 Zehan Li1
Zehan Li1 Jingdan Zhang
Jingdan Zhang Jing Yang
Jing Yang Jinbo Cheng
Jinbo Cheng Xuewu Zhang
Xuewu Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 February 2025
Sec. Multiple Sclerosis and Neuroimmunology
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1555124
This article is part of the Research Topic Remedying the injured brain in cognitive impairment: potential neuroimmune communication signaling and therapeutic opportunities View all 5 articles
Introduction: Alzheimer’s disease (AD), a prevalent neurodegenerative disease, is primarily characterized by progressive neuron loss and memory impairment. NOD-like receptors (NLRs) are crucial for immune regulation and maintaining cellular homeostasis. Recently, NLRs have been identified as important contributors to neuroinflammation, thus presenting a potential approach for reducing inflammation and slowing AD progression.
Methods: We use quantitative RT-PCR to detect levels of NLR family members in AD mouse model. Additionally, we use immunofluorescence to detect NLRP3 expressions in microglia surrounding Aβ plaques in AD mouse model and human AD patients.
Results: In this study, we examined the expression of NLR family members in the human AD database, and found increased levels of CIITA, NOD1, NLRC5, NLRP1, NLRP3, NLRP7, NLRP10, NLRP12, and NLRP13 in hippocampus tissue in patients with AD, along with increased levels of NOD1, NLRC5, NLRX1, NLRP3, and NLRP7 levels in frontal cortex tissue. Furthermore, through detecting their levels in AD mouse model, we found that NLRP3 levels were significantly increased. Additionally, we found that NLRP3 expressions were mainly elevated in microglia surrounding Aβ plaques in AD mouse model and human AD patients.
Discussion: These findings highlight the potential important role of NLRP3 in AD pathology, offering new therapeutic targets and interventions.
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized with by gradual neuron loss and cognition and memory impairment, impacting millions of individuals around the world. The defining characteristics of AD involve the formation of amyloid-beta (Aβ) plaques, the neurofibrillary tangles (NFTs), resulting from hyperphosphorylated tau protein, and persistent neuroinflammation (1, 2). In recent years, the role of the immune system, particularly neuroinflammation, has garnered high attention in AD research.
Inflammation plays a pivotal role in the development of AD, in which microglia, astrocytes and a variety of immune molecules collectively shapes the inflammatory environment in the brain. NOD-like receptors (NLRs) are crucial regulators of inflammatory pathways (3–5), and microglia express various PRRs, including NLRs, which allow them to detect endogenous and exogenous danger signals. When these receptors are activated, these receptors initiate a cascade of inflammatory responses that could result in neuronal damage. Moreover, several factors, including Aβ deposition, tau hyperphosphorylation, and oxidative stress, can trigger the inflammatory response in AD (6, 7). The accumulation of Aβ plaques plays a significant role in the process of neuroinflammation, as it triggers the activation of glial cells such as microglia and astrocytes. This activation results in the release of chemokines, pro-inflammatory cytokines, and reactive oxygen species (ROS). While acute inflammation might be beneficial for the brain, as it facilitates the clearance of Aβ and helps maintain homeostasis, persistent or chronic inflammation can have detrimental effects. In fact, prolonged inflammatory responses contribute to increased neuronal damage and have been associated with an acceleration in the progression of neurodegenerative diseases. Thus, it is critical to understand the dual role of inflammation in the context of Aβ plaque deposition, where the initial protective response may eventually lead to harmful consequences if it becomes chronic activation.
NLRs belong to the larger family of PRRs, which also encompasses Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), and play a crucial role in the innate immune response by detecting pathogen-associated molecular patterns (PAMPs) as well as danger-associated molecular patterns (DAMPs) (8, 9). Typically, NLRs are characterized by a conserved architecture comprising three essential domains: a central nucleotide-binding oligomerization domain (NOD), a C-terminal leucine-rich repeat (LRR) domain that is essential for ligand detection, and an N-terminal effector domain that varies according to the specific NLR (10). The NLR family can be classified into several subgroups based on their domain architecture and functional roles: (a) NLRA: Represented by CIITA, which regulates the expression of major histocompatibility complex class II (MHC II) genes and primarily functions as a transcriptional regulator of adaptive immunity (11). (b) NLRB: Represented by NAIP, which recognize bacterial components such as flagellin and elements of type III secretion systems, usually partners with NLRC4 to form the NLRC4 inflammasome (12). (c) NLRC: Includes receptors such as NOD1 and NOD2, whose functions are to recognize bacterial molecules. Activation of these receptors activate nuclear factor κB (NF-κB) signaling and induce pro-inflammatory cytokines production (13). Other members include NLRC3 that inhibits TLR signaling, and NLRC4 that recognizes bacterial components to form inflammasomes that drive the release of IL-1β and IL-18 (14–17). NLRC5 is crucial in modulating the body’s inflammatory responses through its ability to regulate the expression of MHC Class I and II expression (16), and NLRX1 negatively regulates interferon secretion and promotes viral replication by modifying mitochondrial function (19, 20). (d) NLRP: Includes receptors NLRP1 to NLRP14. The NLRP subfamily is closely associated with inflammasome formation, with NLRP3 being the most extensively studied in AD. Studies have shown that two signals play crucial roles in the NLRP3 inflammasome activation: a priming signal activated by PRRs such as TLRs, which upregulate the expression levels of NLRP3 and pro-IL-1β, and an activation signal, which may involve Aβ, mitochondrial dysfunction, or ROS (18). Once activated, the NLRP3 inflammasome triggers robust inflammatory responses, contributing to AD pathology (6, 7, 19). In addition, NLRP1, NLRP6, and NLRP12, as members of the NLRP subfamily, are also involved in regulating immune responses and inflammation (15, 20–24). Emerging evidence suggests roles for NLRP2, NLRP4, NLRP5, and NLRP7 in embryonic development, although the functions of some members, like NLRP7, NLRP8, and NLRP10, remain unclear and warrant further investigation (25–28).
In this study, we detected the expression levels of NLR family members in the AD mouse model and human AD patient’s database, and found that the NLRP3 levels had a significantly increasing in AD mouse models and human AD patients, especially in microglia surrounding Aβ plaques, suggesting NLRP3 is closely associated with microglial activation and AD pathology.
To evaluate the potential contribution of NLRs in the development of AD, we analyzed the levels of NLR family members in brain samples from AD patients group and control groups in AlzData web server (www.alzdata.org, GSE36980, GSE48350, and GSE5281 datasets). As the results, we found that the NLRA subfamily member CIITA, the NLRC subfamily member NOD1 and NLRC5, and the NLRP subfamily member NLRP1, NLRP3, NLRP7, NLRP10, NLRP12, and NLRP13 levels significantly increased in hippocampus tissue samples of AD patients, as compared to their levels in the healthy controls (Figures 1A–C), suggesting these members might involve in the neuroinflammation and in the development of AD.
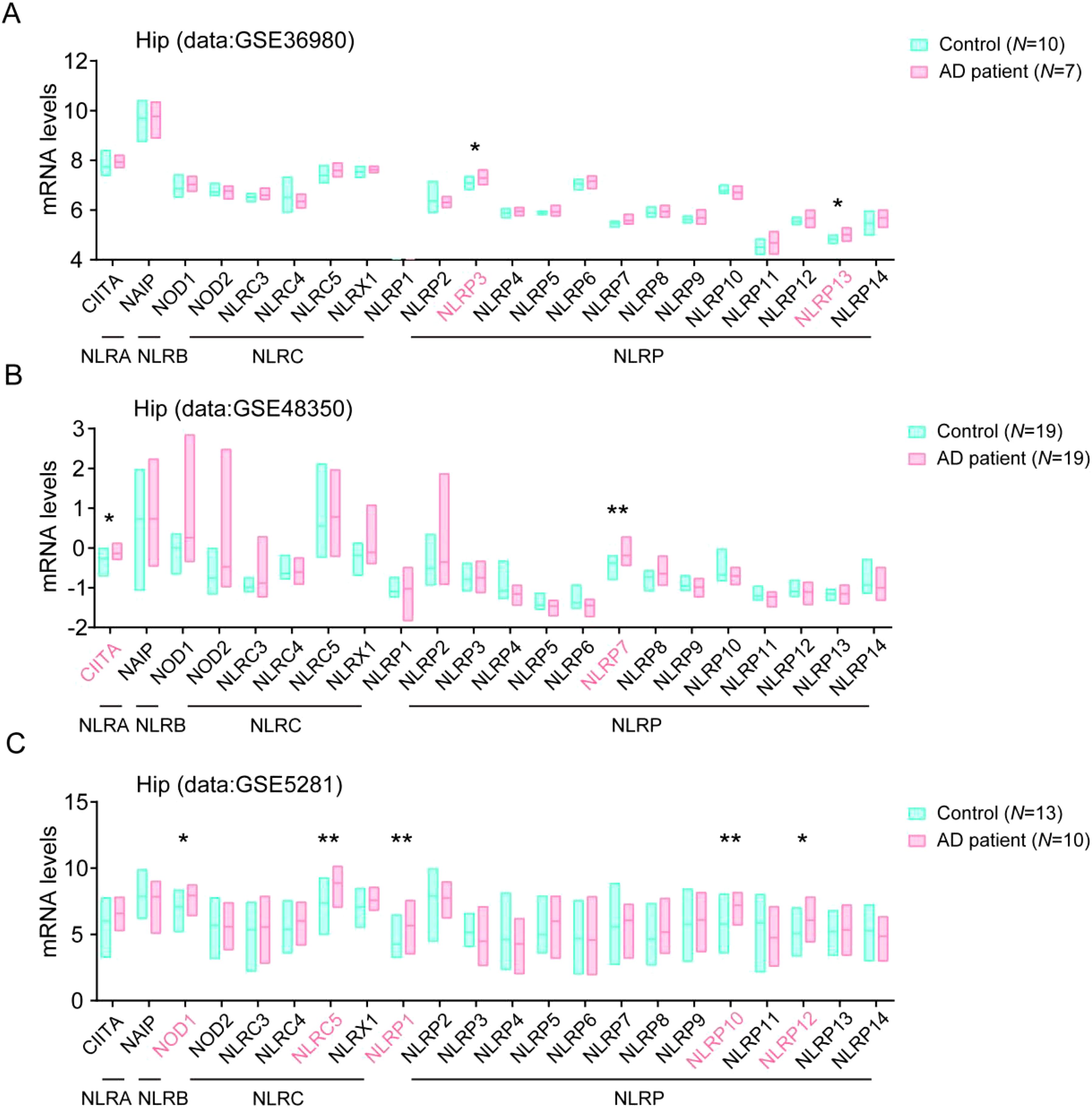
Figure 1. Multiple NLRs members are higher expressed in hippocampus tissue in AD patients. (A) The levels of all NLRs members in hippocampal tissue of AD patients in database of GSE36980 were analyzed (Control, N = 10; AD patient, N = 7). (B) The levels of all NLRs members in hippocampal tissue of AD patients in database of GSE48350 were analyzed (Control, N = 19; AD patient, N = 19). (C) The levels of all NLRs members in hippocampal tissue of AD patients in database of GSE5281 were analyzed (Control, N = 13; AD patient, N = 10). Student’s t-test was used and all values are presented as mean ± SEM. *P < 0.05, **P < 0.01.
To further conform the changes of NLR family protein levels in human AD samples, we then analyzed their levels in frontal cortex (FC) tissue in the GSE36980, GSE48350 and GSE5281 datasets. Consistently, we found the levels of NOD1 and NLRC5, NLRP3, and NLRP7 also significantly enhanced in FC tissue in AD patients brain, compared with their levels in healthy controls samples. In addition, we observed that the levels of NLRX1, one member from NLRC subfamily, also significantly increased in FC tissue in AD patients brain, compared with healthy controls samples (Figures 2A–C). The changes of these NLRs members expression in AD patients might play important roles in the development of AD.
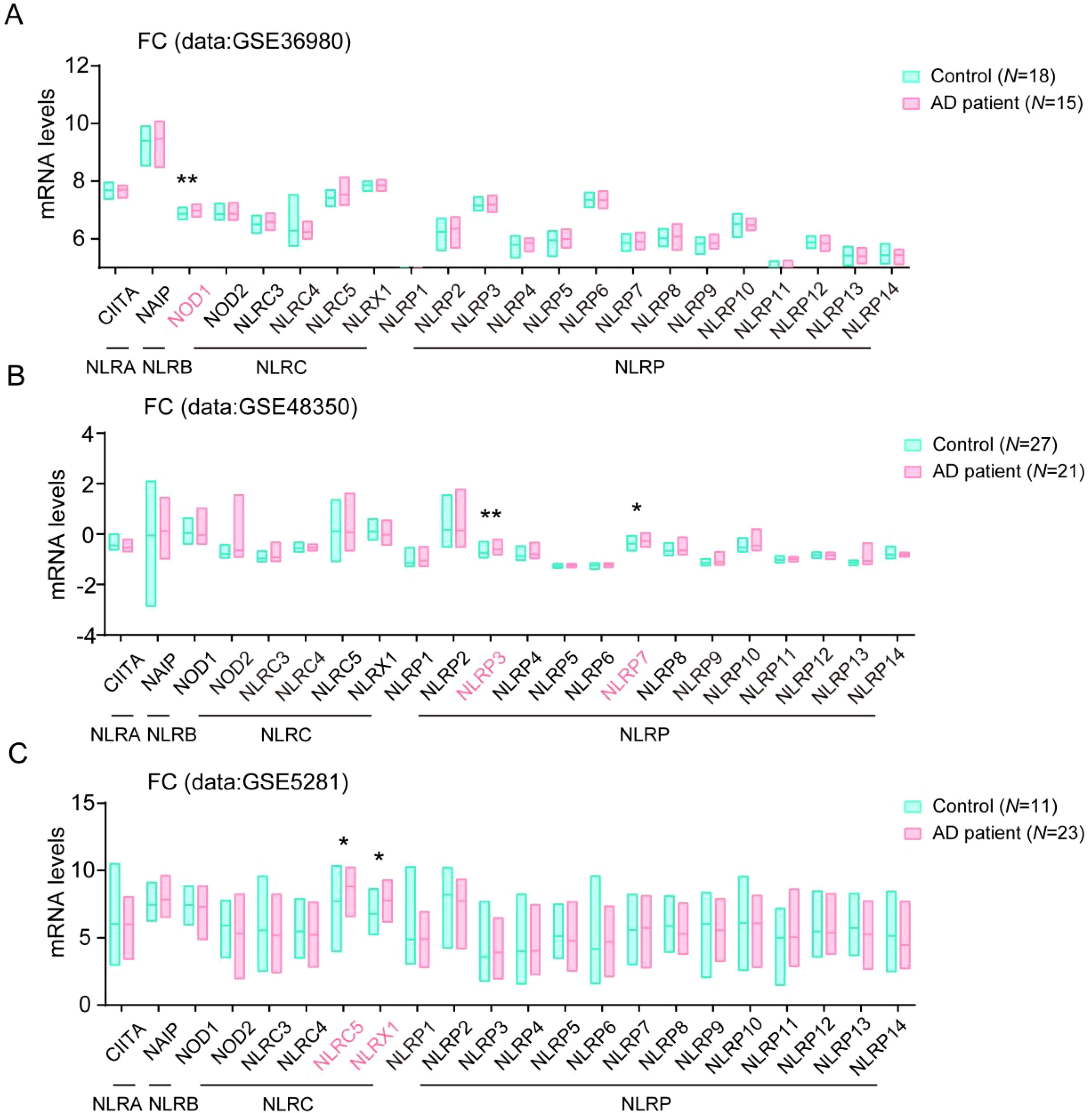
Figure 2. Multiple NLRs members changed in frontal cortex tissue in AD patients. (A) The levels of all NLRs members in frontal cortex tissue of AD patients in database of GSE36980 were analyzed (Control, N = 18; AD patient, N = 15). (B) The levels of all NLRs members in frontal cortex tissue of AD patients in database of GSE48350 were analyzed (Control, N = 27; AD patient, N = 21). (C) The levels of all NLRs members in frontal cortex tissue of AD patients in database of GSE5281 were analyzed (Control, N = 11; AD patient, N = 23). Student’s t-test was used and all values are presented as mean ± SEM. *P < 0.05, **P < 0.01.
To further investigate the changes of these members in AD, we then performed qPCR assay to test the expression levels of CIITA, NOD1, NLRP3, NLRC5, NLRP10, and NLRP12 in the hippocampus tissue in 6-month aged 5XFAD mouse model (Figure 3A). We found that the expressions of CIITA, NOD1, NLRP3, and NLRP10 were significantly elevated, which consistent with the results in the human AD database (Figures 3B–D, F). Moreover, NLRC5 and NLRP12 levels had no significant difference in the hippocampus tissue between WT mouse group and AD model mouse group (Figures 3E, G). Interestingly, the increase of NLRP3 level was more pronounced in comparison, which was consistent with the importance of NLRP3 in AD in previous studies. Taken together, the expressions of CIITA, NOD1, NLRP3, and NLRP10 were significantly elevated in AD mouse model, and the higher expression of NLRP3 indicates the importance of NLRP3 in AD pathology.
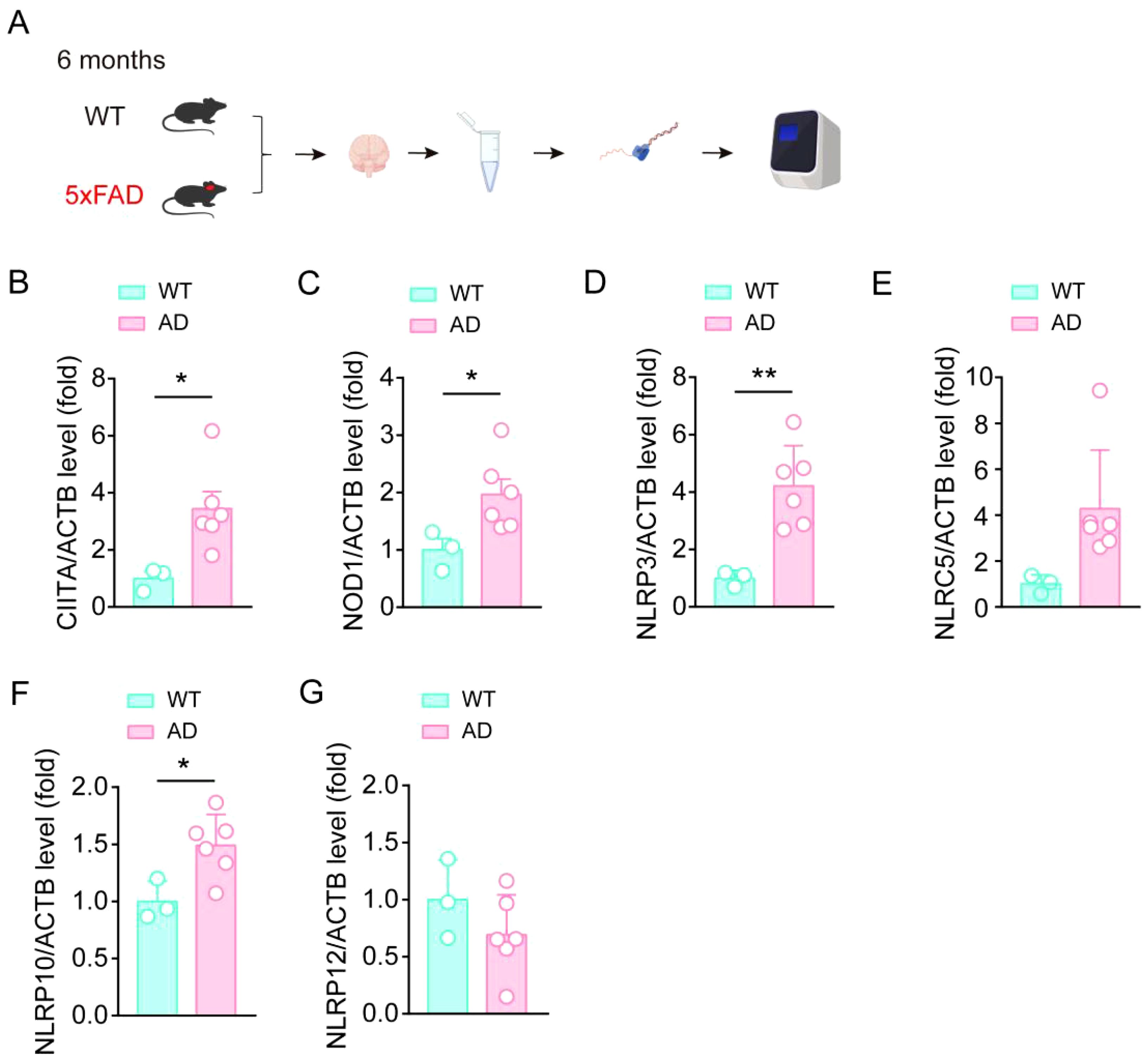
Figure 3. CIITA, NOD1, NLRP3, and NLRP10 are increased in the brain samples in AD mouse. (A) Schematic for hippocampal tissue isolation, RNA extraction, RNA-seq, and qPCR assay to test the expression levels of indicated protein. (B-G) qPCR assay to test the expression levels of CIITA, NOD1, NLRP3, NLRC5, NLRP10, and NLRP12 in isolated hippocampal tissue from 6-month-old WT mice and 6-month-old 5×FAD mice (n = 3 in WT group, n = 6 in 5×FAD group). Student’s t-test was used and all values are presented as mean ± SEM. *P < 0.05, **P < 0.01.
To further validate the up-regulation of NLRP3 levels in AD mouse model, we performed the immunofluorescence staining assay using NLRP3 antibody together with Thioflavin S (TS) staining, and found that NLRP3 immunofluorescence density were significantly increased in the DG region of hippocampus and the cortex in AD mouse, compared to the WT mouse (Figures 4A–D), suggesting NLRP3 levels significantly increase in AD mouse model brain. Considering that the increase of NLRP3 may be a driver of microglial activation in AD pathology, we then stained NLRP3 and analyzed their levels in microglia, and found that NLRP3 levels in microglia around Aβ plaques were significantly increased in the cortex in AD mouse, compared to the WT mouse (Figures 4E, F). This result suggests that NLRP3 might contribute to microglial activation in AD pathology.
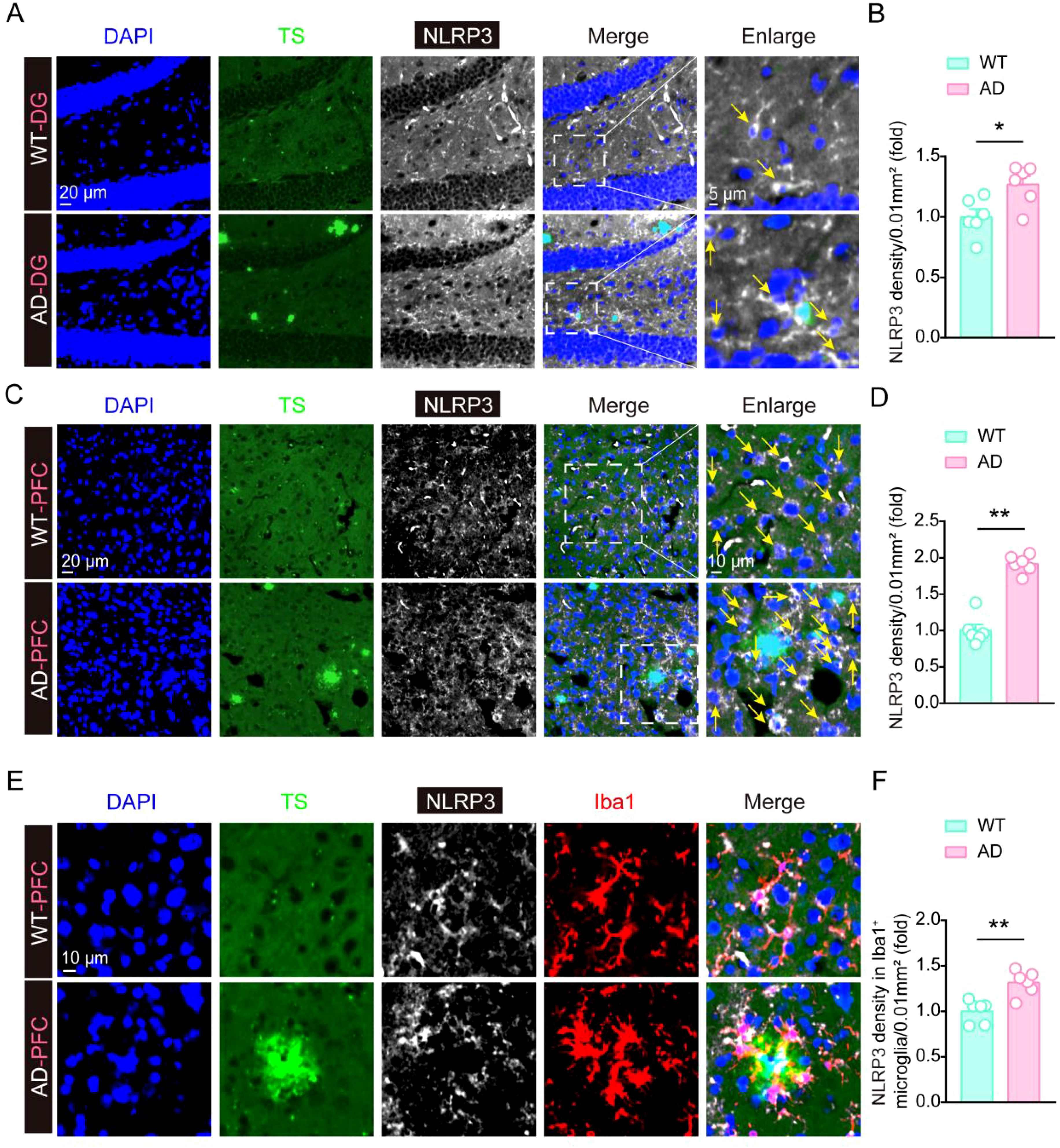
Figure 4. The expression of NLRP3 is significantly increased in AD mouse brain. (A, B) Thioflavin S staining, immunofluorescent staining of NLRP3, and statistical analysis of NLRP3 immunofluorescent density in hippocampal tissue of 6-month-old WT mice and 6-month-old 5×FAD mice (n = 6 mice per group). (C, D) Thioflavin S staining, immunofluorescent staining of NLRP3, and statistical analysis of NLRP3 immunofluorescent density in frontal cortex tissue of 6-month-old WT mice and 6-month-old 5×FAD mice (n = 6 mice per group). (E, F) Thioflavin S staining, immunofluorescent staining of NLRP3 and Iba1, and statistical analysis of NLRP3 immunofluorescent density in Iba1-positive microglia in frontal cortex tissue of 6-month-old WT mice and 6-month-old 5×FAD mice (n = 6 mice per group). Student’s t-test was used and all values are presented as mean ± SEM. *P < 0.05, **P < 0.01.
To further test whether NLRP3 expression is also elevated in AD patients, we preformed immunofluorescent staining experiments to analyze the changes of NLRP3 in human AD patients (Figures 5A, B). Five pairs of age and gender matched control brain sample and AD patients’ samples were obtained from the National Human Brain Bank for Development and Function. Consistently, NLRP3 levels were also increased in microglia around Aβ plaques in AD patients, compared with their levels in control groups, suggesting NLRP3 might also involve in microglial activation and Aβ pathology in the development of AD in clinic.
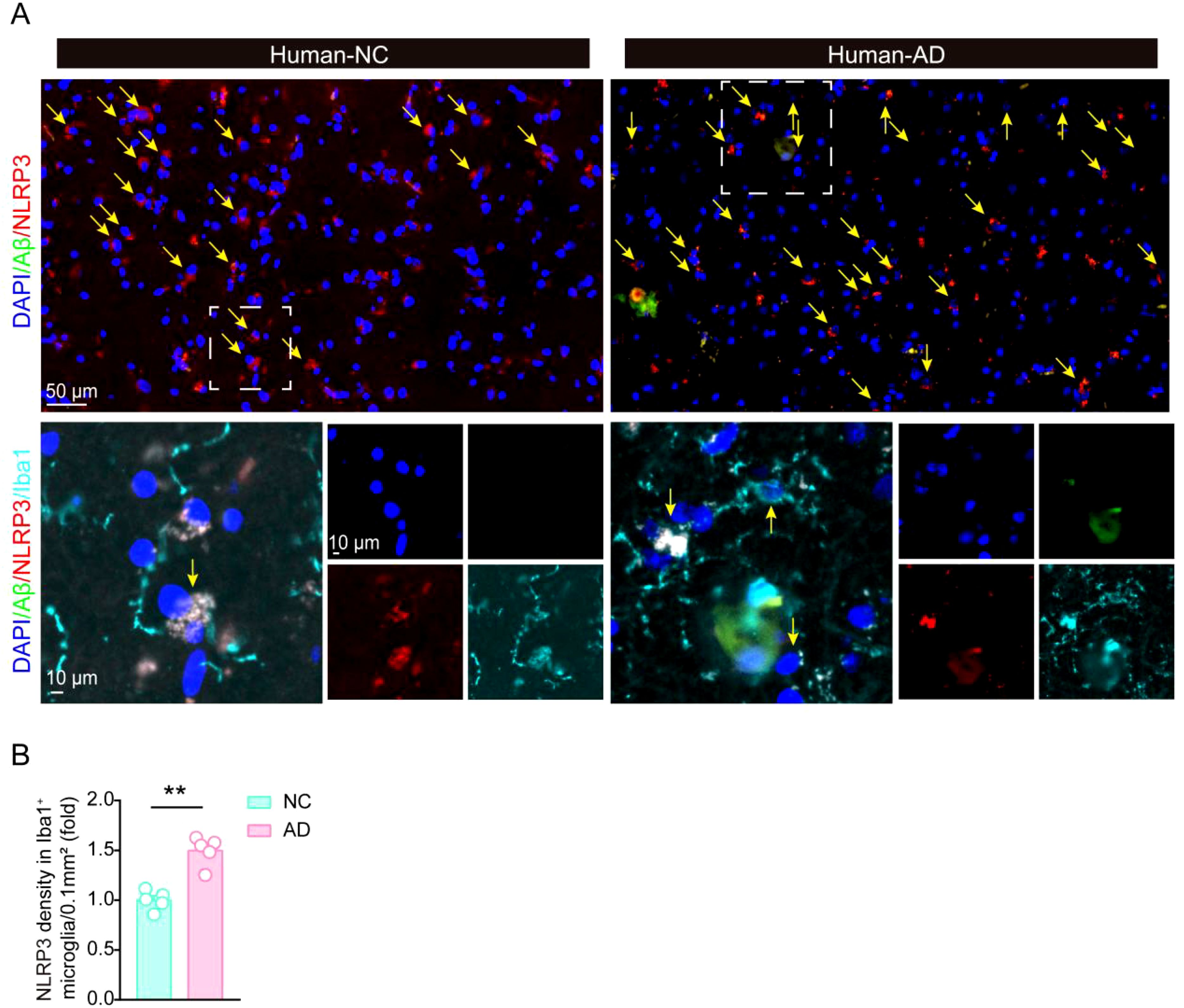
Figure 5. The expression of NLRP3 is significantly increased in AD patients brain. (A, B) Immunofluorescent staining of Aβ and NLRP3, and statistical analysis of NLRP3 immunofluorescent density in Iba1-positive microglia in hippocampal tissue in control brain and AD patients’ brain (N = 5 per group). Student’s t-test was used and all values are presented as mean ± SEM. **P < 0.01.
NLRs are crucial regulators of inflammatory pathways, but their roles in central nervous system are still largely unknown, especially the roles in neuroinflammation and the development of AD. NLRP3 was one of the most studied member in this family, and genetic deletion of NLRP3 or its downstream factor caspase-1 in AD mouse model resulted in reduced Aβ deposition, alleviated cognitive impairment, and decreased neuroinflammation (6). Moreover, further studies have revealed that Tau pathology could activate the NLRP3 inflammasome activation in microglia, resulting in the release of IL-1β and the recruitment of more microglia to Tau deposition sites (7).
Until now, lots of RNA-seq databases analyzing gene changes in human AD patients’ samples have been published. However, there are many factors such as sample diversity, age, gender, geographic location, and other demographic or environmental variables that impair the data, resulting in inconsistent results across different populations or subgroups. For example, genetic data from individuals from different ethnic backgrounds may reveal differences due to population-specific genetic characteristics, while age differences may influence health-related data because certain conditions or biomarkers may change with age. Geographic factors, such as environmental exposure or access to health care, can also contribute to differences in health outcomes or disease prevalence. Similarly, gender can influence disease presentation, drug response, and health status. Therefore, the interpretation of data from the human sample database must take into account these differences, ensuring that the findings are properly contextually and generalized across different populations. Moreover, the expression of 84 inflammasome pathway-related genes was detected in LPS and Aβ42 stimulated monocytes of individuals diagnosed with severe AD, moderate Alzheimer’s disease (MILD), or mild cognitive impairment (MCI), and in age- and sex-matched healthy controls (HC), in which NLRP3 was significantly increased with the degree of AD (29). In this study, not only analyzing multiple RNA-seq database, we also detected their levels in AD mouse model. Moreover, five pairs of human samples (5 sample in Control group, and 5 sample in AD patient group) were chosen and studied in this study. All these samples were gender and age matched, and all from Hippocampus tissue, to avoid differences caused by sample backgrounds. The detailed information was included in Table 1. Interestingly, we found that not only NLRP3 levels increased in AD samples, the levels of CIITA, NOD1, NLRC5, and NLRP10 also elevated in AD samples, suggesting these members might also take important roles in neuroinflammation and AD pathology.
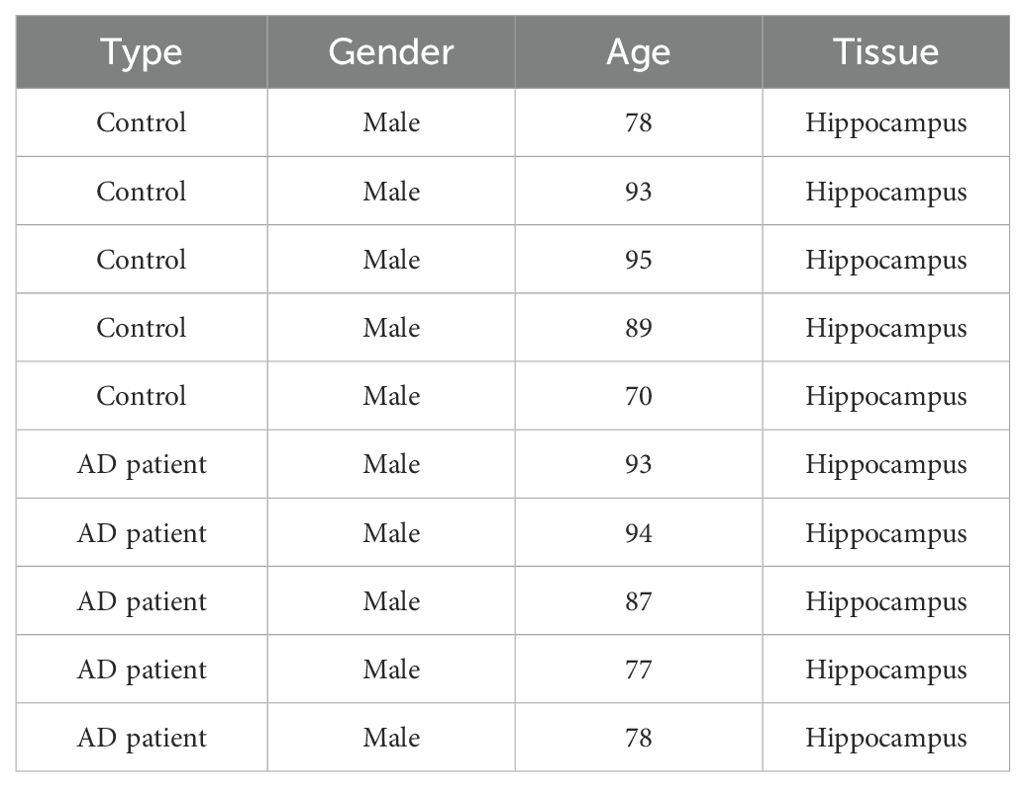
Table 1. The information of brain samples of control and patients with AD.
Microglia and astrocyte are important immune cells in the brain. Our previous study indicated that microglial Calhm2 was crucial for the activation of microglia during chronic neuroinflammation in AD and acute inflammatory responses induced by LPS injection (30). Deletion of Calhm2 in microglia significantly decreased the NLRP3 inflammasome activation. On a mechanistic level, Calhm2 modulated the interaction between P2X7 and NLRP3, and P2X7 could regulate NLRP3 inflammasome activation (31–33). Moreover, the NLRP3 inflammasome had a significantly role primarily in microglia and astrocytes in AD. In the APP/PS1 mutant transgenic AD mouse model, NLRP3 deficiency rendered microglia toward the M2 phenotype, resulting in reduced amyloid deposition (6). Furthermore, studies have found that reactive astrocyte is associated with AD, and specifically knocking out NLRP3 in microglia could inhibit the activation of reactive astrocyte (34). Therefore, the effect of NLRP3 in glia provides a potential target for future AD treatment and early diagnosis marker of neuroinflammation. However, the specific cell in which the NLRP3 inflammasome activation has an important effect in the pathology of AD remains unclear. In this study, our results demonstrated that NLRP3 expression was significantly up-regulated in microglia around Aβ plaques in both AD mouse and AD patients, suggesting NLRP3 activation in microglia play a vital role in Aβ pathology in AD. This study provides new ideas and evidence for further research on the role of NLRP3 in microglia in AD.
Given the central role of NLRs, particularly the NLRP3 in driving neuroinflammation, several strategies have been proposed to inhibit NLR activation, including small-molecule inhibitors and gene-editing approaches. Until now, many NLRP3 inhibitors have been reported to have therapeutic effects both in vitro and in vivo. Parthenolide and Oridonin could ameliorate behavioral impairments in AD mice (35, 36). Liquiritigenin and Baicalin could accelerate Aβ clearance by inhibiting NLRP3 inflammasome (37, 38). MCC950 is one of the most well-studied inhibitors of NLRP3, meanwhile other small-molecule inhibitors targeting components of the inflammasome pathway, such as caspase-1 inhibitors, are also being explored (39, 40). Furthermore, JC124 and BAY 11-7082 are sulfonamide/sulfone-based NLRP3 inhibitors. JC124 administration reduced Aβ deposition and improved memory impairments (41). Administration of BAY 11-7082 limited NLRP3 inflammasome activation and improved neuronal damage and cognitive dysfunction (42). Gene-editing technologies like CRISPR/Cas9 could selectively knockout or modify genes involved in NLR activation. In experimental models, knockout of NLRP3 has been shown to reduce neuroinflammation and slow the progression of neurodegenerative diseases (43). While these approaches are still in early stages, it still holds promise for future therapeutic development. Targeting NLRP3 has great potential as a therapeutic strategy for AD, because it plays a central role in driving neuroinflammation. Since neuroinflammation is known to exacerbate disease progression, inhibiting NLRP3 may help mitigate this inflammatory response, reduce neuronal damage, and slow cognitive deterioration in AD patients. In addition, NLRP3 inhibition could complement other treatments used in clinic, potentially improving the overall effectiveness in clinical AD treatment.
The human samples were obtained from the National Human Brain Bank for Development and Function, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. The detailed information is included in Table 1.
Briefly, brain slides were incubated in 0.002% TS (Sigma-Aldrich, catalog no. T1892-25G) in 50% ethanol for 8 min. After that, co-staining with other antibodies were carried out after PBS washing (×3).
Immunofluorescence staining was performed as described before (30). Mice were perfused with saline, and whole brain was harvested freshly and fixed by 4% paraformaldehyde (w/v) immediately. Fixed tissue was dehydrated in 30% sucrose, then brain tissue was cut into slides into 40 μm, then stained using related antibodies (The information of these antibody was listed in Table 2. The images were taken by confocal microscope with a rotating disk (Andor, England, Oxford), and analyzed by Image-Pro 6.0, ImarisViewer 9.8.0.
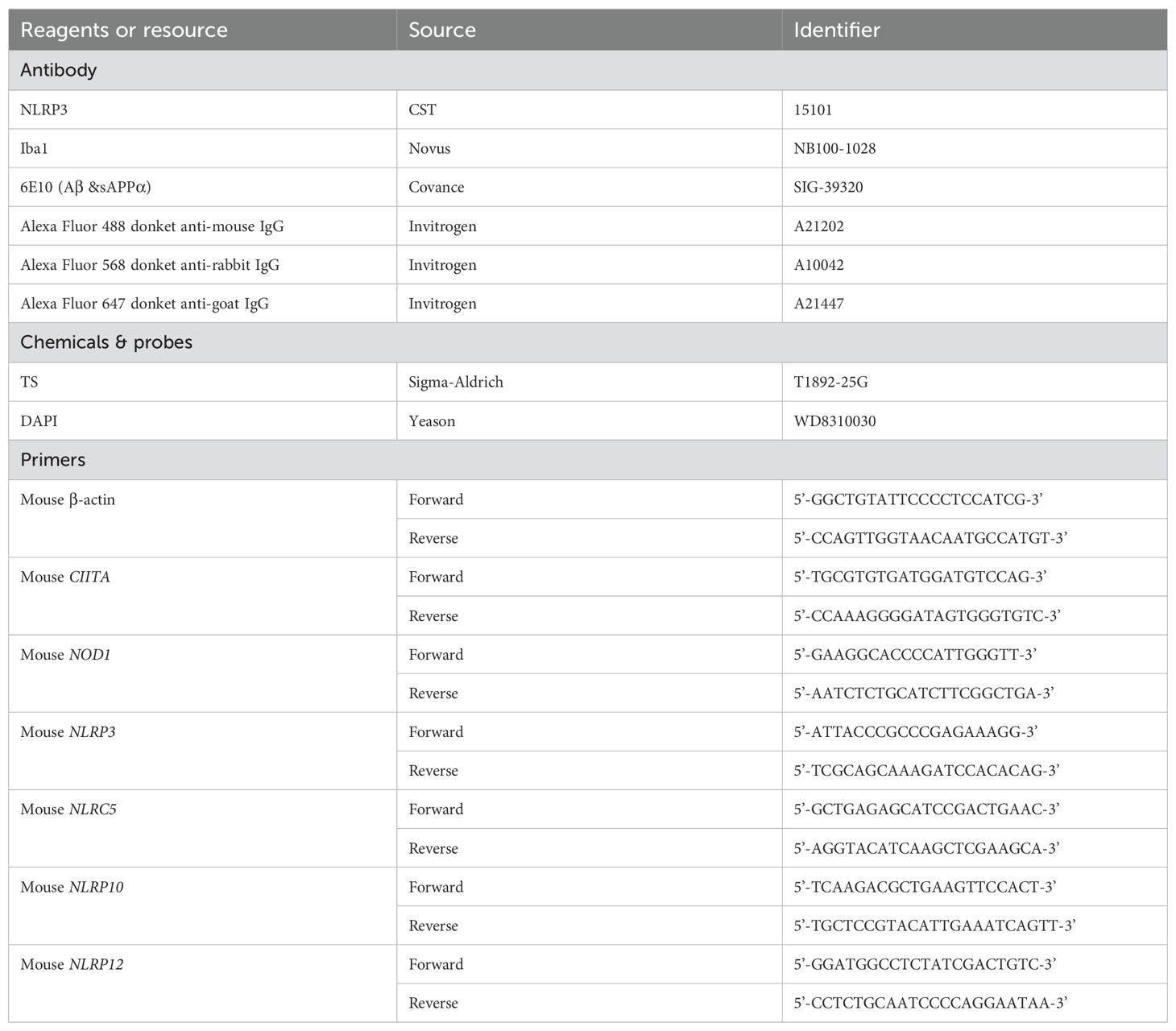
Table 2. The information of reagents or resource.
We conducted the quantitative assay of real-time (RT) PCR as previous described (30). TRIzol reagent (Invitrogen, catalog no. 15596018) was used to extract the total RNA, and a total of 1 μg of RNA was utilized to synthesis the first-strand cDNA using a cDNA synthesis kit (TIANGEN, catalog no. KR118). Finally, quantitative RT-PCR was performed using an Agilent Mx3005P RT-PCR system, and all primers in this process were listed in Table 2.
Data were analyzed using GraphPad Software (Prism GraphPad Software) with Student’s t-test for comparisons between two groups. The detailed comparison test information was described in the related figure legends. All values are presented as mean ± SEM. A P < 0.05 was considered significant.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Development and Function, Chinese Academy of Medical Sciences and Peking Union Medical College. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal studies were approved by Institutional Animal Care and Use Committee at the Beijing Institute of Basic Medical Sciences. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
ZL: Investigation, Writing – review & editing. YH: Investigation, Writing – review & editing. JZ: Investigation, Validation, Writing – review & editing. JY: Resources, Writing – review & editing. JC: Funding acquisition, Supervision, Writing – review & editing. XZ: Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Nature Science Foundation of China (No. 82271237), and the Foundation of Key Laboratory of Neurology (Hebei Medical University, Ministry of Education, China) (No.2023001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. (2019) 179:312–39. doi: 10.1016/j.cell.2019.09.001
2. Graeber MB, Kösel S, Egensperger R, Banati RB, Müller U, Bise K, et al. Rediscovery of the case described by Alois Alzheimer in 1911: historical, histological and molecular genetic analysis. neurogenetics. (1997) 1:73–80. doi: 10.1007/s100480050011
3. Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and central nervous system–associated macrophages—From origin to disease modulation. Annu Rev Immunol. (2021) 39:251–77. doi: 10.1146/annurev-immunol-093019-110159
4. Thion MS, Ginhoux F, Garel S. Microglia and early brain development: An intimate journey. Science. (2018) 362:185–9. doi: 10.1126/science.aat0474
5. Davis BK, Wen H, Ting JP-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. (2011) 29:707–35. doi: 10.1146/annurev-immunol-031210-101405
6. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. (2013) 493:674–8. doi: 10.1038/nature11729
7. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
9. Basset C, Holton J, O’Mahony R, Roitt I. Innate immunity and pathogen–host interaction. Vaccine. (2003) 21:S12–23. doi: 10.1016/S0264-410X(03)00195-6
10. Kong X, Yuan Z, Cheng J. The function of NOD-like receptors in central nervous system diseases. J Neurosci Res. (2017) 95:1565–73. doi: 10.1002/jnr.24004
11. Devaiah BN, Singer DS. CIITA and its dual roles in MHC gene transcription. Front Immunol. (2013) 4:476. doi: 10.3389/fimmu.2013.00476
12. Paidimuddala B, Cao J, Nash G, Xie Q, Wu H, Zhang L. Mechanism of NAIP—NLRC4 inflammasome activation revealed by cryo-EM structure of unliganded NAIP5. Nat Struct Mol Biol. (2023) 30(2):159–66. doi: 10.1038/s41594-022-00889-2 Erratum in: Nat Struct Mol Biol. (2023 30(2):233. doi: 10.1038/s41594-023-00927-7
13. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. (2014) 14:9–23. doi: 10.1038/nri3565
14. Mangan MSJ, Latz E. NLRC3 puts the brakes on STING. Immunity. (2014) 40:305–6. doi: 10.1016/j.immuni.2014.02.007
15. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol. (2012) 13(9):823–31. doi: 10.1038/ni.2378
16. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A. (2010) 107(31):13794–9. doi: 10.1073/pnas.1008684107
17. Guo H, König R, Deng M, Riess M, Mo J, Zhang L, et al. NLRX1 sequesters STING to negatively regulate the interferon response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host Microbe. (2016) 19:515–28. doi: 10.1016/j.chom.2016.03.001
18. Man SM, Kanneganti T-D. Regulation of inflammasome activation. Immunol Rev. (2015) 265:6–21. doi: 10.1111/imr.12296
19. Shi J-Q, Wang B-R, Jiang T, Gao L, Zhang Y-D, Xu J. NLRP3 inflammasome: A potential therapeutic target in fine particulate matter-induced neuroinflammation in Alzheimer’s disease. J Alzheimers Dis. (2020) 77:923–34. doi: 10.3233/JAD-200359
20. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/s1097-2765(02)00599-3
21. Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. (2012) 488:389–93. doi: 10.1038/nature11250
22. Chen GY, Liu M, Wang F, Bertin J, Núñez G. A functional role for nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (Baltimore Md : 1950). (2011) 186:7187. doi: 10.4049/jimmunol.1100412
23. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. (2011) 145:745–57. doi: 10.1016/j.cell.2011.04.022
24. Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A. (2008) 105:1614–9. doi: 10.1073/pnas.0708616105
25. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
26. Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. (2006) 38:300–2. doi: 10.1038/ng1740
27. Peng H, Lin X, Li W, Zhang W. Expression and localization of Nlrp4g in mouse preimplantation embryo. Zygote. (2015) 23:846–51. doi: 10.1017/S0967199414000525
28. Zhang P, Dixon M, Zucchelli M, Hambiliki F, Levkov L, Hovatta O, et al. Expression analysis of the NLRP gene family suggests a role in human preimplantation development. PloS One. (2008) 3:e2755. doi: 10.1371/journal.pone.0002755
29. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegeneration. (2016) 11:23. doi: 10.1186/s13024-016-0088-1
30. Cheng J, Dong Y, Ma J, Pan R, Liao Y, Kong X, et al. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci Adv. (2021) 7:eabe3600. doi: 10.1126/sciadv.abe3600
31. Franceschini A, Capece M, Chiozzi P, Falzoni S, Sanz JM, Sarti AC, et al. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. (2015) 29:2450–61. doi: 10.1096/fj.14-268714
32. Chiozzi P, Sarti AC, Sanz JM, Giuliani AL, Adinolfi E, Vultaggio-Poma V, et al. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci Rep. (2019) 9:6475. doi: 10.1038/s41598-019-42931-2
33. Lee HG, Won SM, Gwag BJ, Lee YB. Microglial P2X 7 receptor expression is accompanied by neuronal damage in the cerebral cortex of the APP swe/PS1dE9 mouse model of Alzheimer’s disease. Exp Mol Med. (2011) 43:7. doi: 10.3858/emm.2011.43.1.001
34. Li S, Fang Y, Zhang Y, Song M, Zhang X, Ding X, et al. Microglial NLRP3 inflammasome activates neurotoxic astrocytes in depression-like mice. Cell Rep. (2022) 41:111532. doi: 10.1016/j.celrep.2022.111532
35. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
36. Wang S, Yu L, Yang H, Li C, Hui Z, Xu Y, et al. Oridonin attenuates synaptic loss and cognitive deficits in an Aβ1-42-induced mouse model of Alzheimer’s disease. PLoS One. (2016) 11:e0151397. doi: 10.1371/journal.pone.0151397
37. Liquiritigenin decreases Aβ Levels and ameliorates cognitive decline by regulating microglia M1/M2 transformation in AD mice. Available online at (Accessed January 31, 2025).
38. Jin X, Liu M-Y, Zhang D-F, Zhong X, Du K, Qian P, et al. Baicalin mitigates cognitive impairment and protects neurons from microglia-mediated neuroinflammation via suppressing NLRP3 inflammasomes and TLR4/NF-κB signaling pathway. CNS Neurosci Ther. (2019) 25:575–90. doi: 10.1111/cns.13086
39. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1457–61. doi: 10.1161/ATVBAHA.117.309575
40. Jiao J, Zhao G, Wang Y, Ren P, Wu M. MCC950, a selective inhibitor of NLRP3 inflammasome, reduces the inflammatory response and improves neurological outcomes in mice model of spinal cord injury. Front Mol Biosci. (2020) 7:37. doi: 10.3389/fmolb.2020.00037
41. Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, et al. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflamm. (2019) 16:81. doi: 10.1186/s12974-019-1471-y
42. Ruan Y, Qiu X, Lv Y-D, Dong D, Wu X-J, Zhu J, et al. Kainic acid Induces production and aggregation of amyloid β-protein and memory deficits by activating inflammasomes in NLRP3- and NF-κB-stimulated pathways. Aging (Albany NY). (2019) 11:3795–810. doi: 10.18632/aging.102017
Keywords: Alzheimer’s disease, NLRs, NLRP3, microglia, Aβ plaques
Citation: Li Z, He Y, Zhang J, Yang J, Cheng J and Zhang X (2025) The changes of NLRs family members in the brain of AD mouse model and AD patients. Front. Immunol. 16:1555124. doi: 10.3389/fimmu.2025.1555124
Received: 03 January 2025; Accepted: 05 February 2025;
Published: 24 February 2025.
Edited by:
Linlin Yin, Capital Medical University, ChinaReviewed by:
Hongyan Li, Capital Medical University, ChinaCopyright © 2025 Li, He, Zhang, Yang, Cheng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinbo Cheng, Y2hlbmdfamluYm9AMTI2LmNvbQ==; Xuewu Zhang, emhhbmd4dWV3dUB5YnUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.