 Boris Klimovich1,2†
Boris Klimovich1,2† Leonard Anton1†
Leonard Anton1† Jinwon Jung3
Jinwon Jung3 Yangmi Lim3
Yangmi Lim3 Bora Lee3
Bora Lee3 Jonghwa Won3
Jonghwa Won3 Latifa Zekri1,2
Latifa Zekri1,2 Anna Chashchina1,2Martin Pflügler1
Anna Chashchina1,2Martin Pflügler1 Jonas S. Heitmann1,2Gundram Jung4
Jonas S. Heitmann1,2Gundram Jung4 Helmut R. Salih1,5*
Helmut R. Salih1,5*- 1Clinical Collaboration Unit Translational Immunology, Department of Internal Medicine, University Hospital Tübingen, Tübingen, Germany
- 2Cluster of Excellence iFIT (EXC 2180) ‘Image-Guided and Functionally Instructed Tumor Therapies’, Eberhard Karls University of Tübingen, Tübingen, Germany
- 3ABL Bio Inc., Seongnam-si, Gyeonggi-do, Republic of Korea
- 4Department of Immunology, Institute for Cell Biology, Eberhard Karls Universität Tübingen, Tübingen, Germany
- 5German Cancer Consortium Deutsche Konsortium für Translationale Krebsforschung (DKTK), partner site Tübingen, a partnership between Deutsche Krebsforschungszentrum (DKFZ) and University Hospital Tübingen, Tübingen, Germany
Despite recent advancements, acute myeloid leukemia (AML) remains a therapeutic challenge. While monoclonal antibodies (mAbs) leveraging natural killer (NK) cells through antibody-dependent cellular cytotoxicity show great potential, none have gained clinical approval for AML. Immunocytokines have emerged as a promising strategy to overcome the limited efficacy of therapeutic antibodies. IL-15 stimulates activation, proliferation cytotoxic activity of NK cells, but its clinical use is prevented by short half-life, poor accumulation in the tumor, and toxicity due to systemic off-target immune activation. Here we report on the generation and preclinical characterization of modified immunocytokines consisting of an Fc-optimized CLEC12A (CLL-1) antibody fused to an IL-15 moiety with E46K mutation. The mutation abrogates binding to IL-15Rα, thereby enabling substitution of physiological trans-presentation by target binding and thus conditional IL-15Rβ/γ stimulation to reduce systemic toxicity. An optimal CLEC12A binder was selected from a range of murine mAbs, based on analysis of AML cell lines and leukemic cells from patients. This antibody was then used to construct an immunocytokine (MIC12) that subsequently was characterized functionally. Analysis of NK cell activation, cytokine release, proliferation and anti-leukemia reactivity demonstrated that MIC12 induced superior target cell killing and NK cell expansion compared to Fc-optimized CLEC12A antibody, with efficacy being dependent on target antigen binding. Our results show that novel immunocytokines with conditional IL-15 activity are capable of inducing potent NK cell responses against AML cells and identify MIC12 as promising therapeutic candidate for leukemia treatment.
1 Introduction
Acute myeloid leukemia (AML) is an aggressive disease characterized by clonal expansion of myeloid progenitor cells with impaired differentiation (1). It is the most common form of acute leukemia in adults, with a median age at diagnosis of 68 years (2). Although various immunotherapeutic approaches such as immune checkpoint inhibitors (3), chimeric antigen receptor (CAR) T cells (4) or bispecific antibodies (5) are currently evaluated in clinical trials, the standard of care for AML patients remains chemotherapy, targeted therapy with small molecules and allogeneic stem cell transplantation. In particular elderly patients face a poor prognosis and a high risk of relapse (6). This emphasizes the pressing need for new therapeutic targets and advanced treatment options.
Monoclonal antibodies (mAb) have revolutionized the treatment of several types of cancer. Rituximab, the first clinically available mAb targeting CD20, and further optimized constructs directed to CD20 have become a mainstay in the treatment of B-cell lymphomas. Despite their remarkable efficacy in lymphoid malignancies, attempts to develop effective mAbs for treating AML were so far not overly successful (7). Efficacy of Rituximab is largely driven by induction of antibody-dependent cellular cytotoxicity (ADCC) mediated by NK cells (8, 9). Enhancing mAb affinity for FcγRIIIa/CD16a through Fc domain modifications, including glycoengineering (10) or mutagenesis, is a common strategy to strengthen ADCC-inducing capabilities. A common example of the latter are the mutations S239D and I332E (hereafter referred to as SDIE modification) (11, 12). As an example, a SDIE-optimized mAb targeting FLT3 in patients with AML in complete remission with persisting minimal residual disease of AML has demonstrated promising results in a Phase I clinical trial (13). However, achieving remission in AML patients with high leukemic burden requires to further enhance mAb efficacy in stimulating NK cells, and combining mAbs with immunostimulatory cytokines as so called immunocytokines offers an attractive approach. Particularly IL-2 and IL-15 are promising in this regard, since they not only boost anti-leukemic activity but also promote NK cell proliferation, which is essential for combating larger tumor burden (14–18). However, the administration of recombinant cytokines is associated with severe side effects, and this would also hold true upon combined application with mAbs. Likewise, most immunocytokines developed so far face the same fundamental problem: in contrast to mAbs, their cytokine moiety lacks conditional target-restricted activity, as the cytokine moieties act independently of target binding. As a consequence, off-target activation of immune cells leads to substantial side effects that constrain dosing and thereby reduce efficacy (19–21).
The unique mechanism of action of IL-15 offers a solution to this fundamental issue: IL-15 effectively engages the IL-15Rβ/γ receptor on cytotoxic lymphocytes only when presented in trans upon binding to the IL-15Rα chain expressed on monocytes and dendritic cells (22–24).
Our group recently introduced a conceptually novel format of target cell-restricted IL-15- “modified immunocytokines” (MIC), designed to deliver conditional cytokine activity to effector cells after the antibody component binds to its target (25). MIC are composed of an SDIE-optimized mAb fused to an IL-15E46K mutein with abrogated IL-15Rα interaction (26). Antibody engagement with the target replaces the binding to IL-15Rα and then conditionally stimulates IL-15Rβ/γ-expressing effector cells. MICs directed against CD20 and CD19 were shown to be superior to Fc-optimized antibodies in inducing NK cell-mediated ADCC and displayed encouraging efficacy in preclinical models while producing substantially weaker off-target effects than classical IL-15-immunocytokines (25).
To extend this strategy to the therapy of AML we developed MIC directed against the CLEC12A antigen (CLL-1). CLEC12A is reportedly expressed on leukemic cells in 90% of AML cases but absent on healthy hematopoietic stem cells (27, 28). This expression pattern makes CLEC12A an attractive target for AML immunotherapy as exemplified by a large body of preclinical data and ongoing clinical trials (29). We here characterized 3 different CLEC12A antibodies to identify an optimal target binder, generated an Fc-optimized MIC construct and functionally characterized it with regard to efficacy using cell lines and primary AML cells.
2 Material and methods
2.1 Antibodies and immunocytokines
Sequences of murine CLEC12A mAbs 16C6, 33C2 and 84A2 were obtained from ABL Bio. Humanization was performed by CDR grafting. Variable domains of heavy and light chains were synthesized as double-strand DNA fragments (Thermo Fischer Scientific) and cloned in frame with Igγ1 or Igκ1 constant regions respectively downstream of the CMV promoter. Vectors carrying wild-type or optimized (S239D and I332E mutations) Fc domains were used for corresponding constructs. MIC constructs were generated by fusing the IL-15E46K mutein to the C-terminus of the heavy chain using a (G4S)4 linker, a widely adopted flexible and low-immunogenic linker that has been validated in several clinical-stage biologics.
Plasmids were purified using NucleoBond Xtra Maxi kit (Macherey-Nagel). Antibodies were produced in Expi-CHO expression system (Thermo Fischer Scientific) according to the manufacturer’s instructions. Purification from culture supernatants was conducted via affinity chromatography on Protein A Sepharose (Mabselect; Cytiva) and subsequent size exclusion chromatography (HiLoad 16/60 Superdex200; Cytiva). Subsequently, analytical size exclusion chromatography (Superdex 200R PC3.2/30 column; Cytiva) and SDS-PAGE in 4-12% gradient gels (Invitrogen) were performed to assess antibody size, aggregation and integrity. Endotoxin content was determined using Endonext Endozyme II (Biomérieux). Preparations containing less than the detection limit of the assay (0.001 EU endotoxin) were considered endotoxin free and used in experiments. When necessary, the Endotrap HD kit (Lionex) was used to remove endotoxins.
To account for differences in molecular weight, equimolar concentrations of MIC and Fc-optimized mAb 1,2 µg/ml and 1 µg/ml, respectively were used if not otherwise indicated.
2.2 Cell culture
Peripheral blood mononuclear cells (PBMC) of healthy donors and AML patients were isolated by FICOLL density gradient centrifugation (Pancoll human; PAN-Biotech) and frozen in liquid nitrogen. Informed consent was obtained from all patients and healthy donors in accordance with the Helsinki protocol and the guidelines of the local ethics committee (approval number 13/2007V).
Isolation of human NK and T cells was performed using the human NK cell isolation kit and the human pan T cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions.
Human cell lines U937, EOL-1, TF-1 and M07e were obtained from German Collection of Microorganisms and Cell Cultures (Deutsche Sammlung von Mikroorganismen und Zellkulturen, DSMZ) and cultured in RPMI 1640 medium (Thermo Fischer Scientific) supplemented with 10% heat-inactivated fetal bovine serum (PAN-Biotech) and 1% penicillin-streptomycin (Lonza). Cultures of TF-1 and M07e cells were supplemented with 2 and 10 ng/µl GM-CSF (Miltenyi Biotec) respectively.
The respective immunophenotype described by the provider was validated after thawing using flow cytometry. Culture supernatants were monthly PCR-tested for mycoplasma contamination.
U937 cells were labeled with GFP using lentiviral infection. A second-generation lentiviral vector containing the EGFP-T2A-Puro cassette under the CMV promoter was cloned by Vector Builder. Lentiviral particles were produced by co-transfecting HEK293T cells with the vector and packaging plasmids (psPAX2 and pMD2.G, Addgene #12260 and #12259, gifts from Didier Trono) using a standard calcium-phosphate transfection protocol. The lentiviral supernatants were then concentrated using the Lenti-X concentrator (Takara) according to the manufacturer’s instructions. U937 cells were infected with the concentrated lentiviral particles via spinoculation (1 hour at 600g, 37°C) and selected with 1 µg/ml puromycin.
2.3 Flow cytometry
CLEC12A expression and binding characterization of CLEC12A antibodies were determined by incubation of CLEC12A-expressing cells with the indicated constructs or isotype controls, followed by goat-anti-mouse-PE or donkey-anti-human PE conjugates (Jackson ImmunoResearch Europe Ltd) staining for murine and human antibodies respectively and analyzed using flow cytometry. BD LSRFortessa (BD Biosciences) was used for data acquisition, data analysis was performed in FlowJo v9 (FlowJo LCC). The following fluorescent antibody conjugates were used: CD3-APC-Fire 750 (SK7, Biolegend), CD33-APC (WM53, Biolegend), CD34-APC (clone 581, BD Bioscience), CD38 and CD56-PE/Cy7 (HCD56, Biolegend); life-dead staining was performed using 7-aminoactinomycin (7-AAD) (Biolegend).
Leukemic cells in patient samples were first selected by FSC/viability gating (staining with 7-AAD (BD Biosciences) and then gates were applied dependent on surface expression of CD33, CD34 or CD38. Gating strategy is shown in Supplementary Figure S4. Specific fluorescence intensity (SFI) were calculated by dividing mean fluorescence intensities measured with specific antibody by mean fluorescence intensities obtained with isotype control.
2.4 Human NK and T cell activation and proliferation
To analyze NK and T cell activation, upregulation of CD69 on the surface of CD3-CD56+, CD3+CD4+ and CD3+CD8+ subpopulation was analyzed by flow cytometry using an CD69-PE (FN50, BD Bioscience) staining.
For analysis of proliferation, PBMC of healthy donors were labelled with CellTrace™ Violet Cell Proliferation Kit (CTV) according to the manufacturer’s instructions (Thermo Fisher). After 6 days of co-incubation with target cells and respective antibodies, dye dilution was analyzed by flow cytometry in NK and T cells identified by counterstaining for CD3, CD4, CD8 and CD56.
2.5 Cytokine quantification
IFN-γ secretion was quantified using enzyme-linked immunosorbent assay (ELISA). For ELISA anti-human IFN-γ monoclonal antibody clones 2G1 and biotinylated B133.5 (Thermo Fisher), as well as poly-HRP 20 Streptavidin (Fitzgerald) were used. The TMB 2-Component Microwell Peroxidase Substrate Kit (Seracare) was used as substrate.
2.6 Cytotoxicity assays
For analysis of NK cell-mediated cytotoxicity, target cells were labelled with CTV. After co-incubation with PBMC of healthy donors and the indicated treatment, living target cells (CTV+7-AAD-) were quantified using flow cytometry. Analysis of equal assay volumes was ensured by using standard calibration beads (3 µm latex beads; Sigma-Aldrich).
Co-cultures of primary AML samples were supplemented with 50 ng/ml G-CSF, IL-3, IL-6, TPO, Flt3L and 100 ng/ml SCF (Peprotech). Due to the higher resistance of primary AML cells to NK cell-mediated lysis compared to cell lines, effector-to-target ratios were adapted based on preparative titration experiments.
Long-term cytotoxicity was analyzed with the IncuCyte S3 Live-Cell Analysis System (Sartorius). GFP-expressing U937 cells were co-cultured with PBMC of healthy donors and treated as indicated. Cells were imaged every 4h and after Top-Hat background subtraction, living target cells were identified with a predefined mask (size and fluorescence intensity) and counted.
2.7 Statistical analysis
GraphPad Prism 9 was used to generate all plots and perform statistical analysis. Statistical analyses included the Friedman test with Dunn’s multiple comparison test for comparing related groups, the Kruskal-Wallis test with Dunn’s test for independent group comparisons, and one-way ANOVA with Sidak’s test for normally distributed data with equal variances.
The statistical tests performed are detailed in the figure legends. Graphs show mean values obtained with n technical or biological replicates, and error bars in all figures represent standard deviation (SD), unless indicated otherwise. A p-value <0.05 has been used as level of significance.
3 Results
3.1 Identification of an optimal target binder
As a first step, we compared the binding properties of 3 murine CLEC12A mAbs termed 33C2, 16C6 and 84A2. Flow cytometric analysis using the CLEC12A-expressing cell lines U937 and EOL-1 revealed that all three mAbs reached saturated binding at approximately 1 nM (0,15 µg/ml) (Figure 1A). Saturation was also observed at comparable concentrations on the surface of patient leukemic blasts (Figure 1B). Antibody 84A2 always showed the weakest binding, clones 33C2 and 16C6 displayed similar binding characteristics using the cell line U937 and primary AML samples (Figures 1A, B). 33C2 achieved slightly higher binding with EOL-1 cells (Figure 1A) and had a lower half-maximal binding concentration in titration experiments with the primary samples AML1, 2 and 3 (Figure 1B). To ascertain selection of a target binder with optimal characteristics, we expanded our analysis to a panel of 22 AML patient samples representing different FAB-subtypes. The clinical characteristics of the patients are given in Table 1. Leukemic cells in all samples were found to be CLEC12A-positive as defined by a specific fluorescence intensity (SFI) level (mean fluorescence intensities measured with specific antibody divided by mean fluorescence intensities obtained with isotype control) of >1.5 (Figures 1C, D). Our results confirm earlier data on widespread expression of the antigen in AML. In agreement with our titration experiments, use of mAb 84A2 reproducibly resulted in lower SFI values than the two others mAbs, especially with leukemic cells expressing low CLEC12A levels (Figure 1C, AML1 and AML2). The analysis with more than 20 patient samples further revealed that 16C6 showed intermediate binding intensity, whereas 33C2 displayed the highest SFI values (mean SFI=35, 42.9 and 53.9 respectively) (Figure 1D). Based on these results, 33C2 was selected as lead candidate.
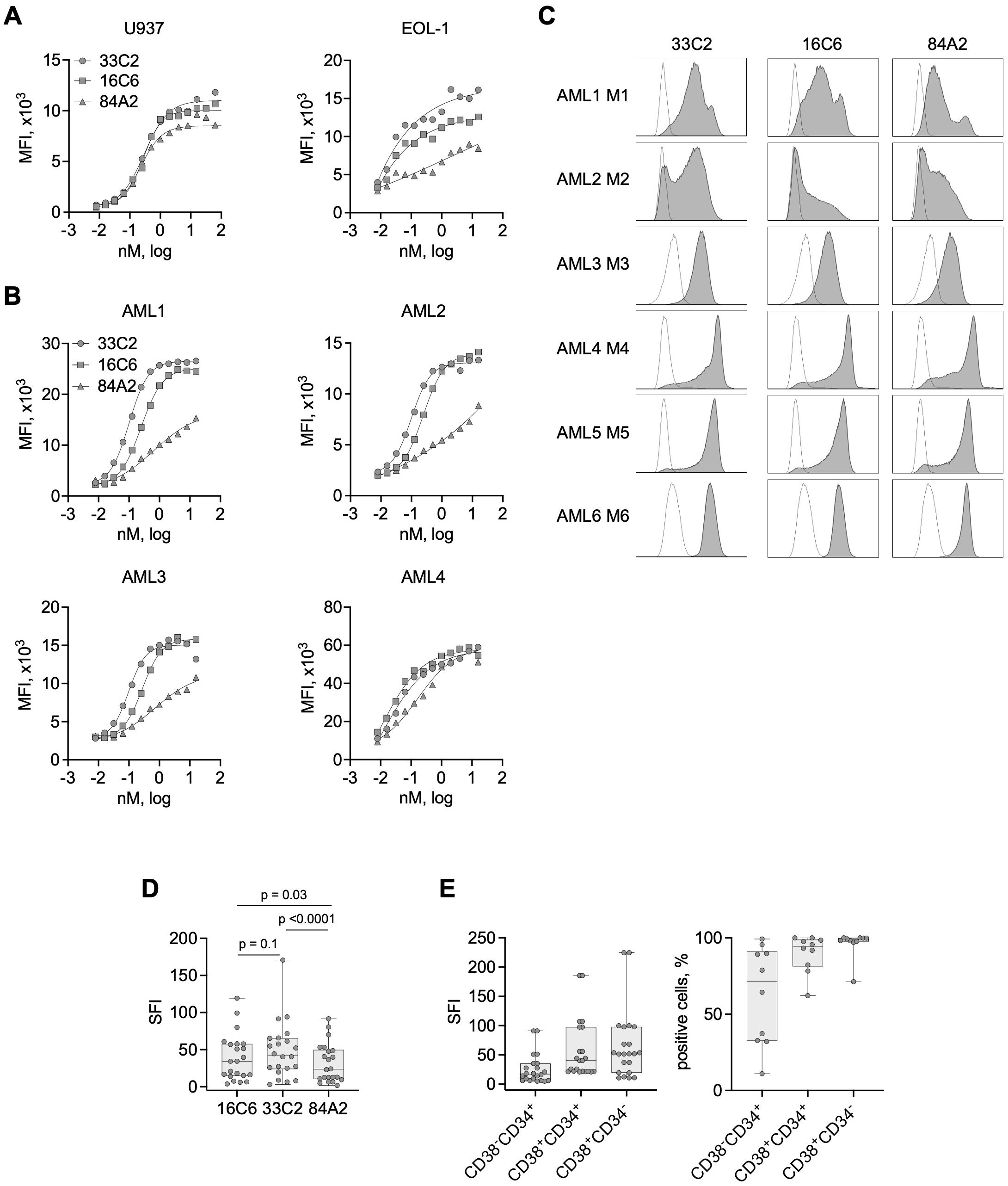
Figure 1. Binding characteristics of CLEC12A antibodies with cell lines and primary AML cells. (A) The indicated AML cell lines were incubated with increasing concentrations of three murine CLEC12A mAbs (33C2, 16C6 and 84A2) followed by anti-mouse PE conjugate and flow cytometric analysis. Surface expression is shown as median fluorescence values (MFI). (B) Four primary AML patient samples (UPN 11, 20, 10, 15) were stained and analyzed as described in (A). (C) Exemplary histograms obtained upon analyzing primary AML cells (UPN 4, 20, 21, 6, 5, 8) of different FAB-subtypes with the mAb clones 33C2, 16C6 and 84A2 (1 µg/ml each) followed by anti-mouse PE and flow cytometric analysis (grey peaks, CLEC12A; outline peaks, isotype control). (D) CLEC12A expression on blasts of AML patients (n = 22) as determined with the indicated mAbs. SFI values were calculated by dividing median fluorescence values obtained with specific mAb by median fluorescence values obtained with isotype control; dots: results of individual patients, box: first to third quartiles, whiskers: min-max. P values were determined with Friedman test with Dunn’s multiple comparison test. (E) CLEC12A expression on leukemic subpopulations: LSC (CD38-CD34+), progenitors (CD38-CD34+) and mature blasts (CD38-CD34+) (n=10) detected using the 33C2 mAb. Data are summarized as SFI values (left) and percent of positive cells.
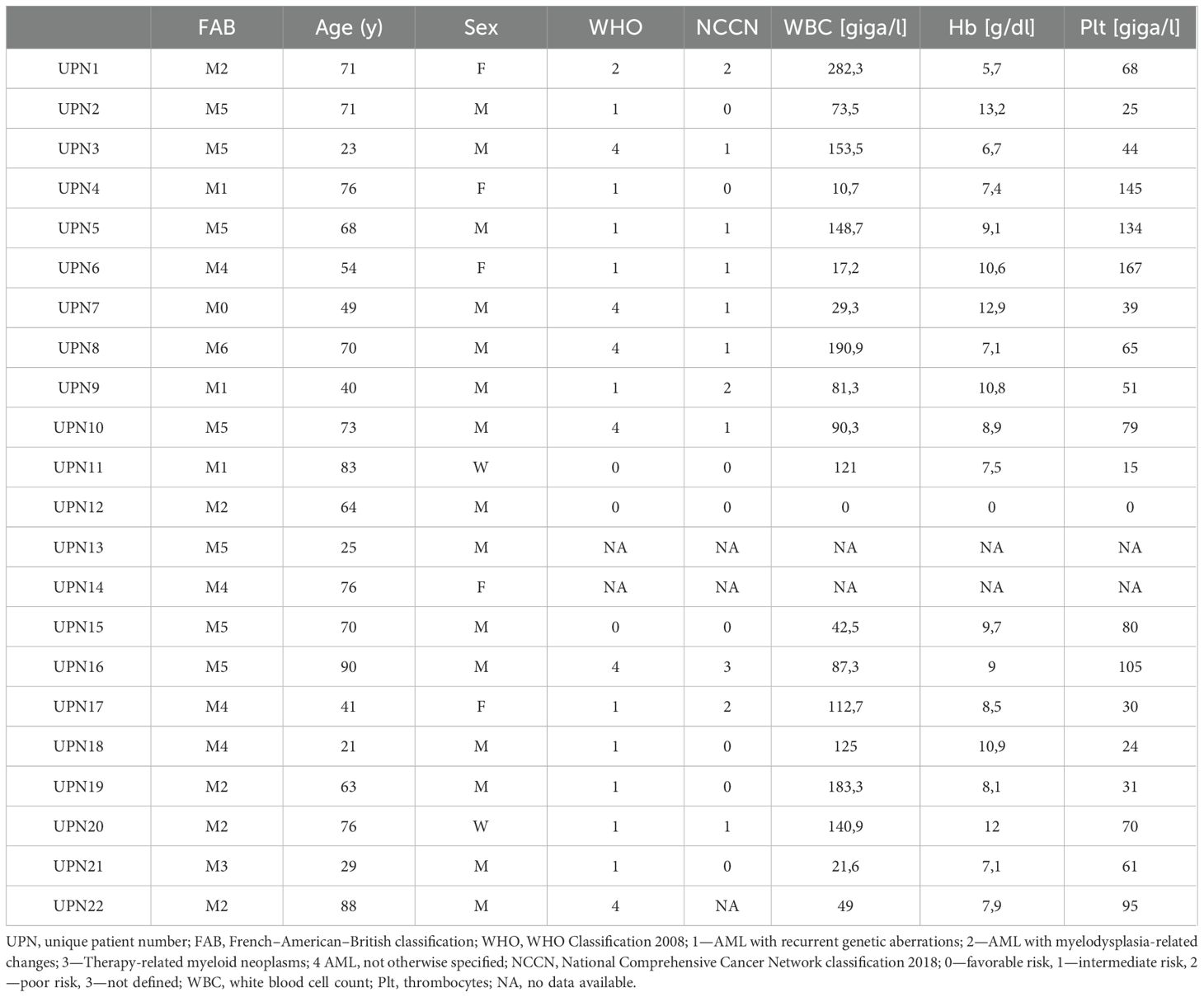
Table 1. Clinical characteristics of the patient cohort.
Next, we tested binding of 33C2 to different leukemia cell subpopulations, i.e. leukemic stem cells (LSC, CD34+CD38-), progenitor cells (CD34+ CD38+) and CD34-, CD38+ leukemic blasts. We detected substantial CLEC12A positivity over all subpopulations in all 22 patient samples (SFI>5), albeit the LSC population displayed lower SFI values and less positive cells when compared with the two other populations (Figure 1E).
3.2 Generation of IL-15 immunocytokines and induction of NK cell reactivity against CLEC12A-expressing cell lines
To generate a MIC protein targeting CLEC12A (MIC12), the construct was composed of humanized VH and VL domains of the 33C2 antibody, the Fc domain of human IgG1 with SDIE modification and a human IL-15E46K mutein with abrogated binding to IL-15Rα as described in the Methods section (Figure 2A). In addition, an Fc-optimized antibody without IL-15 moiety (33C2SDIE) was cloned as a control. In order to gain a first understanding regarding the producibility of the MIC constructs, we evaluated purity and aggregate content using SDS-PAGE and size exclusion chromatography, which documented that our MIC construct displayed the expected molecular weight and showed high purity and no aggregates (Figures 2B, C). Staining of U937 and EOL-1 cells with MIC12 confirmed that target binding was not affected by humanization and immunocytokine construction (Figures 2D, E). Next, we validated binding of the MIC construct to IL-15 receptors. As expected, our MIC12 construct containing the E46K-mutein that abrogates IL15Rα-binding exhibited very weak interaction with IL-15Rα-expressing TF-1 cells compared to an IL-15WT control construct, but retained binding to IL15Rβ/γ-expressing M07e cells (Figure 2F).
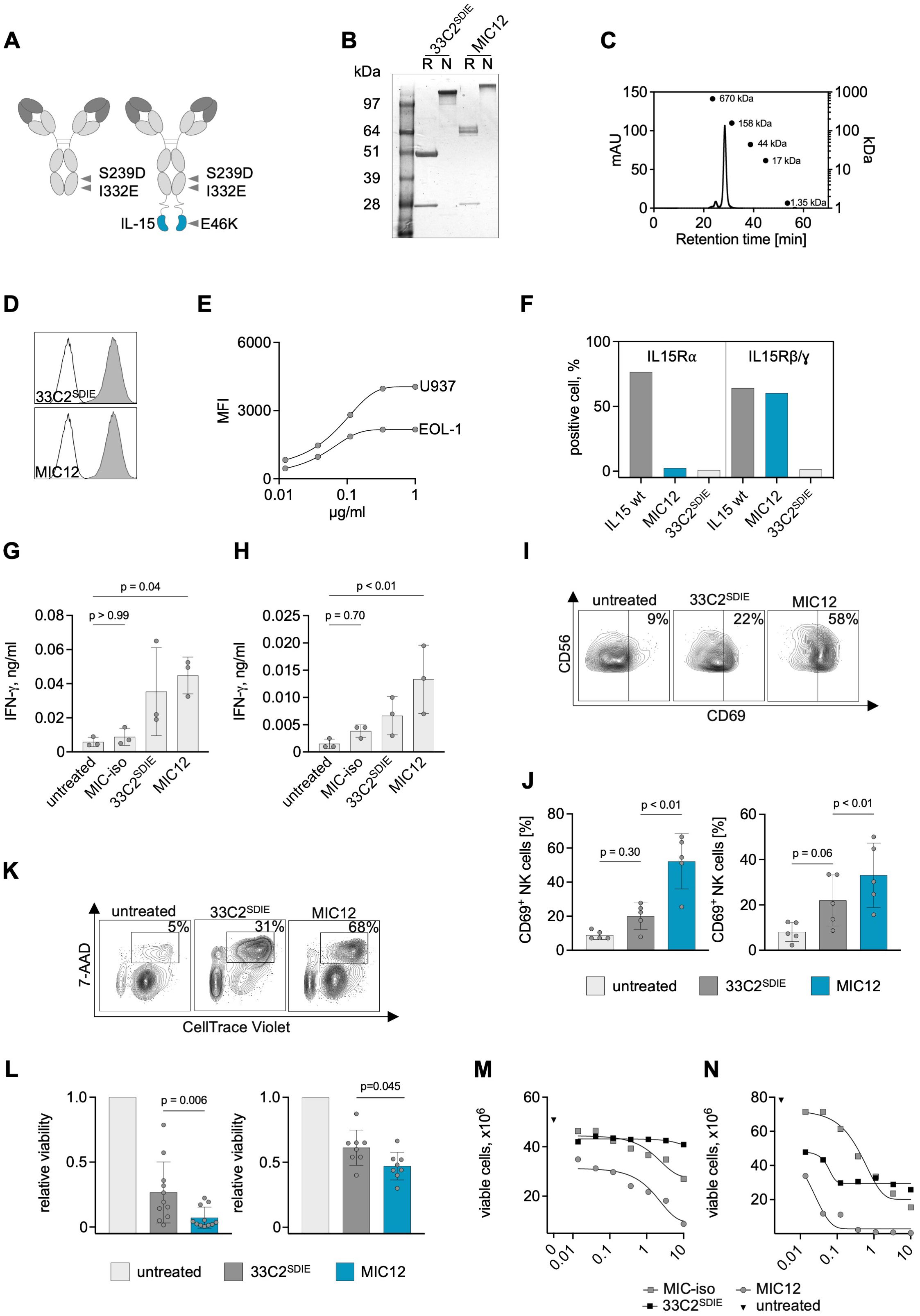
Figure 2. Production of anti-CLEC12A IL-15 immunocytokines and characterization of on-target and off-target activity using leukemia cell lines. (A) Schematic illustrations of the MIC12 immunocytokine consisting of the humanized VH and VL domains of the 33C2, human IgG1 constant domains containing the SDIE modification (S239D; I332E), (G4S)4 linker and a C-terminal IL15E46K mutein (right) and a control Fc-optimized mAb without IL-15 moiety (33C2SDIE) (left). (B) SDS-PAGE electrophoresis of the humanized Fc-optimized 33C2SDIE antibody and MIC12 in reducing (R) and non-reducing (N) conditions. Expected molecular weights: 23 kDa for the light chain, 63 kDa for the heavy chain of the MIC construct and 49 kDa for the heavy chain of the antibody. (C) Exemplary results of a size exclusion chromatography of the MIC12. Expected size of the full construct 172 kDa. (D) Binding of MIC12 and 33C2SDIE to U937 cells at equimolar concentrations detected with donkey anti-human IgG-PE conjugate and flow cytometric analysis (grey peaks, anti-CLEC12A; outline peak, donkey anti-human IgG-PE conjugate). (E) Dose titration of MIC12 on U937 and EOL-1 cells detected with donkey anti-human IgG-PE conjugate and flow cytometric analysis. (F) Binding of MIC12 to IL-15Rα-expressing TF-1 (left) and IL15Rβ/γ-expressing M-07e cells (right). Binding was detected as described in (D). A MIC construct with irrelevant specificity containing wild-type IL-15 was used as a positive control, 33C2SDIE was used as a negative control. (G, H) PBMC from healthy donors (n=3) were cocultured with U937 (G) or EOL-1 (H) cells (E:T 2.5:1) with the indicated constructs at equimolar concentrations (1 µg/ml for the antibody and 1.2 µg/ml for the MIC). IFN-γ secretion in culture supernatants was determined after 24 h using ELISA. MIC-iso, – MIC construct with irrelevant specificity. P values were determined with Kruskal-Wallis test, followed by Dunn’s multiple comparison test. (I, J) PBMC from healthy donors were incubated with U937 and EOL-1 cells at an E:T ratio of 2.5:1 in the presence of constructs for 72h (1 µg/ml for the antibody and 1.2 µg/ml for the MIC). CD69 expression on CD3-CD56+ NK cells was determined by flow cytometry. (I) Exemplary flow cytometry plots derived with U937 target cells; the percentage of CD69+ NK cells is indicated. (J) Combined results obtained with 5 PBMC donors using U937 (left) and EOL-1 cells (right). Data are shown as means of triplicate measurements. P values were determined with Kruskal-Wallis test, followed by Dunn’s multiple comparison test. (K, L) PBMC from healthy donors were incubated for 72 h with CellTrace Violet (CTV)-labelled U937 or EOL-1 cells at an E:T ratio of 2.5:1 in the presence of the indicated constructs at equimolar concentration (1 µg/ml for the antibody and 1.2 µg/ml for the MIC). Target cell lysis was determined by flow cytometry. (K) Exemplary flow cytometry plots obtained with U937 target cells; the percentage of dead target cells (CTV+7AAD+) is indicated. (L) Combined results obtained with U937 (left) and EOL-1 (right) cells using 11 and 7 independent PBMC donors, respectively. Cell viability was normalized to the untreated control (mean of duplicate wells). P values were determined with Friedmans test, followed by Dunn’s multiple comparison test. (M, N) PBMC (M) or purified NK cells (N) from healthy donors were incubated for 72 h with CellTrace Violet (CTV)-labelled U937 cells at an E:T ratio of 2.5:1 (PBMC) or 1:1 (NK cells) in the presence of the indicated constructs titrated in a broad concentration range. Target cell lysis was determined by flow cytometry. Representative results from a total of three donors with similar outcomes are presented.
Given that exogenous cytokines and so far available immunocytokines suffer from drawbacks like Treg induction in case of IL-2 and systemic toxicity upon higher dosing in case of both IL-2 and Il-15 (15–17), we have developed an IL-15 moiety with targeted activity to reduce nonspecific effector cell activation and cytokine release, a key feature of our recently published MIC platform (25). To evaluate this property for MIC12, we analyzed how our construct influenced IL-15-mediated NK cell activation. As control, we used a construct in which the 33C2 variable domain was replaced by an irrelevant target binder (MOPC-21, MIC-iso). We performed a comparative analysis of MIC-iso with MIC12 regarding their ability to stimulate IFN-γ secretion in cocultures of PBMC from healthy donors (PBMC) using U937 and EOL-1 cells as targets. This experiment documented that MIC-iso induced no relevant IFN-γ secretion, whereas target recognition by 33C2SDIE and MIC12 induced significant NK cell reactivity (Figures 2G, H), confirming previous findings on the target cell-restricted mode of action of MIC constructs (25).
To assess the benefits of the MIC12 construct over the Fc-optimized CLEC12 mAb, we first cocultured PBMC with U937 and EOL-1 cells and quantified NK cell activation and cytotoxicity. MIC12 was clearly superior to 33C2SDIE in inducing CD69 expression on CD3-CD56+ NK cells with both target cell lines (Figures 2I, J). This was mirrored by significantly more pronounced target cell lysis as detected in flow cytometry-based cytotoxicity assays with multiple PBMC donors (Figure 2K, L). Of note, effects of both, 33C2SDIE and MIC12, against EOL-1 were generally weaker compared to U937. Analysis of target antigen expression revealed that U937 express profoundly more CLEC12A, which correlates with substantially higher efficacy upon therapeutic targeting (Figure 2E).
Dose titration experiments using both PBMC and isolated NK cells confirmed the superior therapeutic efficacy of MIC12 compared to the Fc-optimized CLEC12A mAb over a broad range of concentrations and also documented a wide therapeutic window upon comparison of MIC12 and MIC-iso (Figures 2M, N).
Next, we aimed to exclude that MIC12 altered NK cell viability, e.g. by inducing fratricide. To this end, we cultured purified NK cells without target cells in the presence of the MIC12 or the 33C2SDIE antibody and employed flow cytometric analysis with 7-AAD to confirm that treatment did not affect NK cell viability (Supplementary Figure S1).
3.3 MIC12 induces NK cell proliferation and long-term clearance of target cells
Since ADCC-inducing antibodies suffer from a low capacity to induce NK cell proliferation, we aimed to overcome this limitation with our MIC construct. To this end we compared NK cell proliferation upon treatment with Fc-optimized mAb and MIC12. Flow cytometric dye dilution assays showed progressively dimmer CTV peaks in the CD3-CD56+ NK cell population upon treatment with MIC12 compared to Fc-optimized mAb after 7 days of coculture with target cells, which is clearly indicative of NK cell proliferation (Figure 3A). Quantification of NK cells in cultures revealed that, unlike the corresponding Fc-optimized mAb, MIC12 promoted pronounced NK cell proliferation, as revealed by more than doubling the NK cell population within PBMC (Figures 3B, C).
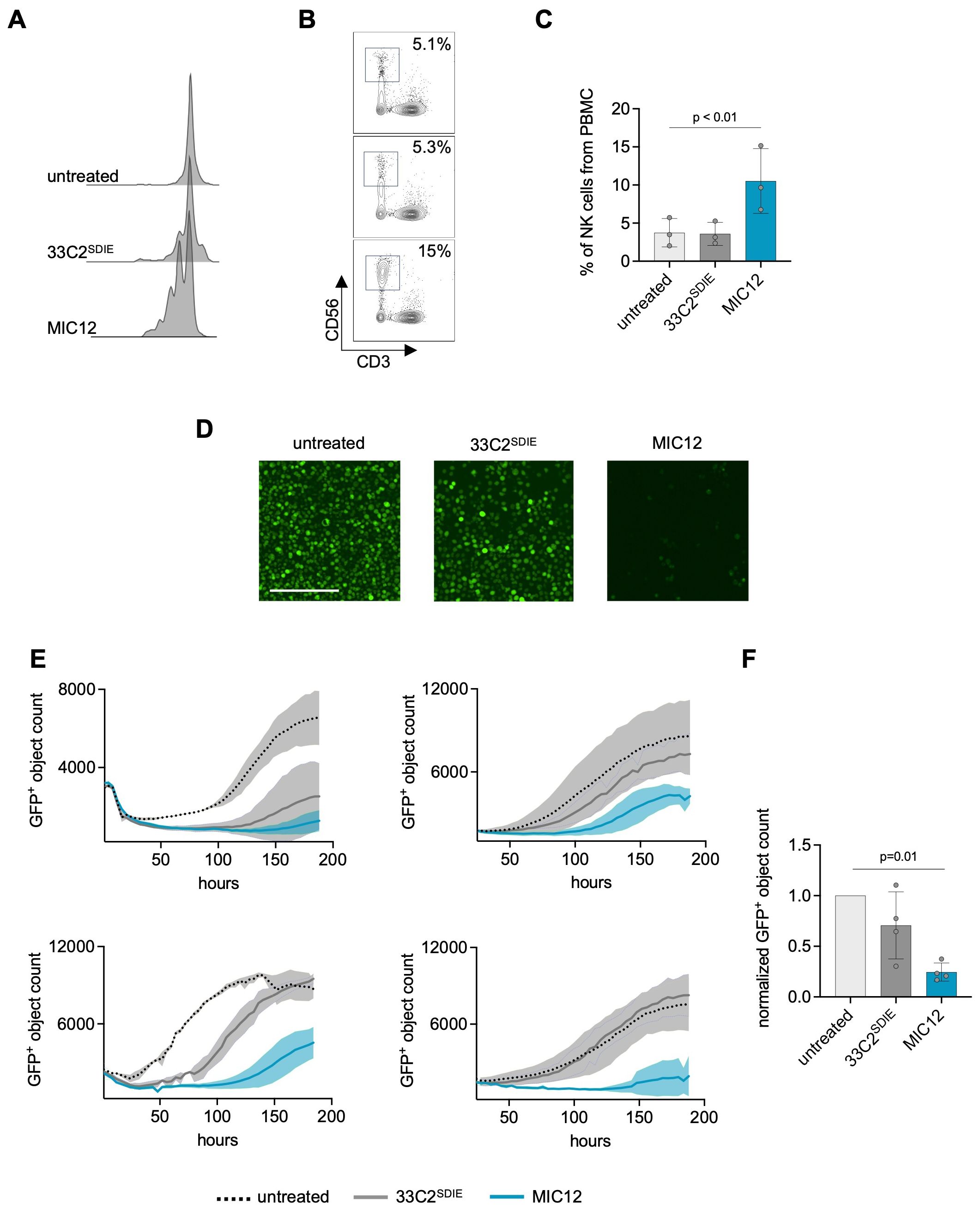
Figure 3. MIC12 induces NK cell proliferation and mediates superior long-term efficacy against U937 cells. (A-C) CTV-labelled PBMC were cultured with U937 cells (E:T 2.5:1) and the indicated constructs at equimolar concentrations. Fresh target cells and constructs were added to the wells after 3 days followed by continued incubation for additional 4 days. (A) CTV dye dilution was analyzed by flow cytometry. Exemplary histogram plots of one representative experiment are shown. (B) Exemplary dot plots demonstrating expansion of CD56+ NK cells. Top, untreated; middle, 33C2SDIE; bottom, MIC12. (C) Quantification of NK cell expansion after 8 days of incubation with the indicated constructs at equimolar concentrations, presented as percentage of the total PBMC population. Each data point represents data obtained with a single PBMC donor. (D, E) Long-term cytotoxicity assay. PBMC were incubated 8 days with GFP+ U937 cells (E:T 2.5:1) and indicated treatments (1 µg/ml). Target cell viability was quantified using Incucyte S3. (D) Exemplary microscopic images, scale bar: 200 µm. (E) Growth curves representing data from cocultures with four independent PBMC donors. Shown are GFP+ object counts at each timepoint with mean values derived from two wells. Standard deviations are shown as shaded areas. (F) Combined results obtained with 4 PBMC donors, 120 hours after assay initiation. Viable cell counts were normalized to untreated control (mean of triplicate measurements). P-values were determined using the Kruskal-Wallis test followed by Dunn’s multiple comparison test.
Next, we determined whether the MIC12-induced expansion of effector cells over time translated into improved long-term efficacy. To this end, we cocultured GFP-expressing U937 cells with PBMC in the presence of the constructs and monitored growth of target cells over one week by live cell imaging. As evident from the exemplary images (Figure 3D), the Fc-optimized antibody achieved only marginal effects, whereas treatment with MIC12 resulted in almost complete depletion of target cells. This was mirrored in time course quantification of target cell density using PBMC of four independent donors (Figure 3E) and in the cumulated data of all 4 donors after 120h (Figure 3F). Despite a substantial donor variation, in all cases target cells treated with 33C2SDIE demonstrated either outgrowth after initial short-term depletion (donors 1-3) or were not affected at all (donor 4). In stark contrast, MIC12 mediated prolonged suppression of target cell growth. Notably, all effects observed following MIC12 treatment were NK cell-dependent, as no T cell activation (Supplementary Figure S2A), cytokine secretion (Supplementary Figure S2C), T cell-mediated cytotoxicity (Supplementary Figures S2D, E), or proliferation (Supplementary Figure S2G) were detected.
These findings showcase the remarkable superiority of our MIC construct over Fc-optimized mAb in eliciting NK cell activation, proliferation, and sustained antitumor efficacy.
3.4 MIC12 induces potent NK cell reactivity against primary AML cells
Finally, we aimed to determine whether superior efficacy of MIC12 could also be demonstrated with primary AML cells from leukemia patients. To this end, we analyzed NK cell activation and IFN-γ release in cocultures of PBMC with primary AML blasts. Flow cytometry analysis revealed profound CD69 upregulation as marker for activation upon treatment with MIC12, but not with 33C2SDIE, and this held true for combinations of leukemic cells of three AML patients and four PBMC donors (Figures 4A, B). Furthermore, MIC12 promoted IFN-γ secretion to levels markedly exceeding those induced by 33C2SDIE (Figure 4C).
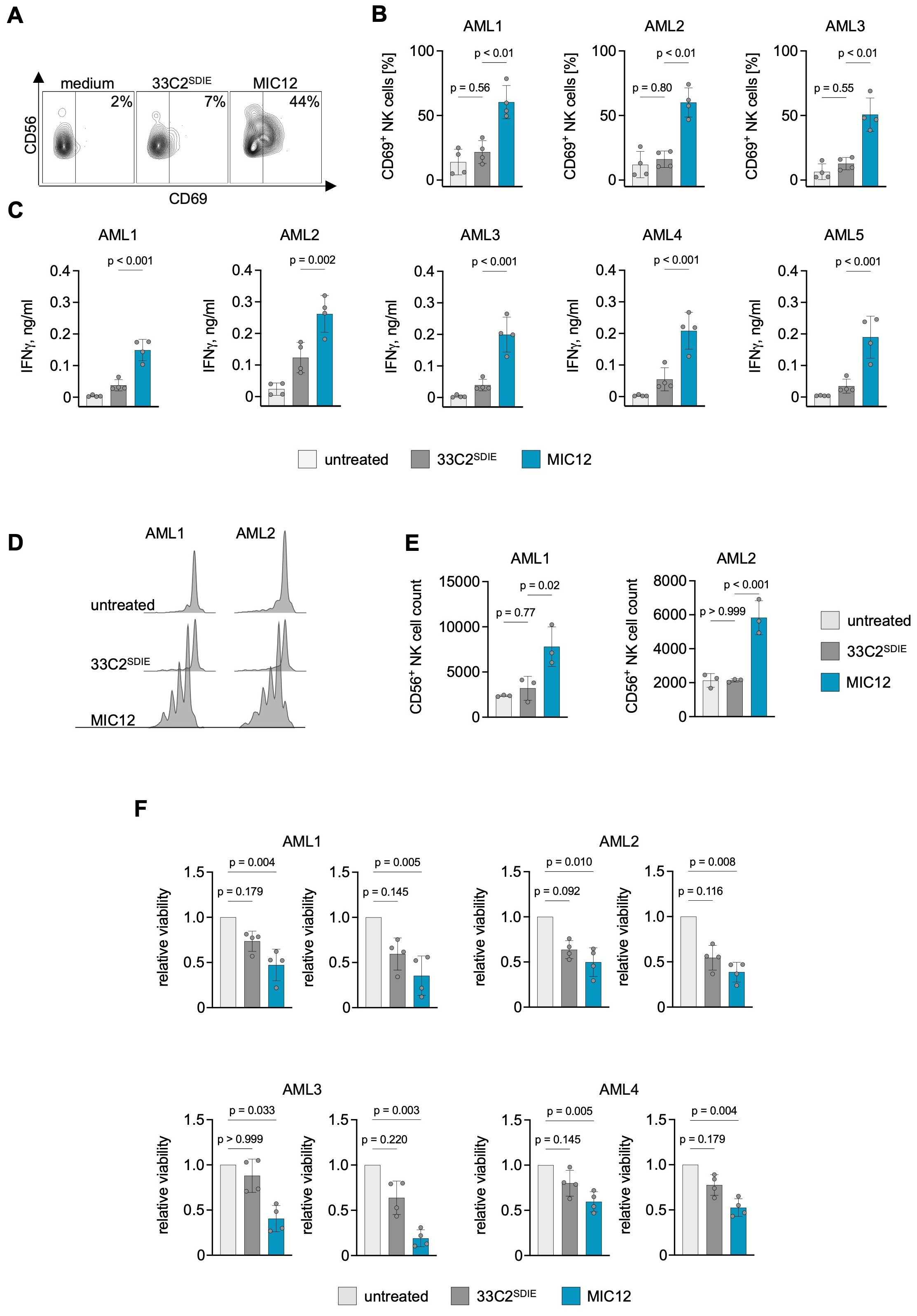
Figure 4. MIC12 induces superior NK cell reactivity against patient AML cells. (A, B) PBMC of healthy donors were incubated with patient AML cells (E:T 2.5:1) in the presence or absence of the indicated at equimolar concentrations. After 3 days, CD69 expression on CD3-CD56+ NK cells was assessed using flow cytometry. (A) Exemplary flow cytometry plots, the percentage of CD69+ NK cells is indicated. (B) Combined results obtained with 4 independent PBMC donors incubated with 3 different AML patient samples. P values were determined with One way ANOVA, followed by Sidak’s multiple comparison test. (C) PBMC from 4 donors were each incubated with leukemic cells of 5 different AML patients. After 24 hours, IFN-γ levels in culture supernatants were measured by ELISA P values were determined with One way ANOVA, followed by Sidak’s multiple comparison test. (D, E) Proliferation of CTV-labelled NK cells incubated for 6 d with patient AML cells in the presence of the indicated constructs at equimolar concentrations (0,1 µg/ml for the antibody and 0,12 µg/ml for MIC) was assessed by flow cytometry. (D) Exemplary dye dilution curves are shown with 2 AML samples. (E) Quantification of CD3-CD56+ NK cells in cocultures combining PBMC from three independent donors with samples from two different AML patients. P values were determined with one-way ANOVA with Sidak’s multiple comparison test. (F) PBMC of 4 different donors were cultured with CTV-labelled patient AML cells at E:T ratios of 20:1 (left) and 10:1 (right) for 6 days. Viability of AML cells was determined by flow cytometry. Shown are viable cell counts normalized to untreated control. P values were determined with Kruskal-Wallis test, followed by Dunn’s multiple comparison test.
To ensure that the key advantage of our MIC constructs – induction of NK cell proliferation – was also achieved when using primary patient samples, we employed dye dilution assays in six-day cocultures of CTV-labeled PBMC and patient AML cells. The observed multiple CTV intensity peaks revealed robust NK cell proliferation induced by MIC12, contrasting with the unchanged CTV staining intensity in cultures treated with 33C2SDIE (Figure 4D). Quantitative analysis revealed a pronounced increase in NK cell proliferation following MIC12 treatment, whereas cultures treated with an Fc-optimized antibody remained unaltered (Figure 4E, Supplementary Figure S3). Within the PBMC, a 3- to 10-fold NK cell expansion clearly exceeding the range of physiological NK cell percentages in healthy individuals was detected, which emphasizes the biological relevance of the induced proliferation (Supplementary Figure S3). Similarly to the experiments with cell lines, MIC12 did not induce T cell activation (Supplementary Figure S2B) or proliferation (Supplementary Figure S2G) in cocultures with primary AML cells. Finally, we studied the anti-leukemic activity of the MIC12 and 33C2SDIE in long-term cytotoxicity assays with the patient cells. We cultured 4 primary AML samples with 4 PBMC donors and quantified surviving AML cells by flow cytometry (Figure 4F). MIC12 consistently induced profoundly stronger NK cell cytotoxicity against CLEC12A-positive AML blasts as compared to the Fc-optimized antibody which did not achieve statistically significant effects in any combination. This supports the essential role of IL-15 in boosting anti-leukemic reactivity.
4 Discussion
Here we report on the development and preclinical characterization of immunocytokines with conditional IL-15 activity targeting CLEC12A for AML treatment (MIC12). This molecule expands our previously described immunocytokine platform, characterized by NK cell-focused IL-15 activity, which has shown superior efficacy compared to Fc-optimized antibodies while reducing systemic cytokine effects to AML.
CLEC12A (CLL-1) emerges as a promising antigen for AML immunotherapy. Preclinical studies have demonstrated the efficacy of CLEC12A bispecific antibodies (30, 31), antibody-drug conjugates (32, 33), antibody-cytokine fusion proteins (34) and CLEC12A-targeted CAR-T cells (35) in eliminating AML cells in vitro and in vivo. Several clinical trials evaluating safety and efficacy of CLEC12A-targeting bispecific antibodies (NCT03038230), CAR-T cells (NCT04219163, NCT06128044, NCT06017258) and antibody-drug conjugates a have been recently completed or are currently ongoing and show manageable toxicity and signs of efficacy (36, 37). A major obstacle of antibody-based immunotherapy is the unintended targeting of healthy tissues expressing the antigen, leading to side effects. CLEC12A is an attractive target for immunotherapeutic approaches due to its limited expression on normal hematopoietic stem cells and other healthy tissues but widespread prevalence on AML blasts and leukemic stem cells (38), which is confirmed by our data obtained in a cohort of 22 AML patients (Figures 1D, E). Our MIC molecules with IL-15 activity being restricted to target antigen binding, combined with CLEC12A’s rather tumor-restricted expression, provides a promising and potentially safe approach for AML therapy.
In recent years many immunocytokines demonstrated remarkable preclinical efficacy, including one CLEC12A-targeting IL15-based NK cell engager (34). However, in all these immunocytokines, the activity of the cytokine moieties is independent of antibody target binding. As a result, these molecules fail to exclusively focus the cytokine effects to the tumor site, leading to profound systemic off-target effects and dose-limiting toxicity (19–21). This is due to the fact that only a minor fraction of the applied immunocytokine is bound to the target at any given time, while the majority is distributed systemically where it mediates undesired effects (39). Furthermore, the cytokine moiety can drive localization of the drug to the receptor-expressing cells irrespectively of antibody specificity (40). So far only few attempts were undertaken to limit the stimulatory activity of the cytokine to the tumor site. For example, an attenuated IL-21 mutein was fused to an PD-1 antibody to restrict cytokine activity to PD-1-expressing cells (41). An IL-2 variant temporally inactivated by a linker that is cleavable exclusively by tumor-specific proteases was used to restrict cytokine function to tumor microenvironment enriched with specific proteolytic enzymes (42). Finally, de novo designed IL2/15Rβγ agonists without binding to IL2Rα showed improved activation of CD8+ T cells, reduced expansion of CD4+ Tregs and improved targeting to the tumor site (43). The design of our MIC immunocytokines incorporates an IL-15E46K mutein with disrupted IL-15Rα binding, allowing for substitution of trans-presentation by target antigen binding, which facilitates target-restricted IL-15Rβγ activation (25). Similar to other MIC of our platform, MIC12 exhibited pronounced target specificity, as indicated by CLEC12A-dependent IFN-γ release (Figure 2H). Although further studies in animal models and ultimately patients are needed to fully validate our concept with regard to potential toxicity/safety, our data provide support for IL-15E46K-based Fc-optimized MIC as a promising immunocytokine format with conditional IL-15 activity.
The pivotal role of NK cells in mediating ADCC upon application of antitumor mAbs is well established. Current efforts are focused on enhancing efficacy by optimizing interactions with activating Fc receptors. The SDIE modification represents a key advance in boosting NK cell-mediated ADCC, and several Fc-optimized mAbs with this modification are either in clinical evaluation or have recently gained approval, such as the CD19-targeting tafasitamab. Combining Fc optimization with IL-15 activity further amplifies the therapeutic potential. Alike our IL-15E46K-based Fc-optimized immunocytokine constructs targeting CD20 and CD19 (25), MIC12 achieved superior NK cell activation and killing of cancer cell lines and primary AML cells when compared with Fc-optimized antibody. Notably, NK cells are generally scarce within PBMC and have been reported to be functionally impaired in MDS and AML patients due to an immunosuppressive microenvironment and chemotherapy (44–46). Therefore and given the unfavorable effector to target ratios that prevail in most patients, NK cell-stimulating therapeutic mAbs so far fail to achieve disease control. Thus, expansion of NK cells and maintenance of their functional capacity is key for successful therapy. Administration of IL-15 leads to massive NK cell expansion in animal models and cancer patients (47–49). In comparative analyses with MIC12, an Fc-optimized control mAb was unable to trigger NK cell expansion, emphasizing the combinatorial effect of Fc receptor engagement and IL-15 signaling in a targeted manner achieved by our constructs. The observed synergy is in agreement with the enhancement of IL-15-mediated NK cell expansion in vivo by FcR signaling (50). This effect can be explained by the recently identified crosstalk between IL-15-induced STAT-5 signaling and ITAM-dependent activating receptors (such as FcγRIII) (51, 52). The NK cell proliferation induced by MIC12 translated into sustained long-term control of U937 cell growth, contrasting with the limited impact of 33C2SDIE. This was mirrored in long-term killing assays with primary AML cells, where 33C2SDIE failed to produce statistically significant effects, but MIC12 effectively depleted AML blasts, further emphasizing the potential of MIC12 to improve leukemia cell elimination in AML patients.
Of note, NK cells demonstrated promising therapeutic potential in AML upon adoptive transfer (44, 53, 54), but the transferred NK cells reportedly become functionally impaired (55). It is tempting to speculate that, beyond being applied as single agent, MIC12 may ensure maintenance of transplanted NK cell upon combinatorial application.
In summary, our findings establish MIC12SDIE as a reagent with superior antileukemic activity, enabled by NK cell proliferation induced by target cell-restricted IL-15 activity that surpasses the potency of Fc-optimized monoclonal antibodies and thus constitutes a promising novel compound for AML therapy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by IRB (ethics committee of the Faculty of Medicine of the Eberhard Karls University Tübingen). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
BK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. LA: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. JJ: Resources, Writing – review & editing. YL: Resources, Writing – review & editing. BL: Resources, Writing – review & editing. JW: Resources, Writing – review & editing. LZ: Conceptualization, Resources, Writing – review & editing. AC: Data curation, Formal analysis, Investigation, Writing – review & editing. MP: Investigation, Resources, Writing – review & editing. JH: Data curation, Formal analysis, Writing – review & editing. GJ: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. HS: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from ABL Bio, Inc., German Cancer Aid (70114180) and Wilhelm-Sander-Stiftung (2017.100.1-3). We acknowledge support from the Open Access Publishing Fund of the University of Tübingen.
Acknowledgments
The authors thank Celine Derrix and Michael Beller for excellent technical assistance, Suyoun Lee, Byeong Min Yoo, and Byungje Sung for MIC12 purification and quality/stability analysis. Flow cytometry sample acquisition was performed on shared instruments of the Flow Cytometry Core Facility Tuebingen.
Conflict of interest
JJ, BL, YL and JW are employees of ABL Bio, Inc. Research related to this work was supported by a grant from ABL Bio, Inc. HS, LZ, BK, GJ and MP are listed as inventors on the patent application “IL-15 fusion proteins with improved properties” EP2024/053601, applicant is German Cancer Research Center, Heidelberg, and Medical Faculty University of Tuebingen, Germany.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from ABL Bio Inc. The funder had the following involvement in the study: provided critical reagents (antibodies).
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1561823/full#supplementary-material
Abbreviations
SFI, Specific fluorescence intensity; CTV, CellTrace Violet.
References
1. De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. (2016) 6:e441–1. doi: 10.1038/bcj.2016.50
2. Boscaro E, Urbino I, Catania FM, Arrigo G, Secreto C, Olivi M, et al. Modern risk stratification of acute myeloid leukemia in 2023: integrating established and emerging prognostic factors. Cancers (Basel). (2023) 15:1–27. doi: 10.3390/cancers15133512
3. Gómez-Llobell M, Peleteiro Raíndo A, Climent Medina J, Gómez Centurión I, Mosquera Orgueira A. Immune checkpoint inhibitors in acute myeloid leukemia: A meta-analysis. Front Oncol. (2022) 12:882531. doi: 10.3389/fonc.2022.882531
4. Saito S, Nakazawa Y. CAR-T cell therapy in AML: recent progress and future perspectives. Int J Hematol. (2024) 120. doi: 10.1007/s12185-024-03809-w
5. Kassner J, Abdellatif B, Yamshon S, Monge J, Kaner J. Current landscape of CD3 bispecific antibodies in hematologic Malignancies. Trends Cancer. (2024) 10:708–32. doi: 10.1016/j.trecan.2024.06.001
6. Kantarjian H, Kadia T, Dinardo C, Daver N, Borthakur G, Jabbour E, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. (2021) 11:41. doi: 10.1038/s41408-021-00425-3
7. Williams BA, Law A, Hunyadkurti J, Desilets S, Leyton JV, Keating A. Antibody therapies for acute myeloid leukemia: Unconjugated, toxin-conjugated, radio-conjugated and multivalent formats. J Clin Med. (2019) 8. doi: 10.3390/jcm8081261
8. Weiner GJ. Rituximab: mechanism of action. Semin Hematol. (2010) 47:115. doi: 10.1053/j.seminhematol.2010.01.011
9. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
10. Wang LX, Tong X, Li C, Giddens JP, Li T. Glycoengineering of antibodies for modulating functions. Annu Rev Biochem. (2019) 88:433–59. doi: 10.1146/annurev-biochem-062917-012911
11. Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. (2006) 103:4005–10. doi: 10.4161/mabs.2.2.11158
12. Awan FT, Lapalombella R, Trotta R, Butchar JP, Yu B, Benson DM, et al. CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain-engineered monoclonal antibody. Blood. (2010) 115:1204–13. doi: 10.1182/blood-2009-06-229039
13. Kayser S, Heitmann JS, Dörfel D, Thol F, Heuser M, Märklin M, et al. Interim results of a first in man study with the fc-optimized FLT3 antibody flysyn for treatment of acute myeloid leukemia with minimal residual disease. Blood. (2019) 134:3928. doi: 10.1182/blood-2019-125327
14. Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat Commun. (2019) 10:1–12. doi: 10.1038/s41467-019-11782-w
15. Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. (2006) 6:595–601. doi: 10.1038/nri1901
16. Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol Res. (2015) 3:219–27. doi: 10.1158/2326-6066.CIR-15-0009
17. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
18. Waldmann TA. IL-15 in the life and death of lymphocytes: Immunotherapeutic implications. Trends Mol Med. (2003) 9:517–21. doi: 10.1016/j.molmed.2003.10.005
19. Albertini MR, Hank JA, Gadbaw B, Kostlevy J, Haldeman J, Schalch H, et al. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol Immunother. (2012) 61:2261–71. doi: 10.1007/s00262-012-1286-5
20. King DM, Albertini MR, Schalch H, Hank JA, Gan J, Surfus J, et al. Phase I clinical trial of the immunocytokine EMD 273063 in melanoma patients. J Clin Oncol. (2004) 22:4463–73. doi: 10.1200/JCO.2004.11.035
21. Osenga KL, Hank JA, Albertini MR, Can J, Sternberg AG, Eickhoff J, et al. A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: A study of the Children’s Oncology Group. Clin Cancer Res. (2006) 12:1750–9. doi: 10.1158/1078-0432.CCR-05-2000
22. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med. (2009) 206:25–34. doi: 10.1084/jem.20082013
23. Stonier SW, Schluns KS. Trans-presentation: A novel mechanism regulating IL-15 delivery and responses. Immunol Lett. (2010) 127:85–92. doi: 10.1016/j.imlet.2009.09.009
24. Giron-Michel J, Giuliani M, Fogli M, Brouty-Boýe D, Ferrini S, Baychelier F, et al. Membrane-bound and soluble IL-15/IL-15Rα complexes display differential signaling and functions on human hematopoietic progenitors. Blood. (2005) 106:2302–10. doi: 10.1182/blood-2005-01-0064
25. Zekri L, Hagelstein I, Märklin M, Klimovich B, Christie M, Lindner C, et al. Immunocytokines with target cell–restricted IL-15 activity for treatment of B cell Malignancies. Sci Transl Med. (2024) 16:1988. doi: 10.1126/scitranslmed.adh1988
26. Quéméner A, Bernard J, Mortier E, Plet A, Jacques Y, Tran V. Docking of human interleukin-15 to its specific receptor alpha chain: correlation between molecular modeling and mutagenesis experimental data. Proteins. (2006) 65:623–36. doi: 10.1136/jitc-2023-007494
27. Bill M B, van Kooten Niekerk P, Woll P S, Laine Herborg L, Stidsholt Roug A, Hokland P, et al. Mapping the CLEC12A expression on myeloid progenitors in normal bone marrow; implications for understanding CLEC12A-related cancer stem cell biology. J Cell Mol Med. (2018) 22:2311–8. doi: 10.1080/14712598.2019.1623200
28. Bakker ABH, Van Den Oudenrijn S, Bakker AQ, Feller N, Van Meijer M, Bia JA, et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. (2004) 64:8443–50. doi: 10.1158/0008-5472.CAN-04-1659
29. Ma H, Padmanabhan IS, Parmar S, Gong Y. Targeting CLL-1 for acute myeloid leukemia therapy. J Hematol Oncol. (2019) 12(1):41. doi: 10.1186/s13045-019-0726-5
30. Lee E, Lee S, Park S, Son YG, Yoo J, Koh Y, et al. Asymmetric anti-CLL-1×CD3 bispecific antibody, ABL602 2 + 1, with attenuated CD3 affinity endows potent antitumor activity but limited cytokine release. J Immunother Cancer. (2023) 11:7494. doi: 10.1136/jitc-2023-007494
31. van Loo PF, Hangalapura BN, Thordardottir S, Gibbins JD, Veninga H, Hendriks LJA, et al. MCLA-117, a CLEC12AxCD3 bispecific antibody targeting a leukaemic stem cell antigen, induces T cell-mediated AML blast lysis. Expert Opin Biol Ther. (2019) 19:721–33. doi: 10.1080/14712598.2019.1623200
32. Zheng B, Yu SF, Del Rosario G, Leong SR, Lee GY, Vij R, et al. An anti–CLL-1 antibody–drug conjugate for the treatment of acute myeloid leukemia. Clin Cancer Res. (2019) 25:1358–68. doi: 10.1158/1078-0432.CCR-18-0333
33. Jiang YP, Liu BY, Zheng Q, Panuganti S, Chen R, Zhu J, et al. CLT030, a leukemic stem cell-targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv (2018) 2(14):1738–49. doi: 10.1182/bloodadvances.2018020107
34. Arvindam US, van Hauten PMM, Schirm D, Schaap N, Hobo W, Blazar BR, et al. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia. (2021) 35(6):1586–96. doi: 10.1038/s41375-020-01065-5
35. Wang XY, Bian MR, Lin GQ, Yu L, Zhang YM, Wu DP. Tandem bispecific CD123/CLL-1 CAR-T cells exhibit specific cytolytic effector functions against human acute myeloid leukaemia. Leukemia (2024) 35(6):1586–1596. doi: 10.1111/ejh.v112.1
36. Liu F, Cao Y, Pinz K, Ma Y, Wada M, Chen K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. (2018) 132:901–1. doi: 10.1182/blood-2018-99-110579
37. Daver N, Salhotra A, Brandwein JM, Podoltsev NA, Pollyea DA, Jurcic JG, et al. A Phase I dose-escalation study of DCLL9718S, an antibody-drug conjugate targeting C-type lectin-like molecule-1 (CLL-1) in patients with acute myeloid leukemia. Am J Hematol. (2021) 96:E175–9. doi: 10.1002/ajh.26136
38. Van Rhenen A, Van Dongen GAMS, Le Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The novel AML stem cell-associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. (2007) 110:2659–66. doi: 10.1182/blood-2007-03-083048
39. Liu Y, Zhang W, Cheung LH, Wu Q, Li C, Van Pelt CS, et al. Pharmacokinetics, tissue distribution, efficacy and toxicity studies of the antimelanoma immunocytokine scFvMEL/TNF. Cancer Res. (2006) 66. doi: 10.1016/j.xcrm.2024.101531
40. Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A. (2015) 112:3320–5. doi: 10.1038/s41586-018-0830-7
41. Shen S, Sckisel G, Sahoo A, Lalani A, Den Otter D, Pearson J, et al. Engineered IL-21 cytokine muteins fused to anti-PD-1 antibodies can improve CD8+ T cell function and anti-tumor immunity. Front Immunol. (2020) 11:832. doi: 10.3389/fimmu.2020.00832
42. Shi W, Liu N, Liu Z, Yang Y, Zeng Q, Wang Y, et al. Next-generation anti-PD-L1/IL-15 immunocytokine elicits superior antitumor immunity in cold tumors with minimal toxicity. Cell Rep Med. (2024) 5:101531. doi: 10.1016/j.xcrm.2024.101531
43. Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labão-Almeida C, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. (2019) 565:186. doi: 10.1038/s41586-018-0830-7
44. Carlsten M, Järås M. Natural killer cells in myeloid Malignancies: immune surveillance, NK cell dysfunction, and pharmacological opportunities to bolster the endogenous NK cells. Front Immunol. (2019) 10:2357. doi: 10.3389/fimmu.2019.02357
45. Kiladjian JJ, Bourgeois E, Lobe I, Braun T, Visentin G, Bourhis JH, et al. Cytolytic function and survival of natural killer cells are severely altered in myelodysplastic syndromes. Leukemia. (2006) 20:463–70. doi: 10.1038/sj.leu.2404080
46. Stringaris K, Sekine T, Khoder A, Alsuliman A, Razzaghi B, Sargeant R, et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica. (2014) 99:836–47. doi: 10.3324/haematol.2013.087536
47. Desbois M, Desbois M, Desbois M, Béal C, Charrier M, Charrier M, et al. IL-15 superagonist RLI has potent immunostimulatory properties on NK cells: Implications for antimetastatic treatment. J Immunother Cancer. (2020) 8:e000632. doi: 10.1136/jitc-2020-000632
48. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
49. Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HEK, Thompson JA, et al. A first-in-human phase I study of subcutaneous outpatient recombinant human IL15 (rhIL15) in adults with advanced solid tumors. Clin Cancer Res. (2018) 24:1525–35. doi: 10.1158/1078-0432.CCR-17-2451
50. Dubois S, Patel HJ, Zhang M, Waldmann TA, Müller JR. Preassociation of IL-15 with IL-15Rα-igG1-fc enhances its activity on proliferation of NK and CD8+/CD44high T cells and its antitumor action. J Immunol. (2008) 180:2099–106. doi: 10.4049/jimmunol.180.4.2099
51. Luu TT, Schmied L, Nguyen NA, Wiel C, Meinke S, Mohammad DK, et al. Short-term IL-15 priming leaves a long-lasting signalling imprint in mouse NK cells independently of a metabolic switch. Life Sci Alliance. (2021) 4. doi: 10.26508/lsa.202000723
52. Sarhan D, Brandt L, Felices M, Guldevall K, Lenvik T, Hinderlie P, et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. (2018) 2:1459–69. doi: 10.1182/bloodadvances.2017012369
53. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. (2014) 123:3855–63. doi: 10.1182/blood-2013-10-532531
54. Bjorklund AT, Carlsten M, Sohlberg E, Liu LL, Clancy T, Karimi M, et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin Cancer Res. (2018) 24:1834–44. doi: 10.1158/1078-0432.CCR-17-3196
Keywords: IL-15, NK cells, AML, immunocytokines, Fc-optimized antibodies, Clec12A, CLL-1
Citation: Klimovich B, Anton L, Jung J, Lim Y, Lee B, Won J, Zekri L, Chashchina A, Pflügler M, Heitmann JS, Jung G and Salih HR (2025) CLEC12A-directed immunocytokine with target cell–restricted IL-15 activity for treatment of acute myeloid leukemia. Front. Immunol. 16:1561823. doi: 10.3389/fimmu.2025.1561823
Received: 16 January 2025; Accepted: 06 March 2025;
Published: 27 March 2025.
Edited by:
Jin S. Im, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Stefan Zielonka, Merck, GermanyRoman Shapiro, Dana–Farber Cancer Institute, United States
Kajal Chaudhry, Children’s National Hospital, United States
Copyright © 2025 Klimovich, Anton, Jung, Lim, Lee, Won, Zekri, Chashchina, Pflügler, Heitmann, Jung and Salih. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helmut R. Salih, aGVsbXV0LnNhbGloQG1lZC51bmktdHVlYmluZ2VuLmRl
†These authors have contributed equally to this work