 Carolina Prado
Carolina Prado Andrés A. Herrada
Andrés A. Herrada Daniel Hevia
Daniel Hevia Lorna Galleguillos Goiry5
Lorna Galleguillos Goiry5 Noelia Escobedo
Noelia Escobedo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 17 February 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1540263
This article is part of the Research Topic The Role of Innate Immunity in the Pathogenesis of Autoimmune and Autoinflammatory Diseases View all articles
Multiple sclerosis (MS) is a chronic autoimmune, inflammatory and neurodegenerative disease affecting the central nervous system (CNS). MS is associated with a complex interplay between neurodegenerative and inflammatory processes, mostly attributed to pathogenic T and B cells. However, a growing body of preclinical and clinical evidence indicates that innate immunity plays a crucial role in MS promotion and progression. Accordingly, preclinical and clinical studies targeting different innate immune cells to control MS are currently under study, highlighting the importance of innate immunity in this pathology. Here, we reviewed recent findings regarding the role played by innate immune cells in the pathogenesis of MS. Additionally, we discuss potential new treatments for MS based on targets against innate immune components.
Multiple sclerosis (MS) is an autoimmune disorder associated with significant neurodegeneration and neuroinflammation. This disease is triggered by an autoimmune response directed against myelin producing demyelinated areas in both the white and gray matter of the brain and spinal cord. These lesions indicate loss of myelin and myelin-producing oligodendrocytes, resulting in disrupted conduction of electrical impulses (1). MS can be divided into 3 subtypes: relapse remitting MS (RMSS), primary progressive MS, and secondary progressive MS (2). The most common type of MS is RRMS, which is characterized by recurring episodes of neurological dysfunction, followed by clinical recovery. The disease symptoms are heterogeneous and depend on the location of the lesions in the CNS and range from sensory disturbances, bladder dysfunction, cognitive deficits, limb weakness, ataxia, and fatigue (3).
Even though the pathogenesis of MS is not fully understood, there are many pieces of the puzzle that are starting to shape up, with factors that may induce a primary inflammatory disease or a primary oligodendroglial pathology followed by inflammation (4). The multifactorial and complex interaction between genetic and environmental factors plays an important role in triggering the disease. The prevalence of the MS-risk allele HLA-DR15 and many single nucleotide polymorphisms of genes that are important for the differentiation or effector function of pathogenic T cells strengthens the concept of immune-mediated disease with the contribution of different risk factors: childhood obesity (3, 5), cigarette smoking (6–8), Epstein-Barr virus infection (9–11), vitamin D deficiency (12–14) and night shift work at young age (15–18).
If we recapitulate in the actual biological knowledge, MS is thought to be caused by an autoimmune response towards central nervous system (CNS) self-antigens in genetically susceptible individuals, where autoreactive T cells are supposed to be the disease-initiating immune cells (19). In the last decades, B cells were recognized as crucial immune cells in this process, including antibody-dependent and independent effects in the compartmentalized inflammation (20–22). Also, myeloid cells are important contributors to the pathology, being central actors in the disease progression, cortical atrophy, neurodegeneration, and disability (23).
The acute inflammation consists of an invasion of monocytes and lymphocytes into white matter (WM), with a lesser degree into deep gray matter, with concomitant activation of microglia and astrocytes. This demyelination leads to the formation of acute focal WM lesions, formed by dense infiltration of myelin-laden macrophages, lymphocytes, and important axonal loss followed by neurological disorders and physical disabilities (24). Chronic inflammation results from diffuse glial activation at the rim of chronic active lesions, which can reach considerable distances into normal- appearing white matter, with predominant lymphocytic inflammation in the meninges and perivascular spaces. Meningeal inflammation is typically diffuse but may also form follicle-like structures (25).
The available therapies for MS aim to shift the immune cell repertoire from a pro-inflammatory towards an anti-inflammatory phenotype, involving regulatory T (Treg) and B cells (mainly) and anti-inflammatory macrophages (in clinical trials). Despite the clear association of dysfunctional T and B cells in MS, during the last years, mounting evidence of different innate immune cell types involved in the pathogenesis of MS has emerged. This review gives a conceptual overview on the different innate immune cell types involved in MS pathology, discussing potential new targets for treatment (26).
Dendritic cells (DCs) are highly specialized antigen presenting cells (APCs) with the unique ability to stimulate naïve T cells. First discovered by Steinman et al. in 1973 and named because of their dendritic shapes (27), DCs are constantly sensing pathogen signals or damage-associated molecular patterns, patrolling in different anatomical locations (28). In this immature state, DCs have the ability to capture and process antigens in a very efficient way, while their capacity to activate naïve T cells is weak, because of the lower expression of costimulatory molecules and cytokines (29). Once DCs capture and process pathogens or damage-associated molecular patterns, DCs go through a maturation process, increasing the expression of costimulatory molecules and loading antigen fragments into the major histocompatibility complex (MHC), while they migrate into lymph nodes where they efficiently activate and differentiate effector T cells (29). Additionally, mature DCs in the presence of Interleukin (IL)-10 and IL-27 can block T cell activation and promote Treg expansion, inducing immune tolerance (30). According to the developmental origin, surface markers and transcriptome profiles, DCs can be divided into three major subsets: Conventional DCs (cDCs), which can be further divided into cDC1s and cDC2s, plasmacytoid DCs (pDCs) and monocyte-derived DCs (moDCs) (31, 32). During steady state, cDCs are distributed in lymph and non-lymphoid tissues, and they are the main APC among DCs subsets, with cDC1 activating CD8+ T cells and cDC2 mainly activating CD4+ T cells (33, 34). pDCs are mostly confined to lymphoid tissues in homeostatic conditions and are capable of rapidly responding to virus infection by producing high levels of type I interferons (IFNs) (33, 34). moDCs develop from monocyte DCs progenitor under inflammatory conditions, working together with cDCs in response to inflammation or infection (35). Because DCs act as a bridge between innate and adaptive immunity, they are key players in autoimmune processes such as MS.
Even though it was first believed that the CNS was an immunoprivileged site, early studies during 1990´s in rats showed the presence of DCs in the meninges and the choroid plexus in healthy conditions, and infiltration of DCs into the brain after inflammatory conditions, suggesting that DCs could be playing an important role in neuroinflammatory processes (36, 37). The use of transgenic animals expressing enhanced yellow fluorescent protein downstream of the DCs-associated CD11c promoter, confirmed the presence of a small population of DCs in the CNS in homeostasis and during different pathological conditions (38–40). An animal model commonly used to study MS immunopathology is the experimental autoimmune encephalomyelitis (EAE) mouse model. As with MS, EAE is accompanied by lesion formation and paralysis caused by immune cells invading the CNS. By using this model, Matyszaki et al. found infiltration of DCs, characterized by the expression of CD103 and MHC class II (MHCII), in different lesions, mostly in perivascular regions but some DCs were also found in the brain parenchyma (41). Soon after, DCs infiltration into perivascular regions and in lesser extent into parenchymal regions of brain and spinal cords were also observed in mice at the peak stage of EAE (42). Although it was clear that DCs are capable of invading the CNS during EAE, the contribution of DCs to induce neuroinflammation during EAE was unclear, with contradictory studies suggesting from one side that DCs inhibit T cell activation reducing neuroinflammation (43, 44), while other studies showing direct contribution of DCs to the induction and maintenance of neuroinflammation in EAE (45, 46), although different DCs maturation stages analyzed could in part explain these contradictory results. In fact, the intracerebral microinjection of DCs cultured in different medium conditions showed that fully mature DCs exacerbated the onset and clinical course of EAE, while intracerebral microinjection of semi-mature DCs delayed EAE symptoms (47). To try to clarify the role of DCs in EAE, the use of constitutively or inducible DCs-depleted mice models were developed, but again, contradictory results were obtained, with some groups showing that DCs depletion leads to the loss of tolerance to self-antigens and increased EAE symptoms while other studies showed no major effect of DCs depletion over EAE symptoms and progression (47–50). However, the finding that CD11c is expressed in other APCs such as microglia, monocytes and macrophages, makes it very difficult to distinguish the real contribution of DCs in EAE versus the other immune cell types in these CD11c-expressing cell ablation systems. To overcome this problem, single cell mapping technology and the use of new transgenic mice has been developed in the last years, to specifically identify different DCs subsets and to interrogate the contribution of each of these subsets in the development of EAE. Thus, by mass cytometry technique together with high-dimensional data mining, Mundt et al. found different APC populations, including cDC1, cDC2 and pDCs, specifically in the outer membrane of the meninges, the dura mater, at steady-state (51). Next, by using the Cx3cr1CreERT2 strain, that allows specific targeting of macrophages, monocytes or DCs depending on the time that tamoxifen is given, where early tamoxifen treatment target long-lived, self-maintaining cells such as microglia and macrophages, whereas late tamoxifen injection target also DCs and monocytes, this group found that cDCs, particularly cDC2 subset, but not microglia or macrophages, are necessary for the activation of T cells in the CNS and to promote EAE pathogenesis (51). By using a different approach based on single-cell RNA sequencing analysis (scRNAseq) of different CNS compartments, Jordão et al. analyzed multiple myeloid cell populations in steady-state as well as during different stages of EAE (52). Again, although DCs numbers are low at homeostatic CNS, this population increase during EAE, and reduction of MHCII levels in DCs decreased EAE severity, confirming a critical role of DCs during EAE (52). Mechanistically, C-X-C chemokine receptor type 1 (CXCR1) expression on DCs seems to be important in this process, since specific ablation of CXCR1 on DCs reduces EAE severity in part by reducing proinflammatory cytokine production by DCs (53). All these results highlight the role of DCs, specifically cDCs, as the main APC in the CNS during EAE and suggest that targeting DC function could be a good strategy to treat MS. In fact, a recent study showed that reducing cDC1 subset by the use of CXCR1-specific chimeric antigen receptor (CAR)-T cells, decreased EAE symptoms in a CD4+ T cell-induced passive EAE (54). Thus, targeting DCs subsets could be a good therapeutic strategy to treat MS, although more studies are needed to confirm these observations.
Soon after the observation of DCs in the CNS of different animal models of MS, two populations of DCs, myeloid and pDCs, were observed in the cerebrospinal fluid (CSF) from healthy volunteers with increased numbers of pDCs in the CSF of people with MS (pwMS) (55). In fact, pwMS during relapses showed an increased number of pDCs in the CSF (56). Increased number of DCs inside of spinal cord and brains from pwMS were observed, specifically in perivascular regions of MS lesions where they contained myelin components, potentially presenting antigens to CD8+ T cells (57). The increased number of DCs in MS lesions could be explained in part by the increased concentration of the chemokines monocyte chemotactic protein (MCP) -1, -2 and -3 and CXCL10 (58, 59). Additionally, increased expression of the chemokine regulated on activation normal T cell expressed and secreted (RANTES) and macrophage inflammatory protein (MIP)-1α/β in the CSF of pwMS have been detected, together with increased expression of their receptor CCR5 in peripheral cDCs of pwMS (60–62). Moreover, scRNAseq analysis of cells from CSF of RMSS patients showed the presence of cDCs and pDCs (63). A different single-cell analysis study showed an increased proportion of cDCs in the CSF compared to the blood from untreated relapsed pwMS (64). All these data suggest that the accumulation of different DCs subsets in perivascular regions of the CNS from pwMS could be contributing to the increase of neuroinflammation by actively presenting myelin antigens to T cells leading to their activation and the perpetuation of the damage and suggest that, targeting DCs could be a good strategy to reduce neuroinflammation in pwMS.
Macrophages represent a heterogeneous group of immune cells that can phagocytose, playing a key role in the initiation, triggering, and resolution of an immune response, as well as repairing inflammation-damaged tissues (65). Macrophages are found in almost all the tissues in the body, where they have specific and different functions, depending on specific stimuli within their microenvironment (66). Based on in vitro experiments, macrophages can be broadly divided into two different functional and metabolic states: M1 and M2 macrophages (67). While M1 or pro-inflammatory macrophages are involved in inflammation and tissue destruction, M2 or anti-inflammatory macrophages are related with inflammation resolution and tissue repair (68). Although it is now clear that M1 and M2 macrophage classification is an oversimplification and represents the extremes of a heterogeneous cells with a very high level of plasticity, it is still useful today to evaluate the role of macrophages in different inflammatory context such as MS (69).
Under homeostatic conditions, macrophages in the CNS can be found in the meninges, perivascular space, and choroid plexus, which are referred to as CNS-associated macrophages (CAMs) (also known as border-associated macrophages (BAMs)) (70). CAMs are highly heterogeneous and can be further divided depending on their anatomical positions into meningeal macrophages (mμΦ), choroid plexus macrophages (cpμΦ), and perivascular macrophages (pvμΦ) (71). CAMs are constantly monitoring the CSF, searching for harmful antigens, and also contributing to the drainage of CNS-derived antigens (70, 72, 73). In pathological conditions, CAMs expand and secrete pro-inflammatory cytokines and chemokines that promote the recruitment of different immune cell population leading to neuroinflammation (52).
The first evidence that macrophages are important during the development of EAE came from a study showing that macrophage depletion with silica, a treatment that preferably depletes macrophages but not DCs, reduces severity and delays the onset of clinical symptoms when is administrated prior to EAE induction, and reduces EAE symptoms when is injected after the appearance of the first clinical signs (74, 75). By using mannosylated liposomes containing dichloromethylene diphosphonate to deplete macrophages, Huitinga et al. showed that intraperitoneal injection of the liposomes after the appearance of clinical symptoms efficiently depletes macrophages, drastically reducing EAE symptoms and diminishing the number of infiltrating macrophages into the CNS (76). Moreover, intraventricular injection of mannosylated clodronate liposomes, to specifically deplete CAMs, showed reduction of clinical symptoms in the EAE mouse model (77). However, these treatments failed to show specific macrophage depletion, since other APCs could also be affected (78, 79). Thus, new strategies were developed to overcome these difficulties. By using a combination of parabiosis experiment where two mice are joined together resulting in a shared circulatory system, and myeloablation, Ajami et al. found that monocyte infiltration and differentiation into macrophages are essential for the EAE progression and pathogenesis (80). Matrix metalloproteinase [MMP]-2 and MMP-9 are two proteins mainly produced by macrophages involved in leukocyte transmigration into the CNS (81). By using the MMP-2 and MMP-9 double knockout mice, Agrawal et al. showed that decreasing leukocyte transmigration and macrophage accumulation in the CNS reduces EAE symptoms, suggesting an active role of macrophages in neuroinflammation (81). Specific subsets of macrophage infiltration occur at different times during EAE progression. Thus, CNS is infiltrated with high levels of M1 macrophages during the onset of the disease, but there is a gradual increase in M2 macrophages during the recovery phase that is associated with improved neurological impairment (82). By using different strategies to either induce M2 macrophage polarization or directly injecting M2 macrophages, it has been showed that this anti-inflammatory macrophage population can reduce EAE symptoms (83–86). Due to the high heterogeneity of macrophage populations (CAMs and monocyte-derived macrophages) that infiltrate the CNS during EAE, new experimental technologies were developed to study the changes and contribution of these macrophage subsets during EAE progression. By using scRNA-seq analysis of the different immune cell types at different stages of EAE, Jordão et al. showed that during EAE, CAMs are transcriptionally distinct from their counterparts during homeostasis, demonstrating the plasticity of macrophages (52). Moreover, local proliferation of CAMs, with increased expression of MHCII, were evident during the onset of the disease, reaching the highest proliferation at the peak of EAE (52). However, MHCII ablation specifically in CAMs did not affect the development of EAE, suggesting that CAMs are redundant at least for antigen presentation in the CNS during EAE. All these results highlight the high heterogenicity of macrophage populations, with different involvement during EAE, but also suggest that treatments that favor M2 macrophage infiltration could be a good therapeutic target to treat neuroinflammation.
Macrophage infiltration into MS lesions was described over almost forty years ago (87). These macrophages express the inducible nitric oxide synthase (iNOS), a M1 marker, suggesting a proinflammatory phenotype (88). In an attempt to further characterize the phenotype of macrophages in MS lesions, Vogel et al, using a panel of typical M1 and M2 markers showed that myelin-laden macrophages in the demyelinated lesion area express high levels of the M1 markers CD40, CD86, CD64 and CD32 (89). Interestingly, M2 markers CD206 and CD163 were also strongly expressed by pvμΦ. Moreover, co-expression of CD40 and CD206 showed close to 70% of infiltrating macrophages positive for both markers, indicating an intermediate activation status (89). By using iron-sensitive magnetic resonance imaging, it was recently confirmed the increase in CD163 expression in myeloid cells from chronic brain active lesion of postmortem in pwMS (87). Applying imaging mass cytometry, Park et al. found the presence of macrophages phagocyting in active lesion of pwMS with differential phenotype depending on the position, from highly activated macrophages in the edges into less activated macrophages in the lesion center, that is in line with the simultaneous expression of pro- and anti-inflammatory markers by macrophages in MS lesions described by Vogel et al. (89). Thus, macrophages in the MS lesions are an heterogenous population with mixed pro- and anti-inflammatory phenotypes, capable of phagocyting myelin and interacting with other immune cells. Whether targeting macrophage population could alleviate MS symptoms needs to be addressed.
Microglia are highly specialized parenchymal-resident macrophages, important in mediating inflammatory and immune responses inside the CNS. While in developmental stages microglia regulates synaptic plasticity by modulating synaptic formation and elimination and shaping embryonic brain circuits, in adult stages microglia contributes to maintain homeostasis by participating in myelination and pruning processes or responding to pathological threats, acting as a first line of defense against pathogens or tissue injury, actively phagocyting and presenting antigens to T cells (90–92). Under homeostatic conditions, microglia are found in a resting state, characterized by a rod-shaped soma, several ramifications and decreased phagocytic conditions, while in an activated state microglia acquire an amoeboid shape, retracting their ramifications and increased their phagocytic and migratory capabilities (93). Microglia, in a similar way than macrophages, can be divided into a M1 and M2 population, although a continuum of intermediate phenotypes can be found (94). M1 microglia produces proinflammatory cytokines such as TNF-α, IL-6 or IL-1β and is involved in inflammation, while M2 microglia produces mainly IL-4 and IL-13 and is related to anti-inflammatory and healing processes (95). Because this dual role of microglia in inflammation and healing processes, these cells are considered to be a double-edged sword, where M1 microglia are necessary to fight against infection, but later M2 microglia need to expand to reduce inflammation and start healing processes, suggesting that a subtle balance in timing and expansion of M1 and M2 microglia are important for keeping the CNS homeostasis, and, as a corollary, a disbalance of both cell populations could contribute to neuroinflammatory and neurodegenerative disorders (96).
A relationship between microglia and MS came from early studies in animal models showing the presence of microglia in spinal cord lesions, together with the capacity of purified microglia to support an effector response by an encephalitogenic myelin basic protein-reactive CD4+ T cell line (97, 98). In fact, microglia activation occurs prior to the development of symptoms, suggesting that microglia are necessary for the development of EAE (99). First attempt to dissect the role of microglia in EAE came with a study by Heppner et al. where, by using a CD11b-HSVTK transgenic mice, all CD11b+ cells, including macrophages and microglia, express the herpes simplex thymidine kinase and after ganciclovir administration, it blocks cell activation, generating a “paralysis” of microglia and macrophages, that ameliorates EAE symptoms (100). However, because of the similarities between microglia and macrophages markers, it was difficult to specifically dissect the role of microglia during EAE progression. In line with this observation, the inhibition of the Colony-Stimulating Factor 1 Receptor (CSF1R) by the PLX5622, reduced both microglia and infiltrating-macrophage population and reduced EAE pathogenesis (101, 102). The use of scRNA-seq analysis as an alternative to overcome this limitation has shown heterogenicity of microglial populations both at homeostasis and during EAE (52). At the peak of the disease, microglia expand dramatically, although reducing MHCII expression in this population did not affect EAE progression, suggesting a redundant function of microglia on T cell activation (52). Recently, it has been shown that CD83 expression in microglia is important for regulating their function, by a study showing that specific deletion of CD83 expression in microglia promoted an over-activated phenotype, increasing the production of TNF and exacerbating EAE symptoms and neuroinflammation, suggesting that microglia, by modulating the microenvironment, could participate in the propagation of the neuro-inflammatory damage in the CNS during EAE (103). However, more studies are necessary to specifically dissect the role of microglia during EAE. Additionally, to the possible role in promoting inflammation and damage during EAE, some studies have suggested that microglia could be playing protective roles during the development of MS, by the production of immunosuppressive factors that could mediate myelin regeneration or myelin clearance, that ultimately leads to better recovery (104–106). An explanation for this dichotomous behavior could be attributed in part to different activation states; the rapid expansion of different microglia populations during disease; or the interaction with other immune cell types during neuroinflammation. The search of new specific markers to differentiate microglia from infiltrating macrophages, together with the development of new genetic or pharmacological approaches to specifically target microglia will be necessary to have a clear picture of the specific role of microglia during EAE.
Clusters of microglia, called microglia nodules, in brain lesions of pwMS have been described for over 30 years (107, 108). The appearance of these microglia nodules occurs before the MS lesion formation and persist throughout the entire course of the disease (109–112). Microglia nodules are highly phagocytic, produce inflammatory cytokines and radical oxygen species (ROS), which could contribute with axonal damage and degeneration (113–115). A recent study has tried to characterize the microglia nodules in MS lesions by using different genetic, molecular, and cellular approaches (116). Strikingly, microglia nodules are in areas with active axon demyelination; they have increased pro-inflammatory and ROS-related gene expression; they are in close contact with other immune cell types such as infiltrating macrophages; and correlated with severe MS pathology, suggesting that microglia nodules are participating in the initiation of lesions and the promotion of neuroinflammation in pwMS (116). Further studies considering the time course of the disease are necessary to confirm these findings, to consider microglia as a therapeutic target for MS.
Neutrophils are the most abundant leukocyte in the blood, representing around 40% to 70% of all white blood cells in humans and 10-25% in mice (117). Neutrophils are generated in the bone marrow and released into the blood, where they can live for some hours, around 10 h in mice and 18 h in humans, and they can be rapidly recruited to sites of infection, by following chemoattractant gradients to reach compromised tissues, working as a first line of defense against invading microorganisms (118–120). ROS production, phagocytosis, and the formation of neutrophil extracellular traps (NETs) are among the main mechanisms used by neutrophils to fight pathogens (121, 122).
Although it was thought that the antimicrobial activity was the main function of neutrophils, emerging evidence from the last 15 years has showed that neutrophils are able to produce cytokines, to interact with other immune cell types and also to express MHCII, suggesting that they can activate CD4+ T cells, modulating adaptive immunity (123–126). In fact, neutrophils can be found in non-inflamed tissues and in lymph nodes during homeostasis, suggesting that neutrophils can be also contributing to tissue homeostasis (125, 127). Due to its varied functions, several lines of evidence suggest that neutrophils dysfunction could be implicated in the pathogenesis of different autoimmune disorders including MS (128).
First evidence suggesting a role of neutrophils in EAE came from studies showing neutrophils accumulation in blood, spleen, peripheral lymph nodes, meninges and CNS during different stages of EAE progression (129–133). Blockade of neutrophil accumulation in circulation or neutrophil depletion, particularly at early stages, reduces EAE severity, suggesting a direct involvement of neutrophils in EAE progression (132, 133). Glutamic acid-leucine-arginine-positive (ELR+) chemokines (CXCL1, CXCL2 and CXCL6), which are produced by Th17 cells, and granulocyte-colony stimulating factor (G-CSF), produced by fibroblasts and epithelial cells after stimulation by Th17 cells, are important chemokines that mediate neutrophil recruitment into the CNS (133, 134). In the CNS, neutrophils seem to be involved in the blood-brain barrier (BBB) breakdown, and to support the activation of microglia and CNS-infiltrating macrophages, amplifying neuroinflammation (135, 136). NETs formation in the brain and spinal cords of animals suffering EAE has been also recently reported, which could facilitate the recruitment of Th1 and Th17 cells into the CNS (137). In the periphery, neutrophils seem to be important in the clonal expansion of autoreactive T cells in peripheral lymph nodes, as suggested by a recent study showing that reducing neutrophil accumulation in lymph nodes attenuate CD4+ T cell expansion and decreases EAE clinical score, by a mechanism dependent on TLR9 (132). Thus, neutrophils seem to contribute to EAE pathogenesis in different anatomical location; in the periphery, by activating and expanding autoreactive CD4+ T cells in peripheral lymph nodes and possibly the meninges, and in the CNS, by directly affecting BBB permeability, together with supporting the activation of microglia and CNS-infiltrating macrophages and promoting the recruitment of autoreactive T cells into the brain and spinal cord, all of which leads to neuroinflammation.
Initial studies about the role of neutrophils in pwMS were conflicting, with some studies showing increased neutrophil priming and neutrophil-producing molecules while other papers showed unaltered or even reduced neutrophil activity in peripheral blood from pwMS (138, 139). The difficulty of specifically analyzing neutrophils from other leukocytes in the blood could explain these contradictory results. In fact, the development of better neutrophil markers has confirmed elevated neutrophil count during MS clinical relapses compared to remission (140). Moreover, the neutrophil-to-lymphocyte ratio (NLR), has been found to be higher in blood from pwMS compared to healthy controls, although no differences between RRMS and progressive MS patients were found (141). A recent retrospective study confirmed the potential use of NLR as predictor of increased relapse rate and severity in MS (142). Blood neutrophils from RRMS patients have higher inflammatory markers, produce more pro-inflammatory cytokines and ROS and are resistant to apoptosis (143). Detection of neutrophils in the CSF of pwMS, particularly at early stages, suggests a direct involvement of neutrophils in the autoimmune damage (144). Additionally, increased levels of NETs and neutrophil elastase have been found in blood from pwMS compared to healthy volunteers, specifically in RRMS patients, suggesting an active role of neutrophils during inflammation (145). All these results suggest that neutrophils have a more activated phenotype in pwMS, and by different modes of action, involving increased production of pro-inflammatory cytokines, NETs formation, and ROS production, could contribute to the autoimmune damage in pwMS.
Innate lymphoid cells (ILCs) represent a subset of immune cells that share characteristics with classical lymphoid T cell subsets but are devoid of the antigenic presentation requirement for their activation. These cells express transcriptional regulators and effector cytokines similar to helper T cell subpopulations and, based on their molecular expression profile, are classified into three groups. ILC1s, which consists in conventional natural killer (NK) cells and so-called helper ILC1; both expressing the transcription factor T-bet and promoting type 1 immunity, are critical for controlling intracellular microbial infections and restraining tumor development. Functionally, NK cells have strong cytotoxic potential, expressing granzymes and perforins, whereas helper ILC1s produce much higher concentrations of IFN-γ than NK cells (146). Group 2 ILCs are dependent on the transcription factors GATA3 and RORα and produce the type 2 cytokines IL-4, IL-5, IL-9, and IL-13 (147–149). ILC2s plays protective roles in the expulsion of intestinal parasites and tissue repair, but also mediate detrimental host immune responses depending on timing, location, and physiological context. Interestingly, ILC2s, despite its scarcity, is the dominant innate lymphoid cell population in the lung, indicating a key role as first responder and amplifier upon immune challenge at this site (150). Group 3 ILCs includes fetal lymphoid tissue-inducer (LTi) as well as adult ILC3s, both depend on RORγt, but their distribution and functions are distinct. Specifically, LTi cells mediate the development of lymphoid tissues during embryogenesis via the production of lymphotoxin, while ILC3s is highly enriched in the gut, where they sense and integrate a wide range of cell-derived signals and environmental cues coming from microbiota and diet, shaping their phenotype and functions (151). ILC3s secrete different effector cytokines including IL-22, IL-17A, IL-17F, GM-CSF and lymphotoxin-α3 (LTα3) (147, 149, 152, 153). Intestinal ILC3s participates in the proper expression of tight junctions on gut epithelial cells, preventing the activation of innate immune cells through the stimulation of pathogen-associated molecular pattern receptors and avoiding the activation of autoreactive T cells by molecular mimicry (154–156). ILC-T cell interactions can contribute to immune tolerance by depleting commensal bacteria-specific T cells during homeostasis (157, 158), but also by stimulating antigen-specific T cells and pathogenic Th1 cell expansion in inflammatory conditions (158–161). Finally, ILCs are not only present on mucosal surfaces but indeed can be found in CNS during steady-state and inflammation. Even when ILC subsets were initially described as tissue-resident cells that proliferate locally, recent studies demonstrated that, in response to inflammation, gut ILCs acquire migratory patrolling attributes (162). ILCs can be recruited into extraintestinal tissues under inflammatory conditions, such as mesenteric lymph nodes, lung and CNS to promote an inflammatory T cell activation (163–165); however, the exact mechanism driving the recruitment of ILCs into the brain are still not well understood. Additionally, the role of ILCs in MS is still not completely understood as controversial findings have been reported assigning them either a protective or disease-accelerating role, depending on the analyzed subset.
Studies in mice models have shown that in homeostatic conditions, the three groups of ILCs reside both in the meninges and choroid plexus (166), giving CNS-resident ILCs an advantageous anatomical site to act as cerebral immune gatekeepers. Like their adaptive counterparts, Th2 cells have been described as having a neuroprotective role for CNS-resident ILC2s. Meningeal ILC2s has emerged as a novel regulator of microglial activation and BBB stability mediated by their IL-10 production (167). As MS pathology is well known to be sexually dimorphic, hormone differences between genders are associated to decreased levels of IL-33 and restricted ILC2 activation in a female transgenic mouse model of EAE, thus promoting increased susceptibility to developing the disease (168). Activation of ILC2s through IL-33 stimulation limits the Th17-dominated response characteristic of susceptible females and drives a non-pathogenic Th2 anti-myelin response. This evidence indicates that increased ILC2 function is associated with improved neurological outcomes in EAE. On the other hand, during EAE, both ILC1s and ILC3s are able to infiltrate the brain parenchyma, accumulating into the CNS. In particular, NK cells are directly involved in the process of demyelination (3). Nonetheless, the major fraction of CNS-infiltrating ILCs during disease belongs to RORγt+ ILC3s and this accumulation is largely due to cell recruitment rather than local proliferation, indicating migration from peripheral tissues (169, 170). ILC3s in the CNS uniquely expresses the trafficking receptors CCR6, CCR5, α4β7 and CXCR3, that are critical for the entry of lymphocytes into the inflamed CNS through the circulation (163). These receptors respond to ligands CCL20, CCL3/4, V-CAM1 and CXCL11, respectively, which are released from the CNS upon neuroinflammation (171). Interestingly, T bet-dependent NKp46+ ILCs (a group that includes NK, ILC1 and ILC3) controls the CNS parenchymal infiltration of myelin-reactive Th17 cells by generating a proinflammatory-cytokine environment in the meninges that is necessary for the reactivation and maintenance of IL-17A-producing CD4+ T cells in the CNS (172), consequently contributing to the propagation of neuroimmune response to CNS injuries. In addition to pro-inflammatory cytokines IL-17 and GM-CSF, meningeal ILC3s constitutively express CD30L and OX40L, denoting that ILC3s sustain neuroinflammation by supporting T cell survival and reactivation in the meninges (169). Inflammatory ILC3s derived from circulation infiltrates the CNS, are located in the proximity of T cells, and works as APCs that restimulate autoreactive T cells, performing complementary but non-redundant roles with conventional DCs that also act restimulating T cells after entry into the CNS (163). This complementary role might be regulated by the distinct localization of these cell types; ILC3s are enriched within focal lesions of the CNS parenchyma, while DCs are mostly concentrated at border-associated brain dura meninges and spinal cord leptomeninges. Collectively, this evidence indicates that ILCs are essential in CNS inflammation and reveals the potential of harnessing peripheral tissue ILCs for the prevention of MS.
Human studies have showed that NK cells participate in the process of demyelination, supporting the notion of a disease-accelerating role in this cell type. Saikali et al, showed that oligodendrocytes from pwMS express ligands for the activating NK cell receptor NKG2D that were not detected in healthy control samples, and that blocking NKG2D on NK cells significantly inhibited the killing of oligodendrocytes, suggesting a NKG2D-mediated killing mechanism for tissue injury in MS (173). Accordingly, EAE symptoms are reduced in NKG2D-deficient mice (174). In fact, NKG2D ligands, particularly UL16-binding protein 4 (ULBP4), was found to be highly expressed in active and chronic active lesions and normal-appearing white matter of pwMS compared to healthy controls (175). NKG2D ligands can also be shedding and soluble NKG2D ligands are elevated in the serum of pwMS (176). NK cells, CD4+ and CD8+ T cells seem to be the main immune cells involved in the killing mechanism mediated by NKG2D, although more experiments are needed to specifically dissect the contribution of each of these cell types in this process (177).
Epstein-Barr virus (EBV), a herpesvirus present in 90% of adults worldwide, has been considered to be an important factor in MS pathogenesis, mainly because molecular mimicry of EBV antigens and proteins expressed in glial cells that generate autoimmune responses (178, 179). However, only a small percentage of EBV-infected people develop MS, indicating that additional mechanisms should be compromised to develop MS in this population. Recently, it was shown that NK cells, particularly NKG2C+ and NKG2D+ NK cells, are capable of controlling autoimmune damage by killing autoreactive glial cells, and this process is dampened in pwMS (180). This study highlights a protective role of a subset of NK cells in MS pathogenesis, although more studies are necessary to clarify the specific role of different NK cell subsets in MS.
Cytomegalovirus (CMV), a herpesvirus, has a controversial role regarding MS pathogenesis, with a study associating CMV with lower MS susceptibility (181), while other study suggesting CMV infection with enhanced MS symptoms (182). NK cells, together with CD8+ T cells, are the main cells capable of eliminating CMV-infected cells (183). Accordingly, pwMS have increased NKG2C expression in NK cells compared to healthy donors, with the highest expression in the CMV-seropositive pwMS compared to CMV-seronegative pwMS, suggesting that CMV could promote NKG2C expression in NK cells, although the mechanism involved or the impact over NK cell function and MS pathogenesis are currently unknown (184). Thus, the relationship between CMV, NK cells and MS requires further studies.
Recent investigations showed a higher frequency of CD56bright NK cells during relapse, indicating an immediate response to disease reactivation, while CCR6-related shifts among this population suggest altered ILC migration to the CNS during MS (185). CD56bright NK cells are enriched in MS lesions and the choroidal tissue from MS donors and display an activated and migratory phenotype, similar to that of CD56bright NK cells in the circulation (186). As such, the enrichment of CD56bright NK cells in the CNS of pwMS could result from selective infiltration from the blood towards the brain. NK are not the only ILC subset with the ability to migrate into the CNS during MS, since ILC3s have also been detected in the cerebrospinal fluid of pwMS and the frequency of these cells positively correlates with the presence of higher lesions (163). Interestingly, both ILC1 and ILC3 subsets expressed Sphingosine-1-phosphate receptor 1 (S1PR1), which explains their reduced absolute numbers in the peripheral blood of RRMS patients treated with Fingolimod, a sphingosine 1-phosphate receptor modulator used as a first-line therapy for RRMS, compared with treatment-free pwMS (187), suggesting that S1PR1 agonist sequesters peripheral blood human ILCs in lymph nodes. Collectively, these data suggest that ILC1 and ILC3 subsets migrate to the CNS and promote cytotoxicity and inflammation in pwMS.
Mast cells (MCs), first described by Paul Ehrlich in 1878, are multifunctional and heterogeneous cells characterized by the expression of CD117, ST2 (also known as Interleukin 1 receptor-like 1) and the high affinity receptor for immunoglobulin (Ig) E (FcϵRI), and classically involved in a IgE-dependent allergic responses against parasitic invasion. They circulate as progenitor cells and populate different tissues, where they become mature MCs by interacting with local stem cell factors. Thus, MCs are located at sites of contact with antigens and allergens: skin, around blood vessels, bronchioles, mucous glands, and gut (188, 189). Upon activation, MCs release granules containing histamine, proteases (tryptase and chymase), prostaglandins, heparin, leukotrienes, cytokines, chemokines, and growth factors (190, 191). MCs are also located in the CNS, particularly in the leptomeninges, dura mater, choroid plexus, parenchyma of the thalamic-hypothalamic region, and cerebral side of BBB, suggesting that they could be involved in CNS pathologies (192, 193). Under physiological conditions, MCs maintain bidirectional communication with neurons so that MCs-heparin secretion blocks calcium flow, reducing neuronal communication, while neurons, through the secretion of neuropeptides, stimulate adhesion, degranulation, and secretion of cytokines and chemokines by MCs (191).
The role of MCs in the pathogenesis of EAE has been challenging due to their non-circulating state in the blood, low representation in tissues, and manipulation. Initially, a deleterious role of MCs in EAE arose from studies using the mast-cell-deficient KitW/W-v mouse model, that showed reduced clinical symptoms and delayed onset of EAE, effect that was reverted by the transplantation of bone-marrow derived MCs (194). Interestingly, bone marrow derived MCs transplantation failed to repopulate MCs in the CNS, suggesting that peripheral MCs are important mediators in EAE (195). Even though this early evidence, the use of a different MC-deficient mouse model, the Kitw-sh mice, showed exacerbated EAE disease and increased T cell response against myelin (196). The discrepancies between these two models of MCs deficiency, could be explained in part by the different immunization protocols, since the same group showed that KitW/W-v animals showed reduced EAE symptoms at high, but not low doses of antigen and adjuvants (196). Further studies showed a pathogenic role of MCs located in the meninges, by contributing to the infiltration of T cells into the CNS, in a process dependent on the production of TNF by MCs (197). High numbers of MCs are found in the meninges, and KitW/W-v mice showed reduced T cell and neutrophil infiltration into the meninges and brain parenchyma, a process that can be reverted by intracranial mast cell reconstitution (197). Other studies have shown that MCs could be involved in the activation of different immune cells through the production of proinflammatory cytokines and chemokine secretion such as MCP-1, IL-6, and IL-33 (190, 198). MCs could also act as APCs, establishing a fine bidirectional communication with T cells, where MCs degrade myelin through their proteases and present antigens to T cells, which in turn stimulate MCs to secrete MMP-9, IL-6, and proteases, enhancing myelin destruction (199). Moreover, it has been demonstrated that MCs, upon activation of the NLRP3 inflammasome, secrete IL-1β, which induces the release of GM-CSF by T cells, a cytokine that enhances EAE pathogenesis (200). Failure in NLRP3 function reduces demyelination and promotes resistance to the development of EAE (193). Additionally, MCs, by secreting IL-4, IL-10, IL-13, TGF-β, TNF-α, and IL-6, can promote T cell differentiation into Th1, Th2, and Th17, while in an OX40L-dependent way, MCs can suppress Treg function (190, 193, 201). It has also been demonstrated that MCs interact with Th9 cells, which have been implicated in the pathogenesis of EAE, and that through the secretion of IL-9, they recruit MCs from the spleen to the CNS, indicating that not only resident MCs but also peripheral MCs are associated with EAE (202).
It seems that MCs have a dual role inside CNS in humans, since the secretion of IL-6 by MCs at low concentrations is neuroprotective and stimulates the proliferation of neuronal stem cells; however, at high concentrations, it promotes neurotoxicity (203). Similarly, the binding of TNF-α to its Tumor necrosis factor receptor (TNF-R) 2 receptor induces neurogenesis, while TNF-R1 activation is associated with neurodegeneration (191). In addition to producing cytokines, MCs produce approximately 20-40% of cerebral serotonin, a neurotransmitter that may play a dual role in MS depending on which receptor it binds to, since H2Rs and H3Rs are neuroprotective, while H1Rs and H4Rs are inflammatories (204). MCs also promote angiogenesis through the secretion of VEGF, FGF-2, and Osteopontin (OPN), which in turn stimulates VEGF secretion establishing synergy. Thus, MCs promote increased recruitment of immune cells to the CNS (205). Therefore, depending on the immune context and inflammatory environment, MCs can act as detrimental or protective agents in MS. In summary, the use of new MCs-deficient mice models is necessary to help clarify the exact role of MCs during MS.
Basophils are short-lived innate immune cells characterized by the presence of basophilic granules in the cytoplasm, representing less than 1% of all leukocytes in the peripheral blood. Basophils share several features with mast cells, including the expression of the FcϵRI, but basophils express high level expression of IL-3 R alpha/CD123, and they do not express CD117/c-kit (206). Basophils, by expressing MHCII and co-stimulatory molecules, are capable of presenting antigens to CD4+ T cells and are believed to be important in promoting Th2 cell differentiation through the production of IL-4 (207). However, additionally to IL-4, basophils can produce IL-6, a cytokine important in mediating a Th17 response, suggesting that they are important mediators in Th17 cell-mediated immune responses (208). The role of basophils in EAE has been first drawn by a seminal study showing that basophils, by actively secreting IL-6, support Th17 polarization by DCs (209). Because Th17 cells are important mediators in EAE, this study suggests that basophils could contribute to the autoimmune damage by indirectly supporting Th17 cell polarization. In fact, by using basophil-deficient mice, the authors found reduced EAE clinical score together with a reduction in the number of CD4+ T cells that infiltrate CNS and decreased levels of IL-17 production by these cells (209). This study suggests a direct role of basophils in promoting autoimmune damage in EAE, although more research is necessary to confirm and expand these findings. Moreover, elevated number of basophils have been observed in the blood of pwMS compared to healthy controls, although the significance of this observation over the development or progression of MS remains unknown (210). Thus, more studies are necessary to understand the role of basophils during MS. A summary of innate immune components involved in MS pathogenesis is shown in Figure 1.
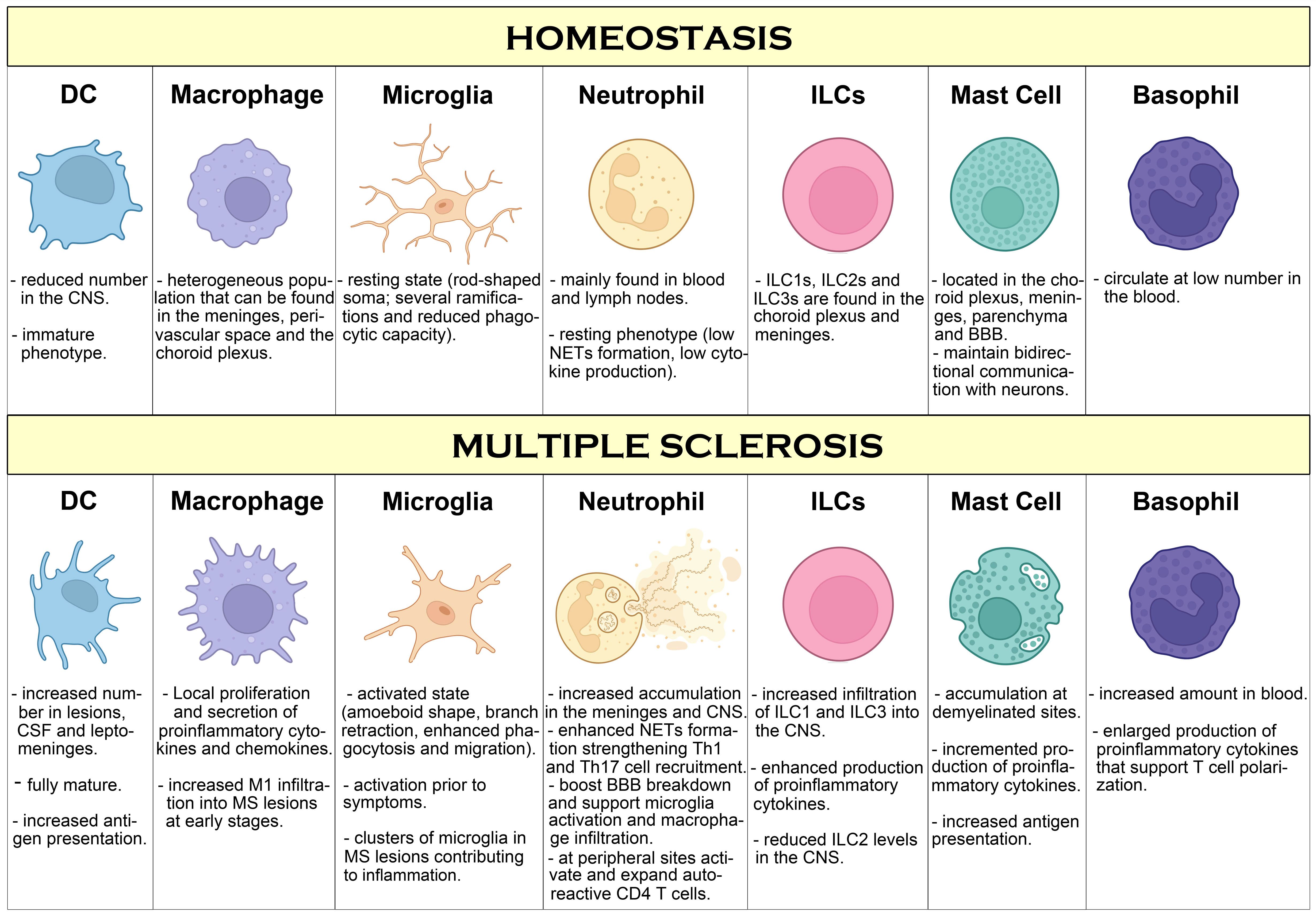
Figure 1. Overview of innate immune cells and pathways compromised during MS. During homeostasis conditions, besides microglia covering the brain and spinal cord in a resting state, few innate immune cell types can be found in the CNS, mostly in the meninges and choroid plexus where they work sensing and controlling possible threats such as pathogen infection. However, during MS, increased infiltration of innate immune cells in perivascular regions and in the brain parenchyma is observed, promoting inflammation, immune cell infiltration and T cell activation that leads to neuroinflammation, neurodegeneration and myelin damage. Additionally, innate immune cells in the periphery support inflammation by activation and polarization of autoreactive T cells, that further contribute to MS pathogenesis.
MS is a complex disease not only because there is no cure for it, but also because its cause is unknown. MS is therefore a disease that presents several challenges in its diagnosis, treatment and management. Some of the key challenges and future directions lie not only in effective and personalized treatment, but in early and accurate diagnosis. While there are treatments to manage the symptoms of MS, there is no known cure. Hence, it is necessary to have a better understanding of the inflammatory processes that contribute to the development of this disease, as well as the modulation of these processes. In this sense, a complex interplay between immune cells coordinates the cascade of inflammatory events that contributes to development of the disease in MS. Adaptive immunity has been studied extensively over the years, but less emphasis has been placed on innate immune changes that occur in MS. As a result of this, current approved MS therapies primarily target peripheral lymphocytes and thus are mainly effective in treating RRMS. In progressive MS, where the adaptive immune response plays a less prominent role, microglia and CNS-associated macrophages are activated in a pro-inflammatory phenotype that promotes demyelination and neurodegeneration. However, to date, there are no specific therapies targeting primarily innate immune cells in MS. As the role of innate immune cells in MS becomes better described (see Table 1), it will be possible to design novel approaches to therapeutically target both central and peripheral innate immunity to promote remyelination, reduce neuroinflammation and increase CNS repair in pwMS.
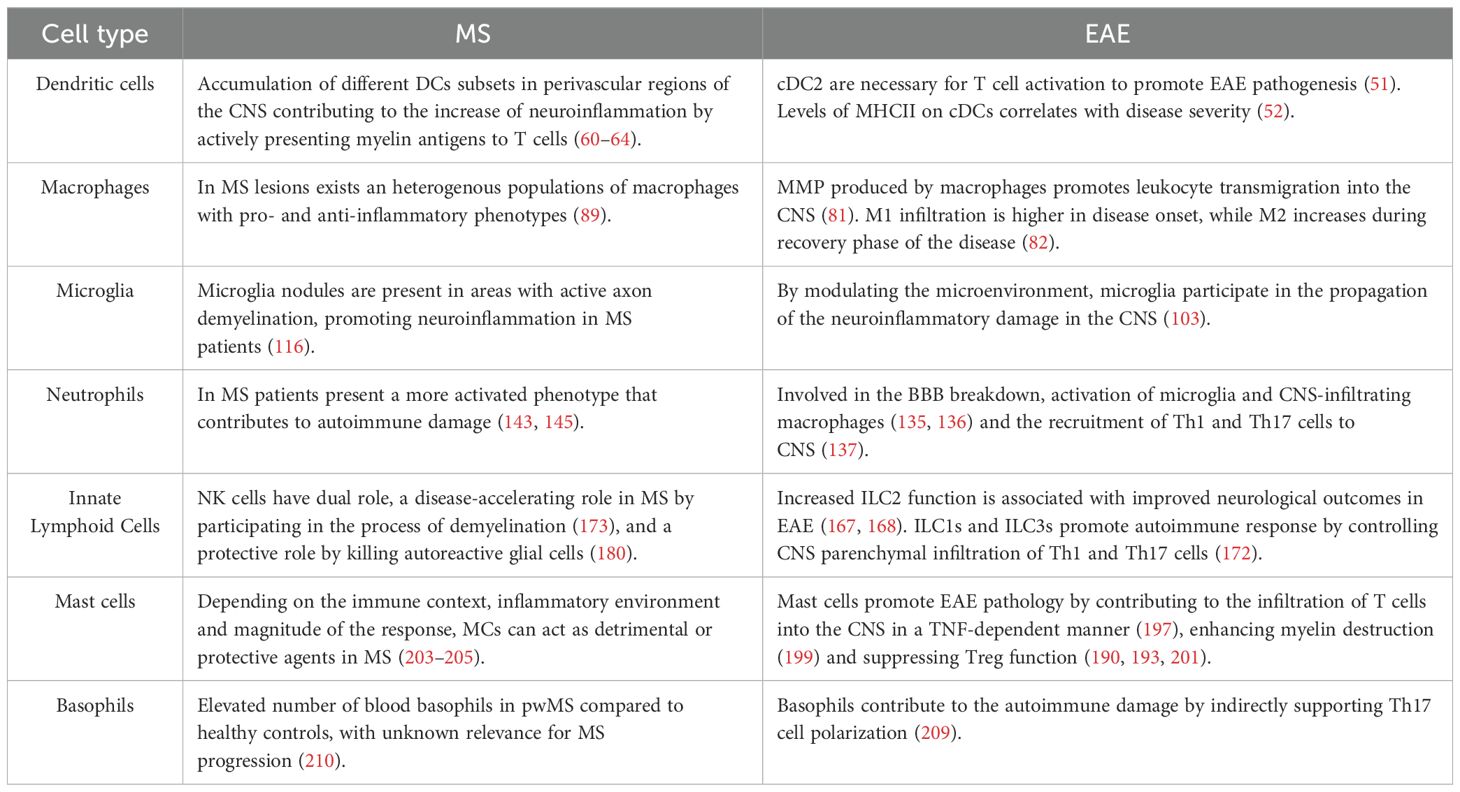
Table 1. Pathogenic and protective roles of innate immune cells in multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE).
Finally, a multidisciplinary approach incorporating collaboration between neurologists, immunologists, geneticists and other experts could certainly help advance the understanding of MS and develop more effective treatments that involve not only neuroinflammation but also neurodegeneration and repair.
CP: Writing – original draft. AH: Conceptualization, Writing – original draft, Writing – review & editing. DH: Writing – original draft. LG: Writing – original draft. NE: Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the following grants: FONDECYT Regular N° 1231431 (AH), FONDECYT Regular N° 1240944 (NE) and FONIS SA23I0038 (NE), and Programa de Apoyo a Centros con Financiamiento Basal AFB-170004) (CP), all from Agencia Nacional de Investigación y Desarrollo de Chile (ANID). AH is a Latin American Fellow in the Biomedical Sciences, supported by The Pew Charitable Trusts.
Parts of the figures were created with the help of BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/NRI3871
2. Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol. (2022) 18:40–55. doi: 10.1038/S41582-021-00581-X
3. Sadeghi Hassanabadi N, Broux B, Marinović S, Gotthardt D. Innate lymphoid cells - neglected players in multiple sclerosis. Front Immunol. (2022) 13:909275. doi: 10.3389/FIMMU.2022.909275
4. Lassmann H. The changing concepts in the neuropathology of acquired demyelinating central nervous system disorders. Curr Opin Neurol. (2019) 32:313–9. doi: 10.1097/WCO.0000000000000685
5. Schreiner TG, Genes TM. Obesity and multiple sclerosis-A multifaceted association. J Clin Med. (2021) 10(12):2689. doi: 10.3390/JCM10122689
6. Arneth B. Multiple sclerosis and smoking. Am J Med. (2020) 133:783–8. doi: 10.1016/J.AMJMED.2020.03.008
7. Oturai DB, Bach Søndergaard H, Koch-Henriksen N, Andersen C, Laursen JH, Gustavsen S, et al. Exposure to passive smoking during adolescence is associated with an increased risk of developing multiple sclerosis. Mult Scler. (2021) 27:188–97. doi: 10.1177/1352458520912500
8. Bazmi E, Behnoush AH, Talebian MT, Afrooghe A, Sahraian MA. Waterpipe tobacco smoking and multiple sclerosis: a systematic review and meta-analysis. Neuroepidemiology. (2024) 25:1–11. doi: 10.1159/000540087
9. Bjornevik K, Münz C, Cohen JI, Ascherio A. Epstein-Barr virus as a leading cause of multiple sclerosis: mechanisms and implications. Nat Rev Neurol. (2023) 19:160–71. doi: 10.1038/S41582-023-00775-5
10. Aloisi F, Giovannoni G, Salvetti M. Epstein-Barr virus as a cause of multiple sclerosis: opportunities for prevention and therapy. Lancet Neurol. (2023) 22:338–49. doi: 10.1016/S1474-4422(22)00471-9
11. Soldan SS, Lieberman PM. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol. (2023) 21:51–64. doi: 10.1038/S41579-022-00770-5
12. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. (2006) 296:2832–8. doi: 10.1001/JAMA.296.23.2832
13. Smolders J, Menheere P, Kessels A, Damoiseaux J, Hupperts R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult Scler. (2008) 14:1220–4. doi: 10.1177/1352458508094399
14. Fatima M, Lamis A, Siddiqui SW, Ashok T, Patni N, Fadiora OE. Therapeutic role of vitamin D in multiple sclerosis: an essentially contested concept. Cureus. (2022) 14(6):e26186. doi: 10.7759/CUREUS.26186
15. Buenafe AC. Diurnal rhythms are altered in a mouse model of multiple sclerosis. J Neuroimmunol. (2012) 243:12–7. doi: 10.1016/J.JNEUROIM.2011.12.002
16. Gustavsen S, Søndergaard HB, Oturai DB, Laursen B, Laursen JH, Magyari M, et al. Shift work at young age is associated with increased risk of multiple sclerosis in a Danish population. Mult Scler Relat Disord. (2016) 9:104–9. doi: 10.1016/J.MSARD.2016.06.010
17. Hedström AK, Åkerstedt T, Olsson T, Alfredsson L. Shift work influences multiple sclerosis risk. Mult Scler. (2015) 21:1195–9. doi: 10.1177/1352458514563592
18. Hedström AK, Akerstedt T, Hillert J, Olsson T, Alfredsson L. Shift work at young age is associated with increased risk for multiple sclerosis. Ann Neurol. (2011) 70:733–41. doi: 10.1002/ANA.22597
19. Haase S, Linker RA. Inflammation in multiple sclerosis. Ther Adv Neurol Disord. (2021) 14:17562864211007687. doi: 10.1177/17562864211007687
20. Rahmanzadeh R, Weber MS, Brück W, Navardi S, Sahraian MA. B cells in multiple sclerosis therapy-A comprehensive review. Acta Neurol Scand. (2018) 137:544–56. doi: 10.1111/ANE.12915
21. Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis - from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. (2021) 17:399–414. doi: 10.1038/S41582-021-00498-5
22. Comi G, Bar-Or A, Lassmann H, Uccelli A, Hartung HP, Montalban X, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol. (2021) 89:13–23. doi: 10.1002/ANA.25927
23. Park C, Ponath G, Levine-Ritterman M, Bull E, Swanson EC, De Jager PL, et al. The landscape of myeloid and astrocyte phenotypes in acute multiple sclerosis lesions. Acta Neuropathol Commun. (2019) 7:130. doi: 10.1186/S40478-019-0779-2
24. Alakhras NS, Kaplan MH. Dendritic cells as a nexus for the development of multiple sclerosis and models of disease. Adv Biol. (2023) 7(7):e2300073. doi: 10.1002/ADBI.202300073
25. Pitt D, Lo CH, Gauthier SA, Hickman RA, Longbrake E, Airas LM, et al. Toward precision phenotyping of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2022) 9(6):e200025. doi: 10.1212/NXI.0000000000200025
26. Dubey D, Forsthuber T, Flanagan EP, Pittock SJ, Stüve O. B-cell-targeted therapies in relapsing forms of MS. Neurology(R) neuroimmunology Neuroinflamm. (2017) 4(6):e405. doi: 10.1212/NXI.0000000000000405
27. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. (1973) 137:1142–62. doi: 10.1084/JEM.137.5.1142
28. Balan S, Saxena M, Bhardwaj N. Dendritic cell subsets and locations. Int Rev Cell Mol Biol. (2019) 348:1–68. doi: 10.1016/BS.IRCMB.2019.07.004
29. Tong C, Liang Y, Han X, Zhang Z, Zheng X, Wang S, et al. Research progress of dendritic cell surface receptors and targeting. Biomedicines. (2023) 11(6):1673. doi: 10.3390/BIOMEDICINES11061673
30. Zhou F, Zhang GX, Rostami A. Distinct role of IL-27 in immature and LPS-induced mature dendritic cell-mediated development of CD4+ CD127 + 3G11+ Regulatory T cell subset. Front Immunol. (2018) 9:2562. doi: 10.3389/FIMMU.2018.02562
31. Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity. (2016) 45:669–84. doi: 10.1016/J.IMMUNI.2016.08.015
32. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571. doi: 10.1038/NRI3712
33. Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira Da Costa M, Reis E Sousa C. Dendritic cells revisited. Annu Rev Immunol. (2021) 39:131–66. doi: 10.1146/ANNUREV-IMMUNOL-061020-053707
34. Ginhoux F, Guilliams M, Merad M. Expanding dendritic cell nomenclature in the single-cell era. Nat Rev Immunol. (2022) 22:67–8. doi: 10.1038/S41577-022-00675-7
35. Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. (1999) 11:753–61. doi: 10.1016/S1074-7613(00)80149-1
36. Hanly A, Petito CK. HLA-DR-positive dendritic cells of the normal human choroid plexus: a potential reservoir of HIV in the central nervous system. Hum Pathol. (1998) 29:88–93. doi: 10.1016/S0046-8177(98)90395-1
37. McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol. (1999) 405:553–62. doi: 10.1002/(SICI)1096-9861(19990322)405:4<553::AID-CNE8>3.0.CO;2-6
38. Gottfried-Blackmore A, Kaunzner UW, Idoyaga J, Felger JC, McEwen BS, Bulloch K. Acute in vivo exposure to interferon-gamma enables resident brain dendritic cells to become effective antigen presenting cells. Proc Natl Acad Sci U.S.A. (2009) 106:20918–23. doi: 10.1073/PNAS.0911509106
39. Felger JC, Abe T, Kaunzner UW, Gottfried-Blackmore A, Gal-Toth J, McEwen BS, et al. Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun. (2010) 24:724–37. doi: 10.1016/J.BBI.2009.11.002
40. Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, et al. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol. (2008) 508:687–710. doi: 10.1002/CNE.21668
41. Matyszak MK, Perry VH. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience. (1996) 74:599–608. doi: 10.1016/0306-4522(96)00160-1
42. Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. (2001) 166:5168–75. doi: 10.4049/JIMMUNOL.166.8.5168
43. Kleindienst P, Wiethe C, Lutz MB, Brocker T. Simultaneous induction of CD4 T cell tolerance and CD8 T cell immunity by semimature dendritic cells. J Immunol. (2005) 174:3941–7. doi: 10.4049/JIMMUNOL.174.7.3941
44. Suter T, Biollaz G, Gatto D, Bernasconi L, Herren T, Reith W, et al. The brain as an immune privileged site: dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol. (2003) 33:2998–3006. doi: 10.1002/EJI.200323611
45. Weir CR, Nicolson K, Bäckström BT. Experimental autoimmune encephalomyelitis induction in naive mice by dendritic cells presenting a self-peptide. Immunol Cell Biol. (2002) 80:14–20. doi: 10.1046/J.1440-1711.2002.01056.X
46. Dittel BN, Visintin I, Merchant RM, Janeway CA. Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J Immunol. (1999) 163:32–9. doi: 10.4049/jimmunol.163.1.32
47. Zozulya AL, Ortler S, Lee JE, Weidenfeller C, Sandor M, Wiendl H, et al. Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci. (2009) 29:140–52. doi: 10.1523/JNEUROSCI.2199-08.2009
48. Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity. (2012) 37:264–75. doi: 10.1016/J.IMMUNI.2012.05.025
49. Isaksson M, Lundgren BA, Ahlgren KM, Kämpe O, Lobell A. Conditional DC depletion does not affect priming of encephalitogenic Th cells in EAE. Eur J Immunol. (2012) 42:2555–63. doi: 10.1002/EJI.201142239
50. Luu T, Cheung JF, Baccon J, Waldner H. Priming of myelin-specific T cells in the absence of dendritic cells results in accelerated development of Experimental Autoimmune Encephalomyelitis. PloS One. (2021) 16(4):e0250340. doi: 10.1371/JOURNAL.PONE.0250340
51. Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol. (2019) 4(31):eaau8380. doi: 10.1126/SCIIMMUNOL.AAU8380
52. Jordão MJC, Sankowski R, Brendecke SM, Sagar LG, Tai YH, Tay TL, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. (2019) 363(6425):eaat7554. doi: 10.1126/SCIENCE.AAT7554
53. Zhuang W, Zhou J, Zhong L, Lv J, Zhong X, Liu G, et al. CXCR1 drives the pathogenesis of EAE and ARDS via boosting dendritic cells-dependent inflammation. Cell Death Dis. (2023) 14(9):608. doi: 10.1038/S41419-023-06126-Y
54. Moorman CD, Yu S, Briseno CG, Phee H, Sahoo A, Ramrakhiani A, et al. CAR-T cells and CAR-Tregs targeting conventional type-1 dendritic cell suppress experimental autoimmune encephalomyelitis. Front Immunol. (2023) 14:1235222. doi: 10.3389/FIMMU.2023.1235222
55. Pashenkov M, Huang YM, Kostulas V, Haglund M, Söderström M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. (2001) 124:480–92. doi: 10.1093/BRAIN/124.3.480
56. Longhini ALF, von Glehn F, Brandão CO, de Paula RFO, Pradella F, Moraes AS, et al. Plasmacytoid dendritic cells are increased in cerebrospinal fluid of untreated patients during multiple sclerosis relapse. J Neuroinflamm. (2011) 8(1):2. doi: 10.1186/1742-2094-8-2
57. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. (2006) 65(8):124–41. doi: 10.1093/JNEN/65.2.124
58. Blandford SN, Fudge NJ, Moore CS. CXCL10 is associated with increased cerebrospinal fluid immune cell infiltration and disease duration in multiple sclerosis. Biomolecules. (2023) 13:1204. doi: 10.3390/BIOM13081204
59. McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2 and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. (1998) 86:20–9. doi: 10.1016/S0165-5728(98)00002-2
60. Boven LA, Montagne L, Nottet HSLM, De Groot CJA. Macrophage inflammatory protein-1alpha (MIP-1alpha), MIP-1beta, and RANTES mRNA semiquantification and protein expression in active demyelinating multiple sclerosis (MS) lesions. Clin Exp Immunol. (2000) 122:257–63. doi: 10.1046/J.1365-2249.2000.01334.X
61. Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. (1999) 103:807–15. doi: 10.1172/JCI5150
62. Pashenkov M, Teleshova N, Kouwenhoven M, Kostulas V, Huang YM, Söderström M, et al. Elevated expression of CCR5 by myeloid (CD11c+) blood dendritic cells in multiple sclerosis and acute optic neuritis. Clin Exp Immunol. (2002) 127:519–26. doi: 10.1046/J.1365-2249.2002.01779.X
63. Esaulova E, Cantoni C, Shchukina I, Zaitsev K, Bucelli RC, Wu GF, et al. Single-cell RNA-seq analysis of human CSF microglia and myeloid cells in neuroinflammation. Neurology(R) neuroimmunology Neuroinflamm. (2020) 7(4):e732. doi: 10.1212/NXI.0000000000000732
64. Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, Lautwein T, et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun. (2020) 11(1):247. doi: 10.1038/S41467-019-14118-W
65. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/J.IMMUNI.2014.06.013
66. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. (2014) 262:36–55. doi: 10.1111/IMR.12223
67. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/JIMMUNOL.164.12.6166
68. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/J.IT.2004.09.015
69. Strizova Z, Benesova I, Bartolini R, Novysedlak R, Cecrdlova E, Foley LK, et al. M1/M2 macrophages and their overlaps - myth or reality? Clin Sci (Lond). (2023) 137:1067–93. doi: 10.1042/CS20220531
70. Kierdorf K, Masuda T, Jordão MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. (2019) 20:547–62. doi: 10.1038/S41583-019-0201-X
71. Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. (2011) 14:1227–35. doi: 10.1038/NN.2923
72. Mundt S, Greter M, Flügel A, Becher B. The CNS immune landscape from the viewpoint of a T cell. Trends Neurosci. (2019) 42:667–79. doi: 10.1016/J.TINS.2019.07.008
73. Mato M, Ookawara S, Sakamoto A, Aikawa E, Ogawa T, Mitsuhashi U, et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc Natl Acad Sci U.S.A. (1996) 93:3269–74. doi: 10.1073/PNAS.93.8.3269
74. Bowers WE, Ruhoff MS, Goodell EM, Stoltenborg JK. The effect of silica treatment on accessory cell-dependent rat T lymphocyte proliferation. Immunobiology. (1988) 176:179–94. doi: 10.1016/S0171-2985(88)80052-4
75. Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J Immunol. (1981) 126:614–20. doi: 10.4049/jimmunol.126.2.614
76. Huitinga I, Van Rooijen N, De Groot CJA, Uitdehaag BMJ, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. (1990) 172:1025–33. doi: 10.1084/JEM.172.4.1025
77. Polfliet MMJ, van de Veerdonk F, Döpp EA, van-Kesteren-Hendrikx EML, van Rooijen N, Dijkstra CD, et al. The role of perivascular and meningeal macrophages in experimental allergic encephalomyelitis. J Neuroimmunol. (2002) 122:1–8. doi: 10.1016/S0165-5728(01)00445-3
78. Xi L, Lin Z, Qiu F, Chen S, Li P, Chen X, et al. Enhanced uptake and anti-maturation effect of celastrol-loaded mannosylated liposomes on dendritic cells for psoriasis treatment. Acta Pharm Sin B. (2022) 12:339–52. doi: 10.1016/J.APSB.2021.07.019
79. Kang K, Lim J-S. Induction of functional changes of dendritic cells by silica nanoparticles. Immune Netw. (2012) 12:104. doi: 10.4110/IN.2012.12.3.104
80. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. (2011) 14:1142–50. doi: 10.1038/NN.2887
81. Agrawal S, Anderson P, Durbeej M, Van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. (2006) 203:1007–16. doi: 10.1084/JEM.20051342
82. Denney L, Kok WL, Cole SL, Sanderson S, McMichael AJ, Ho L-P. Activation of invariant NKT cells in early phase of experimental autoimmune encephalomyelitis results in differentiation of Ly6Chi inflammatory monocyte to M2 macrophages and improved outcome. J Immunol. (2012) 189:551–7. doi: 10.4049/JIMMUNOL.1103608
83. Chu F, Shi M, Lang Y, Chao Z, Jin T, Cui L, et al. Adoptive transfer of immunomodulatory M2 macrophages suppresses experimental autoimmune encephalomyelitis in C57BL/6 mice via blockading NF-κB pathway. Clin Exp Immunol. (2021) 204:199–211. doi: 10.1111/CEI.13572
84. Wang C, Zhou W, Su G, Hu J, Yang P. Progranulin suppressed autoimmune uveitis and autoimmune neuroinflammation by inhibiting th1/th17 cells and promoting treg cells and M2 macrophages. Neurology(R) neuroimmunology Neuroinflamm. (2022) 9(2):e1133. doi: 10.1212/NXI.0000000000001133
85. Jiang HR, Milovanović M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-γ production and inducing alternatively activated macrophages. Eur J Immunol. (2012) 42:1804–14. doi: 10.1002/EJI.201141947
86. Tierney JB, Kharkrang M, La Flamme AC. Type II-activated macrophages suppress the development of experimental autoimmune encephalomyelitis. Immunol Cell Biol. (2009) 87:235–40. doi: 10.1038/ICB.2008.99
87. Hofmann A, Krajnc N, Dal-Bianco A, Riedl CJ, Zrzavy T, Lerma-Martin C, et al. Myeloid cell iron uptake pathways and paramagnetic rim formation in multiple sclerosis. Acta Neuropathol. (2023) 146:707. doi: 10.1007/S00401-023-02627-4
88. De Groot CJA, Ruuls SR, Theeuwes JWM, Dijkstra CD, van der Valk P. Immunocytochemical characterization of the expression of inducible and constitutive isoforms of nitric oxide synthase in demyelinating multiple sclerosis lesions. J Neuropathol Exp Neurol. (1997) 56:10–20. doi: 10.1097/00005072-199701000-00002
89. Vogel DYS, Vereyken EJF, Glim JE, Heijnen PDAM, Moeton M, van der Valk P, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflamm. (2013) 10:35. doi: 10.1186/1742-2094-10-35
90. Borst K, Dumas AA, Prinz M. Microglia: Immune and non-immune functions. Immunity. (2021) 54:2194–208. doi: 10.1016/J.IMMUNI.2021.09.014
91. Paolicelli RC, Gross CT. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol. (2011) 7:77–83. doi: 10.1017/S1740925X12000105
92. McNamara NB, Munro DAD, Bestard-Cuche N, Uyeda A, Bogie JFJ, Hoffmann A, et al. Microglia regulate central nervous system myelin growth and integrity. Nature. (2023) 613:120–9. doi: 10.1038/s41586-022-05534-y
93. Carta AR, Pisanu A. Modulating microglia activity with PPAR-γ agonists: a promising therapy for Parkinson’s disease? Neurotox Res. (2013) 23:112–23. doi: 10.1007/S12640-012-9342-7
94. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
95. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/ANNUREV-IMMUNOL-051116-052358
96. Liu J, Liu L, Wang X, Jiang R, Bai Q, Wang G. Microglia: A double-edged sword in intracerebral hemorrhage from basic mechanisms to clinical research. Front Immunol. (2021) 12:675660. doi: 10.3389/FIMMU.2021.675660
97. Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J Exp Med. (1996) 184:1737–45. doi: 10.1084/JEM.184.5.1737
98. McCombe PA, de Jersey J, Pender MP. Inflammatory cells, microglia and MHC class II antigen-positive cells in the spinal cord of Lewis rats with acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. (1994) 51:153–67. doi: 10.1016/0165-5728(94)90077-9
99. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:39–48. doi: 10.4049/JIMMUNOL.178.1.39
100. Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hövelmeyer N, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. (2005) 11:146–52. doi: 10.1038/NM1177
101. Montilla A, Zabala A, Er-Lukowiak M, Rissiek B, Magnus T, Rodriguez-Iglesias N, et al. Microglia and meningeal macrophages depletion delays the onset of experimental autoimmune encephalomyelitis. Cell Death Dis. (2023) 14(1):16. doi: 10.1038/s41419-023-05551-3
102. Nissen JC, Thompson KK, West BL, Tsirka SE. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp Neurol. (2018) 307:24–36. doi: 10.1016/J.EXPNEUROL.2018.05.021
103. Sinner P, Peckert-Maier K, Mohammadian H, Kuhnt C, Draßner C, Panagiotakopoulou V, et al. Microglial expression of CD83 governs cellular activation and restrains neuroinflammation in experimental autoimmune encephalomyelitis. Nat Commun. (2023) 14(1):4601. doi: 10.1038/S41467-023-40370-2
104. Voß EV, Škuljec J, Gudi V, Skripuletz T, Pul R, Trebst C, et al. Characterisation of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol Dis. (2012) 45:519–28. doi: 10.1016/J.NBD.2011.09.008
105. Kotter MR, Li WW, Zhao C, Franklin RJM. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. (2006) 26:328–32. doi: 10.1523/JNEUROSCI.2615-05.2006
106. Lampron A, Larochelle A, Laflamme N, Préfontaine P, Plante MM, Sánchez MG, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. (2015) 212:481. doi: 10.1084/JEM.20141656
107. Li H, Newcombe J, Groome NP, Cuzner ML. Characterization and distribution of phagocytic macrophages in multiple sclerosis plaques. Neuropathol Appl Neurobiol. (1993) 19:214–23. doi: 10.1111/J.1365-2990.1993.TB00431.X
108. Sanders V, Conrad AJ, Tourtellotte WW. On classification of post-mortem multiple sclerosis plaques for neuroscientists. J Neuroimmunol. (1993) 46:207–16. doi: 10.1016/0165-5728(93)90251-S
109. Michailidou I, Naessens DMP, Hametner S, Guldenaar W, Kooi EJ, Geurts JJG, et al. Complement C3 on microglial clusters in multiple sclerosis occur in chronic but not acute disease: Implication for disease pathogenesis. Glia. (2017) 65:264–77. doi: 10.1002/GLIA.23090
110. Sato F, Martinez NE, Stewart EC, Omura S, Alexander JS, Tsunoda I. Microglial nodules” and “newly forming lesions” may be a Janus face of early MS lesions; implications from virus-induced demyelination, the Inside-Out model. BMC Neurol. (2015) 15:219. doi: 10.1186/S12883-015-0478-Y
111. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. (2013) 125:595–608. doi: 10.1007/S00401-013-1082-0
112. van Noort J M, van den Elsen P, van Horssen J, J.g. Geurts J, van der Valk P, Amor S. Preactive multiple sclerosis lesions offer novel clues for neuroprotective therapeutic strategies. CNS Neurol Disord Drug Targets. (2011) 10:68–81. doi: 10.2174/187152711794488566
113. van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflamm. (2012) 9:156. doi: 10.1186/1742-2094-9-156
114. Burm SM, Peferoen LAN, Zuiderwijk-Sick EA, Haanstra KG, t. HBA, van der Valk P, et al. Expression of IL-1β in rhesus EAE and MS lesions is mainly induced in the CNS itself. J Neuroinflamm. (2016) 13:138. doi: 10.1186/S12974-016-0605-8
115. Hendrickx DAE, Koning N, Schuurman KG, Van Strien ME, Van Eden CG, Hamann J, et al. Selective upregulation of scavenger receptors in and around demyelinating areas in multiple sclerosis. J Neuropathol Exp Neurol. (2013) 72:106–18. doi: 10.1097/NEN.0B013E31827FD9E8
116. van den Bosch AMR, van der Poel M, Fransen NL, Vincenten MCJ, Bobeldijk AM, Jongejan A, et al. Profiling of microglia nodules in multiple sclerosis reveals propensity for lesion formation. Nat Commun. (2024) 15(1):1667. doi: 10.1038/S41467-024-46068-3
117. Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol. (2004) 172:2731–8. doi: 10.4049/JIMMUNOL.172.5.2731
118. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/NRI3399
119. Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. (2002) 100:854–61. doi: 10.1182/BLOOD.V100.3.854
120. Lord BI. Myeloid cell kinetics in response to haemopoietic growth factors. Baillieres Clin Haematol. (1992) 5:533–50. doi: 10.1016/S0950-3536(11)80006-5
121. Brinkmann V. Neutrophil extracellular traps in the second decade. J Innate Immun. (2018) 10:414–21. doi: 10.1159/000489829
122. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. (2017) 7:373. doi: 10.3389/FCIMB.2017.00373
123. Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. Neutrophils acquire the capacity for antigen presentation to memory CD4+ T cells in vitro and ex vivo. Blood. (2017) 129:1991–2001. doi: 10.1182/BLOOD-2016-10-744441
124. Culshaw S, Millington OR, Brewer JM, McInnes IB. Murine neutrophils present Class II restricted antigen. Immunol Lett. (2008) 118:49–54. doi: 10.1016/J.IMLET.2008.02.008
125. Lok LSC, Dennison TW, Mahbubani KM, Saeb-Parsy K, Chilvers ER, Clatworthy MR. Phenotypically distinct neutrophils patrol uninfected human and mouse lymph nodes. Proc Natl Acad Sci U.S.A. (2019) 116:19083–9. doi: 10.1073/PNAS.1905054116
126. Tecchio C, Cassatella MA. Neutrophil-derived chemokines on the road to immunity. Semin Immunol. (2016) 28:119–28. doi: 10.1016/J.SMIM.2016.04.003
127. Casanova Acebes M, Nicolás Ávila JA, Yao Li JL, García Silva S, Balachander A, Rubio Ponce A, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med. (2018) 215:2778–95. doi: 10.1084/JEM.20181468
128. Fu X, Liu H, Huang G, Dai SS. The emerging role of neutrophils in autoimmune-associated disorders: effector, predictor, and therapeutic targets. MedComm (Beijing). (2021) 2:402–13. doi: 10.1002/MCO2.69
129. Christy AL, Walker ME, Hessner MJ, Brown MA. Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE. J Autoimmun. (2013) 42:50–61. doi: 10.1016/J.JAUT.2012.11.003
130. Nygårdas PT, Määttä JA, Hinkkanen AE. Chemokine expression by central nervous system resident cells and infiltrating neutrophils during experimental autoimmune encephalomyelitis in the BALB/c mouse. Eur J Immunol. (2000) 30(7):1911–8. doi: 10.1002/1521-4141(200007)30:7
131. Määttä JA, Sjöholm UR, Nygårdas PT, Salmi AA, Hinkkanen AE. Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. (1998) 90(2):162–75. doi: 10.1016/S0165-5728(98)00135-0
132. Shen P, Rother M, Stervbo U, Lampropoulou V, Calderon-Gomez E, Roch T, et al. Toll-like receptors control the accumulation of neutrophils in lymph nodes that expand CD4+ T cells during experimental autoimmune encephalomyelitis. Eur J Immunol. (2023) 53:e2250059. doi: 10.1002/EJI.202250059
133. Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X, et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. (2015) 212:23–35. doi: 10.1084/JEM.20141015
134. Grist JJ, Marro BS, Skinner DD, Syage AR, Worne C, Doty DJ, et al. Induced CNS expression of CXCL1 augments neurologic disease in a murine model of multiple sclerosis via enhanced neutrophil recruitment. Eur J Immunol. (2018) 48:1199–210. doi: 10.1002/EJI.201747442
135. Steinbach K, Piedavent M, Bauer S, Neumann JT, Friese MA. Neutrophils amplify autoimmune central nervous system infiltrates by maturing local APCs. J Immunol. (2013) 191:4531–9. doi: 10.4049/JIMMUNOL.1202613
136. Carlson T, Kroenke M, Rao P, Lane TE, Segal B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med. (2008) 205:811–23. doi: 10.1084/JEM.20072404
137. Byun DJ, Lee J, Ko K, Hyun YM. NLRP3 exacerbates EAE severity through ROS-dependent NET formation in the mouse brain. Cell Commun Signal. (2024) 22(1):96. doi: 10.1186/S12964-023-01447-Z
138. Podikoglou DG, Lianou PE, Tsakanikas CD, Papavassiliou JT. Polymorphonuclear leukocyte functions and multiple sclerosis. Neurology. (1994) 44:129–32. doi: 10.1212/WNL.44.1.129
139. NœSs A, Nyland H, Glette J, Larsen JP, Rygh T, Solberg CO. Granulocyte function in patients with multiple sclerosis. Acta Pathol Microbiol Immunol Scand C. (1986) 94:89–90. doi: 10.1111/J.1699-0463.1986.TB02095.X
140. Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. (2012) 242:60–71. doi: 10.1016/J.JNEUROIM.2011.11.009
141. Bisgaard AK, Pihl-Jensen G, Frederiksen JL. The neutrophil-to-lymphocyte ratio as disease actvity marker in multiple sclerosis and optic neuritis. Mult Scler Relat Disord. (2017) 18:213–7. doi: 10.1016/J.MSARD.2017.10.009
142. Fathy SE, AbdAllah AM, Helal RY. Neutrophil–lymphocyte ratio and platelet–lymphocyte ratio as predictors of MS severity: a retrospective cohort study. Egyptian J Neurology Psychiatry Neurosurg. (2024) 60:1–10. doi: 10.1186/S41983-024-00802-2/FIGURES/1
143. Hertwig L, Pache F, Romero-Suarez S, Stürner KH, Borisow N, Behrens J, et al. Distinct functionality of neutrophils in multiple sclerosis and neuromyelitis optica. Mult Scler. (2016) 22:160–73. doi: 10.1177/1352458515586084
144. Kostic M, Dzopalic T, Zivanovic S, Zivkovic N, Cvetanovic A, Stojanovic I, et al. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol. (2014) 79:181–6. doi: 10.1111/SJI.12147
145. Tillack K, Naegele M, Haueis C, Schippling S, Wandinger KP, Martin R, et al. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J Neuroimmunol. (2013) 261:108–19. doi: 10.1016/J.JNEUROIM.2013.05.004
146. Artis D, Spits H. The biology of innate lymphoid cells. Nature. (2015) 517:293–301. doi: 10.1038/NATURE14189
147. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/J.CELL.2018.07.017
148. Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. (2018) 48:1104–17. doi: 10.1016/J.IMMUNI.2018.05.013
149. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/NRI3365
150. Mindt BC, Fritz JH, Duerr CU. Group 2 innate lymphoid cells in pulmonary immunity and tissue homeostasis. Front Immunol. (2018) 9:840. doi: 10.3389/FIMMU.2018.00840
151. Melo-Gonzalez F, Hepworth MR. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology. (2017) 150:265–75. doi: 10.1111/IMM.12697
152. Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. (2016) 17:765–74. doi: 10.1038/NI.3489
153. Eberl G, Colonna M, Santo JPD, McKenzie ANJ. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. (2015) 348(6237):aaa6566. doi: 10.1126/SCIENCE.AAA6566
154. Domingues RG, Hepworth MR. Immunoregulatory sensory circuits in group 3 innate lymphoid cell (ILC3) function and tissue homeostasis. Front Immunol. (2020) 11:116. doi: 10.3389/FIMMU.2020.00116
155. Wekerle H. Brain autoimmunity and intestinal microbiota: 100 trillion game changers. Trends Immunol. (2017) 38:483–97. doi: 10.1016/J.IT.2017.03.008
156. Campos-Acuña J, Elgueta D, Pacheco R. T-cell-driven inflammation as a mediator of the gut-brain axis involved in parkinson’s disease. Front Immunol. (2019) 10:239. doi: 10.3389/FIMMU.2019.00239
157. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science. (2015) 348:1031–5. doi: 10.1126/SCIENCE.AAA4812
158. Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/NATURE12240
159. Rao A, Strauss O, Kokkinou E, Bruchard M, Tripathi KP, Schlums H, et al. Cytokines regulate the antigen-presenting characteristics of human circulating and tissue-resident intestinal ILCs. Nat Commun. (2020) 11(1):2049. doi: 10.1038/S41467-020-15695-X
160. Lehmann FM, von Burg N, Ivanek R, Teufel C, Horvath E, Peter A, et al. Microbiota-induced tissue signals regulate ILC3-mediated antigen presentation. Nat Commun. (2020) 11(1):1794. doi: 10.1038/S41467-020-15612-2
161. Von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci U.S.A. (2014) 111:12835–40. doi: 10.1073/PNAS.1406908111
162. Jarade A, Garcia Z, Marie S, Demera A, Prinz I, Bousso P, et al. Inflammation triggers ILC3 patrolling of the intestinal barrier. Nat Immunol. (2022) 23:1317–23. doi: 10.1038/S41590-022-01284-1
163. Grigg JB, Shanmugavadivu A, Regen T, Parkhurst CN, Ahmed A, Joseph AM, et al. Antigen-presenting innate lymphoid cells orchestrate neuroinflammation. Nature. (2021) 600:707–12. doi: 10.1038/S41586-021-04136-4
164. Mathä L, Romera-Hernández M, Steer CA, Yin YH, Orangi M, Shim H, et al. Migration of lung resident group 2 innate lymphoid cells link allergic lung inflammation and liver immunity. Front Immunol. (2021) 12:679509. doi: 10.3389/FIMMU.2021.679509
165. Kästele V, Mayer J, Lee ES, Papazian N, Cole JJ, Cerovic V, et al. Intestinal-derived ILCs migrating in lymph increase IFNγ production in response to Salmonella Typhimurium infection. Mucosal Immunol. (2021) 14:717–27. doi: 10.1038/S41385-020-00366-3
166. Wang S, van de Pavert SA. Innate lymphoid cells in the central nervous system. Front Immunol. (2022) 13:837250. doi: 10.3389/FIMMU.2022.837250
167. Derecki NC, Aleman-Muench GR, Lewis G, Banie H, Eckert W, He Y, et al. Meningeal type-2 innate lymphoid cells emerge as novel regulators of microglial activation and blood-brain barrier stability: A central role for IL-10. SSRN Electronic J. (2019). doi: 10.2139/SSRN.3414004
168. Brown MA, Weinberg RB. Mast cells and innate lymphoid cells: underappreciated players in CNS autoimmune demyelinating disease. Front Immunol. (2018) 9:514. doi: 10.3389/FIMMU.2018.00514
169. Hatfield JK, Brown MA. Group 3 innate lymphoid cells accumulate and exhibit disease-induced activation in the meninges in EAE. Cell Immunol. (2015) 297:69–79. doi: 10.1016/J.CELLIMM.2015.06.006
170. Mair F, Becher B. Thy1+ Sca1+ innate lymphoid cells infiltrate the CNS during autoimmune inflammation, but do not contribute to disease development. Eur J Immunol. (2014) 44:37–45. doi: 10.1002/EJI.201343653
171. Heng AHS, Han CW, Abbott C, McColl SR, Comerford I. Chemokine-driven migration of pro-inflammatory CD4+ T cells in CNS autoimmune disease. Front Immunol. (2022) 13:817473. doi: 10.3389/FIMMU.2022.817473
172. Kwong B, Rua R, Gao Y, Flickinger J, Wang Y, Kruhlak MJ, et al. T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat Immunol. (2017) 18:1117–27. doi: 10.1038/NI.3816
173. Saikali P, Antel JP, Newcombe J, Chen Z, Freedman M, Blain M, et al. NKG2D-mediated cytotoxicity toward oligodendrocytes suggests a mechanism for tissue injury in multiple sclerosis. J Neurosci. (2007) 27:1220–8. doi: 10.1523/JNEUROSCI.4402-06.2007
174. Guerra N, Pestal K, Juarez T, Beck J, Tkach K, Wang L, et al. A selective role of NKG2D in inflammatory and autoimmune diseases. Clin Immunol. (2013) 149:432–9. doi: 10.1016/J.CLIM.2013.09.003
175. Moratalla AC, Solorio YC, Lemaitre F, Farzam-Kia N, Levert A, Zandee SEJ, et al. Stress signal ULBP4, an NKG2D ligand, is upregulated in multiple sclerosis and shapes CD8+ T-cell behaviors. Neurology(R) neuroimmunology Neuroinflamm. (2021) 9(1):e1119. doi: 10.1212/NXI.0000000000001119
176. Fernández-Morera JL, Rodríguez-Rodero S, Lahoz C, Tuñon A, Astudillo A, Garcia-Suarez O, et al. Soluble MHC class I chain-related protein B serum levels correlate with disease activity in relapsing-remitting multiple sclerosis. Hum Immunol. (2008) 69:235–40. doi: 10.1016/J.HUMIMM.2008.01.021
177. Legroux L, Moratalla AC, Laurent C, Deblois G, Verstraeten SL, Arbour N. NKG2D and its ligand MULT1 contribute to disease progression in a mouse model of multiple sclerosis. Front Immunol. (2019) 10:154. doi: 10.3389/FIMMU.2019.00154
178. Dunmire SK, Verghese PS, Balfour HH. Primary Epstein-Barr virus infection. J Clin Virol. (2018) 102:84–92. doi: 10.1016/J.JCV.2018.03.001
179. Levin LI, Munger KL, Rubertone MV, Peck CA, Lennette ET, Spiegelman D, et al. Temporal relationship between elevation of epstein-barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. (2005) 293:2496–500. doi: 10.1001/JAMA.293.20.2496
180. Vietzen H, Berger SM, Kühner LM, Furlano PL, Bsteh G, Berger T, et al. Ineffective control of Epstein-Barr-virus-induced autoimmunity increases the risk for multiple sclerosis. Cell. (2023) 186:5705–5718.e13. doi: 10.1016/J.CELL.2023.11.015
181. Alari-Pahissa E, Moreira A, Zabalza A, Alvarez-Lafuente R, Munteis E, Vera A, et al. Low cytomegalovirus seroprevalence in early multiple sclerosis: a case for the “hygiene hypothesis”? Eur J Neurol. (2018) 25:925–33. doi: 10.1111/ENE.13622
182. Vanheusden M, Broux B, Welten SPM, Peeters LM, Panagioti E, Van WB, et al. Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci Rep. (2017) 7(1):663. doi: 10.1038/S41598-017-00645-3
183. Lopez-Sejas N, Campos C, Hassouneh F, Sanchez-Correa B, Tarazona R, Pera A, et al. Effect of CMV and aging on the differential expression of CD300a, CD161, T-bet, and eomes on NK cell subsets. Front Immunol. (2016) 7:476. doi: 10.3389/FIMMU.2016.00476
184. Perri V, Zingaropoli MA, Pasculli P, Ciccone F, Tartaglia M, Baione V, et al. The impact of cytomegalovirus infection on natural killer and CD8+ T cell phenotype in multiple sclerosis. Biol (Basel). (2024) 13(3):154. doi: 10.3390/BIOLOGY13030154
185. Aglas-Leitner F, Juillard P, Juillard A, Byrne SN, Hawke S, Grau GE, et al. Circulating CCR6+ILC proportions are lower in multiple sclerosis patients. Clin Transl Immunol. (2022) 11(12):e1426. doi: 10.1002/CTI2.1426
186. Rodríguez-Lorenzo S, van Olst L, Rodriguez-Mogeda C, Kamermans A, van der Pol SMA, Rodríguez E, et al. Single-cell profiling reveals periventricular CD56bright NK cell accumulation in multiple sclerosis. Elife. (2022) 11:e73849. doi: 10.7554/ELIFE.73849
187. Eken A, Yetkin MF, Vural A, Okus FZ, Erdem S, Azizoglu ZB, et al. Fingolimod alters tissue distribution and cytokine production of human and murine innate lymphoid cells. Front Immunol. (2019) 10:217. doi: 10.3389/FIMMU.2019.00217
188. Nelissen S, Vangansewinkel T, Geurts N, Geboes L, Lemmens E, Vidal PM, et al. Mast cells protect from post-traumatic spinal cord damage in mice by degrading inflammation-associated cytokines via mouse mast cell protease 4. Neurobiol Dis. (2014) 62:260–72. doi: 10.1016/j.nbd.2013.09.012
189. Kritasl SK, Sagginf A, Cerullp G, Caraffa4 A, Antinolfi P, Pantalonp •A, et al. Impact of mast cells on multiple sclerosis: inhibitory effect of natalizumab. Int J Immunopathol Pharmacol. (2014) 27(3):331–5. doi: 10.1177/039463201402
190. Xu Y, Chen G. Mast cell and autoimmune diseases. Mediators Inflammation. (2015) 2015:246126. doi: 10.1155/2015/246126
191. Hendriksen E, van Bergeijk D, Oosting RS, Redegeld FA. Mast cells in neuroinflammation and brain disorders. Neurosci Biobehav Rev. (2017) 79:119–33. doi: 10.1016/j.neubiorev.2017.05.001
192. Hart DA. Curbing inflammation in multiple sclerosis and endometriosis: should mast cells be targeted? Int J Inflam. (2015) 2015:452095. doi: 10.1155/2015/452095
193. Russi AE, Walker-Caulfield ME, Brown MA. Mast cell inflammasome activity in the meninges regulates EAE disease severity. Clin Immunol. (2018) 189:14–22. doi: 10.1016/j.clim.2016.04.009
194. Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. (2000) 191:813–21. doi: 10.1084/JEM.191.5.813
195. Tanzola MB, Robbie-Ryan M, Gutekunst CA, Brown MA. Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J Immunol. (2003) 171:4385–91. doi: 10.4049/JIMMUNOL.171.8.4385
196. Piconese S, Costanza M, Musio S, Tripodo C, Poliani PL, Gri G, et al. Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient Kit W-sh/W-sh mice. Lab Invest. (2011) 91:627–41. doi: 10.1038/LABINVEST.2011.3
197. Russi AE, Walker-Caulfield ME, Guo Y, Lucchinetti CF, Brown MA. Meningeal mast cell-T cell crosstalk regulates T cell encephalitogenicity. J Autoimmun. (2016) 73:100–10. doi: 10.1016/j.jaut.2016.06.015
198. Theoharides TC, Petra AI, Taracanova A, Panagiotidou S, Conti P. Targeting IL-33 in autoimmunity and inflammation. J Pharmacol Exp Ther. (2015) 354:24–31. doi: 10.1124/jpet.114.222505
199. Conti P, Kempuraj D. Important role of mast cells in multiple sclerosis. Mult Scler Relat Disord. (2016) 5:77–80. doi: 10.1016/j.msard.2015.11.005
200. Duncker PC, Stoolman JS, Huber AK, Segal BM. GM-CSF promotes chronic disability in experimental autoimmune encephalomyelitis by altering the composition of central nervous system-infiltrating cells, but is dispensable for disease induction. J Immunol. (2018) 200:966–73. doi: 10.4049/JIMMUNOL.1701484
201. Elieh-Ali-Komi D, Cao Y. Role of mast cells in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Rev Allergy Immunol. (2017) 52:436–45. doi: 10.1007/s12016-016-8595-y
202. Yin JJ, Hu XQ, Mao ZF, Bao J, Qiu W, Lu ZQ, et al. Neutralization of interleukin-9 decreasing mast cells infiltration in experimental autoimmune encephalomyelitis. (Engl). (2017) 130:964–71. doi: 10.4103/0366-6999.204110
203. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. (2008) 123:398. doi: 10.1111/J.1365-2567.2007.02705.X
204. Jadidi-Niaragh J, Mirshafiey A. Histamine and histamine receptors in pathogenesis and treatment of multiple sclerosis. Available online at: https://sci-hub.se/https://www.sciencedirect.com/science/article/pii/S0028390810001309?via%3Dihub (Accessed November 26, 2024).
205. Ribatti D, Tamma R, Annese T. Mast cells and angiogenesis in multiple sclerosis. Inflammation Res. (2020) 69:1103–10. doi: 10.1007/s00011-020-01394-2
206. Schwartz C, Voehringer D. Identification of murine basophils by flow cytometry and histology. Methods Mol Biol. (2014) 1192:229–37. doi: 10.1007/978-1-4939-1173-8_17
207. Maddur MS, Kaveri SV, Bayry J. Basophils as antigen presenting cells. Trends Immunol. (2010) 31:45–8. doi: 10.1016/J.IT.2009.12.004
208. Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. (2008) 9:310–8. doi: 10.1038/NI1558
209. Yuk CM, Park HJ, Kwon BI, Lah SJ, Chang J, Kim JY, et al. Basophil-derived IL-6 regulates TH17 cell differentiation and CD4 T cell immunity. Sci Rep. (2017) 7:41744. doi: 10.1038/SREP41744
Keywords: innate immune cells, experimental autoimmune encephalomyelitis, multiple sclerosis, macrophages, dendric cells
Citation: Prado C, Herrada AA, Hevia D, Goiry LG and Escobedo N (2025) Role of innate immune cells in multiple sclerosis. Front. Immunol. 16:1540263. doi: 10.3389/fimmu.2025.1540263
Received: 05 December 2024; Accepted: 28 January 2025;
Published: 17 February 2025.
Edited by:
José Jiram Torres-Ruiz, National Institute of Medical Sciences and Nutrition Salvador Zubirán, MexicoReviewed by:
Xin Zang, Capital Medical University, ChinaCopyright © 2025 Prado, Herrada, Hevia, Goiry and Escobedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Noelia Escobedo, bm9lbGlhLmVzY29iZWRvQHVhdXRvbm9tYS5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.