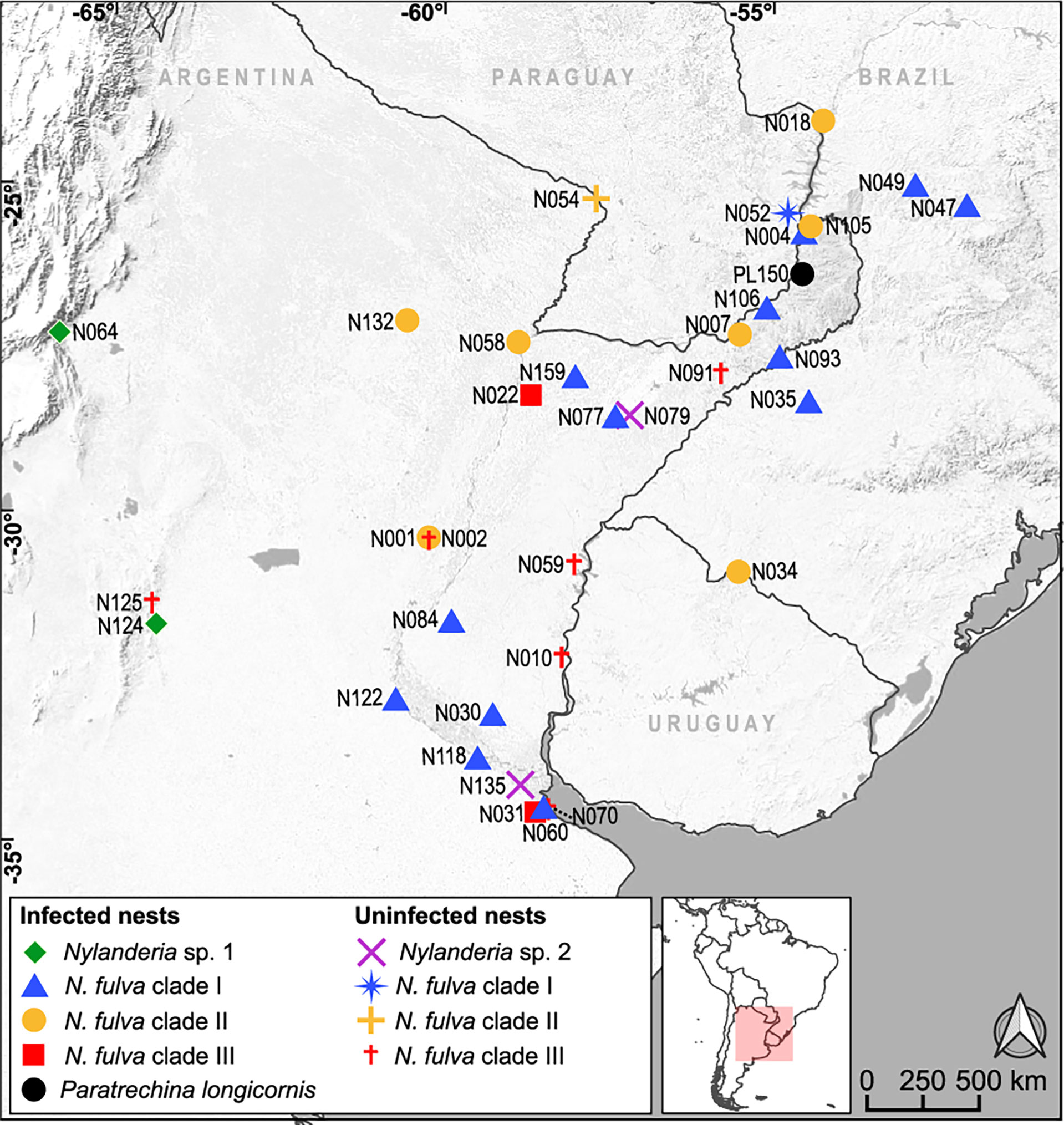
Figure 1 Geographic location of the nests of Nylanderia sp.1, Nylamderia sp. 2, Nylanderia fulva and Paratrechina longicornis infected and uninfected with Wolbachia. Colors in N. fulva correspond to the clades determined in this work (Figure 2).
Phylogenetic relationships of Nylanderia spp. were estimated using a fragment of the mitochondrial gene COI (Figure 2). Nylanderia fulva was recovered as paraphyletic, with N. pubens as a sister species. Within N. fulva, we differentiate three monophyletic clades, which were identified as clades I, II and III. The sequence belonging to an invasive population found in the US falls within clade I of N. fulva. Nucleotide diversity is lower in clades I and II, than in clade III (0.0035, 0.0024, and 0.0203, respectively), although sampling size for the latter was smaller. We tested the hypothesis of a selective sweep in mitochondrial DNA of N. fulva clades I and II (clade III was excluded due to its small sample size). All statistical tests of departure from neutral expectation were negative in both clades I and II (Tajima’s D, Fu and Li’s D* and F*, Table S4), but not significant. Wolbachia infection status was different for each N. fulva clade: clade I had the highest number of infected nests (93%), clade II had 75%, and clade III had the lowest infection rate with only 25% of positive nests (Table 1). Nylanderia sp. 1 was recovered as a sister species to N. fulva. Phylogenetic position of N. fulva and N. sp. 1 within other Nylanderia species is uncertain, due to the low support value of this branch, but both are closely related to another invasive species, Nylanderia steinheili.
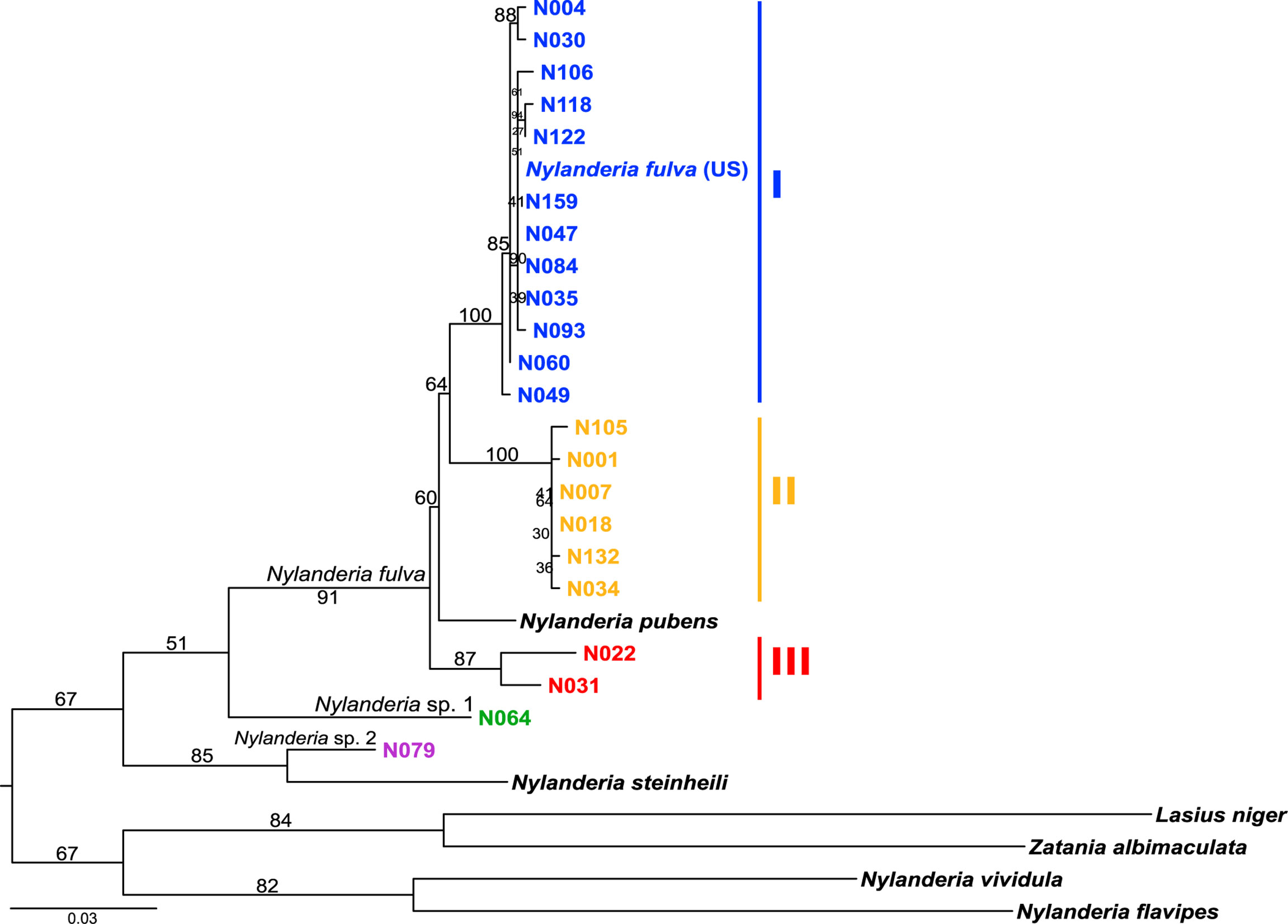
Figure 2 Nylanderia fulva maximum likelihood phylogeny based on mtDNA gene COI. Monophyletic lineages within N. fulva are color coded: clade I (blue), II (yellow) and III (red).
Almost all of the Wolbachia infected samples could be successfully sequenced for the five Wolbachia MLST genes, with exception of gatB, which could only be amplified for three samples (N031, N060, and PL150). For sequencing with the pair of primers for the glycine-tRNA ligase subunit alpha (glyQ) gene, we implemented similar PCR protocols as stated in Wolbachia MLST database and successfully amplified all of the infected samples for a fragment of 333 bp.
By comparing these sequences with published Wolbachia MLST strain database (https://pubmlst.org/organisms/wolbachia-spp/), we determined the sequence type (ST) for our samples, when possible, and defined strain names for 13 unique MLST profiles, wNyla1 to wNyla13 (Table 2). Within N. fulva clade I nests, we found the highest number of Wolbachia strains (eight), while in clades II and III there are only two different strains infecting each. One of the Wolbachia strains infecting N. fulva clade II, wNyla1, is highly prevalent, infecting six out of seven inspected nests. In Nylanderia sp. 1, the same Wolbachia strain infects both nests. We would like to point out, however, that since gatB sequences are lacking for most of the samples, different results are possible and could show higher variability. We found no identical sequence variants infecting different Nylanderia clades. Variability in wsp gene was high, with eight different alleles found in Nylanderia spp., from which two are novel. Diversity estimates for the sequenced gene fragments show that the most variable gene was ftsZ, presenting eleven different haplotypes, and the highest nucleotide diversity was found in wsp gene (Table 3). Compared with the MLST loci all together, the glyQ fragment had an overall low diversity, with only five different haplotypes for Nylanderia spp. and only nine segregating sites. However, the fragment presents similar percentage of segregating sites (about 3%) and nucleotide diversity (0.01) to other MLST genes alone, such as coxA and ftsZ. Additionally, if glyQ gene is considered when establishing sequence types, the number of different haplotypes within Nylanderia spp. rises from 13 to 14 (Table S5).
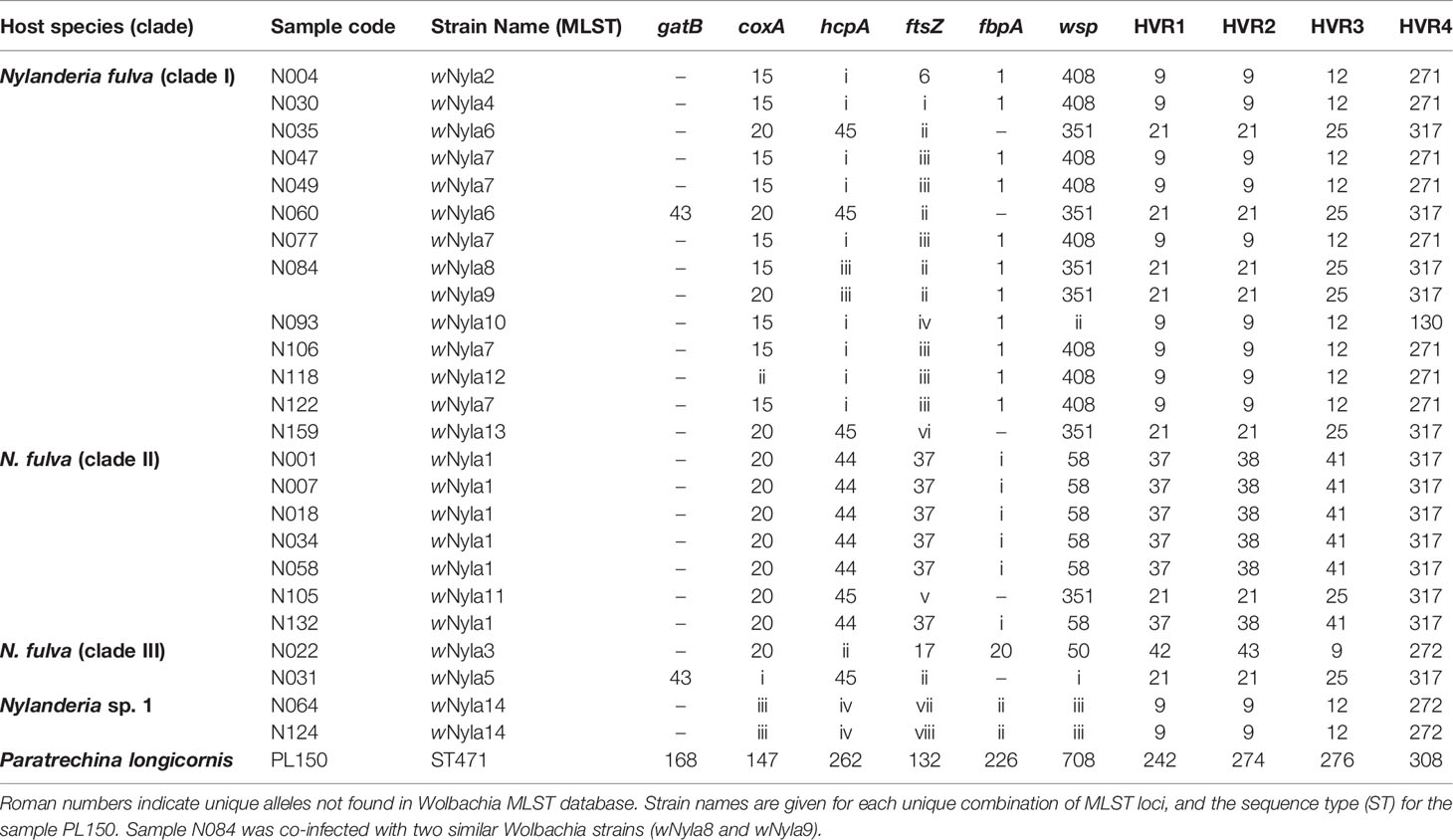
Table 2 Allelic profiles, wsp and HVR allele number identifiers for Wolbachia strains in Nylanderia ants.
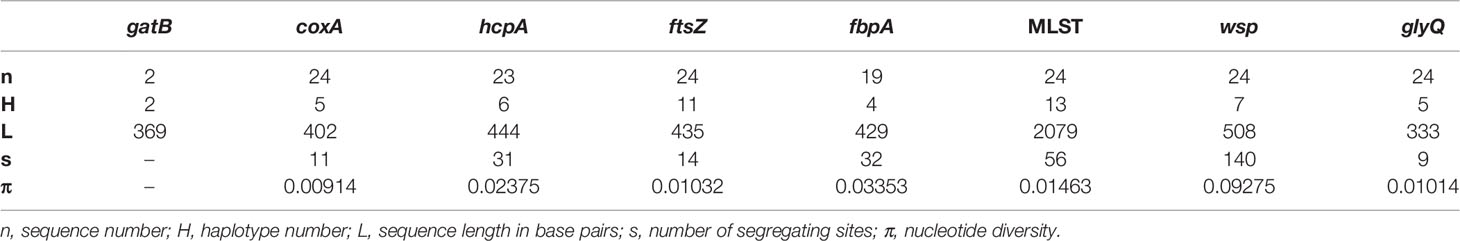
Table 3 DNA variation estimates for each sequenced Wolbachia gene in Nylanderia spp. infected ants.
In the MLST phylogeny of Wolbachia strains infecting Nylanderia spp., all of the strains belong to supergroup A, while P. longicornis was infected with the strain wLonF (ST471), belonging to supergroup F (Figure 3). Most nests of N. fulva clades I and II are infected each with a unique lineage of Wolbachia strains, while clade III harbors two distantly related Wolbachia strains. There is one group of N. fulva nests belonging to clades I, II and III and from various geographic locations that share infection of the same lineage of Wolbachia strains (samples N031, N035, N060, N105 and N159). The four Wolbachia lineages infecting N. fulva are distantly related with each other and do not form a single monophyletic unit. Nylanderia sp. 1 is also infected with a unique Wolbachia strain that is closely related to those infecting other ants.
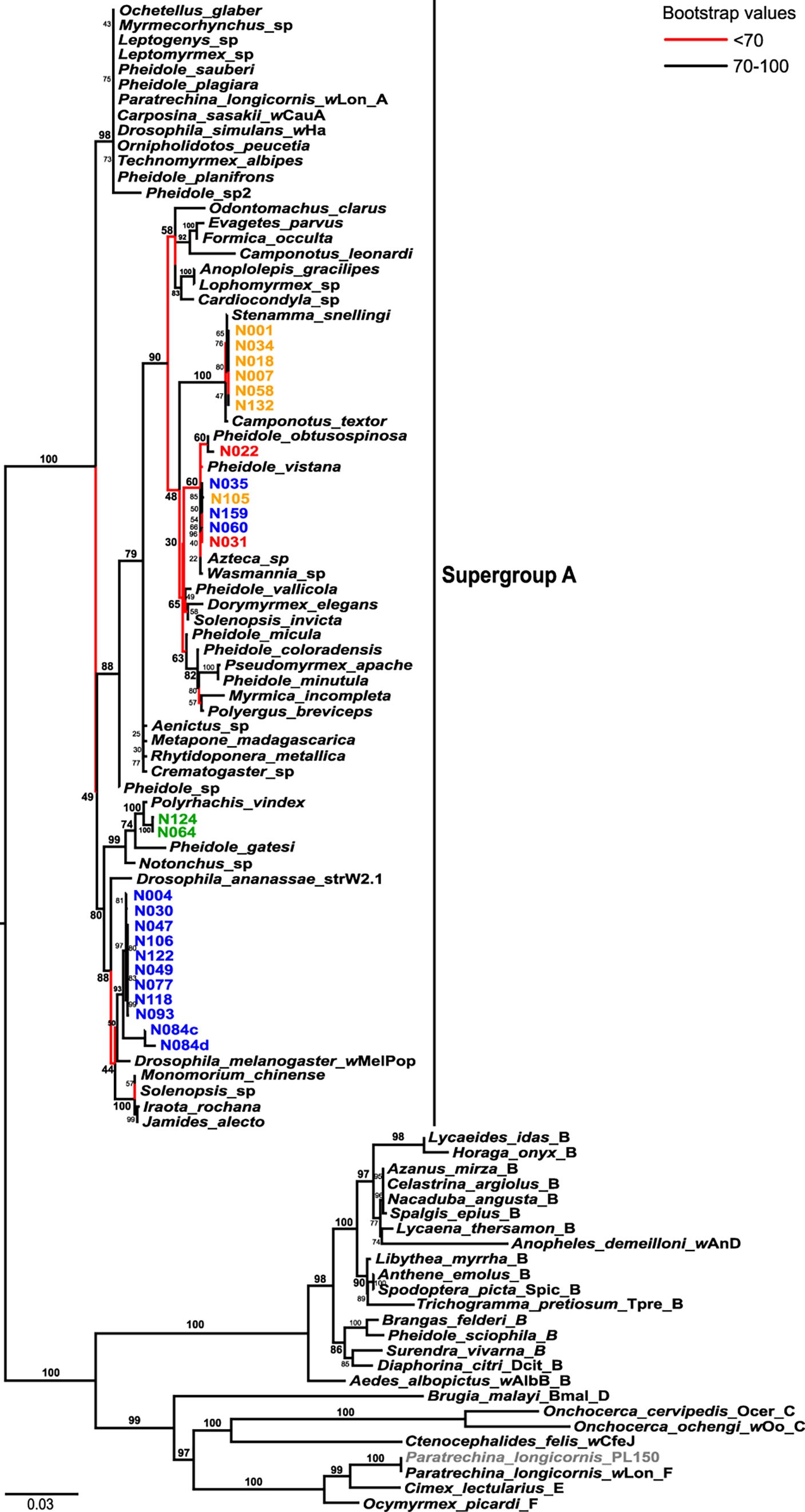
Figure 3 Wolbachia maximum likelihood phylogeny based on multi locus sequence typing (MLST) data with host species as labels. Colors highlight the sequences produced in this work belonging to different Nylanderia mitochondrial DNA clades: clade I (blue), clade II (yellow), clade III (red), and Nylanderia sp. 1 (green). All sequences belong to Wolbachia supergroup A unless stated with its corresponding letter (B-F). Branches with bootstrap support values lower than 70 are indicated in red.
The Wolbachia phylogeny inferred with the glyQ fragment shows a similar pattern as that of the MLST tree (Figure S1). Nylanderia fulva clades appear infected by three monophyletic Wolbachia lineages: one infecting N. fulva clade I nests, another infecting clade II nests, and the third infecting nests that belong to clades I, II and III. On the other hand, these lineages show a different relationship with each other as that seen in the MLST tree. Nylanderia sp. 1 Wolbachia strain is more closely related to that infecting N. fulva clade II in the glyQ phylogeny, but it is more related to clade I when considering the MLST dataset. Plus, comparison with sequences of Wolbachia infecting other insects is limited by available complete genomes, and thus, limits the possibility to compare the relationship of the strain lineages with those of other hosts. Variants found in the sample N084 were distantly related, with variant N084a closely related to those infecting mostly clade I ants, while variant N084b is placed within the group of Wolbachia infecting samples from every N. fulva clade.
Discussion
Nylanderia Phylogeny
We investigated the phylogenetic relationships of an invasive ant species, Nylanderia fulva, by sampling nests across the southern limit of its native distribution. Using the mitochondrial DNA marker COI, we found that N. fulva was conformed of three distinct monophyletic clades (clades I to III) from which clade I included an invasive population found in the US. We proposed a possible Wolbachia influence on N. fulva population structure based on the pattern of reciprocal monophyletic lineages observed in mtDNA. A wide range of causes can explain this observation, such as a speciation process, a recent population expansion or a selective sweep, or a combination of processes affecting each clade differently.
In addition, N. pubens was placed within the boundaries of what we considered the species N. fulva, and thus supports its position as a sibling species. This is in line with a recent work suggesting that N. fulva and N. pubens form a species complex (29). These two species have been commonly mistaken due to the identical aspect of their workers, but are distinguishable inspecting male genital characters (18). More sampling of N. fulva and N. pubens throughout their native ranges with a detailed examination of male genitalia should provide useful evidence to understand the limits of these two close species.
Furthermore, we found another two Nylanderia species present in the surveyed region. Nylanderia sp. 1, which we consider could be the only other common Nylanderia species historically reported for the region, Nylanderia silvestrii (30), and N. sp. 2, which possibly belongs to the N. guatemalensis/N. steinheili species complex. This would be a novel location record for this invasive species in Argentina, in Corrientes province, being previously recorded in the city of Buenos Aires (31). Further analyses of morphological characters and sequencing of type material could be of aid in confirming the identity of these species.
Wolbachia Infection and Diversity in Nylanderia spp.
We have detected Wolbachia infection for the first time in the invasive ant Nylanderia fulva in its native range. Additionally, infection of this endosymbiont was found in other two species, N. sp. 1, and the closely related invasive ant species, P. longicornis. Strains infecting Nylanderia species belonged to supergroup A, while that of P. longicornis was identified as wLonF, from supergroup F. These are the first records of Wolbachia infection for the three species in this region, as well as the first confirmed record of invasion of P. longicornis in Argentina. The strain infecting P. longicornis, wLonF, is primarily horizontally transmitted and widely distributed in this ant around the globe (14). Wolbachia infecting ants mostly belong to supergroup A, secondly to B and a few exceptions to supergroup F, namely Paratrechina longicornis and Ocymyrmex picardi (32, 33). We found that all Wolbachia infecting Nylanderia species belonged to supergroup A, which is consistent with those infecting most of other insect hosts, particularly ants (12). We found no infection of Wolbachia in Nylanderia sp. 2, a putatively invasive species belonging to the N. guatemalensis/N. steinheili species complex. This result coincides with that seen for other invasive ant species, whose introduced populations are usually uninfected (e.g. 34, 35). Furthermore, we found no distinctive distributional pattern of infected/uninfected Nylanderia nests in the surveyed area; Wolbachia infected nests were found throughout northeastern Argentina, southeastern Brazil and in some locations of Paraguay and Uruguay.
Several MLST alleles for the infected Nylanderia samples did not match those in the Wolbachia MLST database. We reported 14 new MLST sequence types, which include 17 new alleles: three for coxA, four for hcpA, eight for ftsZ, and two for fbpA. Additionally, we found two new alleles for the wsp gene to be uploaded to the MLST database.
We designed a new set of primers for a fragment of the glycine-tRNA ligase subunit alpha (glyQ) gene and a PCR protocol that is readily available for use. This fragment presented five different haplotypes for the sampled Wolbachia strains, and overall it matches diversity estimates of other widely used gene fragments such as coxA and ftsZ. When combined with the MLST loci, we retrieved one additional haplotype or sequence type, for a total of 15 different haplotypes in Nylanderia spp. This suggests there is at least some degree of variation in Wolbachia strains still not uncovered by the MLST system, at least for the evaluated species. With the availability of assemblies of more than 20 Wolbachia genomes, more and more information become available to perform genome-wide Wolbachia phylogenies. Chung et al. (4) found more Wolbachia species using a core genome alignment than established supergroups. In other bacteria, such as Staphylococcus aureus or Haemophilus influenzae, the MLST system is comprised of more than five genes, so the incorporation of a new gene could be an interesting addition for Wolbachia MLST scheme (36, 37). Furthermore, given that we were not able to amplify most samples with one of the MLST genes, gatB, and that the protocols and primers established here for glyQ were successful, the possibility to include more genes to the MLST system can be useful in these cases for a more thorough determination of Wolbachia haplotypes.
Though our results show a high rate of Wolbachia infection within some lineages of N. fulva, there is a limitation to our findings regarding the small number of ants sampled by nest. Diversity estimates as well as prevalence values should be taken as a first approximation since more individuals per nest should be inspected to fully understand the prevalence of Wolbachia within populations.
Incongruence Between Wolbachia and Nylanderia fulva Phylogenies
We explored the relationship of Wolbachia strains infecting N. fulva according to their three host clades, clades I to III. Three Wolbachia lineages were found infecting N. fulva clades, although with different prevalence; clade I had the most infected number of nests, second was clade II and clade III had the lowest number of infected nests. We found low nucleotide diversity in N. fulva clades I and II, which can be related to a putative population expansion or population bottleneck, but also to recent symbiont invasions, where selective sweeps cause lower mtDNA diversity, and so can easily be mistaken for the effects of the former events (21). In other insect species with partial Wolbachia infection, as was the case in N. fulva, mitochondrial polymorphism tends to be lower in the infected lineages; additionally, in sister species, Wolbachia infection has been related to reduction in the effective population size (10). Recent studies suggest Wolbachia could cause speciation in its host via induced parthenogenesis; for example, in the weevil Pantomorus postfasciatus, all non-sexual populations harbor Wolbachia while the sexual ones are not infected, and both behave as independent evolutionary units (38). In the invasive ant P. longicornis, it was suggested that an ancestral Wolbachia infection could be associated with a recent speciation in the ants’ clades (8, 14). High prevalence of Wolbachia within clade I may imply an intimate association between the symbiont and its host, but empirical tests would be needed to explore this relationship. Furthermore, Eyer et al. (19) found no evidence of parthenogenetic reproduction in N. fulva. We found no evidence of departure from neutrality in all the tested statistics within clades I or II that could be associated with a selective sweep, although values for the statistics were negative, which can be related to a selection process. We do not fully discard the possibility of a Wolbachia induced selective sweep due to the low sampling size within nests that can underestimate intraspecific variability. There were no clades of N. fulva completely uninfected with Wolbachia, as was the case in other species. Altogether, these results compete with the hypothesis of a selective sweep affecting one particular clade. It has been hypothesized that horizontal transfer as well as a degree of admixture between sexual and parthenogenetic populations, can alter the effects of a vertically transmitted strain, resulting in a hindered Wolbachia fixation and, hence, obscuring the effects of the selective sweep (2). Reproductive isolation experiments could provide useful information to validate this possibility.
The pattern of monophyletic lineages of Wolbachia infecting different groups of N. fulva is incongruent with that observed in the host phylogeny. While in the host phylogeny the three N. fulva clades are closely related and form a monophyletic unit (together with N. pubens), in the Wolbachia phylogeny the lineages are paraphyletic. Also, some of the N. fulva nests belonging to different clades are infected with the same lineage of Wolbachia strains. There are some cases, such as that of Culex pipiens, in that identical strains based on MLST sequence types (STs) and wsp alleles in fact present differences in other inspected genes and significant structural variation that in turn produce different phenotypic effects (39). All of the Wolbachia lineages found in Nylanderia nests are closely related to strains found infecting other insects, such as other ants and Drosophila flies. In light of these results, the discordance between phylogenies of Wolbachia and N. fulva suggests that infection mechanisms of Wolbachia could involve various independent horizontally transmitted Wolbachia infections within N. fulva. Similar results have been obtained for another invasive species, the little fire ant Wasmannia auropunctata (40), who’s native distribution range also overlaps with that of N. fulva. Close relationship of the different Wolbachia infecting Nylanderia with other insects suggests horizontal transmission (HT), possibly through sharing of some aspect of their ecological niches. There are many ways in which Wolbachia can be horizontally transferred between closely related species, for example, if they share parasitoids or parasites. Parasitic phorid flies have been considered a potential source of HT in fire ants, Solenopsis spp., but there is yet no data supporting a shared Wolbachia strain with their hosts (41, 42). The only phorid fly species reported parasitizing N. fulva up to now, Pseudacteon convexicauda (43), could be acting as a vector of Wolbachia between N. fulva clades, thus facilitating its transmission. Ant guests can also present an opportunity for Wolbachia to be horizontally transmitted to the ants, as was the case for P. longicornis and its host-specific ant cricket Myrmecophilus americanus; although in the invasive ant Anoplolepis gracilipes, HT was not present between the ants and their associated cricket species M. albicinctus (8). Moreover, other relationships, such as prey-predator interactions, can also be a way of horizontal transmission (1, 7, 44). Ant workers interact directly with brood by tending and feeding them and frequently with sister workers through trophallaxis, a mechanism in which workers transfer food or other fluids through mouth-to-mouth (45). These interactions could be a means or perpetrating a horizontally transmitted strain within the nest.
An invasive population of N. fulva in the US was found to be closely related to native populations from Argentina belonging to clade I. Native populations of N. fulva clade I showed the highest prevalence of infection and number of Wolbachia strains. In another invasive ant sympatric with N. fulva in Argentina, Solenopsis invicta, it was suggested that possible loss of Wolbachia infection in invasive populations may have a relationship with its invasiveness, and that re-introduction of Wolbachia might be a means of biological control of the ants, if its effect is indeed deleterious (11, 34). Data from invasive populations is needed to confirm if they are infected with Wolbachia.
We found no correlation in phylogenetic relationships of Wolbachia strains infecting N. fulva and the host mtDNA phylogeny. Our research suggests that horizontal transmission could be the most prominent way of Wolbachia transmission in these ants, and that there is no evidence that supports an influence of Wolbachia in Nylanderia mtDNA phylogeny. However, it could be interesting to further investigate prevalence of Wolbachia infection in the introduced N. fulva populations and in other Nylanderia species due to the small sampling size included in this study for Nylanderia sp. 1 and Nylanderia sp. 2. By incorporating more genetic markers, such as nuclear DNA for the ants, and larger sampling, it may be possible to reveal even more variability than we observed in this work, and to have a better understanding of the patterns and consequences of Wolbachia infection in these ants.
Data Availability Statement
The dataset containing sequence alignments used in this paper can be found in Zenodo repository under the DOI address: 10.5281/zenodo.6570990.
Author Contributions
MBF and LC collected the samples. MBF performed the experiments and analyses. MBF, LC, and CB analyzed the data and contributed to writing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MBF was funded by fellowships from Consejo Nacional de Investigaciones Cientı́ficas y Técnicas (CONICET), Universidad de Buenos Aires (UBA) and Potsdam University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We appreciate the constructive comments on the manuscript from Dr. Marcela Rodriguero.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/finsc.2022.905803/full#supplementary-material
References
1. Duron O, Bouchon D, Boutin S, Bellamy L, Zhou L, Engelstädter J, et al. The Diversity of Reproductive Parasites Among Arthropods: Wolbachia Do Not Walk Alone. BMC Biol (2008) 6:1–12. doi: 10.1186/1741-7007-6-27
2. Kaur R, Shropshire JD, Cross KL, Leigh B, Mansueto AJ, Stewart V, et al. Living in the Endosymbiotic World of Wolbachia: A Centennial Review. Cell Host Microbe (2021) 29(6):879–93. doi: 10.1016/j.chom.2021.03.006
3. Werren JH, Baldo L, Clark ME. Wolbachia: Master Manipulators of Invertebrate Biology. Nat Rev Microbiol (2008) 6(10):741–51. doi: 10.1038/nrmicro1969
4. Chung M, Munro JB, Tettelin H, Dunning Hotopp JC. Using Core Genome Alignments to Assign Bacterial Species. mSystems (2018) 3:e00236–18. doi: 10.1128/mSystems.00236-18
5. Asimakis ED, Doudoumis V, Hadapad AB, Hire RS, Batargias C, Niu C, et al. Detection and Characterization of Bacterial Endosymbionts in Southeast Asian Tephritid Fruit Fly Populations. BMC Microbiol (2019) 19(Suppl 1):1–18. doi: 10.1186/s12866-019-1653-x
6. Rodriguero MS. Wolbachia, a Pandemic With Potential. In: Revista De La Sociedad Entomológica Argentina. (2013), 117–37.
7. Sanaei E, Charlat S, Engelstädter J. Wolbachia Host Shifts: Routes, Mechanisms, Constraints and Evolutionary Consequences. Biol Rev (2021) 96(2): 433–53. doi: 10.1111/brv.12663
8. Lee CC, Lin CY, Tseng SP, Matsuura K, Yang CCS. Ongoing Coevolution of Wolbachia and a Widespread Invasive Ant, Anoplolepis Gracilipes. Microorganisms (2020) 8(10):1–17. doi: 10.3390/microorganisms8101569
9. Schuler H, Köppler K, Daxböck-Horvath S, Rasool B, Krumböck S, Schwarz D, et al. The Hitchhiker’s Guide to Europe: The Infection Dynamics of an Ongoing Wolbachia Invasion and Mitochondrial Selective Sweep in Rhagoletis Cerasi. Mol Ecol (2016) 25(7):1595–609. doi: 10.1111/mec.13571
10. Cariou M, Duret L, Charlat S. The Global Impact of Wolbachia on Mitochondrial Diversity and Evolution. J Evol Biol (2017) 30:2204–10. doi: 10.1111/jeb.13186
11. Russell JA. The Ants (Hymenoptera: Formicidae) are Unique and Enigmatic Hosts of Prevalent Wolbachia (Alphaproteobacteria) Symbionts. Myrmecol News (2012) 16(January):7–23.
12. Russell JA, Goldman-Huertas B, Moreau CS, Baldo L, Stahlhut JK, Werren JH, et al. Specialization and Geographic Isolation Among Wolbachia Symbionts From Ants and Lycaenid Butterflies. Evolution (2009) 63(3):624–40. doi: 10.1111/j.1558-5646.2008.00579.x
13. Ramalho MO, Moreau CS. The Evolution and Biogeography of Wolbachia in Ants (Hymenoptera: Formicidae). Diversity (2020) 12(11):1–12. doi: 10.3390/d12110426
14. Tseng SP, Wetterer JK, Suarez AV, Lee CY, Yoshimura T, Shoemaker DW, et al. Genetic Diversity and Wolbachia Infection Patterns in a Globally Distributed Invasive Ant. Front Genet (2019) 10:838. doi: 10.3389/fgene.2019.00838
15. LaPolla JS, Brady SG, Shattuck SO. Phylogeny and Taxonomy of the Prenolepis Genus-Group of Ants (Hymenoptera: Formicidae). Systematic Entomol (2010) 35(1):118–31. doi: 10.1111/j.1365.3113.2009.00492.x
16. Williams JL, Lucky A. Non-Native and Invasive Nylanderia Crazy Ants (Hymenoptera: Formicidae) of the World: Integrating Genomics to Enhance Taxonomic Preparedness. Ann Entomol Soc America (2019) 113(4):318–36. doi: 10.1093/aesa/saz039
17. Wang Z, Moshman L, Kraus EC, Wilson BE, Acharya N, Diaz R. A Review of the Tawny Crazy Ant, Nylanderia Fulva, an Emergent Ant Invader in the Southern United States: Is Biological Control a Feasible Management Option? Insects (2016) 7(4):77. doi: 10.3390/insects7040077
18. Gotzek D, Brady SG, Kallal RJ, LaPolla JS. The Importance of Using Multiple Approaches for Identifying Emerging Invasive Species: The Case of the Rasberry Crazy Ant in the United States. PloS One (2012) 7(9):1–10. doi: 10.1371/journal.pone.0045314
19. Eyer PA, McDowell B, Johnson LNL, Calcaterra LA, Fernandez MB, Shoemaker D, et al. Supercolonial Structure of Invasive Populations of the Tawny Crazy Ant Nylanderia Fulva in the US. BMC Evol Biol (2018) 18(1):2007. doi: 10.1186/s12862-018-1336-5
20. LeBrun EG, Plowes RM, Folgarait PJ, Bolazzi M, Gilbert LE. Ritualized Aggressive Behavior Reveals Distinct Social Structures in Native and Introduced Range Tawny Crazy Ants. PloS One (2019) 14(11):e0225597. doi: 10.1371/journal.pone.0225597
21. Hurst GDD, Jiggins FM. Problems With Mitochondrial DNA as a Marker in Population, Phylogeographic and Phylogenetic Studies: The Effects of Inherited Symbionts. Proc R Soc B: Biol Sci (2005) 272(1572):1525–34. doi: 10.1098/rspb.2005.3056
22. Baldo L, Hotopp JCD, Jolley KA, Bordenstein SR, Biber SA, Choudhury RR, et al. Multilocus Sequence Typing System for the Endosymbiont Wolbachia Pipientis. Appl Environ Microbiol (2006) 72(11):7098–110. doi: 10.1128/AEM.00731-06
23. Bleidorn C, Gerth M. A Critical Re-Evaluation of Multilocus Sequence Typing (MLST) Efforts in Wolbachia. FEMS Microbiol Ecol (2018) 94(1):1–11. doi: 10.1093/femsec/fix163
24. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis Across Computing Platforms. Mol Biol Evol (2018) 35:1547–9. doi: 10.1093/molbev/msy096
25. Edgar RC. MUSCLE: Multiple Sequence Alignment With High Accuracy and High Throughput. Nucleic Acids Res (2004) 32(5):1792–7. doi: 10.1093/nar/gkh340
26. Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP V6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol Biol Evol (2017) 34:3299–302. doi: 10.1093/molbev/msx248
27. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat Methods (2017) 14:587–9. doi: 10.1038/nmeth.4285
28. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol (2020) 37:1530–4. doi: 10.1093/molbev/msaa015
29. Williams JL, Zhang YM, Lloyd MW, LaPolla JS, Schultz TR, Lucky A. Global Domination by Crazy Ants: Phylogenomics Reveals Biogeographical History and Invasive Species Relationships in the Genus Nylanderia (Hymenoptera: Formicidae). Systematic Entomol (2020) 45:730–44. doi: 10.1111/syen.12423
30. Kusnezov N. Hormigas Argentinas: Clave Para Su Identificacion. Golbach R, editor. Tucumán, Argentina: Fundacion Miguel Lillo (1978).
31. Josens R, Sola FJ, Marchisio N, Di Rienzo MA, Giacometti A. Knowing the Enemy: Ant Behavior and Control in a Pediatric Hospital of Buenos Aires. SpringerPlus (2014) 3:229. doi: 10.1186/2193-1801-3-229
32. Kelly M, Price SL, de Oliveira Ramalho M, Moreau CS. Diversity of Wolbachia Associated With the Giant Turtle Ant, Cephalotes Atratus. Curr Microbiol (2019) 76(11):1330–7. doi: 10.1007/s00284-019-01722-8
33. Lefoulon E, Foster JM, Truchon A, Carlow CKS, Slatko BE. The Wolbachia Symbiont: Here, There and Everywhere. In: Kloc M, editor. Symbiosis: Cellular, Molecular, Medical and Evolutionary Aspects, 1st ed, vol. 69. Cham, Switzerland: Springer (2020), p. 423–51. doi: 10.1007/978-3-030-51849-3
34. Shoemaker DD, Ross KG, Keller L, Vargo EL, Werren JH. Wolbachia Infections in Native and Introduced Populations of Fire Ants (Solenopsis Spp.). Insect Mol Biol (2000) 9(6):661–73. doi: 10.1046/j.1365-2583.2000.00233.x
35. Tsutsui ND, Kauppinen SN, Oyafuso AF, Grosberg RK. The Distribution and Evolutionary History of Wolbachia Infection in Native and Introduced Populations of the Invasive Argentine Ant (Linepithema Humile). Mol Ecol (2003) 12:3057–68. doi: 10.1046/j.1365-294X.2003.01979.x
36. Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. Multilocus Sequence Typing for Characterization of Methicillin-Resistant and Methicillin-Susceptible Clones of Staphylococcus Aureus. J Clin Microbiol (2000) 38(3):1008–15. doi: 10.1128/JCM.38.3.1008-1015.2000
37. Meats E, Feil EJ, Stringer S, Cody AJ, Goldstein R, Kroll JS, et al. Characterization of Encapsulated and Noncapsulated Haemophilus Influenzae and Determination of Phylogenetic Relationships by Multilocus Sequence Typing. J Clin Microbiol (2003) 41(4):1623–36. doi: 10.1128/JCM.41.4.1623-1636.2003
38. Elias-Costa AJ, Confalonieri VA, Lanteri AA, Rodriguero MS. Game of Clones: Is Wolbachia Inducing Speciation in a Weevil With a Mixed Reproductive Mode? Mol Phylogenet Evol (2019) 133:42–53. doi: 10.1016/J.YMPEV.2018.12.027
39. Atyame CM, Labbé P, Dumas E, Milesi P, Charlat S, Fort P, et al. Wolbachia Divergence and the Evolution of Cytoplasmic Incompatibility in Culex Pipiens. PloS One (2014) 9(1):e87336. doi: 10.1371/journal.pone.0087336
40. Chifflet L., Guzmán N. V., Rey O., Confalonieri V. A., Calcaterra L. A. (2018). Southern Expansion of the Invasive Ant Wasmannia auropunctata Within Its Native Range and Its Relation With Clonality and Human Activity. PLoS ONE 13(11):L1–16. doi: 10.1371/journal.pone.0206602
41. Dedeine F, Ahrens M, Calcaterra L, Shoemaker DD. Social Parasitism in Fire Ants (Solenopsis Spp.): A Potential Mechanism for Interspecies Transfer of Wolbachia. Mol Ecol (2005) 14(5):1543–8. doi: 10.1111/j.1365-294X.2005.02499.x
42. Tolley SJA, Nonacs P, Sapountzis P. Wolbachia Horizontal Transmission Events in Ants: What do We Know and What can We Learn? Front Microbiol (2019) 10:296(MAR). doi: 10.3389/fmicb.2019.00296
43. Sánchez-Restrepo AF, Chifflet L, Confalonieri VA, Tsutsui ND, Pesquero MA, Calcaterra LA. A Species Delimitation Approach to Uncover Cryptic Species in the South American Fire Ant Decapitating Flies (Diptera: Phoridae: Pseudacteon). PloS One (2020) 15(7):e0236086. doi: 10.1371/journal.pone.0236086
44. Tseng SP, Hsu PW, Lee CC, Wetterer JK, Hugel S, Wu LH, et al. Evidence for Common Horizontal Transmission of Wolbachia Among Ants and Ant Crickets: Kleptoparasitism Added to the List. Microorganisms (2020) 8(6):1–11. doi: 10.3390/microorganisms8060805
Keywords: Wolbachia, ants, invasive insects, horizontal transfer, phylogeny
Citation: Fernández MB, Bleidorn C and Calcaterra LA (2022) Wolbachia Infection in Native Populations of the Invasive Tawny Crazy Ant Nylanderia fulva. Front. Insect Sci. 2:905803. doi: 10.3389/finsc.2022.905803
Received: 27 March 2022; Accepted: 06 May 2022;
Published: 06 June 2022.
Edited by:
Erin Wilson-Rankin, University of California, Riverside, United StatesReviewed by:
Thomas Wolfe, University of Natural Resources and Life Sciences Vienna, AustriaŁukasz Kajtoch, Institute of Systematics and Evolution of Animals (PAN), Poland
Copyright © 2022 Fernández, Bleidorn and Calcaterra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Belén Fernández, mariabelenfernandez@live.com